

Abstract
Regulated cell death (RCD) is essential for successful systemic cancer therapy. Yet, the engagement of RCD pathways does not inevitably result in cell death. Instead, RCD pathways can take part in diverse biological processes if the cells survive. Consequently, these surviving cells, for which we propose the term ‘flatliners’, harbor important functions. These evolutionarily conserved responses can be exploited by cancer cells to promote their own survival and growth, with challenges and opportunities for cancer therapy.
Introduction
The term regulated cell death (RCD), occurring as a result of a molecular pathway, has replaced “programmed cell death,” as the latter was specifically coined to refer to cell death that occurs at defined times during development1. RCD shapes the physiological development of tissues and organs, maintains homeostasis, and plays various roles in multiple disease processes2,3. The molecular machineries of RCD can be initiated by diverse mechanical, biological, physical, and chemical stresses, and are influenced by various cell intrinsic and extrinsic signals. This is opposed to “accidental” cell death, which is an immediate fatal response to severe physical, mechanical, or chemical damage, and is often referred to as “necrosis”4. The molecular events of RCD pathways and the resulting phenotypic changes have been related to various clinical outcomes, particularly in the context of cancer.
While evading cell death has been identified as a hallmark of cancer5, this is often mis-interpreted as suggesting that cancer cells are resistant to RCD. This is incorrect, as highlighted by studies showing that cancer cells can be “primed for death,” and the response to conventional therapy correlates with such priming6. It is therefore understood that a hallmark of cancer is an ability of the cell to evade those RCD mechanisms that are engaged to suppress the oncogenic process, not necessarily RCD in general.
The engagement of RCD does not inevitably result in cell death. Cells that survive the activation of an RCD pathway can undergo changes that influence their behavior and/or that of surrounding cells. These may include genomic instability leading to high mutational burden7,8 and pro- or anti-tumor immune responses9. Moreover, preclinical research provides evidence that sublethal engagement of RCD leads to phenotypic adaptations including epithelial mesenchymal transition (EMT) and altered interaction within the cell’s microenvironment10–13. Consequently, sublethal engagement of cell death contributes to metastasis, invasiveness, and therapy-unresponsiveness, but can also present vulnerabilities that might be harnessed for cancer treatment13–16.
Herein, we introduce the term “flatliner” to represent a cell that has engaged a core RCD mechanism but manages to survive, in analogy to a patient who “flatlines” but is resuscitated. For cells, this is distinct from resistance to or evasion from signals that normally induce cell death. We propose that engaged cell death pathways do not always lead to cell death, and flatliners that survive may have altered properties. We elaborate how cancer cells resist therapy, with particular focus on the molecular mechanisms that underly the evasion of cell death following activation of an RCD pathway, and how survival of flatliners may account for phenomena associated with cancer persistence. Further, we assess how our knowledge and recent advances in the cell death field translate into our understanding of cancer progression and relapse, and how this may undercover novel therapeutic opportunities.
Finally, we discuss the potential relationship of flatliners to drug-tolerant persister cells, characterized as cancer cells without resistance-associated mutations that survive treatment14,17. These definitions are distinct: while both processes are transient (cells revert to the parental drug sensitivity over time), flatliners have demonstrably engaged a core cell death pathway and survived, while persister cells are defined only by their transient drug-tolerant state. While we argue that, at least in some cases, engagement of a core cell death machinery can induce the persister cell phenotype, the distinct definitions of each are important.
RCD pathways in cancer
In the following section we briefly survey four common RCD pathways: apoptosis, necroptosis, pyroptosis and ferroptosis, and discuss the molecular mechanisms that underlie their evasion.
Apoptosis
Apoptosis refers to cell death associated with the activation of Cysteine-Aspartate proteases (caspases) mediating cleavage of target proteins, leading to fragmentation of cellular DNA, nuclear condensation, membrane blebbing, and rapid clearance prior to loss of plasma membrane integrity18. The biochemical and morphological changes associated with apoptosis are orchestrated by the activity of the executioner caspases, caspase-3 and −7, which cleave hundreds of substrates, including those responsible for the changes mentioned above19,20. For example, inter-nucleosomal double strand DNA breaks during apoptosis are caused by the caspase-activated nuclease (CAD). Another protein, inhibitor of CAD (iCAD) acts as a chaperone to bind and inhibit CAD while folding it into an active nuclease. The executioner caspases cleave iCAD, allowing the active CAD to cause DNA fragmentation21.
The executioner caspases are activated by initiator caspases (e.g., caspase-8 and −9), which cleave the inactive, dimeric executioners to activate them. The initiator caspases are not activated by cleavage, but instead, by binding and oligomerization of the inactive monomers on activated adapter proteins. These adapters and initiator caspases define the apoptotic pathways (Figure 1).
Figure 1: Cell death pathways.
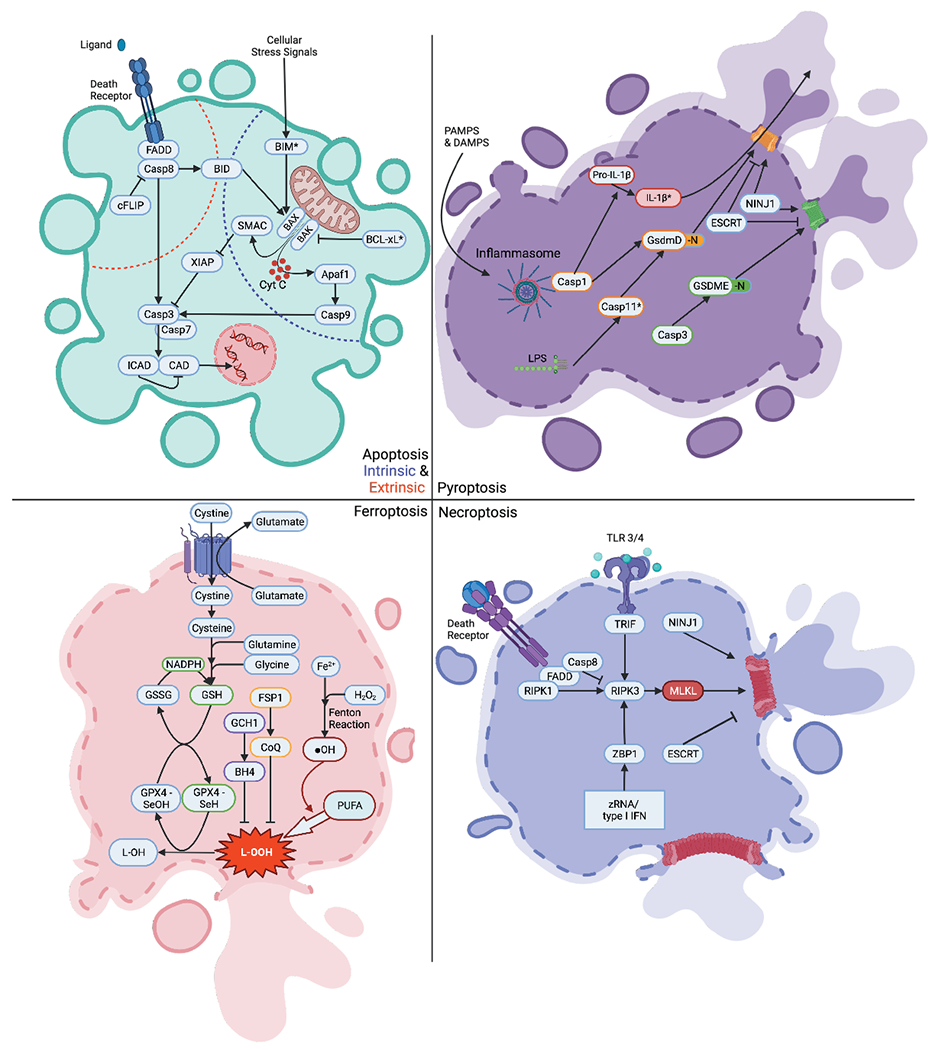
Intrinsic Apoptosis: Upon cellular stress, Bax & Bak permeabilize the outer membrane of the mitochondria releasing many proteins including cytochrome c and SMAC into the cytoplasm. Pro-survival BCL-2 family members (represented by BCL-XL*) can prevent MOMP. Upon the release of cytochrome c, Apaf-1 binds to initiator caspase (caspase-9) forming the “apoptosome”. This apoptosome then cleaves the executioner caspases (caspase-3 and −7) leading the proteolytic cleavage of many substrates including the ICAD which activates CAD. CAD then cleaves nuclear DNA and induces apoptotic cell death. Additionally, MOMP-induced release of SMAC inhibits XIAP and thereby releases the break on caspase-3. Extrinsic Apoptosis: After activation of a death receptor (e.g. FAS), the FADD adaptor recruits and activates caspase-8. Activated caspase-8, if not inhibited by cFLIP binding, then directly cleaves caspase-3 and induces cell death as described above. Additionally, caspase-8 can also cleave BID which induces MOMP directly or by activation of BAX. Pyroptosis: Direct binding of LPS to inflammatory caspase-11 (caspase 4,5 in humans) leads to cleavage of GsdmD releasing the N-terminus (GsdmD-N). The GsdmD-N then forms an oligomer on phospholipid-membranes, permeabilizing it and inducing pyroptotic cell death. Alternatively, PAMPs and DAMPs can activate the inflammasome which can mediate inflammatory caspase-1 cleavage of GsdmD termed the “canonical inflammasome” pathway. Caspase-1 can also process Pro-IL-1β into IL-1β which is released via GsdmD pores. In addition to GsdmD-dependent pyroptosis, GsdmE, after cleavage from executioner caspase-3, can also lead to pore formation via it’s released N-terminal fragment (GsdmE-N). Both GsdmD and GsdmE pores can be repaired via the ESCRT complex. NINJ1 mediates cell rupture. Necroptosis: Upon stimulation of a death receptor, RIPK1 is activated and forms an oligomer with RIPK3, activating it (if Caspase 8 is inhibited). RIPK3 then phosphorylates MLKL which oligomerizes and forms a pore in the plasma membrane. Alternatively, activation of ZBP1 or TRIF can also activate RIPK3 leading to necroptosis. ESCRT proteins can repair MLKL pores. NINJ1 mediates cell rupture. Ferroptosis: Fe2+ reacts with hydrogen peroxide during fenton reaction, thereby generating hydroxyl radicals (•OH), which oxidize PUFAs. The resulting toxic oxidized lipids (L-OOH) can integrate into phospholipid membranes and thereby lead to necrotic cell death. The selenoenzyme GPX4 can directly detoxify L-OOH. Glutathione reduces GPX4 to replenish the antioxidant pool, thereby getting oxidized itself into glutathione disulfate (GSSG). NADPH from the pentose phosphate pathway reduces GSH. The xc– transporter exports glutamate and imports cystine into the cytosol. Cystine is then converted into cysteine, and then synthesized together with glutamine and glycine to glutathione. BH4, as produced by GCH1, is also able to decrease levels of oxidized lipids. FSP1 is able to generate CoQ which acts as an antioxidant capable of inhibiting ferroptosis.
* Bcl-xL is shown as a representative of “pro-survival” BCL-2 family members
* Caspase-11 in mice, Caspase-4,5 in humans.
In the mitochondrial, or intrinsic pathway of apoptosis, the adapter is apoptotic protease activating factor-1 (APAF1), which binds and thereby activates the initiator caspase, caspase-9, which in turn cleaves and thereby activates the executioner caspases19. The latter are inhibited, however, by X-linked inhibitor of apoptosis (XIAP). The activation of APAF1 occurs following mitochondrial outer membrane permeabilization (MOMP), releasing cytochrome c from the mitochondrial inter-membrane space, which induces the activation and oligomerization of APAF1. In addition, proteins that interfere with XIAP are also released upon MOMP, de-repressing the executioner caspases and allowing apoptosis to proceed22. Extensive MOMP in a cell can result in a mitochondrial energetic catastrophe that usually ends in cell death even if the executioner caspase activation is insufficient23.
MOMP is caused by the action of the pro-apoptotic effectors of the BCL-2 family, e.g. BAX and BAK. These are antagonized by the anti-apoptotic BCL-2 proteins, e.g., BCL-2, BCL-XL, and MCL-1. A third type of BCL-2 protein, the BH3-only proteins function to inhibit the anti-apoptotic proteins and/or activate the pro-apoptotic effectors. The functions of the BCL-2 proteins have been reviewed elsewhere24. Engagement of apoptosis in cancer cells is triggered by many chemotherapeutic drugs and radiation therapies, which results in the activation of BAX and BAK through increased function of BH3-only proteins and decreased anti-apoptotic BCL-2 protein function25. Cells that are poised to undergo MOMP (“primed for death”) are associated with better prognosis in response to conventional therapy6 and dynamic BH3 profiling, in which drugs are tested for their ability to prime cells for induction of MOMP by BH3 peptides, has shown promise in predicting therapeutic efficacy26.
One way for cancer cells to survive in the face of chemotherapeutic insult is to prevent engagement of the cell death pathways via mutation or other mechanisms that permanently disrupt the action of the drug, referred to as resistance. This is distinct from survival following engagement of such a pathway and is not considered further herein. Previously, MOMP was widely considered a “point of no return” for cells, but we now know that cells can survive a degree of MOMP and sublethal caspase activation16,27 (Figure 2). It has been observed that not all mitochondria necessarily release cytochrome c upon stimulation with chemotherapeutic agents (incomplete MOMP; iMOMP). iMOMP allows for repopulation of cells with healthy mitochondria and increased clonogenic survival once the death-inducing stress is removed. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) overexpression has also been shown to promote clonogenic survival in cells displaying iMOMP promoting increased glycolysis and a transient increase in mitochondrial mass28. In another study, the repair of double-strand DNA breaks induced upon caspase-mediated activation of CAD was noted as a requirement for cancer cell survival8. Widespread MOMP, followed by extensive apoptotic caspase activation normally is lethal, but cells that engage the mitochondrial apoptosis pathway can survive.
Figure 2: Apoptosis induction and survival.
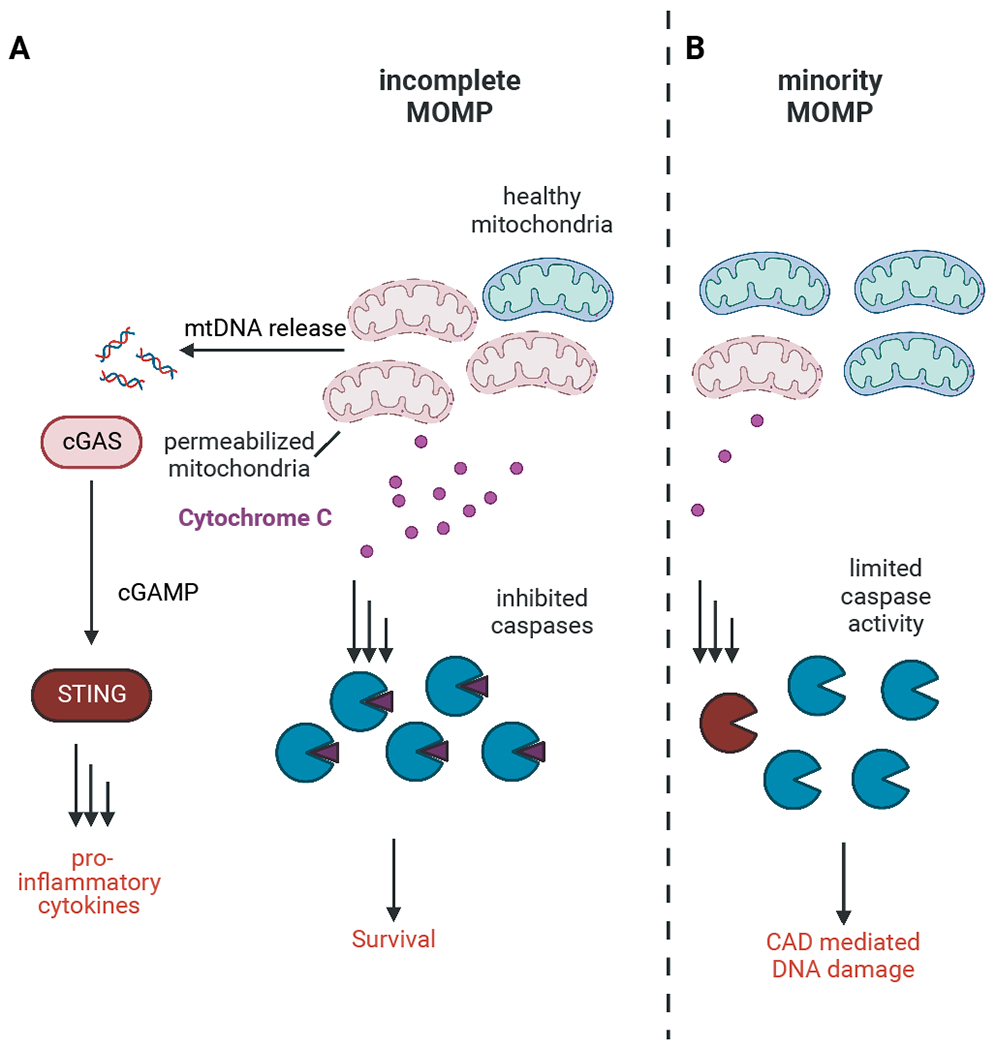
A If apoptotic caspases are inhibited induction of incomplete MOMP leads to release of mitochondrial DNA from permeabilized mitochondria. This stimulated activation of the cGAS-STING pathway and production of pro-inflammatory cytokines. Cells harboring enough healthy mitochondria can expand these mitochondria and survive even after engagement of apoptosis.
B Low levels of MOMP (miMOMP) engage sublethal levels of caspase activation, which can mediate DNA damage through activation of CAD.
A second pathway of apoptosis involves the activation of the initiator caspase, caspase-8, by its adapter, FADD. This can occur upon ligation of death receptors of the TNF receptor family, e.g. TNFR1, FAS (CD95), and TRAIL receptors, and is often referred to as the death receptor, or extrinsic pathway of apoptosis (although other, intrinsic mechanisms, including non-death receptor processes exist to activate FADD-caspase-8 29). Active caspase-8 cleaves and thereby activates the executioner caspases.
Flice-like inhibitor of apoptosis, cFLIPL, (herein, FLIP) resembles caspase-8 but lacks a catalytic cysteine. If FLIP is present, it binds to a monomer of FADD-bound caspase-8, preventing oligomerization of the latter and thus prevents apoptosis30. However, the caspase-8-FLIP heterodimer is proteolytically active and performs other functions, as discussed below. The extent to which caspase-8 and death receptor signaling contribute to cancer is not well understood.
The death receptor pathway of apoptosis appears to be an important mechanism for anti-tumor immunity, as cytotoxic lymphocytes deploy death receptor ligands (FAS/CD95, TRAIL) as one way they kill cancer cells. Caspase-8 is mutated or silenced in some cancers31. However, whether this represents an immune evasion mechanism or an escape from a tumor suppressor mechanism is not clear. Intriguingly, most cancers express FAS32 and many express receptors for TRAIL33, suggesting that such receptors may have roles beyond cell death that are important in cancer maintenance34.
Cells that activate the death receptor pathway can survive, suggesting that low levels of executioner caspase activation are tolerated35. Survival following engagement of apoptosis and activation of executioner caspases has been termed anastasis (defined as cell survival despite activation of executioner caspases)36. Using a fluorescent marker of caspase-mediated cleavage, studies in flies indicated that many cells in the developing animals display evidence of caspase activation without apparent cell death37. Studies in primary and transformed mammalian cells revealed features of anastasis following induction of apoptosis, including DNA damage, oncogenic transformation, and induced gene signatures36. Although MOMP was not assessed in these studies, evidence (discussed above) strongly suggests that cells can survive MOMP, and thus might be considered to have undergone anastasis15,16.
Necroptosis
While necrosis often refers to uncontrolled cell death, we now recognize that there are regulated forms of necrosis. Among these is necroptosis38, in which receptor-interacting kinase-3 (RIPK3) phosphorylates mixed-lineage kinase-like (MLKL) (Figure 1), which then oligomerizes and incorporates into the cell membrane forming a large pore, inducing necroptosis. Three proteins are known to bind and thereby activate RIPK3: RIPK1, TRIF, and ZBP139. These, in turn, are activated by ligation of death receptors, some toll-like receptors, and interferon receptors, respectively. RIPK1 however, associates not only with RIPK3 but also with FADD, and in the absence of FLIP, can cause apoptosis via activation of caspase-8 40. If FLIP is present, apoptosis is blocked, while the catalytic activity of FADD-caspase-8-FLIP cleaves RIPK1 and associated RIPK3, preventing necroptosis. Inhibition of caspase-8 or genetic ablation of either FADD or caspase-8 allows necroptosis to proceed. Such inhibition can occur due to viral caspase inhibitors41. That necroptosis proceeds in the presence of intact caspase-8 signaling has been noted in pathological settings, but the precise mechanisms remain unclear.
Multiple key necroptotic proteins are downregulated in various cancers. MLKL expression is often decreased in AML42, while RIPK3 protein levels are decreased in breast cancer and colorectal cancer and low levels correlate with worse overall survival in colorectal cancer43,44. Further, RIPK1, RIPK3, and phosphorylated MLKL levels correlate with better overall survival and CD8+ T cell infiltration in hepatocellular carcinoma45. Conversely, cancer cells can apparently induce necroptosis in host endothelial cells via DR6 and thereby promote metastasis46.
Negative regulation of necroptosis occurs via post-translational modifications on necroptotic proteins, reviewed elsewhere47. In addition, cells can delay or prevent necroptosis through membrane repair mechanisms. One membrane repair mechanism is mediated by endosomal sorting complexes required for transport (ESCRT) proteins. The ESCRT subcomplexes (ESCRT-0, -I, -II, -III) are the only machinery known in eukaryotic cells that can deform membrane on the distal side48. This machinery has multiple functions in the cell in addition to membrane repair49. Recruitment of the ESCRT-III machinery mediates abscission of the damaged plasma membrane to restore plasma membrane integrity. Cells undergoing necroptosis were able to survive in the presence of ESCRT-mediated membrane repair when the necroptotic stimulus was removed (Figure 3), whereas if ESCRT activity was blocked, cells which had activated MLKL were not able to survive (i.e., become necroptotic flatliners)50. This prolonged survival was necessary for production of cytokines and allowed for enhanced CD8 T-cell cross-priming by necroptotic tumor cells in vivo10,12.
Figure 3: ESCRT mediated membrane repair of plasma membrane damage.
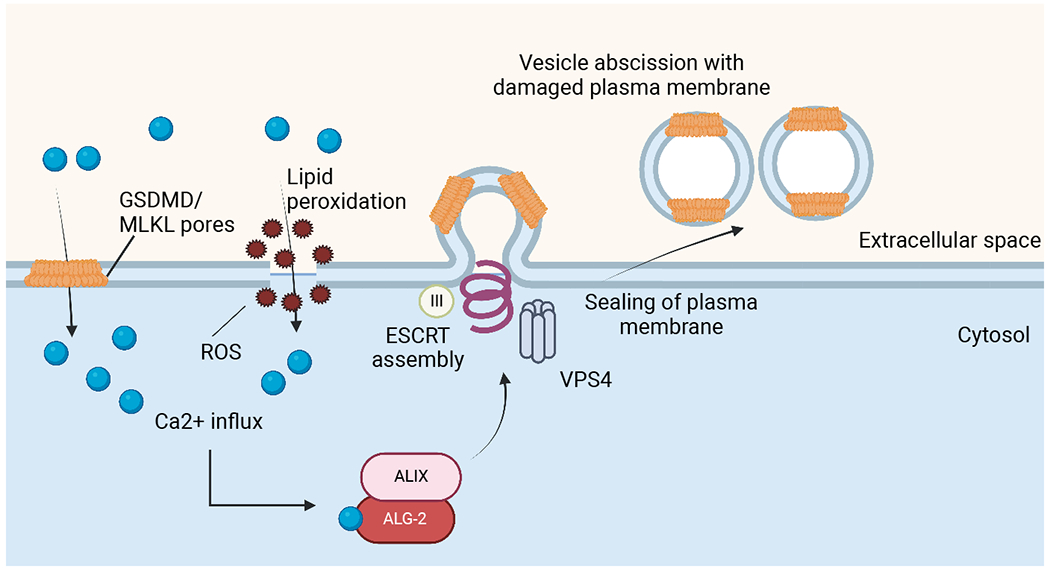
Calcium influx through gasdermin D or MLKL pores as well as plasma membrane (PM) damaged by lipid peroxidation leads to recruitment of ALIX through ALG-2. This promotes assembly of the ESCRT-III machinery at the site of calcium influx. Subsequently, VPS4 mediates abscission of the damaged pieces of PM, restoring its integrity.
Pyroptosis
Pyroptosis refers to a lytic cell death driven by inflammatory caspase activation leading to gasdermin-dependent pore formation in the plasma membrane, mostly in the context of infection and inflammation51 (Figure 1). Certain bacterial components such as cytosolic LPS can directly activate the inflammatory caspase 11 (caspase-4 and caspase-5 in humans), while other stimuli can cause the formation of various inflammasomes that can activate caspase-152. Gasdermin D was identified as the crucial cleaved substrate downstream of caspase-11, and caspase-1, which mediates pore formation in the plasma membrane and initiates lytic cell death51. Other gasdermin family members have been reported to be activated by caspase-3 and −751, caspase-853, Granzyme A54, or a streptococcal exotoxin55,56. The cleaved gasdermin then inserts into the plasma membrane creating pores57,58 which, in the case of Gasdermin D, selectively release mature IL-1 cytokines, which are cleaved by active caspase-151,58.
Gasdermin E, which is activated by caspase-3 and -7, was identified as a tumor suppressor downregulated in many tumors59–61. Chemotherapeutic drugs are able to activate Gasdermin E via caspase-3-dependent cleavage to induce cancer cell death60. Induction of pyroptosis in tumor cells promoted induction of a robust anti-tumor immune response and protective immunity against a re-challenge with the same tumor cells61,62. While gasdermin-mediated pore formation leads to water influx and collapse of the ionic gradient across the plasma membrane, rupture of the cells and the release of large cytosolic molecules is mediated by ninjurin-1 (NINJ1)63. Plasma membrane rupture (PMR) occurs downstream of gasdermin or MLKL activation as well as late after apoptosis induction upon cleavage of Gasdermin E by executioner caspases (also called secondary necrosis). Deletion of NINJ1 delays PMR after MLKL activation but does not block it. The mechanisms of how NINJ1 is activated and induces PMR are unknown63. It is possible that cells lacking NINJ1 can survive the activation of gasdermins (and thus, pyroptosis), although this has not been formally tested.
Gasdermin D-mediated pore formation during pyroptosis is also regulated by ESCRT-mediated membrane repair. Calcium flux through gasdermin pores recruits the ESCRT machinery to the site of pore formation where membrane pieces containing pores are shed to repair the plasma membrane, prolonging survival. ESCRT-deficient macrophages secreted more pro-inflammatory cytokines upon activation of caspase-1 compared to membrane repair proficient cells64. This mechanism can be exploited to increase anti-tumor immunity. Co-delivery of gasdermin and a calcium-chelating agent (to inhibit membrane repair) markedly increased efficacy of PD1-blocking antibodies in various murine tumor models65.
The induction of pyroptosis may be an avenue towards therapeutic treatment of AML66. Further, evidence suggests that the activation of Gasdermin E promotes effective anti-cancer immunity62. One study suggests that one of the inflammasomes (NLRP3) is active in glucocorticoid-resistant B-ALL67. The active Caspase-1 cleaves and thereby inactivates the glucocorticoid receptor, rendering the cells resistant to glucocorticoid treatment. Inhibition of Caspase-1 or disruption of the NLRP3 inflammasome restored glucocorticoid sensitivity in these cells, yet how they remain alive despite demonstrable Caspase-1 activity is unknown.
Ferroptosis
Another form of RCD is ferroptosis, resulting from accumulation of toxic lipid peroxides as a consequence of the iron-dependent Fenton reaction68,69. Most of the regulation of ferroptosis centers around activity of the selenoenzyme, glutathione peroxidase 4 (GPX4)70. GPX4 detoxifies oxidized lipid species (L-OOH) into nontoxic lipid alcohols (L-OH), a mechanism to prevent lipid peroxidation in the plasma membrane and subsequent cell death. Thus, GPX4 is a potent inhibitor of ferroptosis, and GPX4 inhibition drives ferroptosis. One mechanism of GPX4 inhibition occurs via depletion of its regenerative substrate, glutathione (GSH)70 which occurs through inhibition of the upstream cysteine/glutamate transporter (xc-)71. In addition to GPX4, tetrahydrobiopterin (BH4), synthesized by GTP-cyclohydrolase-1 (GCH1), serves as an antioxidant which synergizes with vitamin E to prevent lipid oxidation and subsequent cell death from ferroptosis72. Another negative regulator of ferroptosis, ferroptosis suppressor protein 1 (FSP1) can protect from GPX4 inhibition by regenerating CoQ10, allowing suppression of peroxy radicals in lipid bilayers 73,74.
Many experimental and approved cancer therapeutics are thought to kill cancer cells via ferroptosis75, although in the majority of such reports, the term is invoked when lipid peroxidation is observed, and reactive oxygen scavengers reduce killing in vitro.
While ferroptosis does not involve a dedicated membrane perforation mechanism, ESCRT-mediated membrane repair delays the kinetics of ferroptotic cell death76. Although little is known about sub-lethal activation of ferroptosis in cancer, various reports (elaborated below) suggest that cancer cells which survive activation of other cell death pathways have increased sensitivity to ferroptosis13,77. Thus, the interconnectivity between RCD pathways (reviewed in78) allows for acquired vulnerabilities.
Drug Tolerant Persister cells
The concepts described for antibiotic-tolerant bacterial ‘persisters’ 79 translate to drug-tolerant persister cancer cells (DTPs), defined as a population of genetically identical cells that survive a cell death-inducing, therapeutic treatment. The phenomenon of persistence is distinct from that of drug resistance, in that drug tolerance in persister cells is transient and reverts to the parental, drug-sensitive state over time. These DTPs have been described in cell lines representing a wide variety of cancers in response to a variety of treatments, primarily targeted therapies and chemotherapies80. For example, DTPs have been described in AML patient samples and mouse models in response to BET inhibitors81,82, and in patient melanoma samples and cell lines following treatment with BRAF and/or MAP kinase inhibitors83–85.
There are several characteristics displayed by DTPs, including decreased cell proliferation, a change in cell identity (usually defined by gene expression changes), adaptation of cellular metabolism, and modification of the tumor microenvironment. For most settings, it is unknown if the persisting population was preexistent (i.e. developed independent of the selective pressure) or if they are a response to drug treatment. Slow cycling persister cells which survive treatment with an EGFR inhibitor are pre-existing in a population of PC9 lung cancer cells. Using a single cell barcode tracking system coupled to a fluorescent cell cycle tracker, the proliferative capacity was largely predetermined in the same population of PC9 lung cancer cells and the size of clones surviving and expanding upon treatment was similar across experiments. This suggests the presence of a pre-existing state in a population of cells, existing independently of selection pressure, which allows for persistence in the face of therapy14. It is important to note, however, that this does not preclude an inductive effect of the treatment on the persister phenotype; a pre-existing population may be in a state that is responsive to such an effect. This idea is further supported by considering another feature of DTPs, sensitivity to ferroptosis.
Some cancer cells are constitutively sensitive to ferroptosis induction by inhibition of GPX4, while others are not. Studies of DTPs derived from a wide variety of human cancer cell lines showed that DTPs generated by a variety of therapeutics become dependent on GPX4 for survival70,86. A GPX4 inhibitor induced ferroptosis in these DTPs, however, pretreatment of the parental cells with a GPX4 inhibitor had no effect on their generation87. Therefore, the GPX4-dependent DTPs were not a pre-existing population in the parental cells, but instead, were induced by the therapeutic treatment. Validation of such an effect from patient samples is lacking, however.
Changes in metabolism are another feature of DTPs that survive anti-cancer therapies. DTPs change their metabolism towards mitochondrial oxidative respiration rather than glycolysis, which resembles respiration in untransformed cells88. KRASG12D-mutated mouse pancreatic ductal adenocarcinoma cancer persister cells that survive ablation of the oncogene increase mitochondrial biogenesis and oxidative phosphorylation89. Similar results were obtained in BRAFV600E-mutated melanoma cells that persist following treatment with cisplatin or the BRAF inhibitor, vemurafenib90 or AML cells that persist following cytarabine treatment91. Utilization of alternate metabolic pathways such as fatty acid oxidation has been described in triple negative breast cancer cells92,93, HER2 positive breast cancer cells94 and melanoma cells95. For different cells, upregulation of the fatty acid transporter CD36 seems to be a key mechanism to allow changes in metabolism94,95. These results suggest that a switch in metabolism towards a more oxidative state is associated with dormancy and chemotherapy resistance in DTPs.
In general, all treatments that result in DTPs induce apoptosis via the mitochondrial pathway. This raises the possibility that DTPs are a result of non-lethal effects of engaging this apoptosis pathway. However, activation of apoptosis in surviving cells was not measured in any of these studies. A recent review provided evidence in support of the hypothesis that anastasis may contribute to tumor relapse following therapy, suggesting that drug persistent cancer cells may represent cells that had undergone anastasis96. Cells that “recover” from apoptosis induced by ethyl alcohol can display stem-like properties97, and cells that survive MOMP following treatment with BH3 mimetic drugs display transient, increased drug tolerance in vitro, and increased invasiveness and metastasis in vivo13.
The consequences of sublethal cell death engagement: Hallmarks of flatliners
In the following section we discuss the features of flatliners and their possible relationship to DTPs.
Genomic instability
A frequently described consequence of flatliner survival is genetic alteration that can take place upon sublethal activation of caspases, mainly via the caspase-activated nuclease, CAD (Figure 4). Emerging evidence revealed that limited caspase activation, initiated by death receptor signaling98,99 or by iMOMP16,100 can result in the sublethal activation of CAD and cause mutagenesis. As noted above, survival following activation of CAD has been shown to depend on the repair of double-stranded DNA breaks8.
Figure 4: Non-lethal outcomes for apoptosis flatliners.
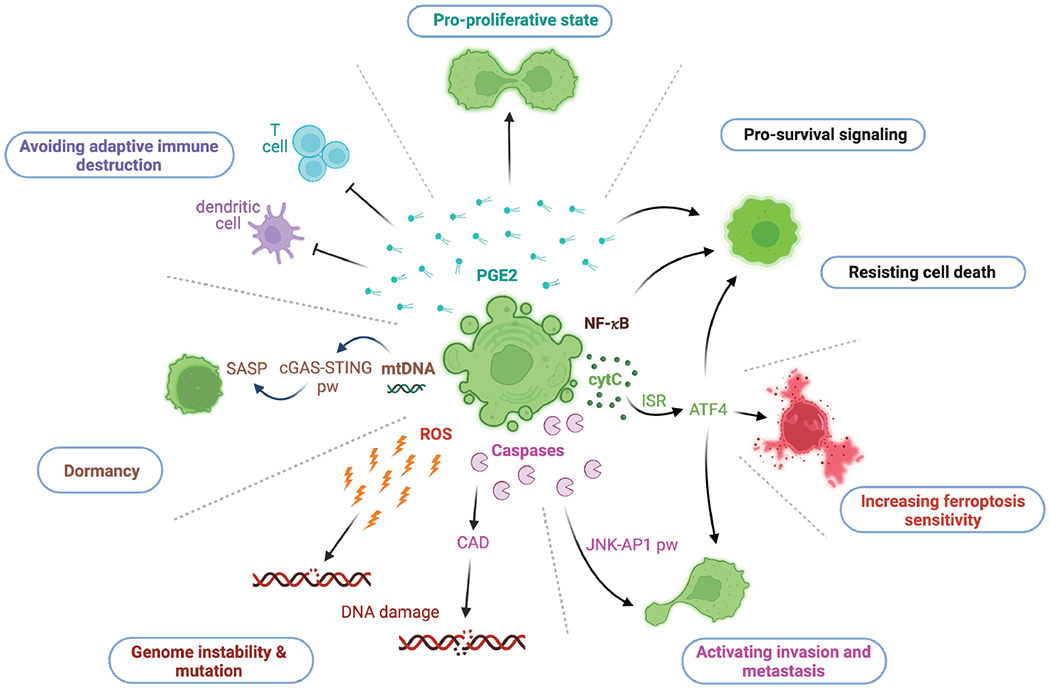
Illustration of evident consequences of the sublethal engagement of the intrinsic pathway of apoptosis and their main molecular players. The release of PGE2 relays inhibitory effects on T cells and dendritic cells. PGE2 can promote a pro-proliferative state and cell survival. NF-κB engages pro-survival signaling. Sublethal cytochrome c release can activate the ISR and thereby ATF4 translation. ATF4 impacts on various signaling pathways that lead to an escape from cell death, an increased sensitivity towards ferroptosis and a metastatic phenotype. Latter can also be promoted by sublethal caspase activation followed by subsequent initiation of the JNK-AP1 pathway. ROS and CAD can cause DNA damage, leading to genome instability and mutation. The cytosolic release of mtDNA can activate the cGAS-STING pathway, which promotes senescence and a senescence-associated secretory phenotype (SASP).
Reactive oxygen species (ROS) are potential contributors to DNA damage-induced genomic alterations during sublethal mitochondrial cell death. Mechanistically, MOMP and the activation of executioner caspases can result in the cleavage of NADH–ubiquinone oxidoreductase 75 kDa subunit (NDUFS1), an essential part of complex I, resulting in a drop in ΔΨm, a rapid reduction in ATP synthesis and an increase in ROS101,102. The tumorigenic effects of mitochondrial ROS have been demonstrated in mouse models with heterogeneous deletion of genes that encode crucial mitochondrial proteins, causing an increase in ROS103–105.
It is likely that a combination of DNA damage by activation of CAD and/or ROS production, together with an impaired DNA-damage response (DDR) increases the risk for oncogenic genomic changes106. Proteins involved in nonhomologous end joining (NHEJ) and homologous recombination (HR) are potential substrates of caspases and might be degraded during limited caspase activity107. Tumor-specific expression of transcription factors that are involved in both DDR and downregulation of apoptosis-associated genes can also potentially promote flatliners with high mutational burden. For example, the transcription factor BRN2 reprograms the DDR, promoting the more error-prone Ku-dependent NHEJ at the expense of HR, and simultaneously suppresses apoptosis in malignant melanoma cells upon various treatments such as UVB, chemotherapy, and vemurafenib108. If BRN2 contributes to the survival of some flatliners, reprogramming could contribute to genomic instability in these cells, a possibility that has not yet been tested.
Proliferation versus dormancy
In vitro and ex vivo data point to distinct heterogeneous subpopulations in untreated and treated cancers, including fast cycling/proliferative, slow cycling/quiescent, or non-cycling/senescent cells109. Apoptosis-induced flatliners can display a pro-proliferative state which relies on the caspase 3-dependent upregulation of prostaglandin E2 (PGE2)110,111. Conversely, patient-derived xenografts of colorectal cancer which were treated with chemotherapy revealed cancer cells that entered the DTP state, characterized by slow cycling tumor cells112. Other evidence suggests that subpopulations of DTP cells with different proliferative states exist, which mainly differ in their antioxidant gene programs and fatty acid oxidation14.
Cellular senescence can be evoked by diverse intra- or extracellular stresses, including mitochondrial damage, oxidative stress, irreparable DNA damage, and oncogene activation113. The permeabilization of the mitochondrial inner membrane in some mitochondria undergoing MOMP114,115 can lead to release of mitochondrial DNA (mtDNA) to the cytoplasm. This causes activation of the cytosolic cGAS-STING pathway, which connects DNA damage and cytosolic DNA-sensing to senescence, and the senescence associated secretory phenotype (SASP)116. Apoptosis-inducing treatments, such as radiation and chemotherapy, often result in a cGAS -dependent release of cytokines and chemokines involved in SASP, such as IFN-β, IL1β, IL-6 and IL-8117 and induce type I interferon (IFN) responses (Figure 4). The removal of mitochondria abrogates the senescent phenotype induced by various drugs or irradiation118. In addition, cleavage and inactivation of mediators in the IFN pathway by caspases can prevent such activation, and cells induced to undergo the mitochondrial pathway of apoptosis activate STING and IFN responses if caspases are inhibited119–122. It is therefore possible that flatliners with limited caspase activation engage STING signaling. The sublethal engagement of MOMP via BH3-mimetics can engage cytokine production by human epithelial cells123 and the inhibition of caspase activation enabled type I IFN responses and anti-tumoral immunity in in vivo experiments of cancer treatment11,124.
Metastatic potential
An increased metastatic potential of flatliners following engagement of the death receptor pathway of apoptosis has been studied in the context of diverse cancers125–127. Commonly, a crucial role of canonical NF-κB signaling for invasion and migration has been found in this setting. Death receptor-induced and cellular IAP (cIAP)-mediated activation of RIPK1 leads to the stabilization and activation of the NFκB1/RelA complex (Figure 5).
Figure 5: Activation of NF-κB signaling.
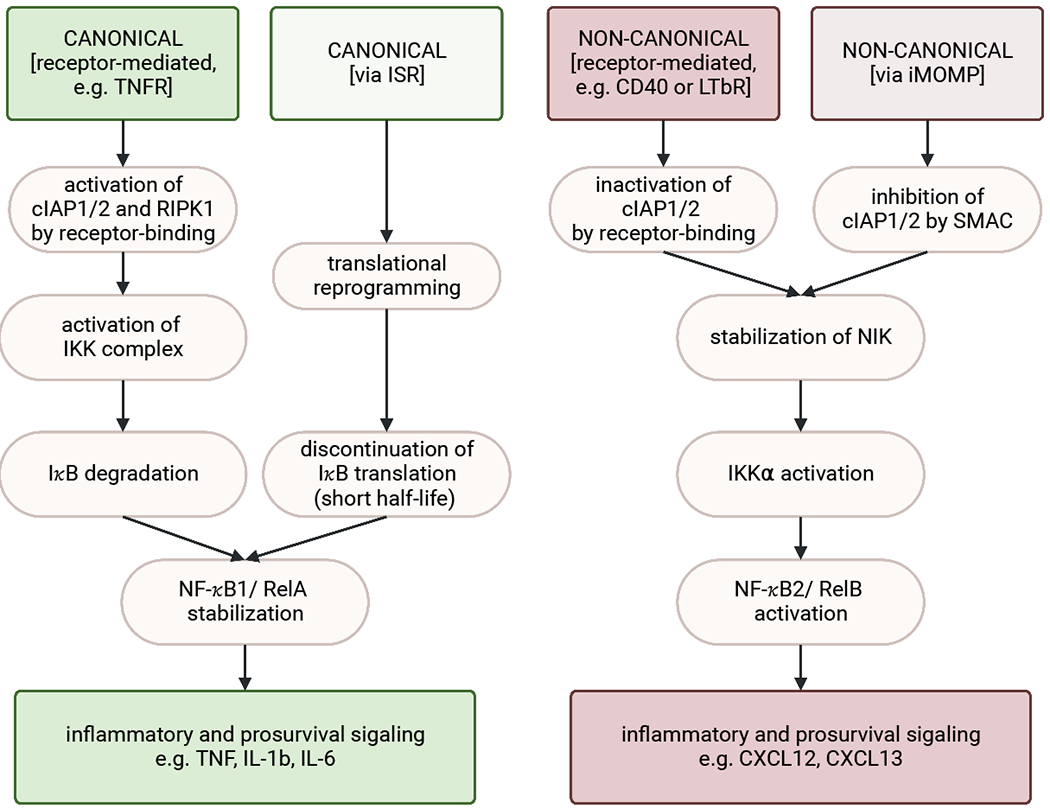
The canonical NF-κB signaling pathway can be promoted via receptor-mediated activation (e.g. via TNF-receptor) or engagement of the ISR. Receptor activation leads to the recruitment of adaptor proteins to form and activate a complex including cIAP and RIPK1. In turn, RIPK1 engages the IKK complex, which induces the proteasomal degradation of IkB. This then allows for the stabilization and nuclear localization of the NF-κB1/RelA heterodimer. Nuclear localization leads to the transcription of proinflammatory cytokines such as TNF, IL-1b, and IL-6. Activation of the ISR followed by translational reprogramming can lead to NF-κB stabilization due to discontinuation of IκB generation, which is a short-lived NFκB-inhibitor.
The non-canonical NF-κB signaling pathway can be engaged by the inhibition of cIAPs, which occurs either upon receptor engagement (e.g. via CD40 or LTbR) or by SMAC, which is released during MOMP. This results in the stabilization of NIK, which is then free to activate IKKα, resulting in NF-κB2/RelB activation and transcription of noncanonical NF-κB associated genes, such as CXCL12 and CXCL13.
Conclusively, upon specific receptor activation cIAPs engage the canonical NF-κB signaling pathway while inhibiting the non-canonical. Vice versa, inactivation of cIAPs promotes non-canonical NF-κB signaling through NIK stabilization while inhibiting the canonical pathway.
Interestingly, highly metastatic apoptosis flatliners engage a pro-invasion program that is promoted by a sublethal caspase-activation15,100,128 (Figure 4). An aggressive phenotype of apoptosis flatliners with increased metastatic capacity can also be promoted by iMOMP independently of caspase activation via the activation of the induced stress response (ISR) and ATF413. The ISR pathway generates transcription signatures consistent with invasiveness and metastasis or that have a documented impact on cell motility.
Intriguingly, there are indications that sublethal apoptosis engagement has a role in tissue regeneration and wound healing for metazoans129,130. This feature of apoptosis flatliners can rely not only on the auto- and paracrine activity of factors released, such as FGF2 or PGE2, but also on the increased motility of cells that survived the engagement of apoptosis15,129–131. It is possible that at the limits of damage in a wounded tissue, cells that engage apoptosis but survive play important roles in the repair process.
Inflammatory outcomes
The three forms of regulated necrosis considered herein (necroptosis, pyroptosis and ferroptosis) have been recognized for their role in inflammatory signaling132. In the absence of pathogens, dying cells release pro-inflammatory damage-associated molecular patterns (DAMPs, which can act as an adjuvant for adaptive immune responses when combined with neo-epitope mediated antigenicity133. This might elicit superior anti-tumor immunity, as combinations of immune checkpoint blockade and chemotherapies or targeted therapies indicate134,135. However, little is known about the impact of sublethal engagement of these pathways on inflammatory signaling, without cell death.
The role of ferroptosis in cancer immunity remains hypothetical in the context of flatliners. If, indeed, apoptotic flatliners and/or persister cells (which may often be the same thing, see above) are more dependent on GPX4 for survival, perturbations in GPX4 function may induce ferroptosis in such cells in vivo. One consideration is that a decrease in GPX4 activity leads to an upregulation of prostaglandins, including prostaglandin E2 (PGE2)136. While PGE2 has not been studied in the context of immunogenicity of ferroptotic cancer cells, there is a body of evidence for its immunosuppressive functions137–139. Ferroptotic cancer cells suppress dendritic cell function and adaptive immune responses despite the release of DAMPs140, and while the molecular events that shape this response have yet to be defined, it is possible that these are mediated via PGE2 or other prostaglandins.
More evident is the contribution of sublethal necroptosis to anti-cancer immunity. RIPK1 activates NF-κB signaling within a dying cell, leading to transcriptional upregulation of inflammatory cytokines, including IL6 and CXCL1, and promoting anti-tumor immunity12 (Figure 5). For efficient cross-priming cells relied on both RIPK1 and NF-κB activation. The ESCRTIII machinery allows cells that have engaged necroptosis to continue this intracellular signaling, leading to production and release of immune mediators and inducing adaptive immunity50.
In stark contrast to regulated necrosis, apoptosis has been considered as an immunologically silent cell death program141. This dogmatic view has been challenged in recent years, owing to evolved experimental approaches and increasing refinement of the characteristics of immunogenic cell death133. Early upstream initiation of the pro-inflammatory and pro-survival signals via the TNFR death receptor family offers an intriguing opportunity to prevent lethal events, and instead to function as a messenger to its environment. Interestingly, FAS/CD95-induced cytokine production requires RIPK1 as well as the downstream XIAP142. Cell survival and tumorigenesis have been widely associated with cIAPs, but this function is mostly attributed to their potential to activate the canonical NF-κB pathway143,144.
In contrast to their activating role in the canonical NF-κB pathway, cIAPs negatively regulate non-canonical NF-κB signaling145, which was demonstrated in the context of pharmaceutical IAP antagonism using SMAC-mimetic drugs 146 and as a consequence of MOMP in the absence of caspases11. In the latter setting, the release of IAP antagonists (such as SMAC/DIABLO) upon MOMP act to inhibit not only XIAP, but also cIAPs, and can thereby engage non-canonical NF-κB signaling (Figure 5). In this pathway, inhibition of cIAPs (e.g. via CD40 or LTbR-receptor) leads to NF-κB-inducing kinase (NIK) stabilization, followed by its binding and activation of the kinase IKKα and thereby initiation of the NFκB2/RelB complex.
NF-κB stabilization can also be a direct effect of translation reprogramming in apoptosis flatliners, where upon eIF2α phosphorylation the short-lived NFκB inhibitor IκB is no longer synthesized147–149 (Figure 5). In addition, cells also trigger inflammatory signaling mediated directly by ATF4, which drives the transcription of several cytokines including IL-8 and CCL213,150,151. However, in vivo experiments so far indicate that a pro-inflammatory role for flatliners of the intrinsic pathway of apoptosis can only be achieved in the absence of caspase activity152 or if apoptosis is followed by secondary necrosis via Gasdermin E60,153 (Figure 1). Yet, Gasdermin E is frequently downregulated in cancers154–156.
Acquired sensitivities of flatliners enable therapies
Does the survival of flatliners contribute to minimal residual disease in treated cancer patients? If so, then acquired vulnerabilities of such cells may invigorate cancer therapies. In a study of BH3 mimetic-induced DTPs, a BAX, BAK, and ATF4 gene signature in these surviving flatliners was identified in a publicly available data set from lung cancers with minimal residual disease, but not in those that were treatment naïve or progressed post-treatment13. Therefore, targeting flatliner survival, may impact minimal residual disease.
In this regard, the expression of ATF4 as a consequence of the ISR may represent a therapeutic target. Studies have indicated that ATF4 is required for the DTP phenotypes13,157,158 and an inhibitor of the ISR, by stabilizing eIF2B, was effective in preventing DTP generation in vitro13. Activators of eIF2B are in preclinical development and may have benefit to limit persistence and residual disease159–161.
The observation that repair of double-strand DNA breaks is necessary for survival of cells that have engaged caspase activation and CAD function8 suggests an approach to limiting the emergence of DTPs. Studies of EGFR mutant non-small cell lung cancer cell lines and patient xenografts showed that the generation of DTPs following targeted therapies was blocked by an inhibitor of ataxia-telangiectasia mutated (ATM). Rare patients who harbor ATM mutations in such cancers showed better prognosis than those whose cancers had functional ATM, highlighting the possible application of ATM inhibitors to prevent relapse8.
Another approach is to harness the increased sensitivity towards ferroptosis in DTPs, which has been established in experimental settings for kinase inhibitor-treated cancer cells86,87,162. While the regulation of ferroptosis appears to be an attractive target to treat susceptible cancer types70 and metastatic outburst163, several hurdles, including the bioavailability and drug stability of specific ferroptosis-inducing small molecules164, must be addressed before clinical use is possible.
Preclinical research on organoids and xenografts revealed that diapause-like adaptation of DTP cancer cells is associated with suppressed MYC activity and can be targeted by CDK9 inhibition, a cyclin-dependent kinase that is involved in transcriptional control165. Diverse CDK9 inhibitors are currently in clinical trials166 and combinational therapy with the BH3-mimetic Venetoclax showed promising results in certain murine tumor models167.
Inhibition of the PGE2 signaling pathway, which may be increased in surviving flatliners (see above) appears to be promising due to its involvement in many processes, including tumor cell proliferation and repopulation as well as chemoresistance and pro-tumoral immunity. In murine experimental settings, neutralizing antibodies against PGE2 or the administration of the cyclooxygenase-2 (COX2) inhibitor celecoxib could successfully abrogate chemoresistance-associated, early tumor repopulation111. Celecoxib has been beneficial in the context of sporadic colorectal adenomas168 and its value in combinational therapies for cancer treatment is under clinical investigation169.
CONCLUSIONS
Flatliner cells that survive the engagement of RCD pathways in response to therapy have the properties ascribed to cancer DTPs, and thus, the goal of most cancer therapies (to induce RCD in cancer cells) directly brings about the cellular changes that we seek to avoid. By recognizing this relationship and how it occurs, we can identify ways to prevent flatliners and thus limit DTP generation and, at least in some cases, minimal residual disease and relapse in cancer.
Acknowledgements
This work was supported by grants from the US National Institutes of Health R35 CA231620 to D.R.G., the German Research Foundation (DFG) KA 4830/1-1, the Advanced Clinician Scientist Programm UMEA2 (Medical Faculty, University Duisburg-Essen) and the Federal Ministry of Education and Research 01EO2104 (BMBF) to H.K, the Swiss National Science Foundation (SNSF) Post.Doc Mobility Fellowship P400PB_194393 to S.R., the US National Cancer Institute T32CA272387 to J.S. and the American Lebanese Syrian Associated Charities (ALSAC, SJCRH). Figures have been created with BioRender.com.
Footnotes
Competing interests
D.R.G. consults for Ventus Therapeutics, Inzen Therapeutics, and Horizon Therapeutics. The other authors declare no competing interests.
REFERENCES
- 1.Lockshin RA & Williams CM Programmed cell death—II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 10, 643–649 (1964). [Google Scholar]
- 2.Boada-Romero E, Martinez J, Heckmann BL & Green DR The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Bio 21, 398–414 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strasser A & Vaux DL Viewing BCL2 and cell death control from an evolutionary perspective. Cell Death Differ 25, 13–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang D, Kang R, Berghe TV, Vandenabeele P & Kroemer G The molecular machinery of regulated cell death. Cell Res 29, 347–364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D & Weinberg R Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Letai A Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. (2008) doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 7.Ichim G & Tait SWG A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer 16, 539–548 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Ali M et al. Small-molecule targeted therapies induce dependence on DNA double-strand break repair in residual tumor cells. Sci Transl Med 14, eabc7480 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fucikova J et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis 11, 1013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yatim N, Cullen S & Albert ML Dying cells actively regulate adaptive immune responses. Nat Rev Immunol 17, 262–275 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Giampazolias E et al. Mitochondrial permeabilisation engages NF-κB dependent anti-tumour activity under caspase deficiency. Nat Cell Biol 19, 1116–1129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yatim N et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350, 328–334 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalkavan H et al. Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 185, 3356–3374.e22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oren Y et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 596, 576–582 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berthenet K et al. Failed Apoptosis Enhances Melanoma Cancer Cell Aggressiveness. Cell Reports 31, 107731 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Ichim G et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Molecular cell 57, 860–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cabanos HF & Hata AN Emerging Insights into Targeted Therapy-Tolerant Persister Cells in Cancer. Cancers 13, 2666 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lockshin RA & Zakeri Z Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Bio 2, 545–550 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Li P et al. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 91, 479–489 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Lakhani SA et al. Caspases 3 and 7: Key Mediators of Mitochondrial Events of Apoptosis. Science 311, 847–851 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bedoui S, Herold MJ & Strasser A Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Bio 21, 678–695 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Du C, Fang M, Li Y, Li L & Wang X Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 102, 33–42 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Tait SWG & Green DR Caspase-independent cell death: leaving the set without the final cut. Oncogene 27, 6452–6461 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalkavan H & Green DR MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25, 46–55 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh R, Letai A & Sarosiek K Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Bio 20, 175–193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhola PD et al. High-throughput dynamic BH3 profiling may quickly and accurately predict effective therapies in solid tumors. Sci Signal 13, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao R et al. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metast Rev 37, 227–236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colell A et al. GAPDH and Autophagy Preserve Survival after Apoptotic Cytochrome c Release in the Absence of Caspase Activation. Cell 129, 983–997 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Estornes Y et al. RIPK1 promotes death receptor-independent caspase-8-mediated apoptosis under unresolved ER stress conditions. Cell Death Dis 5, e1555–e1555 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes MA et al. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol Cell 61, 834–849 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fulda S Caspase-8 in cancer biology and therapy. Cancer Lett 281, 128–133 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Peter ME et al. The role of CD95 and CD95 ligand in cancer. Cell Death Differ 22, 549–559 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alves LC, Corazza N, Micheau O & Krebs P The multifaceted role of TRAIL signaling in cancer and immunity. Febs J 288, 5530–5554 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Cullen SP & Martin SJ Fas and TRAIL ‘death receptors’ as initiators of inflammation: Implications for cancer. Semin Cell Dev Biol 39, 26–34 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Flusberg DA, Roux J, Spencer SL & Sorger PK Cells surviving fractional killing by TRAIL exhibit transient but sustainable resistance and inflammatory phenotypes. Mol Biol Cell 24, 2186–2200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang HL et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell 23, 2240–2252 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang HL, Tang HM, Fung MC & Hardwick JM In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci Rep-uk 5, 9015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Degterev A et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1, 112–119 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Vandenabeele P, Galluzzi L, Berghe T & Kroemer G Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature reviews. Molecular cell biology 11, 700–14 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Tenev T et al. The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol Cell 43, 432–448 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Tummers B & Green DR The evolution of regulated cell death pathways in animals and their evasion by pathogens. Physiol Rev 102, 411–454 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X et al. MLKL promotes cellular differentiation in myeloid leukemia by facilitating the release of G-CSF. Cell Death Differ 28, 3235–3250 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koo G-B et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 25, 707–725 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng X et al. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 62, 592–601 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Nicolè L et al. Necroptosis-driving genes RIPK1, RIPK3 and MLKL-p are associated with intratumoral CD3+ and CD8+ T cell density and predict prognosis in hepatocellular carcinoma. J Immunother Cancer 10, e004031 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strilic B et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Meng Y, Sandow JJ, Czabotar PE & Murphy JM The regulation of necroptosis by post-translational modifications. Cell Death Differ 28, 861–883 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCullough J, Frost A & Sundquist WI Structures, Functions, and Dynamics of ESCRT-III/Vps4 Membrane Remodeling and Fission Complexes. Annu Rev Cell Dev Bi 34, 85–109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vietri M, Radulovic M & Stenmark H The many functions of ESCRTs. Nat Rev Mol Cell Bio 21, 25–42 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Gong Y-N et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Broz P, Pelegrín P & Shao F The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol 20, 143–157 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Broz P & Dixit VM Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Zhang J et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res 31, 980–997 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Z et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (New York, N.Y.) 146, eaaz7548 (2020). [DOI] [PubMed] [Google Scholar]
- 55.LaRock DL et al. Group A Streptococcus induces GSDMA-dependent pyroptosis in keratinocytes. Nature 605, 527–531 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng W et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature 602, 496–502 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruan J, Xia S, Liu X, Lieberman J & Wu H Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xia S et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 1–5 (2021) doi: 10.1038/s41586-021-03478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Wang Q et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 1–7 (2020) doi: 10.1038/s41586-020-2079-1. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Z et al. Gasdermin E suppresses tumor growth by activating anti-tumor immunity. Nature 579, 415–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kayagaki N et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Rühl S et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Li Z et al. Enhancing Gasdermin-induced tumor pyroptosis through preventing ESCRT-dependent cell membrane repair augments antitumor immune response. Nat Commun 13, 6321 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson DC et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med 24, 1151–1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paugh SW et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat Genet 47, 607–614 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dixon SJ et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Conrad M & Pratt DA The chemical basis of ferroptosis. Nat Chem Biol 15, 1137–1147 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Yang WS et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dixon SJ et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soula M et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 16, 1351–1360 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doll S et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Bersuker K et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu Y et al. Ferroptosis in Cancer Treatment: Another Way to Rome. Frontiers Oncol 10, 571127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pedrera L et al. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ 28, 1644–1657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rodriguez R, Schreiber SL & Conrad M Persister cancer cells: Iron addiction and vulnerability to ferroptosis. Mol Cell 82, 728–740 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bedoui S, Herold MJ & Strasser A Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Bio 21, 678–695 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Bigger JosephW. TREATMENT OF STAPHYLOCOCCAL INFECTIONS WITH PENICILLIN BY INTERMITTENT STERILISATION. Lancet 244, 497–500 (1944). [Google Scholar]
- 80.Cabanos HF & Hata AN Emerging Insights into Targeted Therapy-Tolerant Persister Cells in Cancer. Cancers 13, 2666 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bell CC et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat Commun 10, 2723 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fong CY et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Menon DR et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 34, 4448–4459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rambow F et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 174, 843–855.e19 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Roesch A et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1Bhigh Cells. Cancer Cell 23, 811–825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Viswanathan VS et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hangauer MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhu J & Thompson CB Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Bio 20, 436–450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Viale A et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roesch A et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1Bhigh Cells. Cancer Cell 23, 811–825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Farge T et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov 7, 716–735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park JH et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Reports 14, 2154–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Camarda R et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med 22, 427–432 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Feng WW et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Reports 29, 3405–3420.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aloia A et al. A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF-mutated Melanoma to MAPK Inhibitors. Clin Cancer Res 25, 6852–6867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mirzayans R & Murray D Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int J Mol Sci 21, 1308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu Y, So C, Lam H-M, Fung M-C & Tsang S-Y Apoptosis Reversal Promotes Cancer Stem Cell-Like Cell Formation. Neoplasia New York N Y 20, 295–303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miles MA & Hawkins CJ Executioner caspases and CAD are essential for mutagenesis induced by TRAIL or vincristine. Cell Death Dis 8, e3062–e3062 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lovric MM & Hawkins CJ TRAIL treatment provokes mutations in surviving cells. Oncogene 29, 5048–5060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haimovici A et al. Spontaneous activity of the mitochondrial apoptosis pathway drives chromosomal defects, the appearance of micronuclei and cancer metastasis through the Caspase-Activated DNAse. Cell Death Dis 13, 315 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ricci J-E et al. Disruption of Mitochondrial Function during Apoptosis Is Mediated by Caspase Cleavage of the p75 Subunit of Complex I of the Electron Transport Chain. Cell 117, 773–786 (2004). [DOI] [PubMed] [Google Scholar]
- 102.Tait SWG & Green DR Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Bio 11, 621–632 (2010). [DOI] [PubMed] [Google Scholar]
- 103.Remmen HV et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics 16, 29–37 (2003). [DOI] [PubMed] [Google Scholar]
- 104.Woo DK et al. Mitochondrial Genome Instability and ROS Enhance Intestinal Tumorigenesis in APCMin/+ Mice. Am J Pathology 180, 24–31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen P-L et al. Mitochondrial genome instability resulting from SUV3 haploinsufficiency leads to tumorigenesis and shortened lifespan. Oncogene 32, 1193–1201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Negrini S, Gorgoulis VG & Halazonetis TD Genomic instability — an evolving hallmark of cancer. Nat Rev Mol Cell Bio 11, 220–228 (2010). [DOI] [PubMed] [Google Scholar]
- 107.Fischer U, Jänicke RU & Schulze-Osthoff K Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 10, 76–100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herbert K et al. BRN2 suppresses apoptosis, reprograms DNA damage repair, and is associated with a high somatic mutation burden in melanoma. Gene Dev 33, 310–332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Santos-de-Frutos K & Djouder N When dormancy fuels tumour relapse. Commun Biology 4, 747 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huang Q et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 17, 860–866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kurtova AV et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517, 209–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rehman SK et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 184, 226–242.e21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Coppé J-P, Desprez P-Y, Krtolica A & Campisi J The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Pathology Mech Dis 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McArthur K et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, (2018). [DOI] [PubMed] [Google Scholar]
- 115.Riley JS et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. Embo J 37, e99238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li T & Chen ZJ The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Medicine 215, 1287–1299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dou Z et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Correia-Melo C et al. Mitochondria are required for pro-ageing features of the senescent phenotype. Embo J 35, 724–742 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ning X et al. Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol Cell 74, 19–31.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 120.Kazama H et al. Induction of Immunological Tolerance by Apoptotic Cells Requires Caspase-Dependent Oxidation of High-Mobility Group Box-1 Protein. Immunity 29, 21–32 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.White MJ et al. Apoptotic Caspases Suppress mtDNA-Induced STING-Mediated Type I IFN Production. Cell 159, 1549–1562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rongvaux A et al. Apoptotic Caspases Prevent the Induction of Type I Interferons by Mitochondrial DNA. Cell 159, 1563–1577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brokatzky D et al. A non-death function of the mitochondrial apoptosis apparatus in immunity. Embo J 38, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Han C et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat Immunol 21, 546–554 (2020). [DOI] [PubMed] [Google Scholar]
- 125.Ishimura N, Isomoto H, Bronk SF & Gores GJ Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am J Physiol-gastr L 290, G129–G136 (2006). [DOI] [PubMed] [Google Scholar]
- 126.Trauzold A et al. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 25, 7434–7439 (2006). [DOI] [PubMed] [Google Scholar]
- 127.Li Z, Xu X, Bai L, Chen W & Lin Y Epidermal Growth Factor Receptor-mediated Tissue Transglutaminase Overexpression Couples Acquired Tumor Necrosis Factor-related Apoptosis-inducing Ligand Resistance and Migration through c-FLIP and MMP-9 Proteins in Lung Cancer Cells*. J Biol Chem 286, 21164–21172 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu X et al. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res 27, 764–783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bock FJ et al. Apoptotic stress-induced FGF signalling promotes non-cell autonomous resistance to cell death. Nat Commun 12, 6572 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li F et al. Apoptotic Cells Activate the “Phoenix Rising” Pathway to Promote Wound Healing and Tissue Regeneration. Sci Signal 3, ra13 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fanfone D et al. Confined migration promotes cancer metastasis through resistance to anoikis and increased invasiveness. Elife 11, e73150 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Davidovich P, Kearney CJ & Martin SJ Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol Chem 395, 1163–1171 (2014). [DOI] [PubMed] [Google Scholar]
- 133.Galluzzi L, Buqué A, Kepp O, Zitvogel L & Kroemer G Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17, 97–111 (2017). [DOI] [PubMed] [Google Scholar]
- 134.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Vafaei S et al. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int 22, 2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Angeli JPF, Krysko DV & Conrad M Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer 19, 405–414 (2019). [DOI] [PubMed] [Google Scholar]
- 137.Zelenay S et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162, 1257–1270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kalinski P Regulation of Immune Responses by Prostaglandin E2. J Immunol 188, 21–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Göbel C et al. Functional expression cloning identifies COX-2 as a suppressor of antigen-specific cancer immunity. Cell Death & Disease 5, e1568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wiernicki B et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun 13, 3676 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kerr J, Wyllie A & Currie A Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British journal of cancer 26, 239–57 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cullen SP et al. Fas/CD95-Induced Chemokines Can Serve as “Find-Me” Signals for Apoptotic Cells. Mol Cell 49, 1034–1048 (2013). [DOI] [PubMed] [Google Scholar]
- 143.Gyrd-Hansen M & Meier P IAPs: from caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat Rev Cancer 10, 561–574 (2010). [DOI] [PubMed] [Google Scholar]
- 144.Eckelman BP, Salvesen GS & Scott FL Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. Embo Rep 7, 988–994 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Michie J, Kearney CJ, Hawkins ED, Silke J & Oliaro J The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy. Cells 9, 207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Varfolomeev E et al. IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-κB Activation, and TNFα-Dependent Apoptosis. Cell 131, 669–681 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Jiang H-Y et al. Phosphorylation of the α Subunit of Eukaryotic Initiation Factor 2 Is Required for Activation of NF-κB in Response to Diverse Cellular Stresses. Mol Cell Biol 23, 5651–5663 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Deng J et al. Translational Repression Mediates Activation of Nuclear Factor Kappa B by Phosphorylated Translation Initiation Factor 2. Mol Cell Biol 24, 10161–10168 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Abdel-Nour M et al. The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 365, eaaw4144 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Püschel F et al. Starvation and antimetabolic therapy promote cytokine release and recruitment of immune cells. Proc Natl Acad Sci USA 117, 9932–9941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gargalovic PS et al. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA 103, 12741–12746 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ahmed A & Tait SWG Targeting immunogenic cell death in cancer. Mol Oncol 14, 2994–3006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rogers C et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8, 14128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Beeck K. O. de et al. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur J Hum Genet 19, 965–973 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Thompson DA & Weigel RJ Characterization of a gene that is inversely correlated with estrogen receptor expression (ICERE-1) in breast carcinomas. Eur J Biochem 252, 169–177 (2017). [DOI] [PubMed] [Google Scholar]
- 156.Ibrahim J, Beeck K. O. de, Fransen E, Peeters M & Camp GV The Gasdermin E Gene Has Potential as a Pan-Cancer Biomarker, While Discriminating between Different Tumor Types. Cancers 11, 1810 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Figarol S et al. Farnesyltransferase inhibition overcomes the adaptive resistance to osimertinib in EGFR-mutant NSCLC. Biorxiv 2022.04.01.486707 (2022) doi: 10.1101/2022.04.01.486707. [DOI] [Google Scholar]
- 158.Vendramin R et al. Activation of the Integrated Stress Response in drug-tolerant melanoma cells confers vulnerability to mitoribosome-targeting antibiotics. Biorxiv 2020.06.26.173492 (2020) doi: 10.1101/2020.06.26.173492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Knaap MS van der et al. Therapy Trial Design in Vanishing White Matter. Neurology Genetics 8, e657 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wong Y et al. eIF2B activator prevents neurological defects caused by a chronic integrated stress response. Elife 8, e42940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hetz C, Axten JM & Patterson JB Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol 15, 764–775 (2019). [DOI] [PubMed] [Google Scholar]
- 162.Yang WS & Stockwell BR Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem Biol 15, 234–245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ubellacker JM et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang L, Chen X & Yan C Ferroptosis: An Emerging Therapeutic Opportunity for Cancer. Genes Dis 9, 334–346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dhimolea E et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 39, 240–256.e11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mandal R, Becker S & Strebhardt K Targeting CDK9 for Anti-Cancer Therapeutics. Cancers 13, 2181 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Phillips DC et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia 34, 1646–1657 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bertagnolli MM et al. Celecoxib for the Prevention of Sporadic Colorectal Adenomas. New Engl J Medicine 355, 873–884 (2006). [DOI] [PubMed] [Google Scholar]
- 169.Li S, Jiang M, Wang L & Yu S Combined chemotherapy with cyclooxygenase-2 (COX-2) inhibitors in treating human cancers: Recent advancement. Biomed Pharmacother 129, 110389 (2020). [DOI] [PubMed] [Google Scholar]