

Abstract
A visible-light photoredox-catalyzed method is reported that enables the coupling between benzylic C–H substrates and N–H azoles. Classically, medicinally relevant N-benzyl azoles are produced via harsh substitution conditions between the azole and a benzyl electrophile in the presence of strong bases at high temperatures. Use of C–H bonds as the alkylating partner streamlines the preparation of these important motifs. In this work, we report the use of N-alkoxypyridinium salts as a critically enabling reagent for the development of a general C(sp3)–H azolation. The platform enables the alkylation of electron-deficient, -neutral, and -rich azoles with a range of C–H bonds, most notably secondary and tertiary partners. Moreover, the protocol is mild enough to tolerate benzyl electrophiles, thus offering an orthogonal approach to existing SN2 and cross-coupling methods.
N-Benzyl azoles are an abundant motif in drug discovery,1 with key examples including letrozole,2 bifonazole,3 and carboetomidate4 (Figure 1). Such motifs exhibit widespread utility as active compounds across a number of disease areas and medical uses. Generally, azoles, such as pyrazoles, possess tempered nucleophilicity relative to halides or carboxylates.5 Thus, their alkylation via substitution reactions typically requires harsh conditions. Indeed, the preparation of the C–N bond of letrozole relies on an SN2-type protocol with triazole and a benzyl electrophile (halide/tosylate) at 100 °C with strong base.6 Additionally, benzyl electrophiles are prone to hydrolysis and often require extra preparatory steps. Thus, a need exists for new mild and streamlined protocols to be developed. In addition, a strategy that differs from classical transition metal cross-coupling conditions would also allow for an expanded substrate scope to include electron-rich azoles, organoboranes, and alkyl, aryl, and benzyl/allyl halides to maximize synthetic route opportunities.7
Figure 1.
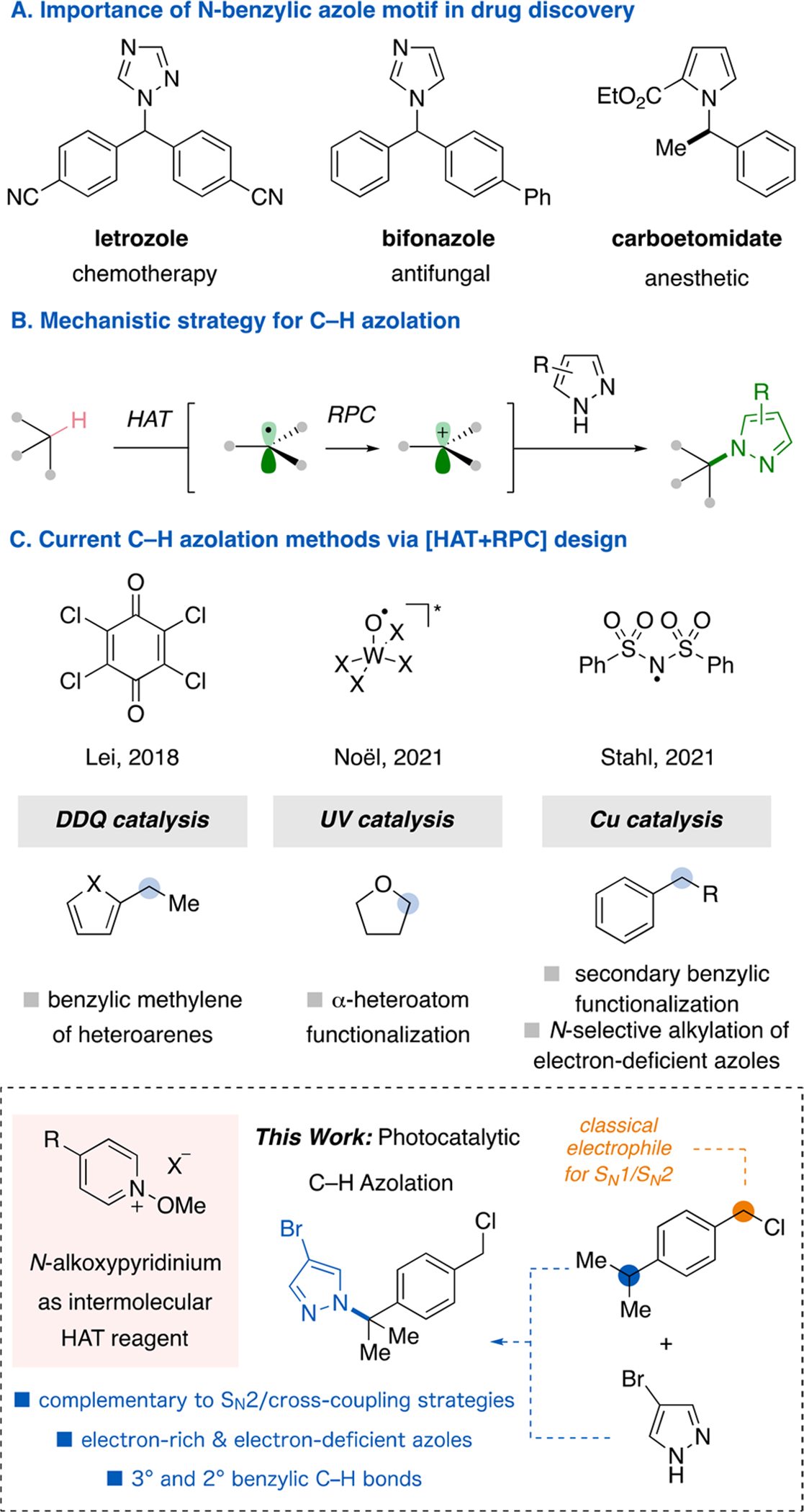
Background on C–H azolation methods. (A) N-benzyl azoles are attractive pharmaceutical motifs. (B) Recent mechanism for C–H functionalization. (C) Current C–H azolation methods utilizing [HAT+RPC].
Recently, alternative alkylating partners have been investigated, with carboxylic acids being a particularly attractive option.8 Although they are abundant and provide a handle for accessing radicals and carbocations, their activation requires acyl group manipulations or strong oxidants to facilitate the decarboxylation. By contrast, C–H bonds represent the most prevalent functionality in organic compounds; accordingly, rendering them reactive for C–X bond formation would be greatly advantageous toward the goal of a streamlined reaction platform.9 The primary advantage of C–H functionalization methods is a decrease in preparatory steps of reagents and eventual use for the rapid diversification of late-stage targets.
A handful of methods have demonstrated that single-electron oxidization of arenes can activate the benzylic C–H positions toward subsequent functionalization, albeit requiring high oxidation potentials.10 In the past few years, a mechanistic strategy for expanding the C–H scope employs a hydrogen-atom transfer (HAT) event prior to a (radical) oxidation (i.e., radical–polar crossover, RPC).11 Notably, the combination of HAT and RPC has been engineered into one catalytic cycle utilizing different HAT species: Lei and co-workers used the phenoxy radical of 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ);12 the Noël group employed a UV-activated decatungstate catalyst;13 and the Stahl group leveraged a sulfonimide radical derived from N-fluorobenzenesulfonimide (NFSI) in conjugation with copper catalysis.14 While all three methods proceed via a carbocation intermediate, noticeably lacking in the scope of each protocol is the formation of fully substituted centers arising from in situ-generated tertiary carbocations. Recently, our group,15 concurrently with the Doyle group,16 published a visible-light photoredox-catalyzed [HAT+RPC] mechanism that can engage classically weak nucleophiles, including fluoride, and readily forge fully substituted centers from tertiary C–H precursors. We hypothesized that the established platform could be extended to the formation of an array of N-benzyl azoles, including those bearing fully substituted centers. In addition, these works demonstrated the power of photoredox catalysis to mediate a formal hydride abstraction with two different types of HAT reagents: tert-butyl peroxybenzoate (TBPB) and N-acyloxyphthalimide. Thus far, only photoredox platforms have exhibited such modularity in the examination of stereoelectronically diverse HAT reagents for the [HAT+RPC] process. Herein we report the implementation of N-alkoxypyridinium salts in this process. Simple N-alkoxypyridinium salts can be readily prepared in one step and offer an electronically tunable HAT scaffold.17,18 Finally, mechanistic evidence suggests that an electron-donor–acceptor (EDA) complex may be operable for activating the pyridinium reagents for certain electron-rich benzylic partners.19
Our efforts toward the development of a C–H azolation protocol initiated with the use of TBPB to facilitate the HAT event. Although successful in our prior work for C–H fluorination with nucleophilic fluoride (N = 10.8–13.2),20 switching to less nucleophilic coupling partners such as pyrazoles (N = 8.9–9.6) resulted in competitive trapping of the carbocation by both benzoate (N = 16.8) and tert-butanol (N = 5.4)5 byproducts (Figure 2A). Work by Hong,21 Li,22 and Lakhdar23 has demonstrated that N-alkoxypyridinium reagents can facilitate intermolecular HAT processes at P–H, C(sp2)–H, Si–H, and α-oxy C(sp3)–H bonds.24 We hypothesized that this reagent could also be used for the intermolecular abstraction of H· at benzylic C(sp3)–H bonds in the desired transformation (Figure 2B). While N-alkoxypyridiniums bearing long-chain alkyl groups have been used for intramolecular 1,5-HAT processes,25 they have not been used widely for intermolecular efforts at C(sp3)–H bonds.22 The pyridinium reagents would be particularly attractive in the desired transformation, as reductive fragmentation would reduce the generation of competitive nucleophilic byproducts (Figure 2B). Accordingly, we rapidly synthesized a suite of N-methoxy- and N-ethoxypyridinum reagents.26 With indane and 4-bromopyrazole as our model substrates, we delightfully observed the desired C–H azolation product (Figure 2C).
Figure 2.
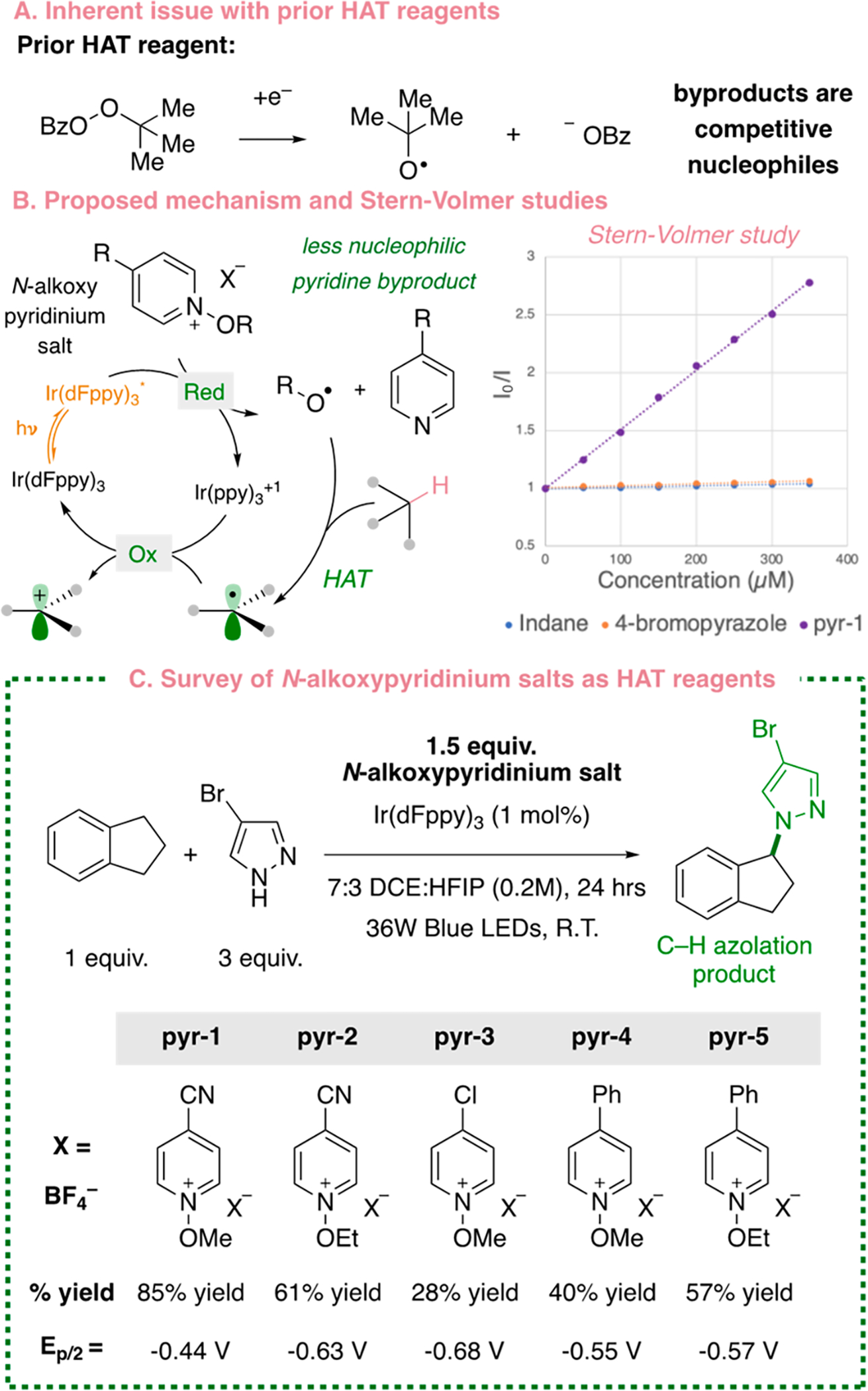
Optimization with N-alkoxypyridinium reagents. (A) Use of TBPB can lead to competitive nucleophiles. (B) Proposed mechanism and Stern–Volmer experiment. (C) Optimization of the N-alkoxypyridinium scaffold.
Electron-withdrawing p-cyanopyridinium pyr-1 was discovered to result in higher product formation, possibly due to a lower reduction potential (see the Supporting Information). Stern–Volmer experiments corroborated an interaction between the excited state of the photocatalyst and pyr-1. Next, we explored the generality of the substrate scope.
Starting with an exploration of azoles (Figure 3), an array of electron-withdrawing groups at the 4-position of pyrazole were well-tolerated, including other halides (2–4), esters (5), and trifluoromethyl (6), cyano (7), and nitro groups (8). Substitution at the 3-position of pyrazole also resulted in good to excellent yields (9–13). Difunctionalized pyrazoles afforded high yields of the products (14–17), and an extended heterocycle was also successful in the protocol (18). Excitingly, more electron-rich pyrazoles were successfully alkylated with our system, representing a class of substrates that are not compatible with base-metal-catalyzed strategies due to potential catalyst poisoning (19–22). Furthermore, 1,2,3-and 1,2,4-triazoles (N ≈ 7.7) were also viable nucleophiles, as was benzotriazole (23–25).26 Substituted tetrazoles, imidazoles, and benzimidazoles were also viable substrates (26–33).
Figure 3.
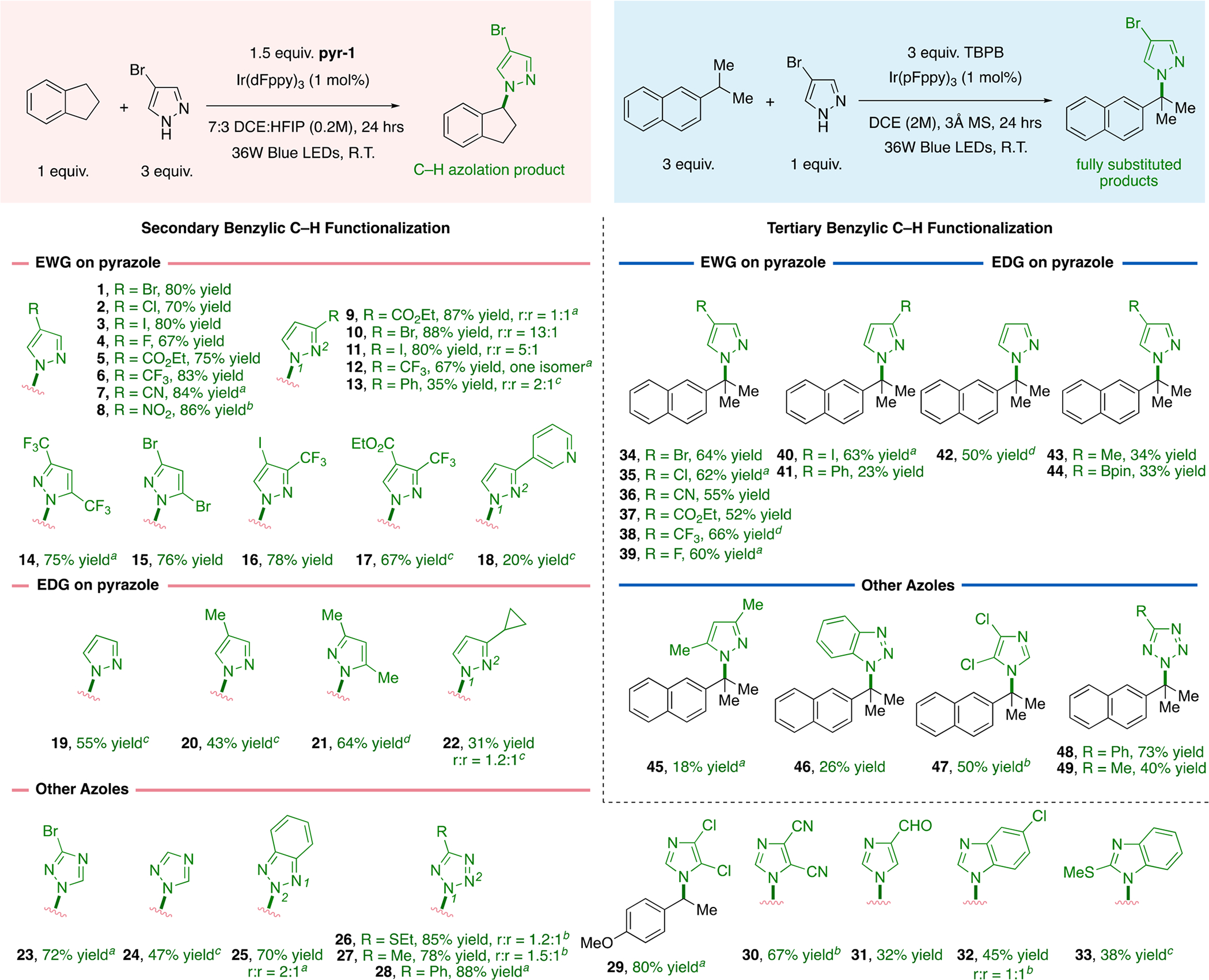
Azole scope for secondary and tertiary benzylic C–H substrates. Reactions were run on a 0.5 mmol scale. DCE = 1,2-dichloroethane. HFIP = hexafluoroisopropanol. a48 h. b72 h. c60 °C. d96 h.
Given the minimal effect of steric hindrance observed, we questioned whether the photoredox-catalyzed [HAT+RPC] platform could enable the functionalization of tertiary benzylic C–H bonds. To the best of our knowledge, the synthesis of fully substituted carbon centers has not been reported in prior C–H azolation methods with the [HAT+RPC] formula, despite the enhanced carbocation stability. Classically, tertiary benzyl halides/tosylates are unstable and/or prepared with harsh reagents (strong acids) and expensive oxidants.27 Moreover, the carboxylic acid equivalent of 2-isopropylnaphthalene is not widely available from commercial vendors. The use of C–H alternatives thus represents an advantage in terms of synthetic ease and available resources. Gratifyingly, 2-isopropylnaphthalene could be readily functionalized with a wide array of azoles using TBPB as the HAT reagent. Currently, we postulate that a methyl radical could be the most effective H-atom abstractor for tertiary benzylic C–H sites, which can be derived from ·OtBu via a facile β-scission.16,28 Various 4- and 3-substituted pyrazoles were successfully alkylated (34–41), with the latter being functionalized at the less sterically hindered nitrogen. Electron-rich pyrazoles also afforded the products in modest yields (42–44). Notably, 44 contains a nucleophilic arylboron functionality that would not be tolerated by transition metal approaches. Moreover, the strength of the carbocation strategy was highlighted with the alkylation of 3,5-dimethylpyrazole to give sterically congested adduct 45, albeit in low yield. Finally, the protocol was also successful at producing fully substituted tetrazole, imidazole, and (benzo)triazole C–N adducts (46–49).29
Next, we examined the generality of the benzylic C–H scope. Methylene sites on both cyclic and acyclic precursors afforded appreciable yields of C–N products (50–57). Diphenylmethane, a common motif in drug targets (Figure 1a), worked in good yield (51). Electron-deficient functional groups at the para position did not significantly hinder the reaction efficiency (52–56); nevertheless, higher yields were observed with electron-donating groups (57). Notably, our carbocation-generating protocol is permissible of aryl bromide and chloride motifs, allowing for the retention of functional handles for further derivatization via classical cross-coupling catalysis. Meta substitution was also well-tolerated (58). The primary benzylic substrate leading to 59 represents another class of substrates not demonstrated in other [HAT+RPC] strategies for azolation. C–H functionalization of allylic positions was achieved, giving 60 and 61 in 18% and 49% yield, respectively. Lastly, α-oxy C–H sites were also viable substrates for the visible-light-mediated azolation (62).
Next, we sought to apply the method to late-stage functionalization of pharmaceutical scaffolds. The seven-membered cyclic core of ivabradine, used in the treatment of heart failure, was successfully elaborated at the α-benzylic site in 48% yield (63). Celestolide (64, 70% yield) and the core of donepezil (65, 45% yield) also underwent C–H azolation with the [HAT+RPC] protocol in appreciable yields. Lastly, we demonstrated that azole derivatives of the antifungal agent bifonazole (66) can be readily prepared in one step from commercially available 4-benzylbiphenyl. These examples demonstrate the utility of direct functionalization of benzylic C–H bonds as opposed to the established multistep processes involving conversion of a benzyl alcohol to a benzyl chloride followed by harsh conditions.30
Next, an array of other tertiary benzylic C–H partners were translated to fully substituted products (67–71). γ-Phenyl-lactone gave sterically congested 68 in good yield, as did phenylcyclohexane (69). Monofunctionalization of compounds containing two tertiary benzylic sites was successful (70), and last, installation of a congested C–N center on 9-methylfluorene was realized (71). Notably, for substrates 68–71, the corresponding benzyl chlorides or carboxylic acids are either commercially unavailable or prohibitively expensive. Excitingly, compound 72 was successfully prepared, demonstrating potential for the azolation of heteroarene C–H substrates.
To probe regioselectivity, a competition experiment was conducted. Using pyr-1, a preference for azolation at indane over 2-isopropylnaphthalene was observed. High selectivity for the secondary position of 73 (16.3:1) was congruent with this finding. The protocol appears to be selective for secondary benzylic C–H sites over primary benzylic (74) and aliphatic tertiary (75) sites and is completely selective for α-oxy benzylic positions over secondary benzylic ones (76; see the Supporting Information).
Finally, we sought to evaluate the specificity and orthogonality of our photocatalytic C–H azolation. Classically, letrazole and bifonazole are prepared via SN2-type reactions on benzyl chlorides with heat and strong bases.6,30 As depicted in Figure 4, this platform is sufficiently mild to tolerate the preparation of 77 in 40% yield with no detection of the SN2 product (>20:1 regioselectivity). Subsequently, high yields were achieved for SN2 azidation (78), thiolation (79), and esterification (80) substitution reactions. Furthermore, the benzyl chloride functionality could also serve as an electrophile in a Pd-catalyzed cross-coupling (81).31
Figure 4.
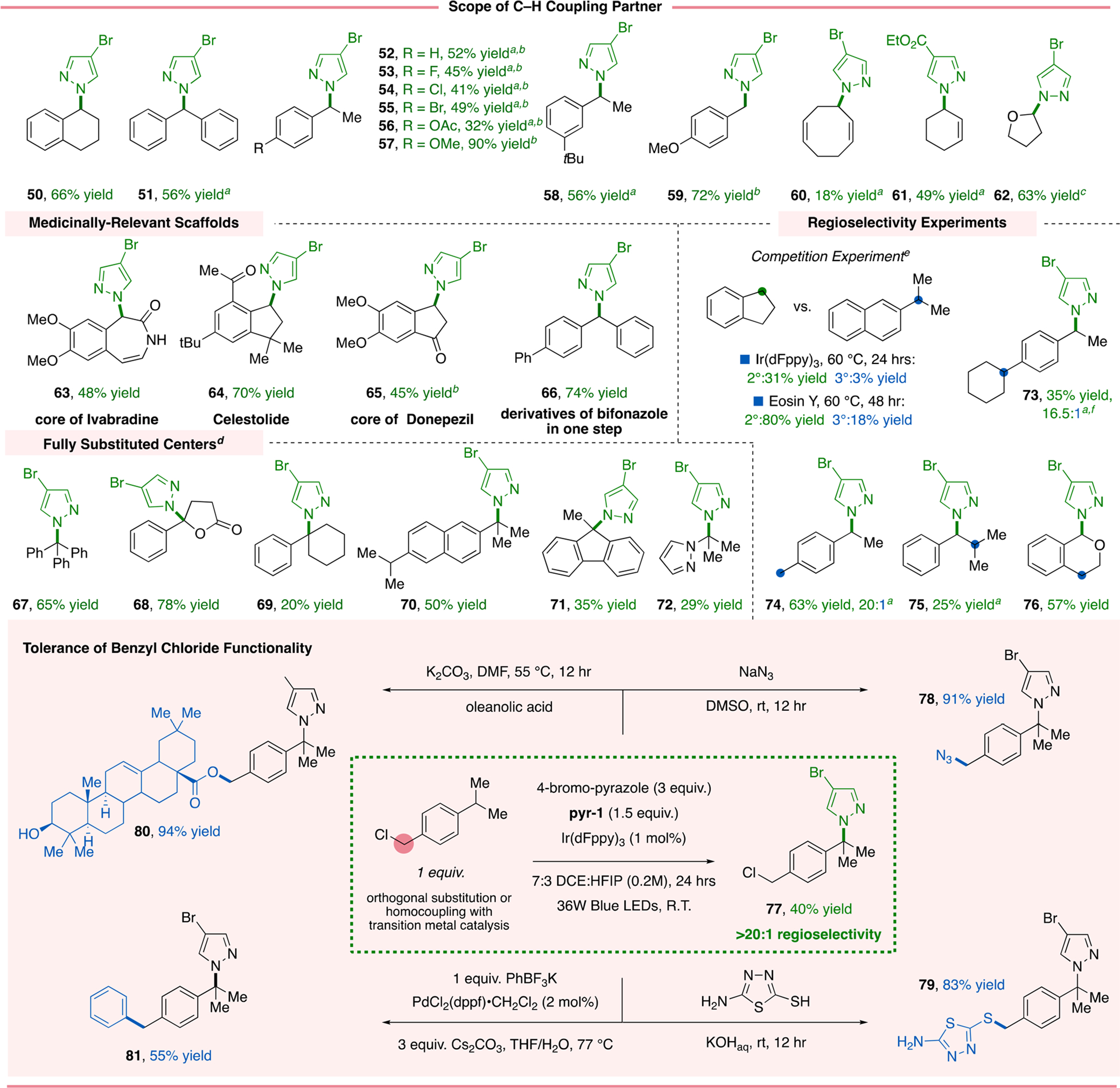
Scope of the C–H reaction partner. a60 °C. b48 h. cTetrahydrofuran (5 equiv). dConditions: hydrocarbon (3 equiv), azole (1 equiv), and TBPB (3 equiv) as indicated in Figure 3. e1 equiv of C–H precursor, azole (3 equiv), and pyr-1 (1.5 equiv). fEosin Y (5 mol %).
Lastly, pyridinium salts have been reported to participate in EDA complexes.18,23b,c,32 Control experiments suggested that when HFIP is added as a cosolvent, an EDA complex could be operable, on the basis of the observation of product without photocatalyst and an observed bathochromic shift in the UV–vis spectra (Figure 5b). Presumably, the EDA complex facilitates oxidation of the arene, which triggers fragmentation of pyr-1 to release a methoxy radical. Subsequent HAT at the benzylic position of the resultant arene radical cation can generate the benzylic carbocation.33 Further UV–vis studies indicated that only electron-rich substrates form an EDA complex (see the Supporting Information), suggesting that other substrates may follow a photocatalyst-mediated mechanism.
Figure 5.

Mechanistic studies. (a) Control experiments for optimized conditions. (b) UV–vis experiments to probe for EDA complex formation and yields of substrates without photocatalyst (PC). a48 h.
In conclusion, we have successfully developed a benzylic C–H azolation reaction via a photoredox-catalyzed formal hydride abstraction mechanism. An N-methoxypyridinium salt was used as an effective intermolecular HAT reagent at benzylic C(sp3)–H bonds. We have demonstrated the generality of the method, as it includes the alkylation of a plethora of azoles. Additionally, a broad C–H partner scope was established, including the alkylation of secondary and tertiary benzylic C–H sites, the latter of which afford a direct and simplified route to N-tert-alkyl azole motifs. Importantly, we showcase the complementary nature of our reaction conditions to classical SN2 and transition metal cross-coupling conditions, which are two current state-of-the-art technologies for forging alkylated azole products. Lastly, mechanistic studies suggest that a plausible EDA mechanism is operable for certain C–H substrates.
Supplementary Material
ACKNOWLEDGMENTS
C.B. thanks the NSF REU Program for summer support. We thank C. C. Le, J. M. Lipshultz, and D. A. Nagib for helpful conversations about the manuscript, Y. Zhang (WPI) and A. Ali (UMass Medical) for NMR assistance, and M. Xatse and C. Olsen (WPI) for HRMS assistance.
Funding
Research reported in this publication was supported by a startup grant from Worcester Polytechnic Institute and the National Institute of Health (R35GM147021). C.B. was supported by the National Science Foundation for summer research through the REU Program (Grant CHE-1950512).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c12850.
Additional experimental details, characterization data, optimization, and relevant spectra (PDF)
Contributor Information
Mrinmoy Das, Department of Chemistry, Worcester Polytechnic Institute, Worcester, Massachusetts 01609, USA.
Leila Zamani, Department of Chemistry, Worcester Polytechnic Institute, Worcester, Massachusetts 01609, USA.
Christopher Bratcher, Department of Chemistry, Worcester Polytechnic Institute, Worcester, Massachusetts 01609, USA.
Patricia Z. Musacchio, Department of Chemistry, Worcester Polytechnic Institute, Worcester, Massachusetts 01609, USA
REFERENCES
- (1).(a) Bennani FE; Doudach L; Cherrah Y; Ramli Y; Karrouchi K; Ansar M; Faouzi MEA Overview of Recent Developments of Pyrazole Derivatives as an Anticancer Agent in Different Cell Line. Bioorg. Chem. 2020, 97, 103470. [DOI] [PubMed] [Google Scholar]; (b) Baumann M; Baxendale IR; Ley SV; Nikbin N An Overview of the Key Routes to the Best Selling 5-Membered Ring Heterocyclic Pharmaceuticals. Beilstein J. Org. Chem. 2011, 7, 442–495. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vitaku E; Smith DT; Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (d) Küçükgüzel ŞG; Şenkardes,¸ S Recent Advances in Bioactive Pyrazoles. Eur. J. Med. Chem. 2015, 97, 786–815. [DOI] [PubMed] [Google Scholar]; (e) Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem. 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (2).Bhatnagar AS The discovery and mechanism of action of letrozole. Breast Cancer Res. Treat. 2007, 105, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).El Hage S; Lajoie B; Feuillolay C; Roques C; Baziard G Synthesis, Antibacterial and Antifungal Activities of Bifonazole Derivatives. Arch. Pharm. Pharm. Med. Chem. 2011, 344, 402–410. [DOI] [PubMed] [Google Scholar]
- (4).Ode K Intravenous Anaesthetic Agents. Anaesth. Intensive Care Med. 2019, 20, 118–125. [Google Scholar]
- (5).Mayr H Mayr’s Database of Reactivity Parameters. https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/fe/ (accessed 2022–12–01).
- (6).(a) Palle VRA; Kalaria AJ; Shelke SA Process for Preparing Letrozole. US 2007/0100149 A1, May 3, 2007. [Google Scholar]; (b) Friedman O; Freger B; Etlin O; Ditkovitch J; Danon E; Seryi Y; Davidi G; Arad O; Kaspi J Letrozole production process. US 7,538,230 B2, May 26, 2009. [Google Scholar]; (c) Doiron J; Soultan AH; Richard R; Touré MM; Picot N; Richard R; Čuperlović- Culf M; Robichaud GA; Touaibia M Synthesis and structure–activity relationship of 1- and 2-substituted-1,2,3-triazole letrozole-based analogues as aromatase inhibitors. Eur. J. Med. Chem. 2011, 46, 4010–4024. [DOI] [PubMed] [Google Scholar]; (d) Long Y; Zheng Y; Xia Y; Qu L; Yang Y; Xiang H; Zhou X Nickel-Catalyzed Synthesis of an Aryl Nitrile via Aryl Exchange between an Aromatic Amide and a Simple Nitrile. ACS Catal. 2022, 12, 4688–4695. [Google Scholar]
- (7).For transition-metal-catalyzed approaches toward N-benzyl/allyl azole synthesis, see: Kong D; Moon PJ; Bsharat O; Lundgren RJ Direct catalytic decarboxylative amination of aryl acetic acids. Angew. Chem., Int. Ed. 2020, 59, 1313–1319.Jiu AY; Slocumb HS; Yeung CS; Yang X-H; Dong VM Enantioselective Addition of Pyrazoles to Dienes. Angew. Chem., Int. Ed. 2021, 60, 19660–19664.Ye Y; Kim S-T; Jeong J; Baik M-H; Buchwald SL CuH-Catalyzed Enantioselective Alkylation of Indole Derivatives with Ligand-Controlled Regiodivergence. J. Am. Chem. Soc. 2019, 141, 3901–3909.Chen H; Yang Y; Li N; Xue X; He Z; Zeng Q Palladium Catalyzed C–N Cross-Coupling of NH-Heteroarenes and Quaternary Ammonium Salts via C–N Bond Cleavage. Org. Process Res. Dev. 2019, 23, 1679–1685.Lacker CR; Delano TJ; Chen EP; Kong J; Belyk KM; Piou T; Reisman SE Enantioselective Synthesis of N-Benzylic Heterocycles by Ni/Photoredox Dual Catalysis. J. Am. Chem. Soc. 2022, 144, 20190–20195.
- (8).For decarboxylation strategies for azole alkylation, see: Sheng T; Zhang H-J; Shang M; He C; Vantourout JC; Baran PS Electrochemical Decarboxylative N-Alkylation of Heterocycles. Org. Lett. 2020, 22, 7594–7598.Liang Y; Zhang X; MacMillan DWC Decarboxylative sp3 C-N coupling via dual copper and photoredox catalysis. Nature 2018, 559, 83–88.Kobayashi R; Shibutani S; Nagao K; Ikeda Z; Wang J; Ibáñez I; Reynolds M; Sasaki Y; Ohmiya H Decarboxylative N-alkylation of azoles through visible-light-mediated organophotoredox catalysis. Org. Lett. 2021, 23, 5415–5419.Li P; Zbieg JR; Terrett JA The Direct Decarboxylative N-Alkylation of Azoles, Sulfonamides, Ureas, and Carbamates with Carboxylic Acids via Photoredox Catalysis. Org. Lett. 2021, 23, 9563–9568.Zhao W; Wurz RP; Peters JC; Fu GC Photoinduced, Copper-Catalyzed Decarboxylative C-N Coupling to Generate Protected Amines: An Alternative to the Curtius Rearrangement. J. Am. Chem. Soc. 2017, 139, 12153–12156.Shao X; Zheng Y; Tian L; Martín-Torres I; Echavarren AM; Wang Y Decarboxylative Csp3–N Bond Formation by Electrochemical Oxidation of Amino Acids. Org. Lett. 2019, 21, 9262–9267.Tang Z-L; Ouyang X-H; Song R-J; Li J-H Decarboxylative C(sp3)–N Cross-Coupling of Diacyl Peroxides with Nitrogen Nucleophiles. Org. Lett. 2021, 23, 1000–1004.Nguyen VT; Nguyen VD; Haug GC; Vuong NTH; Dang HT; Arman HD; Larionov OV Visible-light-enabled direct decarboxylative N-alkylation. Angew. Chem., Int. Ed. 2020, 59, 7921–7927.Merchant RR; Lang SB; Yu T; Zhao S; Qi Z; Suzuki T; Bao J A General One-Pot Protocol for Hindered N-Alkyl Azaheterocycles from Tertiary Carboxylic Acids. Org. Lett. 2020, 22, 4180–4184.
- (9).For selected reviews on C–H functionalization, see: Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-Like Molecules. Chem. Soc. Rev. 2016, 45, 546–576.Abrams DJ; Provencher PA; Sorensen EJ Recent applications of C-H functionalization in complex natural product synthesis. Chem. Soc. Rev. 2018, 47, 8925–8967.Govaerts S; Nyuchev A; Noel T Pushing the boundaries of C–H bond functionalization chemistry using flow technology. J. Flow Chem. 2020, 10, 13–71.Chu JCK; Rovis T Complementary Strategies for Directed C(sp3)–H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem., Int. Ed. 2018, 57, 62–101.
- (10).For examples relying on arene oxidation, see: Pandey G; Laha R; Singh D Benzylic C(sp3)–H Functionalization for C– N and C–O Bond Formation via Visible Light Photoredox Catalysis. J. Org. Chem. 2016, 81, 7161–7171.Ruan Z; Huang Z; Xu Z; Zeng S; Feng P; Sun P-H Late-Stage Azolation of Benzylic C–H Bonds Enabled by Electrooxidation. Sci. China. Chem. 2021, 64, 800–807.Hou Z-W; Liu D-J; Xiong P; Lai X-L; Song J; Xu H-C Site Selective Electrochemical Benzylic C–H Amination. Angew. Chem., Int. Ed. 2021, 60, 2943–2947.Shao X; Tian L; Wang Y C–N Coupling of Azoles or Imides with Carbocations Generated by Electrochemical Oxidation. Eur. J. Org. Chem. 2019, 2019, 4089–4094.Yang Y-Z; Song R-J; Li J-H Intermolecular Anodic Oxidative Cross-Dehydrogenative C(sp3)–N Bond-Coupling Reactions of Xanthenes with Azoles. Org. Lett. 2019, 21, 3228–3231.
- (11).For early examples of the [HAT+RPC] mechanism applied to C–H functionalization, see: Dai C; Meschini F; Narayanam JMR; Stephenson CRJ Friedel-Crafts Amidoalkylation via Thermolysis and Oxidative Photocatalysis. J. Org. Chem. 2012, 77, 4425–4431.Pandey G; Laha R Visible-Light-Catalyzed Direct Benzylic C(sp3)–H Amination Reaction by Cross-Dehydrogenative Coupling. Angew. Chem., Int. Ed. 2015, 54, 14875–14879.Li G-X; Morales-Rivera CA; Gao F; Wang Y; He G; Liu P; Chen G A unified photoredox-catalysis strategy for C(sp3)–H hydroxylation and amidation using hypervalent iodine. Chem. Sci. 2017, 8, 7180–7185.Hu H; Chen S-J; Mandal M; Pratik SM; Buss JA; Krska SW; Cramer CJ; Stahl SS Copper-catalysed benzylic C–H coupling with alcohols via radical relay enabled by redox buffering. Nat. Catal. 2020, 3, 358–367.
- (12).Song C; Dong X; Yi H; Chiang C-W; Lei A DDQ-Catalyzed Direct C(sp3)–H Amination of Alkylheteroarenes: Synthesis of Biheteroarenes Under Aerobic and Metal-Free Conditions. ACS Catal. 2018, 8, 2195–2199. For an electrochemical version, see: Hou, Z.-W.; Li, L.; Wang, L. Organocatalytic electrochemical amination of benzylic C–H bonds. Org. Chem. Front. 2021, 8, 4700–4705.
- (13).Wan T; Capaldo L; Laudadio G; Nyuchev AV; Rincón JA; García-Losada P; Mateos C; Frederick MO; Nuño M; Noël T Decatungstate-Mediated C(sp3)–H Heteroarylation via Radical-Polar Crossover in Batch and Flow. Angew. Chem., Int. Ed. 2021, 60, 17893–17897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chen S-J; Golden DL; Krska SW; Stahl SS Copper-Catalyzed Cross-Coupling of Benzylic C–H Bonds and Azoles with Controlled N-Site Selectivity. J. Am. Chem. Soc. 2021, 143, 14438–14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Zhang Y; Fitzpatrick NA; Das M; Bedre I; Yayla HG; Lall MS; Musacchio PZ A photoredox-catalyzed approach for formal hydride abstraction to enable Csp3–H functionalization with nucleophilic partners (F, C, O, N, and Br/Cl). Chem Catal. 2022, 2, 292–308. [Google Scholar]; (b) Fitzpatrick NA; Zamani L; Das M; Yayla HG; Lall MS; Musacchio PZ A SN1 mechanistic approach to the Williamson ether reaction via photoredox catalysis applied to benzylic C(sp3)–H bonds. Tetrahedron 2022, 125, 132986. [Google Scholar]
- (16).Leibler IN-M; Tekle-Smith M; Doyle AG A general strategy for C(sp3)–H functionalization with nucleophiles using methyl radical as a hydrogen atom abstractor. Nat. Commun. 2021, 12, 6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).He F-S; Ye S; Wu J Recent Advances in Pyridinium Salts as Radical Reservoirs in Organic Synthesis. ACS Catal. 2019, 9, 8943–8960. [Google Scholar]
- (18).For another example of an electronically tunable pyridinium system, see: McClain EJ; Wortman AK; Stephenson CRJ Radical generation enabled by photoinduced N–O bond fragmentation. Chem. Sci. 2022, 13, 12158.
- (19).Crisenza GEM; Mazzarella D; Melchiorre P Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).The nucleophilicity parameter as established by Mayr is represented as N.
- (21).(a) Kim I; Min M; Kang D; Kim K; Hong S Direct Phosphonation of Quinolinones and Coumarins Driven by the Photochemical Activity of Substrates and Products. Org. Lett. 2017, 19, 1394–1397. [DOI] [PubMed] [Google Scholar]; (b) Kim I; Kang G; Lee K; Park B; Kang D; Jung H; He YT; Baik M-H; Hong S Site-Selective Functionalization of Pyridinium Derivatives via Visible-Light-Driven Photocatalysis with Quinolinone. J. Am. Chem. Soc. 2019, 141, 9239–9248. [DOI] [PubMed] [Google Scholar]
- (22).Zheng M; Hou J; Zhan L-W; Huang Y; Chen L; Hua L-L; Li Y; Tang W-Y; Li B-D Visible-Light-Driven, Metal-Free Divergent Difunctionalization of Alkenes Using Alkyl Formates. ACS Catal. 2021, 11, 542–553. [Google Scholar]
- (23).(a) Rammal F; Gao D; Boujnah S; Hussein AA; Lalevée J; Gaumont A-C; Morlet-Savary F; Lakhdar S Photochemical C-H Silylation and Hydroxymethylation of Pyridines and Related Structures: Synthetic Scope and Mechanisms. ACS Catal. 2020, 10, 13710–13717. [Google Scholar]; (b) Quint V; Morlet-Savary F; Lohier J-F; Lalevée J; Gaumont A-C; Lakhdar S Metal-Free, Visible Light-Photo-catalyzed Synthesis of Benzo[b]phosphole Oxides: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2016, 138, 7436–7441. [DOI] [PubMed] [Google Scholar]; (c) Quint V; Chouchene N; Askri M; Lalevée J; Gaumont A-C; Lakhdar S Visible-light-mediated α-phosphorylation of N-aryl tertiary amines through the formation of electron-donor–acceptor complexes: synthetic and mechanistic studies. Org. Chem. Front. 2019, 6, 41–44. [Google Scholar]
- (24).Chang L; An Q; Duan L; Feng K; Zuo Z Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis. Chem. Rev. 2022, 122, 2429–2486. [DOI] [PubMed] [Google Scholar]
- (25).(a) Bao X; Wang Q; Zhu J Remote C(sp3)–H Arylation and Vinylation of N-Alkoxypyridinium Salts to d-Aryl and d-Vinyl Alcohols. Chem. - Eur. J. 2019, 25, 11630–11634. [DOI] [PubMed] [Google Scholar]; (b) Kim I; Park B; Kang G; Kim J; Jung H; Lee H; Baik M-H; Hong S Visible-Light-Induced Pyridylation of Remote C(sp3)–H Bonds by Radical Translocation of N-Alkoxypyridinium Salts. Angew. Chem., Int. Ed. 2018, 57, 15517–15522. [DOI] [PubMed] [Google Scholar]; (c) Bao X; Wang Q; Zhu J Dual Photoredox/CopperCatalysisfor the Remote C(sp3)–H Functionalization of Alcohols and Alkyl Halides by N-Alkoxypyridinium Salts. Angew. Chem., Int. Ed. 2019, 58, 2139–2143. [DOI] [PubMed] [Google Scholar]
- (26).Barthelemy AL; Tuccio B; Magnier E; Dagousset G Alkoxyl Radicals Generated under Photoredox Catalysis: A Strategy for anti-Markovnikov Alkoxylation Reactions. Angew. Chem., Int. Ed. 2018, 57, 13790–13794. [DOI] [PubMed] [Google Scholar]
- (27).(a) Olah GA; Comisarow MB; Kim CJ Stable carbonium ions. LXXIII. 3-Aryl-2,3-dimethyl-2-butyl cations and factors controlling phenonium ion-phenethyl cation-benzyl cation equilibria. J. Am. Chem. Soc. 1969, 91, 1458–1469. [Google Scholar]; (b) Canestrari D; Lancianesi S; Badiola E; Strinna C; Ibrahim H; Adamo MFA Desulfurative Chlorination of Alkyl Phenyl Sulfides. Org. Lett. 2017, 19, 918–921. [DOI] [PubMed] [Google Scholar]; (c) Cantillo D; de Frutos O; Rincon JA; Mateos C; Kappe CO A Scalable Procedure for Light-Induced Benzylic Brominations in Continuous Flow. J. Org. Chem. 2014, 79, 223–229. [DOI] [PubMed] [Google Scholar]
- (28).It is plausible that a methyl radical operates as the HAT reagent. When TBPB is used with a polar solvent, β-scission of the tBuO· radical is likely (see ref 16).
- (29). Teders M; Gómez-Suárez A; Pitzer L; Hopkinson MN; Glorius F Diverse Visible-Light-Promoted Functionalizations of Benzotriazoles Inspired by Mechanism-Based Luminescence Screening. Angew. Chem., Int. Ed. 2017, 56, 902–906. Jian Y; Chen M; Huang B; Jia W; Yang C; Xia W Visible-Light-Induced C(sp2)–P Bond Formation by Denitrogenative Coupling of Benzotriazoles with Phosphites. Org. Lett. 2018, 20, 5370–5374. Xie T; Zhou L; Shen M; Li J; Lv X; Wang X Diastereoselective synthesis of cis-1,2-disubstituted cyclopropanols and cyclopent-3-enols via SmI2 mediated C–N(Bt) bond cleavage. Tetrahedron Lett. 2015, 56, 3982–3987.
- (30).Franckowiak G; Hess C; Weidemann K Preparation of bifonazole, useful as antimycotic agent, comprises reacting biphenyl-4-yl(phenyl) methanol with chlorinating agent in cyclohexane and then with imidazole. DE 10332684 B3, April 21, 2005. [Google Scholar]
- (31).Molander GA; Elia MD Suzuki-Miyaura Cross-Coupling Reactions of Benzyl Halides with Potassium Aryltrifluoroborates. J. Org. Chem. 2006, 71, 9198–9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).For selected examples of pyridinium salts in EDA complexes, see: Wu J; Grant PS; Li X; Noble A; Aggarwal VK Catalyst Free Deaminative Functionalizations of Primary Amines by Photoinduced Single-Electron Transfer. Angew. Chem., Int. Ed. 2019, 58, 5697–5701.Katritzky AR; de Ville GZ; Patel RC The SRN2 Mechanism of Nucleophilic Substitution. Tetrahedron Lett. 1980, 21, 1723–1726.Katritzky AR; de Ville GZ; Patel RC Carbon Alkylation of Simple Nitronate Anions by N-Substituted Pyridiniums. Tetrahedron 1981, 37, 25–30.Katritzky AR; Chen J-L; Marson CM; Maia A; Kashmiri MA The Non-Chain Radicaloid C-Alkylation of Nitronate Anions: Further Evidence for the Mechanism. Tetrahedron 1986, 42, 101–108.Yang M; Cao T; Xu T; Liao S Visible-Light-Induced Deaminative Thioesterification of Amino Acid Derived Katritzky Salts via Electron Donor-Acceptor Complex Formation. Org. Lett. 2019, 21, 8673–8678.James MJ; Strieth-Kalthoff F; Sandfort F; Klauck FJR; Wagener F; Glorius F Visible-Light-Mediated Charge Transfer Enables C-C Bond Formation with Traceless Acceptor Groups. Chem. - Eur. J. 2019, 25, 8240–8244.Wu J; He L; Noble A; Aggarwal VK Photoinduced Deaminative Borylation of Alkylamines. J. Am. Chem. Soc. 2018, 140, 10700–10704.Sandfort F; Strieth-Kalthoff F; Klauck FJR; James MJ; Glorius F Deaminative Borylation of Aliphatic Amines Enabled by Visible Light Excitation of an Electron Donor-Acceptor Complex. Chem. - Eur. J. 2018, 24, 17210–17214.Fu M-C; Shang R; Zhao B; Wang B; Fu Y Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide. Science 2019, 363, 1429–1434.
- (33).See the Supporting Information for the mechanism scheme with EDA complex involvement.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.