

SUMMARY
Intestinal colonization of the oral bacterium Haemophilus parainfluenzae has been associated with Crohn’s disease (CD) severity and progression. This study examines the role of periodontal disease (PD) as a modifier for colonization of H. parainfluenzae in patients with CD and explores the mechanisms behind H. parainfluenzae-mediated intestinal inflammation. Fifty subjects with and without CD were evaluated for the presence of PD, and their oral and fecal microbiomes were characterized. PD is associated with increased levels of H. parainfluenzae strains in subjects with CD. Oral inoculation of H. parainfluenzae elicits strain-dependent intestinal inflammation in murine models of inflammatory bowel disease, which is associated with increased intestinal interferon-γ (IFN-γ)+ CD4+ T cells and disruption of the host hypusination pathway. In summary, this study establishes a strain-specific pathogenic role of H. parainfluenzae in intestinal inflammation and highlights the potential effect of PD on intestinal colonization by pathogenic H. parainfluenzae strains in patients with CD.
Graphical Abstract
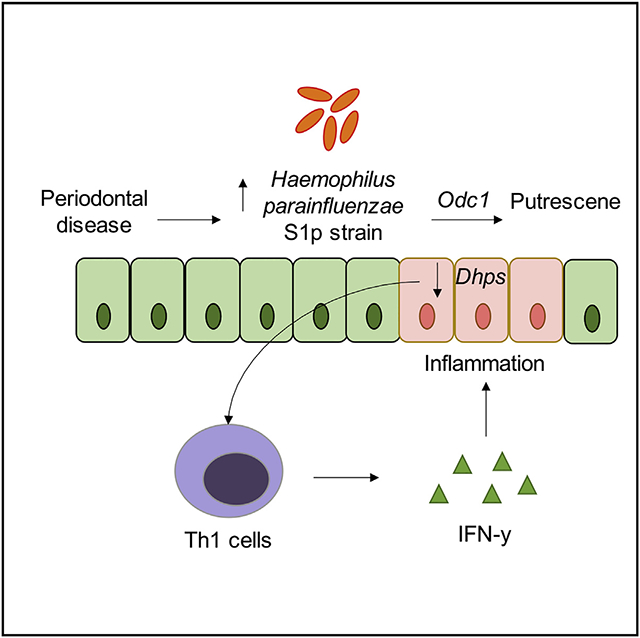
In brief
Sohn et al. show differential pathogenicity of H. parainfluenzae strains and its potential association with periodontal disease in patients with Crohn’s disease.
INTRODUCTION
Crohn’s disease (CD) is a major subtype of inflammatory bowel disease (IBD), which causes relapsing and remitting inflammation in the gastrointestinal tract. Previous studies have suggested that intestinal colonization of Haemophilus parainfluenzae, a Gram-negative oral commensal, is associated with IBD activity.1-6 In fact, the presence of H. parainfluenzae in the intestinal mucosal tissue was one of the most influential features for predicting disease activity in treatment-naive patients with CD.1 Similarly, H. parainfluenzae was highly associated with the severity, progression, and treatment response of ulcerative colitis (UC), another major subtype of IBD.2 Moreover, H. parainfluenzae has been shown to have a substantially higher immunoglobulin A (IgA) coating than other gut bacteria in patients with IBD, suggesting its role in immunopathogenesis.6 While these findings provide evidence for the association between H. parainfluenzae and CD, studies that explore and establish a causal relationship are lacking.
Periodontal disease (PD) is a chronic inflammatory disease of the tissue that supports the tooth. Recent studies have suggested that PD is associated with CD.7-13 In fact, patients with CD have a higher prevalence, severity, and extent of PD compared with the normal population.13 H. parainfluenzae is an abundant oral commensal that is enriched in periodontally healthy individuals compared with subjects with PD.14,15 Intriguingly, studies have shown that there is increased gut colonization of H. parainfluenzae in subjects with PD.16-18 However, there is limited information on the potential effect of PD in the gut colonization of H. parainfluenzae in patients with CD. Similarly, although the species-level population of H. parainfluenzae is genetically diverse,19 no study has investigated the gut dynamics of H. parainfluenzae strains, their associations with PD, and their ability to induce intestinal inflammation.
The aim of this study is to provide a better understanding of the potential effect of PD on the oral and gut colonization of H. parainfluenzae and assess its influence on intestinal inflammation during CD. To this end, we characterized via 16S rRNA sequencing the oral and gut microbiome of subjects with CD and control subjects with and without PD (Table 1). Strain-level analysis of H. parainfluenzae was performed using oligotyping and confirmed using strain-specific qPCR assay.
Table 1.
Demographics
| Healthy controls |
Crohn’s disease |
p | |
|---|---|---|---|
| Sex | – | – | 1 |
| Male | 10 | 10 | – |
| Female | 15 | 15 | – |
| Mean age | 58.28 | 53.48 | 0.36 |
| Weight | 83.59 | 74.9548 | 0.16 |
| Height | 1.70 | 1.66 | 0.13 |
| BMI | 28.50 | 27.08 | 0.45 |
| Bristol stool index | 3.92 | 4.24 | 0.49 |
| Periodontal disease status | – | – | 0.18 |
| Healthy | 4 | 4 | – |
| Moderate | 7 | 13 | – |
| Severe | 14 | 8 | – |
| Diet | – | – | 0.73 |
| Non-restricted | 20 | 19 | – |
| Restricted | 5 | 6 | – |
| Keto diet | 1 | 2 | – |
| Weight loss diet | 3 | 0 | – |
| Vegetarian diet | 1 | 2 | – |
| Restricted Crohn’s dieta | 0 | 2 | – |
| Alcohol | – | – | 0.56 |
| Drinks alcohol | 16 | 14 | – |
| Does not drink alcohol | 9 | 11 | – |
| CD condition | – | – | – |
| Montreal classification | – | – | – |
| Age at onset | – | – | – |
| A1 | – | 10 | – |
| A2 | – | 7 | – |
| A3 | – | 8 | – |
| Disease location | – | – | – |
| L1 | – | 4 | – |
| L2 | – | 6 | – |
| L3 | – | 15 | – |
| Disease behavior | – | – | – |
| B1 | – | 10 | – |
| B2 | – | 9 | – |
| B3 | – | 6 | – |
| Perianal disease | – | 5 | – |
| Hx of bowel surgery | – | 13 | – |
| Drug | – | – | – |
| Biologics | – | – | – |
| Anti-TNF | – | 11 | – |
| Anti-integrin | – | 3 | – |
| Anti-IL12 IL23 | – | 3 | – |
| Mesalamine | – | 3 | – |
Demographics of recruited study participants. Montreal classification definition: A1, below 16 years; A2, between 17 and 40 years; A3, above 40 years; L1, ileal; L2, colonic; L3, ileocolonic; B1, non-structuring and non-penetrating; B2, structuring; B3, penetrating.
Highly individualized diet for Crohn’s disease. p values for quantitative variables were calculated using Student’s t test; categorical variables were calculated using the χ2 test.
Next, we utilized mouse models of IBD to test the pathogenicity of the H. parainfluenzae strains. Finally, comparative genomics analysis was performed on whole-genome sequencing data of isolated H. parainfluenzae strains to elucidate potential mechanistic insight underlying their pathogenicity.
RESULTS
Severe PD in subjects with CD is associated with decreased salivary microbiome alpha diversity
There was a significant reduction of salivary alpha diversity, a measure of species abundance within a community, in subjects with CD compared with non-CD controls (Figure 1A). Prior intestinal resection surgery was not associated with changes in salivary alpha diversity (Figure 1B). There were no significant differences in salivary alpha diversity across groups with different PD statuses (Figure 1C). A subgroup analysis showed that the decreased salivary alpha diversity associated with CD was mainly driven by CD subjects with severe PD (Figure 1D). This result was confirmed by a correlation analysis, which showed a trend for decreased salivary alpha diversity metrics with increased PD severity in subjects with CD, while there was a trend for higher salivary alpha diversity with greater PD severity in non-CD controls (Figure 1E). There were no significant changes in salivary beta diversity, a measure of taxonomic composition variability between two communities, associated with CD or PD measured by unweighted-unique fraction metric (UNiFrac) distance (Figures S1A and S1B). Alteration in salivary microbiota in patients with CD was mainly driven by decreased Leptotrichia wadei and Porphyromonas endodontalis (Table S2). There were no significant salivary taxonomy changes associated with PD status. Salivary H. parainfluenzae was not significantly associated with either CD or PD status (Figures S2A-S2C).
Figure 1. PD is associated with decreased salivary and fecal microbiome richness in subjects with CD.

Salivary or fecal alpha diversity based on (A and F) CD (n = 25 in non-CD and n = 25 in CD); (B and G) CD surgery status (n = 25 in non-CD, n = 12 in CD no surgery, n = 13 in CD surgery); (C and H) PD (n = 8 in healthy, n = 21 in moderate, and n = 21 in severe); or (D and I) stratified by CD and PD status (n = 4 in healthy PD and non-CD, n = 4 in healthy PD and CD, n = 7 in moderate PD and non-CD, n = 13 in moderate PD and CD, n = 14 in severe PD and non-CD, and n = 8 in severe PD and CD) represented by observed OTUs. (E and H) Correlation matrix of salivary and fecal alpha diversity measures and whole-mouth periodontal clinical measurements (n = 25 each group). pielou_e, Pielou’s evenness index; faith_pd, Faith’s phylogenetic diversity; shannon, Shannon diversity index; Brillouin_d, Brillouin index; chao1, Chao1 richness estimate; PI, plaque index; GI, gingival index; PD, mean pocket depth; GR, gingival recession; CAL, mean clinical attachment loss; BOP, bleeding on probing. Statistical testing was performed using (A and F) Mann-Whitney U, (B, C, G, and H) Kruskal-Wallis followed by Dunn’s multiple comparisons test, (D and I) two-way ANOVA followed by Sidak’s multiple comparisons test, and (E and H) Pearson correlation tests with coefficients shown in color scale. A two-tailed test was used to calculate p values. Data are represented as mean ± SEM.
PD status is associated with altered subgingival beta diversity
There were no significant differences in subgingival alpha diversity based on CD or PD status (Figures S3A-S3C). Prior intestinal resection surgery was not associated with changes in subgingival alpha diversity (Figure S3D). A subgroup correlation analysis showed a trend for higher subgingival alpha diversity with increasing PD severity in both patients with CD and non-CD controls (Figure S3E). Beta diversity, measured by unweighted-UNiFrac distance, showed a significant alteration in the whole community composition according to PD status but not CD status (Figures S3F and S3G). No significant subgingival taxonomy changes, including H. parainfluenzae (Figures S2D-S2F), were associated with CD or PD status.
PD in patients with CD is associated with decreased fecal alpha diversity
There was a significant reduction in fecal alpha diversity in patients with CD compared with non-CD controls measured by observed operational taxonomic units (OTUs) (Figure 1F). Prior intestinal resection surgery was the main driver of the observed reduction in fecal alpha diversity (Figure 1G). Furthermore, a significant reduction in alpha diversity was detected in those with moderate PD compared with those with severe PD (Figure 1H). A subgroup analysis showed that the presence of moderate or severe PD in patients with CD decreased fecal alpha diversity (Figure 1I). A subgroup correlation analysis showed a trend for decreased fecal alpha diversity with increasing PD severity in patients with CD; however, a trend for increased fecal alpha diversity with increased PD severity was observed in non-CD controls (Figure 1H). Beta diversity analysis of fecal samples showed a significant difference associated with CD and PD status measured by unweighted-UNiFrac distance (Figures S1C and S1D). CD-mediated fecal microbiota changes were mainly driven by a decrease in Dorea formicigenerans (Table S3). There were no significant salivary taxonomy changes associated with PD status. Fecal H. parainfluenzae was not significantly associated with either CD or PD status (Figures S2G-S2I).
Genomic analysis reveals high diversity and variability of H. parainfluenzae strains
The relative abundance of H. parainfluenzae at species level was not statistically different based on CD or PD status in saliva, fecal, or subgingival samples (Figures S2A-S2I). Shannon entropy analysis of 16S rRNA amplicon sequences mapping to H. parainfluenzae revealed several highly variable nucleotide positions, indicating the existence of multiple strains. Oligotyping, which utilizes subtle variations within 16S rRNA reads to classify distinct taxa, was performed to further investigate the strain diversity of H. parainfluenzae.20 There was an increased fecal H. parainfluenzae oligotype 7 in subjects with CD with severe PD compared with those without PD, suggesting that PD may influence intestinal colonization of select H. parainfluenzae strain in subjects with CD (Figure S4A). There were no differences in identified H. parainfluenzae oligotypes in saliva (Figure S4B) and subgingival (Figure S4C) samples based on CD or PD status.
Isolation of ornithine metabolizing H. parainfluenzae strain
H. parainfluenzae strains were isolated from the collected saliva and fecal samples using a selective medium to further investigate H. parainfluenzae strain-level variation in vitro. Isolated H. parainfluenzae strains were classified into biotypes based on their biochemical profiles.21 H. parainfluenzae biotypes II and V were the two major isolated biotypes (Figure S5A). Biotype II metabolized ornithine, while biotype V did not metabolize ornithine. Two biotypes from saliva (strains S1p and S2) and fecal (strains F2 and F3p) samples were selected to be fully sequenced for whole-genome characterization (Figure S5B). Phylogenetic tree analysis based on whole genomes showed that strains clustered based on the isolation site regardless of the assigned biotype (Figure S5C).
PD is associated with increased gut colonization of H. parainfluenzae S1p strain in subjects with CD
Strain-specific qPCR assay was performed on the collected saliva, subgingival, and fecal samples to accurately quantify the isolated H. parainfluenzae strain levels and find their association with clinical phenotypes. Salivary, but not subgingival or fecal, H. parainfluenzae F3p strains were increased in severe patients with PD compared with the non-PD controls (Figures S6A-S6C). There were no differences in saliva, subgingival, and fecal H. parainfluenzae strains based on CD status (Figures 2A-2C). However, subgroup analysis showed an increase in fecal H. parainfluenzae S1p strains (Figure 2D) but not S2, F2, or F3p strains (Figures S7A-S7C), in patients with CD with severe PD. A correlation analysis showed an increasing fecal H. parainfluenzae S1p strain level with an increasing PD CAL score in patients with CD (Figure 2F; p < 0.0001, R = 0.79) but not in non-CD controls (Figure 2E; p = 0.3085, R = −0.45). No significant association was seen in S2, F2, and F3p strains (Figures S7D-S7I). These results suggest that PD may potentiate the selective gut colonization of H. parainfluenzae S1p strain in patients with CD.
Figure 2. Quantification of H. parainfluenzae strains.

(A–C) qPCR quantification of H. parainfluenzae strains in (A) fecal, (B) saliva, and (C) subgingival samples based on periodontal disease status (n = 25 for non-CD and n = 25 for CD).
(D) Fecal H. parainfluenzae S1p strain level in patients with CD vs. non-CD controls based on periodontal disease status (n = 4 in healthy PD and non-CD, n = 4 in healthy PD and CD, n = 7 in moderate PD and non-CD, n = 13 in moderate PD and CD, n = 14 in severe PD and non-CD, and n = 8 in severe PD and CD).
(E and F) Pearson correlation analysis of fecal H. parainfluenzae S1 strain log (DNA copies/mL) with clinical attachment loss in (E) non-CD (n = 25) and (F) CD (n = 25).
Statistical testing was performed using two-way ANOVA and Sidak’s multiple comparison test and a two-tailed statistical test. Data are represented as mean ± SEM.
Oral administration of H. parainfluenzae S1p strain induces intestinal inflammation
To test the pathogenicity of oral H. parainfluenzae strains, 109 CFU S1p strain (biotype II) or S2 strain (biotype V) isolated from saliva were administered every other day into antibiotic-conditioned DSS-treated mice via oral gavage (Figure 3A). Histopathological analysis showed significant colonic inflammation in mice that received the S1p strains (Figure 3B). Decreased colon length, a marker for intestinal inflammation, was also observed in mice that received the H. parainfluenzae S1p strain (Figure 3D). Intestinal myeloperoxidase (MPO) activity, a quantitative marker of intestinal inflammation, was significantly elevated in mice receiving the S1p strain (Figure 3C). Overall, the S1p strain showed consistent pathogenic properties, whereas the S2 strain did not elicit any pathological response.
Figure 3. Oral inoculation of H. parainfluenzae S1p strain induces intestinal inflammation in an acute model of IBD.

(A) Study design: 4-week-old wild-type Balb/c mice were given daily gavage of antibiotic mixture (see STAR Methods for detail) for 3 days. 5% DSS solution was given via drinking water for 5 days after the antibiotic treatment. 109 CFU H. parainfluenzae strain S1p (n = 10) or S2 (n = 10) or PBS (n = 9) was inoculated every other day until the sacrifice date.
(B) Histopathological analysis of colonic tissue samples.
(C) Intestinal myeloperoxidase activity.
(D) Colon length (cm)/weight (g).
Statistical testing was performed using one-way ANOVA and Tukey’s multiple comparison test. Data are represented as mean ± SEM.
Gut colonization of H. parainfluenzae S1p strain induces intestinal interferon γ (IFN-γ)+ CD4+ T cell and inflammation
To investigate the long-term effect of intestinal colonization by oral H. parainfluenzae strains, a single dose of 109 CFU S1p strain (biotype II) or S2 strain (biotype V) isolated from saliva was administered into antibiotic-conditioned Il10-deficient mice via oral gavage and observed for 8 weeks for disease development (Figure 4A). PCR analysis of the cecal content showed successful intestinal colonization of the H. parainfluenzae S1p strain, but not the S2 strain, 8 weeks after a single-dose inoculation (Figure 4B); since the long-term effect of intestinal colonization by S2 strain could not be determined without the successful colonization, S2 strain data were excluded from the subsequent analyses. Intestinal colonization by the S1p strain was associated with an increased level of intestinal inflammation measured by histopathological score (p = 0.111) (Figure 4C) and MPO activity (p = 0.0317) (Figure 4D). Flow cytometry analysis of the small intestinal lamina propria showed a significant increase in IFN-γ+ CD4+ T cells in mice colonized with the S1p strain, suggesting its role in immunopathogenesis (Figure 4E).
Figure 4. Gut colonization of H. parainfluenzae S1p strain induces intestinal IFN-γ+ CD4+ T cell proliferation and inflammation in a chronic model of IBD.

(A) Study design: 4-week-old Il10-deficient Balb/c mice were given an antibiotic cocktail via autoclaved drinking water for 4 weeks before the oral administration of 109 CFU H. parainfluenzae strain S1p (n = 5) or S2 (n = 5) or PBS (n = 4). Mice were maintained in positively pressured cages for 6 weeks during the intestinal disease development experimental period.
(B) PCR amplification of H. parainfluenzae from mouse fecal samples collected in the 12th week of the experiment shown in gel electrophoresis.
(C) Intestinal myeloperoxidase activity.
(D) Histopathological analysis of colonic tissue samples.
(E) Flow cytometry analysis of intestinal lamina propria CD4+ T helper cells.
Statistical testing was performed using the Mann-Whitney U test and two-way ANOVA with Sidak’s multiple comparison test. Data are represented as mean ± SEM.
Gut colonization of H. parainfluenzae S1p strain is associated with decreased host intestinal hypusination
Biochemical analysis showed that S1p strain metabolizes ornithine (Figure S5A). Comparative genomics analysis also revealed that the H. parainfluenzae S1p strain contains multiple genes related to polyamine synthesis and export pathways, such as ornithine decarboxylase (Odc1) and putrescine-ornithine antiporter (PotE). Polyamine metabolism regulates intestinal helper T cell lineage fidelity and inflammation.22 To investigate the role of polyamine in H. parainfluenzae S1p strain-mediated intestinal inflammation, polyamines in luminal content were measured using ultra-performance liquid chromatography (UPLC) assay. The result showed an approximately 100-fold increase in luminal putrescine level in the mice colonized with the S1p strain (Figure 5B). There were no significant differences in luminal spermidine or spermine levels (Figures 5C and 5D). Ornithine decarboxylase, Odc1, is the rate-controlling enzyme for the host’s biosynthesis of putrescine (Figure 5A). There was no significant difference in host Odc1 expression at the transcription level (Figure 5E), indicating that the increased luminal putrescine level is likely due to increased synthesis of putrescine from H. parainfluenzae S1p strain. Excess putrescine accumulation has been shown to inhibit the formation of eukaryotic initiation factor 5A (eIF5A).23 Deoxyhypusine synthase, Dhps, is the first rate-limiting step of hypusination that catalyzes the cleavage of spermidine and the transfer of the 4-aminobutyl moiety to a lysine residue on the eIF5A. There was a significant decrease in host intestinal Dhps transcription level (Figure 5F) in the mice colonized with S1p strain, indicating that the observed proliferation of intestinal IFN-γ+ CD4+ T cells may be due to excess accumulation of S1p-derived putrescine leading to suppression of hypusination pathway. Spermine oxidase, Smox, converts spermine to spermidine and produces hydrogen peroxide as a byproduct, which could contribute to intestinal inflammation. However, there was no difference in Smox expression level (Figure 5G).
Figure 5. The potential role of polyamines in H. parainfluenzae S1p strain pathogenicity.

(A) Polyamine metabolism pathway. Cecal content polyamine concentrations in mice inoculated with S1p strain (n = 5) or PBS (n = 4) were measured using UPLC assay.
(B–D) Concentrations of polyamines were determined based on standard curves and normalized to protein concentrations of (B) putrescine (PUT), (C) spermidine (SPD), and (D) spermine (SPM).
(F and G) Probe-base qPCR assay was performed on proximal colon samples to measure the transcription activity of ornithine decarboxylase 1 (Odc1), (F) deoxyhypusine synthase (Dhps), and (G) SPM oxidase (Smox) relative to TATA box binding protein (Tbp) housekeeping gene.
Statistical testing was performed using a two-sample t test. Data are represented as mean ± SEM.
Intestinal hypusination pathway and luminal polyamine concentrations are altered in subjects with IBD
To investigate the role of polyamine in the pathogenesis of human IBD, the NIH integrative human microbiome project (iHMP) dataset, which contains comprehensive transcriptomics, metagenomics, metatranscriptomics, and metabolomics data of patients with IBD, was analyzed.3 There was a significant increase in fecal putrescine levels in both subjects with CD and UC compared with non-IBD controls (Figure 6A), while there were no differences in fecal spermidine levels (Figure 6B). DHPS gene expression was significantly decreased in subjects with CD (Figure 6C) and UC (Figure 6D) compared with non-IBD controls. Metatranscriptomic analyses showed a significant increase in gut microbial ornithine decarboxylase transcript in patients with CD but not in patients with UC (Figure 6E). There was no difference in polyamine-transporting ATPase transcript (Figure 6F). Interestingly, ornithine decarboxylase transcriptional activity had the highest association with H. parainfluenzae (false discovery rate [FDR] q = 9.08 × 10−80, R = 0.63) among all organisms detected by metagenomics analysis (Table S4). Moreover, the majority of the bacteria significantly associated with ornithine decarboxylase transcriptional activity, such as Veillonella spp., Streptococcus spp., and Klebsiella pneumoniae, were bacteria of oral origin, highlighting the potential role of oral bacteria in the pathogenesis of CD (Table S4).
Figure 6. Polyamine in patients with IBD.

(A and B) Fecal PUT (A) and SPD (B) concentration of patients with non-IBD (n = 127), UC (n = 146), and CD (n = 261) from iHMP cohort measured by liquid chromatography-mass spectrometry (LC-MS) methods.
(C and D) Ileal (n = 21 in non-IBD and n = 46 in CD) (C) and rectum/sigmoid colon (D) (n = 23 in non-IBD and n = 35 in UC) DHPS normalized counts measured by RNA sequencing (RNA-seq).
(E and F) Gut microbial genetic transcripts of (E) ornithine decarboxylase and (F) polyamine-transporting ATPase from patients with non-IBD (n = 199), UC (n = 232), and CD (n = 382) in iHMP cohort measured by whole metatranscriptome sequencing.
Statistical testing was performed using the Kruskal-Wallis test and Dunn’s multiple comparison test. Data are represented as mean ± SEM.
DISCUSSION
In this study, we explored the colonization dynamics of H. parainfluenzae in patients with CD with and without PD and established a strain-specific pathogenic role of H. parainfluenzae in intestinal inflammation. While 16S rRNA analysis of oral and gut microbiome did not show alteration in H. parainfluenzae at the species level, it interestingly revealed the distinct oral and gut microbiome characteristics of patients with CD with PD.
CD status was associated with a decrease in salivary alpha diversity, mainly driven by those with severe PD. Previous studies that investigated the salivary microbiome of patients with CD found no difference in the alpha diversity measurements compared with controls.24,25 This discrepancy is likely due to the confounding effect of PD on salivary alpha diversity. The progression of PD was directly correlated with increased alpha diversity in the oral cavity,15 which was also observed in our non-CD control group. By stratifying the PD severity, we were able to determine the isolated effect of CD on the saliva microbiome. PD in patients with CD may have unique pathogenesis that is driven by systemic inflammation from CD, which manifests with disruption of salivary microbiota. Patients with CD indeed suffer from more severe forms of PD and other oral manifestations, such as aphthous ulcers.26 Further studies are needed to understand the pathogenesis of PD in the context of CD, which could lead to the development of more targeted therapy for PD in patients with CD.
The gut microbiota of patients with CD have been extensively studied.1,3 However, only a few studies investigated the effect of PD on the gut microbiome of patients with CD.10 As previously reported, we observed a reduced fecal alpha diversity and an alteration in fecal beta diversity associated with CD.1 Interestingly, subgroup correlation analysis showed that fecal alpha diversity decreased with worsening PD in patients with CD, whereas fecal alpha diversity increased with worsening PD in non-CD controls. Decreased alpha diversity is a hallmark of CD progression.27 These results suggest that PD may exacerbate intestinal inflammation in CD by decreasing gut microbial diversity. However, these findings need to be further validated in larger CD populations.
H. parainfluenzae was not associated with CD or PD at the species level. However, oligotyping analysis of the H. parainfluenzae 16S rRNA amplicon sequences revealed several highly variable nucleotide positions, suggesting a diverse number of H. parainfluenzae strains present in the oral and gut samples. Whole-genome sequencing and phylogenetic analysis showed that H. parainfluenzae strains clustered based on isolated locations and not biotypes. This is consistent with a recent study that employed a metapangenomic approach to study the microbial communities of the three different oral habitats—tongue dorsum, buccal mucosa, and supragingival plaque—and showed that H. parainfluenzae strains cluster into three distinct groups based on the location.19 It therefore appears that the oral habitat determines large genomic differences in the H. parainfluenzae population.
To find the clinical relevance of the H. parainfluenzae strains, a strain-specific qPCR assay was performed. Interestingly, the salivary H. parainfluenzae F3p strain was increased in PD. Prior studies consistently showed that oral H. parainfluenzae is decreased in PD.28,29 The strain-level analysis allowed for the detection of biologically relevant strains that could play an essential role in the disease pathogenesis. However, whether the F3p strain affects the pathogenesis of PD remains debatable and more studies are needed to test the clinical relevance of salivary H. parainfluenzae F3p strains.
Strain-level analysis of fecal samples showed that CD or PD status alone did not change the composition of H. parainfluenzae strains. However, subgroup analysis revealed that PD was associated with an increased fecal H. parainfluenzae S1p strain in patients with CD, suggesting that PD may selectively increase gut colonization of H. parainfluenzae strains in subjects with CD. Although a recent study has shown that PD does not further enhance the overall gut colonization by oral bacteria in patients with CD,10 PD may selectively increase the gut transmission of virulent bacterial strains that are capable of evading the host immune system.30 The subgingival microbiota in PD constantly interacts with the host circulatory system in the periodontal pocket, allowing ectopic colonization through the bloodstream.7 Fusobacterium nucleatum, an oral bacterium, and a PD pathogen has been shown to colonize the gut through the blood-stream and influence colorectal cancer progression.31 Biochemical and comparative genomics analysis has shown that clinically relevant H. parainfluenzae S1p strains metabolize ornithine and contain genes involved in polyamine biosynthesis pathways such as ornithine decarboxylase and putrescine-ornithine antiporter. Studies have shown that polyamines play a critical role in microbial pathogenesis by regulating biofilm formation, evasion from phagolysosomes, toxin activity, and protection from oxidative and acid stress.32 The observed selective increase in the fecal H. parainfluenzae S1p strain in patients with CD with PD may be due to the increased pathogenicity related to polyamine production. Further studies are needed to understand how PD increases gut colonization of ornithine metabolizing H. parainfluenzae strain.
To test the role of oral H. parainfluenzae strains in intestinal pathology, saliva-derived H. parainfluenzae S1p or S2 strains were orally administered into DSS-induced colitis mice and Il10-deficient mice. DSS is a sulfated polysaccharide that elicits acute intestinal inflammation by disrupting mucosal barrier function. Il10-deficient mice have characteristic histological findings of discontinuous and transmural inflammatory intestinal lesions similar to those found in human IBD.33 Moreover, Il10-deficient mice develop progressive stages of chronic intestinal inflammation, which is dependent on intestinal microbiota, and therefore allows for evaluating the chronic effect of H. parainfluenzae intestinal colonization in disease pathogenesis.34 In both acute and chronic models of IBD, the S1p strain elicited intestinal inflammation. Interestingly, the S1p strain, but not the S2 strain, was able to colonize the intestine of Il10-deficient mice, indicating that only certain H. parainfluenzae strains may colonize the intestinal tract. This result is consistent with human data, which showed a higher number of gut colonization of the S1p strain than the S2 strain. Studies have shown that Il10-deficient mice have distinct enteric microbiota compared with conventional mice.35 Il10 deficiency could have affected outcomes of H. parainfluenzae strain colonization of the intestine. Moreover, human-to-mouse microbiota transfer is imperfect due to the inherent difference in intestinal anatomy.36 Additional studies are needed to explore factors affecting the selective intestinal colonization of H. parainfluenzae strains.
The CD4+ T helper cell subset population is key in mediating intestinal inflammation in IBD. IFN-γ-producing CD4+ T cells, which play an important role in the elimination of intracellular bacteria and viruses, induce macrophages and cytotoxic CD8+ T cells to promote a proinflammatory response in patients with IBD.37 Other studies also highlight the function of IFN-γ- and interleukin-17 (IL-17)-producing CD4+ T cells, which propagate the differentiation of pathogenic T helper cells in patients with IBD.38 IL-4 is essential in modulating regulatory T cells.39 S1p strain colonization led to a proliferation of intestinal IFN-γ+ CD4+ T cells, while other CD4+ T helper cell populations remained stable. Interestingly, intestinal colonization of Klebsiella strains isolated from salivary microbiota has also been shown to induce intestinal IFN-γ+ CD4+ T cells and cause intestinal inflammation.40
Polyamine metabolism is critical in regulating intestinal T cell lineage fidelity.22 Gut colonization of the H. parainfluenzae S1p strain led to an approximately 100-fold elevation in luminal putrescine level. Excess accumulation of putrescine has been shown to inhibit the hypusination pathway by inhibiting eIF5A.23 Dhps, which mediates the first rate-limiting step of hypusination pathway, was significantly decreased in the intestine of mice colonized with the S1p strain. Previous study has shown that suppression of the polyamine-hypusination axis by deleting the Dhps gene leads to the proliferation of IFN-γ+ CD4+T cells.22 Our results indicate that the observed proliferation of intestinal IFN-γ+ CD4+ T cells in S1p colonized mice may be due to excess production of S1p-derived putrescine leading to suppression of hypusination pathway.
The role of polyamines in intestinal health is not yet fully understood. A recent study identified putrescine to disrupt tight junction integrity.41 Administration of putrescine into DSS-treated mice exacerbated colon inflammation, increased gut permeability, and decreased colon length.41 In a Citrobacter rodentium-induced intestinal inflammation mouse model, putrescine increased fecal shedding of viable C. rodentium, bacterial attachment to the colonic epithelium, and levels of intestinal inflammatory cytokine.41 However, multiple studies have also shown the beneficial effects of polyamines on intestinal health. Putrescine alleviated intestinal inflammatory response to lipopolysaccharide (LPS).42 Colonization of the intestine by Escherichia coli containing ornithine decarboxylase promoted intestinal epithelial proliferation and anti-inflammatory macrophage differentiation in the colon.43 Therefore, more studies are needed to elucidate how H. parainfluenzae elicits intestinal inflammation through the modulation of the polyamine-hypusination pathway.
Finally, to understand the role of polyamine in human IBD, multi-omics data from the NIH iHMP cohort were analyzed.3 Similar to the observation made in our mouse study, an increased putrescine level was observed in the fecal contents of both patients with CD and UC. The transcription level of the DHPS gene was significantly decreased in both CD and UC, indicating that disruption of the polyamine-hypusination pathway may play a role in the pathogenesis of IBD. Moreover, in support of our findings, bacterial ornithine decarboxylase activity was highly upregulated in patients with CD and UC. Interestingly, the observed increased bacterial ornithine decarboxylase activity was highly associated with the presence of H. parainfluenzae, indicating that the increased putrescine level seen in human patients with IBD may be due to H. parainfluenzae ornithine decarboxylase activity. Further studies are warranted to investigate the role of polyamines in human patients with IBD.
Previous studies investigating the mechanistic role of PD in IBD have established the pathogenic role of PD-associated oral pathobionts in intestinal inflammation.40,44,45 Moreover, seminal work by Kitamoto et al. has shown that Th17 cells that develop during oral inflammation can migrate to the gut and contribute to gut inflammation.45 PD likely exerts multi-factorial effects involving immunological and microbiological influence on the pathogenesis of IBD. Further studies exploring the mechanism behind the relationship between PD and IBD are needed. Interventional studies where CD activity is measured after PD treatment would provide invaluable information for the clinical implication of oral treatment for the management of patients with CD with PD.
In summary, this study fills in critical gaps in knowledge by (1) showing that PD is associated with increased gut colonization of pathogenic H. parainfluenzae strains in patients with CD, (2) establishing the differential pathogenicity of H. parainfluenzae strains, and (3) demonstrating potential mechanisms that underly strain-specific H. parainfluenzae-mediated intestinal inflammation.
Limitations of the study
Although this study shows evidence for increased gut colonization of pathogenic H. parainfluenzae strains in CD patients with PD, it needs to be further validated in a larger CD population. Moreover, while our results suggest that increased luminal putrescine produced by H. parainfluenzae may have a pro-inflammatory role, the findings must be further tested and validated.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jiho Sohn (jihosohn@buffalo.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The data generated in this study are available in the NCBI SRA:PRJNA804169, PRJNA831448. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Recruitment of study participants
The study protocol was approved by the University at Buffalo Intuitional Review Board. Written informed consent was received from all study participants. 50 participants, 25 CD and 25 non-CD, were recruited. CD subjects, who had been diagnosed with CD by board-certified gastroenterologists, were first screened from University at Buffalo outpatient clinics. CD phenotype was characterized according to the Montreal classification, which is based on disease location, disease behavior, and age at diagnosis.46 The screened CD patients were brought to University at Buffalo Periodontal Disease Research Center, where certified periodontists/calibrated hygienists examined this cohort for PD, caries, and other oral pathologies, including mucous membrane lesions. Age and sex matched non-Crohn’s controls were directly recruited from the University at Buffalo Periodontal Disease Research Center. PD status was determined based on Centers for Disease Control and Prevention/American Academy of Periodontology criteria using the following measurements: plaque index (PI), gingival index (GI), pocket depth (PD), clinical attachment loss (CAL), bleeding on probing (BOP), gingival recession (GR), and the number of remaining teeth.47 Anthropomorphic measurements, including resting blood pressure, pulse, weight, and, height were obtained during the visit. Medical, dental, and social histories were also taken using a questionnaire assessment, which included: age, race, sex, dietary pattern, medications, stool consistency, oral hygiene habits, general well-being, past medical history, life stresses, occupation, and education level. In order to minimize the variability within subjects, the study design excluded CD patients with extraintestinal manifestations involving the oral cavity. Other exclusion criteria included recent antibiotic use within the past 6 months and the presence of infectious oropharyngeal diseases such as tonsillitis, pharyngitis, endodontic diseases, and candidiasis. Post-clinical examination, each patient provided non-stimulated saliva collected in 15 mL tube on ice. Subgingival plaque samples were collected by trained periodontists or dental hygienists from the deepest interproximal pockets in each of the four quadrants using a periodontal curette. Participants were asked to collect a fecal sample 24 h prior to the study visit in a provided collection tube containing RNAprotect Tissue Reagent (Qiagen) and to transport it on an ice pack to the clinic. Demographic data can be found in Table 1.
Animal experiments
All animal experiments were performed in accordance with the study protocol approved by Institutional Animal Care and Use Committee (IACUC) of the University at Buffalo.
Dextran sodium sulfate (DSS) mouse model for establishing pathogenicity of H. parainfluenzae strains
Four-week-old wild-type BALB/cJ male mice (Jackson Laboratory) were given an antibiotic cocktail of 1 mg mL−1 each of Ampicillin, Neomycin sulfate, Metronidazole, and 0.5 mg mL−1 of Vancomycin hydrochloride via oral gavage using the stainless steel feeding tube for three days to suppress host microbiota. Mice were raised in positive ventilated cages and given autoclaved standard chow diet, beddings, and enrichments such as nestlets and enviro-dry. All interventions were performed during the light cycle. Approximately 1 × 109 CFU of H. parainfluenzae strain was administered per group (n = 9–10) every other day until the end of the experiment (day 5) via oral gavage using the stainless steel feeding tube. 5% DSS in autoclaved drinking water solution was given to mice to induce intestinal inflammation. DSS solution was changed every day.
Interleukin 10 (Il10) deficient mice for determining the mechanism underlying H. parainfluenzae-mediated intestinal inflammation
Four-week-old Il10 deficient BALB/cJ male mice (Jackson Laboratory) were given an antibiotic cocktail of 1 mg mL−1 each of Ampicillin, Neomycin sulfate, Metronidazole, and 0.5 mg mL−1 of Vancomycin hydrochloride via drinking water for four weeks (refilled every 96 h) to ablate host microbiota. Mice were raised in positive ventilated cages and given autoclaved standard chow diet, beddings, and enrichments such as nestlets and enviro-dry. All interventions were performed during the light cycle. A single dose of 1 × 109 CFU of H. parainfluenzae strain was administered per group (n = 4–5) via oral gavage using the stainless steel feeding tube. At week 12, intestinal tissue and contents were harvested for analysis.
METHOD DETAILS
Microbiome sequencing and analysis
DNA from fecal samples was extracted using DNeasy PowerSoil Kit (Qiagen). Subgingival samples from the four quadrants were first pooled and then extracted using QIAamp DNA Mini kit (Qiagen). DNA from the saliva sample was extracted using QIAamp DNA Mini kit (Qiagen). 16S rRNA gene amplicon sequencing libraries of the V3-V4 hypervariable region were prepared and sequenced at University Buffalo’s Genomics and Bioinformatics Core as described in a previous study.44 16S rRNA gene data was analyzed using QIIME2 2020.2.48 DADA2 was used to demultiplex raw sequence data.49 Taxonomic classification was performed against the genome taxonomy database r86 reference sequence using classify-sklearn. Alpha and beta diversity was calculated using the q2-diversity plugin.50 Statistical testing for alpha and beta diversity were performed using Kruskal-Wallis and PERMANOVA, respectively. Differential abundance analysis was performed with ANCOM.51 Shannon entropy analysis and oligotyping was performed for the evaluation of H. parainfluenzae 16S rRNA gene sequences intra-species variability.20
Isolation of Haemophilus parainfluenzae strains
H. parainfluenzae was isolated from the collected fecal and saliva samples using Remel Haemophilus Isolation Agar w/Bacitracin and Horse Blood (Thermo Scientific) and subsequently subcultured in Trypticase soy agar (BD) containing factor V (Sigma). Isolated organisms were first rapidly identified using biochemical assay, RapID™ NH System (Thermo Scientific), and confirmed using whole genome sequencing (see below for methods).
H. parainfluenzae cultivation
H. parainfluenzae strains were cultivated overnight in 5% CO2 chamber at 37°C in Tryptic Soy Broth (BD) medium supplemented with factor V, centrifuged, and washed with sterile PBS solution once.
Bacterial whole genome sequencing
Bacterial DNA was extracted using QIAamp DNA Mini kit (Qiagen), and a whole genome sequencing library was constructed using the Nextera DNA Library Preparation Kit (Illumina). 150-cycle paired-end DNA sequencing was performed on MiSeq platform using MiSeq Reagent Kit v2 (300-cycles) (Illumina) in University Buffalo’s Genomics and Bioinformatics Core. Raw sequencing reads were first processed and trimmed using Trim Galore.52 Genome assembly was performed using SPAdes53 and annotation was performed using Prokka.54 Finally, comparative genomics analysis against all publicly available H. parainfluenzae genomes was performed using Sybil.55
Histology
Intestinal tissue samples were collected and formalin-fixed for 24 h. Formalin-fixed samples were parafilm-embedded and horizontally sectioned into 5 μm slide. Samples were subsequently stained with Hematoxylin and eosin. The Histology score was based on inflammation severity, inflammation extent, epithelial changes, and mucosal architecture.56 Samples were blinded and scored by two pathologist-trained individuals.
Isolation and analysis of intestinal lamina propria lymphocytes
Intestinal cells were dissociated using the lamina propria dissociation kit (Miltenyi Biotec) in combination with gentleMACS™ Octo Dissociator with Heaters (Miltenyi Biotec). Percoll gradient separation was performed using 40% Percoll underlaid with 80% Percoll and lymphocytes at interphase were collected. Lymphocytes were stimulated using the Cell Activation Cocktail with Brefeldin A (Biolegend). The following stains were used for extracellular staining: LIVE/DEAD Fixable Dead Cell Stain Kits (Invitrogen; Cat#L34955), Rat Anti-Mouse CD16/CD32 (BD; Cat#553141, Clone: 2.4G2), anti-CD3 (Miltenyi Biotec; Cat#130-102-943, Clone: REA641), and anti-CD4 (Invitrogen; Cat#25-0041-82, Clone: GK1.5), and anti-CD8 (Miltenyi Biotec; Cat#130-120-806, REA601). Foxp3/Transcription Factor Staining Buffer Set (Invitrogen; Cat#00-5523-00) was used for permeabilizing cells for intracellular staining. The following stains were used for intracellular staining: anti-IL17 (Miltenyi Biotec; Cat#130-102-262, Clone: TC11-18H10), anti-IL4 (Miltenyi Biotec; Cat#130-102-435, Clone: BVD4-1D11), and anti-IFN-y (Miltenyi Biotec; Cat#130-102-388, Clone: AN.18.17.24), examined by LSR II cell analyzer (Becton, Dickinson and Company, USA) and analyzed by FlowJo v10.6 software (Becton, Dickinson and Company, USA).
Myeloperoxidase assay
Myeloperoxidase assay was performed using the methods previously described.57 Briefly, intestinal tissues in hexadecyltrimethylammonium bromide (HTAB) buffer was homogenized using Fast Prep-24 (MP). Diluted H2O2 and o-dianisidine dihydrochloride solution were added to the supernatant of homogenized tissue, and absorbance at 450 nm were measured in triplicates using a spectrophotometer, taking three readings at 30-s intervals.
Design of strain-specific primers for H. parainfluenzae strains
Strain-specific primers were designed following the workflow previously described.58 Briefly, raw sequencing reads from whole genome sequencing of H. parainfluenzae strains were aligned to a reference genome (Haemophilus parainfluenzae ATCC 33392) using Bowtie2.59 The unmapped reads were assembled de novo using SPAdes.53 Open reading frames (ORFs) were extracted from the contigs using EMBOSS getorf.60 The extracted ORFs were searched against all bacterial nucleotide sequences in GenBank using BLAST.61 ORFs with BLAST hits were discarded. Unique strain-specific ORFs with no BLAST hits were used to design primers using BatchPrimer3.62 The candidate primer pairs were submitted to PrimerBLAST63 to verify that there were no off-target bacterial sequences. Finally, primers were verified in the wet lab for functionality and specificity through strain cross-validation and melt-curve analysis. The final primer and probe sequence used for this experiment can be found in Table S1.
Quantitative polymerase chain reaction (qPCR)
All reactions were performed on CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad).
Quantification of H. parainfluenzae strains
Probe-based qPCR was performed using PrimeTime™ Gene Expr Master Mix (IDT) using cycling protocol of 1 × 95°C polymerase activation step for 3 min, 45 × 95°C denaturation step for 15 s, and 60°C annealing/extension step for 1 min. Custom gBlocks Gene Fragments (IDT) containing amplicon sequences were designed and used to create a qPCR standard curve for absolute quantification.
Gene expression analysis
RNA was extracted from the proximal colon and distal ileum using Qiagen RNeasy Mini Kit (Qiagen) and treated with DNase (Qiagen) according to the manufacturer’s protocol. Probe-base qPCR assay was performed using Reliance One-Step Multiplex RT-qPCR Supermix (Biorad) and predesigned qPCR primers and probes targeting Dhps (IDT; Assay ID: Mm.PT.58.5299492), Smox (IDT; Assay ID: Mm.PT.58.11011908), and Odc1 (IDT; Assay ID: Mm.PT.58.7815184.g). Tbp (IDT; Assay ID: Mm.PT.39a.22214839) was used for the reference gene. Cycling protocol of 1 × 50°C reverse transcription step for 10 min, 1 × 95°C DNA polymerase activation and template denaturation step for 10 min, 40 × 95°C template denaturation for 10 s and 60°C annealing and extension for 30 s.
Ultra-performance liquid chromatography (UPLC) assay for polyamines
25 mg of homogenized tissue was sonicated in 250 μL of 0.6 M perchloric acid. A Fisher Scientific Sonic Dismembrator model 100 was used to sonicate samples for 2 rounds of 15 s on output 3. Lysed samples were centrifuged at 4°C for 15 min at 10,000 x g, the remaining supernatant was analyzed on the UPLC machine following dansylation. Pellets were stored at −80°C for further protein analysis for normalization of polyamine levels. All polyamine measurements were carried out using an Acquity UPLC BEH Shield RP18 1.7 μm 2.1 × 100 mm column with an RP18 VanGuard Pre-column, 130 Å, 1.7 μm, 2.1 mm × 5 mm on an Acquity UPLC machine in the Bioanalytics, Metabolomics, and Pharmacokinetics Core Facility, at Roswell Park Comprehensive Cancer Center. The column was stored in 65% acetonitrile. A constant flow rate was held at 0.17 mL per minute, with a column temperature of 50°C and sample temperature of 5° C. Buffer A contained 55% 10 mM ammonium acetate at pH 4.4, and 45% HPLC grade acetonitrile. Buffer B was 100% acetonitrile. Dansylated polyamines were eluted with a linear gradient from 100% Buffer A to 18% Buffer A and 82% Buffer Bfor 6 min, which was then held for 3 min. By 10.6 min the conditions returned to 100% Buffer A, which also served to equilibrate the column for the next sample. Standards (polyamines and acetylpolyamines) were also eluted using the same conditions and ranged in concentration from 0.01 to 100 μM. Spike-ins of each standard confirmed the location of the peaks within the samples. Concentrations of polyamines were determined based on standard curves and normalized to protein concentrations.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed in Prism 9 (GraphPad). The statistical test used is indicated in the relevant figure legend.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD16/CD32 Antibody, anti-mouse | BD | Cat#553141; RRID: AB_394656 |
| CD3 Antibody, anti-mouse | Miltenyi Biotec | Cat# 130-120-826; RRID: AB_2752207 |
| CD4 Antibody, anti-mouse | Invitrogen | Cat#25-0041-82; RRID: AB_469576 |
| CD8a Antibody, anti-mouse | Miltenyi Biotec | Cat#130-120-806; RRID: AB_2752203 |
| IL-17A Antibody, anti-mouse | Miltenyi Biotec | Cat#130-102-262; RRID: AB_2660786 |
| IL-4 Antibody, anti-mouse | Miltenyi Biotec | Cat#130-102-435; RRID: AB_2660586 |
| IFN-γ Antibody, anti-mouse | Miltenyi Biotec | Cat#130-102-388; RRID: AB_2659990 |
| Bacterial and virus strains | ||
| Haemophilus parainfluenzae S1p | Isolated from saliva of study participant | N/A |
| Haemophilus parainfluenzae S2 | Isolated from saliva of study participant | N/A |
| Haemophilus parainfluenzae F2 | Isolated from feces of study participant | N/A |
| Haemophilus parainfluenzae F3p | Isolated from feces of study participant | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dextran Sulfate Sodium Salt | Thermo Scientific | AAJ1448922 |
| Critical commercial assays | ||
| Remel Haemophilus Isolation Agar w/Bacitracin and Horse Blood | Thermo Scientific | Cat# R01470 |
| RapID™ NH System | Thermo Scientific | Cat# R8311001 |
| Deposited data | ||
| Whole genome sequences of Haemophilus parainfluenzae strains | This study | NCBI SRA:PRJNA804169 |
| 16S rRNA sequence of Crohn disease patients | This study | NCBI SRA:PRJNA831448 |
| Experimental models: Organisms/strains | ||
| BALB/cJ | Jackson Laboratory | RRID:IMSR_JAX:000651 |
| C.129P2(B6)-Il10 < tm1Cgn>/J | Jackson Laboratory | RRID:IMSR_JAX:004333 |
| Oligonucleotides | ||
| Dhps | IDT | Mm.PT.58.5299492 |
| Smox | IDT | Mm.PT.58.11011908 |
| Odc1 | IDT | Mm.PT.58.7815184.g |
| Tbp | IDT | Mm.PT.39a.22214839 |
| Primer for Haemophilus parainfluenzae S1p strain (see Table S1) | This study | N/A |
| Primer for Haemophilus parainfluenzae S2 strain (see Table S1) | This study | N/A |
| Primer for Haemophilus parainfluenzae F2 strain (see Table S1) | This study | N/A |
| Primer for Haemophilus parainfluenzae S3p strain (see Table S1) | This study | N/A |
| Software and algorithms | ||
| QIIME2 2020.2 | https://qiime2.org/ | N/A |
| Oligotyping | https://merenlab.org/software/oligotyping/ | N/A |
| Sybil | https://sybil.sourceforge.net/ | N/A |
Highlights.
Haemophilus parainfluenzae elicits strain-dependent intestinal inflammation
Gut colonization of H. parainfluenzae is associated with increased IFN-γ+ CD4+ T cells
Periodontitis is associated with increased H. parainfluenzae in patients with Crohn’s disease
ACKNOWLEDGMENTS
The authors would like to first acknowledge the contributions of Dr. Robert J. Genco, who initially led the project but sadly passed away before completion. The authors would also like to thank the staff members of the University at Buffalo’s Periodontal Disease Research Center, Optical Imaging and Analysis Facility, Laboratory Animal Facilities, and Genomics and Bioinformatics Core. Finally, the authors would like to thank the study participants. This work was supported by National Institutes of Health (NIH) grants R01DE028258 (K.L.K.), R01DE028258S1 (K.L.K.), K18DE029526 (K.L.K.), R01AI125982 (Y.S.), and R01CA241123 (Y.S.) and the Sunstar Group (P.I.D.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112120.
REFERENCES
- 1.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. (2014). The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392. 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schirmer M, Denson L, Vlamakis H, Franzosa EA, Thomas S, Gotman NM, Rufo P, Baker SS, Sauer C, Markowitz J, et al. (2018). Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe 24, 600–610.e4. 10.1016/j.chom.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kansal S, Catto-Smith AG, Boniface K, Thomas S, Cameron DJ, Oliver M, Alex G, Kirkwood CD, Wagner J, and Wagner J (2019). The microbiome in paediatric Crohn’s disease—a longitudinal, prospective, single-centre study. J. Crohns Colitis 13, 1044–1054. 10.1093/ecco-jcc/jjz016. [DOI] [PubMed] [Google Scholar]
- 5.Putignani L, Oliva S, Isoldi S, Del Chierico F, Carissimi C, Laudadio I, Cucchiara S, and Stronati L (2021). Fecal and mucosal microbiota profiling in pediatric inflammatory bowel diseases. Eur. J. Gastroenterol. Hepatol 33, 1376–1386. 10.1097/meg.0000000000002050. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro JM, de Zoete MR, Palm NW, Laenen Y, Bright R, Mallette M, Bu K, Bielecka AA, Xu F, Hurtado-Lorenzo A, et al. (2021). Immunoglobulin A targets a unique subset of the microbiota in inflammatory bowel disease. Cell Host Microbe 29, 83–93.e3. 10.1016/j.chom.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sohn J, Sun Y, Genco RJ, and Kirkwood KL (2020). The periodontal microenvironment: a potential reservoir for intestinal pathobionts in Crohn’s disease. Curr. Oral Health Rep 7, 37–44. 10.1007/s40496-020-00251-9. [DOI] [Google Scholar]
- 8.She Y.-y., Kong X.-b., Ge Y.-p., Liu Z.-y., Chen J.-y., Jiang J.-w., Jiang H.-b., and Fang S.-l. (2020). Periodontitis and inflammatory bowel disease: a meta-analysis. BMC Oral Health 20, 67. 10.1186/s12903-020-1053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Qiao D, Chen R, Zhu F, Gong J, and Yan F (2021). The association between periodontitis and inflammatory bowel disease: a systematic Review and meta-analysis. BioMed Res. Int 2021, 6692420. 10.1155/2021/6692420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imai J, Ichikawa H, Kitamoto S, Golob JL, Kaneko M, Nagata J, Takahashi M, Gillilland MG III, Tanaka R, Nagao-Kitamoto H, et al. (2021). A potential pathogenic association between periodontal disease and Crohn’s disease. JCI Insight 6, e148543. 10.1172/jci.insight.148543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan CXW, Brand HS, Kalender B, De Boer NKH, Forouzanfar T, and de Visscher JGAM (2021). Dental and periodontal disease in patients with inflammatory bowel disease. Clin. Oral Investig 25, 5273–5280. 10.1007/s00784-021-03835-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brito F, de Barros FC, Zaltman C, Carvalho ATP, Carneiro A.J.d.V., Fischer RG, Gustafsson A, and Figueredo C.M.d.S. (2008). Prevalence of periodontitis and DMFT index in patients with Crohn’s disease and ulcerative colitis. J. Clin. Periodontol 35, 555–560. 10.1111/j.1600-051X.2008.01231.x. [DOI] [PubMed] [Google Scholar]
- 13.Habashneh RA, Khader YS, Alhumouz MK, Jadallah K, and Ajlouni Y (2012). The association between inflammatory bowel disease and periodontitis among Jordanians: a case-control study. J. Periodontal. Res 47, 293–298. 10.1111/j.1600-0765.2011.01431.x. [DOI] [PubMed] [Google Scholar]
- 14.Liu G, Luan Q, Chen F, Chen Z, Zhang Q, and Yu X (2018). Shift in the subgingival microbiome following scaling and root planing in generalized aggressive periodontitis. J. Clin. Periodontol 45, 440–452. 10.1111/jcpe.12862. [DOI] [PubMed] [Google Scholar]
- 15.Genco RJ, LaMonte MJ, McSkimming DI, Buck MJ, Li L, Hovey KM, Andrews CA, Sun Y, Tsompana M, Zheng W, et al. (2019). The subgingival microbiome relationship to periodontal disease in older women. J. Dent. Res 98, 975–984. 10.1177/0022034519860449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawamoto D, Borges R, Ribeiro RA, de Souza RF, Amado PPP, Saraiva L, Horliana ACRT, Faveri M, and Mayer MPA (2021). Oral dysbiosis in severe forms of periodontitis is associated with gut dysbiosis and correlated with salivary inflammatory mediators: a preliminary study. Front. Oral Health 2, 722495. 10.3389/froh.2021.722495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lourenςo TGB, Spencer SJ, Alm EJ, and Colombo APV (2018). Defining the gut microbiota in individuals with periodontal diseases: an exploratory study. J. Oral Microbiol 10, 1487741. 10.1080/20002297.2018.1487741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Lu H, Wu H, Huang S, Chen L, Gui Q, Zhou W, Yang Y, Wu Y, Zhang H, et al. (2020). Periodontitis in elderly patients with type 2 diabetes mellitus: impact on gut microbiota and systemic inflammation. Aging 12, 25956–25980. 10.18632/aging.202174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Utter DR, Borisy GG, Eren AM, Cavanaugh CM, and Mark Welch JL (2020). Metapangenomics of the oral microbiome provides insights into habitat adaptation and cultivar diversity. Genome Biol. 21, 293. 10.1186/s13059-020-02200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eren AM, Borisy GG, Huse SM, and Mark Welch JL (2014). Oligotyping analysis of the human oral microbiome. Proc. Natl. Acad. Sci. USA 111, E2875–E2884. 10.1073/pnas.1409644111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor DC, Cripps AW, Clancy RL, Murree-Allen K, Hensley MJ, Saunders NA, and Sutherland DC (1992). Biotypes of Haemophilus parainfluenzae from the respiratory secretions in chronic bronchitis. J. Med. Microbiol 36, 279–282. 10.1099/00222615-36-4-279. [DOI] [PubMed] [Google Scholar]
- 22.Puleston DJ, Baixauli F, Sanin DE, Edwards-Hicks J, Villa M, Kabat AM, Kamiński MM, Stanckzak M, Weiss HJ, Grzes KM, et al. (2021). Polyamine metabolism is a central determinant of helper T cell lineage fidelity. Cell 184, 4186–4202.e20. 10.1016/j.cell.2021.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tome ME, Fiser SM, Payne CM, and Gerner EW (1997). Excess putrescine accumulation inhibits the formation of modified eukaryotic initiation factor 5A (eIF-5A) and induces apoptosis. Biochem. J 328, 847–854. 10.1042/bj3280847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Said HS, Suda W, Nakagome S, Chinen H, Oshima K, Kim S, Kimura R, Iraha A, Ishida H, Fujita J, et al. (2014). Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 21, 15–25. 10.1093/dnares/dst037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu S, Png E, Gowans M, Ong DEH, de Sessions PF, Song J, and Nagarajan N (2021). Ectopic gut colonization: a metagenomic study of the oral and gut microbiome in Crohn’s disease. Gut Pathog. 13, 13. 10.1186/s13099-021-00409-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vavricka SR, Manser CN, Hediger S, Vögelin M, Scharl M, Biedermann L, Rogler S, Seibold F, Sanderink R, Attin T, et al. (2013). Periodontitis and gingivitis in inflammatory bowel disease: a case-control study. Inflamm. Bowel Dis 19, 2768–2777. 10.1097/01.MIB.0000438356.84263.3b. [DOI] [PubMed] [Google Scholar]
- 27.Kowalska-Duplaga K, Gosiewski T, Kapusta P, Sroka-Oleksiak A, Wędrychowicz A, Pieczarkowski S, Ludwig-Słomczyńska AH, Wołŗkow PP, and Fyderek K (2019). Differences in the intestinal microbiome of healthy children and patients with newly diagnosed Crohn’s disease. Sci. Rep 9, 18880. 10.1038/s41598-019-55290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu C, Zhao Q, Deng J, Chen K, Jiang X, Ma F, Ma S, and Li Z (2022). Salivary microbiome profile of diabetes and periodontitis in a Chinese population. Front. Cell. Infect. Microbiol 12, 933833. 10.3389/fcimb.2022.933833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diao J, Yuan C, Tong P, Ma Z, Sun X, and Zheng S (2021). Potential roles of the free salivary microbiome dysbiosis in periodontal diseases. Front. Cell. Infect. Microbiol 11, 711282. 10.3389/fcimb.2021.711282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Read E, Curtis MA, and Neves JF (2021). The role of oral bacteria in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol 18, 731–742. 10.1038/s41575-021-00488-4. [DOI] [PubMed] [Google Scholar]
- 31.Abed J, Maalouf N, Manson AL, Earl AM, Parhi L, Emgård JEM, Klutstein M, Tayeb S, Almogy G, Atlan KA, et al. (2020). Colon cancer-associated fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front. Cell. Infect. Microbiol 10, 400. 10.3389/fcimb.2020.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shah P, and Swiatlo E (2008). A multifaceted role for polyamines in bacterial pathogens. Mol. Microbiol 68, 4–16. 10.1111/j.1365-2958.2008.06126.x. [DOI] [PubMed] [Google Scholar]
- 33.Keubler LM, Buettner M, Häger C, and Bleich A (2015). A multihit model: colitis lessons from the interleukin-10-deficient mouse. Inflamm. Bowel Dis 21, 1967–1975. 10.1097/mib.0000000000000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kühn R, Löhler J, Rennick D, Rajewsky K, and Müller W (1993). Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274. 10.1016/0092-8674(93)80068-. [DOI] [PubMed] [Google Scholar]
- 35.Maharshak N, Packey CD, Ellermann M, Manick S, Siddle JP, Huh EY, Plevy S, Sartor RB, and Carroll IM (2013). Altered enteric microbiota ecology in interleukin 10-deficient mice during development and progression of intestinal inflammation. Gut Microb. 4, 316–324. 10.4161/gmic.25486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, and Gordon JI (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med 1, 6ra14–16ra14. 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imam T, Park S, Kaplan MH, and Olson MR (2018). Effector T helper cell subsets in inflammatory bowel diseases. Front. Immunol 9, 1212. 10.3389/fimmu.2018.01212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harbour SN, Maynard CL, Zindl CL, Schoeb TR, and Weaver CT (2015). Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 112, 7061–7066. 10.1073/pnas.1415675112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakajima-Adachi H, Shibahara K, Fujimura Y, Takeyama J, Hiraide E, Kikuchi A, Murakami H, Hosono A, Nochi T, Wakatsuki Y, et al. (2017). Critical role of intestinal interleukin-4 modulating regulatory T cells for desensitization, tolerance, and inflammation of food allergy. PLoS One 12, e0172795. 10.1371/journal.pone.0172795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, Kiguchi Y, Yasuma K, Watanabe E, Tanoue T, et al. (2017). Ectopic colonization of oral bacteria in the intestine drives T(H)1 cell induction and inflammation. Science 358, 359–365. 10.1126/science.aan4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grosheva I, Zheng D, Levy M, Polansky O, Lichtenstein A, Golani O, Dori-Bachash M, Moresi C, Shapiro H, Del Mare-Roumani S, et al. (2020). High-Throughput screen identifies host and microbiota regulators of intestinal barrier function. Gastroenterology 159, 1807–1823. 10.1053/j.gastro.2020.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Liu B, Jiang X, Cai L, Zhao X, Dai Z, Wu G, and Li X (2019). Putrescine mitigates intestinal atrophy through suppressing inflammatory response in weanling piglets. J. Anim. Sci. Biotechnol 10, 69. 10.1186/s40104-019-0379-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura A, Kurihara S, Takahashi D, Ohashi W, Nakamura Y, Kimura S, Onuki M, Kume A, Sasazawa Y, Furusawa Y, et al. (2021). Symbiotic polyamine metabolism regulates epithelial proliferation and macrophage differentiation in the colon. Nat. Commun 12, 2105. 10.1038/s41467-021-22212-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sohn J, Li L, Zhang L, Settem PR, Honma K, Sharma A, Falkner KL, Novak JM, Sun Y, and Kirkwood KL (2022). Porphyromonas gingivalis indirectly elicits intestinal inflammation by altering the gut microbiota and disrupting epithelial barrier function through IL9-producing CD4+ T cells. Mol. Oral Microbiol 37, 42–52. 10.1111/omi.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG 3rd, Hayashi A, Imai J, Sugihara K, Miyoshi M, Brazil JC, Kuffa P, et al. (2020). The intermucosal connection between the mouth and gut in commensal pathobiont-driven colitis. Cell 182, 447–462.e14. 10.1016/j.cell.2020.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Satsangi J, Silverberg MS, Vermeire S, and Colombel JF (2006). The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut 55, 749–753. 10.1136/gut.2005.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eke PI, Page RC, Wei L, Thornton-Evans G, and Genco RJ (2012). Update of the case definitions for population-based surveillance of periodontitis. J. Periodontol 83, 1449–1454. 10.1902/jop.2012.110664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37, 852–857. 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, and Gregory Caporaso J (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90. 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, and Peddada SD (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis 26, 27663. 10.3402/mehd.v26.27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krueger F, and Trim Galore. A Wrapper Tool Around Cutadapt and FastQC to consistently Apply Quality and Adapter Trimming to FastQ Files. 2015. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/. [Google Scholar]
- 53.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol 19, 455–477. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seemann T (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 55.Riley DR, Angiuoli SV, Crabtree J, Dunning Hotopp JC, and Tettelin H (2012). Using Sybil for interactive comparative genomics of microbes on the web. Bioinformatics 28, 160–166. 10.1093/bioinformatics/btr652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Erben U, Loddenkemper C, Doerfel K, Spieckermann S, Haller D, Heimesaat MM, Zeitz M, Siegmund B, and Kühl AA (2014). A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol 7, 4557–4576. [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JJ, Shajib MS, Manocha MM, and Khan WI (2012). Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp, 3678. 10.3791/3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hernández I, Sant C, Martínez R, and Fernández C (2020). Design of bacterial strain-specific qPCR assays using NGS data and publicly available resources and its application to track biocontrol strains. Front. Microbiol 11, 208. 10.3389/fmicb.2020.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rice P, Longden I, and Bleasby A (2000). EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277. 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 61.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 62.You FM, Huo N, Gu YQ, Luo M.-c., Ma Y, Hane D, Lazo GR, Dvorak J, and Anderson OD (2008). BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 9, 253. 10.1186/1471-2105-9-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, and Madden TL (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134. 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available in the NCBI SRA:PRJNA804169, PRJNA831448. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.