

Significance
The availability of increasing large genomic datasets, combined with advances in artificial intelligence, allows large-scale analysis of protein sequences. Here, we studied the sequences of roughly 280’000 J-domain proteins, revealing their staggering functional diversity, apparent from the large number of different domains they comprise. Using artificial neural networks, we found that the sequences of the J-domains alone carry orthogonal signatures of their phylogenetic, cellular, and functional origin and that these fingerprints mark the interaction surface with their obligatory partner, the Hsp70 chaperone. From a general perspective, our work highlights how the combination of genomic data and machine learning approaches can reveal otherwise inaccessible information.
Keywords: J-domain proteins, protein homeostasis, large-scale data analysis, Hsp40 co-chaperones, artificial neural networks
Abstract
The 70-kD heat shock protein (Hsp70) chaperone system is a central hub of the proteostasis network that helps maintain protein homeostasis in all organisms. The recruitment of Hsp70 to perform different and specific cellular functions is regulated by the J-domain protein (JDP) co-chaperone family carrying the small namesake J-domain, required to interact and drive the ATPase cycle of Hsp70s. Besides the J-domain, prokaryotic and eukaryotic JDPs display a staggering diversity in domain architecture, function, and cellular localization. Very little is known about the overall JDP family, despite their essential role in cellular proteostasis, development, and its link to a broad range of human diseases. In this work, we leverage the exponentially increasing number of JDP gene sequences identified across all kingdoms owing to the advancements in sequencing technology and provide a broad overview of the JDP repertoire. Using an automated classification scheme based on artificial neural networks (ANNs), we demonstrate that the sequences of J-domains carry sufficient discriminatory information to reliably recover the phylogeny, localization, and domain composition of the corresponding full-length JDP. By harnessing the interpretability of the ANNs, we find that many of the discriminatory sequence positions match residues that form the interaction interface between the J-domain and Hsp70. This reveals that key residues within the J-domains have coevolved with their obligatory Hsp70 partners to build chaperone circuits for specific functions in cells.
Hsp70 is a crucial and highly ubiquitous chaperone involved in a wide variety of constitutive and stress-related cellular processes, including the promotion of protein folding (1, 2), the prevention of formation of cytotoxic aggregates (3–6), the import of polypeptides into cell compartments (7, 8), the (dis)assembly of oligomeric protein complexes (9–11), the regulation of protein activity (12), and the targeting of terminally damaged proteins for degradation (13–15). Underlying these diverse functions is the fundamental ability of Hsp70 to bind substrate proteins upon a conformational change that depends on the nature of the bound nucleotide, which alternates between ATP and ADP. This biochemical cycle is stringently regulated by J-domain containing proteins (JDPs) that, together with the substrate, accelerate ATP hydrolysis in Hsp70s by orders of magnitude (16, 17), resulting in ultra-affinity binding, namely a significant, nonequilibrium enhancement of the Hsp70 affinity for its substrates (18). Subsequently, Hsp70 nucleotide exchange factors (NEFs) bind and accelerate ADP release, thus allowing rebinding of ATP and client protein release (19) (Fig. 1A).
Fig. 1.

Structural views of the J-domain and domain architectures of JDPs. (A) Schematic view of the canonical Hsp70 cycle. (B) Structural view of the Hsp70-J-domain complex [PDB ID: 5NRO (20)]. NBD denotes the Hsp70 nucleotide-binding domain, and SBD the Hsp70 substrate-binding domain. The four helices (α1–α4) forming the J-domain and the characteristic HPD motif are highlighted. (C) Cytosolic class A JDP of Saccharomyces cerevisiae (Ydj1) in its constitutive homodimeric form [the structure is a combination of the separate J-domain, PDB ID 5VSO (21), CTDs PDB ID 1NLT (22), and dimerization domains, PDB ID 1XAO (23)]; in lack of a known experimentally determined structure, the G/F-rich linker between the J-domain and the first C-terminal domain has been hand-drawn on one of the two protomers. The different domains have been colored only on one protomer. The general architecture of class A JDPs is highlighted below the structure (G/F: glycine/phenylalanine rich linker; CTD: C-terminal, substrate-binding domain; ZFLR: cysteine-rich, zinc-finger-like region; DD: dimerization domain). (D) Class B JDP of Thermus thermophilus in its constitutive homodimeric form [PDB ID 1C3G (24)]. The different domains have been colored only on one protomer. The general architecture of class B JDPs is highlighted below the structure. (E) Escherichia coli HscB [class C; PDB ID 1FPO (25)] with its architecture (HSCB_C: C-terminal domain of HscB). (F) Mus musculus Erdj5 [class C; PDB ID 5AYK (26)] and its architecture.
All members of the JDP family invariably contain the namesake J-domain, which is ~63 residue-long. The J-domain is formed by a well-defined arrangement of four alpha helices (Fig. 1B) with a highly-conserved histidine–proline–aspartate (HPD) motif between helices 2 and 3. The helix II–HPD–helix III motif forms the primary interface for the interaction and stimulation of ATP hydrolysis in Hsp70 (16, 17, 20, 27–29). The remaining domains of JDPs help target them to specific locations, client proteins, or protein conformations (e.g., unfolded, misfolded, or aggregated polypeptides), thereby recruiting the action of Hsp70s to selected substrates (30, 31).
A classification scheme for JDPs has been suggested based on the overall domain architecture, resulting in three main classes (30, 32–34). Class A JDPs share the domain architecture of the prototypical DnaJ member of E. coli. Members of this class consist of an N-terminal J-domain, followed by a glycine and phenylalanine (G/F) rich region and two successive C-terminal β-sandwich domains (CTDs), with a zinc-finger-like region (ZFLR) domain protruding from the first CTD, and are terminated by a short dimerization domain at the C-terminal (Fig. 1C). Class B JDPs have in common only the presence of an N-terminal J-domain, followed by a G/F region, like in class A. This loose grouping includes both JDPs that are architecturally, as well as structurally, very similar to the members of class A with the notable absence of the ZFLR (henceforth called “canonical” class B, Fig. 1D), alongside more diverse ones (33). The G/F-rich region of both classes is typically considered largely unstructured. All the JDPs belonging to class A and some of the ones belonging to class B constitutively form homodimers through the C-terminal dimerization domain (Fig. 1 C and D) and bind a large variety of unfolded, misfolded, or aggregated substrates through one or multiple interaction sites located on their CTDs and, possibly for class A, on the ZFLR (35, 36). These dimeric JDPs are also known collectively as Hsp40s owing to their approximate 40-kDa molecular mass. Class A and most class B JDPs are known to recruit Hsp70s to misfolded/aggregated proteins and to promote their crowding on these substrates (4). Class C JDPs, in contrast, display a much higher variability in domain architectures (two examples are shown in Fig. 1 E and F) and have been historically classified by what they are not, namely neither A nor B, rather than by what they are. Indeed, while members of class C represent the vast majority of JDPs, and intervene in a wide range of unrelated biological pathways (e.g., auxilin, which is involved in clathrin cage disassembly; Hsc20, which regulates FeS cluster biogenesis; Sec63, which promotes posttranslational protein import in the ER; Zuotin, which assists protein translation), they share with each other little or no common functional or phylogenetic features, except for the presence of a J-domain.
Here, we leveraged the recent exponential growth in genomic data and the improvements in homology detection tools to explore the relations between JDP sequences and their phylogenetic and functional classification at various resolutions. We then used artificial neural networks (ANNs) to show that even the relatively short and simple sequences of the J-domains alone carry sufficient phylogenetic and functional signatures that may modulate cellular JDP-Hsp70 chaperone circuits. These unique signatures reliably identify the architecture of the corresponding full-length JDP, its cell localization, and the taxonomic clade. This machine learning approach allowed us to use amino acid sequence variations embedded in the J-domain to curate existing and new JDP sequences reported in various organisms with high precision. By identifying the residues that are most important for classification, we further pinpointed the structural positions where the class- and organism-specific signatures are located.
From a more general perspective, we highlighted how the combination of interpretable large-scale sequence analysis techniques and coarse-grained annotations can unlock the wealth of information that would otherwise remain hidden in exponentially growing, and thus manually unmanageable, sequence databases.
Results
Construction of an Extensive Annotated J-domain Protein Sequence Dataset.
The number of deposited JDP sequences in the UniProt repository (37) has been exponentially growing over the past two decades, increasing by an order of magnitude every 6 to 7 y (Fig. 2A), from 103 in year 2003 to 105 in year 2018, and projected to reach 106 sequences in 2024 to 2025 and 107 in the early 2030s. Using stringent homology search criteria and annotations, we extracted a total of 279’565 JDP sequences from existing genome databases (Methods). The associated annotations allowed us to assign all JDPs to different phylogenetic classes at various taxonomic levels. Using sequence domain models from the InterPro database (38), we further characterized the domain composition of full-length sequences of each JDP. Protein localization prediction algorithms were used to infer the most probable organelle of residence of eukaryotic JDPs (Methods). This compound procedure allowed us to assign to each JDP sequence in the dataset a set of ground-truth attributes comprising phylogenetic, architectural, and cellular localization features. With this procedure, the characteristics that describe, for instance, human DNAJA1 were a) its full amino acid sequence, b) its domain composition (J-domain, G/F-rich region, substrate-binding domains, and ZFLR), c) its class (class A), d) its phylogeny (eukaryotic/metazoan/mammalian protein), and e) its cellular localization (predominantly cytoplasmic).
Fig. 2.

Characterization of the JDP dataset. (A) Growth of the number of JDP sequences found in the UniProt Database over the last three decades. The red dashed line shows an exponential fit. (B) Class proportions in the dataset. (C) Amino acid frequencies of the G/F linker region of class A JDPs. (D) Amino acid frequencies of the G/F linker region of class B JDPs. (E) Number of JDPs in the different kingdoms, highlighted by their class (A: green; B: blue; C: red). (F) The 12 most abundant J-protein domain architectures. Numbers on the left denote the number of sequences with this architecture in our dataset, and, on the right, we report the corresponding human, yeast, plant, or bacterial proteins. (G) Highlight of the evolutionary conservation of the cysteine-rich proto-ZFLR in ER-resident JDPs in six metazoans and two plants.
Overall Characterization of the Diversity of the J-domain Protein Dataset.
Based on the overall domain composition of JDPs, the three established functional classes were clearly identifiable: class A and canonical class B (members with the canonical Hsp40 domain architecture, Fig. 1 C and D) represented 21.0% and 10.7% of the dataset, respectively (Fig. 2B), with the remaining JDPs (68.3%, corresponding to ~190’000 sequences) assigned to class C or noncanonical class B. Approximately half of them (100’483) have a domain architecture that, according to the InterPro-based domain annotation, consists only of the J-domain (henceforth referred to as JD-only JDPs, Fig. 2F). These were slightly enriched in eukaryotic organisms (SI Appendix, Fig. S1A) but were found uniformly throughout all predicted subcellular compartments (SI Appendix, Fig. S1B). The full protein length distribution for JD-only JDPs was significantly shorter than for JDPs having multidomain architectures (SI Appendix, Fig. S1C). However, a significant number of JD-only JDPs had rather long protein sequences. To further characterize the architectures of these JDPs, we leveraged the structural predictions available in the AlphaFold Protein Structure Database (39). We collected the 94’252 available structural models for the JDPs in our dataset and analyzed their uncertainty metrics (pLDDT and PAE, SI Appendix, Fig. S2) as proxies for the existence of unannotated structured domains. We found that the predicted structures of the J-domains had, on average, low uncertainty (high pLDDT/low PAE) for all JDPs. Instead, the structural predictions for the non-J-domain region of well-annotated JDPs were on average less uncertain than for JD-only JDPs. Furthermore, the non-J-domain regions of JD-only JDPs comprised both highly unstructured polypeptides (low pLDDT/high PAE) and highly structured ones, potentially hinting at structured domains not annotated in InterPro rather than at a systematic lack of structure of JD-only JDPs.
Collectively, the 12 most populated domain architectures in our dataset (including classes A and B) covered approximately 85% of the total dataset (238’217 sequences). Visual inspection of class A and B JDP sequences showed that, beyond the clear difference due to the presence of the ZFLR, they could also be distinguished by their G/F-rich regions, which showed a more marked segregation of phenylalanine, arginine, glycine, and glutamate residues in class A than in class B JDPs (Fig. 2 C and D). This was quantitatively confirmed by a simple linear classification model which showed that the (unaligned) amino acid sequences of the G/F region alone were able to successfully discriminate JDPs of class A and B (classification accuracy of ~93%, see Methods for details). These significant differences in amino acid compositions suggest that at least parts of the G/F-rich region could have different functions beyond being a simple flexible linker. This is in keeping with recent findings showing that in some class B members a small helix in the G/F region docks with the J-domain, likely with functional consequences (40–43). For example, it has been recently shown that G/F regions could facilitate some class A and B JDPs to form cellular condensates (44).
In agreement with previous observations (31), class C members dominated the dataset (Fig. 2B) mainly due to their overwhelming abundance in eukaryotes (Fig. 2E and SI Appendix, Fig. S3 for a more detailed analysis of class C abundance in the dataset). There were 2,712 different architectures present in class C, which were built from 1,725 distinct domains, several of which were Domain of Unknown Function (DUFs). Overall, we found little to no co-occurrence of the most common domains (SI Appendix, Fig. S4). The only notable exceptions are JDPs containing multiple repeats of tetratricopeptides (TPR) associated with several different InterPro families (in Fig. 2F and SI Appendix, Fig. S4 all these TPR domains are collected in a single group). Inspection of the architectures of the most frequently observed JDP domains within class C revealed very disparate phylogenetic and/or localization signatures (Fig. 2F). For instance, the DjlA family, which formed the most abundant class C-subtype architecture, is present only in bacteria. The HscB family (human DNAJC20, Fig. 1F), which participates in iron-sulfur cluster assembly pathway, is present in both prokaryotes and eukaryotes. Others, such as the Sec63 (human DNAJC23) are eukaryotic and localized to the membrane of the endoplasmic reticulum (ER); DNAJC18 (which shares almost the same domain composition as DNAJB12 and DNAJB14), DNAJC11, and DNAJC3/DNAJC7 were found only in eukaryotes. Some highly represented architectures obeyed an even finer phylogenetic distribution, with Djp1/Caj1, Jjj1, and Jjj3 present only in yeast (e.g., in S. cerevisiae). These findings across all organisms mirror and greatly expand recent observations made on bacterial and phage JDPs that also revealed the diversity of architectures among class C JDPs, often with yet unknown cellular functions (45).
While the vast majority of JDPs were characterized by the conserved HPD motif, the presence of an HPE triad was detected in 2,921 J-domains (~1% of the total), with roughly 1,700 prokaryotic and 1,200 eukaryotic sequences exhibiting this variation. Approximately 1,600 of the total HPE JDPs lacked any other domains, such as DjlB and DjlC, while most of the remaining ones were combined with various domains mostly involved in Sodium/Calcium exchange. Among these, the NAC1 gene, coding for a sodium/calcium exchanger found primarily in mammals, exhibits an HPD in humans and an HPE in about 90% of other organisms (e.g., Bos taurus and Canis lupus familiaris). The relatively low prevalence of this variation in the HPD motif (approximately in 1% of all JDPs analyzed) appears to suggest additional fine-tuning of binding and stimulation of ATP hydrolysis in Hsp70s only in a few selected cases.
As mentioned above, our analysis highlighted several JDP families where the J-domain is paired with DUFs in specific clades. For instance, DUF3444 (IPR024593, SI Appendix, Fig. S4) is exclusive to Viridiplantae and predominantly found in Streptophyta. Approximately two thirds of the proteins containing this domain are associated with a J-domain, while approximately 30% lack any other bound domain. Another example is the presence of myb/SANT domains (IPR001005, SI Appendix, Fig. S4) in proteins such as DNAJC1 or DNAJC2, which are mostly found in animals and plants. These domains belong to the homeobox-like domain superfamily and can be categorized into three groups: the myb-like HTH domain, which binds DNA; the SANT domain, which functions as a protein–protein interaction module; and the myb-like domain, which can participate in either of these functions. The identification of associations between JDPs and uncharacterized domains may offer a promising avenue for uncovering interactions and processes requiring the intervention of Hsp70s.
We then analyzed the predicted subcellular localization of JDPs to assess whether the classes are differently represented in various cellular compartments. Analysis of the eukaryotic organellar makeup of the dataset revealed that mitochondrial JDPs were mainly of class A and C (SI Appendix, Fig. S3) with only 10% of class B. In contrast, ER-localized JDPs showed an intriguing class distribution. First, they were significantly enriched in class C JDPs compared to mitochondria (90% vs. 68%, P < 0.0001, two-sided χ2 test). Interestingly, the ER contains a resident class A/B-type JDP, which in fungi (and some other unicellular eukaryotes such as amoebas) is a canonical class A (yeast Scj1), while in metazoan and plants, it is customarily assigned to class B (DNAJB11/Erdj3) due to the lack of a ZFLR. In fact, instead of the customary ZFLR, a noncanonical cysteine-rich region (which we called here pseudoZFLR) was found in these class B-like members displaying regularity and high degree of similarity in plants and metazoan (Fig. 2G), with only two Cys pairs. The first Cys pair showed a highly conserved Cys–Asp–Cys (CNC) motif, while the second pair has the two cysteines separated by two residues. This motif represents an interesting evolutionary conundrum, given that the most accepted view of the evolution of eukaryotes posits that the divergence in opisthokonts, between fungi and metazoan, took place after the split from plants.
J-domain Sequences Contain Functional and Phylogenetic Information at Multiple Scales.
Organisms ranging from viruses to prokaryotes to eukaryotes rely on multiple pairs of Hsp70s and JDPs to support various biological activities. What largely remains unclear is how a particular JDP recognizes its partner Hsp70, an interaction that is largely mediated by the J-domain. It has been speculated for many years that the specificity should thus be encoded in the sequences of both partners, calling into question the degree to which the J-domains can be interchanged between different JDPs with minimal consequences for their primary function. In fact, not all J-domains interact indifferently with Hsp70s. Exchanges of J-domains between two JDPs frequently result in nonfunctional chimeras, suggesting that there is selectivity in the pairing of JDPs and Hsp70s (34, 46–50). Evolutionary and functional factors such as phylogeny, interactions with different Hsp70 paralogs, as well as the colocalization of J- and non-J-domains in the same polypeptide, may have left traces in the sequence of the J-domain. If this was the case, the sequence of a J-domain alone should carry relevant information about the overall JDP to which it belongs.
To test this hypothesis and provide evidence to support that the sequences of J-domains carry a rich source of disparate evolutionary and functional signatures, we first extracted and aligned 279'565 J-domain sequences from our JDP repertoire (see Methods for detailed procedure). We then used the unsupervised UMAP algorithm (51) to provide a (nonlinear) projection of the sequences in two dimensions, allowing for their direct visual inspection (Fig. 3 and Methods). The projections revealed that the J-domain sequences are gathered in clusters and that the different groups, highlighted by different colors, often associated well with various classification schemes, such as their i) A, B, or C class (Fig. 3A), ii) bacteria rather than eukaryotes (Fig. 3B) iii) various C subclasses (only the five more abundant ones are represented in Fig. 3C for the sake of clarity). To highlight the finer signatures that are encoded in the J-domain sequences, we highlighted HscB, whose sequences formed well-defined cluster in the global landscape (circles in Fig. 3 A –C). Furthermore, zooming in on this cluster, we observed that HscB sequences from metazoan and plants were well-grouped (Fig. 3D). In contrast, fungal HscB showed a higher spread within the HscB cluster. This indicates that different and nondependent layers of information are simultaneously encoded in the sequences of the J-domains, both to finer phylogenetic levels and for other criteria, such as subcellular localization (SI Appendix, Fig. S5).
Fig. 3.
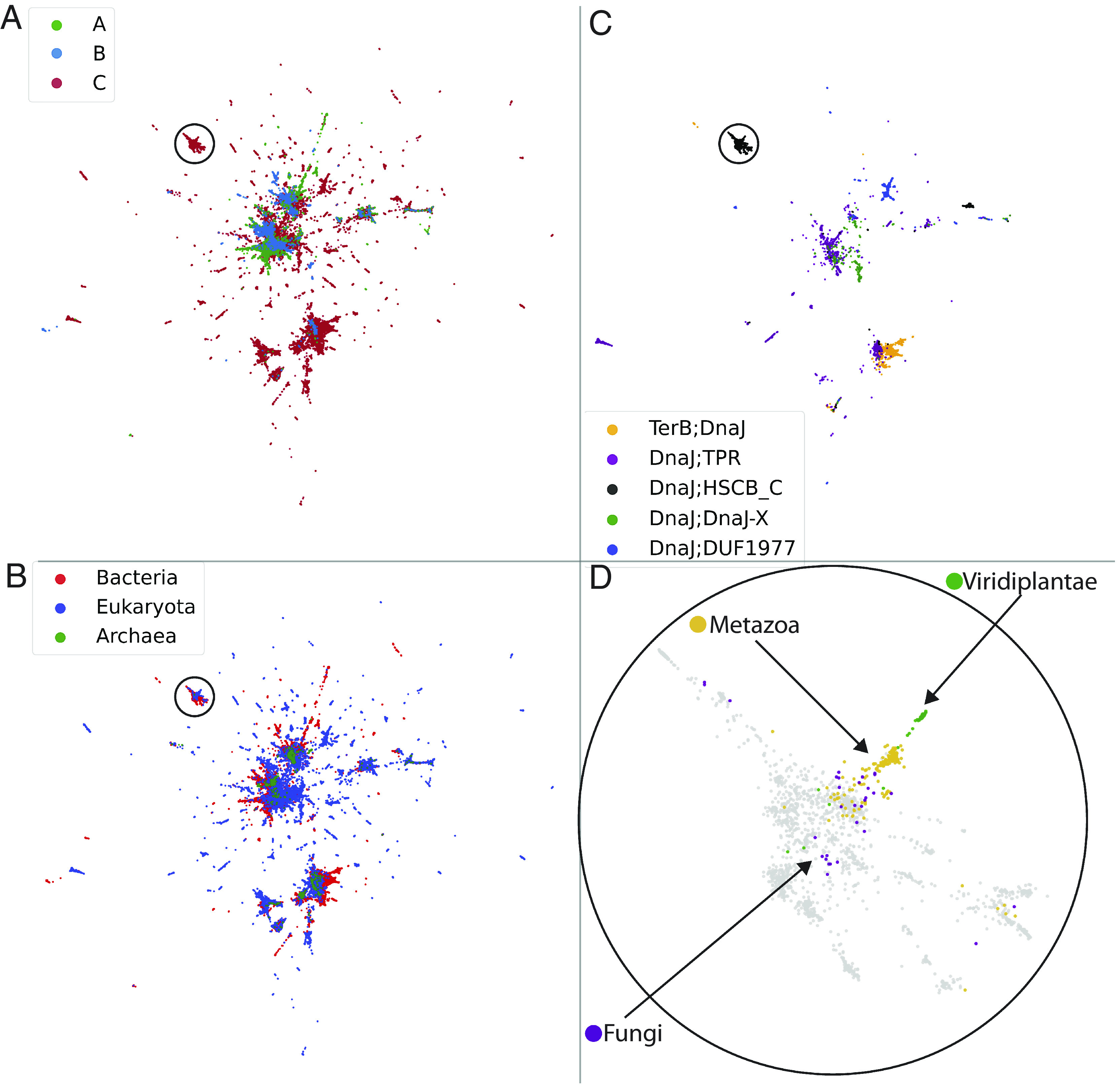
Two-dimensional representations of J-domains. JDP sequences are projected in two dimensions by a nonlinear transformation (UMAP) and colored according to different schemes. (A) J-domains are identified as being of class A (green), B (blue), or C (red) JDPs. (B) J-domains are identified as being of bacterial (red), eukaryotic (blue), or archaea (green) origin. (C) The five most abundant C subclasses are highlighted (see Fig. 2F for representative members). The circles in each panel focus on the group identified as HscB-like JDPs. (D) Zoom on the encircled region in the three other panels, highlighting the further differentiation of the eukaryotic ones into fungi, metazoans, and plants (bacterial HscB sequences are shown in grey background). For visual clarity, a random subset of 50% of all points are displayed.
To assess to what extent the different signatures imprinted in the J-domain sequences could be ascribed to evolution, we constructed maximum likelihood (ML) phylogenetic trees for human and yeast JDPs using both full-length sequences and only the J-domain sequences (Methods and SI Appendix, Fig. S6 A and B). Our analysis revealed that certain functional properties, such as ER localization of JDPs, appear independently on multiple branches. Furthermore, JDPs such as DNAJB1, DNAJB6, and DNAJC7, which have partially overlapped functions [they all prevent poly-Q aggregation, but through different mechanisms (52)], evolved on distant branches in the human tree. These results suggest that the acquisition of JDP functional properties follows a mixture of phylogenetic drift and convergent evolution. We found similar results for yeast JDPs (SI Appendix, Fig. S6 C and D). Thus, a phylogenetic tree-based analysis based solely on J-domain sequences does not optimally correlate with the full ground truth for each JDP (i.e., phylogeny, functional class, domain architecture, and cellular localization). To address this issue, we turned to a machine learning strategy for a more precise classification.
Classification of J-domains by ANNs.
To find the detailed determinants in J-domain sequences that underpin the differentiation of JDPs according to various criteria (phylogenetic, functional, and localization), we used an approach based on ANN to explicitly classify JDPs based solely on the J-domain sequence. For each task (e.g., predicting the A/B/C class of a JDP), we built a separate ANN-based classifier and analyzed their implicit sequence embeddings and feature importance. We focused on a few selected classification tasks, which cover the complexity of the JDPs at various scales. The analyzed classification tasks are as follows:
Class A vs. class B (henceforth referred to as A/B);
Class A vs. class B vs. class C (henceforth referred to as A/B/C);
Classification of 12 different architectural classes, including the architectures of classes A and B JDP, the 9 most populated C subclasses (excluding JDPs containing only the DnaJ domain), and an additional “other” class generically comprising all remaining JDPs (again not including JDPs containing only DnaJ domain) (henceforth referred to as Arch12). The 12 architectural classes in the Arch12 task are thus “ClassA (DnaJ-DnaJ_C-DnaJ_Cys-rich,” “Class B (DnaJ-DnaJ_C),” “TerB-DnaJ,” “DnaJ-TPR,” “DnaJ-HSCB_C,” “DnaJ-DUF1977,” “DnaJ-DnaJ-X,” “DnaJ_Sec63,” “DnaJ-DnaJ-C11,” “DnaJ-DPH-MB,” “Class B (DnaJ-DnaJ_C-PseudoZFLR),” “other” (see methods for domain naming convention).
Bacteria vs. Eukaryotes vs. Archaea vs. Viruses (henceforth referred to as Bac/Euk/Arch/Vir);
Classification of Eukaryotes: Metazoan vs. Fungi vs. Viridiplantae vs. Other Eukaryotes
Classification of Bacteria: Terrabacteria vs. Proteobacteria vs. FCB (Fibrobacterota, Chlorobiota and Bacteroidota) vs. PVC (Planctomycetota, Verrucomicrobiota, and Chlamydiota) vs. Other bacteria.
Classification of Metazoan: Protostomia vs. Mammalia vs. Sauropsida vs. Other Metazoan
Subcellular localization: Endoplasmic reticulum vs. mitochondria vs. plastids (henceforth referred to as Localization).
The taxonomic splits were based on the largest groups identified in the UniProt database. Although these different groupings are far from exhaustive, we chose them as representatives of both complementary and orthogonal criteria.
To visually inspect how the ANNs internally represented the JDP repertoire according to the various tasks, we projected the ANN embeddings into 2D and colored the JDPs according to the ground truth labels of the corresponding task (Methods). As shown in SI Appendix, Fig. S7A for the classification task A/B/C, the ANN had learned an implicit task-specific embedding of the JDP repertoire, based solely on the J-domain sequences.
To move beyond the visual appraisal of the ANN inner workings and quantify their performance, we computed their confusion matrices (SI Appendix, Fig. S7B), which report the misclassification of each category with respect to their ground truth. An example is shown in SI Appendix, Fig. S7B where J-domains belonging to JDPs of class A were correctly classified 98.4% of the times and misclassified as class B or C members only 1.1% and 0.5% of the times, respectively. Similarly, class B J-domains were classified according to their ground truth 94.7% of the times. Overall, the performance of the ANNs for all classification tasks was excellent (SI Appendix, Fig. S8). The two classification tasks that were slightly less accurate were localization and bacterial subgroup discrimination. In the case of localization, it is important to recall that the accuracy in this case was compatible with the prediction accuracy of the algorithms used to define the localization ground truth (Methods). The relative difficulty for classifying JDPs into bacterial subgroups is likely indicative either of a high degree of lateral-gene transfer or of heterogeneity in bacterial groups, or both.
To compare the ANN performance to a baseline classifier, we used a standard Hidden Markov model approach (HMM) to establish a baseline performance (Methods). For each task, we built individual HMM models for the sequences in the various subgroups on the training set and then evaluated the E-values on each subclass HMM (53). Sequences were then assigned to the class with the lowest E-value for each task individually. Comparison of the HMM and ANN performance (SI Appendix, Table S12) shows that ANNs systematically and largely outperformed the HMM-based approach. Thus, ANNs could extract intricate information from J-domains, beyond the capability of simple profile models. Overall, the success of our ANN classification approach highlights that a myriad of phylogenetic and functional signatures is indeed encoded in J-domain sequences alone and that this methodology can be used to tell J-domains apart from each other depending on different, possibly orthogonal, criteria such as phylogeny, subcellular localization, and domain organization.
We then used our trained ANNs to predict the A, B, and C classes, as well as the Arch12 labels of the 100’483 JD-only JDPs, none of which was used during network training. The A/B/C ANN correctly classified the vast majority of these as belonging to class C. Interestingly, more than 60% of them were classified as “Other” in the Arch12 task, implying that the network had probably recognized that their J-domain sequence signatures were dissimilar from the other classes used to learn this task. The second and third most predicted sequence architectures corresponded to that of J-domains in class A and B (~9% and ~17% respectively).
The ANN-based approach introduced here has a broad applicability, beyond the JDP family, as we have shown by evaluating its performance for the different and large family of proteins containing SH2 domains, finding similar results (see Supplementary Information).
Structural Characterization of the Most Relevant Discriminative Sequence Positions.
Next, we used the interpretability of the ANNs to identify the sequence positions that were most relevant for each individual classification task by assigning them a relevance score (54). We found that although several of the sequence positions were expectedly task-specific, some were ubiquitously important for all tasks (Fig. 4A). Upon close inspection, sequence positions at the contact interface between Hsp70 and the J-domain (20) displayed significantly higher average relevance scores (P = 0.0011, two-sided Mann–Whitney U test, Fig. 4 A and B and Methods). The detailed analysis of the task-specific relevance scores confirmed an enrichment of interface positions among the strongly relevant positions (Fig. 4C). Furthermore, this also revealed a mixture of positions highly relevant for multiple tasks (multicolored rings, e.g., S13, R22, F47, K51, and E52 in E. coli DnaJ numbering) and of positions primarily relevant for single tasks (single-colored rings). Structural mapping of the five positions with the highest relevance score on the structure of the J-domain of E. coli DnaJ in the DnaJ-DnaK complex provided a visual confirmation of these findings (Fig. 4D) (20). Modulating the interaction of the J-domain by directly controlling interface residues is likely a dominant factor in determining the relevant positions across multiple classification schemes.
Fig. 4.
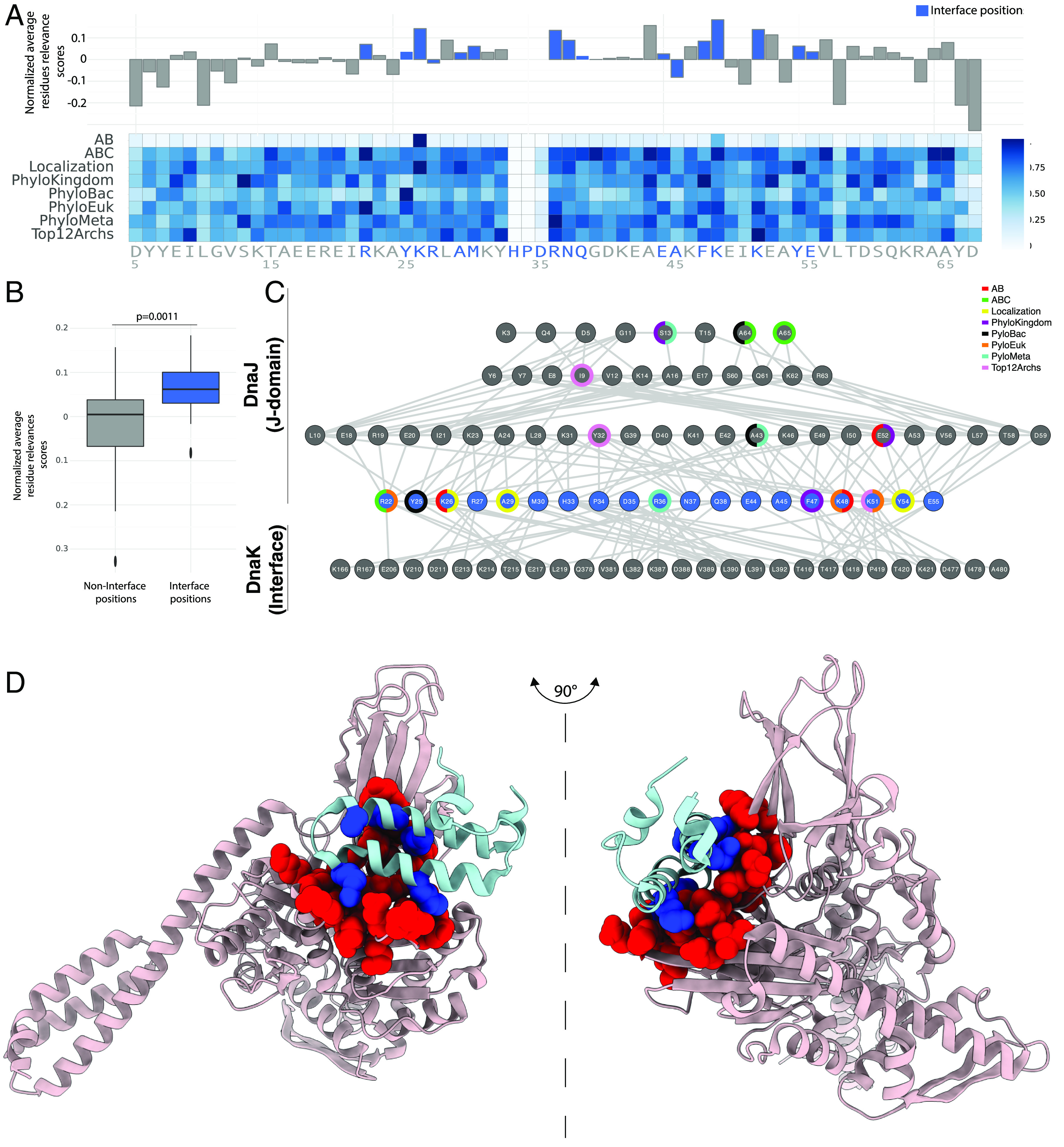
Identification of the sequence and structure positions most relevant for the classification. (A) The relevance score for each position and for different classification tasks is represented in a heatmap. For each position, the relevance score averaged over the different tasks is represented, relative to the average over all positions, above the heatmap (HPD are not considered as due to their characteristic conservation they have negligible contributions to classification). Interface positions in the bacterial J-domain/DnaK complex (20) dimer are highlighted in blue. (B) Comparison of the relevance scores between interface and noninterface positions in the J-domain/DnaK hetero (20). The P-value was computed by the two-sided Mann–Whitney test with continuity correction. (C) Interresidue interaction network between bacterial J-domain/DnaK highlighting the 3 most relevant residues for each classification task. Bottom row: Only the interface residues for DnaK are shown. Blue nodes: Interface J-domain residues. Top 3 rows: Noninterface J-domain residues. The colored rings around the nodes indicate which J-domain residues are most relevant for each classification task. Links in the network are structural contacts between pairs of residues, measured on the crystal structure (20) (Methods). (D) The five top scores identified in the top panel are highlighted on the structure of the complex (20); blue: five most relevant residues on the J-domain; red: the interaction surface on DnaK. The five overall most relevant positions on the J-domain participate in the interaction surface between the two proteins.
These are intriguing results because at no step of our analysis we used information about the binding of J-domains to Hsp70. It is thus tempting to hypothesize that the various, possibly orthogonal, classifications were successful because, among other factors, J-domains coevolved together with Hsp70 following a concerted phylogenetic and functional divergence. We presume that these positions likely facilitate modulation of the interaction between J-domains and their disparate Hsp70 partners and the consequent HPD-dependent tuning of the ATPase activity of Hsp70.
Classification of J-domain Proteins according to Their J-domains.
We then employed the ANNs as class predictors for the human JDPs. We excluded from the training set all human JDPs, together with those of other model organisms (Methods), to prevent any bias during the learning process. In SI Appendix, Table S1, we report the probabilities attributed to each human J-domain with respect to a given class or architectural group.
The J-domains of human class A (DNAJA1-4) and “canonical” class B (DNAJB1, DNAJB4, and DNAJB5) were correctly classified according to the tasks A/B, A/B/C, and Arch12 (SI Appendix, Table S1 and S2 for the task A/B), confirming that the ANNs were able to reliably learn their characteristics during the training phase. A more specific discussion is warranted for DNAJB11 and DNAJB13. While DNAJB11 was predicted as class C in the A/B/C task, it was correctly assigned its domain architecture containing a class B-like domains with a pseudoZFLR domain in the Arch12 task. In contrast, DNAJB13, which possesses a domain architecture of a canonical class B JDP, but without the classical HPD motif, was correctly classified by the A/B/C task but misclassified by the Arch12 task as TPR-containing JDP. These results suggest that DNAJB11 and DNAJB13, despite a likely class B origin, have further diverged enough that their sequences do not fully conform to the majority of class B JDPs.
Class B JDPs with noncanonical architectures (DNAJB2, DNAJB3, DNAJB6, DNAJB7, DNAJB8, DNAJB9, DNAJB12, and DNAJB14) have historically been attributed to class B due to the presence of an N-terminal J-domain followed by a G/F-rich region. We used both the A/B/C and Arch12 classification tasks to try and reclassify these proteins. According to the A/B/C task, only J-domains of DNAJB3, DNAJB6, and DNAJB9 were predicted to carry the sequence signatures of class Bs, albeit with significantly lower probability compared to J-domains of bona fide class B JDPs. We then cross-checked these predictions with the Arch12 task. We found that J-domains of DNAJB12 and DNAJB14 were predicted to be class C and to belong to the same architectural class as DNAJC18, with whom they share the DUF1977 domain. According to the Arch12 task, J-domains of DNAJB2, DNAJB7, and DNAJB9 were predicted to be like that of genuine class B JDPs, while J-domains of DNAJB3, DNAJB6, and DNAJB8 were predicted to belong to class A. The phylogenetic tree in SI Appendix, Fig. S6B, based only on J-domain sequences, was similarly unable to place canonical and noncanonical class B JDPs on the same subbranch. Taken together, these results suggest a complex evolutionary origin of the noncanonical class B JDPs, with some of them possibly descending from class A JDPs, some from class B JDPs, and some possibly directly from pre-existing class C JDPs (the case of DNAJB12, DNAJB14, and DNAJC18 would warrant a more in-depth study to decide which of the three appeared first). Next, we classified class C JDPs into subclasses. All original class C members were correctly predicted by the A/B/C classification task. The Arch12 task also correctly classified most human class C JDPs.
We repeated this analysis for several other model organisms covering a wide phylogenetic range, including S. cerevisiae, M. musculus, R. norvegicus, M. mulatta, D. rerio, D. melanogaster, C. elegans, A. thaliana, and E. coli (SI Appendix, Tables S3–S11).
We used the trained classifiers also on JD-only JDPs, which were excluded from the training set. Most of them were identified as belonging to class C (SI Appendix, Fig. S1D) and, by and large, to none of the 12 most abundant architectural classes (SI Appendix, Fig. S1E).
Discussion
In the course of evolution, the JDP family expanded both in number and structural diversity, and in eukaryotic organisms, there is a more than two-fold increase in the number of JDPs relative to Hsp70s, indicating that the limited repertoire of Hsp70 paralogs is utilized by multiple different JDPs to generate various chaperoning activities (e.g., unfolding of misfolded protein, disassembly of functional or aggregated protein oligomers, protein translocation into organelles and target proteins for degradation). The assembly of different JDP-Hsp70 chaperone machines is initiated by J-domains. Genetic, biochemical, and structural data accumulated over the years provide a relatively comprehensive map of the J-domain-Hsp70 interface (20, 27, 55–60), which serves a single task, i.e., to establish the vital physical interaction that promotes ATP hydrolysis in Hsp70 (16, 17, 20, 27–29), thus allowing the chaperone to capture client substrates (18). Proper docking of the J-domain onto ATP-bound Hsp70 is ensured by a core set of intermolecular contacts occurring along the helix II–loop (HDP motif)–helix III region of the J-domain, which is structurally conserved from bacteria to human (20, 27). However, there is very little knowledge of the regulatory elements in the J-domains that critically fine-tune this interaction (46) despite such modulation having been hypothesized over the years. Our ANN-based observations showed the presence of phylogenetic and functional signatures buried within the amino acid sequences of J-domains, which appear to modulate the pairing of JDPs with Hsp70s to form chaperone machines with different activities.
The interpretable ANN approach we used in this work allowed the identification of the sequence positions that most contribute to the success of each classification task. The most discriminative positions structurally coincided with the J-domain-Hsp70 binding interface, strongly hinting that some amino acids have a regulatory role to optimize the selection of unique functionally specific Hsp70-JDP pairs. These in silico observations are strongly supported by indirect experimental evidence. For example, measurements of JDP-stimulated ATPase activity in Hsp70s show that J-domains are not fully interchangeable with respect to their collaboration with different Hsp70s within a given species. The yeast cytosol contains seven Hsp70 paralogs (Ssa1-4, Ssb1-2, and Ssz1). Both Ssa’s and Ssb’s bind to nascent polypeptide chains to promote de novo protein folding. However, only the ATPase activity of Ssa’s, but not of Ssb’s, is stimulated in vitro by the two major cytosolic yeast JDPs Ydj1 (class A) and Sis1 (class B) (61). This indicates that J-domains of Ydj1 and Sis1 have evolved to differentially recognize these Hsp70 family members. Similarly, the ATPase activity of ER-localized yeast Hsp70 Bip/Kar2 (HSPA5) was shown to be induced by the J-domain of Sec63 (ER; class C) 16 times higher than the J-domain of ERdj3 (ER; class C) (62) suggesting that, though both JDPs function with this Hsp70, there are differences in how the two JDPs stimulate Bip for specific activities. Recent results have also highlighted that, for example, the J-domain of human DNAJC27, involved in ciliogenesis, cannot be substituted by almost any other human J-domain (63). In addition, as expected, speciation also has left a clearly discernible signature on the J-domain sequence, which points towards some incompatibilities in pairing JDP-Hsp70 from different organisms, resulting in poor chaperoning activities. For example, bacterial class A JDP DnaJ stimulates the ATPase activity of bacterial DnaK and human HSPA8 (Hsc70) very differently (64). Similarly, mouse DNAJC7 (DjC7; class C JDP) stimulates the ATPase activity of bacterial DnaK and murine Hsp70s (HSPA8 and HSPA5) at varying levels (65) possibly leading to differences in function. Our in silico findings strongly suggest that these incompatibilities arise from discriminatory signatures present within the amino acid sequences of J-domains and open the possibility to selectively fine-tune JDP-Hsp70 interactions to support specific chaperoning activities in cells.
Taken together, the great diversity in domain architecture of the noncanonical class B JDPs (DNAJB2, DNAJB3, DNAJB6-9, DNAJB12, and DNAJB14) and the inability of our ANN analysis to unambiguously group them together with the canonical ones (DNAJB1, DNAJB4, and DNAJB5) suggests a complex evolutionary origin of these JDPs. The presence of a G/F-rich region following the J-domain, which was the rationale to define them as class B, might just suggest that these JDPs may have evolved from class A or class B ancestors, followed by some extensive sequence divergence. Further work might in the future elucidate this interesting issue. The most abundant groups of class C JDPs, which represent the largest subfamily (45), could be clearly recognized according both to their overall domain architecture and to the ANN-based sequences of their J-domains alone, indicating a strong coevolution between their J-domain sequences and their function.
From a more general perspective, the recent advent of ANN-based approaches in structural biology, epitomized by the spectacular success of AlphaFold for protein structure prediction (66, 67), highlights that the exponentially growing number of sequences available in public repositories holds a yet unexplored wealth of information that can, and must, be exploited. At the same time, this data abundance also creates a tension between the indiscriminate use of sequences from all sources and the cautious desire to use carefully curated sequences and calls for an intermediate approach, where stringent, conservative automated criteria go hand-in-hand with manual curation at higher scales (e.g., domain composition). In this respect, the present work can be considered an important example where this strategy allows for an unbiased, semiunsupervised coupled functional/phylogenetic–structural investigation: It is supervised for the classification task but unsupervised for the emergence of the relevant structural features. The successful application of our method to the SH2 family highlights the generality of the approach presented in this work.
Limitations of the Study.
We show here that the proposed automated classification is already possible, although likely limited mostly by two factors. On the one hand, several architectural C subclasses are too poorly represented in the dataset, making it difficult to train a network to tell them apart from more abundant subclasses. However, this problem is likely to be solved thanks to the exponential growth of the number of reported JDP sequences. On the other hand, a large swath of class C JDPs comprise domains that have not yet been annotated in InterPro, or just some disordered regions of various lengths. Yet, the advent of deep learning approaches on scales even larger than the present ones is recognizing domains that were previously uncharacterized (68), paving the way for a better assignment of class C JDPs to precise, well-populated architectural classes.
Methods
Construction of a Curated J-protein Dataset.
We constructed a JDP sequence database by querying the UniProt database (union of SwissProt and TrEMBL, release 2021_04) (37) to extract all sequences annotated to have a J-domain according to the InterPro domain annotation, i.e., all sequences containing InterPro domain IPRO01623) (69). For each JDP sequence, we recovered its full-length sequence as well as the full taxonomic lineage from UniProt. We then extracted the J-domain sequence of each JDP by aligning the full-length sequences to the HMM of the Pfam J-domain (PF0226) family using the HMMalign utility of the HMMer package (53, 70). The full domain architecture was obtained by querying the InterPro database for each JDP entry in our database and the domain order validated using the domain boundaries reported in InterPro. For convenience, we use the following compact domain naming convention for the discussed InterPro domains: {DnaJ: IPR001623, DnaJ_C: IPR002939, DnaJ_Cys-rich: IPR001305, TerB: IPR007791, TPR: IPR0019734, HSCB_C: IPR009073, DUF1977: IPR015399, DnaJ-X: IPR026894, Sec63: IPR004179, DnaJ-C11: IPR024586, DPH-MB: IPR007872}.
The domain architectures were used to classify all sequences as either belonging to canonical class A (architecture “DnaJ-DnaJ_C-DnaJ_Cys-rich”), canonical class B (architecture “DnaJ-DnaJ_C;”), and class C (all other sequences).
We further identified the G/F-rich region in all JDPs as being the stretch of 40 amino acids directly following the JD sequence, if at least 25% of its amino acids are either glycines or phenylalanines.
For ease of analysis, we also grouped all domains composing TPR repeat proteins (annotated as TPR_XX) into a single virtual TPR domain (as shown in Fig. 2).
Subcellular localizations were predicted for eukaryotic sequences using the TargetP software (71), version 2.0. This resulted in an initial, but not final, dataset containing 297’398 annotated JDP sequences that we further curated at a large scale. We first removed all sequences for which the UniProt and InterPro annotations were not mutually consistent and also removed all sequences which did not have a full characteristic HPD motif (sequences with HPE, or other variations of the triad, were separately analyzed). Furthermore, we also removed sequences containing multiple J-domains. Finally, all sequences associated to unclassified organisms were also removed. This resulted in a more reliably annotated final dataset comprising 279’565 JDP sequences.
Annotation of a Pseudo Zinc-Finger-Like Region (pseudoZFLR) Domain in JDPs.
We created a domain definition to annotate the pseudo zinc-finger-like domains similar to the ones observed in DnaJB11 and ErdJ3. These domains are characterized by one CNC and one CxxC motifs, in contrast to the four pairs of CxxC motifs of the canonical zinc finger motif corresponding to the canonical InterPro DnaJ_CXXCXGXG domain. We thus built a multiple sequence alignment containing 35 manually selected and curated JDPs containing a pseudoZFLR domain, from organisms including vertebrates, invertebrates, and plants. We then aligned this seed using MAFFT (v7.487) (72) and manually identified a region comprising 55 positions defining the pseudoZFLR domain. We then constructed a HMM profile of the domain using the HMMbuild utility from the HMMer suite (70) and used to profile to query all the 279’565 JDP sequences in our curated dataset to identify JDPs containing a pseudoZFLR domain. Because of the similarity between this domain and the canonical ZFLR domain, we used a more stringent E-value = 10−4 cutoff to identify pseudoZFLR domains (SI Appendix, Fig. S9). This resulted in a total of 1,810 JDPs identified as containing a pseudoZFLR domain, of which 1,746 possess a DnaJ-DnaJ_C-pseudoZFLR domain architecture.
Dimensionality Reduction of JDP Amino Acid Sequences by UMAP.
To apply the UMAP algorithm (51) to the amino acid sequences of our J-domain repertoire for visualization, we precomputed a pairwise distance metric using the Blosum62 substitution matrix to quantify amino acid similarities. The metric defining a distance between sequences a and b was defined as
where the sum runs over the 63 positions of the aligned sequences (index i), S(x,y) denotes the Blosum62 substitution matrix between amino acids x and y, and the constant 3 corresponds to the highest off-diagonal element of S and ensures positivity of the metric. Gap–amino acid pairs were assigned a −5 score, and gap-gap pairs were scored as 0.
We then applied the UMAP dimensionality reduction technique using the precomputed metric, with standard parameters as provided by the original publication.
Preparation of JDP Datasets for Training Neural Networks.
To avoid training the neural networks on sequences for which we wanted to predict and interpret the class labels, and thus avoid biases in the training phase, sequences from model organisms (H. sapiens, M. musculus, S. cerevisiae, R. norvegicus, melanogaster, D. rerio, C. elegans, and A. thaliana) were removed from the dataset prior to training. To interpret the predictions of the trained models, we also removed noncanonical class B proteins from our training set. These were identified as JDPs containing a DnaJ domain, an annotated G/F region (see above) but no DnaJ_C domain. This procedure resulted in a reduced set of 260’204 sequences used for building the classification models.
To reduce the effects of sampling bias during the training phase (i.e., to take into account that sequences that are too similar should not be considered as independent samples), we used a simple sequence reweighting scheme previously used in several sequence analysis pipelines (73, 74). We assigned to each sequence i a weight wi of 1/n80, where n80 denotes the number of sequences with sequence identity greater than 80% from it. These weights were task-specific because some tasks were trained only on subsets of sequences. Each sequence is then weighted in the final loss function minimized during training, by weighting the per-sample loss by the sample weight, i.e.
where i is an index running over all sequences in the training set, are the computed sequence weights, and si the categorical cross-entropy loss for each sample i.
We randomly split the processed dataset into training, validation, and test set in such a way that sequences from the same organism always belonged to the same set. This ensured that the performance of the ANN on the test and validation datasets was biased by organism-specific knowledge acquired during the training phase. The sum of the weights of each dataset was respectively 70%, 15%, and 15% of the total weight of the dataset.
Class A vs. B JDP Classification Using Unaligned G/F Region Sequences.
To test the discrimination power of the G/F regions of class A and B JDPs, we built an L2-regularized logistic regression model to classify JDPs as either belonging to class A or B. The unaligned G/F regions (comprising the 40 amino acids following the J-domain) where one-hot encoded and fed into a standard logistic regression model. The regularization parameter C has been chosen by scanning the validation set accuracy (SI Appendix, Fig. S10). The resulting best model (C = 3.0) achieved classification accuracy of ~95%.
Feed-Forward ANN Classification.
To feed the sequences to a neural network, we encoded them into binary vectors (one-hot encoding): Every sequence position is expanded to 21 positions, one for each amino acid species and the gap. Only the one corresponding to the residue actually present in a given sequence had value 1, while the others had value 0. Thus, each sequence resulted in a 1323-dimensional binary vector (63 × 21 entries), characterized by the presence of 63 unitary elements and 1,260 null ones (in SI Appendix, Fig. S11A, the sequence passed to the network is still represented by the residue symbols for sake of clarity).
A Dropout (75) layer was used between the input and first hidden layer to randomly mask a fraction of inputs and has been found to improve the generalization accuracy of the network.
For each task described in the main text, we trained a feed-forward neural network to predict the corresponding label of each sequence (SI Appendix, Fig. S11B). The networks were trained by minimizing the categorical cross entropy between the output layer and the categorical encoding of the label. All weights have been regularized using an L2 regularizer, and we used the Adam (76) protocol during the minimization procedure and a learning decay rate equal to 0.01. A batch size of 1,024 was used, and SoftPlus (77) activation was used for all hidden layers. The remaining hyperparameters (number of layers, units per layer, dropout rate, weight regularizations, and initial learning rate) were optimized by performing a grid search over parameter space, testing 288 combinations of hyperparameters per task. The best-performing model was selected based on the maximum accuracy on the validation set (SI Appendix, Table S13). The model performances are reported on the held-out test set.
After the training completed, we analyzed the trained networks using the technique described in ref. 54 to obtain 21 × 63 coefficients describing how the networks use every bit in the input in order to classify the sequence. The relevance scores for each residue were finally computed as the Frobenius norm (sum of the squares) of their corresponding elements in the coupling matrix (see ref. 54 for details on the relevance score calculations).
Generation of JDP Repertoire for Model Organisms.
We built the JDP repertoires for the following model organisms: H. sapiens, S. cerevisiae, M. musculus, R. norvegicus, M. mulatta, D. rerio, D. melanogaster, C. elegans, A. thaliana, and E. coli. All entries in the human, yeast, and bacterial organisms were manually curated and cross-referenced with the JDP repertoires of these organisms. For the remaining model organisms, we extracted all entries in our dataset corresponding to the target organism, removed entries marked as fragments and then reduced redundancy by only retaining entries with both unique full-length sequences and gene names. To select which UniProt entry to keep in the tables, we prioritized entries in the annotated SwissProt repository of UniProt and then selected the ones with the highest UniProt annotation score.
Phylogenetic Tree.
Maximum likelihood phylogenetic trees were generated by MEGA X (78), using the JTT distance matrix and NJ/BioNJ initial tree with 200 bootstraps replicate. Trees were made with iTOL (79).
Interresidue Contacts.
The interresidue contact network presented in Fig. 4 was computed using the Arpeggio (80) software with standard parameters. All interresidue contact types are reported.
HMM-Based Classification of Protein Sequences.
For each classification task, we first split the training set sequences into individual groups of the same category (e.g., for A/B/C, we created three sequence files containing only class A, B, or C sequences). We then built a separate HMM profile for each category using the HMMer software suite. Any sequence to be classified was then scored against all HMMs for the given task (e.g., A.HMM, B.HMM, and C.HMM for the A/B/C task), and the sequence was then assigned to the class with the lowest E-value.
Construction and Analysis of the SH2 Dataset.
The construction and analysis of the SH2 dataset followed the same steps as the JDP dataset construction. We extracted all UniProt sequences containing the annotated IPR000980 InterPro domain. We then focused on the PhyloMeta and Arch12 classification tasks. As SH2 displays a much broader architecture distribution compared to JDPs (SI Appendix, Fig. S12), we trained the classifier to distinguish the top 11 Architectures, excluding sequences containing only an SH2 annotated domain and all sequences grouped as “Other,” as in contrast to JDPs, this heterogeneous class was the most populated in the case of SH2.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
P.D.L.R. and D.M. thanks the Swiss National Science Foundation for financial support under grant number 200020_163042. D.M. thanks the Swiss National Science Foundation for financial support under grant number P2ELP3_181910. D.M. thanks ASLAC for support on this project. N.B.N. thanks National Health and Medical Research Council of Australia Investigator Grant APP1197021 and Recruitment Grant from Monash University Faculty of Medicine Nursing and Health Sciences with funding from the State Government of Victoria and the Australian Government.
Author contributions
D.M., S.Z., A.B., N.B.N., and P.D.L.R. designed research; D.M. and S.Z. performed research; D.M., S.Z., M.E.R., and P.D.L.R. analyzed data; and D.M., S.Z., M.E.R., N.B.N., and P.D.L.R. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Nadinath B. Nillegoda, Email: nadinath.nillegoda@monash.edu.
Paolo De Los Rios, Email: paolo.delosrios@epfl.ch.
Data, Materials, and Software Availability
The codebase required to generate the dataset and perform the ANN classification analysis is freely available at https://gitlab.com/LBS-EPFL/papers/jdp_phyloann_2023 (81).
Supporting Information
References
- 1.Mandal A. K., Nillegoda N. B., Chen J. A., Caplan A. J., Ydj1 protects nascent protein kinases from degradation and controls the rate of their maturation. Mol. Cell Biol. 28, 4434–4444 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Behnke J., Mann M. J., Scruggs F. L., Feige M. J., Hendershot L. M., Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol. Cell 63, 739–752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nillegoda N. B., et al. , Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 524, 247–251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wentink A. S., et al. , Molecular dissection of amyloid disaggregation by human HSP70. Nature 587, 483–488 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Hageman J., et al. , A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–69 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Kumar J., Kline N. L., Masison D. C., Human DnaJB6 antiamyloid chaperone protects yeast from polyglutamine toxicity separately from spatial segregation of aggregates. Mol. Cell Biol. 38, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha D., Srivastava S., D’Silva P., Functional diversity of human mitochondrial J-proteins is independent of their association with the inner membrane presequence translocase. J. Biol. Chem. 291, 17345–17359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassdenteufel S., et al. , Chaperone-mediated Sec61 channel gating during ER import of small precursor proteins overcomes Sec61 inhibitor-reinforced energy barrier. Cell Rep. 23, 1373–1386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li K., et al. , Tetrameric assembly of K+ channels requires ER-located chaperone proteins. Mol. Cell 65, 52–65 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Fotin A., et al. , Structure of an auxilin-bound clathrin coat and its implications for the mechanism of uncoating. Nature 432, 649–653 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Walsh P., Bursać D., Law Y. C., Cyr D., Lithgow T., The J-protein family: Modulating protein assembly, disassembly and translocation. EMBO Rep. 5, 567–571 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez F., et al. , Molecular basis for regulation of the heat shock transcription factor σ32 by the DnaK and DnaJ chaperones. Mol. Cell 32, 347–358 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Westhoff B., Chapple J. P., Van Der Spuy J., Höhfeld J., Cheetham M. E., HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr. Biol. 15, 1058–1064 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Grove D. E., Fan C. Y., Ren H. Y., Cyr D. M., The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRΔF508. Mol. Biol. Cell 22, 301–314 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto Y. H., et al. , ERdj8 governs the size of autophagosomes during the formation process. J. Cell Biol. 219, e201903127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suh W. C., Lu C. Z., Gross C. A., Structural features required for the interaction of the Hsp70 molecular chaperone DnaK with its cochaperone DnaJ. J. Biol. Chem. 274, 30534–30539 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Jiang J., et al. , Structural basis of J cochaperone binding and regulation of Hsp70. Mol. Cell 28, 422–33 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Los Rios P., Barducci A., Hsp70 chaperones are non-equilibrium machines that achieve ultra-affinity by energy consumption. Elife 3, e02218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bracher A., Verghese J., The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kityk R., Kopp J., Mayer M. P., Molecular mechanism of J-domain-triggered ATP hydrolysis by Hsp70 chaperones. Mol. Cell 69, 227–237.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Schilke Brenda B. A., et al. , Broadening the functionality of a J-protein/Hsp70 molecular chaperone system. PLoS Genet. 13, 1–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J., Qian X., Sha B., The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure 11, 1475–1483 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Wu Y., Li J., Jin Z., Fu Z., Sha B., The crystal structure of the C-terminal fragment of yeast Hsp40 Ydj1 reveals novel dimerization motif for Hsp40. J. Mol. Biol. 346, 1005–1011 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Sha B., Lee S., Cyr D. M., The crystal structure of the peptide-binding fragment from the yeast Hsp40 protein Sis1. Structure 8, 799–807 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Cupp-Vickery J. R., Vickery L. E., Crystal Structure of Hsc20, a J-type Co-chaperone from Escherichia coli. J. Mol. Biol. 304, 835–845 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Maegawa K., et al. , The highly dynamic nature of ERdj5 is key to efficient elimination of aberrant protein oligomers through ER-associated degradation. Structure 25, 846–857.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Malinverni D., Jost Lopez A., De Los Rios P., Hummer G., Barducci A., Modeling Hsp70/Hsp40 interaction by multi-scale molecular simulations and co-evolutionary sequence analysis. Elife 6, e23471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomiczek B., et al. , Two-step mechanism of j-domain action in driving hsp70 function. PLoS Comput. Biol. 16, 1–29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai J., Douglas M. G., A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding§. J. Biol. Chem. 271, 9347–9354 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Kampinga H., Craig E., The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Craig E. A., Marszalek J., How do J-proteins get Hsp70 to do so many different things? Trends Biochem. Sci. 42, 355–368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajan V. B. V., D’Silva P., Arabidopsis thaliana J-class heat shock proteins: Cellular stress sensors. Funct. Integr. Genomics 9, 433–446 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Cheetham M. E., Caplan A. J., Structure, function and evolution of DnaJ: Conservation and adaptation of chaperone function. Cell Stress Chaperones 3, 28–36 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang R., Malinverni D., Cyr D. M., Rios P. D. L., Nillegoda N. B., J-domain protein chaperone circuits in proteostasis and disease. Trends Cell Biol. 3, 30–47 (2023), 10.1016/j.tcb.2022.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Y., Rossi P., Kalodimos C. G., Structural basis for client recognition and activity of Hsp40 chaperones. Science 365, 1313–1319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baaklini I., et al. , The DNAJA2 substrate release mechanism is essential for chaperone-mediated folding. J. Biol. Chem. 287, 41939–41954 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bateman A., et al. , UniProt: The universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paysan-Lafosse T., et al. , InterPro in 2022. Nucleic Acids Res. 51, D418–D427 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varadi M., et al. , AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryder B. D., et al. , Regulatory inter-domain interactions influence Hsp70 recruitment to the DnaJB8 chaperone. Nat. Commun. 12, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faust O., et al. , HSP40 proteins use class-specific regulation to drive HSP70 functional diversity. Nature 587, 489–494 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Yu H. Y., Ziegelhoffer T., Craig E. A., Functionality of Class A and Class B J-protein co-chaperones with Hsp70. FEBS Lett. 589, 2825–2830 (2015), 10.1016/j.febslet.2015.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karamanos T. K., Tugarinov V., Clore G. M., Unraveling the structure and dynamics of the human DNAJB6b chaperone by NMR reveals insights into Hsp40-mediated proteostasis. Proc. Natl. Acad. Sci. U.S.A. 116, 21529–21538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.J. G, et al. , Hsp40 proteins phase separate to chaperone the assembly and maintenance of membraneless organelles. Proc. Natl. Acad. Sci. U.S.A. 117, 31123–31133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barriot R., Latour J., Castanié-Cornet M. P., Fichant G., Genevaux P., J-domain proteins in bacteria and their viruses. J. Mol. Biol. 432, 3771–3789 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Hennessy F., Nicoll W. S., Zimmermann R., Cheetham M. E., Blatch G. L., Not all J domains are created equal: Implications for the specificity of Hsp40-Hsp70 interactions. Protein Sci. 14, 1697–709 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu X.-B., Shao Y.-M., Miao S., Wang L., The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 63, 2560–2570 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kluck C. J., et al. , Structure-function analysis of HscC, the Escherichia coli member of a novel subfamily of specialized Hsp70 chaperones. J. Biol. Chem. 277, 41060–41069 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Schlenstedt G., Harris S., Risse B., Lill R., Silver P. A., A yeast DnaJ homologue, Scj1p, can function in the endoplasmic reticulum with BiP/Kar2p via a conserved domain that specifies interactions with Hsp70s. J. Cell Biol. 129, 979–988 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sullivan C. S., Cantalupo P., Pipas J. M., The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol. Cell Biol. 20, 6233–6243 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McInnes L., Healy J., Melville J., UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv [Preprint] (2018). https://arxiv.org/abs/1802.03426 (Accessed 30 September 2020).
- 52.Hou Z., et al. , DnaJC7 binds natively folded structural elements in tau to inhibit amyloid formation. Nat. Commun. 12, 5338 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mistry J., Finn R. D., Eddy S. R., Bateman A., Punta M., Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zamuner S., De Los Rios P., Interpretable neural networks based classifiers for categorical inputs. arXiv [Preprint] (2021). https://arxiv.org/abs/2102.03202 (Accessed 5 February 2021).
- 55.Gässler C. S., et al. , Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Biochemistry 95, 15229–15234 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greene M. K., Maskos K., Landry S. J., Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc. Natl. Acad. Sci. U.S.A. 95, 6108–6113 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suh W. C., et al. , Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc. Natl. Acad. Sci. U.S.A. 95, 15223–8 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wittung-Stafshede P., Guidry J., Horne B. E., Landry S. J., The J-domain of Hsp40 couples ATP hydrolysis to substrate capture in Hsp70. Biochemistry 42, 4937–4944 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Landry S. J., Structure and energetics of an allele-specific genetic interaction between dnaJ and dnaK: Correlation of nuclear magnetic resonance chemical shift perturbations in the J-domain of Hsp40/DnaJ with binding affinity for the ATPase domain of Hsp70/DnaK. Biochemistry 42, 4926–4936 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Barducci A., De Los Rios P., Non-equilibrium conformational dynamics in the function of molecular chaperones. Curr. Opin. Struct. Biol. 30, 161–169 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Lopez-Buesa P., Pfund C., Craig E. A., The biochemical properties of the ATPase activity of a 70-kDa heat shock protein (Hsp70) are governed by the C-terminal domains. Proc. Natl. Acad. Sci. U.S.A. 95, 15253–15258 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vembar S. S., Jin Y., Brodsky J. L., Hendershot L. M., The mammalian Hsp40 ERdj3 requires its Hsp70 interaction and substrate-binding properties to complement various yeast Hsp40-dependent functions. J. Biol. Chem. 284, 32462–32471 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Piette B. L., et al. , Comprehensive interactome profiling of the human Hsp70 network highlights functional differentiation of J domains. Mol. Cell 81, 2549–2565.e8 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Minami Y., Höhfeld J., Ohtsuka K., Hartl F. U., Regulation of the heat-shock protein 70 reaction cycle by the mammalian DnaJ homolog, Hsp40. J. Biol. Chem. 271, 19617–19624 (1996). [DOI] [PubMed] [Google Scholar]
- 65.Kroczynska B., Blond S. Y., Cloning and characterization of a new soluble murine J-domain protein that stimulates BiP, Hsc70 and DnaK ATPase activity with different efficiencies. Gene 273, 267–274 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021), 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tunyasuvunakool K., et al. , Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bileschi M. L., et al. , Using deep learning to annotate the protein universe. Nat. Biotechnol. 40, 932–937 (2022), 10.1038/s41587-021-01179-w. [DOI] [PubMed] [Google Scholar]
- 69.Paysan-Lafosse T., et al. , InterPro in 2022. Nucleic Acids Res. 51, D418–D427 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finn R. D., Clements J., Eddy S. R., HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, 29–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armenteros J. J. A., et al. , Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katoh K., Misawa K., Kuma K., Miyata T., MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morcos F., Hwa T., Onuchic J. N., Weigt M., “Direct coupling analysis for protein contact prediction” in Protein Structure Prediction, Kihara D. Ed. (Springer, New York, 2014), pp. 55–70. 10.1007/978-1-4939-0366-5_5. [DOI] [PubMed] [Google Scholar]
- 74.Ekeberg M., Lövkvist C., Lan Y., Weigt M., Aurell E., Improved contact prediction in proteins: Using pseudolikelihoods to infer Potts models. Phys. Rev. E 87, 1–16 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Srivastava N., Hinton G., Krizhevsky A., Salakhutdinov R., Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 15, 1929–1958 (2014). [Google Scholar]
- 76.Kingma D. P., Ba J., “Adam: A method for stochastic optimization” in Proceedings of the International Conference on Learning Representations, arXiv:1412.6980 (2015). [Google Scholar]
- 77.Zheng H., Yang Z., Liu W., Liang J., Li Y., “Improving deep neural networks using softplus units” in 2015 International Joint Conference on Neural Networks (IJCNN) (Killarney, 2015), pp. 1–4. 10.1109/IJCNN.2015.7280459. [DOI] [Google Scholar]
- 78.Kumar S., Stecher G., Li M., Knyaz C., Tamura K., MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Letunic I., Bork P., Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jubb H. C., et al. , Arpeggio: A web server for calculating and visualising interatomic interactions in protein structures. J. Mol. Biol. 429, 365–371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Malinverni D., et al. , Codebase for manuscript “Data-driven large-scale genomic analysis reveals an intricate phylogenetic and functional landscape in J-domain proteins”. Gitlab repository. https://gitlab.com/LBS-EPFL/papers/jdp_phyloann_2023. Accessed 13 July 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
The codebase required to generate the dataset and perform the ANN classification analysis is freely available at https://gitlab.com/LBS-EPFL/papers/jdp_phyloann_2023 (81).