

SUMMARY
To improve the understanding of chemo-refractory high-grade serous ovarian cancers (HGSOCs), we characterized the proteogenomic landscape of 242 (refractory and sensitive) HGSOCs, representing one discovery and two validation cohorts across two biospecimen types (formalin-fixed paraffin-embedded and frozen). We identified a 64-protein signature that predicts with high specificity a subset of HGSOCs refractory to initial platinum-based therapy and is validated in two independent patient cohorts. We detected significant association between lack of Ch17 loss of heterozygosity (LOH) and chemo-refractoriness. Based on pathway protein expression, we identified 5 clusters of HGSOC, which validated across two independent patient cohorts and patient-derived xenograft (PDX) models. These clusters may represent different mechanisms of refractoriness and implicate putative therapeutic vulnerabilities.
In brief
Patients with high-grade serous ovarian cancers (HGSOCs) have a poor outcome, with the standard of care not having changed over the decades. A detailed characterization of the proteogenomic landscape of HGSOCs across multiple cohorts and validation studies identifies a distinct signature that predicts with high specificity a subset of patients with chemotherapy-refractory cancers and implicates potential therapeutic vulnerabilities.
Graphical Abstract
INTRODUCTION
Epithelial ovarian cancer accounts for >185,000 deaths/year worldwide.1 The most common subtype, high-grade serous ovarian cancer (HGSOC), accounts for 60% of deaths. Despite improvements in surgical and chemotherapeutic approaches, HGSOC mortality has not changed in decades.2 The 5-year survival rate remains ~30% for the majority of patients.1,2
Standard of care involves surgical debulking combined with adjuvant or neoadjuvant chemotherapy with carbo- or cisplatin in combination with a taxane.1,3 At diagnosis, HGSOC is among the most chemo-sensitive of all epithelial malignancies, with initial response rates of ~85%, presumably related to DNA repair defects.4 Platinum is thought primarily to drive the response rate, due to the lower single-agent response rate for taxanes.5
10%–20% of HGSOC patients have treatment-refractory disease at diagnosis, fail to respond to initial therapy, and have a dismal prognosis.6 The poor response to subsequent therapy and median overall survival of ~12 months for these patients has not changed in 40 years.7,8 Even for the ~85% of tumors that respond to initial chemotherapy, most relapse as resistant disease,9 defined as recurrence within 6 months of completing platinum-based chemotherapy.10 This chemo-resistant phenotype, which can occur following a single or multiple rounds of therapy, is also associated with poor responses to subsequent therapies11 and limited survival. Patients whose ovarian cancer has responded with a partial or complete remission are often treated with PARP inhibitors as maintenance therapy, with the greatest benefit in relapse-free survival observed in cancers with BRCA1 or BRCA2 mutations.12 These regimens are supported by level 1 evidence from randomized phase III trials13–16 but unfortunately do not apply to patients with platinum-refractory HGSOC.
Despite >30 years of literature studying platinum resistance in cancer,17 there currently is no way to distinguish refractory from sensitive HGSOCs prior to therapy. Consequently, patients with refractory disease experience the toxicity of platinum-based chemotherapy without benefit. Due to their rapid progression, they are commonly excluded from participating in clinical trials. Accordingly, there is no ongoing clinical research that could identify effective therapeutic agents for these patients or provide insights into molecular mechanisms of refractory disease.
To address this unmet clinical need, we performed proteogenomic analysis of treatment-naive HGSOCs (chemo-sensitive and chemo-refractory) and identified a 64-protein signature that predicts a subset of chemo-refractory HGSOCs with high specificity and is validated in two independent patient cohorts. Additionally, five molecular subtypes are identified, and implicate possible therapeutic vulnerabilities.
RESULTS
Study cohort and proteogenomic profiling
We performed proteogenomic analyses on pre-treatment biopsies from three HGSOC patient cohorts: FFPE (formalin-fixed paraffin-embedded) discovery (n = 158), FFPE validation (n = 20), and frozen validation (n = 64). Biopsies were collected at the time of primary debulking surgery (prior to chemotherapy) and were reasonably balanced between chemo-refractory and sensitive tumors (Figure 1A). Refractory cancers were defined as those that progressed or had stable disease within 6 cycles of initial platinum/taxane therapy after initial debulking surgery (STAR Methods). Sensitive tumors were defined as those that responded to initial platinum/taxane therapy and did not progress within 2 years (STAR Methods). Demographic and clinical annotations are provided in Table S1. Biopsies were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a multiplexed tandem-mass-tag (TMT) isobaric labeling approach coupled with randomization to avoid bias (STAR Methods). Details regarding biospecimens and data quality assessments are provided (Figures S1B–S1J; STAR Methods). Proteogenomic data can be queried, visualized, and downloaded from http://ptrc.cptac-data-view.org/.
Figure 1. Overview of multi-omics datasets.
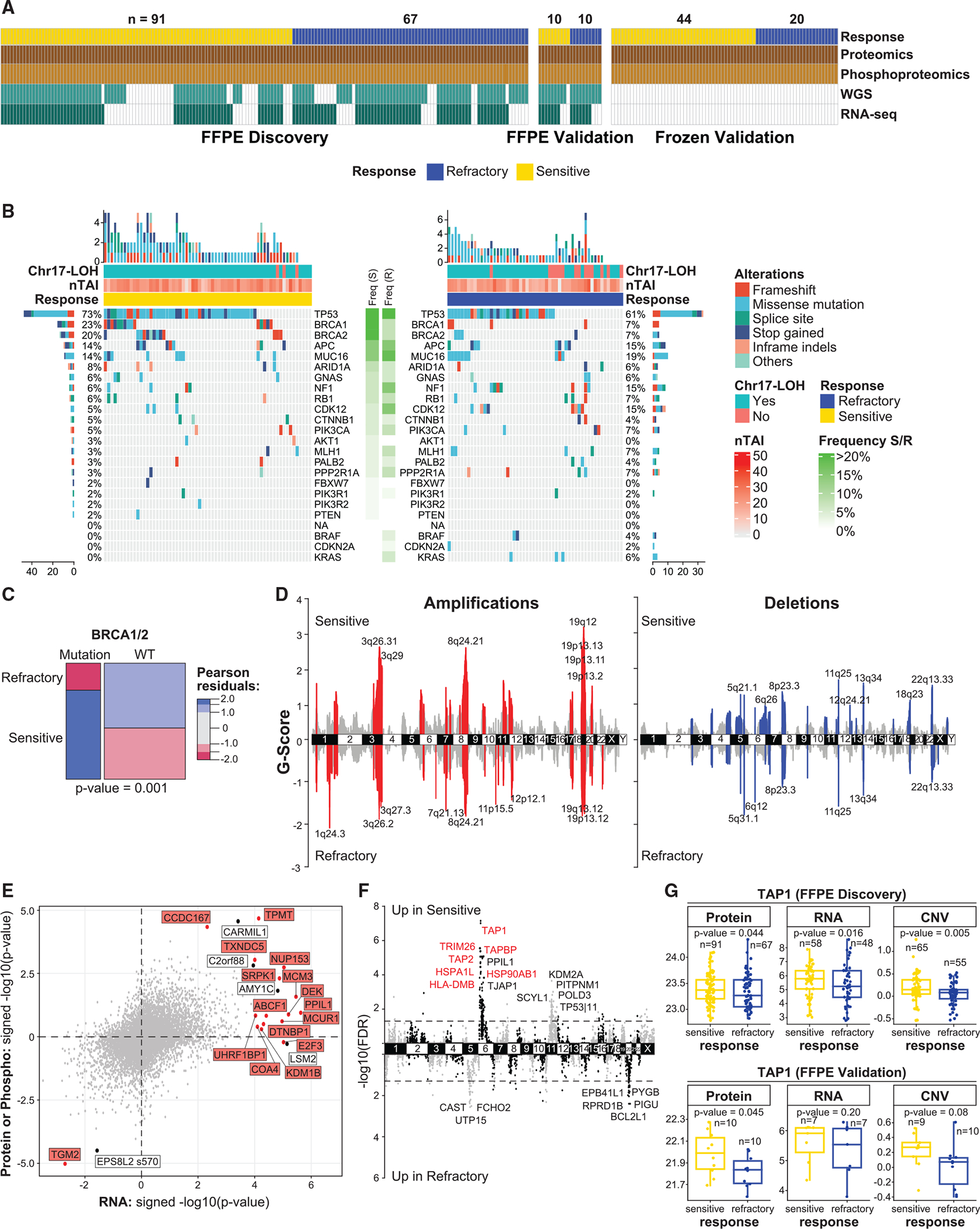
(A) Availability of the proteomic, phospho-proteomic, WGS, and RNA-seq data across the cohorts.
(B) OncoPrint of likely pathogenic genetic variants in sensitive and refractory tumors. Genes are ordered by mutation frequency in sensitive tumors. Chr17-LOH binary variable indicates whether more than 25% of Chr17 has LOH. nTAI denotes the number of chromosome arms with telomeric allelic imbalance.
(C) Association between mutations in BRCA1/2 and tumor response. The p value is based on the chi-square test.
(D) Genomic regions with recurrent focal amplifications (left) and deletions (right) in sensitive (top) and refractory (bottom) tumors. The G score is proportional to −log(probability|background). Cytobands of the most significant peaks are annotated.
(E) Concordance of genes associated with refractoriness across global proteomic (y axis), phospho-proteomic (y axis), and RNA-seq data (x axis) (FFPE discovery cohort). Labeled genes/proteins/phosphosites are significantly associated with refractoriness (FDR < 0.1). Genes/proteins highlighted in red also showed association with refractoriness based on CNV data.
(F) Association between genes and refractoriness from the integrated CNV, RNA, and protein analysis (FFPE discovery cohort). Highlighted genes survivecombined FDR < 0.1 (FFPE discovery cohort) and are validated (marginal p value < 0.05) using the proteomic data from R1 validation cohort.
(G) Protein, RNA, and copy-number levels for TAP1 in the FFPE discovery and validation cohorts. p values are determined by covariate-adjusted regressions(Table S1).
See also Figure S1.
The FFPE discovery cohort consisted of 158 biopsies (91 sensitive and 67 refractory) (Figure 1A) sourced from 3 academic centers and representing a wide range of storage times (Figure S1A). 11,080 proteins and 11,817 phosphosites were quantified by LC-MS/MS. 8,800 proteins and 2,648 phosphosites were observed in >50% of the biospecimens in either sensitive or refractory groups and were included in downstream analyses (STAR Methods, processed data). Whole-genome sequencing (WGS) and RNA-seq were performed on the FFPE discovery cohort samples. Using mutation and copy-number (CN) variation (CNV) information derived from the WGS data (STAR Methods), we delineated sets of likely somatic or germline pathogenic variants (“mutations”) and CN aberrations. After imposing strict quality control (QC) (STAR Methods), genomic data were available for mutations (n = 120 tumors), CNV (n = 120 tumors), loss of heterozygosity (LOH) (n = 118 tumors), and RNA-seq-based gene expression (n = 106 tumors) (Figure 1A).
The FFPE validation cohort was an independent set of 10 sensitive and 10 refractory HGSOCs from a 4th academic center. TMT-based LC-MS/MS analysis of the FFPE validation cohort tumors identified 8,237 proteins and 3,080 phosphosites that were observed in >50% of the biospecimens (in either sensitive or refractory groups) and were included in downstream analyses (STAR Methods, processed data). Additionally, after QC (STAR Methods), somatic mutation data and DNA CN alteration data were derived for 19 of the 20 samples, while RNA-seq-based gene expression data passed QC filters for 14 samples (Figure1A).
The frozen validation cohort included 64 HGSOC tumors (44 sensitive, 20 refractory) (Figure 1A), of which 29 overlapped with the FFPE discovery cohort to assess replicability between biospecimen types (i.e., paired frozen and FFPE biospecimens from the same patient). TMT-based LC-MS/MS analyses in this cohort identified 8,272 proteins and 17,448 phosphosites that were observed in >50% of the biospecimens in either sensitive or refractory groups (STAR Methods, processed data) and were included in the downstream analyses.
In addition to the validation cohorts generated in this study, two external, independent cohorts were used for validating findings: (1) Memorial Sloan Kettering (MSK)-IMPACT18 and (2) the National Cancer Institute’s Clinical Proteomics Tumor Analysis Consortium’s study (“CPTAC-2016”).19
Genomic alterations associated with chemorefractoriness
Consistent with prior reports,20 we found few recurrently mutated genes (FFPE discovery cohort) (Figure 1B; Table S1). Tumors harbored 1–7 (median = 2) mutations in known tumor suppressor genes, with mutations in DNA damage response genes TP53 (67.5%), BRCA1 (15.8%), BRCA2 (15%), and CDK12 (10%) being among the most common (Table S1). As expected, mutations in BRCA1 or BRCA2 were more frequent in sensitive (S) vs. refractory (R) tumors (p = 0.001) (Figure 1C). No other significant associations between simple mutations (including TP53) and response were observed.
We observed a lower frequency of TP53 mutations compared with previous studies,20,21 likely because we used an allele frequency cutoff of 11% for calling mutations, which was considerably higher than that used in The Cancer Genome Atlas (TCGA),20 resulting in a lower mutation call. We chose this cutoff to focus primarily on clonal mutation (driver) events likely to contribute to tumor biology and clinical behavior.
Because HGSOC is characterized by extensive genomic instability exemplified by segmental and chromosome-level alterations, or even whole-genome duplication,22,23 we performed absolute CN analysis (STAR Methods). Both sensitive and refractory tumors were remarkably unstable, with similar levels of aneuploidy and polyploidy, as well as broadly similar frequencies and patterns of gains and losses (Figure S1K). Further comparison of local genomic alterations revealed similar patterns of focal chromosomal gains and losses (Figure 1D).
Transcripts, proteins, and CNVs associated with chemorefractoriness
We screened for individual RNAs/proteins/phosphosites whose abundances were associated with treatment response using linear regression models adjusted for anatomical tumor location, neoadjuvant status, age, institutional sample source site, and tumor purity (STAR Methods). 22 RNAs, 4 proteins, and 1 phosphosite showed association (false discovery rate [FDR] ≤ 0.1) with treatment response (FFPE discovery cohort) (Figure 1E; Table S1). The observed fold differences for significant proteins and phosphosites were small (0.77–1.29), while fold differences for significant RNAs ranged from 0.20 to 10.18. Among the four proteins associated with treatment response, transglutaminase 2 (TGM2) was more abundant in refractory vs. sensitive tumors, while CARMIL1, CCDC167, and TPMT were more abundant in sensitive tumors. Examination of the validation cohorts indicated that only CCDC167 was significantly elevated (marginal p value < 0.05) in sensitive tumors. Moreover, no association between the four proteins and survival was detected using the dataset from the CPTAC-2016 study19 (STAR Methods). The weak association between “individual” RNA/protein species and treatment response could be due to the high heterogeneity among HGSOCs and multitude of mechanisms underlying platinum responses.17
Examining the CNV data, we detected 938 genes associated with treatment response (FDR < 0.1; Table S1). As CNV profiles have strong spatial correlation, many of these genes are likely passengers sitting close to genes affecting treatment response, rather than direct contributors. In fact, of 938 genes, 361 genes were from nearby regions on chromosome 6 (Chr6), 355 genes from chromosome 11 (Chr11), 158 genes from chromosome 5 (Chr5), and 46 genes from chromosome 20 (Chr20). It is reasonable to assume that only a small subset of transcripts/proteins in these big regions might be the real “drivers” of response. To point to potential treatment-related genes over passengers, we performed an integrative analysis, as described below.
Integrative analysis identifies chemotherapy-response-associated genes
We performed an integrative analysis combining CNV, RNA, and global protein data (FFPE discovery cohort) and validated our findings using independent cohorts (STAR Methods). 424 genes with a combined FDR < 0.1 were identified, 303 of which had consistently higher CNV/RNA/protein measurements in sensitive HGSOCs and 121 of which were higher in refractory tumors. Of these 424 genes, 53 were validated (marginal p value < 0.05) in at least one independent validation cohort from this study or the CPTAC-2016 cohort19 (Table S1). Interestingly, a small region on Chr6 (Figure 1F) contains a group of 7 genes (TRIM26, HLA-DMB, TAP1, TAP2, TAPBP, HSP90AB1, and HSPA1L) that were up in sensitive tumors and are members of immune pathways (e.g., antigen presentation/processing, interferon) (Figure 1F). For example, our analysis of both FFPE discovery and FFPE validation cohorts revealed that chemo-sensitive HGSOCs had elevated TAP1 protein, RNA, and CN compared with refractory tumors (Figure 1G). TAP1 is a transporter associated with antigen processing.24 We also identified higher (in refractory vs. sensitive) CN, RNA, and protein abundance of BCL2L1/BCLXL, an antiapoptotic protein associated with treatment resistance25,26 (Figure S1L).
cis-regulation between CNV, RNA, and protein differs between sensitive and refractory tumors
The average gene-level CNV-RNA and RNA-protein correlations in sensitive tumors were significantly higher than in refractory tumors (Figure 2A). When applying iProFun27 (STAR Methods) to screen for genes whose CNVs were significantly associated with their own RNA and protein levels (“CNV-RNA/protein cascade genes”), we observed a higher percentage of cascade genes in sensitive vs. refractory tumors: of the 8,605 genes with both RNA and protein abundances measured, 55% and 42% in sensitive and refractory tumors, respectively, were detected as CNV-RNA-protein cascade genes (p < 2.2e–16, Figure 2B; Table S2). The higher percentage of cascade genes in sensitive vs. refractory tumors was validated in the CPTAC-2016 cohort19: 49% and 41% (of the 4,795 eligible genes) in proxy-sensitive and proxy-refractory tumors (STAR Methods), respectively, had CNV-RNA/protein cascade effects (p < 2.2e–16; Figure S2A; Table S2). For both cohorts, metabolism pathways (e.g., tricarboxylic acid (TCA) cycle, fatty acid metabolism, and heme metabolism) have a higher percentage of cascade genes, while ribosome and translation pathways have lower percentage of cascade genes relative to the genome average.
Figure 2. Association between treatment response and CNV-RNA/protein cis-regulations, chromosome-arm-level alterations, and TP53 signatures.
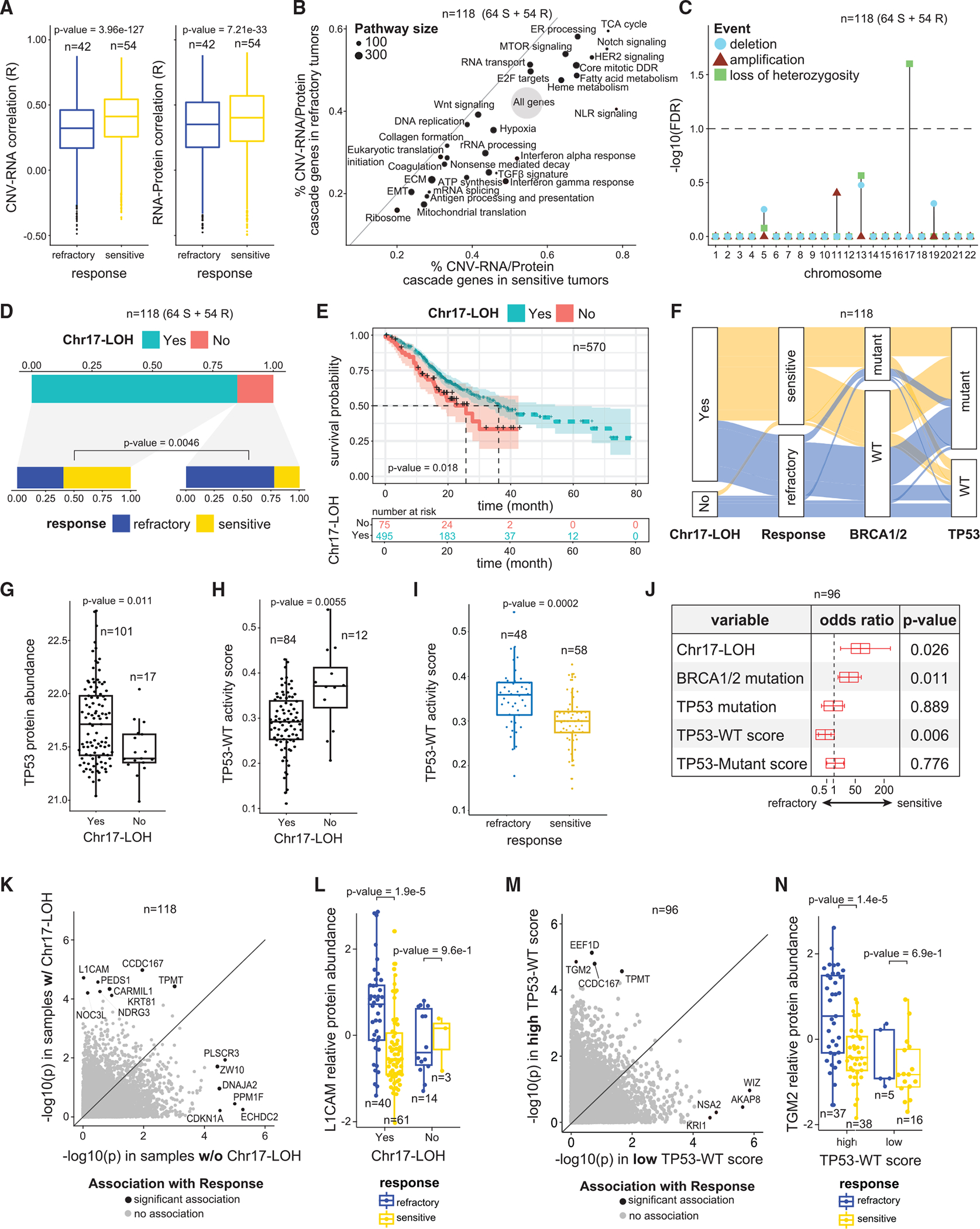
(A) Genome-wide distributions of correlation coefficients between DNA copy-number variations and RNA levels (left), as well as RNA and protein abundances(right) of the same genes. p values are based on Wilcoxon Rank Sum test.
(B) Scatter plot showing the proportion of CNV-RNA/protein cascade genes in each pathway in sensitive (x axis) vs. refractory (y axis) tumors. Pathway sizeindicates the number of genes in the pathway.
(C) Associations between refractoriness and chromosome losses, gains, and LOH. At the chromosome level, only Chr17-LOH is associated with refractoriness (FDR < 0.1).
(D) Association between Chr17-LOH and refractoriness. Among the 15% of samples without Ch17-LOH, more than 75% are refractory, whereas most sampleswith Chr17-LOH are sensitive (p value = 0.0046, Fisher’s Exact test).
(E) MSK-IMPACT cohort18 HGSOC samples stratified by Chr17-LOH status. The group enriched with Chr17-LOH samples shows a better survival than the group without Chr17-LOH (p = 0.018, based on Cox regression model).
(F) Distributions of genomic features and treatment response.
(G and H) Samples with Chr17-LOH have significantly higher p53 protein abundance (p = 0.011, Wilcoxon Rank Sum test) and significantly lower TP53-WT activity scores (p = 0.006, Wilcoxon Rank Sum test), vs. samples without Chr17-LOH.
(I) Refractory samples have higher TP53-WT activity scores (p = 0.0002, Wilcoxon Rank Sum test), vs. sensitive tumors.
(J) Results of the multivariate logistic regression model with refractory status as the response variable and genomic features as predictor variables.
(K) Associations (p values) between protein abundances and treatment response among tumors with (y axis) vs. without (x axis) Chr17-LOH.
(L) L1CAM protein abundances stratified by Chr17-LOH status and treatment response. p values are determined by covariate-adjusted regressions.
(M) Associations (p values) between protein abundances and refractoriness among tumors with high (y axis) vs. low (x axis) TP53-WT activity scores.
(N) TGM2 protein abundances stratified by TP53-WT activity scores and treatment responses. p values are determined by covariate-adjusted regressions.
See also Figure S2.
Chr17-LOH is associated with chemo-sensitivity
Consistent with previous studies,28 we noted significant LOH for most chromosomes (Figure S2B), with chromosome 17 LOH (Chr17-LOH) being the most common (~80% of cases). Among all LOH events, only Chr17-LOH was significantly (FDR < 0.1) associated with chemo-sensitivity (Figures 2C and 2D). No other chromosome-level gain, loss, or LOH event showed association with treatment response (Figure 2C). 75% of tumors heterozygous for Chr17 (i.e., without Chr17-LOH) were refractory, compared with only ~40% of those with Chr17-LOH (Figure 2D). Consistent with these results, we observed in an independent cohort, MSK-IMPACT,18 that tumors enriched for Chr17-LOH have significantly higher overall survival (Figure 2E; STAR Methods), as expected for chemo-responsive disease.
TP53 inactivation is associated with chemo-sensitivity
Chr17 harbors multiple tumor suppressor genes, including TP53. Bi-allelic inactivation of TP53 in cancers typically occurs through missense mutations accompanied by LOH.29 Indeed, we find that mutations in TP53 are significantly associated with Chr17-LOH (odds ratio [OR]: 14.6, p < 6.05e–06) (Figure 2F). We observed significantly higher TP53 RNA expression (p = 0.002) and p53 protein abundance (p = 0.001) in tumors with TP53 missense mutations vs. wild-type (WT) (consistent with the known stabilization of p53 by missense mutations30), and lower TP53 RNA expression (p = 0.024) and p53 protein abundance (p = 0.004) in tumors with TP53-truncating mutations (Figures S2D and S2E). Additionally, we observed elevated p53 levels (p = 0.011) among cases with Chr17-LOH (Figure 2G), in line with the enrichment of TP53 mutations in tumors with Chr17-LOH.
We hypothesized that the association between Chr17-LOH and sensitivity (Figures 2C and 2D) is due to bi-allelic inactivation of TP53. We interrogated treatment responses stratified by genetic aberrations (i.e., TP53 mutations and Chr17-LOH) and by transcriptional signatures of both “WT” and “mutant” p53. Missense mutations abrogate WT p53 transcriptional activity31 and are associated with a mutant transcriptional signature.29 A WT p53 signature based on validated, direct, transcriptional targets of WT p53 has also been reported.32
As predicted, we found that Chr17-LOH was associated with a significantly lower WT p53 signature (Figure 2H) and a significantly higher mutant p53 signature (Figure S2G). The WT p53 signature tended to be higher in tumors without TP53 mutations, although the difference was not significant (Figure S2F). Moreover, tumors without Chr17-LOH have significantly higher expression of MDM2, a gene activated by WT p53, and significantly lower expression of CDC20, a gene repressed by WT p53 (Figures S2H and S2I). These results suggest that tumors without Chr17-LOH retain some p53 activity.
Next, we asked whether WT or mutant p53 transcriptional signatures are associated with chemo-responsiveness. WT p53 activity was higher in refractory tumors (Figure 2I), whereas the mutant p53 signature was higher in sensitive tumors (Figure S2J). Tumors with both high WT TP53 activity and low mutant TP53 activity were predominately refractory (Figure S2K). Figure S2L illustrates selected pathways associated with Chr17-LOH. E2F targets are upregulated in tumors with Chr17-LOH (FDR = 1.47e–20), consistent with their lower WT p53 activity (Figure 2H), since WT p53 (acting through p21/CDKN1A and RB1) represses E2F.33 In addition, Chr17-LOH tumors show increased mTOR pathway expression (FDR = 3.1–21), consistent with repression of the mTOR pathway by WT P5334 (Table S2).
Using a multivariate regression model, we found that Chr17-LOH (OR: 15.76, p < 0.026), BRCA1/2 mutation (OR: 4.75, p < 0.011), and loss of WT TP53 activity (OR: 0.4, p < 0.006) remain independent predictors of response (Figure 2J). These results suggest that WT p53 activity is associated with refractoriness and that tumors with Chr17-LOH lack WT p53 activity, likely due to bi-allelic inactivation of TP53. Conversely, tumors with monoallelic, perhaps dominant-negative TP53 mutations, may retain some p53 WT activity.30
Response-associated proteins stratified by Chr17-LOH, BRCA1/2, and TP53 mutations
We hypothesized that additional biomarkers will be identified when tumors are stratified by Chr17-LOH, BRCA1/2, and TP53 mutations (Table S2). Using multivariate regression models with tumor mutation burden (TMB), patient age, and tumor purity as covariates (STAR Methods), we found that when cases were stratified by Chr17-LOH (Figure 2K), the abundance of L1CAM protein, a key driver of tumor cell invasion and motility35 that is strongly associated with TP53 mutations36 and poor prognosis,37 was positively associated with refractoriness (p = 1.9–5) in tumors with Chr17-LOH but not among cases heterozygous for Chr17 (p = 0.96) (Figure 2L). Conversely, expression of CDKN1A, a protein induced by WT p53,38 was associated with refractoriness in tumors without Chr17-LOH (Figure 2K). Because WT p53 activity was associated with refractoriness, we also dichotomized tumors by p53 activity scores (Figure 2M) and found that the TGM2 protein, a known TP53 target,39 was associated with refractoriness among high p53 activity tumors (Figure 2N).
Genomic scars associated with chemo-response
HGSOC genomes are characterized by “genomic scars” (i.e., summary scores reflecting patterns of genomic instability).40 In the clinical setting, the extent of number of telomeric allelic imbalances (nTAIs), weighted genomic integrity index (wGII), number of large-scale transitions (nLSTs), and number of LOH (nLOH) are used to identify homologous recombination-deficient (HRD) HGSOC as a Food and Drug Administration (FDA)-approved companion diagnostic for selecting patients who benefit from PARP inhibitors.38 Since HRD is associated with chemo-sensitivity,41 we tested whether HRD scores differed between refractory and sensitive tumors (Figure 3A). We found elevated levels of “HRD scars” in sensitive tumors based on nTAI (p = 0.0176, Figure 3A left) and wGII (p = 0.053053, Figure 3A middle). Furthermore, chemo-sensitive tumors are associated with elevated frequencies of focal (short) gains and losses (p value = 0.0011, Figure 3A, right), but not with LSTs.
Figure 3. Proteogenomics-based predictive models.
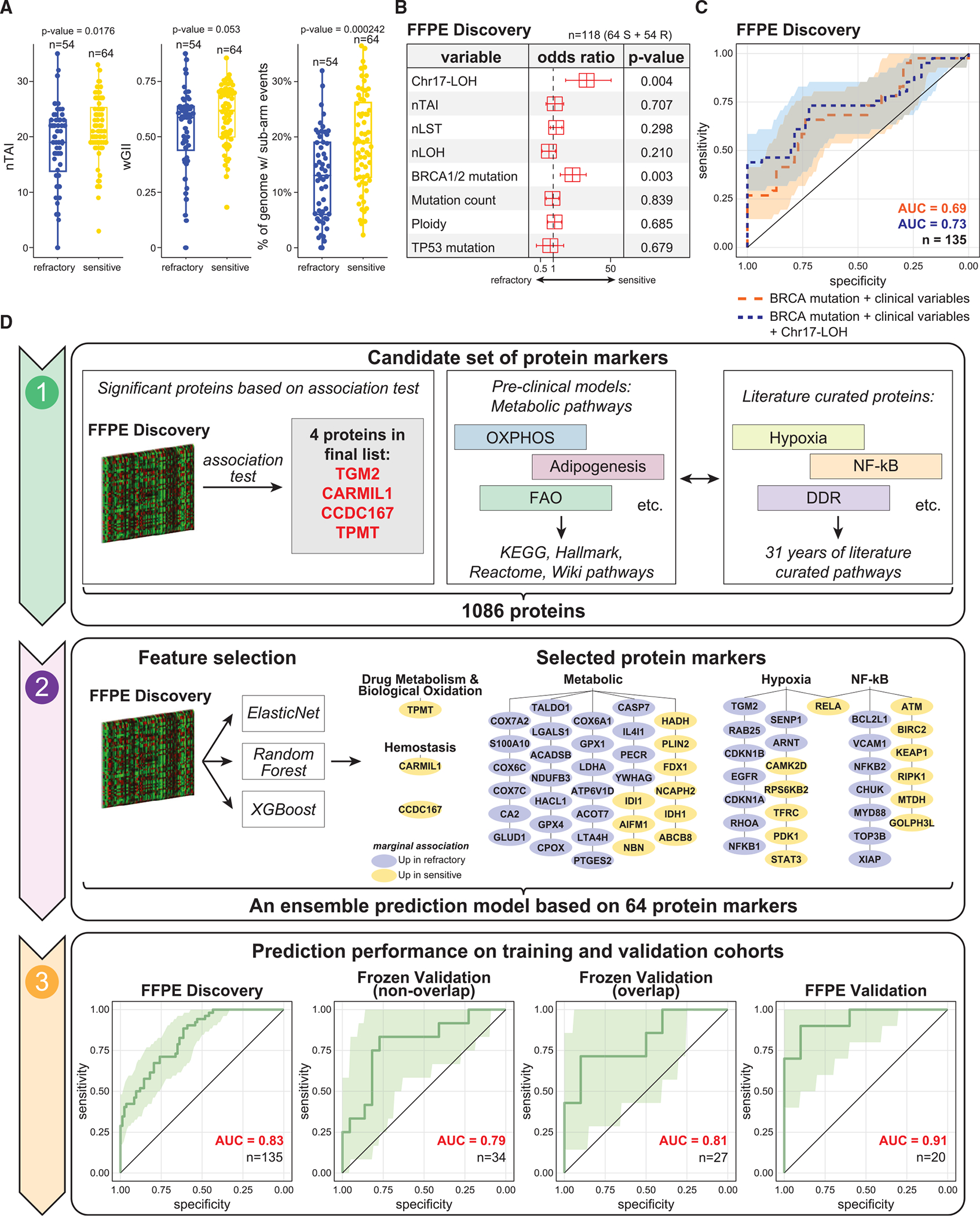
(A) Association between genomic instability scores and chemo-response. p values are determined by Wilcoxon Rank Sum test.
(B) Forest plot of a multivariate logistic regression model predicting refractoriness based on HRD and genetic variables.
(C) ROC curves showing the prediction performance of models based on (1) BRCA1/2 mutation status + clinical variables (patient age and tumor location) and (2) BRCA1/2 mutation status + clinical variables + Chr17-LOH status.
(D) Workflow and results for the proteomic prediction model: (D1) the initial candidate set of protein markers was the union of multiple information sources, including 4 proteins significantly associated with treatment response (based on the FFPE discovery cohort global proteomic data), protein pathways associated with treatment response (based on cell line and PDX models42), and platinum-response-relevant proteins curated from the literature.17 (D2) Feature selection was performed using non-linear machine learning models based on FFPE discovery cohort global proteomic data, and 64 proteins were selected. (D3) An ensemble prediction model based on the 64 protein markers was derived and tested in independent cohorts.
See also Figure S3.
Above, we reported an association between Chr17-LOH and treatment response. Interestingly, tumors lacking Chr17-LOH had lower levels of genomic instability and HRD scores (nTAI and wGII), as well as weaker genome-wide LOH patterns vs. Chr17-LOH tumors (Figures S3A and S3B). Conversely, the genome-wide frequencies and distribution of chromosomal gains and losses were similar between tumors with and without Chr17-LOH (FigureS3B). To interrogate Chr17-LOH asa potential predictor of chemo-sensitivity in the context of the clinically established predictors (HRD score and BRCA1/2 mutation), we tested the association in a multivariate model (Figure 3B) and found that only Chr17-LOH and BRCA1/2 mutation status were predictive of response, both associated with chemo-sensitivity. We also noted an improvement in predictive power when Chr17-LOH is added to a baseline model combining clinical covariates and BRCA1/2 status in two S3H; independent patient cohorts (Figures 3C and STAR Methods).The area under the curve (AUC) of the receiver operating characteristic (ROC) of the resulting model is 0.73 with a 95% confidence interval (CI) of 0.63–0.84 (FFPE discovery cohort) (Figure 3C). In addition, the AUC of the resulting model is 0.66 with a 96% CI: 0.40–0.92 (FFPE validation cohort) (Figure S3H). Despite these encouraging results, better prediction models are needed for clinical practice. This motivated us to explore the proteomic data to determine whether more effective prediction models could be built based on protein markers.
A protein panel predictive of refractoriness
We constructed an ensemble-based prediction model trained using global proteomic data (FFPE discovery cohort). We selected a panel of 1,082 proteins predictive of treatment responses (Figure 3D1; STAR Methods) by leveraging proteogenomic profiles from HGSOC cell lines and patient-derived xenograft (PDX) models,42 31 years of literature on platinum resistance,17 and a machine-learning-based feature selection procedure (STAR Methods). We identified a subset of 64 proteins from the metabolic, hypoxia, and NF-κB pathways (Table S3; STAR Methods) that were associated with treatment response. We built an ensemble prediction model based on the 64 proteins using the ElasticNet,43 Random Forest,44 and XGBoost45 algorithms (Figures 3D2; STAR Methods). Details regarding the 64 protein markers are shown in Figure S3C. For 7 out of the 64 proteins, the RNA-protein correlations were <0.25, and the correlations were negative for COX7A2, COX6C, and BIRC2 (Figure S3D), suggesting post-translational regulation.
We evaluated performance of the prediction model through 5-fold cross validation (CV) using the FFPE discovery cohort proteomic data. The ROC of the ensemble prediction models evaluated on the CV sets showed an AUC of 0.83 (95% CI: 0.77–0.90) (Figures 3D3). At 98% specificity, we observed a sensitivity of 0.35 with 95% CI: 0.19–0.52. Next, we evaluated the performance of the prediction model using two independent patient cohorts (frozen and FFPE validation) (Figures 3D3). For the frozen validation cohort, we separately considered the subsets of tumors that are independent of vs. overlapping with the FFPE discovery cohort. For the frozen-independent and frozen-overlapping subsets, we obtained AUCs of 0.79 (95% CI: 0.63–0.96) and 0.81 (95% CI: 0.61–1.00), respectively. At 98% specificity, we observed a sensitivity of 0.25 (95% CI: 0.08–0.58) and 0.43 (95% CI: 0.14–0.86) for the frozen-independent and the frozen-overlapping subsets, respectively.
Finally, because the FFPE validation data cannot be readily aligned with the FFPE discovery data (due to the lack of a common reference sample and small sample size), we re-estimated and evaluated the prediction model using 3-fold CV. The resulting AUC is 0.91 (95% CI: 0.84–1.00). At 98% specificity, we observed a sensitivity of 0.70 with 95% CI: 0.40–1.00. Based on these results, we conclude that a prediction model based on these 64 proteins detects a subset of refractory HGSOCs with high specificity.
We performed technical validation of the predictor model using a targeted multiple reaction monitoring-mass spectrometry (MRM-MS) assay panel that quantifies 70 peptides representing 22 proteins from the model (STAR Methods). The assay was associated with an AUC of 0.76 (Figure S3G; CV), demonstrating the feasibility of developing a multiplex MRM assay for prediction and paving the way for an expanded assay including all 64 proteins in the model.
We built an ensemble-based prediction model using the phospho-proteomic data (FFPE discovery cohort) (STAR Methods) and obtained 89 phosphosites corresponding to metabolic, ERK, Fanconi, Hippo, and WNT signaling pathways. We evaluated the prediction performance of our model using the selected features in the FFPE discovery and FFPE validation phosphoproteomic data through CV. In the FFPE discovery data we obtained an AUC of 0.78, while in FFPE validation data, we obtained an AUC of 0.76 (Figures S3E and S3F).
A proteogenomic panel predictive of refractoriness
We examined the prediction performance of a joint model using genetic predictors (Chr17-LOH and BRCA1/2 mutation status), the 64 protein markers, and clinical features (Figures S3I and S3J; STAR Methods). The resulting model achieved an AUC of 0.87 (CI: 0.79–0.93) on the training (FFPE discovery) cohort, and an AUC of 0.98 (CI: 0.93–1.00) on the FFPE validation cohort. These results were significantly better than the prediction models based on BRCA1/2 mutation status, Chr17, and clinical features in both the FFPE discovery (p = 0.038) and the FFPE validation cohorts (p = 0.016) (Table S3; STAR Methods). Moreover, at 98% specificity, the combined model had a sensitivity of 0.46 (CI: 0.1–0.66) and 0.8 (CI: 0.60–1.00) in the FFPE discovery and FFPE validation cohorts, respectively.
Pathway analysis shows diverse processes associated with refractoriness
Using enrichment analysis of global proteomic, phospho-proteomic, and RNA-seq data, we searched for pathways associated with sensitivity/refractoriness (Table S4). Figure 4A shows a subset of differentially expressed pathways (FDR < 0.1) in one or more of the omics datasets (FFPE discovery cohort). E2F targets, G2M checkpoint, and DNA replication, etc. are elevated in sensitive tumors in all omics datasets. Conversely, hypoxia, transforming growth factor β (TGF-β), and epithelial-mesenchymal transition (EMT), etc. are elevated in refractory tumors in all datasets. Oxidative phosphorylation (OXPHOS) and tricarboxylic acid (TCA) cycle are elevated in refractory tumors based on protein, but not RNA data, consistent with prior reports that concordance between proteomic and RNA-seq data are low for metabolic pathways.46 The implicated pathways have varying proportions of overlap with reported platinum resistance genes (Figure 4A, left). We validated the association between several pathways and chemo-response using independent cohorts (FFPE, frozen, and CPTAC-201619) (Figure 4A; Table S4; STAR Methods).
Figure 4. Pathway enrichment analysis shows diverse processes associated with refractoriness and reveals sample clusters.
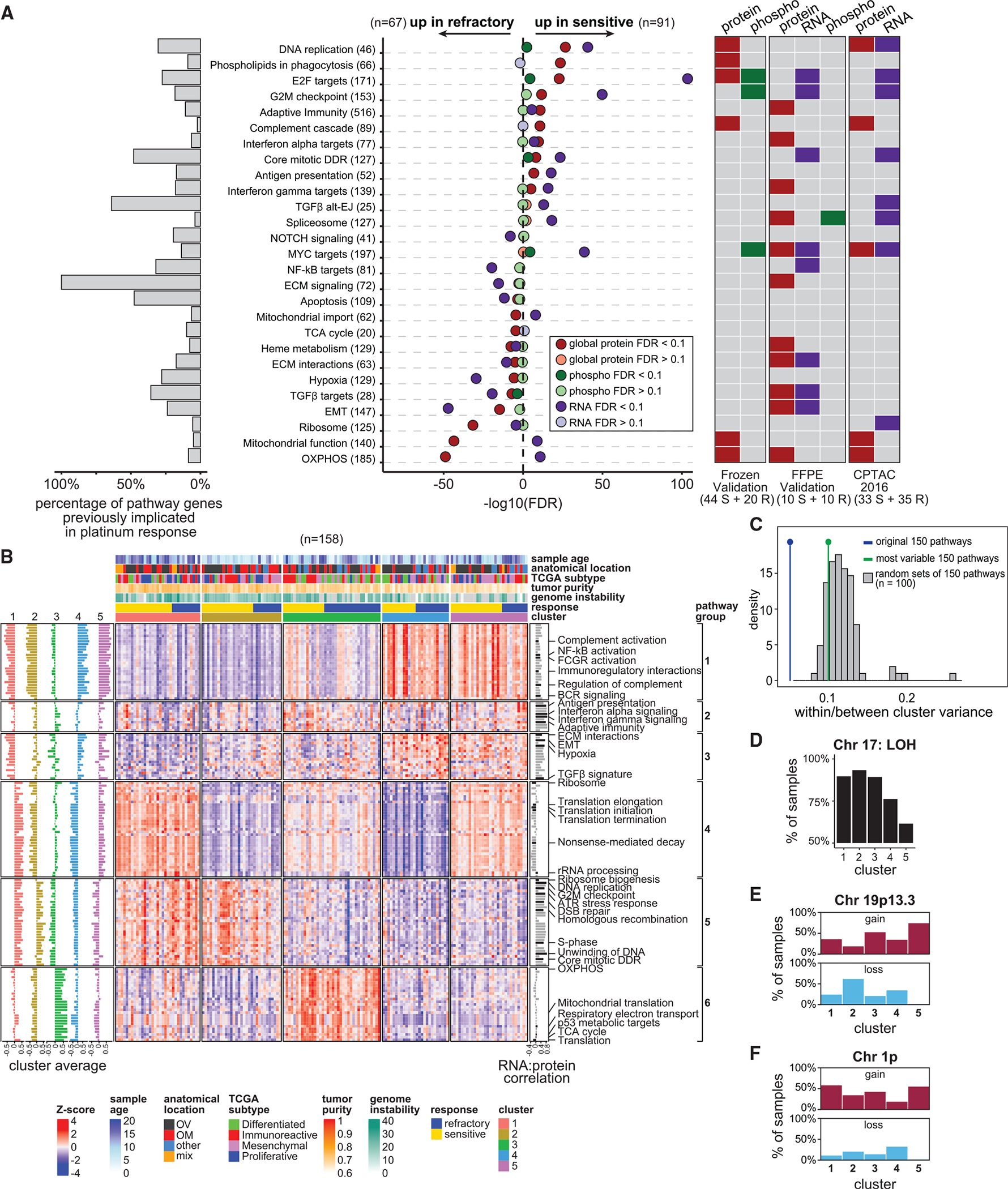
(A) Top pathways associated with treatment response (FDR < 0.1) (FFPE discovery cohort). The left barplot indicates the percentage of genes in each of the pathways overlapping with the literature-curated genes.17 The heatmap on the right indicates if the associations were also observed in the frozen validation, FFPE validation, and the CPTAC-201619 cohorts.
(B) Heatmap showing 5 clusters of tumors based on the 150 pathways associated with refractoriness. Color scale represents the ssGSEA pathway scores derived from global proteomic data. The bars on the left represent mean pathway ssGSEA scores of each cluster. The bar on the right represents the correlation between proteomic- and transcriptomic-based pathway scores.
(C) The tumor clusters (in B) are not likely due to chance. Shown are the within/between variances for the proposed clustering result (blue line), that based on the 150 most variable pathways (green line), and that based on the 100 random sets of 150 pathways (gray histogram).
(D) Percentage of samples with Chr17-LOH among the 5 clusters, with significantly more frequent Chr17-LOH in tumors in clusters 1–3 than in clusters 4 and 5 (p = 0.020, Fisher’s Exact Test).
(E) Percentage of samples with gains/losses in Chr9p13.3 (791681–6199529) in the 5 clusters. Deletions of this region in cluster 2 tumors is significantly lower than that in other samples (p value = 0.0003, Wilcoxon Rank Sum test).
(F) Percentage of samples with gains/losses in Chr1p (32013868–121575702) in the 5 clusters. Deletions of this region in cluster 4 tumors is significantly lower thanthat in other samples (p value = 0.023, Wilcoxon Rank Sum test).
See also Figure S4.
E2F transcription factors have been implicated in platinum response,47 and our study suggests an important role of silencing of this pathway in refractory disease, as significantly lower levels were observed in refractory tumors vs. sensitive tumors (Figure S4A). We also see that a multi-gene proliferation score48 is downregulated (Figure S4A; STAR Methods) in refractory tumors, suggesting a relatively lower proliferation rate. Consistent with these observations, downregulation of the E2F target PRIM2 (Figure S4A), which encodes the large subunit of the DNA primase, is associated with worse prognosis in ovarian cancer.49
Multiple metabolic pathways were associated with chemo-refractory disease in two out of the three independent validation cohorts (Figure 4A), consistent with our previous studies using cell line and PDX models,42 where metabolic pathways were significantly upregulated in resistant cell lines and/or refractory tumors. Figure S4B illustrates the association of 492 proteins from metabolic pathways with response phenotype (FFPE discovery cohort). Among the 492 proteins, 45 were upregulated in refractory tumors (p < 0.05), including COX4I1, COX6A1, COX6C, COX7A2, COX7B, COX7C, MT-CO2, and NDUFA4, which are all part of mitochondrial complex IV (suggested to negatively regulate mitochondria-derived reactive oxygen species50,51). Together with previous findings,42 our observations suggest association of these metabolic pathways with chemo-refractoriness in HGSOC.
Protein-based pathway scores reveal five HGSOC clusters validated in independent patient cohorts and PDX models
To characterize the heterogeneity in mechanisms17 contributing to treatment response in HGSOCs, the top 150 pathways significantly enriched in differentially expressed proteins between chemo-sensitive and chemo-refractory tumors (FDR < 0.01, Table S4) were used to perform consensus clustering analysis (FFPE discovery cohort). Using single sample gene set enrichment analysis (ssGSEA)-based protein pathway scores of the 150 pathways (STAR Methods), we identified 5 tumor clusters (Figure 4B). Cluster 1 was associated with higher expression of translational and rRNA processing pathways (pathway group 4, Figure 4B). Clusters 1 and 2 showed high cell-cycle-related pathways (pathway group 5). Cluster 3 showed higher expression of metabolic pathways (pathway group 6), whereas cluster 4 showed upregulation of hypoxia, EMT, and TGF-β pathways (pathway group 3). Elevated immune pathway scores were observed in cluster 4 and 5 tumors (pathway group 1 and 2). Moreover, cluster 3 was enriched in chemo-refractory tumors (p = 0.027, Figure S4C). We did not observe association between the 5 clusters and tumor anatomical location (Figure S4D). We did observe more frequent Chr17-LOH in tumors in clusters 1–3 vs. clusters 4 and 5 (p = 0.020, Figure 4D). Additionally, one region of chromosome 19p13.3 showed more frequent deletions in cluster 2 vs. other clusters (p < 0.0003) (Figure 4E; STAR Methods), while a large region of chromosome 1p had more frequent deletions in cluster 4 tumors vs. other clusters (p < 0.023, Figure 4F). When comparing this clustering result with that based on random sets of 150 pathways or the 150 pathways exhibiting the most variable scores, the above clustering results achieved the smallest within/between-cluster-variances ratio, indicating that it is not likely due to chance (p = 0; Figure 4B; STAR Methods).
Strikingly, these five proteomic clusters were validated in two independent cohorts (frozen validation and CPTAC-201619) and PDX models42 based on both supervised and unsupervised analyses (Figures 5A–5C, S5A, and S5B; Table S5; STAR Methods).
Figure 5. Validation of the proteomic clusters using independent cohorts.
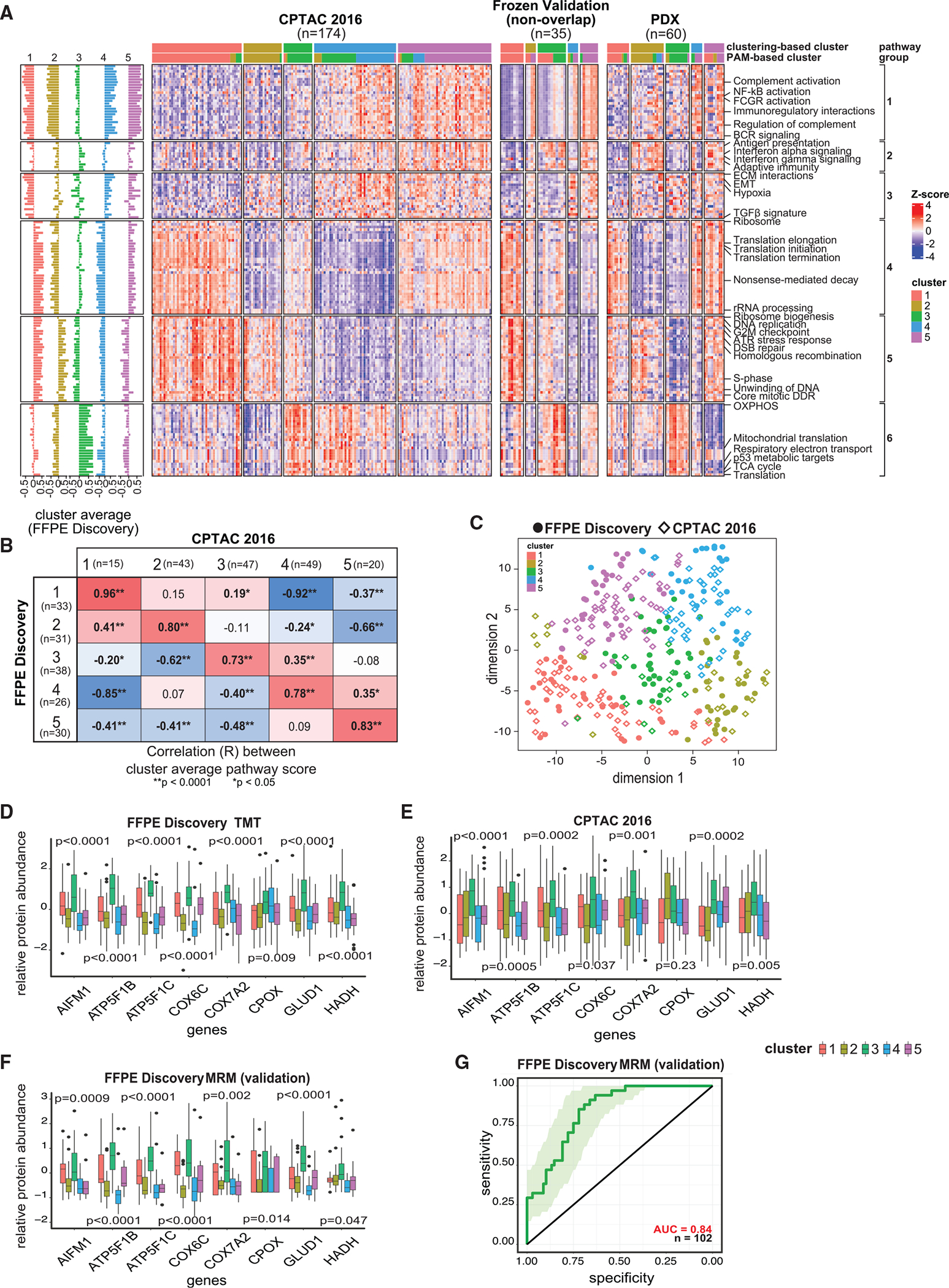
(A) Heatmaps showing proteomic ssGSEA scores of 150 pathways across the 5 clusters for the samples from the CPTAC-2016 cohort,19 frozen validation cohort, and PDX proteomic dataset.42
(B) Heatmap of the Pearson correlations between the average pathway scores of each cluster in the FFPE discovery cohort vs. the CPTAC-2016 study19 (based on consensus clustering). p values are based on R function cor.test.
(C) Concordance among the 5 clusters in the FFPE discovery and the CPTAC-2016 cohorts.19 Sample sizes of the clusters in FFPE discovery cohort are respectively 33, 31, 38, 26, and 30; while those in CPTAC-201619 (based on consensus clustering) are 15, 43, 47, 49, and 20.
(D) Protein abundances of 8 metabolic protein markers showing upregulation in cluster 3 vs. other clusters. The sample sizes of the 5 clusters in FFPE discovery cohort are 33, 31, 38, 26, and 30. p values are based on Student’s t test.
(E) Protein abundances of 8 metabolic protein markers (as in D) showing upregulation in cluster 3 vs. other clusters in the CPTAC-2016 cohort.19 Sample sizes of the 5 clusters in the CPTAC-2016 cohort (based on PAM clustering) are 43, 28, 41, 26, and 36. p values are based on Student’s t tests.
(F) Protein abundances of 8 metabolic protein markers (as in D) showing upregulation in cluster 3 vs. other clusters based on MRM data. The sample sizes of the 5 clusters in FFPE discovery cohort for which MRM experiment was done are respectively 17, 27, 34, 15, and 9. p values are based on R function cor.test.
(G) ROC showing the prediction performance of the 8 metabolic protein markers (as in D) based on the average abundance of their Z scores determined by MRM.
See also Figure S5.
We developed an MRM-based assay to quantify 29 proteotypic peptides representing 10 metabolic proteins that were upregulated in cluster 3 vs. other clusters (STAR Methods). We applied the MRM assay to 102 FFPE discovery cohort (all tumors with sufficient remaining sample material). For 8 of the 10 proteins, we obtained qualified MRM-based abundance measurements (Table S3), which confirmed the upregulation of all 8 metabolic protein markers in cluster 3 (Figures 5D and 5F). These 8 proteins also showed higher abundances in cluster 3 vs. other clusters in the independent CPTAC-2016 cohort19 (Figure 5E). Moreover, we utilized the average abundances (Z scores) of the 8 proteins in the MRM data as a classifier and obtained an ROC-AUC of 0.84 for identifying cluster 3 tumors (Figure 5G).
Comparison with other clustering results
While our gene-level “RNA-seq” data replicate the subtypes reported by TCGA20 and others52 (Figure S4G; STAR Methods), the 5 “protein-based” clusters were distinct from the “RNA”-based subtypes (Figure S4F). This is consistent with previous studies53–55 reporting different results for transcriptomic- vs. proteomic-based pathway activities, likely because proteomic data capture post-transcriptional patterns invisible to RNA-seq data. Indeed, based on our FFPE discovery cohort, proteomic and transcriptomic pathway scores showed low correlation for many pathways (Figure 4B), especially metabolic pathways, suggesting extensive post-transcriptional regulation. Consequently, performing consensus clustering based on the RNA-seq vs. protein pathway data results in very different clusters (Figures S4F and S4H; STAR Methods).
Pathway-based clustering resulted in better separation of tumor clusters vs. protein-based analysis, as shown by significantly lower within/between variances based on pathway activity scores (Figure S4E; STAR Methods). Moreover, individual protein-based results failed to reproduce across validation datasets (Figure S5E), while pathway-based analysis results in more stable/meaningful clusters, consistent with recent literature.56
Immune infiltration
To characterize the tumor microenvironment, we analyzed global proteomic and RNA-seq profiles (FFPE discovery cohort) using two deconvolution methods, XDec57 and BayesDebulk58 (STAR Methods). The estimated proportions of major cell types showed large variation across the 5 tumor clusters (Figures 6A and S6C; Table S6). We observed the highest proportion of epithelial cells in cluster 1 tumors, and increased proportions of stromal cells (fibroblast, adipose) in cluster 2 and 4 tumors. High immune infiltration in cluster 5 was supported by high abundance of CD4, CD8A, and several HLA complex proteins. To verify these computational observations, we performed a blinded pathological review of a subset of the samples (STAR Methods) and found that the pathologist’s estimated proportions of epithelial, stromal, and immune cells showed good concordance with the computational results (Figure S6A). Additionally, multiplex in situ analysis using a fluorescent multiplex immunohistochemistry (IHC) panel for immune cell markers (CD8, CD4, and CCR5) validated the inferred percentage of CD8+ T cells and macrophage from BayesDebulk58 (Figure S6B; STAR Methods).
Figure 6. Tumor microenvironment landscape.
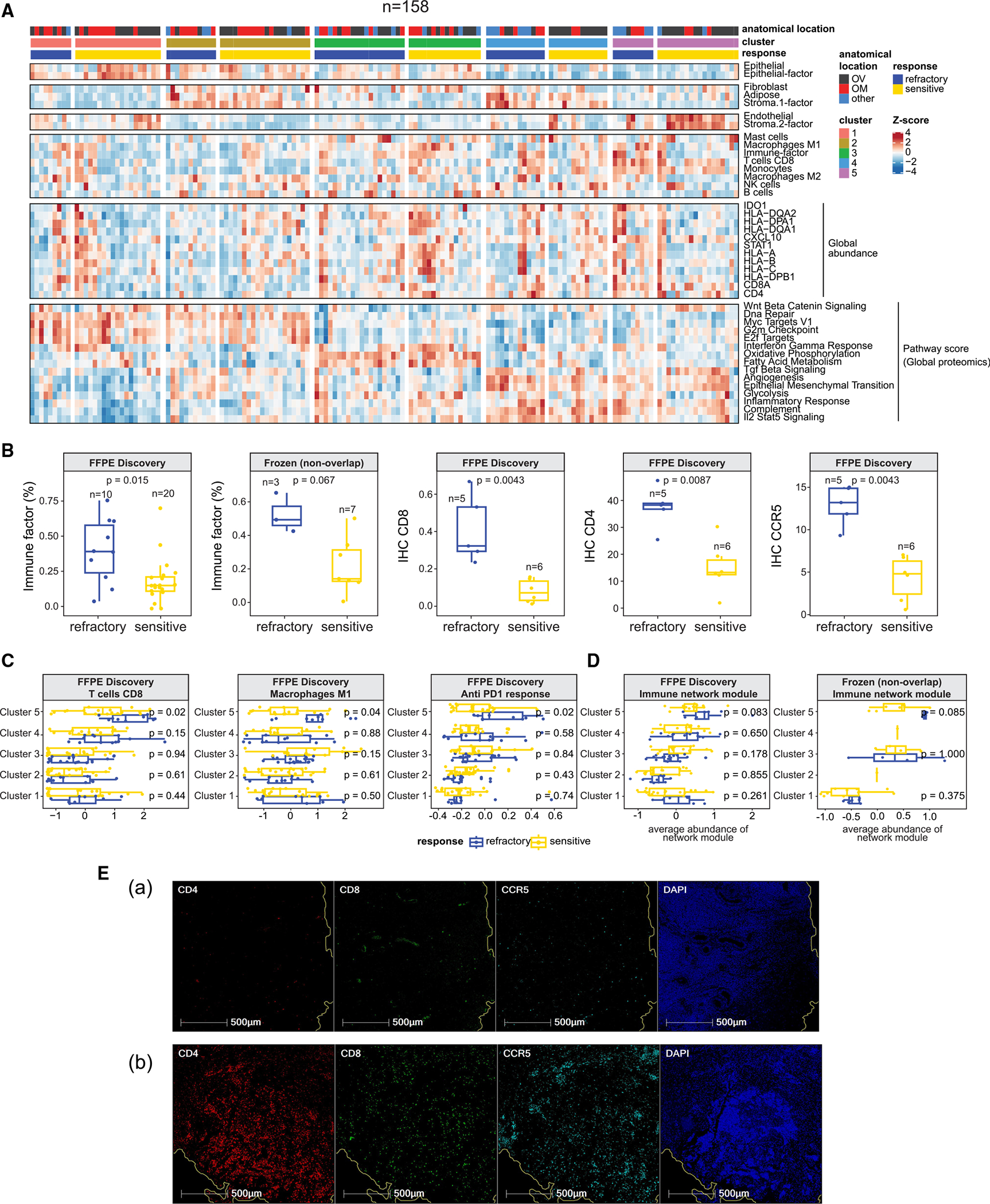
(A) Heatmap illustrating cell-type compositions of the FFPE discovery cohort (n = 158). The first four sections display cell-type proportions estimated by XDec57 and BayesDebulk.58 The fifth section includes the normalized global abundance of key immune cell markers. The last section illustrates ssGSEA pathway scores of pathways that corroborate the estimated cell-type composition.
(B) First two boxplots show the comparison of the estimated immune proportions by XDec57 among cluster 5 refractory and sensitive tumors. p values are based on Student’s t tests. The remaining three boxplots show comparison of IHC staining levels of CD8, CD4, and CCR5 between refractory and sensitive tumors. p values based on two-sided Wilcoxon Rank Sum test.
(C) Comparison of the estimated proportions by BayesDebulk58 for several immune subtypes (CD8 T cells and M1 macrophages), and anti-PD1 (programmed cell death 1) response signature across all 5 clusters and treatment response status (FFPE discovery cohort). p values are based on two-sided Wilcoxon Rank Sum test. The sample sizes of the 5 clusters stratified by response are cluster 1 (22S + 11R), cluster 2 (20S + 11R), cluster 3 (16S + 22R), cluster 4 (13S + 13R), and cluster 5 (20S + 10R).
(D) Module abundance scores (averaged Z scores of proteins mapping to the network module) across different protein clusters and response groups in the FFPE discovery and frozen validation (non-overlapping) cohorts. p values are based on two-sided Wilcoxon Rank Sum test. Sample sizes of the 5 clusters stratified by response in FFPE discovery cohort are cluster 1 (22S + 11R), cluster 2 (20S + 11R), cluster 3 (16S + 22R), cluster 4 (13S + 13R), and cluster 5 (20S + 10R); sample sizes in the frozen validation (non-overlapping) cohort are cluster 1 (10S + 7R), cluster 3 (4S + 3R), and cluster 5 (7S + 3R).
(E) Evaluation of CD8, CD4, and CCR5 expression by fluorescent multiplex immunohistochemistry. A representative chemo-sensitive HGSOC (Ea) and chemo-refractory (Eb) HGSOC were stained using the Akoya Opal Multiplex IHC assay. Tissues were counterstained offline with 4′,6-diamidino-2-phenylindole (DAPI) to identify the nuclei. Representative areas show CD4 (red), CD8 (green), CCR5 (cyan), and nuclei (blue).
See also Figure S6.
We observed significantly higher immune infiltration in the cluster 5 refractory vs. cluster 5 sensitive tumors (Wilcoxon test p = 0.015, Figure 6B). The frozen validation cohort showed a similar trend (Figures 6B and S6D; STAR Methods). Investigation of specific immune cell types (inferred by BayesDebulk) suggested higher CD8 T cell (Wilcoxon test, p = 0.02) and macrophage (Wilcoxon test, p = 0.04) infiltration in the cluster 5 refractory vs. sensitive tumors (Figure 6C). Additionally, IHC data confirmed that the “cluster 5” refractory tumors had significantly higher percentages of cells with positive CD8, CD4, and CCR5 stains vs. cluster 5 sensitive tumors (Wilcoxon test, p < 0.01, Figure 6B). Figure 6E shows substantial staining of CD8, CD4, and CCR5 in a refractory tumor sample, while limited staining of these IHC markers can be seen in a sensitive tumor sample.
It has been reported that CD8+ T cell infiltration predicts response to immunotherapy in multiple cancers.59–61 We evaluated an anti-PD1 response signature62 in cluster 5 tumors (Table S6). This signature was identified using a pan-cancer approach (9,282 patients of 31 different histologies) and has been shown to predict favorable response to PD-L1/PD1 checkpoint inhibitors. We observed a higher (p = 0.02) antiPD1 response signature in the cluster 5 refractory vs. sensitive tumors (Figure 6C). Note, some genes in the signature are related to immune cells, so changes of this signature were likely driven both by cell-type composition differences as well as molecular changes within the same type of cells. These observations suggest that this subset of tumors might respond to immunotherapy.
By studying the protein co-expression network in refractory tumors, we identified a module of highly correlated proteins containing T cell markers, e.g., CD8A, CD7, and CD3D (Figure S6G; STAR Methods). There was a trend toward increased abundance of proteins in this module in cluster 5 refractory vs. sensitive tumors (Wilcoxon test p = 0.083; Figure 6D). This same trend also occurred in the frozen validation cohort (Wilcoxon test p = 0.085; Figure 6D). Leveraging a published ovarian tumor single-cell RNA-seq dataset,63 we were able to annotate most genes in the module as T cell and macrophage markers (Figure S6E; STAR Methods), consistent with increased T cell and macrophage infiltration in cluster 5 refractory tumors (Figures 6B and 6C). The most connected protein in this cluster (hub-protein) was SLAMF6, a regulator of exhausted CD8 T cells.64,65 Based on single-cell RNA data,63SLAMF6 is preferentially expressed in CD8T cells vs. other cell types (Figure S6F; STAR Methods), suggesting that SLAMF6 may play a role in immune regulation in some HGSOCs.
Association of TGF-β, alt-EJ, and βAlt signatures with cluster and response
Among the pathways overexpressed in refractory HGSOCs (Figure 4A), the TGF-β signaling pathway was of interest due to its role in cancer development66–69 and as a therapeutic target.70–72 An inverse correlation between TGF-β and alt-EJ gene expression signatures across tumor types has been reported.73–75 Importantly, for tumors with low TGF-β and high alt-EJ gene expression (i.e., high “βAlt score”), clinical outcomes were better.73,74 We observed similar inverse correlations between TGF-β and alt-EJ gene signatures at both the RNA and proteome levels in four independent cohorts (Figure 7A). We also observed that the TGF-β, alt-EJ, and βAlt scores significantly differ among tumors assigned to different protein-based clusters (Figure 7B; STAR Methods). TGF-β scores were higher in clusters 4 and 5, while alt-EJ scores were higher in clusters 1 and 2. The RNA-seq data suggested similar trends, although the correlation was less significant (Figure S7A). Similar associations between the TGF-β, alt-EJ, βAlt scores and clusters were seen in two independent validation cohorts (Figures S7B and S7C).
Figure 7. Association of TGF-β, alt-EJ, and βAlt scores with treatment response and clusters.
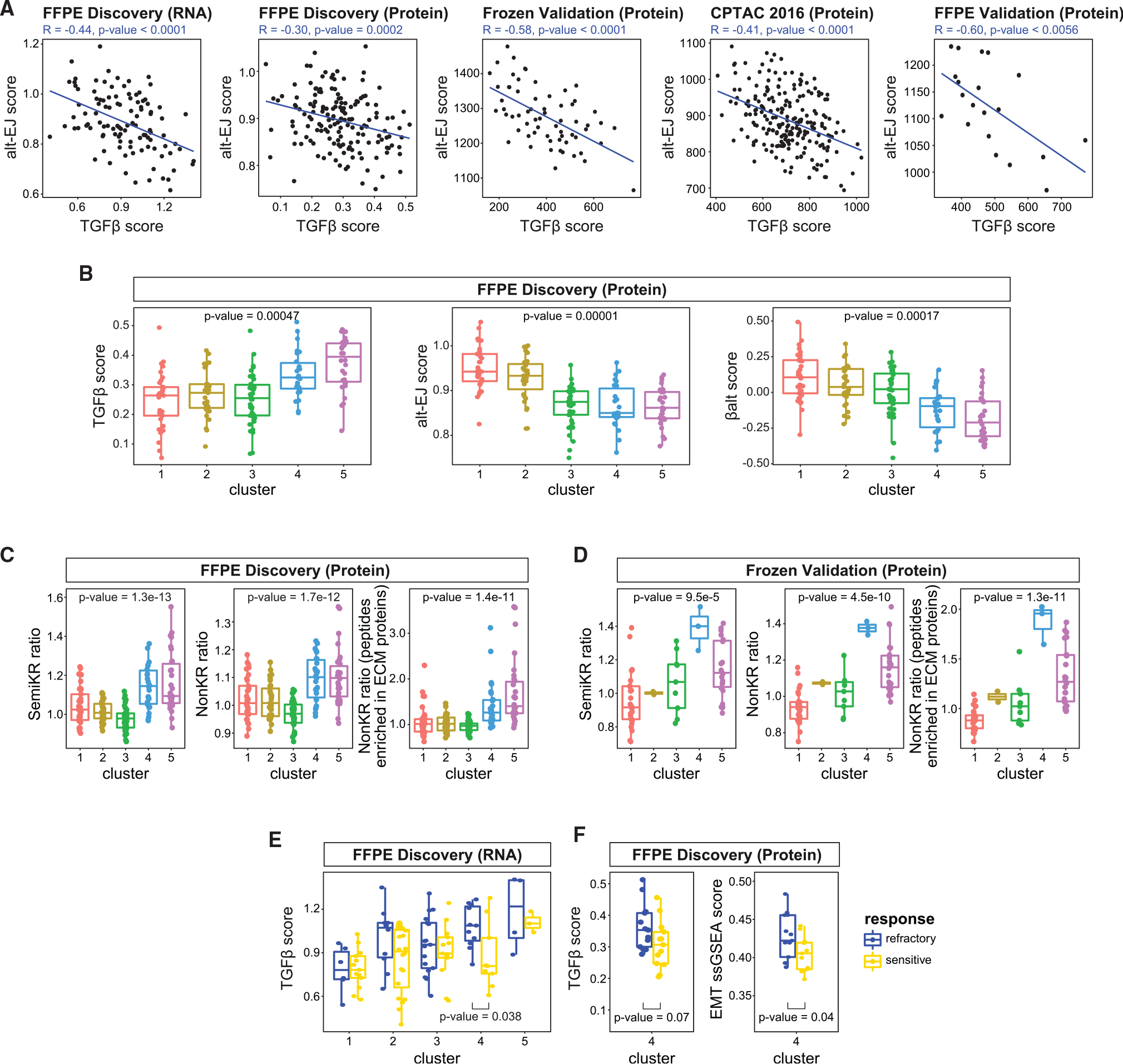
(A) TGF-β and alt-EJ scores are negatively correlated with each other across independent cohorts. p values are for testing the Pearson correlation between the two variables (R function cor.test).
(B) The TGF-β, alt-EJ, and βAlt scores are significantly correlated with proteomic clusters. The sample sizes of the 5 clusters are 33, 31, 38, 26, and 30. p values are derived from an ANOVA test based on regression analyses.
(C and D) SemiKR (semi-tryptic peptides) ratio, NonKR (non-tryptic peptides) ratio, and NonKR ECM-related ratio are elevated in clusters 4 and 5 in both the FFPE discovery (C) and the frozen validation (D) cohorts. Sample sizes of the 5 clusters in FFPE discovery cohort are 33, 31, 38, 26, and 30, while in the frozen validation cohort (based on PAM clustering) are 28, 2, 9, 3, and 22. p values are derived from an ANOVA test based on regression analyses.
(E) Based on the RNA data, the TGF-β score is higher (p = 0.038, Student’s t test) in cluster 4 refractory vs. sensitive tumors. The sample sizes of the 5 clusters in FFPE discovery cohort (based on the 106 samples in RNA data) stratified by response are cluster 1 (13S + 6R), cluster 2 (20S + 10R), cluster 3 (13S + 17R), cluster 4 (9S + 11R), and cluster 5 (3S + 4R).
(F) TGF-β score and EMT pathway ssGSEA scores are significantly higher in cluster 4 refractory vs. cluster 4 sensitive tumors. p values are determined by Student’s t test. The sample size of cluster 4 stratified by response is (13S + 13R).
See also Figure S7.
Bidirectional regulatory loops between TGF-β, proteolytic activity, and matrix metalloproteinase (MMP) expression levels have been associated with increased migration, invasion, and EMT.76–79 To evaluate the possibility that MMP activity is related to TGF-β in our cohorts, we identified peptides resulting from proteases other than trypsin (STAR Methods) to serve as a surrogate phenotype for non-tryptic protease activity.66 When we calculated the semi- and non-tryptic median ratios for each tumor (STAR Methods), these ratios were elevated in clusters 4 and 5 (FFPE discovery cohort) (Figure 7C). This finding was validated in the frozen validation cohort (Figure 7D). Some non-tryptic peptides (79 in the FFPE discovery and 185 in the frozen validation cohort) that showed extremely high ratios belonged to extracellular matrix (ECM) proteins (Figures 7C right and 7D right; Table S7), consistent with increased MMP activity. The postulated increase in non-tryptic enzymatic activity was also supported by the increased protein abundances of several MMPs and TGF-β in cluster 4 and 5 tumors (Figure S7D).
Interestingly, further comparison between the TGF-β, alt-EJ, and βAlt scores and treatment response within each sample cluster revealed elevated TGF-β scores (Figure 7E; p = 0.038) and lower βAlt scores (Figure S7E; p = 0.029) in refractory vs. sensitive tumors in cluster 4. These trends were also seen in an independent validation dataset (Figure S7F). In addition, we observed that the EMT pathway score was significantly higher in cluster 4 refractory vs. sensitive tumors (p = 0.04, Figure 7F, right). The abundances of the non-tryptic peptides enriched in ECM-related proteins (Table S7) showed a similar trend of upregulation in cluster 4 refractory tumors (Figure S7G). Collectively, these observations suggest that MMPs and TGF-β may have increased activity in cluster 4 refractory samples, motivating the hypothesis that these tumors may respond to therapeutic agents targeting TGF-β activity.
DISCUSSION
Implications of the results for precision oncology
Despite >3 decades of research on platinum responses in cancer,17 no predictive biomarker has been translated into clinical use. Predictors of refractory disease could enable a precision oncology approach and provide a means to select patients for clinical trials to identify and implement effective therapies.
In this study, we leveraged technological advances (e.g., LC-MS/MS-based multiplex protein quantification, machine learning algorithms) to employ an approach integrating: (1) decades of studies reporting single-analyte biomarker candidates,17 (2) proteogenomic analyses for dynamic profiling of pre-clinical models,42 and (3) proteogenomic analyses of human HGSOCs to identify an ensemble prediction model of chemo-refractoriness based on 64 proteins. The 64-protein prediction model detects a subset of ~35% refractory tumors with high (98%) specificity and is validated in 2 independent patient cohorts (Figure 3).
Further validation of the 64-protein prediction model in clinical trials requires development of a higher throughput, clinical-grade, multiplexed assay, which is currently underway (Figure S3G) using a clinically translatable platform based on MRM-MS.80–82 Once mature, the assay will be evaluated for performance as a multi-protein panel, which may be algorithmically integrated to generate a single parameter, or score, tied to the likelihood of the patient having chemo-resistant disease (e.g., similar to the way that Oncotype DX83 provides the likelihood of recurrence of breast cancer). Similarly, data from our proteomic signature, evaluation of Chr17-LOH, and the identification of BRCA mutations could be combined into a single output for interpretation by a practicing pathologist. A current example in the clinical laboratory lies in the combination of LC-MS/MS proteomic analysis of amyloidosis samples with histology, germline genetic mutations, and clinical history in the rendering of a diagnosis by the pathologist.84,85 Prior to full clinical deployment, validation of this classifier will need to be performed in additional retrospective and prospective studies, but the data from this study suggest that variability in study site and preanalytical processing procedure had limited effects on the resulting proteomic data, suggesting that the quantitative measurements will be robust in future evaluations.
We identified 5 subtypes of HGSOC (replicated in independent patient cohorts and PDX models; Figure 5) based on pathway protein expression (Figure 5A), possibly reflecting different mechanisms of refractoriness and implicating potential subtype-specific treatment approaches (e.g., immune therapies, TGF-β inhibitors, and metabolic inhibitors). In support of the hypothesis that these subtypes might predict therapeutic vulnerabilities, we demonstrated42 that a treatment-refractory HGSOC PDX model and cell line mapping to cluster 3 and showing upregulation of pathway “group 6” (metabolism, Figure S5C) are sensitized to platinum-based therapy either through pharmacological inhibition (Figure S5D) and/or CRISPR knockout42 of CPT1A, which catalyzes a rate limiting step in fatty acid oxidation. This motivates a hypothesis that refractory tumors in cluster 3 might respond to the addition of drugs targeting metabolic processes and sets the stage for further mechanistic studies, as well as additional in vivo studies to test other possible therapeutic vulnerabilities identified by our data (e.g., immune therapies and TGF-β inhibitors).
Strengths of this study
Strengths of this study include the (1) rigorous definition of refractory disease, requiring objective radiographic progression, (2) inclusion of two independent validation cohorts (and leveraging of two additional, independent published cohorts), in which major findings are confirmed, (3) multi-institutional design with inclusion of samples from 4 academic centers, making the results likely to be generalizable beyond this study, (4) retrospective study design enabling access to clinical responses and enrichment for chemo-refractory tumors, and (5) use of FFPE tissues, which should facilitate clinical validation studies and general applicability.
Limitations of this study
First, the sample size (242 tumors) was insufficient to fully capture the complexity of the heterogeneous mechanisms underlying chemo-response.17 Although the 64-protein predictor model detects a subset (~35%) of refractory tumors with high (98%) specificity, other refractory tumors are not detected. Although the current predictor will need to be refined in a larger study to predict 100% of refractory cases, this study (1) lays out a road map and justification for an expanded study and (2) reports a current predictor that if clinically validated would be a game changer for the 35% of patients with refractory disease who could avoid ineffective chemotherapy. Second, the reliance on archival FFPE biospecimens (vs. frozen) makes genomic profiling challenging and results in a reduced ability to study post-translational modifications. Regardless, we detected a panel of phosphosites predictive of refractoriness using the FFPE phospho-proteomic data (Figures S3E and S3F), suggesting that some clinically relevant signals may be preserved in archival tissues. Third, the lack of germline DNA presents challenges to calling somatic mutations. Fourth, although bulk profiling of cancer tissues has led to many advances, tumor heterogeneity is a complication. Although we employed pathological review, IHC, and deconvolution analyses to address heterogeneity, spatially resolved proteogenomics may add value.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Amanda Paulovich (apaulovi@fredhutch.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw proteomic data files and all processed proteogenomic datasets as well as clinical meta information have been deposited at the Proteomic Data Commons and are publicly available as of the date of this publication. URLs are listed in the key resources table. DNA and RNA sequencing data have been deposited at dbGaP and are publicly available as of the date of this publication. Accession numbers are listed in the key resources table. H&E images for all the tumors analyzed in this study have been deposited at The Cancer Image Archive and are publicly available as of the date of this publication. DOIs are listed in the key resources table. In addition, all processed proteogenomic datasets as well as clinical meta information can be publicly queried, visualized, and downloaded from an interactive ProTrack data portal as of the date of this publication. The URL for ProTrack is listed in the key resources table. All processed data has been deposited at a publicly accessible URL listed in the key resources table. All raw data, manually integrated peak areas, transition information, and retention times generated from these stressor and time course experiments for the LC-MRM peptide target assays have been deposited at Panorama Public86 and are publicly available at the URL is listed in the key resources table. Characterization data for available assays are found in the CPTAC Assay Portal (assays.cancer.gov).
All original code for the data analysis and figures generated for this study has been deposited at this Github repository and is publicly available as of the date of publication: https://github.com/WangLab-MSSM/CPTAC_Ovarian_Chemo_Response. URLs for this repository and for other code used in this study are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD8, Clone C8/144B | DAKO | Cat#M7103; RRID:AB_2075537 |
| CD4, EP204 | Cell Marque | Cat#104R-26; RRID:AB_1516770 |
| CD68, Clone PG-M1 | DAKO | Cat#M0876; RRID:AB_2074844 |
| CD14, EPR3653 | Cell Marque | Cat#114R-14; RRID:AB_2827391 |
| CCR5 | Matthias Mack Lab | N/A |
| PanCK, Clone AE1/AE3 | DAKO | Cat#M3515; RRID:AB_2132885 |
| Opal Polymer HRP Ms+Rb | Akoya Biosciences | Cat#ARH1001EA; RRID:AB_2890927 |
|
| ||
| Biological samples | ||
|
| ||
| Fresh frozen tissue samples | See STAR Methods | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| HEPES (pH8.0) | Alfa Aesar | Cat#J63002 |
| Hydroxylamine | Millipore Sigma | Cat#438227 |
| TMT 11plex reagents | Thermo Fisher Scientific | Cat#A34808 |
| Stable isotope-labeled synthetic peptide standards | Vivitide | N/A |
| Phosphatase Inhibitor Cocktail 2 | Millipore Sigma | Cat#P5726 |
| Phosphatase Inhibitor Cocktail 3 | Millipore Sigma | Cat#P0044 |
| Protease Inhibitor Cocktail | Millipore Sigma | Cat#P8340 |
| Lys-C | Wako Chemicals | Cat#129-02541 |
| Sequencing grade modified trypsin | Promega | Cat#V5113 |
| Urea | Millipore Sigma | Cat#U0631 |
| Trizma base (Tris), pH 8.0 | Millipore Sigma | Cat#T2694 |
| iodoacetamide (IAM) | Millipore Sigma | Cat#A3221 |
| EDTA | Millipore Sigma | Cat#E7889 |
| EGTA | Bioworld | Cat#40520008-1 |
| phosphate buffered saline | Thermo Fisher Scientific | Cat#14190144 |
| tris(2-carboxyethyl)phosphine (TCEP) | Thermo Fisher Scientific | Cat#77720 |
| Acetonitrile | Fisher Chemical | Cat#A955-4 |
| Water | Fisher Chemical | Cat#W64 |
| ammonium bicarbonate | Millipore Sigma | Cat#A6141 |
| ammonium hydroxide solution | Millipore Sigma | Cat#320145 |
| Formic acid | Millipore Sigma | Cat#1116701000 |
| Trifluoroacetic Acid | Millipore Sigma | Cat#302031 |
| RapiGest SF | Waters | Cat#186001861 |
| Bond Dewax Solution | Leica | Cat#AR9222 |
| BOND Epitope Retrieval Solution 2 | Leica | Cat#AR9640 |
| Wash Solution 10X Concentrate | Leica | Cat#AR9590 |
| tertiary TSA-amplification reagent | Akoya Biosciences | Cat#FP1135 |
| Spectral DAPI | Akoya Biosciences | Cat#FP1490 |
| Prolong Gold Antifade | Invitrogen | Cat#P36930 |
| Ni-NTA Magnetic Agarose Beads | Qiagen | Cat#36113 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Micro BCA protein assay | Thermo Fisher Scientific | Cat#23235 |
| TruSeq RNA Sample Prep Kit | Illumina | Cat#FC-122-1001 |
| KAPA Library Preparation Kit | Roche | Cat#KK8201 |
| AllPrep DNA/RNA FFPE kit | Qiagen | Cat#80234 |
| QIAamp® DNA FFPE Tissue Kit | Qiagen | Cat#56404 |
| miRNeasy FFPE kit | Qiagen | Cat#217504 |
| QIAsymphony DSP DNA Midi Kit | Qiagen | Cat#937255 |
| KAPA HyperPrep with RiboErase kit | Roche | Cat#KK8561 |
| KAPA Stranded RNA-Seq with RiboErase kit | Roche | Cat#KK8484 |
| Accel-NGS S2 DNA prep reagents | Swift Biosciences | Cat#210384 |
| Fragment Analyzer RNA kit | Agilent | Cat#DNF-471-1000 |
| AllPrep DNA/RNA FFPE Kit | Qiagen | Cat#80234 |
| Kapa Biosystems library quantification kit | Roche | Cat#KK4854 |
| TapeStation 2200 D1000 screentape | Agilent | Cat#5067-5582 |
|
| ||
| Deposited data | ||
|
| ||
| PTRC HGSOC FFPE Validation - Phosphoproteome | This paper | Proteomic Data Commons:PDC000357 |
| PTRC HGSOC FFPE Validation - Proteome | This paper | Proteomic Data Commons:PDC000358 |
| PTRC HGSOC FFPE Discovery - Phosphoproteome | This paper | Proteomic Data Commons:PDC000359 |
| PTRC HGSOC FFPE Discovery - Proteome | This paper | Proteomic Data Commons:PDC000360 |
| PTRC HGSOC Frozen Validation - Phosphoproteome | This paper | Proteomic Data Commons: |
| PTRC HGSOC Frozen Validation - Proteome | This paper | Proteomic Data Commons:PDC000362 |
| PTRC HGSOC DNA sequencing | This paper | dbGaP:phs003152.v1.p1 |
| PTRC HGSOC RNA sequencing | This paper | dbGaP:phs003152.v1.p1 |
| ProTrack Data Portal: Processed proteogenomic dataset visualization | This paper | http://ptrc.cptac-data-view.org/ |
| Processed LC-MRM-MS data | This paper | https://www.dropbox.com/s/7zul3j1vyrxo40c/processed_data.zip?dl=0 |
| Raw LC-MRM-MS data | This paper | Panoramaweb:Paulovich_PTRC_HGSOC |
| H&E images | This paper | TCIA:doi.org/10.7937/6rda-p940 |
| PhosphoSitePlus | Hornbeck et al.152 | https://www.phosphosite.org |
| MSK-IMPACT | Nguyen et al.18 | N/A |
| CPTAC 2016 | Zhang et al.19 | N/A |
| TCGA – GBM | The Cancer Genomic Atlas Research Network87 | https://portal.gdc.cancer.gov/ |
| TCGA – LGG | The Cancer Genomic Atlas Research Network147 | https://portal.gdc.cancer.gov/ |
| TGFβ and Alternative end-joining pathways | Liu et al.74 | https://pubmed.ncbi.nlm.nih.gov/33568520/ |
| HGSOC RNAseq: GSE154600 | Gene Expression Omnibus | https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/geo/query/acc.cgi?acc=GSE154600 |
| LM22 signature matrix from Cibersort | Chen et al.88 | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5895181/#:~:text=LM22%20is%20a%20signature%20matrix,NK%20cells%2C%20and%20myeloid%20subsets |
| UniProt 2019_06 | reviewed Human Universal Protein Resource sequence database | https://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2019_06/knowledgebase/ |
| UniProt 2020_03_30 | reviewed Human Universal Protein Resource sequence database | https://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2020_03/knowledgebase/ |
| TP53 transcriptional program | Andrysik et al.32 | N/A |
| Mutant TP53 signature | Donehower et al.29 | N/A |
| Genes associated with platinum resistance | Huang et al.17 | http://ptrc-ddr.cptac-data-view.org. |
| Phosphosites related to ischemia | Mertins et al.91, | N/A |
| MSigDB’s database (v 7.5.1) | Liberzon et al.93 | http://software.broadinstitute.org/gsea/msigdb/index.jsp |
| DDR pathway database | Huang et al.17 | http://ptrc-ddr.cptac-data-view.org |
|
| ||
| Software and algorithms | ||
|
| ||
| Github repository for code used for data analysis and figures for this paper | This paper | https://github.com/WangLab-MSSM/CPTAC_Ovarian_Chemo_Response |
| HALO Tissue Classifier machine learning algorithm | Indica Labs | https://indicalab.com/halo-ai/ |
| Philosopher v3.2.8 | Alexey Nesvizhskii Lab; da Veiga Leprevost et al.94 | https://philosopher.nesvilab.org/ |
| MSFragger v3.0 | Alexey Nesvizhskii Lab; Kong et al.95 | https://msfragger.nesvilab.org/ |
| PeptideProphet | Keller et al.96 | http://peptideprophet.sourceforge.net/ |
| PTMProphet | Shteynberg et al.97 | http://www.tppms.org/tools/ptm/ |
| TMT-Integrator | Djomehri et al.98 | http://github.com/huiyinc/TMT-Integrator |
| MaxQuant/Andromeda | Tyanova et al.99 | http://maxquant.org |
| ComBat (v3.20.0) | Johnson et al.100 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| DreamAI | Pei Wang Lab | https://github.com/WangLab-MSSM/DreamAI |
| XDec | Genboree | https://github.com/BRL-BCM/XDec |
| TPO workflow | Michigan Center for Translational Pathology | https://github.com/mctp/tpo |
| Bbduk and bbduk2 | BBMap | https://github.com/BioInfoTools/BBMap/tree/master/sh |
| BWA-mem | Li and Durbin101 | https://github.com/lh3/bwa |
| Seurat | Stuart et al.102 | https://github.com/satijalab/seurat/releases/tag/v3.0.0 |
| BayesDebulk | Petralia et al.58 | https://www.biorxiv.org/content/10.1101/2021.06.25.449763v2 |
| DAGBagM | Chowdhury et al.103 | https://www.biorxiv.org/content/10.1101/2020.10.26.349076v1 |
| GISTIC2.0 | Mermel et al.89 | https://github.com/broadinstitute/gistic2/ |
| iProFun | Song et al.27 | https://github.com/WangLab-MSSM/iProFun |
| ESTIMATE | Yoshihara et al.104 | https://bioinformatics.mdanderson.org/public-software/estimate/ |
| Joint Random Forest | Petralia et al.105 | https://rdrr.io/cran/JRF/man/JRF.html |
| TSNet | Petralia et al.106 | https://github.com/WangLab-MSSM/TSNet |
| xCell | Aran et al.90 | http://xcell.ucsf.edu/ |
| iCAVE | Liluashvili et al.92 | http://labs.icahn.mssm.edu/gumuslab/software |
| ConsensusClusterPlus | Wilkerson and Hayes107 | http://bioconductor.org/packages/release/bioc/html/CancerSubtypes.html |
| Strelka2 v2.9.3 | Kim et al.108 | https://github.com/Illumina/strelka |
| CNVkit v. 2.9.3 | Talevich et al.109 | https://github.com/etal/cnvkit |
| STAR v2.6.1d | Dobin et al.110 | https://github.com/alexdobin/STAR |
| GENCODE v27 | GENCODE consortium | https://www.gencodegenes.org/human/release_27.html |
| RSEM v1.3.1 | Li and Dewey111 | https://github.com/deweylab/RSEM |
| Sumer | Savage et al.112 | https://github.com/bzhanglab/sumer |
| TCGAbiolinks | Colaprico et al.113 | https://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html |
| MoonlightR | Colaprico et al.114 | https://bioconductor.org/packages/release/bioc/html/MoonlightR.html |
| MuSiC | Wang et al.115 | https://github.com/xuranw/MuSiC |
| GATK | Broad Institute | https://gatk.broadinstitute.org/hc/en-us/articles/360035890811-Resource-bundle |
| VEP | McLaren et al.116 | https://useast.ensembl.org/info/docs/tools/vep/index.html |
| vcfAnno | Pedersen et al.117 | https://github.com/brentp/vcfanno |
| CNVEX | Michigan Center for Translational Pathology | https://github.com/mctp/cnvex |
| DNAscope | Sentieon | https://support.sentieon.com/versions/201911/manual/DNAscope_usage/dnascope/ |
| Kallisto | Bray et al.118 | https://pachterlab.github.io/kallisto/download.html |
| Ascore v1.0.6858 | Github | https://github.com/PNNL-Comp-Mass-Spec/AScore |
| MASIC | Monroe et al.119 | https://github.com/PNNL-Comp-Mass-Spec/MASIC |
| MS-GF+ v9981 | Kim and Pevzner120 | https://github.com/MSGFPlus/msgfplus |
| mzRefinery | Gibbons et al.121 | https://pnnl-comp-mass-spec.github.io/MzRefinery |
| pamr | Tibshirani lab | https://CRAN.R-project.org/package=pamr |
| XGBoost | Chen and Guestrin45 | https://cran.r-project.org/web/packages/xgboost/index.html |
| RandomForest | Breiman44 | https://cran.r-project.org/web/packages/randomForest/ |
| glmnet | Tibshirani lab | https://cran.r-project.org/web/packages/glmnet/ |
| Skyline | MacLean et al.122 | https://skyline.ms/project/home/software/Skyline/begin.view |
| GSVA | Hänzelmann et al.123, | https://www.bioconductor.org/packages/release/bioc/html/GSVA.html |
| Ovarian cancer molecular subtype classifier | Chen et al.124 | http://bioconductor.org/packages/release/bioc/html/consensusOV.html |
|
| ||
| Other | ||
|
| ||
| Glass slides | Leica Biosystems | Cat#3800040 |
| Tissue Bags | Covaris | Cat#TT1, 520001 |
| 10 mg Sep-Pak solid-phase extraction | Waters | Cat#186000128 |
| 5 mg Sep-Pak solid-phase extraction | Waters | Cat#186000309 |
| 2 mg Sep-Pak solid-phase extraction | Waters | Cat#186001828BA |
| PicoTip™ emitter, 50 μm ID × 20 cm | New Objective | Cat#FS360-50-15-N-20-C12 |
| 1 mL deep well plate | Thermo Fisher Scientific | Cat#95040450 |
| 4.6 mm × 250 mm Zorbax Extend- C18, 3.5 μm, column | Agilent | Cat#770953-902 |
| ReproSil-Pur, 120 Å, C18-AQ | Dr. Maisch | Cat#r119.aq |
| EvoTip Pure | EvoSep | Cat#EV2011 |
| Endurance OE, 15 cm × 150 μm, 1.9 μm | EvoSep | Cat#EV-1113 |
| epMotion 5075 | Eppendorf | Cat#5075 900.157-13/0411 |
| Agilent 1200 HPLC | Agilent | Cat#G2262-90010 |
| KingFisher Flex | Thermo Fisher Scientific | Cat#N13141 |
| Easy-nLC 1000 | Thermo Fisher Scientific | Cat#LC120 |
| LTQ-Orbitrap Fusion mass spectrometer | Thermo Fisher Scientific | Cat#IQLAAEGAAPFADBMBCX |
| Evosep One LC | EvoSep | Cat#EV1000 |
| 6500+ QTRAP mass spectrometer | Sciex | Cat#5039926 |
| OptiFlow Turbo V source | Sciex | Cat#5028138 |
| Qubit 4 fluorometer | Thermo Fisher Scientific | Cat#Q33238 |
| Agilent Tapestation 2200 | Agilent | Cat# 5067-5582 |
| Illumina NovaSeq 6000 | Illumina | Cat#20012850 |
| Beckman Coulter Biomek i7 | Beckman Coulter | https://www.illumina.com/content/dam/illumina/gcs/assembled-assets/marketing-literature/novaseq-6000-spec-sheet-m-gl-00271/novaseq-6000-spec-sheet-m-gl-00271.pdf |
| BOND Rx autostainer | Leica | Cat#21.2821 |
| PhenoImager HT Automated Imaging System | Akoya Biosciences | Cat#CLS143455 |
| Protrack data portal | This paper | http://pbt.cptac-data-view.org |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Patient Selection and Cohorts
Treatment-naive tissue specimens from Stage III or IV HGSOCs that underwent primary debulking followed by platinum/taxane adjuvant therapy were selected for the study and categorized with respect to response to adjuvant chemotherapy, as defined below. (After the profiling was completed, we learned that a subset of 13 tumors had undergone neoadjuvant chemotherapy, and these are annotated in Table S1. These samples were not included in the construction and evaluation of the prediction model, and neoadjuvant status was included as a covariate in other analyses.)
Refractory tumors were defined as follows: After primary debulking: for R0 disease (no residual/microscopic disease after primary resection), radiographically detectable disease must have been present at the end of 6 cycles of initial platinum/taxane therapy; for R1 disease (radiographically detectable residual disease is present after primary resection), residual disease must either progress or stay stable (radiographically) after 6 cycles of initial platinum/taxane therapy.
Sensitive tumors were defined by R1 or R2 disease that received primary resection followed by platinum/taxane adjuvant therapy and had a progression-free survival of at least 2 years (no R0 disease).
All biospecimens were collected with Institutional Review Board (IRB) approvals: the Mayo Clinic IRB numbers: 08–005749 and 17–010405; Fred Hutchinson Cancer Research Center IRB number 4563; University of Alabama at Birmingham IRB number 131007005; and MD Anderson Cancer Center IRB number 131007005.
The FFPE Discovery cohort consisted of 158 HGOC tissue biopsies (91 sensitive and 67 refractory) from three institutes. The FFPE Validation cohort consists of 20 HGSOC tumors from MD Anderson Cancer Center, an independent set of patients representing 10 sensitive and 10 refractory cases. The Frozen Validation cohort consists of 64 HGSOC tumors (44 sensitive, 20 refractory) from Fred Hutchinson Cancer Research Center. And 29 of these 64 tumors were from overlapping patients from the FFPE Discovery cohort.
In addition to the validation cohorts from this study, two published ovarian cancer datasets were also used as independent cohorts for validating findings: (i) Memorial Sloan Kettering (MSK)-IMPACT18 and (ii) the National Cancer Institute’s Clinical Proteomics Tumor Analysis Consortium’s 2016 study (“CPTAC-2016”).19
Ovarian Cancer Tissues
10 μm sections of the FFPE samples were cut using a microtome and mounted on glass slides (Leica Biosystems Cat# 3800040). The first and last “bookend” sections were 4 μm H&E sections for pathology review. Digital images of the H&E slides were recorded using a ScanScope AT Slide Scanner (Leica Aperio Technologies, Vista, CA, USA) under 20X objective magnification (0.5 μm resolution). Tissue sub-compartment cellularity was reported by a pathologist using the HALO Image Analysis Platform (Indica Labs, NM, USA). Follow-up multiplexed IHC analysis was performed on a subset of the FFPE samples from four 4 μm sections prepared as above.
METHOD DETAILS
Whole Genome and RNA Sequencing of FFPE Samples
Sample Processing
Samples were processed as follows: the protocol adapted from QIAGEN AllPrep DNA/RNA FFPE Kit 80234, QIAamp® DNA FFPE Tissue Kit (56404) and miRNeasy FFPE kit (217504). This protocol is optimized for 150–250 mm2 10-μm sections. All isolations were performed under RNase-free working environment. De-paraffination and re-hydration was performed using Xylene and ethanol as per standard protocols. Tissue was lysed in buffer PKD as per manufacturer’s recommendations. The supernatant was processed for RNA and the pellet for DNA (after Protein K digestion and using QIAamp MinElute spin column). All nucleic acids were quantified by 260/280 determination.
RNA Sequencing Library Preparation
Purified total RNA samples were evaluated for quantity and quality by calculating the percent DV200 with an Agilent Fragment Analyzer RNA kit and reagents (Cat# DNF-471–1000). Samples with <100 ng of RNA and DV200 < 30% were excluded from the study. Sequencing libraries were prepared using Kapa RNA HyperPrep with RiboErase (Cat# KK8561) from 100 ng of RNA according to the manufacturer’s protocol.
Whole Genome Library Preparation
Purified gDNA was quantified by Qubit Fluorometer and sheared to 300 bp using a Covaris M220. Sequencing libraries were prepared using Swift Biosciences Accel-NGS S2 DNA prep reagents (Cat# 210384) from 100 ng of DNA according to the manufacturer’s protocol.
Next-Generation Sequencing
The finished libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200 D1000 screentape (Cat# 5067–5582), and RT-qPCR using the Roche Kapa Biosystems library quantification kit (Cat# KK4854) according to manufacturers’ protocols. Whole genome libraries were sequenced with >400M 150 bp read pairs and RNAseq libraries (Ribo-Erase) were sequenced with >50M 100 bp read pairs on an Illumina NovaSeq 6000 by the Molecular Biology Core Facilities at the Dana-Farber Cancer Institute.
Whole Genome Sequencing (WGS) Data Preprocessing and QC
WGS Data Processing and Alignment
All genomic data processing has been performed using the TPO workflow (https://github.com/mctp/tpo), which implements standardized pipelines for the analysis of DNA and RNA sequencing data.
DNA sequencing paired-end reads were trimmed from adapter sequences using BBMap’s bbduk tool with parameters ‘ktrim=r k=23 mink=11 hdist=1 ignorebadquality=t qin=33 tpe tbo’.125 Sequencing data were aligned to the GRCh38 reference (GRCh38.d1.vd1), according to the functional equivalence standards126 and BWA-mem101 (version 0.7.17-r1188) settings ‘-Y -K 10000000’. The alignments were sorted using Sentieon tools sort, which is equivalent to samtools sort.127 Alignment statistics have been collected using the algorithms MeanQualityByCycle, QualDistribution, GCBias, AlignmentStat, and InsertSize MetricAlgo. Following sorting we followed GATK best-practices, including indel realignment, base quality score recalibration (using algorithmsRealigner and QualCal, respectively), settings ‘-k ‘DBsnp all variants 20180418’, and the ‘known_indels’ and ‘Mills_indels’ from the GATK resource bundle. The resulting alignments were genotyped at common heterozygous SNPs (DNAscope gVCF typing) and all samples from the same individual were verified to have matching genotypes ‘tpo/refs/grch38/custom/genotype_positions_hg38.bed’. Duplicates were removed using Sentieon LocusCollector and Dedup. Since the sequencing was performed on a Novaseq an optical duplicate threshold of 2500 was used to mark ExAmp duplicates ‘--optical_dup_pix_dist 2500’.
WGS Data QC
For the QC of WGS data, we derived multiple quality control measures, such as GC BIAS or duplication percentage, and confirmed that average qualities are not significantly different between the compared clinical patient subsets (Figure S1J). Downstream copy-number analysis factored DNA data quality directly into the procedure of segmentation and inference to minimize the influence of data quality on sensitivity and recall.
WGS Data Variant/Mutation Calling
Tumor-only somatic variant calling has been performed using TNScope, which is a modified version of GATK3 MuTect2 algorithm, with the following settings ‘--max_fisher_pv_active 0.05 --min_tumor_allele_frac 0.0075 --min_init_tumor_lod 2.5 --assemble_mode 4 --trim_soft_clip’ and ‘--dbsnp homo_sapiens_assembly38.dbsnp138.vcf.gz’, which is included in the TPO resource bundle.
Specifically, the ‘trim_soft_clip’ parameter reduces the number FFPE-induced artifacts. Multi-nucleotide variants were merged based on phasing information using an inhouse script ‘tpo/pipe/carat-anno/carat_mnv.R’. The resulting variants were first filtered to protein coding regions (based on Ensemble 97), annotated using VEP,116 and further annotated with vcfAnno,117 using the following sets of parameters: ‘--assembly GRCh38 --species homo_sapiens --cache_version 97 --format vcf --gene_phenotype --symbol --canonical --ccds --hgvs --biotype --tsl --uniprot --domains --appris --protein --variant_class --sift b, --polyphen b --no_stats --total_length --allele_number --no_escape --flag_pick_allele --pick_order canonical,tsl,biotype,rank,ccds,length --buffer_size 20000’. For vcfAnno multiple sources of annotations were used including:
GDC Panel-of-Normal v4136 based on MuTect2
1000 Genomes phase3 variants
GnomAD exomes r301
GnomAD exomes r221
ClinVar release Dec. 2019
Cosmic v87
dbSNP release 20180418
UniProt release Dec. 2019
The resulting annotated variants were subsequently filtered to select for likely somatic and pathogenic variants based on automated rules, gene-specific threshold, and manual filters. We applied filters based on sequencing evidence (variant allele frequency, coverage, mutation likelihood TLOD, strand bias, allele depth, multi-allelic variants), and overlap in problematic regions including regions with low-mappability and repetitive sequence as well as homopolymer repeats. In addition, all long indels (>20 bp) were removed, as these were significantly enriched in this data due to the low quality of the input DNA. All called variants in genes known to be recurrently mutated in HGSOC identified based on COSMIC and literature, were manually reviewed, according to a procedure following applicable recommendations in Barnell et al.128
Tumor Mutation Burden
The tumor mutation burden was determined for each sample by counting the number of genes that have any type of mutations. The mutation call was first determined for each gene by the above pipeline, and then for a subset of known cancer driving genes the mutation call went through additional manual check. For subjects with multiple DNA samples, the mutation of a gene was called if it was observed in any DNA sample.
CNV Analysis
Copy-number analysis on the tumor DNA was performed by using whole-genome sequencing (WGS) coverage data and variant calls (see above). To perform the analysis, we used CNVEX (https://github.com/mctp/cnvex), a comprehensive copy number analysis tool that has been used previously in our ccRCC studies.55 CNVEX uses whole-genome aligned reads to estimate coverage within fixed genomic intervals and variant calls to compute B-allele frequencies (BAFs) at variant positions (as characterized above). Coverages were computed in 10 kb bins, and the resulting log coverage ratios between tumor and normal samples were adjusted for GC bias using weighted LOESS smoothing across mappable and non-blacklisted genomic intervals within the GC range 0.3–0.7, with a span of 0.5 (all target and configuration files are provided within the CNVEX repository). The adjusted log coverage-ratios (LR) and BAFs were jointly segmented by a custom algorithm based on Circular Binary Segmentation (CBS). Alternative probabilistic algorithms were implemented in CNVEX, including algorithms based on recursive binary segmentation (RBS),129 and dynamic programming,130 as implemented in the R-package jointseg.131 For the CBS-based algorithm, first LR and mirrored BAF were independently segmented using CBS (parameters alpha=0.01, trim=0.025) and all candidate breakpoints collected.
The resulting segmentation track was iteratively “pruned” by merging segments that had similar LR and BAFs, short lengths, enrichment in blacklisted regions, and a high variation in coverage among whole cohort germline samples. For the RBS- and DP-based algorithms, joint-break-points were “pruned” using a statistical model selection method.132 For the final set of CNV segments, we chose the CBS-based results because they did not require specifying a prior number of expected segments (K) per chromosome arm, were robust to unequal variances between the LR and BAF tracks and provided empirically the best fit to the underlying data. Given the relatively high variance of the sequencing data, we noticed that some samples were over-segmented using the default settings. To adjust the segmentation algorithm to the variance of each sample individually, we performed an adaptive procedure of pruning. Default pruning: Perform pruning as described above for all samples. Strict pruning: Count the number of segments for each sample, take the samples with the top 20 percentile number of segments and perform pruning with stricter parameters. Relaxed pruning: Take the samples with the lowest 20 percentile number of segments and perform more loose pruning on them. The above procedure was repeated two times, effectively adjusting segmentation pruning parameters to the noise-level of each sample. The quality of the resulting segmentations was manually evaluated.
The resulting segmented copy-number profiles were then subject to joint inference of tumor purity and ploidy and absolute copy number states, implemented in CNVEX, which is most similar to the mathematical formalism of ABSOLUTE28 and PureCN.133 Briefly, the algorithm inputs the observed log-ratios (of 10 kb bins) and BAFs of individual SNPs. LRs and BAFs were assigned to their joint segments and their likelihood is determined given a particular purity, ploidy, absolute segment copy number, and number of minor alleles. To identify candidate combinations with a high likelihood, we followed a multi-step optimization procedure that includes gridsearch across purity-ploidy combinations, greedy optimization of absolute copy numbers, and maximum-likelihood inferences of minor allele counts. Following optimization, CNVEX ranked candidate solutions. Because the copy-number inference problem can have multiple equally likely solutions, further biological insights were necessary to choose the most parsimonious result. The solutions were reviewed by independent analysts following a set of guidelines. Solutions implying whole-genome duplication were supported by at least one large segment that cannot be explained by a low-ploidy solution, inferred purity was consistent with the variant-allele-frequencies of somatic mutations, and large homozygous segments were not allowed.
After selecting the best solution for purity/ploidy of a sample, we then used the method explained in PureCN133 to assign total absolute CN to each CNV segment using LR values. Purity and ploidy estimates also allowed us to use BAF values for calculating the absolute copy number for each allele. We then used the copy number of minor allele to detect LOH for each segment. Any segment that had a minor allele copy number of 0, was considered to harbor an LOH event.
Chr17-LOH Calling
To call samples with Chr17LOH, we used the same method mentioned above to call LOH for all the segments on chr17. Samples that had <25% of their chr17 covered with LOH segments were called chr17HET. Any other samples were placed in the chr17LOH group.
MSK-IMPACT LOH Analysis
To validate the results of our chr17LOH, CNV data from MSK-IMPACT18 (997 patients) was used. Because the LOH values were not provided for this cohort, we used the LogRatio values as a surrogate. In our own cohort, we found that Chr17-HET and Chr17-LOH have significantly different LR values (Wilcox test P=0.0023, Figure S2C). We therefore used our own cohort of patients to define a LR threshold which separates patients into two groups of “Chr17-LOH enriched” and “Chr17-HET enriched”. For this purpose, we defined samples with chr17 average LR > 0.046 as a surrogate for “Chr17-HET” (75 patients) group and samples with chr17 average LR < −0.114 as a surrogate for “Chr17-LOH” (495 patients) group. We then used this stratification to perform Cox proportional hazards survival analysis.
Instability Measures
For measuring overall instability, we used an already published measure of Weighted Genome Instability Index (wGII),134 which was measured as the proportion of each chromosome that has a different copy number compared to the baseline copy number of the sample. Then the average of scores for each chromosome was calculated, weighted by the length of the chromosome such that each chromosome has the same contribution to the overall instability score.
To quantify the HRD for each sample, we reimplemented the measures described in Sztupinszki et al.135 In short, allelic imbalance was defined as the unequal contribution of parental alleles to a region of the genome and contains LOH as well. For nLST we defined “large segment” as a segment with minimum length of 10 Mb, and to count one event of large segment transition, we allowed maximum distance of 3Mb between two “large segments” with allelic imbalances. For nTAI, we counted the allelic imbalances with minimum length of 5Mb that stretch to the telomeric end of each chromosome, which on GRCh38 we used the 10Kb of chromosome start/end as telomere. For nLOH, we counted any segment of minimum length of 15Mb which has Loss of heterozygosity. For all of these HRD measures, if the segment is affecting the whole arm of the chromosome (arm-level event), it is not counted toward the HRD measures.
RNA Sequencing Processing and Alignment
The total RNA-seq stranded paired-end reads were trimmed of adapter sequences using BBMap’s bbduk2 tool with parameters ‘minlength=25 k=31 qskip=3 rieb=t tbo=t tpe=t’, the resulting reads were aligned to a GRCh38 reference including common human oncogenic viruses using STAR, using the following settings:
--alignSJoverhangMin 8 \
--alignSJDBoverhangMin 3 \
--scoreGenomicLengthLog2scale 0 \
--alignIntronMin 20 \
--alignIntronMax 1000000 \
--alignMatesGapMax 1000000 \
--outStd “SAM” \
--outReadsUnmapped “Fastx” \
In addition, trimmed paired-end reads were merged into a synthetic single-end reads using the BBMap bbmerge tool, the resulting merged single-end and not-merged trimmed paired-end reads were aligned a second time with STAR using settings to increase the sensitivity of fusion detection (see below):
--outFilterType Normal \
--alignIntronMax 150000 \
--alignMatesGapMax 150000 \
--chimSegmentMin 10 \
--chimJunctionOverhangMin 1 \
--chimScoreSeparation 0 \
--chimScoreJunctionNonGTAG 0 \
--chimScoreDropMax 1000 \
--chimScoreMin 1
The resulting RNA-seq BAM files were genotyped using DNAscope with the RNASplitReadsAtJunction algorithm and compared to the DNA-based genotypes.
Genotype RNA alignments were used subsequently for gene expression quantification (see below) as well as quality control. The alignments were input into CODAC, a component of TPO designed to detect gene fusions, as well as perform quality control, and realignment using minimap136 and GMAP.137 The following CODAC settings were used:
- Annotation: /tpo/refs/grch38/config/codac-grch38.97.rds
- Stringency: longread-balanced-stranded
RNA-Sequencing Quantification
Total RNA-seq were quantified using Kallisto.118 First, RNA-seq FASTQ files were trimmed of adapter sequences as described above using BBMap bbduk2. Next, Kallisto was run using the default settings with ‘--rf-stranded’ against the Ensembl 97 transcript database. Kallisto estimated counts were summarized (summed) at the gene level based. Both raw counts (optionally adjusted to total sequencing depth) and TPMs were used depending on the downstream analysis.
RNA-sequencing Data Quality Control
To mitigate challenges of analyzing genomic data from archival FFPE specimens, we performed extensive quality control of the RNA sequencing (RNA-seq) to identify low quality samples based on multiple individual control variables specific to the RNA, such as estimates of PCR duplication percentage, reverse transcription template-switching artifacts as well as 5’ / 3’ bias, transcriptome coverage and number of detected splice-junctions. Robust thresholds and cut-point for the control variables were established based on a University of Michigan institutional cohort of FFPE and clinical samples.138 While we did not detect any differences in quality measures of sensitive and refractory samples, we observed variation in duplication percentages and detected splice junction numbers across sample source sites (Figure S1I). Thus, in the downstream analyses, as appropriate, sample source site has been used as a covariate in all association tests.
Preprocessing of FFPE Samples for Proteomics
Deparaffinization and Rehydration of FFPE Samples
FFPE samples were processed as previously described.139 Briefly, slide mounted FFPE tissue sections were incubated three times in xylene for 3 minutes followed by 100% (v/v) ethanol twice for 3 minutes. The tissue was then hydrated twice in 85% (v/v) ethanol for 3 minutes, 70% (v/v) ethanol for 3 minutes, and distilled water for 3 minutes. The tissue was then blotted and scraped off the slide into a screw cap microfuge tube.
Protein Extraction from FFPE Samples
For each sample tube containing 10 μm FFPE tissue sections, extraction buffer (0.2% RapiGest in 50 mM NH4HCO3) was added and incubated at 95°C for 30 minutes with mixing at 1000 RPM (Thermomixer, Eppendorf). The samples were then cooled on ice for 5 minutes and sonicated twice in a cup horn probe (filled with ice water) at 50% power for 30 seconds. The samples were then incubated at 80°C for 120 minutes with mixing at 1000 RPM and then cooled on ice for 5 minutes. 100 μL of 50 mM NH4HCO3, pH 8.0 was added, and the samples were sonicated twice in the cup horn probe (filled with ice water) at 50% power for 30 seconds. Following processing, samples were stored at −80°C until the day of trypsin digestion.
Preprocessing of Frozen Samples for Proteomics
Protein Extraction of Frozen Samples
Frozen tissues were placed in Covaris tissue bags (TT1, 520001) and cryo-pulverized using a cryoPREP CP-02 (Covaris). 1 mL of lysis buffer (25 mM Tris, 6 M Urea, 1 mM EDTA, 1 mM EGTA, 1 mM TCEP, 1% Sigma phosphatase inhibitor cocktail 3, 1% Sigma phosphatase inhibitor cocktail 2) was added to each sample, transferred to a microfuge tube, vortexed for 10–15 seconds and sonicated three times in a cup horn probe (filled with ice water) at 50% power for 30 seconds. The samples were stored in liquid nitrogen until the day of digestion.
Discovery Mass Spectrometry
The protocols below describe the tryptic digestion, tandem mass tag (TMT) labeling of peptides, peptide fractionation by basic reversed-phase liquid chromatography, phosphopeptide enrichment using immobilized metal affinity chromatography, and LC-MS/MS performed for profiling 20 unique tumor samples in the FFPE Validation, 158 in the FFPE Discovery, and 64 in the Frozen Validation cohorts.
Trypsin Digestion
Protein concentration was quantified by Micro BCA Assay (ThermoFisher). 100 μg of protein of FFPE or 500 μg frozen tissue lysates (diluted to 2 mg/mL in lysis buffer) was transferred to a deep-well plate for processing on an epMotion 5075 liquid handler (Eppendorf, Enfield, CT). Lysates were reduced in 16.5 mM TCEP per mg protein for 30 minutes at 37°C with shaking, followed by alkylation with 36 mM IAM per mg protein in the dark at room temperature. Lysates were then diluted with 200 mM TRIS (pH 8.0), to a urea concentration of < 1M before Lys-C (Wako) was added to lysates at 1:50 (enzyme:protein) ratio by mass and incubated for 2 hours at 37°C with mixing at 600 RPM (Thermomixer, Eppendorf). After 2 hours, trypsin (Promega) was added at 1:100 enzyme:protein. Digestion was carried out overnight at 37°C with mixing at 600 RPM. After 16 hours, the reaction was quenched with formic acid (FA; final concentration 1% by volume).
Desalting
Samples were desalted using Oasis HLB 96-well plates (Waters Cat# 186000128 for 500 μg aliquots and Cat# 186000309 for 100 μg aliquots) and a positive pressure manifold (Waters). The plate wells were washed with 3 × 400 μL of 50% MeCN/0.1% FA, and then equilibrated with 4 × 400 μL of 0.1% FA. The digests were applied to the wells, then washed with 4 × 400 μL 0.1% FA before being eluted drop by drop with 3 × 400 μL of 50% MeCN/0.1% FA. The eluates were lyophilized, followed by storage at −80°C until use.
11-plex TMT Experimental Layout
The FFPE Validation cohort was analyzed in two TMT 11-plex groups, the FFPE Discovery was analyzed in 21 TMT 11-plex groups, and the Frozen was arranged in 8 TMT 11-plex groups. For each TMT 11-plex group, 10 individual tumors occupied the first 10 channels, and the 11th channel was a “Bridge Channel” (i.e., common reference sample, used for quantitative comparison across all the TMT 11-plex groups). The common reference sample consisted of a mixed of an equal amount of protein from each HGSOC sample in the experiment, except in the case of the FFPE Discovery experiment, where, due to sample limitations, 58 of the 158 (37%) samples were included in the common reference sample (its composition was checked to ensure that this subset was representative of the whole sample set in terms of collection site and tumor treatment response). Replicate samples were randomly dispersed in the TMT 11-plex groups, with triplicates of five samples included in the FFPE Discovery experiment and triplicates of four samples in the Frozen Validation experiment. A mix of cell lysate was included in each TMT 11-plex group of the FFPE Discovery and Frozen Validation experiments, and the common reference sample from the Frozen Validation cohort was also included in each TMT 11-plex group of the FFPE Discovery experiment. The composition and run order of each TMT group were randomized to avoid bias, ensuring equal distribution of tissue source site, tumor response, age of tumor block, and hemoglobin content across and within the TMT groups.
TMT Isobaric Labeling of Peptides
Individual trypsin digestion samples were labeled with the TMT11plex™ isobaric label reagent set (TMT; ThermoFisher Scientific, Cat# A34808). Desalted peptides were resuspended in 50 mM HEPES at 1 mg/mL based on starting protein mass. TMT reagents were resuspended in 257 μL MeCN and transferred to the peptide sample. Samples were incubated at room temperature for 1 hour with mixing. Labeling reactions were quenched by the addition of 50 μL of 5% hydroxylamine (Sigma) and incubated for 15 minutes at room temperature with mixing. The independent labeling reactions were then pooled together and lyophilized. The labeled peptides were desalted as above and then lyophilized and stored at −80°C.
Peptide Fractionation by Basic Reversed-Phase Liquid Chromatography (bRPLC)
Labeled and mixed samples were fractionated by high-pH reverse phase (bRP) liquid chromatography. Frozen tissue samples were fractionated prior to IMAC enrichment. For the FFPE samples, the IMAC enrichment flow through was fractionated. Each sample was brought up in 0.5 mL mobile phase (A) and loaded onto a LC system consisting of an Agilent 1200 HPLC (Agilent, Santa Clara, CA) with mobile phases of 5 mM NH4HCO3, pH 10 (A) and 5 mM NH4HCO3 in 90% MeCN, pH 10 (B). The peptides were separated by a 4.6 mm × 250 mm Zorbax Extend- C18, 3.5 μm, column (Agilent Cat# 770953–902) over 96 minutes at a flow rate of 1.0 mL/min by the following timetable: hold 0% B for 9 minutes, gradient from 0 to 10% B for 4 minutes, 10 to 28.5% B for 50 minutes, 28.5 to 34% B for 5.5 minutes, 34 to 60% B for 13 minutes, hold at 60% B for 8.5 minutes, 60 to 0% B for 1 minute, re-equilibrate at 0% B for 5 minutes. 1-minute fractions were collected from 0–96 minutes by the shortest path by row in a 1 mL deep well plate (Thermo Cat# 95040450). The high pH RP fractions were concatenated into 24 samples by every other plate column (e.g., sample 1 contained fractions from wells A1, C1, E1, etc.). For the frozen tissue samples, 95% of every 12th fraction of the 24 samples was combined (1,13; 2,14; ...etc.) to generate 12 samples, which were dried down and stored at −80°C prior to phosphopeptide enrichment.
Immobilized Metal Affinity Chromatography (IMAC)
IMAC enrichment was performed on a KingFisher (Thermo Scientific) platform using Ni-NTA-agarose beads (Qiagen Cat# 36113) stripped with EDTA and incubated in a 10mM FeCl3 solution to prepare Fe3+-NTA-agarose beads. For the frozen tissue samples, bRP fractions were reconstituted in 200 μL of 0.1% trifluoroacetic acid (TFA; Sigma Cat# 302031) in 80% MeCN. For the FFPE tissue samples, unfractionated samples were reconstituted in 600 μL of 0.1% TFA in 80% MeCN and split into three aliquots. The samples were incubated for 30 minutes with 100 mL of the 5% bead suspension while mixing at room temperature. After incubation, beads were washed 3 times with 300 μL of 0.1% TFA in 80% MeCN. Phosphorylated peptides were eluted from the beads using 200 μL of 70% MeCN, 1% ammonium hydroxide for 1 minute with agitation at room temperature. 30 μL of 20% FA was added to each sample, and the FFPE aliquots were pooled back to their original samples. The samples were dried down, re-suspended in 0.1% FA, 3% MeCN and frozen at −80°C until analysis. For the FFPE samples, the flow through of each aliquot was pooled back to their original samples, dried down and stored at −80°C prior to bRP fractionation.
Nano-Liquid Chromatography-Tandem Mass Spectrometry (nano LC-MS/MS)
Fractionated samples were analyzed by LC-MS/MS on an Easy-nLC 1000 (Thermo Scientific) coupled to an LTQ-Orbitrap Fusion mass spectrometer (Thermo Scientific) operated in positive ion mode. The LC system consisted of a fused-silica nanospray needle (PicoTip™ emitter, 50 mm ID × 20 cm, New Objective) packed in-house with ReproSil-Pur C18-AQ with mobile phases of 0.1% FA in water (A) and 0.1% FA in MeCN (B) and a flow rate of 300 nL/min. The peptide sample was diluted in 12 μL of 0.1% FA, 3% MeCN and 5 μL was loaded onto the column. A spray voltage of 2200 V was applied to the nanospray tip. MS/MS analysis occurred over a 3 second cycle time consisting of 1 full scan MS from 350–2000 m/z at resolution 120,000 followed by data dependent MS/MS scans using HCD activation with 40% normalized collision energy of the most abundant ions. Selected ions were dynamically excluded for 20 seconds after a repeat count of 1. Global proteome samples were separated over 206 minutes with a gradient from 2 to 5% B for 2 minutes, 5 to 28% B for 180 minutes, 28 to 50% B for 10 minutes, hold 50% B for 1 minute, 50 to 90% B for 2 minutes, hold 90% B for 11 minutes. Phosphoproteome samples were separated over 176 minutes with a gradient from 2 to 5% B for 2 minutes, 5 to 28% B for 150 minutes, 28 to 50% B for 10 minutes, hold 50% B for 1 minute, 50 to 90% B for 2 minutes, hold 90% B for 11 minutes. The unfractionated phosphoproteome samples from the FFPE samples were analyzed by LC-MS/MS on an LTQ-Orbitrap Eclipse mass spectrometer (Thermo Scientific) operated in positive ion mode with the same LC system and setup as on the Fusion. MS/MS analysis occurred over a 1 second cycle time consisting of 1 full scan MS from 350–2000 m/z at resolution 120,000 followed by data dependent MS/MS scans using HCD activation with 38% normalized collision energy of the most abundant ions. Selected ions were dynamically excluded for 20 seconds after a repeat count of 1. Samples were separated over 162 minutes with a gradient from 4 to 9% B for 2 minutes, 9 to 25% B for 80 minutes, 25 to 44% B for 60 minutes, 44 to 63% B for 8 minutes, hold 63% B for 1 minute, 63 to 90% B for 11 minutes.
Mass Spectrometry Data Analysis
Raw MS/MS spectra from the analysis were searched against the reviewed Human Universal Protein Resource (UniProt) sequence database release 2019_06 appended with an equal number of decoy sequences using MSFragger V3.0.95 For the analysis of whole proteome data, MS/MS spectra were searched using a precursor-ion mass tolerance of 10 ppm, allowing C12/C13 isotope errors (−1/0/1/2/3). MS and MS/MS mass calibration, MS/MS spectral deisotoping, and parameter optimization were enabled.140 Cysteine carbamidomethylation (+57.0215), lysine TMT labeling (+229.1629), and peptide N-terminal TMT labeling were specified as fixed modifications. Methionine oxidation (+15.9949) was specified as a variable modification. The search was restricted to tryptic and semi-tryptic peptides, allowing up to two missed cleavage sites. For phosphopeptide enriched data, the set of variable modifications also included phosphorylation (+79.9663) of serine, threonine, and tyrosine residues.
The post-processing of the search results was done using the Philosopher toolkit version v3.2.8.94 MSFragger output files (in pepXML format) were processed using PeptideProphet96 (with the high–mass accuracy binning and semi-parametric mixture modeling options) to compute the posterior probability of correct identification for each peptide to spectrum match (PSM). In the phosphopeptide-enriched dataset, PeptideProphet files were additionally processed using PTMProphet97 to localize the phosphorylation sites. The resulting pepXML files from PeptideProphet (or PTMProphet) from all 25 TMT 11-plex experiments were then processed together to assemble peptides into proteins (protein inference) and to create a combined file (in protXML format) of high confidence protein groups.
The combined protXML file and the individual PSM lists for each TMT 11-plex were further processed using the Philosopher filter command as follows. Each peptide was assigned either as a unique peptide to a particular protein group or set as a razor peptide to a single protein group with the most peptide evidence. The protein groups assembled by ProteinProphet141 were filtered to 1% protein-level False Discovery Rate (FDR) using the best peptide approach (allowing both unique and razor peptides) and applying the picked FDR target-decoy strategy. In each TMT 11-plex, the PSM lists were filtered using a sequential FDR strategy, retaining only those PSMs with PeptideProphet probability of 0.9 or higher (which in these data corresponded to less than 1% PSM-level FDR) and mapped to proteins that also passed the global 1% protein-level FDR filter. For each PSM that passed these filters, the corresponding precursor ion MS1 intensity was extracted using the Philosopher label-free quantification module, using 10 ppm mass tolerance and 0.4 min retention time window for extracted ion chromatogram peak tracing.
For all PSMs corresponding to a TMT-labeled peptide, eleven TMT reporter ion intensities were extracted from the MS/MS scans (using 0.002 Da window). The precursor ion purity scores were calculated using the intensity of the sequenced precursor ion and that of other interfering ions observed in MS1 data (within a 0.7 Da isolation window). All supporting information for each PSM, including the accession numbers and names of the protein/gene selected based on the protein inference approach with razor peptide assignment and quantification information (MS1 precursor-ion intensity and the TMT reporter ion intensities), was summarized in the output PSM.tsv files, one file for each TMT 11-plex experiment.
To generate summary reports on different levels (gene, peptide, and protein for global and phosphopeptide enriched data; additional modification site report for phosphopeptide data), all PSM.tsv files were processed together using TMT-Integrator98 using default parameters described previously.55 In generating the site-level reports (phosphopeptide-enriched data), sites with PTMProphet computed localization probability equal or greater than 0.75 were considered as confidently localized.
Quality Control Metrics for Peptide-level Data
Raw MS/MS spectra from the analysis were searched against the reviewed Human Universal Protein Resource (UniProt) sequence database release 2020_03_30 using MaxQuant/Andromeda.99 The search was performed in unspecific digestion mode, oxidized methionine set as a variable modification, and carbamidomethylated cysteine set as a static modification. Peptide MH+ mass tolerances were set at 20 ppm. The overall PSM-level FDR was set at≤1%.
Peptides were categorized into fully tryptic, semi-tryptic (either tryptic end), and non-tryptic. For the FFPE Discovery Cohort dataset, peptides were filtered to have a ratio in at least one sample. To calculate the non-tryptic ratio per sample, the median ratio of all non-tryptic peptides (n = 2877) was divided by the median ratio of all fully tryptic peptides (n = 126952). To calculate the semi-tryptic ratio per sample, the median ratio of all semi-tryptic peptides (n = 21426) was divided by the median ratio of all fully tryptic peptides (n = 126952). For the Frozen Discovery Cohort dataset, peptides were filtered to have a ratio in at least one sample. To calculate the non-tryptic ratio per sample, the median ratio of all non-tryptic peptides (n = 2190) was divided by the median ratio of all fully tryptic peptides (n = 114620). To calculate the semi-tryptic ratio per sample, the median ratio of all semi-tryptic peptides (n = 23257) was divided by the median ratio of all fully tryptic peptides (n = 114620). To calculate the missed cleavage ratio per sample, the median ratio of fully tryptic peptides with at least one internal K or R (that was not followed by P) was divided by the median ratio of fully tryptic peptides with no internal K or R (or those that are followed by P). To calculate the oxidation ratio, the median ratio of fully tryptic peptides that were identified to contain an oxidized M (regardless of site localization score) was divided by the median ratio of non-oxidized fully tryptic peptides. To calculate the c-term ratio per sample, the median ratio of all tryptic peptides with an K at the c-terminal was divided by the median ratio of all tryptic peptides with an R at the c-terminal.
Proteomics data quality assessment using peptide-level QC metrics
First, distribution of the number of peptides quantified per protein is shown in Figure S1G. In all the three study cohorts (Frozen, FFPE discovery, and FFPE validation), no more than 5% of the proteins were quantified by a single peptide. Proteomic data quality was assessed using peptide-level metrics derived from the raw LC-MS/MS data as described in the previous section (Figures S1D and S1H). No association was detected between these metrics and treatment response or sample storage time, but a substantial variation in the distribution of three metrics (semi-tryptic ends, C-terminal lysines, and missed cleavages) across sample source sites, possibly due to different biospecimen handling. Hence, to account for these variations, sample source sites have been included as covariates in all downstream analysis.
Preprocessing of Proteomics Data
For the global and phospho proteomics data, after removing the reference bridge samples from every TMT, we performed global normalization by aligning the sample median and scaling by median absolute deviation to remove any systematic variation across the samples.
Next, we performed outlier removal at the TMT group level. For every protein, we performed simple t-tests to compare samples in every TMT group with the remaining samples. If the data-points of this protein in one TMT-plex were found to be outliers, i.e., significantly different from the other TMT-plex (p-value cutoff = 10−7), we replaced all data-points in the outlier TMT-plex by NA. In the FFPE Discovery data set, we removed 1334 and 176 outlier data points from the gene level global proteomics and phosphosite-proteomics data, respectively. Next, we filtered proteins based on the missing rate. We removed proteins that were observed in fewer than 50% of the sensitive or refractory samples. We applied batch correction on global and phospho-filtered normalized data to remove the technical difference (batch-effect) between different TMT-plexes. We used an R tool: ComBat to remove batch-effect.100 Finally, we performed imputation of the missing values using the tool DreamAI.142 We also quantified the number of proteins and phosphosites observed per sample in the batch-corrected filtered data based on FFPE discovery, FFPE validation and Frozen cohort (Table S1).
Note that the overall missing rate in the global proteomics data is quite low, as illustrated in Figure S1E. While we used “a missing rate <50% in either the sensitive or refractory group” as the filtering criterion, only 48 out of 8800 proteins had missing rate above 50% in the complete cohort, and the maximum missing rate is 52%. For the majority of the proteins (83%), the imputed data points were less than 10%. In the FFPE phospho data, as expected from archival samples, the missing rates were in general higher than that of the global proteomics data (Figure S1F). As mentioned in the “limitation of this study” section of the manuscript, the reliance on archival FFPE biospecimens results in a reduced ability to study post-translational modifications (compared with frozen biospecimens). Nevertheless, only 67 out of 2648 phosphosites required us to impute 50–54% of datapoints.
Alignment of proteomics data from Frozen samples to FFPE discovery samples
After we processed two data sets separately, we then aligned frozen samples to the protein intensities of FFPE discovery samples. Between frozen and FFPE cohorts, there are 29 overlapping samples. In addition, there are 14 other FFPE-Frozen pairs of samples coming from different tumors (primary and metastasis) of the same patients. We thus utilized all 43 common samples between these two experiments to align the two proteomic data sets. For each protein identified in both experiments, mean value and standard deviation of intensities among the 43 common samples were calculated from both experiments in the observed data sets before missing value imputation. We estimated a linear transform on the 43 frozen samples, in order to align the mean and SD intensities to those of the 43 FFPE discovery samples. And applying the same linear transform to the entire frozen sample experiment data, we obtained the aligned proteomics data of frozen samples to the FFPE discovery samples.
Proteomics data quality assessment using replicate samples
To assess reproducibility in TMT experiments, 5 replicate samples from the FFPE Discovery cohort were distributed across different TMT-plexes. We observed high correlation among global (R > 0.99) and phospho--proteomic (R > 0.975) profiles of the replicate samples (Figure S1B). The Frozen Validation global/phospho-proteomics experiments included a set of 4 replicate samples, and sample-wise correlations were similarly high (R > 0.99 for global; R > 0.98 for phospho-proteomic data). For the 29 tumors with paired FFPE and frozen biospecimens, we also observed high correlation (R = 0.980 – 0.998) between the global and phospho--proteomic profiles of the paired samples (Figure S1C). These replicate data (Table S1) demonstrate well controlled technical variations in the proteomic experiments.
Sample Labeling Check for Proteogenomic Profiles
While integration of the multiple omics data enhances our understanding about complex molecular mechanisms in PTRC, unintended errors in annotations and sample labels often occur in generation or management of large-scale data.143 Sample mislabeling includes swapping, shifting, or duplicating. Therefore, we performed a systematic quality control procedure to confirm whether the samples in RNAseq, CNV, global proteomics, and phosphor proteomics profiles are from the same individuals as annotated. We applied a pairwise alignment procedure144 using samples with the same labels between RNAseq vs CNV, RNAseq vs global, global and phosphor profiles. Each pairwise alignment was performed in following sequences: 1) Cis genes between two data profiles were identified based on correlation coefficient (top 1000 positively correlated & Pearson correlation test FDR < 0.01); 2) The values of the selected cis genes were rank transformed in both profiles; 3) The sample similarity scores of samples by samples are estimated as correlation coefficients of the rank-transformed values; 4) If a sample is matched between two types of data, the sample similarity score is expected to be higher compared to the score with others which has null distribution with mean 0. If the sample similarity score of a sample is above the top 5% in both directions, the sample is determined as “self-aligned”. Or if the score is below the top 5% in either direction, the sample was called as “not-aligned”. Applying the approach to samples with the same labels, we confirmed that all samples were well aligned between RNAseq and CNV (111 samples from the same individuals), RNAseq and global (120 samples), and global and phosphoproteomics (168 samples).
Targeted Mass Spectrometry
We performed a technical validation of the predictor model using an orthogonal assay based on targeted multiple reaction monitoring mass spectrometry (MRM-MS). We developed and characterized an MRM assay panel enabling quantification of 70 peptides representing 22 proteins in our predictor panel (Figure 3). This subset of 22 proteins was selected for assay development because their signals were sufficient for direct detection by mass spec, without antibody enrichment. We applied this 70-plex assay to the 102 FFPE tumors in our study (all tumors with enough remaining sample). The protocols below describe the internal standard addition, tryptic digestion, and LC-MRM performed on 102 samples of the FFPE Discovery cohorts.
LC-MRM-MS assay development
Proteotypic peptides were empirically identified from the mass spectrometry-based datasets from the discovery experiments. Crude synthetic peptides were used to determine the optimum transitions (based on intensity and freedom from noise) and collision energies (to produce the most complete fragmentation and highest intensity) for MRM analysis. The optimized transition ions are reported in Table S1.
Trypsin Digestion
Protein concentration was quantified by Micro BCA Assay (ThermoFisher). 13.5 μg of protein of FFPE was transferred to a deep-well plate for processing on an epMotion 5075 liquid handler (Eppendorf, Enfield, CT). Lysates were reduced in ~20 mM TCEP per mg protein for 30 minutes at 37°C with shaking, followed by alkylation with ~40 mM IAM per mg protein in the dark at room temperature. Lysates were then diluted 10x with 200 mM TRIS (pH 8.0), 400 fmol of stable isotope-containing synthetic peptide standards (Vivitide) were spiked into each sample, and Lys-C (Wako) was added to lysates at 1:35 (enzyme:protein) ratio by mass and incubated for 2 hours at 37°C with mixing at 600 RPM (Thermomixer, Eppendorf). After 2 hours, trypsin (Promega) was added at 1:70 enzyme:protein. Digestion was carried out overnight at 37°C with mixing at 600 RPM. After 16 hours, the reaction was quenched with formic acid (FA; final concentration 1% by volume).
Desalting
Samples were desalted using Oasis HLB 96-well plates (Waters Cat# 186001828BA) and a positive pressure manifold (Waters). The plate wells were washed with 3 × 200 mL of 50% MeCN/0.1% FA, and then equilibrated with 4 × 200 mL of 0.1% FA. The digests were applied to the wells, then washed with 4 × 200 mL 0.1% FA before being eluted drop by drop with 3 × 200 mL of 50% MeCN/0.1% FA. The eluates were lyophilized, followed by storage at −80°C until use.
Nano-liquid chromatography-tandem mass spectrometry
Targeted LC-MRM-MS analysis of 20% of sample loaded onto an EvoTip Pure (EvoSep Cat #EV2011) was performed on an Evosep One LC system (EvoSep, Odense, Denmark) coupled to a 6500+ QTRAP mass spectrometer (Sciex, Foster City, CA). Mobile phases consisted of 0.1% FA in water (A) and 0.1% FA in MeCN (B), and the EvoSep was running the 30 Samples per Day method while connected to an 15 cm × 150 μm analytical column with 1.9 μm beads (Endurance OE. EvoSep Cat. #EV-1113) with the column temperature set to 45 °C. The column was connected to an OptiFlow Turbo V source (Sciex, Foster City, CA) operating in the Nano (< 10μL) configuration. The QTRAP was operated in positive ion MRM mode with a 3000 V ion spray voltage, curtain gas setting of 20, collision gas settings of 10, 350 °C temperature, and ion source gas 1 set to 6. CE was set to optimized values for individual transitions, DP was set to 100, EP was set to 10, CXP was set to 6, Q1 and Q3 set to unit/unit resolution (0.7 Da), and a 0 ms settling time and a 3 ms pause between mass ranges were employed.
Fit-for-purpose assay validation
Performance figures of merit for the multiplexed LC-MRM-MS assay were determined using best practices in a fit-for-purpose method validation approach.145,146 Briefly, three experiments (described below) were performed to characterize the analytical performance of the assays: i) response curves, ii) repeatability, iii) stability.
Response curves
Response curves were used to characterize the linear range, LOD, LLOQ, and ULOQ. Curves were performed in both FFPE and frozen tissue protein lysate matrices. Synthetic unlabeled (“light”) crude peptides were also added at a constant concentration. Varying the heavy peptide amounts enables estimation of the linear range and detection limits directly in the background matrix of interest without interference from the endogenous peptides.145 Quantitative assays were characterized in background matrices consisting of a mix of protein lysates from 6 commercially available HGSOC FFPE tissues. Digestion was performed as described above. Reverse curves were prepared in triplicate by varying SIS peptide concentration over 8 concentration points (5000, 1000, 200, 40, 16, 6.4, 2.56, 1.024 fmol/sample). Blanks contained no SIS peptide. Light peptide was added at a constant concentration of ~400 fmol/sample. The response was measured by evaluating the heavy peptide signals relative to the light endogenous peptides. Linear regression was performed using a 1/x weighting on all points within the linear range, defined as those points such that the R2 > 0.98. The LOD was defined as the average of the blank measurements plus three times the standard deviation of the noise. The LLOQ was defined by the lowest point above the LOD in the linear range of the response with a CV < 20%. The ULOQ was defined as the highest concentration point of the response curve that was maintained in the linear range of the response.
Repeatability
Repeatability was determined using the same pooled lysate matrix used to generate the response curves. Heavy peptides spiked into the digest from 50 μg (FFPE tissue lysate) or 100 μg (frozen tissue lysate) aliquots at three different concentrations: “LoQC” (40 fmol per sample), “MedQC” (~400 fmol/sample) and “HiQC” (4000 fmol/sample). Light peptides were added at 400 fmol/sample. Complete process triplicates (including digestion, capture, and mass spectrometry) were prepared and analyzed on five independent days. Intra-assay variation was calculated as the mean CV obtained within each day. Inter-assay variation was the mean CV calculated from the individual replicates across the five days.
Peptide stability
Stability of the enriched peptides was determined using the same pooled lysate matrix used to generate the response curves. Heavy peptides were spiked in at the medium concentration (400 fmol/sample). Light peptides were added at 400 fmol/sample. Complete process triplicates (including digestion, capture, and mass spectrometry) were prepared and analyzed after storage at 4°C in the autosampler for approximately 6 hours and 24 hours. Other aliquots were analyzed following 2 freeze-thaws.
MRM mass spectrometry data analysis
MRM peak integration was performed by Skyline,122 and the integrations were manually inspected to ensure correct peak detection, absence of interferences, and accurate integration. Reported peak areas are the sum of the peak area and background area reported by Skyline. The sum of all transitions with no interferences was used for quantification. The individual transitions were used to confirm the specificity of the assay for the targeted analyte. Specificity was established by equivalent elution times and equivalent relative peak intensities for transitions for light and heavy peptides. Peak specificity between the light (or endogenous) and heavy (or standard) MRM signal was defined as the detection of ≥1 transition from the endogenous peptide exactly co-eluting with ≥2 transitions from the stable isotope-labeled peptide, with a ratio dot product > 0.9 as reported by Skyline. Integration areas of both the heavy and light peaks were compared to the LLOQ and ULOQ areas of the assay. Light endogenous areas below the LLOQ were reported as “LLOQ” and heavy standard areas below the LLOQ are reported as “NA”. Peptide levels were reported as the peak area ratio of the light and heavy peptides. Protein abundance levels were calculated using the averages peak area ratios of the peptides in the protein. Protein targets with < 20% of the measurements below LLOQ were not considered in further analysis (Table S3).
Multiplexed IHC
Sample processing
Slide-mounted FFPE sections were dewaxed and stained on a Leica BOND Rx autostainer (Leica, Buffalo Grove, IL) using Leica Bond reagents for dewaxing (Dewax Solution), antigen retrieval and antibody stripping (Epitope Retrieval Solution 2) and rinsing after each step (Bond Wash Solution). A high stringency wash was performed after the secondary and tertiary applications using high-salt TBST solution (0.05M Tris, 0.3M NaCl, and 0.1% Tween-20, pH 7.2–7.6).
Multiplexed IHC target panel
Details of antibodies for CD8, CD4, and CCR5 are provided in Table S6
Antigen retrieval and antibody stripping
Antigen retrieval and antibody stripping steps were performed at 100°C with all other steps at ambient temperature. Endogenous peroxidase was blocked with 3% H2O2 for 8 minutes followed by protein blocking with TCT buffer (0.05M Tris, 0.15M NaCl, 0.25% Casein, 0.1% Tween 20, pH 7.6 +/− 0.1) for 30 minutes. The first primary antibody (position 1) was applied for 60 minutes followed by the secondary antibody application for 10 minutes and the application of the tertiary TSA-amplification reagent (OPAL fluor, Akoya Biosciences, Menlo Park, CA) for 10 minutes. The primary and secondary antibodies were stripped with retrieval solution for 20 minutes before repeating the process with the second primary antibody (position 2) starting with a new application of 3% H2O2. The process was repeated until all positions were completed; however, there was no stripping step after the last position. Slides were removed from the autostainer and stained with Spectral DAPI (Akoya) for 5 minutes, rinsed for 5 minutes, and cover-slipped with Prolong Gold Antifade reagent (Invitrogen/Life Technologies, Grand Island, NY).
Image collection
Slides were cured for 24 hours at room temperature in the dark, then representative images from each slide were acquired on the Akoya PhenoImager HT Automated Imaging System. Images were spectrally unmixed using Akoya inForm software and exported as multi-image TIFF’s for use in the HALO Link image management system (Indica Labs, Corrales, NM).
Image analysis
Cellular analysis of the images was then performed with HALO image analysis software. After the cells were visualized based on nuclear and cytoplasmic stains, the software measured mean pixel fluorescence intensity in the applicable compartments of each cell (e.g., Ki67 in the nuclear compartment). A mean intensity threshold above background was used to determine positivity for each fluorochrome, thereby, defining cells as either positive or negative for each marker. The positive cell data was then used to define colocalized populations and to perform spatial analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Multiple Linear Regression Model for Association Tests
We performed a multiple linear regression model to test for the association of the individual gene/protein/phosphosite with platinum response. Specifically, we considered the following model for testing the association:
gene/protein/phosphosite ~ response (platinum sensitive/platinum refractory) + tumor location + source site + age + neoadjuvant treatment status + tumor purity
For identifying the genes/proteins/phosphosites differing between platinum sensitive and platinum refractory samples, we tested for the regression coefficient of the factor platinum response conditional on other covariates and obtained the marginal p-values.
After adjusting for multiple hypothesis testing using Benjamini-Hochberg (BH) method, we obtained the adjusted p-values for each individual gene/protein/phosphosite. We consider a gene/protein/phosphosite significantly associated with treatment response if its adjusted p-value is < 0.1.
To define sensitive and refractory cases from the CPTAC-201619 cohort, we used the patients’ overall survival (OS) information. Specifically, we defined the sensitive patients as those with OS > 5.5 years and refractory cases to be those with OS < 1.5 years.
CNV, RNA and Protein Combined Analysis
We performed an association test to identify if any individual genes/proteins or copy number of genes is associated with sensitive or refractory response. We then combined the p-values from the association analysis based on protein, RNA and copy number data using a Fisher’s test and derived corrected p-values (FDR) after multiplicity correction. For 8590 genes observed in all three omics datasets: protein, RNA and CNV, 3600 genes showed consistent direction (either “up in sensitive” or “up in refractory”) in all three omics data. Out of 3600, 1962 genes were consistently “up in sensitive” based on protein, RNA and CNV; while 1638 genes were consistently “up in refractory” based on all three datasets. Out of 3600 genes, that showed consistent direction in all three datasets, 424 genes satisfy the “combined_FDR” < 0.1, out of which 303 genes were consistently “up in sensitive” and 121 genes were consistently “up in refractory” based on all three omics data. Out of these 424 genes, 53 were validated (marginal p-value < 0.05) in at least one of the independent validation proteomics data sets including the Frozen validation, FFPE validation and CPTAC 201619 proteomics data (Table S1). Based on the Frozen cohort alone, 17 genes were validated, out of which 3 were “up in refractory” while 14 were “up in sensitive”. Based on the FFPE Validation cohort alone, 9 genes were validated, out of which 1 was “up in refractory” while 8 were “up in “sensitive”. Finally, out of the CPTAC2 cohort19,147 alone, 23 genes were validated, out of which 4 were “up in refractory” while 19 were “up in sensitive”.
iProFun-Based Cis Association Analysis
We used iProFun, an integrative analysis tool, to identify multi-omic molecular quantitative traits (QTs) perturbed by cis DNA-level variations.27 iProFun starts with separate regressions for different QTs and integrates their association summaries. We considered three regressions separately for gene expression, global protein and phosphoprotein. For each regression, we evaluated the cis-associations with three DNA-level variations (somatic mutation, CNV (dosage change) and LOH). We controlled age, tumor location and tumor purity as covariates for all regressions and controlled RNA quality control index for regression on gene expression. Most importantly, to understand how associations differ between sensitive and refractory tumors, we included the main effect of tumor response (sensitive vs. refractory) and its interactive effects with three DNA-level alterations (tumor response * somatic mutation, tumor response * CNV, and tumor response * LOH) in each regression. For different data types, the number of genes and samples may be different, and we used all quality-controlled data available. In detail, we performed regression for 19605 genes in 106 samples for gene expression, 8800 genes in 124 samples for protein, and 1278 genes (2648 phosphosites) in 124 samples for phosphoprotein. As phosphoprotein was summarized at site level in the data, all phosphosites were considered in regression and the site most significantly associated with DNA-level variations and refractory tumors was used to indicate the strongest signals for each gene. We considered a total of 542 somatic mutations with mutation in at least 6 subjects, and 19920 CNVs and 19269 LOHs with <= 50% missing rate.
The resulting association summary statistics were used in iProFun to call probabilities of belonging to each of the 23=8 possible configurations. This procedure borrowed information across data types to improve the estimation of association probabilities. For genes missing quantifications on certain data type(s) (e.g., phosphoprotein), iProFun calculates probabilities of belonging to each of the remaining configurations (e.g., 22=4). The resulting probabilities of associating with each QT was calculated by combining probabilities from relevant configurations. An association is identified in iProFun if (1) the empirical false discovery rate (eFDR) is <10%, (2) posterior probability is > 75% and (3) the association direction is consistent with directions of the same DNA-level variation on different data types if they satisfy (1) and (2). As the number of somatic mutations was small and association probabilities could not be accurately estimated, we used family-wise error rate (FWER) 10% to call significance directly from the association p-values (without borrowing information across data types).
TP53 Signatures
Previously published gene sets were used to develop the TP53 wild-type and mutant signatures. For the wild-type TP53 signature, we used the previously identified core TP53 transcriptional program based on Andrysik et al.32 We focused on the key 31 genes with direct bounding and identified in all three cell line experiments (HCT116, MCF7, SJSA). For the mutant TP53 signature, we used the previously published analysis.29 The top 20 upregulated genes across all mutant TP53 cancers were selected for the signature. Using the GSVA package (method = “ggsea”) in R,148,123 we obtained the ssGSEA scores for the TP53 wild-type and TP53 mutant gene sets for all samples in the FFPE Discovery RNA-seq dataset (n = 106). Additionally, we obtained the ssGSEA scores for the TP53 wild-type and TP53 mutant gene sets for all samples in the FFPE Discovery proteomics dataset (n = 158).
We then performed a multiple linear regression model to test for the association of each TP53 signature with platinum response. Specifically, we considered the following model for testing the association: ssGSEA scores (WT or mutant) ~ platinum response + tumor location + source site + sample age + neoadjuvant treatment status + tumor purity.
Identification of Proteins/Pathways Associated with genomic abnormalities
To understand the functional consequences of the genomic abnormality events, we evaluated the proteins and pathways associated with chr17-LOH, nTAI, BRCA1/2 mutation and total mutation burden (TMB). We started by regressing the protein level of each gene on these somatic alterations, adjusting for each other, as well as patient age, tumor location and tumor purity. To ensure a fair comparison of association strength between binary (BRCA1/2 mutation vs WT, chr17-LOH vs no chr17-LOH) and continuous predictors (nTAI and TMB), we dichotomized the nTAI (>15 v.s. <=15) and TMB (>100 vs <=100) into high vs low groups. For each genomic abnormality event, the resulting p-value for each gene was transformed into a significance score S for the association, such as S= signed log10(p-value), where the sign was determined by the direction of the regression coefficients. Pathway analysis was performed using these scores for 2923 pathways from KEGG, Hallmark, Reactome, DDR and DQ databases. For each pathway, we compared the significance scores of genes inside vs outside of the pathway, using Wilcoxon rank score test. We controlled the resulting pathway association p-values using the FDR BY procedure separately for each pathway database. We also calculated the averaged log fold change for each pathway by taking the mean value of the regression coefficients for all genes in the pathway.
Identification of Proteins/Pathways Associated with TP53 wide-type score
Similarly, as running the functional consequences of the genomic abnormality events, we evaluated the proteins and pathways associated with TP53 wide-type score in samples with expression profiles. We started by regressing the protein level of each gene on the TP53 wide-type score, adjusting for patient age, tumor location and tumor purity. The resulting p-values for each gene was transformed into a significance score S for the association, such as S= signed log10(p-value), where the sign was determined by the direction of the regression coefficients. For each of the 2923 pathways under investigation, we compared the significance scores of genes inside vs outside of the pathway, using Wilcoxon rank score test. We controlled the resulting p-values using FDR BY procedure separately for each pathway database. We also calculated the averaged log fold change for each pathway by taking the mean value of the regression coefficients for all genes in the pathway.
Identification of Refractory Tumor Associated proteins in subsets of tumors
To understand how proteins are associated with refractory tumors in subsets of tumors characterized by different genomic abnormalities or TP53 activities, we performed association analysis as follows: Let X denote the binary indicator of TP53 wild-type score (high vs low), chr17-LOH (yes vs now), nTAI (high vs low), BRCA1/2 mutation (yes vs now) and high TMB (high vs low). We regress each protein Y on refractory tumors allowing different coefficients for X=0 vs 1, as such Y = beta_0 + beta_1 I(X==1)*Refractory + beta_2 I(X==0)*Refractory + beta_3 covariates, where summary statistics on beta_1 and beta_2 represent the association patterns in two subsets of the tumors (X=0 vs 1). The covariates include patient age, tumor location and tumor purity. Significance was claimed using 10% FDR. We also contrasted the −log10(p-values) for beta_1 and beta_2 and annotated the significant genes to identify genes with different association patterns in different subsets of genes.
Prediction Model for Chemotherapy Response
Feature Selection
For building the prediction model, we first leveraged the information from our pre-clinical proteomics study based on cell line and PDX models and combined with literature to select pathways and gene sets associated with chemo-response. The cell line molecular profiles revealed extensive responses to carboplatin and differential responses between sensitive and resistant cells.42 A subset of these molecular differences was confirmed in PDX models using global proteomic profiling of 20 HGSOC patient-derived xenograft models (10 platinum-sensitive, 10 platinum-refractory). Consistent with the cell line results, we found that both OXPHOS and fatty acid oxidation (FAO) pathways were increased in the global profiles of platinum refractory PDX tumors. Also, as a resource to the cancer research community, we built a comprehensive overview17 accompanied by a manually curated database of the >900 genes/proteins that were associated with platinum resistance over the last 30 years of literature. The database was annotated with possible pathways through which the curated genes were related to platinum resistance, types of evidence, and hyperlinks to literature sources. The searchable, downloadable database is available online at http://ptrc-ddr.cptac-data-view.org. Specifically, we leveraged the aforementioned manually curated sets of platinum relevant genes from 22 pathways based on 31 years of literature.17 We then identified a significant association between chemo-resistance and activities of 3 metabolic pathways, Oxidative Phosphorylation, Adipogenesis, and Fatty Acid Metabolism, using both the ovarian cancer cell line and PDX proteomics data set.
Focusing on these 25 gene-sets/pathways, we searched for protein markers predictive of chemo response based on the global proteomics data of FFPE samples from 83 sensitive and 52 refractory HGSOC tumors (after removing the samples that were mixture of both primary and metastatic tumors and the samples that received neo-adjuvant therapy). The preprocessed data contained protein abundance measurement for 8800 unique genes, among which 1082 were from the 25 selected pathways/protein-sets. We then performed feature selection among these 1082 genes by aggregating results from multiple machine learning (ML) models trained to predict chemo-response. Specifically, for the 22 literature curated gene sets, which were of low dimension (50 or fewer genes in each set), we assessed the prediction performance of each set using either Elastic Net, RandomForest or XGBoost models through 5-fold cross validation (CV). For each pathway, we selected the ML model with the best CV performance and recorded the corresponding AUC. We then selected the pathway/function categories whose 95% confidence interval of AUC exceeded 0.6. Two out of the 22 gene sets, that correspond to the Hypoxia and NFKB pathways respectively, were selected using this criterion. We did not do additional feature selection within these two sets, as their sizes were small (33 genes in total). As to the three metabolic pathways, we considered the union of the three pathways (492 genes in total after combining the three-pathway members annotated in the KEGG, Hallmark, Reactome and Wiki pathway databases), and performed feature selection through random cross validation. Specifically, we generated 500 bootstrap (resampled without replacement) data sets of 80% sample size. On each of the 500 data sets, we fitted a prediction model using three machine learning (ML) methods. After deriving feature selection frequencies across the 500 models of each method, 35 out of the 492 genes had selection frequencies of 50% or higher by at least one of the three ML methods and were deemed as predictive features. This led to a combined predictive marker panel of 68 genes. On the other hand, among all the 8800 genes in the FFPE global proteomic data, four markers showed significant association with chemo-response at genome-wide FDR level of 0.1 (one of them was already in the selected list). In the end, we filtered away 8 protein markers not detected in the other proteomics data sets from human tumors such as the Frozen and FFPE validation datasets.
The final marker panel then consisted of 64 proteins: 33 from metabolic, 15 from Hypoxia, 14 from NFKB (one protein belongs to both Hypoxia and NFKB), and 4 proteins with significant marginal association with chemo-response based on the proteomic data of the FFPE discovery cohort.
Prediction Model Building
Here we provide the details for building the ensemble prediction model . The input of is the protein abundances of 64 protein markers in a tumor sample. The output of is a score between 0 and 1, indicating the probability of the tumor to be refractory. The prediction function takes the following form:
where , and are the fitted prediction models based on the FFPE discovery data set using ElasticNet, Random Forest and XGBoost respectively. Below we describe the construction parameters and results for each of the three prediction models.
ElasticNe.
We constructed the ElasticNet model (linear additive model) based on the abundances of the selected 64 protein markers in the FFPE discovery data using the “cv.glmnet” function from the R package “glmnet”.43 The mixing parameter (weights between the l1 and l2 penalty) in ElasticNet was set to be 0.2. Other tuning parameters were selected based on cross-validation errors. The resulting model is based on 50 out of 64 proteins.
Random Forest.
We fit the Random Forest prediction model using the R package “randomForest.”44 The total number of trees was set to be 1000. We obtained the “MeanDecreaseGini” values for each of the 64 proteins in the resulting model. The higher the “MeanDecreaseGini” values are, the bigger contribution the variables have towards predicting the refractory status.
XGBoost.
We fit the XGBoost model using the function “xgboost” from R package “xgboost”.45 The maximum number of boosting iterations was set to be 500. There were 58 features selected in the final model. We obtained the gain or the fraction of contributions of each protein in the model.
Prediction Performance Evaluation
For the prediction performance evaluation based on FFPE discovery cohort global proteomics data we further removed the samples with neo-adjuvant therapy. Using the 83 sensitive and 52 refractory HGSOC tumors, we built a prediction model based on the 64 proteins using the ensemble of ElasticNet, RF, and XGBoost. We obtained the AUC using a five-fold cross validation. We then derived a prediction model using the same ensemble strategy based on the complete data set. We evaluated the performance of this prediction model using an independent validation cohort of Frozen Cohort tumors (±29 overlapping samples with the FFPE discovery cohort). Note that from the Frozen cohort too we removed the samples with neo-adjuvant therapy and got 34 in the independent set and 27 in the overlapping set. Finally, we also evaluated the prediction performance of the 64 proteins in the FFPE Validation Cohort data of 20 tumors using the same ensemble strategy. In this case we used a three-fold cross validation to obtain the AUC. We calculated the 95% confidence interval for the AUCs by bootstrap approach. We also compared the AUCs of two ROCs using Delong’s test.
Prediction model based on the phospho-proteomic data
For prediction model building based on the phospho-proteomic data of the FFPE discovery cohort, we focused on the same three metabolic pathways42 and the 22 additional pathways from the literature curated list17 that have been described before in the feature selection section. After removing the ischemia sites91 from the FFPE Discovery cohort phospho data we did the feature selection using the three machine learning models (described earlier) and selected 41 sites corresponding to the metabolic genes and 48 sites corresponding the to the platinum relevant genes in ERK, Fanconi, Hippo signaling and WNT signaling pathways. We evaluated the prediction performance of our model using the selected features in the FFPE Discovery data through 5-fold cross-validation and obtained an AUC of 0.78. Out of the selected phosphosites, 56 were observed in the FFPE validation phospho-proteomic data. We also did similar prediction performance evaluation in the FFPE validation phospho data through 3-fold cross-validation and obtained an AUC of 0.76 (Figures S3E and S3F).
Prediction model based on the MRM data
Using similar strategies, prediction models were fit based on MRM measurements of 22 proteins across 102 tumors. The resulting model was associated with an AUC of 0.76 (Figure S3G; cross-validation).
Pathway Enrichment Analysis using Wilcoxon Test
Pathway enrichment analysis was conducted to characterize the differences between platinum sensitive and platinum refractory samples, based on results from association tests (described in Multiple Linear Regression Model for Association Tests). Gene set enrichment was conducted across a collection of gene sets from MSigDB’s database (v 7.5.1) that includes: KEGG, Hallmark and Reactome. These collections were downloaded from http://software.broadinstitute.org/gsea/msigdb/index.jsp.93 Along with these databases we also considered a literature curated database of platinum relevant pathways17 and a manually curated database of DDR pathways. Finally, we also considered a database of TGFβ and alternative end-joining (Alt-EJ) pathways.74 We performed a Wilcoxon test to compare the distribution of signed p-values (obtained from the linear model-based regression analysis) of the genes within the pathways to that of the remaining genes in the dataset. Gene sets with <5 member genes were excluded. We obtained the marginal p-values based on the Wilcoxon test for each individual pathway and obtained the adjusted p-values after multiplicity correction using the BH method. We consider a pathway to be significantly associated with platinum response if its adjusted p-value is < 0.1. We calculated the difference between the average of the signed p-values (from the linear regression model) of the genes within the pathway and that of the remaining genes in the dataset to determine the direction of the pathway. A positive difference indicates the pathway is upregulated in the sensitive tumors, while a negative value of this difference indicates that the pathway is upregulated in refractory tumors, given that its adjusted p-value is significant (< 0.1). Finally, to help identify pathways distinctly associated with platinum response and to consolidate redundant pathway results, Sumer software was utilized with the default parameters.112
Identification of Proteomic Clusters Through Consensus Clustering
We identified distinct proteomics clusters among the ovarian tumors using outcome-guided clustering analysis. We first obtained the single sample gene set enrichment analysis (ssGSEA) score for the 150 significant pathways that showed differential expression between sensitive and refractory tumors based on FFPE Discovery Cohort global proteomics data (adjusted p-value < 0.01). To obtain the ssGSEA score we used the GSVA package (method = “ggsea”) in R.148,123 We then performed consensus clustering of the 150 pathway scores using the ConsensusClusterPlus package in R.107 Prior to clustering, the data matrix was scaled so that each pathway had a mean 0 and a standard deviation of 1 across samples. K-means clustering based on a Euclidean distance metric was conducted across 100 repetitions for cluster numbers ranging from 2 through 7 using otherwise default parameters. Our clustering analysis identified 5 distinct subtypes of the 158 tumor samples.
For both the FFPE Discovery cohort and the Frozen Validation cohort, we obtained a detailed annotation of the sample storage time. While there is variation among sample storage time across different clusters, the distributions are widespread within each cluster and there are no consistent patterns between the FFPE Discovery and Frozen Validation cohorts. Thus, it is less likely that the differences between the clusters are driven by quality factors due to sample storage time.
Sensitivity Analysis of Proteomic Pathway Clusters
Additionally, we performed consensus clustering using the ssGSEA scores of 100 randomly chosen sets of 150 pathways. We calculated the within/between cluster variance ratios for the original 150 pathways and also for the randomly chosen 150 pathways in each replication. We average this ratio over the original 150 pathways and also the randomly chosen 150 pathways in each replication and thus obtain one within/between cluster variance ratio for each replication for the latter. On the other hand, we also perform consensus clustering using the ssGSEA scores of the most variable 150 pathways chosen from the pathway databases. We then calculate the average of the within/between cluster variance ratios for these variable 150 pathways. We observe that this ratio based on our original 150 pathways is much smaller than that of the most variable pathways, as well as the distribution of the 100 of those ratios based on randomly chosen pathways, indicating more perfect clustering based on our initial 150 pathways (Figure 4C).
Calculating p value of CNV enrichment for clusters
To find regions with significantly different copy number among clusters, we first calculated the baseline copy number of the whole sample (weighted median of segments). Next, for each gene, we calculated a value we called C.dosage as “gene copy number” “baseline copy number”. After that, each gene was assigned a value as 1 if C.dosage > 0 (gene amplified), 0 if C.dosage = 0 (gene CNV not changed) or −1 if C.dosage < 0 (gene has a deleted copy). Then for each gene, we ran a statistical test (Wilcoxon signed- rank test) to compare the amount of gain/loss for each cluster versus all the other 4 clusters. Then we estimated the boundary of significant amplification/deletion regions using the boundary of adjacent genes which were called significant. In the end, to calculate the significance of the found region (and not only genes in it), we performed the same analysis and assigned a 1,0,−1 value to each region for each patient (if a region has different segments with different copy numbers, then we assigned the copy number which covers the majority of that region). Then we perform the statistical test again for these values and report the P value.
Recapitulation of TCGA subtypes based on FFPE RNAseq data
We applied the established ovarian cancer molecular subtype classifier124 on the gene level RNAseq data of our FFPE discovery cohort, and evaluated the subtype-specific genes and pathways reported by TCGA20 respectively. We observed consistent up/down-regulation patterns for these genes/pathways as observed in the previous work (Figure S4G; Table S4). For example, transcription factors SOX11, HMGA2 and proliferation marker MCM2 were significantly upregulated in the proliferative (PRO) subtype. At the same time, the third (green) gene group, which were upregulated in the proliferative subtype compared to other subtypes, were significantly enriched in DNA replication related pathways (Table S4). Also, the mesenchymal subtype (MES) was associated with upregulation of genes such as FAP, ANGPTL1 and ANGPTL2 and ECM interaction related pathways, consistent with what is reported in Zhang et al.19 and Cancer Genome Atlas Research Network20
Also, performing similar consensus clustering using the ssGSEA scores of the same 150 pathways based on the transcriptomic data of the FFPE Discovery Cohort did not reveal similar clusters (Figures S4F and S4H).
Comparison between protein based clustering v.s. protein-pathway based clustering
In contrast to pathway-based clustering, we also performed parallel clustering analysis using individual protein abundances (STAR Methods). First, we observed that based on our FFPE Discovery data, pathway-based analysis resulted in better separation of tumor clusters than the protein-based analysis. As illustrated in Figure S4E, the within/between variances (y-axis) for the clustering results based on the pathway activity scores (red) are significantly lower than that based on the protein abundances (blue). Second, the protein-based result failed to reproduce across the validation datasets. As illustrated in Figure S5E, we observed sporadic correlations when comparing the protein abundance mean vector of each cluster in the FFPE Discovery and CPTAC-201619 cohorts. Our observation that pathway-based analysis results in more stable/meaningful molecular clusters vs gene-based analysis is consistent with recent literature.56
Validation of Proteomic Clusters in Independent Cohorts
For the three validation datasets we did similar consensus clustering. Firstly, ssGSEA scores for 150 pathways were calculated for the Frozen Validation Cohort proteomics dataset (n = 64), a previously published retrospective global proteomics dataset19 (n = 174) and a PDX global proteomic dataset42 (n = 20). After that, consensus clustering was applied to the pathway scores of each dataset (with cluster numbers set to be 5) as described in the previous section (identification of proteomic clusters through consensus clustering). Then, the concordance between the clustering results of these validation cohorts and that from the FFPE discovery cohort were evaluated from multiple aspects.
Correlation among cluster mean vectors of different cohorts
The cohort from Zhang et al.19 contains adequate samples (n > 10) in each of the five clusters (Figure 5A). After calculating the average of the ssgsea scores of each of the 150 pathways in every cluster in the Zhang et al.19 dataset, we derived the Pearson correlation coefficients between these 150-pathway-score-cluster mean vectors and that of the FFPE Discovery cohort, and observed high concordance (Figure 5B). In the Frozen-independent Validation cohort (n=35), due to its relatively small sample size, only Cluster 1, 3, and 5 had adequate representation (n>5). Focusing on these three clusters, we obtained high correlations between the 150-pathway-score mean vectors in the Frozen Validation cohort and that of the FFPE Discovery cohort (Figure S5B).
tSNE visualization of cluster concordance between different cohorts
Using a tSNE (t-distributed stochastic neighbor embedding) dimensionality reduction analysis based on the combined 150-pathway-score matrices of both the cohort from Zhang et al.19 and the FFPE Discovery cohort, we observed a perfect overlay between the five clusters of the two cohorts (Figure 5C). Similarly, we performed tSNE dimensionality reduction analysis as above using the combined ssGSEA score matrices of the 150 pathways of both the Frozen-independent Validation and FFPE Discovery cohorts and observed a perfect overlay between the two cohort samples from clusters 1, 3, and 5 (Figure S5A).
A classification (PAM) based validation
In addition to the above investigation using unsupervised consensus clustering, we also validated the cluster results using supervised analysis. Using the nearest-shrunken-centroid classification method, PAM,149 we trained a classification model to predict cluster labels based on the 150-pathway-score matrix of the FFPE Discovery cohort global proteomic data. We then applied this classification method to the three validation cohorts to predict cluster labels. The results were like those from the unsupervised clustering analysis (Figure 5A), demonstrating the generalizability of these findings.
Specifically, a centroid shrinkage approach based on PAM149 was utilized. With the pamr.train function in the R package ‘pamr150‘, the model was trained using the pathway scores for the FFPE Discovery Cohort samples (n = 158) and their corresponding proteomic subtypes estimated using consensus clustering. Based on the lowest classification error, the model threshold was selected (threshold = 0) which resulted in all 150 pathways being informative for the predictor. The ssGSEA scores were aligned to the FFPE discovery dataset for the Frozen dataset using only the overlapping samples (n = 29). We calculated the mean and standard deviation for the two datasets across the 29 samples. The aligned Frozen dataset was then calculated for all samples:
The ssGSEA scores were also aligned for the CPTAC 2016 dataset19 . We calculated the mean and standard deviation for the two datasets across all samples. The aligned CPTAC 2016 dataset was then calculated for all samples:
Using the pamr.predict function in the R package ‘pamr’, the model was then applied to the aligned pathway scores of each of the testing datasets . The maximum posterior probability estimated from pamr was used to assign the proteomic subtype for each sample.
Identification of Proteomic Cluster Signatures
We performed association tests within each proteomic subtype to identify proteins associated with treatment response within the subtypes. Within each subtype we performed a multiple regression analysis based on the following model:
protein ~ response (platinum sensitive/platinum refractory) + tumor location + source site + age + neoadjuvant treatment status + tumor purity
We tested for the regression coefficient corresponding to the factor response conditional on other covariates to identify proteins associated with treatment response within each subtype. We obtained the marginal p-values and the adjusted p-values after adjusting for multiple hypothesis testing using the BH method for each individual protein within each subtype. We consider a protein to be significantly associated with treatment response within a subtype if its adjusted p-value is < 0.1.
Tumor Purity, Stromal and Immune Scores
We utilized ESTIMATE104 to infer immune and stromal scores based on FFPE discovery global proteomic data. Tumor purity was inferred using TSNet106 based on proteomics data.
XDec
Deconvolution of bulk FFPE tumors was performed using XDec (https://github.com/BRL-BCM/XDec). Stage 0 estimates the informative genes to model the constituent cell types based on single cell sequencing data. Raw data for HGSOC samples was downloaded from GEO (GSE15460063,102) and processed with R package ‘Seurat’.105 Cell profiles were filtered to contain < 5% mitochondrial genes and unique gene counts < 5000. Profiles from the 5 patients were combined and cell type labels were determined based on the original study.63,102 Due to sparsity, profiles of the same cell type were ordered based on total gene count and pseudobulk profiles were generated by summing every 5 profiles. In order to make it equal, the top 40 pseudo-bulk profiles for each cell type were used and normalized to match the highest coverage pseudo-bulk across all cell types. We then performed Student’s t-tests across the 9 cell types (adipose, B cells, endothelial, epithelial, fibroblast, macrophages, monocytes, natural killer cells, CD8 T cells) comparing the cell profiles from one cell type to all others. For those genes with a significant difference (p-value < 1e-10), the 25 most upregulated and 25 most downregulated genes were selected to represent each cell type. Genes that appeared in multiple cell types were excluded. This resulted in 326 genes of which 211 were detected in the FFPE Discovery cohort dataset and 196 were detected in the Frozen Validation cohort dataset.
Stage 1 was performed on the FFPE Discovery cohort (n = 158) using the 211 informative genes modeling 4 cell types based on the stability criteria. The stability criteria is determined by modeling the cell count from 3 to 10 cell types using three randomly chosen subsets of 80% of the data. The cell type number showing the highest correlation across the estimated proportions across the 3 replicates is chosen for the model. Estimated constituent cell types were correlated to the processed single-cell profiles63,102 across the informative genes to determine the cell type identity. Stage 1 was also performed on the Frozen Validation cohort (n = 64) using the 196 informative genes and on the CPTAC 2016 retrospective dataset19 (n = 174) using the 189 informative genes. Cell type proportions were compared (platinum refractory vs. platinum sensitive) using Student’s t-test and determined significant if p-value < 0.05.
BayesDebulk
We performed a multi-omic based deconvolution of bulk FFPE tumors based on the integration of global proteomic and RNAseq data via BayesDebulk.58 For this deconvolution, we considered immune, endothelial, adipose, epithelial and fibroblast cells. In order to estimate the fraction of endothelial, epithelial, adipose and fibroblast cells, we used the gene signature derived from the single cell study63,102 described above; while the LM22 signature matrix from Cibersort was considered in order to derive the list of cell-specific markers of immune cells.124 For BayesDeBulk estimation, 5000 Markov-Chain Monte Carlo (MCMC) iterations were considered. The estimated fractions were derived as the median of the MCMC iterations after discarding a burn-in of 1,000 iterations.
Co-expression Networks Based on Global Proteomic Abundance of FFPE Samples
We estimated co-expression networks capturing the associations between proteins in refractory and sensitive tumors. The two co-expression networks have been estimated via JRF,151 which can perform a simultaneous estimation of multiple networks to better capture the shared structure. In order to reduce the dimensionality of the protein space, first, proteins have been allocated into three different subsets based on the proteomic abundance across refractory/sensitive samples via K-means clustering. This clustering resulted in three different protein sets of size 2838, 4097 and 1865. Then, for each subset of proteins, a co-expression network was estimated via JRF for refractory and sensitive tumors. In order to adjust for tumor location in this analysis, a tumor location node was added to the network. In this way, the model accounted for tumor location when deriving the association across proteins. Significant edges have been identified using permutation techniques.151 Only edges with a 0.1% FDR based on 30 permutations were considered as significant.
Single Cell Analysis
Raw data for HGSOC samples was downloaded from GEO (GSE15460063,102 and processed with R package ‘Seurat’.105 Cell profiles were filtered to contain < 5% mitochondrial genes and unique gene counts < 5000. Profiles from the 5 patients were merged using Seurat and cell type labels were determined based on the original study.63,102 Data was normalized using the function “Normalize-Data” (normalization.method = “LogNormalize”, scale.factor = 10000). Additionally, data was scaled using “ScaleData” using all genes as features.
In order to test each gene for cell type specificity, we used the “FindAllMarkers” function (only.pos = TRUE, min.pct = 0.1, thresh.-use = 0.25). Genes identified in the refractory co-expression network analysis were filtered to those that could be tested (minimum percent cell of cells expression > 10%, log2 fold change > 0.25) and resulted in an adjusted p-value < 0.05. Average expression of the significant genes was calculated across all cell types for visualization. Genes not meeting any of those criteria were excluded from the z-score heatmap.
All single cell expression data (< 5% mitochondrial genes, unique gene counts < 5000) was used to visualize the expression of SLAMF6.
TGFβ Signature
| Pathway | number of genes | FFPE discovery RNA | FFPE discovery Protein |
|---|---|---|---|
|
| |||
| TGFβ | 50 | 49 | 28 |
| Alt-EJ | 36 | 35 | 25 |
To test the significance of the linear trend we fitted a linear model regressing TGFβ/Alt-EJ/β-Alt score on the subtypes adjusting for other covariates including tumor purity, tumor location, study sites etc. We also fitted another model regressing TGFβ/Alt-EJ/β-Alt score only on the covariates and then comparing the two models using an ANOVA test we obtained the significance of the subtypes. For the FFPE discovery data we used the subtype labeling from consensus clustering, while for the Frozen and CPTAC 201619 data we used the subtype labeling from the classification analysis performed on those datasets. In the table above we give the number of genes in TGFβ and Alt-EJ pathways that were observed in the FFPE discovery data.
ADDITIONAL RESOURCES
To facilitate ready exploration of the study results, we have developed a web portal with interactive visualizations of the results, known as ProTrack High Grade Serous Ovarian Cancer.152 Users can enter a gene list to generate an interactive heatmap, which includes clinical annotation, tumor clustering and classification tracks. For each gene, available multi-omic data tracks are shown, including normalized protein abundance, RNA expression, phosphosites, and CNV measurements. Users can sort the generated heatmap by any track to explore trends of interest. The web portal also generates boxplots of the inputted gene list, stratified by the categorical feature of their choosing, including proteomic cluster or tumor grade. Additionally, users can view scatterplots and correlations of two genes across different -omics types. Users can also explore results for iProFun cascade events for any selected gene. Results can be downloaded and exported as .xlsx files. The web portal can be accessed at: http://ptrc.cptac-data-view.org/.
Supplementary Material
Highlights.
A comprehensive proteogenomic analysis of 242 HGSOC tumors was performed
A lack of Chr17-LOH was observed to be associated with refractoriness
A 64-protein signature predicts refractoriness in multiple tumor cohorts
Pathway-based clustering reveals 5 subtypes validated in independent cohorts
ACKNOWLEDGMENTS
This work was done in collaboration with the US National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) and supported by grants U01CA214114 (to A.G.P. and M.J.B.), R50CA211499 (to J.R.W.), U24CA210993 (to P.W.), U24CA271114 (to P.W.), U24CA210954 (to B.Z.), U24CA210967 (to A.I.N.), U24CA210972 (to D.F.), Leidos Biomedical Research contract S21-167 (to D.F.), DOD W81XWH-16-2-0038 (to M.J.B.), P30CA240139 (to V.S. and S.C.S.), as well as a generous donation from the Aven Foundation. Sample collection at Mayo Clinic was supported by P50CA136393 (to S.H.K.) and P30 CA015083. Scientific Computing Infrastructure at Fred Hutch was funded by ORIP grant S10OD028685. Experimental Histopathology Shared Resource at Fred Hutch was funded in part by the Fred Hutch/University of Washington Cancer Consortium grant P30CA015704. The CCR5 antibody was provided by Dr. Matthias Mack at the Universitätsklinikum Regensburg in Germany (http://www.macklab.org/). We would like to honor our co-author and long-time colleague Julia Voytovich, who recently passed away while in Ukraine performing humanitarian work in the service of those affected by the war.
Footnotes
DECLARATION OF INTERESTS
M.J.B. has participated in advisory boards for the following companies: Clovis, Astra Zeneca, and GSK-Tesaro. A.N.H. is the Associate Editor of Clinical Chemistry and has a financial interest in the company Seattle Genetics. A.G.P. is the founder of Precision Assays, LLC. A.B.B. is employed by Synapse. S.Y. is an employee and shareholder of Sema4. B.J.W. has stock interests in Verastem and Exact Sciences.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2023.07.004.
REFERENCES
- 1.Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, and Karlan BY (2016). Ovarian cancer. Nat. Rev. Dis. Prim. 2, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bast RC Jr., Matulonis UA, Sood AK, Ahmed AA, Amobi AE, Balkwill FR, Wielgos-Bonvallet M, Bowtell DDL, Brenton JD, and Brugge JS (2019). Critical questions in ovarian cancer research and treatment: report of an American Association for Cancer Research Special Conference. Cancer 125, 1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vergote I, Coens C, Nankivell M, Kristensen GB, Parmar MKB, Ehlen T, Jayson GC, Johnson N, Swart AM, and Verheijen R (2018). Neoadjuvant chemotherapy versus debulking surgery in advanced tubo-ovarian cancers: pooled analysis of individual patient data from the EORTC 55971 and CHORUS trials. Lancet Oncol. 19, 1680–1687. [DOI] [PubMed] [Google Scholar]
- 4.Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, and Karlan BY (2003). Improved survival in women with BRCA-associated ovarian carcinoma. Cancer 97, 2187–2195. [DOI] [PubMed] [Google Scholar]
- 5.Muggia FM (2003). Sequential single agents as first-line chemotherapy for ovarian cancer: a strategy derived from the results of GOG-132. Int. J. Gynecol. Cancer 13, 156–162. [DOI] [PubMed] [Google Scholar]
- 6.Cannistra SA (2004). Cancer of the ovary. N. Engl. J. Med. 351, 2519–2529. 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 7.McGuire WP, Penson RT, Gore M, Herraez AC, Peterson P, Shahir A, and Ilaria R (2018). Randomized phase II study of the PDGFRα antibody olaratumab plus liposomal doxorubicin versus liposomal doxorubicin alone in patients with platinum-refractory or platinum-resistant advanced ovarian cancer. BMC Cancer 18, 1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sayal K, Gounaris I, Basu B, Freeman S, Moyle P, Hosking K, Iddawela M, Jimenez-Linan M, Abraham J, Brenton J, et al. (2015). Epirubicin, cisplatin, and capecitabine for primary platinum-resistant or platinum-refractory epithelial ovarian cancer: results of a retrospective, single-institution study. Int. J. Gynecol. Cancer 25, 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMullen M, Karakasis K, Madariaga A, and Oza AM (2020). Overcoming platinum and PARP-inhibitor resistance in ovarian cancer. Cancers 12, 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markman M, Rothman R, Hakes T, Reichman B, Hoskins W, Rubin S, Jones W, Almadrones L, and Lewis JL Jr. (1991). Second-line platinum therapy in patients with ovarian cancer previously treated with cisplatin. J. Clin. Oncol. 9, 389–393. 10.1200/JCO.1991.9.3.389. [DOI] [PubMed] [Google Scholar]
- 11.Vaughan S, Coward JI, Bast RC, Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, et al. (2011). Rethinking ovarian cancer: recommendations for improving outcomes. Nat. Rev. Cancer 11, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wethington SL, Wahner-Hendrickson AE, Swisher EM, Kaufmann SH, Karlan BY, Fader AN, and Dowdy SC (2021). PARP inhibitor maintenance for primary ovarian cancer - A missed opportunity for precision medicine. Gynecol. Oncol. 163, 11–13. 10.1016/j.ygyno.2021.08.002. [DOI] [PubMed] [Google Scholar]
- 13.González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, et al. (2019). Niraparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 381, 2391–2402. 10.1056/NEJMoa1910962. [DOI] [PubMed] [Google Scholar]
- 14.Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, Fujiwara K, Vergote I, Colombo N, Mäenpää J, et al. (2019). Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N. Engl. J. Med. 381, 2416–2428. 10.1056/NEJMoa1911361. [DOI] [PubMed] [Google Scholar]
- 15.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, Lisyanskaya A, Floquet A, Leary A, Sonke GS, et al. (2018). Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 379, 2495–2505. 10.1056/NEJMoa1810858. [DOI] [PubMed] [Google Scholar]
- 16.DiSilvestro P, Banerjee S, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, Lisyanskaya A, Floquet A, Leary A, et al. (2023). Overall survival with maintenance olaparib at a 7-year follow-up in patients with newly diagnosed advanced ovarian cancer and a BRCA mutation: The SOLO1/GOG 3004 Trial. J. Clin. Oncol. 41, 609–617. 10.1200/JCO.22.01549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang D, Savage SR, Calinawan AP, Lin C, Zhang B, Wang P, Starr TK, Birrer MJ, and Paulovich AG (2021). A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 40, 6395–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen B, Fong C, Luthra A, Smith SA, DiNatale RG, Nandakumar S, Walch H, Chatila WK, Madupuri R, Kundra R, et al. (2022). Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 185, 563–575.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, et al. (2016). Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765. 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patch A-M, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, et al. (2015). Whole–genome characterization of chemoresistant ovarian cancer. Nature 521, 489–494. [DOI] [PubMed] [Google Scholar]
- 22.Wang ZC, Birkbak NJ, Culhane AC, Drapkin R, Fatima A, Tian R, Schwede M, Alsop K, Daniels KE, Piao H, et al. (2012). Profiles of genomic instability in high-grade serous ovarian cancer predict treatment OutcomeGenomic. Clin. Cancer Res. 18, 5806–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang S, Lheureux S, Karakasis K, Burnier JV, Bruce JP, Clouthier DL, Danesh A, Quevedo R, Dowar M, and Hanna Y (2018). Landscape of genomic alterations in high-grade serous ovarian cancer from exceptional long-and short-term survivors. Genome Med. 10, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tabassum A, Samdani MN, Dhali TC, Alam R, Ahammad F, Samad A, and Karpiński TM (2021). Transporter associated with antigen processing 1 (TAP1) expression and prognostic analysis in breast, lung, liver, and ovarian cancer. J. Mol. Med. (Berl) 99, 1293–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minn AJ, Rudin CM, Boise LH, and Thompson CB (1995). Expression of bcl-xL can confer a multidrug resistance phenotype. Blood 86, 1903–1910. [PubMed] [Google Scholar]
- 26.Simonian PL, Grillot DA, and Nuñez G (1997). Bcl-2 and Bcl-XL can differentially block chemotherapy-induced cell death. Blood 90, 1208–1216. [PubMed] [Google Scholar]
- 27.Song X, Ji J, Gleason KJ, Yang F, Martignetti JA, Chen LS, and Wang P (2019). Insights into impact of DNA copy number alteration and methylation on the proteogenomic landscape of human ovarian cancer via a multi-omics integrative analysis. Mol. Cell. Proteomics 18. S52–S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, Li X, Babur O, Hsu T-K, Lichtarge O, et al. (2019). Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 28, 1370–1384.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frum RA, and Grossman SR (2014). Mechanisms of mutant p53 stabilization in cancer. Mutant p53 and MDM2 in Cancer (Springer; ), pp. 187–197. [DOI] [PubMed] [Google Scholar]
- 31.Pfister NT, and Prives C (2017). Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harbor Perspect. Med. 7, a026054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrysik Z, Galbraith MD, Guarnieri AL, Zaccara S, Sullivan KD, Pandey A, MacBeth M, Inga A, and Espinosa JM (2017). Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 27, 1645–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polager S, and Ginsberg D (2009). p53 and E2F: partners in life and death. Nat. Rev. Cancer 9, 738–748. [DOI] [PubMed] [Google Scholar]
- 34.Feng Z, Zhang H, Levine AJ, and Jin S (2005). The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 102, 8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiefel H, Bondong S, Hazin J, Ridinger J, Schirmer U, Riedle S, and Altevogt P (2012). L1CAM: a major driver for tumor cell invasion and motility. Cell Adh. Migr. 6, 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdel Azim SA, Duggan-Peer M, Sprung S, Reimer D, Fiegl H, Soleiman A, Marth C, and Zeimet AG (2016). Clinical impact of L1CAM expression measured on the transcriptome level in ovarian cancer. Oncotarget 7, 37205–37214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bondong S, Kiefel H, Hielscher T, Zeimet AG, Zeillinger R, Pils D, Schuster E, Castillo-Tong DC, Cadron I, Vergote I, et al. (2012). Prognostic significance of L1CAM in ovarian cancer and its role in constitutive NF-κB activation. Ann. Oncol. 23, 1795–1802. [DOI] [PubMed] [Google Scholar]
- 38.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, Howard TP, Takeda DY, Ly SH, Kim E, et al. (2018). Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 50, 1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeo SY, Itahana Y, Guo AK, Han R, Iwamoto K, Nguyen HT, Bao Y, Kleiber K, Wu YJ, Bay BH, et al. (2016). Transglutaminase 2 contributes to a TP53-induced autophagy program to prevent oncogenic transformation. eLife 5, e07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watkins JA, Irshad S, Grigoriadis A, and Tutt AN (2014). Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 16, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, et al. (2014). Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, Fallopian tube, and peritoneal CarcinomasDNA. Clin. Cancer Res. 20, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang D, Chowdhury S, Wang H, Savage SR, Ivey RG, Kennedy JJ, Whiteaker JR, Lin C, Hou X, Oberg AL, et al. (2021). Multiomic analysis identifies CPT1A as a potential therapeutic target in platinum-refractory, high-grade serous ovarian cancer. Cell Rep. Med. 2, 100471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friedman J, Hastie T, and Tibshirani R (2010). Regularization paths for generalized linear models via coordinate descent. J. Stat. Software 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- 44.Breiman L (2001). Random forests. Mach. Learn. 45, 5–32. [Google Scholar]
- 45.Chen T, and Guestrin C (2016). Xgboost: a scalable tree boosting system. KDD ‘16: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 785–794. [Google Scholar]
- 46.Yang M, Petralia F, Li Z, Li H, Ma W, Song X, Kim S, Lee H, Yu H, Lee B, et al. (2020). Community assessment of the predictability of cancer protein and phosphoprotein levels from genomics and transcriptomics. Cell Syst. 11, 186–195.e9. 10.1016/j.cels.2020.06.013. [DOI] [PubMed] [Google Scholar]
- 47.Wiedemeyer WR, Beach JA, and Karlan BY (2014). Reversing platinum resistance in high-grade serous ovarian carcinoma: targeting BRCA and the homologous recombination system. Front. Oncol. 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ellis MJ, Suman VJ, Hoog J, Goncalves R, Sanati S, Creighton CJ, DeSchryver K, Crouch E, Brink A, Watson M, et al. (2017). Ki67 proliferation index as a tool for chemotherapy decisions during and after neoadjuvant aromatase inhibitor treatment of breast cancer: results from the American College of Surgeons Oncology Group Z1031 trial (alliance). J. Clin. Oncol. 35, 1061–1069. 10.1200/JCO.2016.69.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. (2017). A pathology atlas of the human cancer transcriptome. Science 357, eaan2507. [DOI] [PubMed] [Google Scholar]
- 50.Sipos I, Tretter L, and Adam-Vizi V (2003). Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J. Neurochem. 84, 112–118. [DOI] [PubMed] [Google Scholar]
- 51.Zhen D, Chen Y, and Tang X (2010). Metformin reverses the deleterious effects of high glucose on osteoblast function. J. Diabetes Its Complications 24, 334–344. [DOI] [PubMed] [Google Scholar]
- 52.Verhaak RG, Tamayo P, Yang J-Y, Hubbard D, Zhang H, Creighton CJ, Fereday S, Lawrence M, Carter SL, and Mermel CH (2012). Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest. 123, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, Dou Y, Reva B, Kane MH, Avanessian SC, et al. (2021). A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371.e40. 10.1016/j.cell.2021.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petralia F, Tignor N, Reva B, Koptyra M, Chowdhury S, Rykunov D, Krek A, Ma W, Zhu Y, Ji J, et al. (2020). Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell 183, 1962–1985.e31. 10.1016/j.cell.2020.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih TM, Chang HY, et al. (2019). Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 179, 964–983.e31. 10.1016/j.cell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garofano L, Migliozzi S, Oh YT, D’Angelo F, Najac RD, Ko A, Frangaj B, Caruso FP, Yu K, Yuan J, et al. (2021). Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2, 141–156. 10.1038/s43018-020-00159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murillo OD, Petrosyan V, LaPlante EL, Dobrolecki LE, Lewis MT, and Milosavljevic A (2022). XDec simplex map of breast cancer cell states enables precise modeling and targeting of breast cancer. 10.1101/2022.07.06.498858. [DOI]
- 58.Petralia F, Krek A, Calinawan AP, Feng S, Gosline S, Pugliese P, Ceccarelli M, and Wang P (2022). BayesDeBulk: a flexible bayesian algorithm for the deconvolution of bulk tumor data. 10.1101/2021.06.25.449763. [DOI]
- 59.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, Osokin N, Kozlov I, Frenkel F, Gancharova O, et al. (2021). Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 39, 845–865.e7. 10.1016/j.ccell.2021.04.014. [DOI] [PubMed] [Google Scholar]
- 61.Zaitsev A, Chelushkin M, Dyikanov D, Cheremushkin I, Shpak B, Nomie K, Zyrin V, Nuzhdina E, Lozinsky Y, Zotova A, et al. (2022). Precise reconstruction of the TME using bulk RNA-seq and a machine learning algorithm trained on artificial transcriptomes. Cancer Cell 40, 879–894.e16. 10.1016/j.ccell.2022.07.006. [DOI] [PubMed] [Google Scholar]
- 62.Roelands J, Hendrickx W, Zoppoli G, Mall R, Saad M, Halliwill K, Curigliano G, Rinchai D, Decock J, Delogu LG, et al. (2020). Oncogenic states dictate the prognostic and predictive connotations of intratumoral immune response. J. Immunother. Cancer 8, e000617. 10.1136/jitc-2020-000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geistlinger L, Oh S, Ramos M, Schiffer L, LaRue RS, Henzler CM, Munro SA, Daughters C, Nelson AC, Winterhoff BJ, et al. (2020). Multiomic analysis of subtype evolution and heterogeneity in high-grade serous ovarian carcinoma. Cancer Res. 80, 4335–4345. 10.1158/0008-5472.CAN-20-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hajaj E, Eisenberg G, Klein S, Frankenburg S, Merims S, Ben David I, Eisenhaure T, Henrickson SE, Villani AC, Hacohen N, et al. (2020). SLAMF6 deficiency augments tumor killing and skews toward an effector phenotype revealing it as a novel T cell checkpoint. eLife 9, e52539. 10.7554/eLife.52539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yigit B, Wang N, Ten Hacken E, Chen SS, Bhan AK, Suarez-Fueyo A, Katsuyama E, Tsokos GC, Chiorazzi N, Wu CJ, et al. (2019). SLAMF6 as a regulator of exhausted CD8(+) T cells in cancer. Cancer Immunol. Res. 7, 1485–1496. 10.1158/2326-6066.CIR-18-0664. [DOI] [PubMed] [Google Scholar]
- 66.Fahrner M, Kook L, Fröhlich K, Biniossek ML, and Schilling O (2021). A systematic evaluation of semispecific peptide search parameter enables identification of previously undescribed N-terminal peptides and conserved proteolytic processing in cancer cell lines. Proteomes 9, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jakowlew SB (2006). Transforming growth factor-β in cancer and metastasis. Cancer Metastasis Rev. 25, 435–457. [DOI] [PubMed] [Google Scholar]
- 68.Massagué J (2012). TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Syed V (2016). TGF-β signaling in cancer. J. Cell. Biochem. 117, 1279–1287. [DOI] [PubMed] [Google Scholar]
- 70.Ciardiello D, Elez E, Tabernero J, and Seoane J (2020). Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann. Oncol. 31, 1336–1349. [DOI] [PubMed] [Google Scholar]
- 71.Liu Q, Chen G, Moore J, Guix I, Placantonakis D, and Barcellos-Hoff MH (2022). Exploiting canonical TGFβ signaling in cancer TreatmentTargeting. Mol. Cancer Ther. 21, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, and de Gramont A (2015). Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 147, 22–31. [DOI] [PubMed] [Google Scholar]
- 73.Guix I, Liu Q, Pujana MA, Ha P, Piulats J, Linares I, Guedea F, Mao JH, Lazar A, Chapman J, et al. (2022). Validation of anticorrelated TGFβ signaling and alternative end-joining DNA repair signatures that predict response to genotoxic cancer therapy. Clin. Cancer Res. 28, 1372–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu Q, Palomero L, Moore J, Guix I, Espín R, Aytés A, Mao J-H, Paulovich AG, Whiteaker JR, Ivey RG, et al. (2021). Loss of TGFβ signaling increases alternative end-joining DNA repair that sensitizes to genotoxic therapies across cancer types. Sci. Transl. Med. 13, eabc4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Q, Ma L, Jones T, Palomero L, Pujana MA, Martinez-Ruiz H, Ha PK, Murnane J, Cuartas I, Seoane J, et al. (2018). Subjugation of TGFβ signaling by human papilloma virus in head and neck squamous cell carcinoma shifts DNA repair from homologous recombination to alternative end JoiningHPV. Clin. Cancer Res. 24, 6001–6014. [DOI] [PubMed] [Google Scholar]
- 76.Ashraf TS, Obaid A, Saeed TM, Naz A, Shahid F, Ahmad J, and Ali A (2019). Formal model of the interplay between TGFbeta1 and MMP-9 and their dynamics in hepatocellular carcinoma. Math. Biosci. Eng. 16, 3285–3310. 10.3934/mbe.2019164. [DOI] [PubMed] [Google Scholar]
- 77.Krstic J, and Santibanez JF (2014). Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stromainfiltrating myeloid cells. ScientificWorldJournal 2014, 521754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, and Nishimura SL (2002). The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP–dependent activation of TGF-β1. J. Cell Biol. 157, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Safina A, Ren MQ, Vandette E, and Bakin AV (2008). TAK1 is required for TGF-β1-mediated regulation of matrix metalloproteinase-9 and metastasis. Oncogene 27, 1198–1207. [DOI] [PubMed] [Google Scholar]
- 80.Kennedy JJ, Whiteaker JR, Kennedy LC, Bosch DE, Lerch ML, Schoenherr RM, Zhao L, Lin C, Chowdhury S, Kilgore MR, et al. (2021). Quantification of human epidermal growth factor receptor 2 by immunopeptide enrichment and targeted mass spectrometry in formalin-fixed paraffin-embedded and frozen breast cancer tissues. Clin. Chem. 67, 1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang B, Whiteaker JR, Hoofnagle AN, Baird GS, Rodland KD, and Paulovich AG (2019). Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 16, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shuford CM, Johnson JS, Thompson JW, Holland PL, Hoofnagle AN, and Grant RP (2020). More sensitivity is always better: measuring sub-clinical levels of serum thyroglobulin on a μLC–MS/MS system. Clin. Mass Spectrom. 15, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kaklamani V (2006). A genetic signature can predict prognosis and response to therapy in breast cancer: oncotype DX. Expert Rev. Mol. Diagn. 6, 803–809. 10.1586/14737159.6.6.803. [DOI] [PubMed] [Google Scholar]
- 84.Dasari S, Theis JD, Vrana JA, Rech KL, Dao LN, Howard MT, Dispenzieri A, Gertz MA, Hasadsri L, Highsmith WE, et al. (2020). Amyloid typing by mass spectrometry in clinical practice: a comprehensive review of 16,175 samples. Mayo Clin. Proc. 95, 1852–1864. 10.1016/j.mayocp.2020.06.029. [DOI] [PubMed] [Google Scholar]
- 85.Phipps WS, Smith KD, Yang HY, Henderson CM, Pflaum H, Lerch ML, Fondrie WE, Emrick MA, Wu CC, MacCoss MJ, et al. (2022). Tandem mass spectrometry-based amyloid typing using manual microdissection and open-source data processing. Am. J. Clin. Pathol. 157, 748–757. 10.1093/ajcp/aqab185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sharma V, Eckels J, Schilling B, Ludwig C, Jaffe JD, MacCoss MJ, and MacLean B (2018). Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Mol. Cell. Proteomics 17, 1239–1244. 10.1074/mcp.RA117.000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cancer Genome Atlas Research Network (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen B, Khodadoust MS, Liu CL, Newman AM, and Alizadeh AA (2018). Profiling tumor infiltrating immune cells with CIBERSORT. In Cancer Systems Biology (Springer; ), pp. 243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJ, et al. (2014). Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteomics 13, 1690–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liluashvili V, Kalayci S, Fluder E, Wilson M, Gabow A, and Gümüxs ZH (2017). iCAVE: an open source tool for visualizing biomolecular networks in 3D, stereoscopic 3D and immersive 3D. GigaScience 6, 1–13. 10.1093/gigascience/gix054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang HY, Shanmugam AK, Mellacheruvu D, Kong AT, and Nesvizhskii AI (2020). Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat. Methods 17, 869–870. 10.1038/s41592-020-0912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- 97.Shteynberg DD, Deutsch EW, Campbell DS, Hoopmann MR, Kusebauch U, Lee D, Mendoza L, Midha MK, Sun Z, Whetton AD, et al. (2019). PTMProphet: fast and accurate mass modification localization for the trans-proteomic pipeline. J. Proteome Res. 18, 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Djomehri SI, Gonzalez ME, da Veiga Leprevost F, Tekula SR, Chang H-Y, White MJ, Cimino-Mathews A, Burman B, Basrur V, and Argani P (2020). Quantitative proteomic landscape of metaplastic breast carcinoma pathological subtypes and their relationship to triple-negative tumors. Nat. Commun. 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- 100.Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- 101.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chowdhury S, Wang R, Yu Q, Huntoon CJ, Karnitz LM, Kaufmann SH, Gygi SP, Birrer MJ, Paulovich AG, Peng J, and Wang P (2022. Aug). BMC Bioinformatics 23 (1), 321. 10.1186/s12859-022-04864-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, and Levine DA (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Petralia F, Song WM, Tu Z, and Wang P (2016). New method for joint network analysis reveals common and different coexpression patterns among genes and proteins in breast cancer. J. Proteome Res. 15, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Petralia F, Wang L, Peng J, Yan A, Zhu J, and Wang P (2018). A new method for constructing tumor specific gene co-expression networks based on samples with tumor purity heterogeneity. Bioinformatics 34. i528–i536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 109.Talevich E, Shain AH, Botton T, and Bastian BC (2016). CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12, e1004873. 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Savage SR, Shi Z, Liao Y, and Zhang B (2019). Graph algorithms for condensing and consolidating gene set analysis results. Mol. Cell. Proteomics 18, S141–S152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71. 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Colaprico A, Olsen C, Bailey MH, Odom GJ, Terkelsen T, Silva TC, Olsen AV, Cantini L, Zinovyev A, Barillot E, et al. (2020). Interpreting pathways to discover cancer driver genes with Moonlight. Nat. Commun. 11, 69. 10.1038/s41467-019-13803-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang X, Park J, Susztak K, Zhang NR, and Li M (2019). Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 10, 380. 10.1038/s41467-018-08023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl variant effect predictor. Genome Biol. 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pedersen BS, Layer RM, and Quinlan AR (2016). Vcfanno: fast, flexible annotation of genetic variants. Genome Biol. 17, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Nearoptimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. [DOI] [PubMed] [Google Scholar]
- 119.Monroe ME, Shaw JL, Daly DS, Adkins JN, and Smith RD (2008). MASIC: a software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput. Biol. Chem. 32, 215–217. 10.1016/j.compbiolchem.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim S, and Pevzner PA (2014). MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277. 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gibbons BC, Chambers MC, Monroe ME, Tabb DL, and Payne SH (2015). Correcting systematic bias and instrument measurement drift with mzRefinery. Bioinformatics 31, 3838–3840. 10.1093/bioinformatics/btv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen GM, Kannan L, Geistlinger L, Kofia V, Safikhani Z, Gendoo DMA, Parmigiani G, Birrer M, Haibe-Kains B, and Waldron L (2018). Consensus on molecular subtypes of high-grade serous ovarian carcinoma. Clin. Cancer Res. 24, 5037–5047. 10.1158/1078-0432.CCR-18-0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bushnell B (2014). BBMap: a Fast, Accurate, Splice-Aware Aligner (Lawrence Berkeley National Lab.). [Google Scholar]
- 126.Regier AA, Farjoun Y, Larson DE, Krasheninina O, Kang HM, Howrigan DP, Chen B-J, Kher M, Banks E, and Ames DC (2018). Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat. Commun. 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Barnell EK, Ronning P, Campbell KM, Krysiak K, Ainscough BJ, Sheta LM, Pema SP, Schmidt AD, Richters M, Cotto KC, et al. (2019). Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples. Genet. Med. 21, 972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gey S, and Lebarbier E (2008). Using CART to detect multiple change points in the mean for large sample. https://hal.science/hal-00327146/document.
- 130.Bellman R (1961). On the approximation of curves by line segments using dynamic programming. Commun. ACM 4, 284. [Google Scholar]
- 131.Pierre-Jean M, Rigaill G, and Neuvial P (2015). Performance evaluation of DNA copy number segmentation methods. Brief. Bioinform. 16, 600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lebarbier É (2005). Detecting multiple change-points in the mean of Gaussian process by model selection. Signal Process. 85, 717–736. [Google Scholar]
- 133.Riester M, Singh AP, Brannon AR, Yu K, Campbell CD, Chiang DY, and Morrissey MP (2016). PureCN: copy number calling and SNV classification using targeted short read sequencing. Source Code Biol. Med. 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller M-C, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. (2013). Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sztupinszki Z, Diossy M, Krzystanek M, Reiniger L, Csabai I, Favero F, Birkbak NJ, Eklund AC, Syed A, and Szallasi Z (2018). Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. npj Breast Cancer 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li H (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wu TD, Reeder J, Lawrence M, Becker G, and Brauer MJ (2016). GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. In Statistical Genomics (Springer; ), pp. 283–334. [DOI] [PubMed] [Google Scholar]
- 138.Cobain EF, Wu Y-M, Vats P, Chugh R, Worden F, Smith DC, Schuetze SM, Zalupski MM, Sahai V, Alva A, et al. (2021). Assessment of clinical benefit of integrative genomic profiling in advanced solid tumors. JAMA Oncol. 7, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kennedy JJ, Whiteaker JR, Schoenherr RM, Yan P, Allison K, Shipley M, Lerch M, Hoofnagle AN, Baird GS, and Paulovich AG (2016). Optimized protocol for quantitative multiple reaction monitoring-based proteomic analysis of formalin-fixed, paraffin-embedded tissues. J. Proteome Res. 15, 2717–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu F, Teo GC, Kong AT, Haynes SE, Avtonomov DM, Geiszler DJ, and Nesvizhskii AI (2020). Identification of modified peptides using localization-aware open search. Nat. Commun. 11, 4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- 142.Ma W, Kim S, Chowdhury S, Li Z, Yang M, Yoo S, Petralia F, Jacobsen J, Li JJ, and Ge X (2021). DreamAI: algorithm for the imputation of proteomics data. 10.1101/2020.07.21.214205. [DOI]
- 143.Alyass A, Turcotte M, and Meyre D (2015). From big data analysis to personalized medicine for all: challenges and opportunities. BMC Med. Genomics 8, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yoo S, Huang T, Campbell JD, Lee E, Tu Z, Geraci MW, Powell CA, Schadt EE, Spira A, and Zhu J (2014). MODMatcher: multiomics data matcher for integrative genomic analysis. PLoS Comp. Biol. 10, e1003790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, Hü ttenhain R., Koomen JM., et al. (2014). Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917. 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Grant RP, and Hoofnagle AN (2014). From lost in translation to paradise found: enabling protein biomarker method transfer by mass spectrometry. Clin. Chem. 60, 941–944. 10.1373/clinchem.2014.224840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cancer Genome Atlas Research Network, r DJ., Verhaak RG., Aldape KD., Yung WK., Salama SR, Cooper LA., Rheinbay E., Miller CR., Vitucci M, et al. (2015). Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 372, 2481–2498. 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tibshirani R, Hastie T, Narasimhan B, and Chu G (2002). Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 99, 6567–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hastie T, Tibshirani R, Narasimhan B, and Chu G (2019). Pam: Prediction Analysis for Microarrays R Package Version 1. https://cran.r-project.org/web/packages/pamr/index.html.
- 151.Calinawan AP, Song X, Ji J, Dhanasekaran SM, Petralia F, Wang P, and Reva B (2020). ProTrack: an interactive multi-omics data browser for proteogenomic studies. Proteomics 20, e1900359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43. D512–D520. 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw proteomic data files and all processed proteogenomic datasets as well as clinical meta information have been deposited at the Proteomic Data Commons and are publicly available as of the date of this publication. URLs are listed in the key resources table. DNA and RNA sequencing data have been deposited at dbGaP and are publicly available as of the date of this publication. Accession numbers are listed in the key resources table. H&E images for all the tumors analyzed in this study have been deposited at The Cancer Image Archive and are publicly available as of the date of this publication. DOIs are listed in the key resources table. In addition, all processed proteogenomic datasets as well as clinical meta information can be publicly queried, visualized, and downloaded from an interactive ProTrack data portal as of the date of this publication. The URL for ProTrack is listed in the key resources table. All processed data has been deposited at a publicly accessible URL listed in the key resources table. All raw data, manually integrated peak areas, transition information, and retention times generated from these stressor and time course experiments for the LC-MRM peptide target assays have been deposited at Panorama Public86 and are publicly available at the URL is listed in the key resources table. Characterization data for available assays are found in the CPTAC Assay Portal (assays.cancer.gov).
All original code for the data analysis and figures generated for this study has been deposited at this Github repository and is publicly available as of the date of publication: https://github.com/WangLab-MSSM/CPTAC_Ovarian_Chemo_Response. URLs for this repository and for other code used in this study are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD8, Clone C8/144B | DAKO | Cat#M7103; RRID:AB_2075537 |
| CD4, EP204 | Cell Marque | Cat#104R-26; RRID:AB_1516770 |
| CD68, Clone PG-M1 | DAKO | Cat#M0876; RRID:AB_2074844 |
| CD14, EPR3653 | Cell Marque | Cat#114R-14; RRID:AB_2827391 |
| CCR5 | Matthias Mack Lab | N/A |
| PanCK, Clone AE1/AE3 | DAKO | Cat#M3515; RRID:AB_2132885 |
| Opal Polymer HRP Ms+Rb | Akoya Biosciences | Cat#ARH1001EA; RRID:AB_2890927 |
|
| ||
| Biological samples | ||
|
| ||
| Fresh frozen tissue samples | See STAR Methods | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| HEPES (pH8.0) | Alfa Aesar | Cat#J63002 |
| Hydroxylamine | Millipore Sigma | Cat#438227 |
| TMT 11plex reagents | Thermo Fisher Scientific | Cat#A34808 |
| Stable isotope-labeled synthetic peptide standards | Vivitide | N/A |
| Phosphatase Inhibitor Cocktail 2 | Millipore Sigma | Cat#P5726 |
| Phosphatase Inhibitor Cocktail 3 | Millipore Sigma | Cat#P0044 |
| Protease Inhibitor Cocktail | Millipore Sigma | Cat#P8340 |
| Lys-C | Wako Chemicals | Cat#129-02541 |
| Sequencing grade modified trypsin | Promega | Cat#V5113 |
| Urea | Millipore Sigma | Cat#U0631 |
| Trizma base (Tris), pH 8.0 | Millipore Sigma | Cat#T2694 |
| iodoacetamide (IAM) | Millipore Sigma | Cat#A3221 |
| EDTA | Millipore Sigma | Cat#E7889 |
| EGTA | Bioworld | Cat#40520008-1 |
| phosphate buffered saline | Thermo Fisher Scientific | Cat#14190144 |
| tris(2-carboxyethyl)phosphine (TCEP) | Thermo Fisher Scientific | Cat#77720 |
| Acetonitrile | Fisher Chemical | Cat#A955-4 |
| Water | Fisher Chemical | Cat#W64 |
| ammonium bicarbonate | Millipore Sigma | Cat#A6141 |
| ammonium hydroxide solution | Millipore Sigma | Cat#320145 |
| Formic acid | Millipore Sigma | Cat#1116701000 |
| Trifluoroacetic Acid | Millipore Sigma | Cat#302031 |
| RapiGest SF | Waters | Cat#186001861 |
| Bond Dewax Solution | Leica | Cat#AR9222 |
| BOND Epitope Retrieval Solution 2 | Leica | Cat#AR9640 |
| Wash Solution 10X Concentrate | Leica | Cat#AR9590 |
| tertiary TSA-amplification reagent | Akoya Biosciences | Cat#FP1135 |
| Spectral DAPI | Akoya Biosciences | Cat#FP1490 |
| Prolong Gold Antifade | Invitrogen | Cat#P36930 |
| Ni-NTA Magnetic Agarose Beads | Qiagen | Cat#36113 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Micro BCA protein assay | Thermo Fisher Scientific | Cat#23235 |
| TruSeq RNA Sample Prep Kit | Illumina | Cat#FC-122-1001 |
| KAPA Library Preparation Kit | Roche | Cat#KK8201 |
| AllPrep DNA/RNA FFPE kit | Qiagen | Cat#80234 |
| QIAamp® DNA FFPE Tissue Kit | Qiagen | Cat#56404 |
| miRNeasy FFPE kit | Qiagen | Cat#217504 |
| QIAsymphony DSP DNA Midi Kit | Qiagen | Cat#937255 |
| KAPA HyperPrep with RiboErase kit | Roche | Cat#KK8561 |
| KAPA Stranded RNA-Seq with RiboErase kit | Roche | Cat#KK8484 |
| Accel-NGS S2 DNA prep reagents | Swift Biosciences | Cat#210384 |
| Fragment Analyzer RNA kit | Agilent | Cat#DNF-471-1000 |
| AllPrep DNA/RNA FFPE Kit | Qiagen | Cat#80234 |
| Kapa Biosystems library quantification kit | Roche | Cat#KK4854 |
| TapeStation 2200 D1000 screentape | Agilent | Cat#5067-5582 |
|
| ||
| Deposited data | ||
|
| ||
| PTRC HGSOC FFPE Validation - Phosphoproteome | This paper | Proteomic Data Commons:PDC000357 |
| PTRC HGSOC FFPE Validation - Proteome | This paper | Proteomic Data Commons:PDC000358 |
| PTRC HGSOC FFPE Discovery - Phosphoproteome | This paper | Proteomic Data Commons:PDC000359 |
| PTRC HGSOC FFPE Discovery - Proteome | This paper | Proteomic Data Commons:PDC000360 |
| PTRC HGSOC Frozen Validation - Phosphoproteome | This paper | Proteomic Data Commons: |
| PTRC HGSOC Frozen Validation - Proteome | This paper | Proteomic Data Commons:PDC000362 |
| PTRC HGSOC DNA sequencing | This paper | dbGaP:phs003152.v1.p1 |
| PTRC HGSOC RNA sequencing | This paper | dbGaP:phs003152.v1.p1 |
| ProTrack Data Portal: Processed proteogenomic dataset visualization | This paper | http://ptrc.cptac-data-view.org/ |
| Processed LC-MRM-MS data | This paper | https://www.dropbox.com/s/7zul3j1vyrxo40c/processed_data.zip?dl=0 |
| Raw LC-MRM-MS data | This paper | Panoramaweb:Paulovich_PTRC_HGSOC |
| H&E images | This paper | TCIA:doi.org/10.7937/6rda-p940 |
| PhosphoSitePlus | Hornbeck et al.152 | https://www.phosphosite.org |
| MSK-IMPACT | Nguyen et al.18 | N/A |
| CPTAC 2016 | Zhang et al.19 | N/A |
| TCGA – GBM | The Cancer Genomic Atlas Research Network87 | https://portal.gdc.cancer.gov/ |
| TCGA – LGG | The Cancer Genomic Atlas Research Network147 | https://portal.gdc.cancer.gov/ |
| TGFβ and Alternative end-joining pathways | Liu et al.74 | https://pubmed.ncbi.nlm.nih.gov/33568520/ |
| HGSOC RNAseq: GSE154600 | Gene Expression Omnibus | https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/geo/query/acc.cgi?acc=GSE154600 |
| LM22 signature matrix from Cibersort | Chen et al.88 | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5895181/#:~:text=LM22%20is%20a%20signature%20matrix,NK%20cells%2C%20and%20myeloid%20subsets |
| UniProt 2019_06 | reviewed Human Universal Protein Resource sequence database | https://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2019_06/knowledgebase/ |
| UniProt 2020_03_30 | reviewed Human Universal Protein Resource sequence database | https://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2020_03/knowledgebase/ |
| TP53 transcriptional program | Andrysik et al.32 | N/A |
| Mutant TP53 signature | Donehower et al.29 | N/A |
| Genes associated with platinum resistance | Huang et al.17 | http://ptrc-ddr.cptac-data-view.org. |
| Phosphosites related to ischemia | Mertins et al.91, | N/A |
| MSigDB’s database (v 7.5.1) | Liberzon et al.93 | http://software.broadinstitute.org/gsea/msigdb/index.jsp |
| DDR pathway database | Huang et al.17 | http://ptrc-ddr.cptac-data-view.org |
|
| ||
| Software and algorithms | ||
|
| ||
| Github repository for code used for data analysis and figures for this paper | This paper | https://github.com/WangLab-MSSM/CPTAC_Ovarian_Chemo_Response |
| HALO Tissue Classifier machine learning algorithm | Indica Labs | https://indicalab.com/halo-ai/ |
| Philosopher v3.2.8 | Alexey Nesvizhskii Lab; da Veiga Leprevost et al.94 | https://philosopher.nesvilab.org/ |
| MSFragger v3.0 | Alexey Nesvizhskii Lab; Kong et al.95 | https://msfragger.nesvilab.org/ |
| PeptideProphet | Keller et al.96 | http://peptideprophet.sourceforge.net/ |
| PTMProphet | Shteynberg et al.97 | http://www.tppms.org/tools/ptm/ |
| TMT-Integrator | Djomehri et al.98 | http://github.com/huiyinc/TMT-Integrator |
| MaxQuant/Andromeda | Tyanova et al.99 | http://maxquant.org |
| ComBat (v3.20.0) | Johnson et al.100 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| DreamAI | Pei Wang Lab | https://github.com/WangLab-MSSM/DreamAI |
| XDec | Genboree | https://github.com/BRL-BCM/XDec |
| TPO workflow | Michigan Center for Translational Pathology | https://github.com/mctp/tpo |
| Bbduk and bbduk2 | BBMap | https://github.com/BioInfoTools/BBMap/tree/master/sh |
| BWA-mem | Li and Durbin101 | https://github.com/lh3/bwa |
| Seurat | Stuart et al.102 | https://github.com/satijalab/seurat/releases/tag/v3.0.0 |
| BayesDebulk | Petralia et al.58 | https://www.biorxiv.org/content/10.1101/2021.06.25.449763v2 |
| DAGBagM | Chowdhury et al.103 | https://www.biorxiv.org/content/10.1101/2020.10.26.349076v1 |
| GISTIC2.0 | Mermel et al.89 | https://github.com/broadinstitute/gistic2/ |
| iProFun | Song et al.27 | https://github.com/WangLab-MSSM/iProFun |
| ESTIMATE | Yoshihara et al.104 | https://bioinformatics.mdanderson.org/public-software/estimate/ |
| Joint Random Forest | Petralia et al.105 | https://rdrr.io/cran/JRF/man/JRF.html |
| TSNet | Petralia et al.106 | https://github.com/WangLab-MSSM/TSNet |
| xCell | Aran et al.90 | http://xcell.ucsf.edu/ |
| iCAVE | Liluashvili et al.92 | http://labs.icahn.mssm.edu/gumuslab/software |
| ConsensusClusterPlus | Wilkerson and Hayes107 | http://bioconductor.org/packages/release/bioc/html/CancerSubtypes.html |
| Strelka2 v2.9.3 | Kim et al.108 | https://github.com/Illumina/strelka |
| CNVkit v. 2.9.3 | Talevich et al.109 | https://github.com/etal/cnvkit |
| STAR v2.6.1d | Dobin et al.110 | https://github.com/alexdobin/STAR |
| GENCODE v27 | GENCODE consortium | https://www.gencodegenes.org/human/release_27.html |
| RSEM v1.3.1 | Li and Dewey111 | https://github.com/deweylab/RSEM |
| Sumer | Savage et al.112 | https://github.com/bzhanglab/sumer |
| TCGAbiolinks | Colaprico et al.113 | https://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html |
| MoonlightR | Colaprico et al.114 | https://bioconductor.org/packages/release/bioc/html/MoonlightR.html |
| MuSiC | Wang et al.115 | https://github.com/xuranw/MuSiC |
| GATK | Broad Institute | https://gatk.broadinstitute.org/hc/en-us/articles/360035890811-Resource-bundle |
| VEP | McLaren et al.116 | https://useast.ensembl.org/info/docs/tools/vep/index.html |
| vcfAnno | Pedersen et al.117 | https://github.com/brentp/vcfanno |
| CNVEX | Michigan Center for Translational Pathology | https://github.com/mctp/cnvex |
| DNAscope | Sentieon | https://support.sentieon.com/versions/201911/manual/DNAscope_usage/dnascope/ |
| Kallisto | Bray et al.118 | https://pachterlab.github.io/kallisto/download.html |
| Ascore v1.0.6858 | Github | https://github.com/PNNL-Comp-Mass-Spec/AScore |
| MASIC | Monroe et al.119 | https://github.com/PNNL-Comp-Mass-Spec/MASIC |
| MS-GF+ v9981 | Kim and Pevzner120 | https://github.com/MSGFPlus/msgfplus |
| mzRefinery | Gibbons et al.121 | https://pnnl-comp-mass-spec.github.io/MzRefinery |
| pamr | Tibshirani lab | https://CRAN.R-project.org/package=pamr |
| XGBoost | Chen and Guestrin45 | https://cran.r-project.org/web/packages/xgboost/index.html |
| RandomForest | Breiman44 | https://cran.r-project.org/web/packages/randomForest/ |
| glmnet | Tibshirani lab | https://cran.r-project.org/web/packages/glmnet/ |
| Skyline | MacLean et al.122 | https://skyline.ms/project/home/software/Skyline/begin.view |
| GSVA | Hänzelmann et al.123, | https://www.bioconductor.org/packages/release/bioc/html/GSVA.html |
| Ovarian cancer molecular subtype classifier | Chen et al.124 | http://bioconductor.org/packages/release/bioc/html/consensusOV.html |
|
| ||
| Other | ||
|
| ||
| Glass slides | Leica Biosystems | Cat#3800040 |
| Tissue Bags | Covaris | Cat#TT1, 520001 |
| 10 mg Sep-Pak solid-phase extraction | Waters | Cat#186000128 |
| 5 mg Sep-Pak solid-phase extraction | Waters | Cat#186000309 |
| 2 mg Sep-Pak solid-phase extraction | Waters | Cat#186001828BA |
| PicoTip™ emitter, 50 μm ID × 20 cm | New Objective | Cat#FS360-50-15-N-20-C12 |
| 1 mL deep well plate | Thermo Fisher Scientific | Cat#95040450 |
| 4.6 mm × 250 mm Zorbax Extend- C18, 3.5 μm, column | Agilent | Cat#770953-902 |
| ReproSil-Pur, 120 Å, C18-AQ | Dr. Maisch | Cat#r119.aq |
| EvoTip Pure | EvoSep | Cat#EV2011 |
| Endurance OE, 15 cm × 150 μm, 1.9 μm | EvoSep | Cat#EV-1113 |
| epMotion 5075 | Eppendorf | Cat#5075 900.157-13/0411 |
| Agilent 1200 HPLC | Agilent | Cat#G2262-90010 |
| KingFisher Flex | Thermo Fisher Scientific | Cat#N13141 |
| Easy-nLC 1000 | Thermo Fisher Scientific | Cat#LC120 |
| LTQ-Orbitrap Fusion mass spectrometer | Thermo Fisher Scientific | Cat#IQLAAEGAAPFADBMBCX |
| Evosep One LC | EvoSep | Cat#EV1000 |
| 6500+ QTRAP mass spectrometer | Sciex | Cat#5039926 |
| OptiFlow Turbo V source | Sciex | Cat#5028138 |
| Qubit 4 fluorometer | Thermo Fisher Scientific | Cat#Q33238 |
| Agilent Tapestation 2200 | Agilent | Cat# 5067-5582 |
| Illumina NovaSeq 6000 | Illumina | Cat#20012850 |
| Beckman Coulter Biomek i7 | Beckman Coulter | https://www.illumina.com/content/dam/illumina/gcs/assembled-assets/marketing-literature/novaseq-6000-spec-sheet-m-gl-00271/novaseq-6000-spec-sheet-m-gl-00271.pdf |
| BOND Rx autostainer | Leica | Cat#21.2821 |
| PhenoImager HT Automated Imaging System | Akoya Biosciences | Cat#CLS143455 |
| Protrack data portal | This paper | http://pbt.cptac-data-view.org |