


Abstract
The selenocysteine (Sec) tRNA (tRNA[Ser]Sec) governs Sec insertion into selenoproteins by the recoding of a UGA codon, typically used as a stop codon. A homozygous point mutation (C65G) in the human tRNA[Ser]Sec acceptor arm has been reported by two independent groups and was associated with symptoms such as thyroid dysfunction and low blood selenium levels; however, the extent of altered selenoprotein synthesis resulting from this mutation has yet to be comprehensively investigated. In this study, we used CRISPR/Cas9 technology to engineer homozygous and heterozygous mutant human cells, which we then compared with the parental cell lines. This C65G mutation affected many aspects of tRNA[Ser]Sec integrity and activity. Firstly, the expression level of tRNA[Ser]Sec was significantly reduced due to an altered recruitment of RNA polymerase III at the promoter. Secondly, selenoprotein expression was strongly altered, but, more surprisingly, it was no longer sensitive to selenium supplementation. Mass spectrometry analyses revealed a tRNA isoform with unmodified wobble nucleotide U34 in mutant cells that correlated with reduced UGA recoding activities. Overall, this study demonstrates the pleiotropic effect of a single C65G mutation on both tRNA phenotype and selenoproteome expression.
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Selenium is an essential trace element in mammals that is involved in redox biology. The association of selenium deficiencies with the increased risks of developing several pathologies including cancers, neurodegenerative diseases, cardiovascular disorders and infectious diseases is now well reported (1–7). Most of the role of selenium in living organisms is related to its presence as selenocysteine (Sec) in selenoproteins. Sec is the 21st amino acid from the genetic code and it stands out from other proteinogenic amino acids in that it is synthesized from a peculiar and unique tRNA and is encoded by a UGA stop codon. Therefore, a set of dedicated factors, components of the Sec insertion machinery, is employed to perform what is known as UGA-Sec translational recoding (8–14). These factors include a cis-acting stem–loop–stem–loop RNA structure, named the selenocysteine insertion sequence (SECIS), present in the 3′-untranslated region (UTR) of all selenoprotein mRNAs, and the SECIS-binding protein 2 (SECISBP2) which plays the role of a platform to recruit the complex composed of the tRNA[Ser]Sec and the dedicated elongation factor EFSec. Recently, data obtained with cryo-electron microscopy (cryo-EM) have allowed the visualization of all these constituents within the 80S ribosomes, thereby unveiling fresh structural insights into the interplay amongst the various partners (15). These findings furnish significant structural information about the selenosome and the mechanisms that enable ribosomal decoding of the UGA codon into the Sec amino acid. Nonetheless, we have yet to fathom out the entire molecular mechanism and comprehend how the orchestration of all these elements competes with the translation termination complex. The translational UGA recoding event is a rate-limiting step of selenoprotein expression and its efficiency controls both the steady-state levels of selenoproteins and their expression in response to various stimuli (9). The regulation of selenoprotein synthesis by selenium bioavailability has been studied in many reports (16–24), but other exogenous stimuli such as oxidative stress, replicative senescence and potentially pathophysiological conditions are also key players to modulate the UGA-Sec recoding event (9,10,25–29).
tRNA is an adapter molecule that physically links a codon on the mRNA with a defined proteinogenic amino acid during protein synthesis. The tRNA repertoire of an organism specifies its genetic code. In eukaryotes, the UGA codon is primarily read by the translation termination factor eRF1 as a stop signal for protein synthesis for the vast majority of cellular mRNAs. However, in many organisms, it can also be decoded as a Sec codon by tRNA[Ser]Sec, and this occurs in mRNAs coding for selenoproteins (30–34). This leads to a competition between translation termination and Sec insertion at the UGA codons. tRNA[Ser]Sec displays several features that make it unique compared with other cellular tRNAs (31,34). In fact, tRNA[Ser]Sec plays a crucial role in regulating the expression of a cluster of genes, specifically the 25 selenoproteins that constitute the selenoproteome. In Homo sapiens, there is only one gene coding for tRNA[Ser]Sec and it is located on chromosome 19 (TRNAU1 or TRU-TCA1-1) (35). There is also a pseudogene that carries an inactive acceptor arm which is present on chromosome 22 (TRNAU2 or TRU-TCA2-1), but this gene does not seem to be transcriptionally active (35). In mice, gene inactivation of tRNA[Ser]Sec leads to early embryonic lethality, indicating the importance of selenoproteins in development (36). tRNA[Ser]Sec presents unique structural and functional features such as its size, structure, transcription, modification, aminoacylation and transport. It is by far the largest tRNA in eukaryotes (96 nucleotides in length), mostly due to a particularly large variable arm that folds in a 16 nucleotides stem–loop (see Figure 1). tRNA[Ser]Sec adopts a 9/4 secondary structure in contrast to the canonical 7/5 structure found in other tRNAs. These structural differences prevent it from interacting with the elongation factor EF-1A, but they are also used for recognition by the dedicated elongation factor EFSec. The transcription of the TRNAU1 gene by RNA polymerase III (POLR3) is also original as it uses both intragenic and upstream promoters. This tRNA is called [Ser]Sec because it is first loaded with the amino acid serine which is transformed into Sec by a cascade of enzymatic reactions involving seryl-tRNA synthetase (SARS), phosphoseryl-tRNA kinase (PSTK), selenocysteine synthase (SEPSECS) and selenophosphate 2 synthetase (SEPHS2) (8–10,30). Note that SEPHS2 is a selenoprotein itself and constitutes a regulatory loop for tRNA aminoacylation (37). In addition, tRNA[Ser]Sec exhibits fewer post-transcriptional modifications than most tRNAs. As reviewed in (38,39), methyladenosine (m1A) and pseudouridine (ψ) are both present, at positions 58 and 55, respectively, to ensure proper tRNA folding. The other modifications are found in the anticodon loop and are expected to be critical for anticodon folding and UGA recoding. For instance, the N6-isopentenyladenosine (i6A) is at position 37 which is 3′-adjacent to the anticodon and prevents illicit hydrogen bonding between U33 and A37 in order to preserve the anticodon loop. i6A37 stabilizes the anticodon–codon Watson–Crick base pairing by base stacking (40). The modification of the U34 wobble position in tRNA[Ser]Sec is also of particular interest since it is sensitive to the selenium status. As such, the U34 position is in the form of 5-methoxycarbonylmethyluridine (mcm5U) which can be further methylated to 5-methoxycarbonylmethyluridine-2′-O-methylribose (mcm5Um) (41,42). In mouse models, the expression of a mutant transgene of tRNA[Ser]Sec (A37G) that lacked two base modifications, i6A37 and Um34, resulted in a severe lack of expression of several selenoproteins including GPX1, GPX3, SELENOT, SELENOW and MSRB1. However, several other selenoproteins including TXNRD1, TXNRD2, GPX2, GPX4, SELENOP and SELENOF were, at least, partially expressed in comparison with mice expressing the wild-type (WT) transgene (43). It has been suggested that the hierarchical response of selenoproteins to variations in selenium levels involves the equilibrium between two distinct isoforms of tRNA carrying either the mcm5U34 or mcm5Um34 modification (30,31), but the molecular basis of this mechanism remains unknown.
Figure 1.

In silico prediction of C65G mutation-induced changes in tRNA[Ser]Sec structure. (A) Schematic representation of the 2D structure of the human tRNA[Ser]Sec; the position of the mutated nucleotide is shown in red (C65). The post-transcriptionally modified nucleotides are also indicated: methyladenosine (m1A58), pseudouridine (ψ55), N6-isopentenyladenosine (i6A37), 5-methoxycarbonylmethyluridine (mcm5U34) and 5-methoxycarbonylmethyluridine-2′-O-methylribose (mcm5Um34). (B) Representation of the 3D structure of WT and C65G tRNA[Ser]Sec predicted with RNAComposer based on human and mouse crystallographic structures (PDB: 3A3A and 3RG5) in a ladder and ribbon layout. The mutated C65G residue is indicated in red. Nucleotides located at positions 9 and 65 are depicted in stick format in an enlarged view, allowing for better visualization of the relative arrangement of the nitrogenous base planes in both WT and C65G predictions.
Multiple mutations in mitochondrial- and nuclear-encoded tRNA genes have been linked to human diseases (44). While ∼370 mutations in all 22 mitochondrial tRNAs genes have been reported in humans and associated with multiple diseases (45,46), only three clinical cases have been related to mutations of nuclear-encoded tRNA genes. The first example concerns the isodecoder tRNAArg (anticodon TCT) 4–1 which is specifically expressed in the central nervous system (47). This C50T mutation located in the T-loop interfered with pre-tRNA maturation and aminoacylation levels and led to widespread neurodegeneration. The other two case reports concerned a homozygous single mutation (C65G) in the acceptor arm of the tRNA[Ser]Sec gene (48,49). In the first report, the 8-year-old male patient had thyroid dysfunction and a low plasma selenium level as well as symptoms such as abdominal pain, fatigue and muscle weakness (48). The authors observed a decrease in the expression of several selenoproteins (e.g. TXNRD1, GPX4, SELENON, SELENOP and SELENOT) while others were not detected (e.g. GPX1, GPX3, SELENOH and SELENOS). Although the ratio of the methylated (mcm5Um) versus the unmethylated (mcm5U) form of tRNA[Ser]Sec was slightly lower in the patient, the mechanism for this differential expression was not addressed. In the second case, a 13-year-old male patient was medically followed for 6 years and had thyroid dysfunction, sometimes but not always low selenium levels, and no further clinical outcomes (49). Only few biochemical analyses were performed and the blood level of GPX3 was found to be in the low-normal range. Unlike the first case where both parents were carriers of the mutation, for the second patient only his father was a carrier, his mother being free of the C65G variant. The second patient was found to have a paternal uniparental disomy. In the first patient, the clinical management was limited to alleviating symptoms, but the authors proposed a selenium supplementation to compensate for specific selenoprotein deficiencies as a rational therapeutic approach (50,51).
Interestingly, despite having a central role in the synthesis of the entire selenoproteome, the C65G mutation in tRNA[Ser]Sec leads to a relatively mild clinical outcome compared with mutations in other components of the Sec insertion machinery (50–52). In this study, we have employed a CRISPR/Cas9 [clustered regularly interspaced palindromic repeats (CRISPR)/CRISPR-associated protein 9] strategy to generate the C65G mutation in HAP1 human cell lines, resulting in a significant reduction of tRNA[Ser]Sec production caused by a decrease of POLR3 recruitment on the TRNAU1 gene promoter. Our findings indicate that the mutant tRNA was less functional than its WT counterpart and acted as an inhibitory competitor when overexpressed. This reduction in efficiency was accompanied by a hypomodification of the wobble U34 nucleotide. Furthermore, expression of the selenoproteome in C65G cell lines was no longer sensitive to selenium.
MATERIALS AND METHODS
This manuscript adopts the systematic nomenclature of selenoprotein names (53). Experiments were replicated at least three times, unless otherwise stated. Statistical analyses and graphical illustrations were performed with GraphPad Prism, version 9.1.2. Error bars represent the standard deviation (SD). Mean values were tested for statistical significance by unpaired Student's t-tests.
Cell culture
HAP1 (Horizon Discovery, Cambridge, UK, Cat.# C631) and HEK293T (Life Technologies, Carlsbad, CA, USA, Cat.# R75007) cells were grown and maintained in Dulbecco's modified Eagle’s medium (DMEM; ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS; Merck), 100 μg/ml streptomycin, 100 U/ml penicillin and 2 mM l-glutamine (ThermoFisher Scientific). Selenium concentration in the control (Ctrl) medium was 19.4 nM. For selenium dose–response experiments, cells were grown in DMEM supplemented with 2% FBS; the selenium concentration was then ∼3.9 nM. Cells were cultivated at 37°C in a humidified atmosphere containing 5% CO2. Selenium supplementation was achieved by adding a defined volume of a concentrated 0.1 mM sodium selenite solution (Merck) to the culture medium. For the assessment of tRNA stability, transcription was blocked by actinomycin D (Merck) used at 10 μg/ml, and cells were harvested at the indicated time points. For phenotypic analysis, cells were grown for 48 h in medium supplemented or not with 100 nM sodium selenite before protein and RNA extractions. For luciferase assays and rescue experiments, cells were transfected with the indicated plasmids using JetOptimus Transfection Reagent (Ozyme) according to the manufacturer's instructions.
Generation of HAP1 cell lines carrying the C65G mutation using CRISPR/Cas9 technology
We used CRISPR/Cas9 technology coupled with a cell endogenous repair pathway to introduce the C65G mutation in the TRNAU1 gene in HAP1 cells. CRISPR/Cas9-induced DNA double-strand breaks (DSBs) can be resolved by homology-directed repair (HDR) using a donor single-stranded DNA (ssDNA) repair template. In order to efficiently edit HAP1 cells and limit off-target effects, we used viral-like particles (VLPs) named nanoblades, as previously described (54,55). The variable part of the single guide RNA (sgRNA) that targets the tRNA[Ser]Sec gene (CTTAGTTACTACCGCCCGAA) was cloned into pBLADE and this plasmid was used to produce nanoblade VLPs. HEK293 cells were plated at 3.5 × 106 cells/10 cm diameter plate 24 h before transfection with JetPrime reagent (Ozyme). Plasmids encoding sgRNA (4.5 μg), Gag-PolMLV (3.3 μg), GagMLV–Cas9 fusion (2.0 μg), baboon endogenous retrovirus Rless glycoprotein (BaEVRless) (0.7 μg) and vesicular stomatitis virus glycoprotein (VSV-G) (0.5 μg) were co-transfected. Two days post-tranfection, the culture medium was collected, filtered at 0.45 μm and concentrated by ultracentrifugation at 4°C (1 h 30 min, 69 000 g). After removal of the supernatant, the concentrated particles from one 10 cm plate were resuspended in 100 μl of phosphate-buffered saline (PBS), aliquoted and stored at –80°C for several months or at +4°C for several weeks.
HAP1 cells were electroporated with ssDNA repair template 5′-CATTCTAAATTCCGCGAATCAATGATGGCGGGCTTTGCACCTTAACTTGGTGAGACAGCGATCTGTCGATGTCCAAAC-3′ at 50 pmol oligonucleotide per 100 000 cells according to the manufacturer's instructions (Neon transfection system, ThermoFisher Scientific). A total of 5 × 105 cells were electroporated and plated in a minimal volume and, after 4 h, a volume of VLP corresponding to 20 μg of Cas9 was added to the medium. Seven days later, cellular clones were isolated in 96-well plates by limiting dilution. After clonal expansion, the genotype of each clone was determined. Genomic DNAs were collected using the Nucleospin gDNA extraction kit (Macherey-Nagel) and 150 ng was then used for polymerase chain reaction (PCR) amplification with the forward (5′-CAGGGCTGTCACCCACCGCTGCGTCCTC-3′) and reverse (5′-GTCAACCATCTCACACCTTTCCAAAGG-3′) primers. The PCR products were verified on 2% agarose gels, sequenced and tested for the presence of a MaeII (HpyCH4IV) restriction site that is created by the C65G mutation. We obtained 11/96 heterozygotes clones containing the C65G mutation (11.5%). As the introduction of C65G mutation induces the destruction of the protospacer adjacent motif (PAM) sequence, we used a WT/C65G clone to perform another round of CRISPR/Cas9/HDR. After clonal selection, we obtain 7/48 homozygous clones (14.6%).
HAP1 proliferation assay
HAP1 cells were labeled using the CellTrace™ Violet (CTV) Cell Proliferation Kit (Invitrogen) according to the manufacturer's instructions. A 2 μl aliquot of CTV was incubated with 2 × 106 HAP1 cells in 2 ml of PBS for 20 min at 37°C. To neutralize the unbound CTV, 8 ml of DMEM was added. After two washes with PBS, every cellular clone was plated in a 6-well plate at 3 × 105 cells per well and grown with or without selenium supplementation. Cells were harvested at the indicated time points and analyzed by flow cytometry all at once at the end of the kinetics. Flow cytometry was performed on a MACSQuant® VYB Flow cytometer (Miltenyi Biotec) and the analysis was done with FlowJo software (Treestar, Ashland, OR, USA). The mean fluorescence intensity (MFI) of each condition was plotted as a function of time. Kinetic curves were fitted with y = a.exp(−k.t), where the doubling time t1/2 = [ln(2)/k], and the represented time necessary for a 50% decrease in the MFI signal.
Total RNA extraction and analysis by northern blot and RT–qPCR
As previously described (55), total RNAs were purified with Tri Reagent™ (Molecular Research Center) according to the manufacturer's instructions and resuspended in water. A 15 μg aliquot of purified RNAs was size-fractionated on a 1× TBE, urea (8 M), polyacrylamide (15%) gel and electroblotted onto a Hybond-N nylon membrane (GE Healthcare). The blots were hybridized overnight at 58°C with 5′-terminally IRdye-labeled sequence-specific oligonucleotides (IDT: Integrated DNA Technologies): tRNA-Sec 5′-(IRD800)CCACTGAGGATCATCCGGGC-3′ and tRNA-Ser 5′-(5IRD700)CGTAGTCGGCAGGATTCGAA-3′ in PerfectHyb™ Plus Hybridization Buffer (Sigma-Aldrich). Membranes were washed with 1× standard saline citrate (SSC)/0.1% sodium dodecylsulfate (SDS) buffer twice at 37°C, and then once at room temperature. The infrared signal from the membrane was detected with an Odyssey imaging system CLx (LI-COR Biosciences). An initial scan at a low resolution (337 μm) was performed to locate and verify the quality of the signal. This region was then scanned at a higher resolution (42 μm). Quantifications were performed using ImageStudioLite software.
To assess the levels of selenoprotein mRNAs, we performed quantitative reverse transcription–PCR (RT–qPCR) experiments as described previously (23,25,55). Total RNAs were reverse transcribed using the qScript cDNA Synthesis kit (Quanta Bio) according to the manufacturer's instructions. Real-time PCR was performed in triplicate using FastStart Universal SYBR® Green master mix (Roche Applied Science) on a StepOne Real-Time PCR System (Applied Biosystems). Primers used are listed in Supplementary Table S1. Serial dilutions of a cDNA mixture were used to create a standard curve and determine the efficiency of the amplification for each pair of primers. The quantification of selenoprotein mRNAs was performed using a set of four reference genes.
Isolation and LC/MSMS analysis of tRNA[Ser]Sec
To analyze the post-transcriptional modifications of tRNA[Ser]Sec by mass spectrometry (MS), we harvested cells from twenty 15 cm diameter plates, which represented ∼4 × 108 cells. A 12 mg aliquot of total RNA purified with Tri Reagent was separated by anion exchange chromatography with DEAE Sepharose Fast Flow (GE Healthcare) to remove polysaccharides and rRNA according to (56). The RNA samples were applied to 1 ml of DEAE–Sepharose FF (GE Healthcare) in 5 ml disposable columns (Bio-Rad) equilibrated with 5 ml of buffer A [10 mM HEPES-KOH (pH 7.5), 200 mM NaCl and 2 mM dithiothreitol (DTT)]. The RNA samples on DEAE–Sepharose were washed with buffer A and then eluted with buffer B (10 mM HEPES-KOH pH 7.5, 1 M NaCl and 2 mM DTT) in order to obtain ∼1 mg of crude tRNAs. As described in (57), tRNA[Ser]Sec was further purified using a 3′-biotinylated DNA oligonucleotide (5′-CGCCCGAAAGGTGGAATTGAACCACTCTGTCGCTA-3′) immobilized on streptavidin-conjugated M-280 magnetic Dynabeads according to the manufacturer's instructions (Life Technologies). For the mutant tRNA, another 3′-biotinylated DNA oligonucleotide was used (5′-CGCCCGAAACGTGGAATTGAACCACTCTGTCGCTA-3′). Purified tRNA[Ser]Sec was isolated on a denaturing 10% polyacrylamide gel containing 8 M urea, and liquid chromatography/tandem MS (LC/MSMS) analysis was performed as previously described (58). Gel pieces containing tRNA[Ser]Sec were digested by 20 μl of 0.1 U/μl RNase T1 (ThermoFisher Scientific) for 4 h at 50°C. Using ZipTip C18 (Milllipore), samples were desalted by several washes with 200 mM ammonium acetate, eluted with 50% acetonitrile in milliQ water and dried under vacuum. The pellet containing RNase digestion products was resuspended in 3 μl of milliQ water and separated on an Acquity peptide BEH C18 column (130 Å, 1.7 μm, 75 μm × 200 mm) using a nanoAcquity system (Waters). The column was equilibrated in buffer A containing 7.5 mM TEAA (triethylammonium acetate), 7.0 mM TEA (triethyammonium) and 200 mM HFIP (texafluoroisopropanol) at a flow rate of 300 nl/min. Oligonucleotides were eluted using a gradient from 15% to 35% of buffer B (100% methanol) for 2 min followed by elution with an increase of buffer B to 50% in 20 min. MS and MS/MS analyses were performed using a SYNAPT G2-S from Waters. All experiments were performed in negative mode with the capillary voltage set at 2.6 kV and the sample cone voltage set at 30 V. The source was heated at 130°C. The samples were analyzed over an m/z range from 500 to 1500 for the full scan, followed by fast data direct acquisition scan (Fast DDA). Collision-induced dissociation (CID) spectra were deconvoluted using MassLynx software from Waters and manually sequenced by following the y and/or c series.
ChIP-qPCR experiments
A total of 107 HAP1 cells were cross-linked with 1% formaldehyde for 10 min at room temperature directly in 15 cm plates. As described in (59), cross-linking was quenched by addition of 0.125 M glycine, and cells were harvested and centrifuged at 800 g. Cells were lysed for 30 min in Lysis Buffer (1% Triton X-100, 0.1% SDS, 140 mM NaCl, 50 mM HEPES-KOH pH 7.5, 1 mM EDTA complemented with protease inhibitor cocktail and phosphatase inhibitor). Nuclei were pelleted and resuspended in 1 ml of shearing buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 0.1% SDS). Chromatin was sheared in order to obtain fragments ranging from 200 to 800 bp using Covaris S220 (20 min, Peak Power: 140; Duty Factor: 5; Cycles/burst: 200) and debris were eliminated by a 10 min centrifugation at 13 200 rpm at 4°C. A 25 μg aliquot of chromatin was pre-cleared using 30 μl of Dynabeads® Protein A/G (ThermoFisher) for 30 min. Then 1 ml of Dilution Buffer (1% Triton X-100, 0.01% SDS, 150 mM NaCl, 10 mM Tris–HCl pH 8, 1 mM EDTA) was added to the chromatin and immunoprecipitation was performed overnight at 4 °C with 5 μg of antibodies, either rabbit anti-POLR3A antibody (D5Y2D, Cell Signaling Technology) or control rabbit IgG (Cell Signaling Technologies). Complexes were recoreved by addition of 30 μl of Dynabeads® Protein A/G (ThermoFisher) and incubated for 2 h. Complexes were washed with 1 ml of each of the following buffers: Wash 1 [1% Trition, 0.1% sodium deoxycholate (NaDOC), 150 mM NaCl, 10 mM Tris–HCl pH 8], Wash 2 (1% NP-40, 1% NaDOC, 150 mM KCl, 10 mM Tris–HCl pH 8), Wash 3 (0.5% Triton, 0.1% NaDOC, 500 mM NaCl, 10 mM Tris–HCl pH 8), Wash 4 (0.5% NP-40, 0.5% NaDOC, 250 mM LiCl, 20 mM Tris–HCl pH 8, 1 mM EDTA) and Wash 5 (0.1% NP-40, 150 mM NaCl, 20 mM Tris–HCl pH 8, 1 mM EDTA). Immunoprecipitated complexes were released with 500 μl of elution buffer (1% SDS, 200 mM NaCl, 100 mM NaHCO3) and de-cross-linked by a 16 h incubation at 65°C. The immunoprecipitated chromatin was then purified by phenol–chloroform extraction, and qPCR was performed as described above. We used the tRNALeu gene as a reference to normalize the occupancy of POLR3 at the TRNAU1 gene. Primers used for chromatin immunoprecipitation (ChIP) experiments are listed in Supplementary Table S1.
Protein extraction and analysis by western blot
Cellular protein extracts were harvested in a lysis buffer (25 mM Tris–HCl, pH 7.8, 2 mM DTT, 2 mM EDTA, 1% Triton X‐100 and 10% glycerol). Then, protein concentrations were measured using the DC kit protein assay kit (Biorad) in microplate assays. Equal protein amounts (30 μg) were separated on 4–12% Bis-Tris NuPAGE Novex Midi Gels and transferred onto nitrocellulose membranes using the iBlot® DRy blotting System (ThermoFisher Scientific). Membranes were probed with the indicated primary antibodies and horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse secondary antibodies. Antibodies were purchased from Abcam (GPX1, #ab108427; GPX4, #ab125066; TXNRD1, #ab124954; TXNRD2, #ab180493; SELENOH, #ab151023; SELENOO, #ab172957; SELENOT, #ab176192) and Sigma (SELENOS, #HPA010025; Actin, #A1978, HRP-conjugated goat anti-rabbit IgG, #A6154, HRP-conjugated goat anti-mouse IgG, #A9044). The chemiluminescence signal was detected using an ECL Select detection kit (GE Healthcare) in the Chemidoc Imager (Biorad). Data quantifications were performed with ImageLab Software (Biorad, Version 6.0.1). The top part of the membranes were reblotted with anti-actin antibodies following the same procedure. Values were expressed relative to the signal obtained for actin.
GPX and TXNRD enzymatic assays
GPX and TXNRD activities were measured in an enzymatic coupled assay as described (27). For GPX activity, the reaction mixture was composed of 50 μg of protein extract, 0.25 mM NADPH (Merck), 2 mM reduced l-glutathione and 1.5 IU of glutathione reductase (Merck) adjusted to a total volume of 250 μl with 50 mM potassium phosphate buffer (pH 7.5). The reaction was started by the addition of 300 nM tert-butyl hydroperoxide (t-BHP, Merck), and consumption of NAPDH was followed at 340 nm with the FLUOSTAR OPTIMA microplate reader (BMG Labtech). GPX enzymatic activities (U/mg of protein extracts) were expressed as nmol of glutathione/min/mg of protein extracts. For TXNRD enzymatic activity, the enzymatic reduction of 5,5′-dithiobis(2-nitrobenzoic) acid (DTNB) with NADPH into 5-thio-2-nitrobenzoic acid (TNB2−) generated a strong yellow color at 412 nm. In this assay, the reaction mixture was composed of 50 μg of protein extract, 0.2 mM NADPH, 10 mM EDTA (Merck) and 0.2 mg/ml bovine serum albumin (BSA), adjusted to a total volume of 250 μl with 50 mM potassium phosphate buffer (pH 7.5). The reaction was started by the addition of 25 mM DTNB, and the production of TNB acid was followed at 410 nm with the FLUOSTAR OPTIMA microplate reader. TXNRD enzymatic activities (U/mg) were expressed as nmol of NADPH/min/mg (milliunits per milligram of protein extracts). The enzymatic assays were performed in duplicate for three independent experiments and the background values were subtracted.
Evaluation of selenocysteine insertion efficiency
To analyze Sec insertion efficiency in the different HAP1 cellular clones, we used luciferase-based reporter constructs that were validated for UGA-Sec recoding in transfected cells (25,27,60). The minimal SECIS elements from MSRB1, GPX1, GPX4, TXNRD1 and TXNRD2 were cloned downstream of a luciferase-coding sequence, which was modified to contain an in-frame UGA codon at position 258 (Luc UGA/SECIS). Other luciferase constructs were also generated with the codon UAA or UGU at position 258 from Luc UGA/SECIS MSRB1 (60). In all constructs, UAA is the stop codon of the luciferase-coding sequence. Transfections were performed in 6-well plates with a mix of 0.25 μg of the indicated luciferase construct, 0.25 μg of the GFP (green fluorescent protein) plasmids and 0.5 μg of an empty vector, with 1.5 μl of JetOptimus per well. Cells were harvested 2 days post-transfection in lysis buffer (25 mM Tris–HCl, pH 7.8, 2 mM DTT, 2 mM EDTA, 1% Triton X‐100 and 10% glycerol). GFP levels and luciferase activity (Promega Luciferase Assay system) were measured using the FLUOSTAR OPTIMA and LUMISTAR OPTIMA microplate readers from BMG Labtech. The luciferase/GFP ratio is first used to normalize transfection efficiency. Then the ratio of the indicated construct to the Luc–UGU construct is used to express the percentage of Sec insertion efficiency. As a control of mRNA stability, the levels of luciferase mRNA were assessed by RT–qPCR as described above; primers used are listed in Supplementary Table S1.
RESULTS
In silico prediction of C65G mutation-induced changes in tRNA[Ser]Sec structure
The analysis of the two-dimensional (2D) structure of tRNA[Ser]Sec, as depicted in Figure 1A, suggests that the substitution of the C65 nucleotide with a G may disrupt a Watson–Crick C:G base pairing, and may also impose a steric hindrance due to the presence of two purine residues facing each other. However, it is possible that two G residues may form various types of non-Watson–Crick base pairs, as described previously (61). To determine whether this could also occur in this case and prevent a complete destabilization of the cloverleaf tRNA folding, we conducted in silico analyses to predict possible RNA structures. To accomplish this, we used RNAComposer, an online software that provides 3D structure models based on the sequence and secondary structure topological folding of RNA (62). Since both mouse and human tRNA[Ser]Sec structures were resolved (38,63), a high confidence for RNAComposer-generated models was expected. The generated pdb files were then visualized and analyzed using Chimera UCSF software (64). Our findings indicate that the C65G mutation indeed disrupts one G:C base pair but without significantly affecting the overall architecture of the 3D structure, as illustrated in Figure 1B and Supplementary Movies S1 and S2. However, the loss of a G:C base pairing may impair the global stability of the molecule, and further investigation is required to confirm this hypothesis.
Generation of HAP1 cell lines carrying the C65G mutation
In order to investigate the impact of the C65G mutation on cellular processes, particularly tRNA production and selenoproteome synthesis, we successfully generated heterozygous and homozygous mutant HAP1 cell lines using CRISPR/Cas9 DNA DSBs associated with HDR. CRISPR gene editing provides several strategies to manipulate mammalian genomes in cellular and in vivo models (65,66). Previously, we used VLPs containing Cas9–gRNA ribonucleoprotein (RNP), also called nanoblades, to produce site-specific DNA DSBs in the human tRNA[Ser]Sec gene (TRNAU1) and to induce gene disruption by error-prone non-homologous end joining (NHEJ) (54,55). We validated this strategy in several cell lines, including HAP1, commonly used in CRISPR studies, since these cells are nearly haploid (55). Here, we introduced the C65G mutation in the tRNA gene using HDR after nanoblade-induced DSB. As illustrated in Figure 2A, the repair template had homology to the targeted site but contained the C to G mutation. Interestingly, this single mutation has three distinct effects: (i) it introduces the nucleotide change at position 65 as is found in the patients (48,49); (ii) it disrupts the PAM sequence which is needed for Cas9 recognition which prevents recurrent CRISPR activity after HDR; and (iii) it creates a MaeII restriction site (see Figure 2A) that will be used to evaluate the efficiency of HDR in CRISPR-treated cells and to screen and select for the clones harboring the C65G mutation. Since HDR (10–15% of editing events) was in competition with NHEJ (70% of editing events) after DSB, the CRISPR-treated genomic DNA (gDNA) would not only exhibit the expected mutations but will also contain the INDELs reported in (55) due to NHEJ imperfect repair. In addition, we observed that HAP1 cells became rapidly diploid after CRISPR experiments and clonal selection as previously observed in (67). Therefore, due to this rather low probability of HDR, we employed a two-step approach to achieve homozygous genotypes (C65G/C65G) in HAP1 cells. The first step consisted of the selection of a heterozygous clone (WT/C65G) using a MaeII enzymatic digestion for an initial screen followed by systematic DNA sequencing for the positive clones. A heterozygous (WT/C65G) clone underwent another round of CRISPR and HDR treatment to generate the homozygous mutant cell lines (C65G/C65G). We picked three cellular clones that were validated for the MaeII restriction site and Sanger sequencing of the gDNA (see Figure 2B and C). These three homozygous clones were referred to as C65G #1, C65G #2 and C65G #3.
Figure 2.

CRISPR/Cas9 design and generation of HAP1 cell lines carrying the C65G mutation. (A) The human TRNAU1 gene sequence is presented in black, with the PAM position indicated in red, the sgRNA in orange above and the ssDNA repair template in blue below. The desired C65G mutation is highlighted in red in the repair template. Transduction of the nanoblades induces the cleavage of the TRNAU1 gene by Cas9 RNA-guided nuclease followed by the addition of the DNA repair template-induced HDR and eventually insertion of the C65G mutation. The change of the C65 residue in G65 adds a MaeII digestion site (indicated by two black arrows) and destroys the PAM sequence. (B) Genotypic analysis of selected HAP1 clones and the parental cell line by MaeII digestion. The region of the TRNAU1 gene was amplified from the gDNA by PCR, and the fragments were digested by MaeII and analyzed on a 2% agarose gel. The positions of the undigested and digested products are indicated. (C) The PCR fragments were also analyzed by Sanger sequencing. The sequencing traces are shown with the WT and C65G sequences above and below, respectively. The colors of the chromatograms are as follows: A in green, T in red, G in black and C in blue; the arrows indicate the position of the C65G mutation. (D) The proliferation rate of the different HAP1 clones was evaluated with CellTrace™. Cells were stained with CTV, cultured, harvested at the indicated time and analyzed by flow cytometry. The MFI is plotted as a function of time and fitted as described in the Materials and Methods. (E) The inferred population doubling time (in hours) is indicated for each clone and growth condition. Error bars represent the SD. Mean values were tested for statistical significance by an unpaired Student's t-test (****P ≤ 0.0001; ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05; ns, not significant).
To verify that the C65G mutation of tRNA[Ser]Sec did not alter the proliferation of HAP1 cells, we used the CellTrace™ Violet Cell Proliferation Kit to label and monitor cell divisions by measuring dye dilution with flow cytometry. This dye binds covalently to all free amines on the surface and inside of cells with very little cytotoxicity. The fluorescence signal remained stable over several days in a cell culture environment and was tracked by observing the decrease in fluorescence intensity over time, which provides insight into the time required for a single division. As depicted in Figure 2D, the rate of fluorescent dye dilution was consistent across all cell lines examined in this study, with a doubling time of ∼10 h. In addition, we did not observe any notable effects of selenium supplementation on cell division in any of the cell lines. Subsequent experiments were conducted using all three clones; however, as the proliferation was comparable between clones, only the results of clone C65G #1 are presented in the figures, with data for the other clones provided in the Supplementary figures.
The C65G homozygous cell lines expressed reduced levels of tRNA[Ser]Sec and were no longer sensitive to selenium
We subsequently performed a quantification of the intracellular levels of Sec tRNAs using northern blot, as previously described (55), with serine tRNA used as a reference for normalization. The different cell lines carrying the WT, WT/C65G and C65G genotypes were cultivated in the presence or absence of 100 nM selenium and harvested 48 h later. Interestingly, the steady-state level of tRNA[Ser]Sec under control conditions exhibited a decline of 46% in the heterozygous clone (WT/C65G) and 65% in the homozygous mutant clone (C65G) compared with the WT parental cell line (Figure 3A, B). The expression level of tRNA[Ser]Sec remained consistent across all three isolated C65G clones, as demonstrated in Supplementary Figure S1. These findings were consistent with the decreased levels of tRNA observed previously (48), where a 50% reduction in expression was observed in the homozygous patient. Moreover, as previously documented in human or rodent cells (22,42,68), we observed that supplementation of the growth medium with 100 nM selenium led to a 30% increase in tRNA[Ser]Sec levels in WT cells. This stimulation was also observed with the heterozygous clone (WT/C65G) but not with the mutant cellular clones (C65G) (Figure 3A, B; Supplementary Figure 1). Thus, the C65G mutation not only decreased the overall tRNA[Ser]Sec level, but also abrogated its regulation by selenium supplementation.
Figure 3.

Steady-state levels and stability of tRNA[Ser]Sec in WT, WT/C65G and C65G cell lines. (A) HAP1 cells were grown for 48 h with or without 100 nM selenium in the 10% FBS culture medium, followed by the extraction of total RNAs. A 15 μg aliquot of RNAs was subjected to northern blot analysis using tRNA[Ser]Sec (green) and tRNASer (red) complementary DNA probes for hybridization. (B) The levels of tRNA[Ser]Sec were normalized to those of tRNASer quantified from three independent northern blots, and expressed relative to the first lane. (C) To compare the stabilities of tRNA[Ser]Sec in different clones with parental cell lines, HAP1 cells were treated with actinomycin D (ActD) for 6 h, and the total RNAs were extracted and analyzed by northern blot as mentioned above. The ratio of tRNA[Ser]Sec to tRNASer signal was quantified from three independent northern blots and plotted, with the 0 h condition set as 1 for each histogram. The statistical significance of mean values was assessed by an unpaired Student's t-test, and the error bars represent the SD (****P ≤ 0.0001; ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05; ns, not significant).
In order to determine whether the drop of tRNA[Ser]Sec level in the mutant cells was due to a decrease in its stability, we treated WT and homozygous C65G mutant cells with the transcriptional inhibitor actinomycin D. Since prolonged incubation significantly impaired the viability of HAP1 cell lines, the incubation time was limited to 6 h. Actinomycin D inhibits all the three RNA polymerases, including POLR3 involved in the transcription of tRNAs, by interfering with RNA polymerase elongation. Total RNAs were isolated from the harvested cells and analyzed by northern blotting, with tRNA[Ser]Sec levels expressed relative to that of tRNASer. As shown in Figure 3C, there was a 70% reduction in tRNA[Ser]Sec levels in WT cells following actinomycin D treatment (Figure 3C). Despite lower initial levels of tRNA[Ser]Sec in C65G cells, the relative decrease of 53% in response to actinomycin D treatment was only slightly lower than that of WT cells (Figure 3C). Additionally, we investigated whether selenium supplementation could affect the tRNA stability, as initially noted in rodent cells (68). In our hands, addition of selenium improved the stability of tRNA[Ser]Sec in both WT and homozygous C65G mutant cells (Figure 3C), although the effect was weaker for the latter. In conclusion, our findings suggest that the decrease in tRNA levels in C65G mutant cell lines is not due to reduced tRNA stability.
The recruitment of RNA polymerase III at the tRNA[Ser]Sec promoter site was affected by the C65G mutation
Given that the stability of tRNA[Ser]Sec appeared comparable between WT and homozygous C65G mutant cells, we sought to investigate whether the lower steady-state levels of the tRNA in the mutant cells resulted from a lower transcriptional activity. Analysis of POLR3 transcription of the TRNAU1 gene revealed that the C65G mutation may affect the recruitment of POLR3 at the promoter site (Figure 4A). To confirm this hypothesis, we employed ChIP-qPCR to analyze POLR3 occupancy at the promoter site (Figure 4B). We used the tRNALeu gene as a reference to normalize the occupancy of POLR3. Our data revealed that POLR3 occupancy in the tRNA[Ser]Sec promoter region was reduced by 50% between the WT and the mutant cells, indicating a significant reduction of tRNA[Ser]Sec transcription in mutant cells. Furthermore, the addition of selenium to the growth medium significantly increased POLR3 occupancy for the WT but not for the C65G cells, suggesting an enhanced POLR3 recruitment after selenium supplementation. Overall, our findings suggest that the transcription of the TRNAU1 gene by POLR3 is impaired by the C65G substitution.
Figure 4.

POLR3 occupancy at the WT and C65G tRNA[Ser]Sec gene promoter. (A) Schematic representation of the human TRNAU1 gene. The black box represents the tRNA[Ser]Sec sequence. Gray boxes represent regulatory elements: AE, activator element; PSE, proximal sequence element; and TS, termination signal. The location of qPCR primers used for the ChIP analysis are shown by red arrows. (B) POLR3 occupancy was measured in WT and C65G HAP1 cells with or without selenium supplementation by ChIP-qPCR using an antibody against POLR3. The occupancy was normalized to that of the tRNALeu gene and quantified over three independent experiments. Mean values were tested for statistical significance by an unpaired Student's t-test, and the error bars represent the SD (***P ≤ 0.001; **P ≤ 0.01; ns, not significant).
The C65G mutation led to a hypomodification of tRNA[Ser]Sec at the wobble nucleotide U34
As C65G mutation impairs the transcription of tRNA[Ser]Sec, we aimed to investigate whether it also has an impact on post-transcriptional modifications that are critical for tRNA structure and function. Specifically, two modified bases located in the anticodon loop, mcm5U at position 34 and i6A at position 37, are crucial for UGA recoding (Figure 5A). Furthermore, the mcm5U34 base, which is located at the wobble position in tRNA[Ser]Sec, can be further methylated into mcm5Um. This methylation reaction is not complete, resulting in the presence of both mcm5U34 and mcm5Um34 isoforms in the cytoplasm, with the mcm5Um34 being stimulated by selenium supplementation. The modification status of the U34 residue at the wobble position appears particularly important for the synthesis and regulation of selenoproteins by selenium levels (31). To examine the modifications at position U34 and A37 in the anticodon loop of tRNA[Ser]Sec, we performed MS analysis with tRNA[Ser]Sec purified from the WT and C65G cell lines, cultivated with or without supplementation with 100 nM selenium. The tRNA fraction was enriched from total RNAs using anion exchange chromatography and the tRNA[Ser]Sec was purified from crude tRNAs by hybridization to biotinylated oligonucleotides and fixation to streptavidin magnetic beads. RNase T1 digestion was performed, and the resulting tRNA fragments were subjected to LC/MS and LC/MSMS analysis. We detected three oligoribonucleotides corresponding to the anticodon fragment of tRNA digested by RNase T1 in our samples (Figure 5B), which were identified by the m/z values (Figure 5C) and the MS/MS fragmentation analyses (Figure 5D). Our analysis revealed that while the i6A37 modification was present in all three digested fragments, the U34 position differed. Specifically, we found that the modified bases mcm5U and mcm5Um were present in the fragmentation pattern of RNA fragment II and III, respectively, while RNA fragment I corresponded to the unmodified U34 base (Figure 5D). Quantifying the relative abundance of the isoforms, we observed that the mcm5U (II) and mcm5Um (III) isoforms were the most prevalent in tRNA eluted from WT cells, while the unmodified U34 remained below 10% (Figure 5E). Interestingly, no significant difference was detected in the distribution of the three isoforms in response to selenium supplementation of the WT cells. However, in the mutant cells, the distribution across the isoforms was significantly different compared with the WT, with a significant increase of the unmodified isoform (U34). Between 35% and 40% of the tRNAs were not modified at the wobble position in tRNA[Ser]Sec, suggesting that the C65G mutation impairs the modification of the tRNA at this U34 position (Figure 5E). This hypomodified pattern of tRNA[Ser]Sec at U34 may have consequences for selenoprotein mRNA translation as the wobble position is crucial for codon–anticodon interaction.
Figure 5.

MS analysis of tRNA[Ser]Sec to evaluate the post-transcriptional modification status of U34 and A37. (A) Schematic representation of the anticodon loop region of tRNA[Ser]Sec covering the U34 and A37 residues, with the RNase T1 cleavage sites indicated by black arrows. Nucleotide U34 is sequentially modified in cm5U, mcm5U and mcm5Um by the elongator complex, ALKBH8 and FTSJ1, respectively. (B) Affinity-purified tRNA[Ser]Sec from WT and C65G HAP1 cells grown in the presence or absence of 100 nM selenium were digested by RNase T1 and the resulting oligoribonucleotide fragments were analyzed by nano-ion-pair reversed-phase high-performance liquid chromatography coupled with MS. A representative portion of the total ion count (TIC) elution profile containing the RNase T1-digested anticodon-containing fragments is shown for the WT and mutant tRNA (grown in non-supplemented conditions). Three major elution peaks, named I, II and III, and colored in blue, yellow and orange, respectively, were observed and analyzed by MS (C) and MS/MS (D). (C) MS spectra of elution peaks I, II and III obtained with tRNA[Ser]Sec purified from C65G cells showed the presence of one major oligoribonucleotide per peak. (D) CID spectra for peak I, II and III. A value of 1 m/z was selected for each elution peak, as indicated below each CID spectrum, for oligoribonucleotide fragmentation analysis. A fragment-ion map illustrating the sequence coverage of each oligoribonucleotide is indicated above each CID spectrum for peak I, II and III, in blue, yellow and orange, respectively. The three oligoribonucleotides differ only by the modification of nucleotide U34: peak I, unmodified U; peak II, mcm5U; peak III, mcm5Um. (E) The relative abundance of each fragment in WT and C65G cells was determined by integrating the TIC elution peaks as observed in (B) from multiple ESI-MS analyses, four for the WT tRNA, and three for the mutant tRNA due to its lower expression in cells. Mean values were tested for statistical significance by an unpaired Student's t-test, and the error bars represent the SD (**P ≤ 0.01; *P ≤ 0.05; ns, not significant).
The selenoproteome expression was dramatically altered in C65G cell lines and no longer sensitive to selenium activation
The expression of selenoproteins is primarily dependent on the availability of selenium. Cells cultured with 10% FBS, referred to as Ctrl conditions, have suboptimal selenium content, yet these growth conditions cannot be defined as selenium deficiency. While the optimal level of expression can be achieved by supplementing with 100 nM sodium selenite, selenium deficiency can be mimicked by reducing the serum amount to 2%. We investigated the expression profile of selenoproteins in WT, WT/C65G and C65G cells in response to selenium in two series of experiments. Firstly, we compared the expression of selenoproteins under control conditions and selenium-supplemented conditions (Figure 6; Supplementary Figure S2). Secondly, we examined their regulation across several selenium concentrations ranging from selenium deficiency conditions (2% FBS) to supplementation at 100 nM (Figure 7; Supplementary Figure S3). Cell extracts were analyzed through western blots and enzymatic assays specific for glutathione peroxidase or thioredoxin reductase activities (Figure 6C; Supplementary Figure S2C). Through western blot analysis, we were able to detect expression of eight selenoproteins in HAP1 cell extracts, including SELENOS, TXNRD2, SELENOH, GPX4, GPX1, SELENOO, TXNRD1 and SELENOT. In selenoprotein genes, when the UGA-selenocysteine codon is recognized as a stop codon, the truncated proteins that are produced are rapidly eliminated by the proteasome and the expression level observed by western blot corresponds to the full-length protein level (69). In WT HAP1 cells, selenium supplementation increased the expression of six selenoproteins, with the exception of TXNRD2 and SELENOT. Notably, the majority of selenoproteins investigated here demonstrated a significant alteration in expression levels in C65G mutant cells compared with WT cells, with the perturbation being particularly pronounced in cells grown in selenium-supplemented conditions (Figure 6A; Supplementary Figure S2A). In all cases, the expression of these selenoproteins was no longer sensitive to selenium addition in C65G cells, suggesting that the mutation strongly interferes with the mechanism of response to selenium. Additionally, in WT/C65G cells, the phenotype was attenuated compared with the double mutant, with only a mild impact on selenoprotein levels compared with the WT cells. Heatmaps were created to show the ratio of selenoprotein levels in supplemented conditions compared with the WT cells (Figure 6D) and the ratio of selenoprotein levels in the supplemented conditions compared with control conditions for each cell line (Figure 6E). This heatmap representation indicates that the hierarchy of selenium-regulated selenoproteins in WT cells is not identical to the hierarchy of selenoproteins impacted by the C65G mutation, suggesting distinct molecular mechanisms. Enzymatic assays specific for GPX and TXNRD activities confirmed the lack of activation upon selenium supplementation in C65G cells (Figure 6B; Supplementary Figure S2B, C). Notably, the levels of GPX activities in control conditions were similar between WT and C65G cells, indicating a relatively efficient steady-state level of selenoprotein synthesis for GPX1 and GPX4, and further supporting the observations from western blot analysis. Overall, these results demonstrate the significant impact of the C65G mutation on the expression of selenoproteins in response to selenium supplementation. In order to rule out the possibility that the observed effects were due to modifications in the steady-state levels of selenoprotein mRNAs in the mutant cell lines, RT–qPCRs were conducted using total RNA extracts from WT, C65G/WT and C65G cells. As illustrated in Supplementary Figure S4, the selenoprotein mRNA levels remained unaffected by the presence of the C65G mutation under any circumstances, indicating that this mutation has an impact on selenoprotein expression at the translational level.
Figure 6.

Effect of the C65G mutation on selenoprotein production and its selenium sensitivity. (A) WT, WT/C65G and C65G cells were grown for 48 h with or without 100 nM seleniume in the 10% FBS culture medium. Protein extracts were analyzed for selenoprotein levels by western blots with specific antibodies. The quantifications of three independent western blots were normalized by the signal for actin and expressed relative to the WT condition supplemented with selenium, set as 1. Corresponding values are indicated below each panel. GPX (B) and TXNRD (C) enzymatic activities in HAP1 protein extracts were evaluated for three independent experiments. The statistically significant fold changes are indicated on top of each histogram. Error bars represent the SD. Mean values were tested for statistical significance by an unpaired Student's t-test (****P ≤ 0.0001; ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05; ns, not significant). (D) Heatmap representation of changes in selenoprotein expression levels between WT and mutant cells grown in selenium-supplemented media. (E) Heatmap representation of selenoprotein activation by selenium in the three cell lines. Mean values were tested for statistical significance by an unpaired Student's t-test, where the P-values are denoted by asterisks (****P ≤ 0.0001; ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05); if absent, no significant difference was found.
Figure 7.

Effect of selenium concentrations on selenoprotein production in WT, WT/C65G and C65G HAP1 cells. (A) WT, WT/C65G and C65G cells were grown in the presence of 2% FBS (instead of 10%) supplemented with the indicated selenium concentrations and harvested as previously described. A basal selenium concentration of 3.9 nM was expected in the absence of added selenium. Protein extracts were analyzed for selenoprotein levels by western blots with specific antibodies. The quantifications of three independent western blots were normalized by the signal for actin and expressed relative to the 100 nM selenium condition, set as 1. Fold changes were determined and are indicated below each panel. GPX (B) and TXNRD (C) enzymatic activities in HAP1 protein extracts were also evaluated for three independent experiments. (D) Quantification of the western blot presented in (A). Mean values were tested for statistical significance by an unpaired Student's t-test, and the error bars represent the SD (****P ≤ 0.0001; ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05; ns, not significant).
In the selenium dose–effect experiments illustrated in Figure 7 and Supplementary Figure S3, we covered concentrations ranging from deficiency to optimum. Specifically, by reducing the FBS concentration from 10% to 2%, we decreased the selenium concentration in the first condition by 5-fold to reach 3.9 nM. Although not entirely comparable, the control conditions used in the previous experiments lie between concentrations of 10 and 20 nM. Similar to the previous study, western blot analyses and enzymatic activities were conducted on cellular extracts to compare the selenium activation of selenoproteins among WT, C65G/WT and C65G cells. Among the eight selenoproteins detected in HAP1 cells, all (except for TXNRD2 and SELENOT) were stimulated by selenium supplementation. Interestingly, under selenium-deficient conditions (condition 0 in Figure 7), the expression levels of selenoproteins were very close between all three cell lines, except for TXNRD2, suggesting that the C65G tRNA is as active as the wild-type tRNA under selenium-deficient conditions where only a few selenoproteins were produced. Subsequently, upon the addition of selenium to the culture medium, differences were observed between the cell lines, with low activation levels in C65G cells and an intermediate situation in heterozygous cells. In C65G/WT cells, the activation by selenium was close to that of WT cells for selenoproteins SELENOH, GPX4, GPX1, SELENOO and TXNRD1. In the case of C65G cells, their ability to respond to changes in selenium availability was considerably impaired compared with WT cells, and only a marginal increase in the expression of SELENOH, GPX1 and GPX4 was detected upon selenium supplementation, as depicted in Figure 7 and Supplementary Figure S3. The enzymatic activity measurements confirm the low activation of glutathione peroxidase but not of thioredoxin reductase in C65G. Overall, these results confirm the almost total loss of response to selenium variations in C65G cells compared with the WT.
The UGA-Sec recoding activity was altered in C65G mutant cells
In order to investigate whether the decreased expression levels of selenoproteins in C65G mutant cells were associated with a reduction in the translational efficiency of selenoprotein mRNAs, a luciferase-based reporter system was used (Figure 8). This system is based on a luciferase gene in which there is an in-frame UGA codon at position 258, downstream of which a minimal SECIS element was cloned. Several variants of this construct were used as reference or negative controls (60). The UGA codon was mutated to a UGU codon and used to set the 100% reading of luciferase position 258, while the UAA construct harbored a stop codon at position 258, serving as a negative control. Different SECIS elements isolated from selenoprotein mRNAs were also cloned downstream of the luciferase-coding region (60). These constructs were transfected into WT, WT/C65G and C65G cell lines, and the UGA recoding activities were measured after growth in either control or 100 nM selenium medium for 48 h. The UGA recoding activities for Luc UGA/Msrb1-SECIS reached a value of 3.8% in WT cells, but were significantly reduced to 0.59% in C65G cells (Figure 8A). This reduction was observed without any change in stop codon readthrough activity induced by the mutation (Figure 8B). In addition, as the expression levels of luciferase mRNAs were similar in the different cell lines (Supplementary Figure S5B), this rules out the involvement of nonsense-mediated decay or premature degradation of the transcripts in our experimental system. The inhibition of selenocysteine insertion in the mutant cells was consistent with the decrease in selenoprotein levels observed by western blots (Figures 6 and 7). Selenium supplementation increased the recoding activity in WT cells by 92%, which was reduced in WT/C65G heterozygous cells (+53%) and completely abolished in C65G homozygous mutant cells. This activity was similar in all three C65G cell lines (Supplementary Figure S5A). Furthermore, variations in the recoding efficiency in WT cells due to the nature of the SECIS elements located downstream of the UGA were observed, but a drastic decrease in recoding efficiency in C65G cell lines was found for all SECIS tested in the experimental setting (Figure 8C). Overall, our results confirm that the down-regulation of selenoprotein expression observed in C65G cells is attributable to a decrease in selenoprotein synthesis. Whether this lower activity in cells resulted from a reduced expression of tRNA[Ser]Sec or an inherent decrease in efficacy of the C65G mutant tRNA to decode the UGA codon requires further investigation.
Figure 8.

The UGA-Sec recoding efficiency is dramatically impaired in C65G cells. A series of luciferase constructs were co-transfected with a GFP plasmid to normalize transfection efficiency. The luciferase mRNAs expressed in each experiment are represented on top of each histogram, with gray and black bars corresponding to coding and untranslated sequences, respectively. (A) Evaluation of recoding efficiency in the different WT, WT/C65G and C65G cells grown with or without 100 nM Se supplementation with luciferase constructs containing the SECIS of Msrb1. UGA recoding efficiency was expressed as a percentage of the activity obtained with the UGU construct. (B) Comparison between UGA and UAA readthrough efficiency in WT, WT/C65G and C65G cells. (C) Various SECIS elements (Gpx1, Gpx4 Txrnd2 and Txnrd1) were cloned downstream of the luciferase-coding sequence and evaluated for their ability to drive UGA recoding in WT, WT/C65G and C65G cells. For clarity, only the fold changes in UGA recoding efficiency between WT and C65G cell extracts are indicated. Three independent experiments were used for statistical analysis. Mean values were tested for statistical significance by an unpaired Student's t-test, and the error bars represent the SD (****P ≤ 0.0001; ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05; ns, not significant).
The phenotype was partially rescued by overexpression of WT but not mutant tRNA in C65G cell lines
In order to differentiate between the two potential causes for a defect in selenoprotein expression mentioned above, we employed an approach involving overexpression of a cDNA plasmid encoding either WT or mutant C65G tRNA[Ser]Sec in both WT and C65G cell lines, with or without supplementation of 100 nM selenium. As illustrated in Figure 9, we therefore performed northern blots and western blots against several selenoproteins (SELENOS, SELENOH, GPX4, GPX1, SELENOO and TXNRD1). Previous studies have shown that the introduction of multiple copies of WT tRNA[Ser]Sec resulted in increased tRNA levels but only a moderate effect on selenoprotein expression in mice (70,71). In this study, we faced the challenge of low transfection efficiency of HAP1 cells, which reached a maximum of 40% under optimal conditions. When we transfected the plasmid containing the WT tRNA[Ser]Sec gene under these experimental conditions, we observed only a moderate increase (∼3.5-fold) in the total concentration of tRNA[Ser]Sec (Figure 9A, compare lane 1 with 2, and lane 4 with 5, respectively). Furthermore, transfection of the plasmid containing the mutant tRNA[Ser]Sec gene had a limited effect on the levels of tRNA[Ser]Sec in WT HAP1 cells (Figure 9A, compare lane 1 with 3, and lane 4 with 6, respectively). These findings confirm the difference in transcriptional efficiency between the WT and mutant tRNA genes, as shown in the ChIP-qPCR experiments described in Figure 4B. Interestingly, when we transfected the plasmids containing either the WT or mutant genes into C65G cells (Figure 9A, lanes 7–12), we observed a sharp increase in the global level of tRNA[Ser]Sec in both cases; once again the WT tRNA was better expressed than its mutated counterpart.
Figure 9.

The overexpression of C65G tRNA[Ser]Sec was unable to restore selenoprotein expression in HAP1 C65G cells. WT and C65G HAP1 cells were grown in the presence or absence of 100 nM selenium prior to the transfection of a plasmid expressing either the WT or C65G tRNA[Ser]Sec. A control (Ctl) condition was used as a reference where an empty vector was used instead. GFP was systematically co-transfected to normalize the transfection efficiency. Two days post-transfection, the cells were harvested and split in half for either total RNA or cellular protein extraction. (A) A 15 μg aliquot of total RNAs was employed for the northern blot using tRNA[Ser]Sec (green) and tRNASer (red) complementary DNA probes for hybridization as described earlier. The levels of tRNA[Ser]Sec were normalized over those of tRNASer for three independent northern blots and expressed relative to the first lane. For clarity, only the fold changes between Ctl with WT and Ctl with C65G transfections are indicated. Error bars represent the SD. Mean values were tested for statistical significance by an unpaired Student's t-test (***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05). (B) A 40 μg aliquot of protein extracts was used for the western blots to evaluate the expression levels of SELENOS, SELENOH, GPX4, SELENOO, GPX1, TXNRD1, GFP and actin. The quantifications of three independent western blots were normalized by the signal for actin and expressed relative to those of lane 4, set as 1. The relative selenoprotein levels were measured and mean values were tested for statistical significance by an unpaired Student's t-test (***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05; ns, not significant).
As illustrated in Figure 9B, we confirmed that tRNA[Ser]Sec levels in WT cells were not limiting, as its overexpression did not drastically change the production of selenoproteins. However, in C65G cells, we found that the overexpression of WT tRNA[Ser]Sec was able to restore, at least partially, the levels of selenoprotein synthesis, and this was observed both with and without selenium addition. As previously mentioned, the incomplete transfection of HAP1 cells could be a potential explanation for the partial rescue of selenoprotein expression. Importantly, overexpression of the mutant tRNA[Ser]Sec did not have any significant effect on selenoprotein levels in C65G cells (Figure 9B), even though the levels of mutant tRNA were similar to that of WT tRNA in control WT cells (Figure 9A, compare lane 1 with 7, and lane 4 with 10, respectively). This indicates that the recoding activity of the C65G mutant tRNA[Ser]Sec is significantly less effective than the WT tRNA[Ser]Sec. We also observed that in WT cells, the very weak overexpression of the C65G mutant tRNA induced a low, but significant, decrease of several selenoproteins. This effect was particularly visible in selenium-supplemented conditions and suggests that the C65G mutant tRNA could interfere with the normal function of the WT tRNA (Figure 9B, compare lane 4 with 6). Overall, these experiments demonstrate that the mutant tRNA is less efficient than the WT tRNA for recoding the UGA codon into selenocysteine.
The C65G tRNA is a competitor of WT tRNA when overexpressed in WT cell lines
To investigate the potential inhibitory activity of the mutant tRNA[Ser]Sec, we employed an approach involving tRNA overexpression in HEK293 cells, which were highly susceptible to efficient transfection (>90%). We transfected plasmids containing either the WT or mutant tRNA[Ser]Sec gene into HEK293 cells cultured in the presence or absence of added selenium, following a similar experimental procedure to that used in HAP1 cells. As depicted in Figure 10A, we achieved a 100-fold and 20-fold overexpression of WT and mutant tRNA[Ser]Sec, respectively. Overexpression of the WT tRNA did not significantly impact selenoprotein expression (Figure 10B), while overexpression of the C65G mutant tRNA[Ser]Sec significantly reduced selenoprotein levels (Figure 10B). This effect was more evident in cell extracts supplemented with selenium due to higher levels of selenoproteins under this condition. The quantitative analysis shown below the western blots indicates a significant decrease in the expression of all selenoproteins in selenium-supplemented conditions, even for TXNRD1, known to be insensitive to expression variations (25). These data further confirm results obtained in HAP1 and show that the C65G mutant tRNA[Ser]Sec is less active than the WT tRNA[Ser]Sec in UGA recoding as selenocysteine and can act as a competitive inhibitor in WT cell lines.
Figure 10.
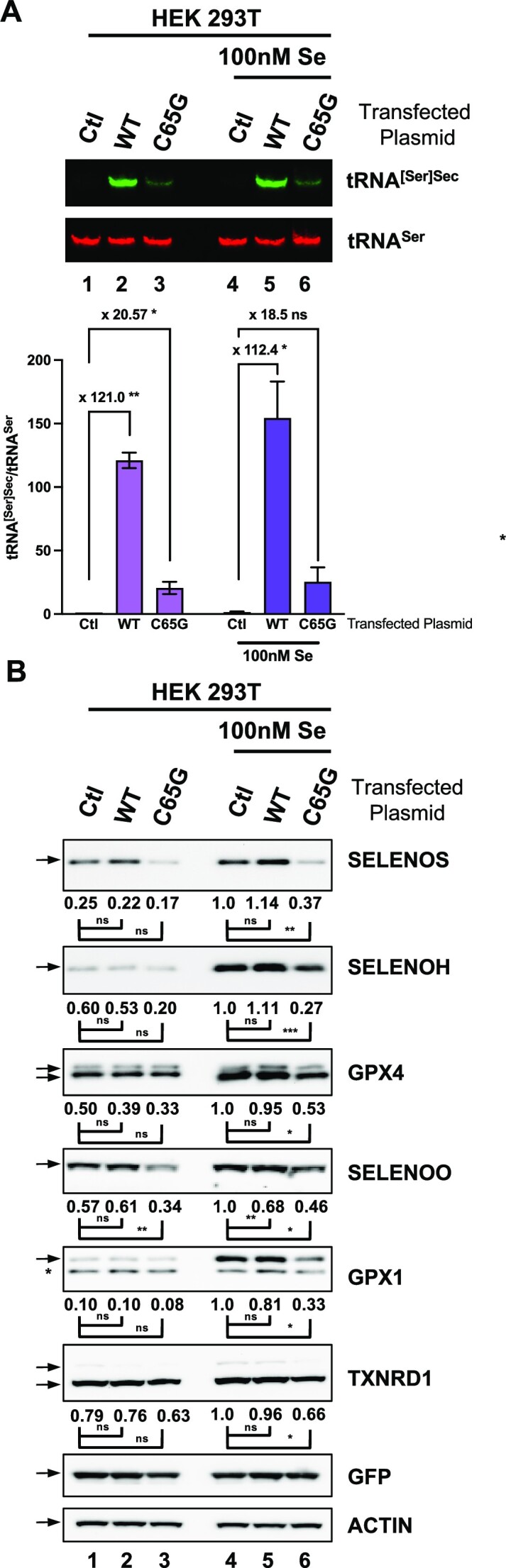
Overexpression of C65G tRNA[Ser]Sec inhibits selenoprotein expression in HEK293T cell lines. HEK293 cells were grown in the presence or absence of 100 nM selenium prior to the transfection of a plasmid expressing either the WT or C65G tRNA[Ser]Sec. GFP was systematically co-transfected to verify the transfection efficiency. Two days post-transfection, the cells were harvested and split in half for either total RNA or cellular protein extraction. (A) Northern blot analyses of total RNA extracts to detect tRNA[Ser]Sec (green) and tRNASer (red). The fold changes between Ctl with WT and Ctl with C65G transfections were calculated for three independent experiments. (B) Protein extracts were analyzed for the expression of different selenoproteins, GFP and actin as described before. The quantifications of three independent western blots were normalized by the signal for actin and expressed relative to those of lane 4, set as 1. The corresponding values are indicated below each panel Mean values were tested for statistical significance by an unpaired Student's t-test (***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05; ns, not significant).
DISCUSSION
In this study, we have delved into the ramifications of a specific genetic mutation that has been observed in two unrelated individuals who presented symptoms of thyroid dysfunction, low levels of selenium in the blood, fatigue and abdominal pain (48,49). This C65G mutation occurs on the human tRNA[Ser]Sec gene (TRNAU1) which is present as a single copy within the genome. This tRNA is essential for the synthesis of the entire family of the 25 selenoproteins that constitute the selenoproteome (31). The recoding of the UGA codon into selenocysteine is a rare but crucial event, as inactivation of the mechanism results in lethality in mice models (36). Furthermore, various genetic diseases have been documented in humans concerning the selenocysteine insertion machinery, each with its own distinct phenotype (50–52). In this study, we have engineered homozygous and heterozygous HAP1-derived cell lines to investigate the impact of the C65G mutation of tRNA[Ser]Sec on its structure, expression, post-transcriptional modifications, UGA-Sec recoding activity and how it affects the level of expression of the selenoproteins.
The C65G mutation in the tRNA[Ser]Sec gene results in reduced tRNA expression
Our study confirms, in a laboratory cell model, the findings of a previous study on a young patient carrying the homozygous C65G mutation of the TRNAU1 gene, i.e. that the level of tRNA[Ser]Sec expression is ∼4 times lower than in WT cells. This observation was initially made on cells taken from the proband and from controls or family members, of various ages and sexes, thus it was important to verify that the effect on the tRNA[Ser]Sec expression in our system was consistent. This observation was made only in the first reported case of C65G mutation (48) but not investigated in the second proband (49). Our results demonstrate that the low level of tRNA[Ser]Sec expression could be explained by a reduced rate of transcription, as evidenced by ChIP analysis showing a lower occupancy level of POLR3 at the mutant TRNAU1 gene (Figure 4). Transfection experiments using plasmids containing the WT or mutant tRNA gene further confirmed a marked difference between the two genes. Specifically, in both HAP1 and HEK293 cell lines, the plasmid carrying the WT gene was expressed 4–5 times more efficiently than the gene carrying the C65G mutation (Figures 9 and 10). Notably, this single C65G mutation is not located within any of the five critical regions for tRNA[Ser]Sec gene transcription described in the literature, i.e. box A, box B, the activator, the proximal sequence element or the transcription termination signal [see Figure 4 and (48)]. However, it is positioned only three bases downstream of box B. The precise mechanism for understanding how this mutation alters the level of expression linked to POLR3 remains to be elucidated, but previous results obtained on other tRNA genes have shown that deletion of regions downstream of box B dramatically impaired the transcription of tRNA (72). Given these results, it is imperative to pay particular attention to any modifications resulting from single nucleotide polymorphisms (SNPs) in the regulatory regions of tRNA[Ser]Sec, as has been previously done with selenoprotein genes (73).
The UGA recoding activity of C65G tRNA[Ser]Sec is significantly reduced
Since our data show that the C to G mutation lowers the expression level of this tRNA by 4-fold, an immediate question was to determine whether the effects of selenoprotein expression were caused by insufficient levels of the tRNA or by a defect in its function. We tackled this by overexpressing the WT and mutant tRNA[Ser]Sec in both HAP1 C65G cells and parental HAP1 cell lines, and measuring the level of expression of a set of selenoproteins. Our findings indicate that tRNA[Ser]Sec containing the C65G mutation is inherently less efficient in the UGA recoding process than the WT form, and can even interfere with the function of the WT tRNA when overexpressed. Furthermore, our in silico analysis suggests that the C65G substitution has minimal impact on the overall 3D structure of tRNA[Ser]Sec, with the structural collision predicted by the 2D structure not being observed (see Figure 1 and Supplementary movies). Our experimental results further confirm that the mutant tRNA structure is not recognized as abnormal by the tRNA surveillance system and should not be too distorted as there were no significant differences between the WT and C65G forms of in vitro transcribed tRNAs in assays of (i) serine aminoacylation, (ii) interaction with SEPSECS or (iii) selenocysteine synthesis by SARS, PSTK, SEPSECS and SEPHS2 (48). Overall, our data demonstrate that the C to G mutation has a major functional impact on the UGA recoding process through expression level and recoding efficiency, despite minimal variations in the overall structural architecture of the tRNA[Ser]Sec.
The hypomodification at position U34 could explain the reduced tRNA activity
During its maturation, the tRNA experiences a plethora of enzyme-catalyzed post-transcriptional modifications that are essential for all key facets of tRNA function, such as folding, stability and decoding (74). One area of particular significance for tRNA modification is position 34, which is the initial position of the anticodon that engages with the third position of the mRNA codon (75). In the case of tRNA[Ser]Sec, the Elongator complex initiates the addition of a carboxymethyl group to the C5-position of U34 (cm5U), thereby preparing this nucleoside for subsequent conversion to mcm5U by the enzyme ALKBH8 and to mcm5Um by FTSJ1 (31,76), as illustrated in Figure 5A. The Elongator complex is a highly conserved protein complex which has been implicated in various cellular processes, including transcription regulation, DNA repair and cell migration. However, its main role is to produce specific chemical side chains, on tRNA uridines at the wobble position (77–80). Contrary to previously held beliefs, it has been demonstrated through MS in mice with an inactivated Trit1 gene that i6A modification at position A37 by TRIT1 is not a prerequisite for the modification of U34 (76). Using LC/MSMS with both WT and C65G purified tRNA[Ser]Sec, it was found that the C65G cells exhibited a defect in U34 modification, with ∼40% of the tRNA no longer being modified, while in WT cells, the presence of unmodified U34 was barely detectable, as previously reported (76). Notably, the presence of this unmodified base could not be detected by the method used in the original work describing the phenotype of this mutation (48), and thus went unreported. Our findings indicate that C65G mutation impairs the first step of U34 post-transcriptional modification, specifically the formation of cm5U by the Elongator complex (Figure 5A). Additionally, it has been shown that the tRNA recognition by the Elongator complex involves not only the anticodon loop, but also other domains such as the acceptor arm, as revealed through the crystal structure of the complex (78). Our data suggest that the C65G mutant may sufficiently impact the recognition of tRNA[Ser]Sec by the Elongator complex, resulting in a reduced rate of cm5U formation despite relatively minor predicted structural changes. However, it appears that this structural alteration in the mutant tRNA does not affect the activity of the enzyme ALKBH8, as no cm5U-containing oligoribonucleotides were detected in our LC/MSMS experiments. Interestingly, in mice with a genetic deletion of ALKBH8, the formation of cm5U34 and 5-carbamoylmethyluridine (ncm5U34) was observed, yet it had only a limited effect on selenoprotein expression (57). Because of its critical role in 11 tRNA U34 modifications, Elongator dysfunction has been shown to alter proteome expression due to a reduction in codon-dependent translation speed and an increase in frameshifting (79,80). In humans, gene mutations in Elongator subunits have been associated with neurodevelopmental diseases, amyotrophic lateral sclerosis, rolandic epilepsy, intellectual disability and scoliosis (77).
Furthermore, using luciferase reporter constructs, we were able to confirm that the defect in recoding activity was not dependent on the nature of the SECIS element, but was specifically linked to an impairment in the decoding properties of tRNA[Ser]Sec. It remains unclear whether the remaining Sec insertion activity is exclusively attributable to the presence of the mcm5U and mcm5Um isoforms, or whether it could also be the result of the unmodified tRNA[Ser]Sec isoform. In any case, the predominant phenotype of the C65G mutant tRNA appears to be the hypomodification of the tRNA at position U34. Additionally, our findings reveal that the overexpression of the C65G tRNA[Ser]Sec has a competitor activity, indicating that the unmodified tRNA impairs the cognate decoding of UGA in selenoprotein mRNAs. Furthermore, this mutation does not impact aminoacylation (48), but instead appears to directly affect the UGA recoding within the ribosome (our study), possibly through the formation of abortive or imperfect codon–anticodon interaction. Our results further underscore the importance of the U34 position of tRNA[Ser]Sec as a subtle control point for the efficient recoding of UGA as Sec.
Localization of tRNA[Ser]Sec within the UGA translating ribosome: potential implications of C65G mutation
Recently, the first structure of the 80S ribosome as it decodes the selenocysteine UGA codon, the selenosome (15), has been solved by cryo-EM (PDB: 7ZJW). This study reveals the structure of various components of the Sec insertion machinery and their respective positioning and interplay within the translation context. The structure of ribosome-bound tRNA[Ser]Sec has been specifically elucidated, wherein the C65 residue, found to be mutated in patients, is located at the contact interface with EFSec (Supplementary Figure S6). The dedicated elongation factor EFSec indeed binds tRNA[Ser]Sec, with its D1, D2 and D3 domains, located at the GTPase-associated center, contacting the acceptor arm, and D4 interacting simultaneously with the variable arm of the tRNA and the apical loop of the SECIS element. Please note that the D4 domain of EFSec is not present in EF-1A, the elongator factor in charge of the other cellular tRNAs. The distinctive interaction between EFSec, the tRNA[Ser]Sec and the SECIS may possess functional significance in initiating the delivery of the cognate tRNA when both the SECIS, delivered by SECISBP2, and the UGA codon are concurrently present in the ribosome. While our in silico structural analysis illustrated in Figure 1 for the C65G mutant suggests a close resemblance to the WT tRNA, the C65G mutation may potentially affect the structure and function of the tRNA–EFSec complex. Further in vitro investigations are required to analyze this interaction, and novel cryo-EM structures of the selenosome with the mutant tRNA would be very informative.
The loss of selenium regulation, an important feature for the treatment of patients
The C65G mutation, also known as the SNP rs879255589 in databases of genetic variations, is an extremely rare alteration (https://www.ncbi.nlm.nih.gov/snp/rs879255589), present in 17 alleles among a total of 427 962 (frequency A = 0.0000397) when compiling data from various sources, i.e. Trans-Omics for Precision Medicine (TOPMED), Genome Aggregation Database (GnomAD), Allele Frequency Aggregator (ALPHA) and Estonian Biobank. As previously observed in the first clinical case and confirmed in our study, the mutation only impacts selenoproteome expression when it is present in the homozygous state, while the phenotype of heterozygotes is much more similar to that of WT cells. A significant finding of our research is that homozygous C65G cells display a poor response to selenium, suggesting that dietary supplementation may not provide a long-term benefit for patients with this genotype. To mitigate the effects of reduced antioxidant defense resulting from reduced selenoprotein expression, antioxidant therapy and limiting exogenous cellular stressors such as smoking, UV light and xenobiotic exposure may alleviate the clinical symptoms. For example, dietary supplementation with alpha tocopherol (vitamin E) has been shown to reduce oxidative damage in cells and tissues of patients with SECISBP2 mutations (81). However, the long-term consequences of such therapy are uncertain. Importantly, although expression levels were close to undetectable for several selenoproteins, certain key members such as TXNRD1, GPX1 and GPX4 were present in almost the same abundance as in WT cells grown under control conditions. This may explain why both selenium deficiencies and symptoms were moderate in the two patients, compared with other clinical cases affecting SECISBP2 or SEPSECS, two other components of the Sec insertion machinery (50,82). Other human SNPs in the TRNAU1 gene, including those in the promoter or coding sequence, warrant further investigation to determine their effects on tRNA expression, UGA recoding activity and selenium sensitivity.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Pierre Ray (CHU de Grenoble, UFR de Biochimie et Génétique Moléculaire, Grenoble, France) for very informative discussions about genetic mutations, and Dr Yann Ponty [Laboratoire d’Informatique de l'École Polytechnique (LIX CNRS UMR 7161), Palaiseau, France] for useful insights about 3D structures.
Contributor Information
Caroline Vindry, CIRI, Centre International de Recherche en Infectiologie, 69007 Lyon, France; INSERM U1111, 69007 Lyon, France; Ecole Normale Supérieure de Lyon, Lyon, France; Université Lyon 1, Lyon, France; CNRS/ENS/UCBL1 UMR5308, 69007 Lyon, France.
Olivia Guillin, CIRI, Centre International de Recherche en Infectiologie, 69007 Lyon, France; INSERM U1111, 69007 Lyon, France; Ecole Normale Supérieure de Lyon, Lyon, France; Université Lyon 1, Lyon, France; CNRS/ENS/UCBL1 UMR5308, 69007 Lyon, France.
Philippe Wolff, Architecture et Réactivité de l’ARN, Institut de Biologie Moléculaire et Cellulaire du CNRS, Université de Strasbourg, F-67084 Strasbourg, France.
Paul Marie, Ecole Normale Supérieure de Lyon, Lyon, France; Université Lyon 1, Lyon, France; LBMC, Laboratoire de Biologie et Modélisation de la Cellule, 69007 Lyon, France; CNRS/ENS/UCBL1 UMR5239, 69007 Lyon, France; INSERM U1210, 69007 Lyon, France.
Franck Mortreux, Ecole Normale Supérieure de Lyon, Lyon, France; Université Lyon 1, Lyon, France; LBMC, Laboratoire de Biologie et Modélisation de la Cellule, 69007 Lyon, France; CNRS/ENS/UCBL1 UMR5239, 69007 Lyon, France; INSERM U1210, 69007 Lyon, France.
Philippe E Mangeot, CIRI, Centre International de Recherche en Infectiologie, 69007 Lyon, France; INSERM U1111, 69007 Lyon, France; Ecole Normale Supérieure de Lyon, Lyon, France; Université Lyon 1, Lyon, France; CNRS/ENS/UCBL1 UMR5308, 69007 Lyon, France.
Théophile Ohlmann, CIRI, Centre International de Recherche en Infectiologie, 69007 Lyon, France; INSERM U1111, 69007 Lyon, France; Ecole Normale Supérieure de Lyon, Lyon, France; Université Lyon 1, Lyon, France; CNRS/ENS/UCBL1 UMR5308, 69007 Lyon, France.
Laurent Chavatte, CIRI, Centre International de Recherche en Infectiologie, 69007 Lyon, France; INSERM U1111, 69007 Lyon, France; Ecole Normale Supérieure de Lyon, Lyon, France; Université Lyon 1, Lyon, France; CNRS/ENS/UCBL1 UMR5308, 69007 Lyon, France.
Data Availability
The data underlying this article are available in the article and in its online supplementary data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Agence Nationale de Recherche sur le SIDA et les hépatites virales (ANRS); the Institut National de la Santé et de la Recherche Médicale (INSERM); the Centre National de la Recherche Scientifique (CNRS); the École Normale Supérieure (ENS) de Lyon ‘Emerging Project’ [L.C. in 2016 and T.O. in 2018]; the CNRS MITI programs ‘Isotop’ and ‘Metallo-Mix’; the ANRS [to C.V.]; the Université Claude Bernard Lyon 1 [to O.G.]; the Fondation pour la Recherche Médicale [to P.M.]; the Agence Nationale de la Recherche [ANR-10-LABX-0036_NETRNA French National Program Investissement d’Avenir (Labex NetRNA)].
Conflict of interest statement. P.E.M. and T.O. are named as inventors on a patent relating to the Nanoblades technology (patent applicants: Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), Ecole Normale Superieure de Lyon, Université Claude Bernard Lyon 1; name of inventors: Theophile Ohlmann, Philippe Mangeot, Emiliano Ricci; application number: WO 2017/068077 Al; patent status: published, 27 April 2017; all aspects of the manuscript are covered by the patent application. The remaining authors declare no competing interests.
REFERENCES
- 1. Guillin O.M., Vindry C., Ohlmann T., Chavatte L.. Selenium, selenoproteins and viral infection. Nutrients. 2019; 11:2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papp L.V., Holmgren A., Khanna K.K.. Selenium and selenoproteins in health and disease. Antioxid. Redox Signal. 2010; 12:793–795. [DOI] [PubMed] [Google Scholar]
- 3. Sonet J., Bulteau A.-L., Chavatte L.. Michalke B. Selenium and selenoproteins in human health and diseases. Metallomics: Analytical Techniques and Speciation Methods. 2016; Hoboken, NJ: Wiley-VCH Verlag GmbH & Co. KGaA; 364–381. [Google Scholar]
- 4. Rayman M.P. Selenium and human health. Lancet. 2012; 379:1256–1268. [DOI] [PubMed] [Google Scholar]
- 5. Hatfield D.L., Tsuji P.A., Carlson B.A., Gladyshev V.N.. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem. Sci. 2014; 39:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hatfield D.L., Carlson B.A., Xu X.M., Mix H., Gladyshev V.N.. Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog. Nucleic Acid Res. Mol. Biol. 2006; 81:97–142. [DOI] [PubMed] [Google Scholar]
- 7. Schweizer U., Fabiano M.. Selenoproteins in brain development and function. Free Radic. Biol. Med. 2022; 190:105–115. [DOI] [PubMed] [Google Scholar]
- 8. Hatfield D.L., Gladyshev V.N.. How selenium has altered our understanding of the genetic code. Mol. Cell Biol. 2002; 22:3565–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vindry C., Ohlmann T., Chavatte L.. Translation regulation of mammalian selenoproteins. Biochim. Biophys Acta. 2018; 1862:2480–2492. [DOI] [PubMed] [Google Scholar]
- 10. Bulteau A.-L., Chavatte L.. Update on selenoprotein biosynthesis. Antioxid. Redox Signal. 2015; 23:775–794. [DOI] [PubMed] [Google Scholar]
- 11. Driscoll D.M., Copeland P.R.. Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 2003; 23:17–40. [DOI] [PubMed] [Google Scholar]
- 12. Shetty S.P., Copeland P.R.. Selenocysteine incorporation: a trump card in the game of mRNA decay. Biochimie. 2015; 114:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Squires J.E., Berry M.J.. Eukaryotic selenoprotein synthesis: mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life. 2008; 60:232–235. [DOI] [PubMed] [Google Scholar]
- 14. Park S.I., Park J.M., Chittum H.S., Yang E.S., Carlson B.A., Lee B.J., Hatfield D.L.. Selenocysteine tRNAs as central components of selenoprotein biosynthesis in eukaryotes. Biomed. Environ. Sci. 1997; 10:116–124. [PubMed] [Google Scholar]
- 15. Hilal T., Killam B.Y., Grozdanovic M., Dobosz-Bartoszek M., Loerke J., Burger J., Mielke T., Copeland P.R., Simonovic M., Spahn C.M.T.. Structure of the mammalian ribosome as it decodes the selenocysteine UGA codon. Science. 2022; 376:1338–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bermano G., Arthur J.R., Hesketh J.E.. Selective control of cytosolic glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase mRNA stability by selenium supply. FEBS Lett. 1996; 387:157–160. [DOI] [PubMed] [Google Scholar]
- 17. Bermano G., Arthur J.R., Hesketh J.E.. Role of the 3′ untranslated region in the regulation of cytosolic glutathione peroxidase and phospholipid-hydroperoxide glutathione peroxidase gene expression by selenium supply. Biochem. J. 1996; 320:891–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bermano G., Nicol F., Dyer J.A., Sunde R.A., Beckett G.J., Arthur J.R., Hesketh J.E.. Tissue-specific regulation of selenoenzyme gene expression during selenium deficiency in rats. Biochem. J. 1995; 311:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wingler K., Bocher M., Flohe L., Kollmus H., Brigelius-Flohe R.. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur. J. Biochem. 1999; 259:149–157. [DOI] [PubMed] [Google Scholar]
- 20. Weiss Sachdev S., Sunde R.A.. Selenium regulation of transcript abundance and translational efficiency of glutathione peroxidase-1 and -4 in rat liver. Biochem. J. 2001; 357:851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lei X.G., Evenson J.K., Thompson K.M., Sunde R.A.. Glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase are differentially regulated in rats by dietary selenium. J. Nutr. 1995; 125:1438–1446. [DOI] [PubMed] [Google Scholar]
- 22. Chittum H.S., Hill K.E., Carlson B.A., Lee B.J., Burk R.F., Hatfield D.L.. Replenishment of selenium deficient rats with selenium results in redistribution of the selenocysteine tRNA population in a tissue specific manner. Biochim. Biophys Acta. 1997; 1359:25–34. [DOI] [PubMed] [Google Scholar]
- 23. Guillin O.M., Vindry C., Ohlmann T., Chavatte L.. Interplay between selenium, selenoproteins and HIV-1 replication in human CD4 T-lymphocytes. Int. J. Mol. Sci. 2022; 23:1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sonet J., Bulteau A.L., Touat-Hamici Z., Mosca M., Bierla K., Mounicou S., Lobinski R., Chavatte L.. Selenoproteome expression studied by non-radioactive isotopic selenium-labeling in human cell lines. Int. J. Mol. Sci. 2021; 22:7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Touat-Hamici Z., Bulteau A.L., Bianga J., Jean-Jacques H., Szpunar J., Lobinski R., Chavatte L.. Selenium-regulated hierarchy of human selenoproteome in cancerous and immortalized cells lines. Biochim. Biophys. Acta. 2018; 1862:2493–2505. [DOI] [PubMed] [Google Scholar]
- 26. Touat-Hamici Z., Legrain Y., Sonet J., Bulteau A.-L., Chavatte L.. Hatfield D.L., Schweizer U., Tsuji P.A., Gladyshev V.N.. Selenium: Its Molecular Biology and Role in Human Health. 2016; 4th ednNY: Springer Science+Business Media, LLC; 539–551. [Google Scholar]
- 27. Touat-Hamici Z., Legrain Y., Bulteau A.-L., Chavatte L.. Selective up-regulation of human selenoproteins in response to oxidative stress. J. Biol. Chem. 2014; 289:14750–14761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Legrain Y., Touat-Hamici Z., Chavatte L.. Interplay between selenium levels, selenoprotein expression, and replicative senescence in WI-38 human fibroblasts. J. Biol. Chem. 2014; 289:6299–6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hammad G., Legrain Y., Touat-Hamici Z., Duhieu S., Cornu D., Bulteau A.L., Chavatte L.. Interplay between selenium levels and replicative senescence in WI-38 human fibroblasts: a proteomic approach. Antioxidants (Basel). 2018; 7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carlson B.A., Lee B.J., Tsuji P.A., Copeland P.R., Schweizer U., Gladyshev V.N., Hatfield D.L.. Selenocysteine tRNA[Ser]Sec, the central component of selenoprotein biosynthesis: isolation, identification, modification, and sequencing. Methods Mol. Biol. 2018; 1661:43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carlson B.A., Lee B.J., Tsuji P.A., Tobe R., Park J.M., Schweizer U., Gladyshev V.N., Hatfield D.L.. Hatfield D.L., Schweizer U., Tsuji P.A., Gladyshev V.N.. Selenium: Its Molecular Biology and Role in Human Health. 2016; 4th ednNY: Springer Science+Business Media, LLC; 3–12. [Google Scholar]
- 32. Carlson B.A. Hatfield D.L., Schweizer U., Tsuji P.A., Gladyshev V.N.. Selenium: Its Molecular Biology and Role in Human Health. 2016; 4th ednNY: Springer Science+Business Media, LLC; 555–566. [Google Scholar]
- 33. Jameson R.R., Diamond A.M.. A regulatory role for Sec tRNA[Ser]Sec in selenoprotein synthesis. RNA. 2004; 10:1142–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Serrao V.H.B., Silva I.R., da Silva M.T.A., Scortecci J.F., de Freitas Fernandes A., Thiemann O.H.. The unique tRNA(Sec) and its role in selenocysteine biosynthesis. Amino Acids. 2018; 50:1145–1167. [DOI] [PubMed] [Google Scholar]
- 35. Santesmasses D., Mariotti M., Guigo R.. Computational identification of the selenocysteine tRNA (tRNASec) in genomes. PLoS Comput Biol. 2017; 13:e1005383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bosl M.R., Takaku K., Oshima M., Nishimura S., Taketo M.M.. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc. Natl Acad. Sci. USA. 1997; 94:5531–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mariotti M., Santesmasses D., Capella-Gutierrez S., Mateo A., Arnan C., Johnson R., D’Aniello S., Yim S.H., Gladyshev V.N., Serras F.et al.. Evolution of selenophosphate synthetases: emergence and relocation of function through independent duplications and recurrent subfunctionalization. Genome Res. 2015; 25:1256–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Itoh Y., Chiba S., Sekine S., Yokoyama S.. Crystal structure of human selenocysteine tRNA. Nucleic Acids Res. 2009; 37:6259–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim L.K., Matsufuji T., Matsufuji S., Carlson B.A., Kim S.S., Hatfield D.L., Lee B.J.. Methylation of the ribosyl moiety at position 34 of selenocysteine tRNA[Ser]Sec is governed by both primary and tertiary structure. RNA. 2000; 6:1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schweizer U., Bohleber S., Fradejas-Villar N.. The modified base isopentenyladenosine and its derivatives in tRNA. RNA Biol. 2017; 14:1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chittum H.S., Baek H.J., Diamond A.M., Fernandez-Salguero P., Gonzalez F., Ohama T., Hatfield D.L., Kuehn M., Lee B.J.. Selenocysteine tRNA[Ser]Sec levels and selenium-dependent glutathione peroxidase activity in mouse embryonic stem cells heterozygous for a targeted mutation in the tRNA[Ser]Sec gene. Biochemistry. 1997; 36:8634–8639. [DOI] [PubMed] [Google Scholar]
- 42. Hatfield D., Lee B.J., Hampton L., Diamond A.M.. Selenium induces changes in the selenocysteine tRNA[Ser]Sec population in mammalian cells. Nucleic Acids Res. 1991; 19:939–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carlson B.A., Moustafa M.E., Sengupta A., Schweizer U., Shrimali R., Rao M., Zhong N., Wang S., Feigenbaum L., Lee B.J.et al.. Selective restoration of the selenoprotein population in a mouse hepatocyte selenoproteinless background with different mutant selenocysteine tRNAs lacking Um34. J. Biol. Chem. 2007; 282:32591–32602. [DOI] [PubMed] [Google Scholar]
- 44. Orellana E.A., Siegal E., Gregory R.I.. tRNA dysregulation and disease. Nat. Rev. Genet. 2022; 23:651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor R.W., Turnbull D.M.. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005; 6:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ruiz-Pesini E., Lott M.T., Procaccio V., Poole J.C., Brandon M.C., Mishmar D., Yi C., Kreuziger J., Baldi P., Wallace D.C.. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007; 35:D823–D828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ishimura R., Nagy G., Dotu I., Zhou H., Yang X.L., Schimmel P., Senju S., Nishimura Y., Chuang J.H., Ackerman S.L.. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014; 345:455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schoenmakers E., Carlson B., Agostini M., Moran C., Rajanayagam O., Bochukova E., Tobe R., Peat R., Gevers E., Muntoni F.et al.. Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J. Clin. Invest. 2016; 126:992–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Geslot A., Savagner F., Caron P.. Inherited selenocysteine transfer RNA mutation: clinical and hormonal evaluation of 2 patients. Eur. Thyroid J. 2021; 10:542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schoenmakers E., Chatterjee K.. Human genetic disorders resulting in systemic selenoprotein deficiency. Int. J. Mol. Sci. 2021; 22:12927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schoenmakers E., Chatterjee K.. Human disorders affecting the selenocysteine incorporation pathway cause systemic selenoprotein deficiency. Antioxid. Redox Signal. 2020; 33:481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fradejas-Villar N. Consequences of mutations and inborn errors of selenoprotein biosynthesis and functions. Free Radic. Biol. Med. 2018; 127:206–214. [DOI] [PubMed] [Google Scholar]
- 53. Gladyshev V.N., Arner E.S., Berry M.J., Brigelius-Flohe R., Bruford E.A., Burk R.F., Carlson B.A., Castellano S., Chavatte L., Conrad M.et al.. Selenoprotein gene nomenclature. J. Biol. Chem. 2016; 291:24036–24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mangeot P.E., Risson V., Fusil F., Marnef A., Laurent E., Blin J., Mournetas V., Massourides E., Sohier T.J.M., Corbin A.et al.. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9–sgRNA ribonucleoproteins. Nat. Commun. 2019; 10:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vindry C., Guillin O., Mangeot P.E., Ohlmann T., Chavatte L.. A versatile strategy to reduce UGA-selenocysteine recoding efficiency of the ribosome using CRISPR-Cas9-viral-like-particles targeting selenocysteine-tRNA[Ser]Sec gene. Cells. 2019; 8:574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoshida M., Kataoka N., Miyauchi K., Ohe K., Iida K., Yoshida S., Nojima T., Okuno Y., Onogi H., Usui T.et al.. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc. Natl Acad. Sci. USA. 2015; 112:2764–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Songe-Moller L., van den Born E., Leihne V., Vagbo C.B., Kristoffersen T., Krokan H.E., Kirpekar F., Falnes P.O., Klungland A.. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol. Cell Biol. 2010; 30:1814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Antoine L., Wolff P.. Mapping of posttranscriptional tRNA modifications by two-dimensional gel electrophoresis mass spectrometry. Methods Mol. Biol. 2020; 2113:101–110. [DOI] [PubMed] [Google Scholar]
- 59. Ameur L.B., Marie P., Thenoz M., Giraud G., Combe E., Claude J.B., Lemaire S., Fontrodona N., Polveche H., Bastien M.et al.. Intragenic recruitment of NF-kappaB drives splicing modifications upon activation by the oncogene Tax of HTLV-1. Nat. Commun. 2020; 11:3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Latreche L., Jean-Jean O., Driscoll D.M., Chavatte L.. Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine. Nucleic Acids Res. 2009; 37:5868–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leontis N.B., Stombaugh J., Westhof E.. The non-Watson–Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002; 30:3497–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Popenda M., Szachniuk M., Antczak M., Purzycka K.J., Lukasiak P., Bartol N., Blazewicz J., Adamiak R.W.. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012; 40:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ganichkin O.M., Anedchenko E.A., Wahl M.C.. Crystal structure analysis reveals functional flexibility in the selenocysteine-specific tRNA from mouse. PLoS One. 2011; 6:e20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 65. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Llargues-Sistac G., Bonjoch L., Castellvi-Bel S.. HAP1, a new revolutionary cell model for gene editing using CRISPR-Cas9. Front. Cell Dev. Biol. 2023; 11:1111488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Beigl T.B., Kjosas I., Seljeseth E., Glomnes N., Aksnes H.. Efficient and crucial quality control of HAP1 cell ploidy status. Biol. Open. 2020; 9:bio057174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jameson R.R., Carlson B.A., Butz M., Esser K., Hatfield D.L., Diamond A.M.. Selenium influences the turnover of selenocysteine tRNA[Ser]Sec in Chinese hamster ovary cells. J. Nutr. 2002; 132:1830–1835. [DOI] [PubMed] [Google Scholar]
- 69. Lin H.C., Ho S.C., Chen Y.Y., Khoo K.H., Hsu P.H., Yen H.C.. Selenoproteins. CRL2 aids elimination of truncated selenoproteins produced by failed UGA/Sec decoding. Science. 2015; 349:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Carlson B.A., Xu X.M., Gladyshev V.N., Hatfield D.L.. Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine tRNA. J. Biol. Chem. 2005; 280:5542–5548. [DOI] [PubMed] [Google Scholar]
- 71. Moustafa M.E., Carlson B.A., El-Saadani M.A., Kryukov G.V., Sun Q.A., Harney J.W., Hill K.E., Combs G.F., Feigenbaum L., Mansur D.B.et al.. Selective inhibition of selenocysteine tRNA maturation and selenoprotein synthesis in transgenic mice expressing isopentenyladenosine-deficient selenocysteine tRNA. Mol. Cell Biol. 2001; 21:3840–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Galli G., Hofstetter H., Birnstiel M.L.. Two conserved sequence blocks within eukaryotic tRNA genes are major promoter elements. Nature. 1981; 294:626–631. [DOI] [PubMed] [Google Scholar]
- 73. Meplan C. Selenium and chronic diseases: a nutritional genomics perspective. Nutrients. 2015; 7:3621–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chujo T., Tomizawa K.. Human transfer RNA modopathies: diseases caused by aberrations in transfer RNA modifications. FEBS J. 2021; 288:7096–7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Agris P.F., Narendran A., Sarachan K., Vare V.Y.P., Eruysal E.. The importance of being modified: the role of RNA modifications in translational fidelity. Enzymes. 2017; 41:1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fradejas-Villar N., Bohleber S., Zhao W., Reuter U., Kotter A., Helm M., Knoll R., McFarland R., Taylor R.W., Mo Y.et al.. The effect of tRNA[Ser]Sec isopentenylation on selenoprotein expression. Int. J. Mol. Sci. 2021; 22:11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dalwadi U., Mannar D., Zierhut F., Yip C.K.. Biochemical and structural characterization of human core elongator and its subassemblies. ACS Omega. 2022; 7:3424–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dauden M.I., Jaciuk M., Weis F., Lin T.Y., Kleindienst C., Abbassi N.E.H., Khatter H., Krutyholowa R., Breunig K.D., Kosinski J.et al.. Molecular basis of tRNA recognition by the Elongator complex. Sci. Adv. 2019; 5:eaaw2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Goffena J., Lefcort F., Zhang Y., Lehrmann E., Chaverra M., Felig J., Walters J., Buksch R., Becker K.G., George L.. Elongator and codon bias regulate protein levels in mammalian peripheral neurons. Nat. Commun. 2018; 9:889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tukenmez H., Xu H., Esberg A., Bystrom A.S.. The role of wobble uridine modifications in +1 translational frameshifting in eukaryotes. Nucleic Acids Res. 2015; 43:9489–9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saito Y., Shichiri M., Hamajima T., Ishida N., Mita Y., Nakao S., Hagihara Y., Yoshida Y., Takahashi K., Niki E.et al.. Enhancement of lipid peroxidation and its amelioration by vitamin E in a subject with mutations in the SBP2 gene. J. Lipid Res. 2015; 56:2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fradejas-Villar N., Zhao W., Reuter U., Doengi M., Ingold I., Bohleber S., Conrad M., Schweizer U.. Missense mutation in selenocysteine synthase causes cardio-respiratory failure and perinatal death in mice which can be compensated by selenium-independent GPX4. Redox Biol. 2021; 48:102188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary data.