

Abstract
Cilia are near ubiquitous small, cellular appendages critical for cell-to-cell communication. As such, they are involved in diverse developmental and homeostatic processes, including energy homeostasis. ARL13B is a regulatory GTPase highly enriched in cilia. Mice expressing an engineered ARL13B variant, ARL13BV358A which retains normal biochemical activity, display no detectable ciliary ARL13B. Surprisingly, these mice become obese. Here, we measured body weight, food intake, and blood glucose levels to reveal these mice display hyperphagia and metabolic defects. We showed that ARL13B normally localizes to cilia of neurons in specific brain regions and pancreatic cells but is excluded from these cilia in the Arl13bV358A/V358A model. In addition to its GTPase function, ARL13B acts as a guanine nucleotide exchange factor (GEF) for ARL3. To test whether ARL13B’s GEF activity is required to regulate body weight, we analyzed the body weight of mice expressing ARL13BR79Q, a variant that lacks ARL13B GEF activity for ARL3. We found no difference in body weight. Taken together, our results show that ARL13B functions within cilia to control body weight and that this function does not depend on its role as a GEF for ARL3. Controlling the subcellular localization of ARL13B in the engineered mouse model, ARL13BV358A, enables us to define the cilia-specific role of ARL13B in regulating energy homeostasis.
Keywords: ARL13B, cilia, hyperphagia, obesity, energy homeostasis
Introduction
Primary cilia are small microtubule-based appendages that extend from the cell membrane of nearly every vertebrate cell. Cilia function as critical signaling hubs implicated in a variety of biological processes1. Cilia dysfunction leads to a variety of symptoms including obesity as occurs in certain childhood obesity syndromes such as Alstro m (ALMS) syndrome and Bardet-Biedl syndrome (BBS)2-5. These syndromic obese ciliopathies are thought to result from altered cilia signaling6-8. For example, BBS mutations disrupt the composition of signaling proteins within the ciliary membrane9. Despite such monogenic syndromes clearly linking cilia and obesity, the exact role of the cilium or of ciliary signaling in regulating obesity is not clear. Indeed, distinguishing the signaling within the cilium from that in the cell is a major challenge. Understanding the role of cilia and ciliary signaling in obesity is likely to uncover important mechanisms driving common forms of obesity.
Genetic mouse models have led to insightful discoveries regarding cilia-associated obesity including identifying critical cell types, understanding how cilia control aspects of feeding behavior, and pinpointing roles of cilia in peripheral tissues and metabolic phenotypes10. For example, tissue-specific genetic ablation of cilia in adult animals causes obesity due to hyperphagia, and leads to elevated levels of leptin, glucose and insulin11. Moreover, loss of primary cilia specifically on POMC or MC4R neuronal populations in the hypothalamus results in obesity, suggesting neuronal primary cilia are critical for food intake and body weight regulation11,12. In addition, loss of primary cilia on β-cells in the pancreas results in reduced insulin secretion, which leads to impaired glucose tolerance and the development of diabetes13. Together these findings indicate that primary cilia are required for regulating energy homeostasis and this complex regulation involves multiple tissues, cell types and signaling pathways. Animal models of cilia-associated obesity recapitulate the obesity phenotype in humans14. To date, these approaches have been unable to reveal the roles of proteins specifically within cilia vs the cellular pool in complex processes like energy homeostasis.
ARL13B, an ADP-ribosylation factor (ARF) protein family member, encodes a regulatory GTPase enriched on the ciliary membrane15,16. Null mutations in mouse Arl13b, called hennin (hnn), lead to embryonic lethality, aberrant Hedgehog (Hh) signaling and defects in ciliogenesis17. The N-terminus of the protein contains ARL13B’s GTPase domain and the C-terminus contains a coiled helical domain, cilia localization sequence, and a proline-rich domain18. ARL13B functions as a guanine nucleotide exchange factor (GEF) for another ARF family member, ARL3, that also localizes to cilia19-21. ARL13B regulates the phospholipid composition of the ciliary membrane as it retains inositol polyphosphate-5-phosphatase E (INPP5E) in cilia22-24. INPP5E removes the 5 phosphate from PI(3,4,5)P and PI(4,5)P to generate PI(3,4)P and PI(4)P; thus INPP5E regulates the PIP composition of the ciliary membrane and ciliary signaling25-27. ARL13B mutations cause Joubert syndrome (JS), a ciliopathy characterized by a hindbrain malformation that presents with additional ciliopathy-associated phenotypes including intellectual disability, seizures, polydactyly and cleft palate28-31. JS-causing mutations in ARL13B disrupt ARL13B’s GEF activity for ARL320,32. Missense mutations in the phosphatase domain of INPP5E cause JS, while C-terminus mutations, that remove the CaaX domain leaving the catalytic activity intact, abolish INPP5E retention in the ciliary membrane and cause MORM (mental retardation, truncal obesity, retinal dystrophy and micropenis) syndrome27,33,34.
To distinguish how ciliary signaling proteins like ARL13B act within the cilium and outside the cilium, we engineered an ARL13B mouse allele that is excluded from cilia, ARL13BV358A yet maintains its known enzymatic functions 35. In contrast to null Arl13b mutant mice, homozygous Arl13bV358A/V358A mice are viable with intact cilia and transduce Hh signaling normally17,35. These findings demonstrate that ARL13B plays distinct functions within and outside cilia. Here we report that Arl13bV358A/V358A mice are obese. We performed physiological assessments of Arl13bV358A/V358A mice including body weight, body composition, food intake, and glucose and insulin metabolism. We tested whether ARL13B’s GEF activity for ARL3 is required to regulate body weight by analyzing the body weight of mice expressing ARL13BR79Q, a variant that lacks ARL13B GEF activity for ARL332,36. The spatial control of ciliary ARL13B in the engineered mouse model, ARL13BV358A, enables us to unravel the cilia-specific function of ARL13B in the obesity phenotype. These findings not only indicate ciliary ARL13B regulates energy homeostasis but also serve as an entry point for understanding the complex roles cilia signaling play in adult energy homeostasis.
Results
Arl13bV358A/V358A mice weigh significantly more than control mice
Unlike Arl13bhnn/hnn mice (null Arl13b allele), homozygous Arl13bV358A/V358A mice survive into adulthood and become heavier than Arl13b+/+ mice. To investigate their weight profile, we generated weekly body weight data from weaning (week 3) to adult (week 10). We found no significant differences in body weight curves among Arl13b+/+, Arl13b+/V358A and Arl13b+/hnn male and female mice over 10 weeks. Early body weight data from Arl13bV358A/V358A mice was indistinguishable from Arl13b+/+, Arl13b+/V358A and Arl13b+/hnn mice in males and females. From week 5 until week 10, male Arl13bV358A/V358A mice were significantly heavier than all other control genotypes: Arl13b+/+, Arl13b+/V358A and Arl13b+/hnn mice, (28.6±3.1 grams vs. 22.8±2.3 grams, 23.4±3.1 grams, 24.4±1.9 grams, respectively) (Fig 1A). At week 10, Arl13bV358A/V358A mice were, on average, 30% heavier than animals in the control groups (Fig 1A). Female Arl13bV358A/V358A mice displayed a similar but slightly delayed body weight phenotype. At week 7, female Arl13bV358A/V358A mice weighed significantly more than Arl13b+/+, Arl13b+/V358A and Arl13b+/hnn mice (26.7±4.9 grams vs. 20.6±1.9 grams, 21.4±2.6 grams, 21.9±1.8 grams, respectively) (Fig 1B). At week 10, Arl13bV358A/V358A mice were on average 33% heavier than animals in the control groups (Fig 1B). Thus, regardless of sex, Arl13bV358A/V358A mice weigh significantly more than their littermate controls by 5-7 weeks, a post-weaning age suggestive that the weight difference emerged when mice self-feed.
Figure 1: Arl13bV358A/V358A mice become obese.

(A-B) Mice maintained on breeder chow diet were weighed weekly from 3 to 10 weeks of age. Body weights of Arl13b+/+ (males n=14 and females n=13), Arl13b+/V358A (males n=22 and females n=22), Arl13+/hnn (males n=10 and females n=9), Arl13bV358A/V358A (males n=24 and females n=26), and Arl13bV358A/hnn (males n=15 and females n=17). Repeated measures ANOVA, *p < 0.01. (C-C’, D-D’) Lean mass and fat mass were measured using EchoMRI (n ≥ 5 per group). (E-E’) Perigonadal fat pads were isolated and weighed in 10-week-old mice (n=7 per group). (F-F’) Energy expenditure was analyzed by CalR ANCOVA, with body weight included as a covariate (n ≥ 5 per group). (G-G’) Food intake was measured daily from week 4 to week 5 (males Arl13b+/+ n=10 and Arl13bV358A/V358A n= 10; females Arl13b+/+ n=10 and Arl13bV358A/V358A n=10). Wild type (+/+), Arl13bV358A/V358A (A/A). Error bars represent standard deviation.
To determine whether the Arl13bV358A/V358A weight phenotype is Arl13b gene dosage sensitive, we bred the Arl13bV358A allele with the null Arl13bhnn allele. We weighed male and female Arl13bV358A/hnn mice from week 3 to 10. We found Arl13bV358A/hnn mice displayed a similar body weight increase as Arl13bV358A/V358A mice, indicating that the overall dosage of ARL13B protein had no impact on the weight phenotype (Fig 1A-B). Taken together, these data argue that ARL13B functions from within cilia to control body weight.
We recently reported that Arl13bV358A/V358A mice develop kidney cysts37. Small cysts are visible at weaning age and become progressively larger by ~4 months of age. The kidney phenotype coinciding with the time course of body weight increase could suggest the increased body weight is due to developing kidney cysts. To evaluate this, we weighed the kidneys and subtracted the kidney weight from the total body weight (data not shown). We found that adjusted body weight in Arl13bV358A/V358A mice was still significant, indicating that kidney cysts did not drive the obesity phenotype.
Arl13bV358A/V358A mice have increased fat mass and are hyperphagic
To determine whether the increased body weight in Arl13bV358A/V358A mice was due to increased lean or fat mass, we assessed body composition using EchoMRI. We found that lean mass was comparable between Arl13bV358A/V358A and Arl13b+/+ animals (Fig. 1B, B’). However, the difference in body weight between the two genotypes was characterized by a significant increase in fat mass (Fig. 1C, C’). Consistent with this, we found the adipose volume in perigonadal fat pads in male and female Arl13bV358A/V358A mice was significantly increased over control mice (Fig. 1D, D’).
To investigate whether the increased body weight in Arl13bV358A/V358A mice was due to reduced activity, we measured energy expenditure using a Comprehensive Lab Animal Monitoring System (CLAMS). We found no difference in the daily energy expenditure between Arl13bV358A/V358A and Arl13b+/+ animals suggesting Arl13bV358A/V358A animals are equally active as Arl13b+/+ animals (Fig. 1E, E’).
To examine whether cilia-excluded ARL13B impacts food consumption, we measured daily food intake in Arl13bV358A/V358A mice between weeks 4 and 5, when we observed the increase in body weight. We found that male and female Arl13bV358A/V358A mice consumed on average ~20% more food per day compared to Arl13b+/+ littermate controls (Fig 1F, F’). The feeding behavior in Arl13bV358A/V358A mice implicates the hypothalamus is involved.
Energy expenditure and food intake are controlled by neuronal circuits within the hypothalamus. To determine whether ARL13B is in primary cilia on neurons within the major feeding centers in the hypothalamus, we performed immunofluorescence staining using antibodies directed against ARL13B and the neuronal cilia marker adenylate cyclase 3 (ACIII). We found that ARL13B co-localizes with ACIII in the arcuate nucleus (ARC), the paraventricular nucleus (PVN) and the ventromedial hypothalamic nucleus (VMH) in Arl13b+/V358A mice (Fig 2A, C, E). However, while ACIII positive cilia were present, we did not detect ARL13B in the cilia in these brain regions in Arl13bV358A/V358A mice (Fig 2B, D, F). In the ARC, appetite-stimulating agouti-related peptide (AgRP)-expressing neurons, and the appetite-suppressing pro-opiomelanocortin (POMC)-expressing neurons are key regulators of food intake. To assess whether loss of ciliary ARL13B disrupted the number or patterning of AgRP and POMC neurons in the ARC, we performed RNAscope in situ hybridization. We found normal frequency and patterning for both AgRP and POMC neurons in the ARC of Arl13bV358A/V358A mice, indicating ciliary ARL13B does not play a role in specifying these cell types (Fig 2G-I). Taken together, these data indicate that the increased body weight in Arl13bV358A/V358A mice is due to an increase in fat mass which is associated with hyperphagia, rather than changes in energy expenditure. Coupled to the absence of ARL13B in neuronal cilia in the hypothalamus, the data suggest that ciliary ARL13B is involved in regulating feeding behavior.
Figure 2: Neuronal cilia staining in hypothalamic feeding centers show ARL13B is usually in cilia.
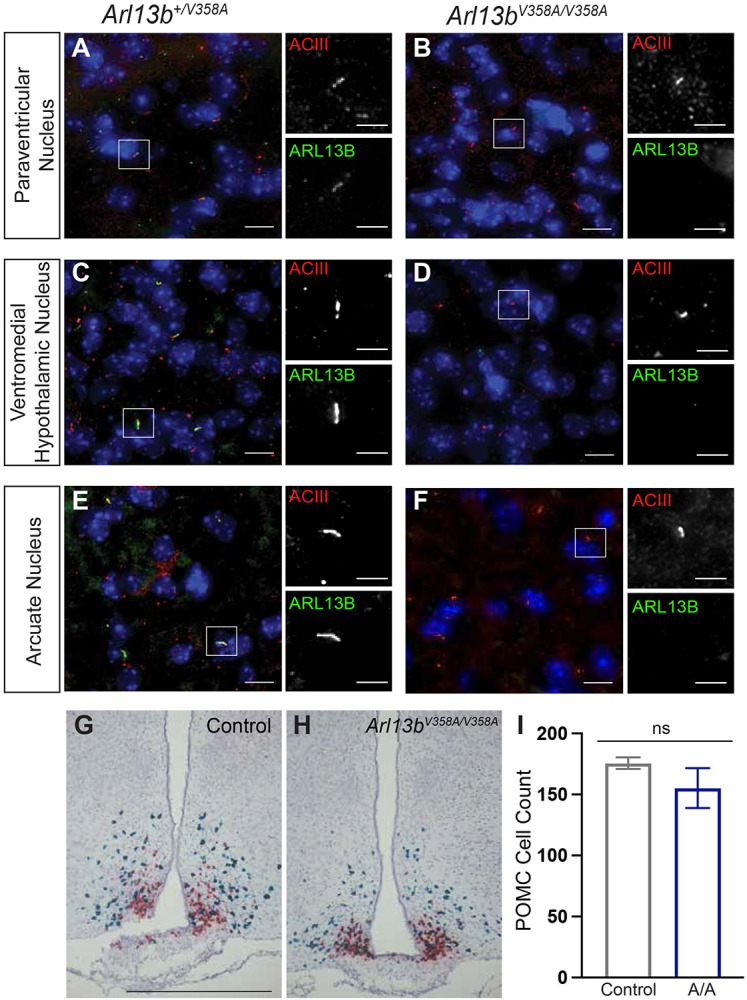
(A-F) Representative immunofluorescence staining of neurons in the hypothalamic feeding centers (PVN, VMH, ARC) in the mouse brain stained with antibodies against ACIII (red) and ARL13B (green). Scale bar for large panels: 20um; scale bar for insets: 5um. (G-H) Representative RNAscope in situ hybridization images of AgRP (red) and POMC (blue) neurons in the ARC. (I) Quantification of POMC neurons (n = 3 per group). Scale bar 100 μm. Error bars represent standard deviation.
Arl13bV358A/V358A mice display impaired glucose and insulin metabolism and have elevated levels of serum hormones
The pancreas maintains glucose homeostasis through the coordinated actions of glucagon and insulin. Impaired glucose homeostasis leads to the development of diabetes commonly associated with obese individuals. To investigate the homeostatic response to glucose and insulin, we performed glucose and insulin tolerance tests in 11 to 14-week-old Arl13bV358A/V358A mice. We grouped Arl13b+/+, Arl13bV358A/+, and Arl13bhnn/+ mice in a control group since we did not observe differences in body weight among these genotypes (Fig 1A-B). Male and female mice displayed no significant differences in fasting blood glucose levels between control and Arl13bV358A/V358A mice in either assay, suggesting that despite their significant increase in weight, Arl13bV358A/V358A mice maintain blood glucose levels within normal range (Fig 3A-D). To assess glucose clearance, we administered D-glucose by intraperitoneal injection. Male Arl13bV358A/V358A mice exhibited elevated blood glucose levels, but levels were not significantly above control animals demonstrating relatively normal glucose clearance (Fig 3A). In contrast, female Arl13bV358A/V358A mice displayed significantly elevated blood glucose levels relative to control mice demonstrating an impairment of glucose clearance (Fig 3B). To assess insulin action, we administered insulin by intraperitoneal injection. As expected, insulin lowered blood glucose levels in control animals. However, insulin did not lower blood glucose levels in male and female Arl13bV358A/V358A mice, revealing an insulin-resistance phenotype (Fig 3C-D). These data show Arl13bV358A/V358A mice possess a significant impairment of homeostatic maintenance of blood glucose levels.
Figure 3: Arl13bV358A/V358A mice have impaired glucose and insulin metabolism.

For the glucose tolerance test, (A) male Arl13bV358A/V358A and control mice did not display a significant difference in blood glucose levels. (B) In contrast, blood glucose levels were significantly increased in female Arl13bV358A/V358A mice over controls beginning at 20 minutes post-glucose administration. For the insulin tolerance test, (C-D) both male and female Arl13bV358A/V358A mice had significantly higher blood glucose levels than controls at all time points post-insulin administration. Baseline blood glucose levels did not differ between the two genotypes in either males or females. The control group is comprised of pooled data from Arl13b+/+, Arl13bV358A/+, and Arl13bhnn/+ mice that did not differ in body weight. Error bars represent standard deviation. p-values < 0.05 *, < 0.01 **, < 0.001 ***, < 0.0001 ****; ns = not significant.
Primary cilia on pancreatic islets are linked to pancreatic function so we examined ARL13B localization in primary cilia of alpha and beta cells. We performed immunofluorescence staining using antibodies directed against ARL13B, glucagon, insulin and acetylated α-tubulin in pancreas tissue from Arl13bV358A/V358A and Arl13b+/V358A mice. We found that ARL13B co-localizes with acetylated α-tubulin on both alpha and beta cells in Arl13b+/V358A mice, but not on pancreatic islets in Arl13bV358A/V358A mice (Fig 4A-D). Paired with the impaired glucose and insulin metabolism we observed in Arl13bV358A/V358A mice, the absence of ARL13B from cilia on alpha and beta cells suggests ciliary ARL13B plays a role in cilia-mediated signaling that regulates glucose homeostasis to prevent the development of diabetes.
Figure 4: Arl13b+/V358A mice have ARL13B in α-cell and β-cell cilia.

(A-D) Representative immunofluorescence staining of cells in the mouse pancreas shows ARL13B (green) localized to cilia (stained with antibody against acetylated α-tubulin, white) of α-cells (stained with antibody against glucagon, red) and β-cells (stained with antibody against insulin, red) in Arl13b+/V358A mice. Arl13bV358A/V358A mice lack ciliary ARL13B in these cells.
Lastly, we performed physiological analysis on serum to measure circulating levels of insulin and leptin in Arl13bV358A/V358A and Arl13b+/V358A mice. We found that male Arl13bV358A/V358A mice had elevated levels of serum insulin and leptin compared to control littermates (Fig 5A-B). Female Arl13bV358A/V358A mice exhibited slightly elevated levels of insulin but levels were not significantly above control animals (Fig 5C). On the other hand, leptin levels were significantly elevated compared to littermate controls (Fig 5D). These results indicate that loss of ciliary ARL13B disrupts homeostatic regulation in the pancreas and causes an increase in hormone levels similar to other models of cilia-associated obesity.
Figure 5: Arl13bV358A/V358A mice have elevated levels of serum insulin and leptin.

(A-B) Nonfasted serum insulin and leptin levels were increased in male Arl13bV358A/V358A mice compared to controls. (C-D) Nonfasted serum insulin levels in female Arl13bV358A/V358A mice were not significantly different compared to controls. In contrast, serum leptin levels were significantly elevated. p-values < 0.05 *, < 0.001 ***; ns = not significant. n=7 animals per genotype. Error bars represent standard deviation
Arl13bV358A/V358A mice become obese, whereas Arl13bR79Q/R79Q mice do not
To determine whether the role of ciliary ARL13B in regulating body weight is mediated via its GEF activity for ARL3, we measured body weight in Arl13bR79Q/R79Q animals, which lack GEF activity for ARL332. We found no difference in the body weight of Arl13bR79Q/R79Q and Arl13b+/+ mice regardless of sex (Fig 6A-B). These findings indicate that the obesity phenotype in Arl13bV358A/V358A mice does not depend on the GEF activity of ciliary ARL13B.
Figure 6: Loss of Arl13b GEF activity does not lead to obesity.

Arl13bR79Q disrupts ARL13B GEF activity for ARL3. (A-B) Body weights of Arl13b+/+ (n=5 animals per sex) and Arl13bR79Q/R79Q (n=5 animals per sex) mice were measured between 14 and 18 weeks of age. Arl13bR79Q/R79Q mice are not obese. ns = not significant.
Discussion
Our study identifies a cilia-specific role of ARL13B as a critical regulator of energy homeostasis and demonstrates this role does not function via ARL13B’s GEF activity for ARL3. Mice expressing the cilia-excluded variant, ARL13BV358A, become hyperphagic and obese soon after weaning. Arl13bV358A/V358A mice displayed defects in glucose metabolism and high levels of insulin and leptin in the blood. Furthermore, we showed that ARL13B GEF activity for ARL3 is not required to regulate body weight. These data suggest that ARL13B functions from within cilia to control food intake and body weight.
The brain and pancreas play distinct yet complementary roles in regulating energy homeostasis and metabolism38. We found that ARL13B is normally in cilia on neurons in multiple hypothalamic feeding centers and on pancreatic islets, but absent from cilia on these cell types in Arl13bV358A/V358A mutants. Each of these cell types are key candidates to contribute to the constitutive mutant phenotype. Analysis of conditional Arl13b mutants (Arl13bV358A/flox) with cell-type specific Cre lines will pinpoint which tissue types require ciliary ARL13B for the regulation of energy homeostasis.
We were surprised to observe obesity in Arl13bV358A/V358A mice as known mutations in ARL13B lead to JS, which is not generally associated with obesity. However, impaired ciliary enrichment of ARL13B is linked to obesity in some patient populations. Mutations in tubby genes are associated with obesity and insulin resistance39-41. TULP3, a member of the tubby family, is required for ciliary enrichment of ARL13B in some cell types42-44. RPGRIP1L encodes a transition zone protein localized near the base of cilia45,46. In the genome, RPGRIP1L resides near the FTO locus, well-established and associated with human obesity47. Hypomorphic mutations in RPGRIP1 lead to hyperphagic obesity caused by a reduction in leptin sensitivity in neurons48,49. When RPGRIP1L is disrupted in mice or patient fibroblasts, ciliary ARL13B is significantly diminished50. These data show a correlation of obesity and loss of ciliary ARL13B, suggestive of a role of ciliary ARL13B in regulating energy homeostasis in humans.
Consistent with JS not being associated with obesity, Arl13bR79Q/R79Q mice, mimicking a JS-causing mutation, were of normal weight28,36. This mutation disrupts ARL13B GEF activity for ARL3 and ARL13BR79Q protein remains enriched in cilia32,51. There are also JS patients with ARL3 mutations who do not present with obesity52. Taken together, these data suggest that JS-causing ARL13B mutations are mechanistically distinct from the engineered V358A mutation that reveals the ciliary role of ARL13B in regulating body weight.
One possible mechanism through which ciliary ARL13B regulates energy homeostasis involves the established interaction between ARL13B and INPP5E in the ciliary membrane. Patients lacking the C-terminal CaaX domain of INPP5E lose INPP5E ciliary localization and exhibit MORM syndrome, which includes obesity33. INPP5E is not detected in the cilia of Arl13bV358A/V358A cells, consistent with data showing the ciliary retention of INPP5E depends on ARL13B24,35. Thus, the loss of ciliary INPP5E in the absence of ciliary ARL13B would change the PIP composition of the ciliary membrane, impacting signaling therein. This would suggest that JS-causing mutations in ARL13B act via ARL3 whereas the obesity-causing ARL13BV358A mutation functions via INPP5E. While a tantalizing model, it is hard to reconcile with INPP5E mutations causing JS or the dogma in the field that INPP5E requires activated ARL3 (ARL3-GTP) to enter cilia23. ARL13BR79Q disrupts ARL13B GEF activity for ARL3, but another GEF could compensate. We do not formally know whether ARL3 is activated in ARL13BR79Q mice.
In other systems, the ARL13B orthologue ARL13 displays a role in the ciliary import and export of membrane-associated proteins20,22,53,54. In C. elegans, arl13 mutants displayed abnormal cilia morphology and defects in ciliary protein localization and transport compared to wildtype animals. Intraflagellar transport (IFT) motility assays showed that anterograde IFT was diminished in arl13 mutants, consistent with the abnormal distribution of transmembrane proteins54. In Chlamydomonas, arl13 mutants accumulated similar proteins in the cilia as bbs mutants, suggesting an overlapping role of ARL13 and the BBSome in ciliary protein trafficking. A defect in BBSome-dependent cargo transport in arl13 mutants suggests a role for ARL13 in regulating BBSome-cargo interactions that are essential for ciliary exit53. Whether these mechanisms of Arl13 are conserved across species needs to be investigated as they may be key in understanding the function of ciliary ARL13B in regulating energy homeostasis.
Our Arl13bV358A/V358A model uncouples the ciliary and cellular roles of the ARL13B protein. This model enables us to directly interrogate, at a subcellular resolution, how ARL13B controls cilia-mediated signaling pathways involved in maintaining energy homeostasis. Our data indicate that ARL13B typically localizes to cilia in tissues that regulate energy homeostasis. The absence of ARL13B from cilia in these tissues in Arl13bV358A/V358A mice suggests a role for ciliary ARL13B. Future studies will identify the mechanism of ARL13B action within cilia to regulate energy homeostasis and define what cell type or combination of cells require ciliary ARL13B function.
Materials and Methods
Mouse lines:
All mice were cared for in accordance with NIH guidelines and Emory University’s Institutional Animal Care and Use Committee (IACUC). Lines used were Arl13bV358A (C57BL/6J-Arl13bem1Tc) [MGI: 6256969] and Arl13bR79Q (C57BL/6J-Arl13bem2Tc) [MGI: 6279301]35,36. Mice were genotyped for the V358A mutation using primers MB11: 5’-CCTATATTCTTCTAGAAAACAGTAAGAAGAAAACCAAGAAACTAAGACTCCTTTTCATTCATCGGGC-3’ and MB12: 5’-GACAGTAAAGGATTCTTCCTCACAACCTGAC-3’ to detect the mutant allele, and primers MB21: 5’-CTTAAGATGACTTTGAGTTTGGAAGAAATACAAGATAGC-3’ and MB22: 5’-GCGTGGGACTCTTTGGAGTAGACTAGTCAATACAGACGGGTTCTA-3’ to detect the wildtype allele. Band sizes were 395bp for wildtype and 273bp for mutant. Mice were genotyped for the R79Q mutation using primers 223_2F: 5’-TCACTTGCAACAGAGCATCC-3’ and 223_2R: 5’-ACAGCTCTGCCCGTGTTTAC-3’ followed by Sanger sequencing the 304bp PCR product.
Body composition analysis and fat mass measurement:
Lean mass and fat mass were measured using the EchoMRI™ system (Houston, TX). Perigonadal fat pads were removed from the mouse carcass manually and weighed. (n ≥ 5 per group).
Mouse metabolism studies:
Energy expenditure was measured by the Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments, Columbus, Ohio). Mice were tested over 96 continuous hours, and the data from the last 48 hours were analyzed. Energy expenditure is expressed in terms of kilocalories per hour and calculated using the Lusk equation: EE = (3.815 + 1.232 × respiratory exchange ratio [RER]) × oxygen consumption rate (VO2), and was analyzed with CalR software55 (ANCOVA with body weight used as a covariate).
Food intake:
Mice were single housed for 7 days and food intake was measured daily by weighing the food in the food hopper. The difference in food weight was calculated each day. The average food weight between groups was taken (n ≥ 6 per group).
Glucose tolerance test:
Mice were fasted for 16 hours and injected with 1 g/kg body weight glucose intraperitoneally (Sigma G7021). Blood glucose was measured at 0, 10, 20, 30, 60, 90 and 120 minutes via tail sampling with the AlphaTrak glucose meter (Abbott Laboratories). A blood sample for fasting glucose levels was taken at time point 0, before glucose was injected (n ≥ 9 per group) (age ≥ 11 weeks).
Insulin tolerance test:
Mice were fasted for 3 hours and subsequently injected with 0.75 U/kg body weight regular human insulin (Humulin R, Lilly USA). Blood glucose levels were measured at 0, 15, 30, 45, and 60 minutes. A blood sample for fasting insulin levels was taken at time point 0, before insulin was injected (n ≥ 10 per group) (age ≥ 11 weeks).
Blood serum analysis:
Blood from animals was allowed to coagulate at room temperature for 30 minutes. After being centrifuged at 3500 rpm for 10 min at room temperature, serum was collected and stored at −80°C for later analysis. Insulin ELISA- insulin levels were determined with Crystal Chem’s ultra-sensitive mouse insulin ELISA kit according to the manufacturer’s protocol (catalog # 90080). The insulin concentration of each sample was determined by interpolation using a standard curve generated by plotting absorbance versus corresponding concentration in Excel (n ≥ 6 per group). Leptin ELISA- leptin levels were determined with Crystal Chem’s mouse leptin ELISA kit according to the manufacturer’s protocol (catalog # 90030). The leptin concentration of each sample was determined by interpolation using a standard curve generated by plotting absorbance versus corresponding concentration in Excel (n ≥ 6 per group). Mean absorbance at 450nm and 630nm was measured on a FLUOstar Omega microplate reader.
Tissue harvesting and preparation:
Mice were euthanized at the indicated ages by isoflurane inhalation followed by perfusion with ice-cold PBS and ice-cold 4% paraformaldehyde (PFA). Brain and pancreas tissues were harvested following perfusion and post-fixed with 4% PFA. Tissues were then incubated in 30% sucrose in 0.1 M phosphate buffer at 4°C until tissues sank in solution. Samples were then placed in optimal cutting temperature compound (Tissue-Tek OCT, Sakura Finetek) and embedded and frozen on dry ice.
Immunohistochemistry:
OCT-embedded tissues were sectioned at 12 μm or 20 μm. Sections were rehydrated and blocked in antibody wash (5% heat inactivated goat serum, 0.1% Triton X-100 in Tris-Buffered Saline) for 45 minutes. Tissues were incubated with primary antibodies overnight at 4°C, washed three times with 0.1% Triton X-100 in Tris-Buffered Saline (TBST), and incubated with secondary antibodies for 1 hour at room temperature. Tissues were washed three times with TBST and incubated with Hoechst 33342 for 5 minutes. Slides were coverslipped with ProLong Gold (ThermoFisher) mounting media and imaged on a BioTek Lionheart FX microscope. Primary antibodies used were: mouse anti-ARL13B (1:1000, NeuroMab, N295B/66); rabbit anti-acetylated α-tubulin (1:1000,Cell Signaling, 5335); mouse anti-acetylated α-tubulin (1:2500, Sigma, T7451); chicken anti-ACIII (1:1000, Encor, CPCA-ACIII); rat anti-insulin (1:1000, R&D systems, MAB1417); rabbit anti-glucagon (1:2000, Abcam, ab92517). Secondary antibodies used were goat anti-mouse AlexaFluor 488, goat anti-chicken AlexaFluor 647, goat anti-rat AlexaFluor 568 and donkey anti-rabbit AlexaFluor 555 (1:500, ThermoFisher).
RNAScope:
Brains from adult mice were harvested, fixed and prepared for RNAscope in situ hybridization56,57. Briefly, 15μm cryosections were mounted on slides then post-fixed with 4% PFA for 16hr at 4°C. Detection of transcripts was performed using the RNAscope 2.5 HD Duplex Assay (Advanced Cell Diagnostics (ACD), Newark, CA). Tissue pretreatment was performed according to technical note 320534 Rev A. Probe hybridization, counterstaining, and mounting of slides was performed according to user manual no. 322500-USM Rev A. Slides were assayed using probes to AgRP (Cat No. 400711-C2) and POMC (Cat No. 314081-C2) transcripts (ACD). Sections were counterstained with hematoxylin, dehydrated, and mounted using VectaMount (Vectorlabs, Burlingame, CA). Slides with positive control probe (PPIB-C1/POLR2A-C2; Cat No. 321651) and negative control probe (DapB; Cat no. 320751) were run with each experiment. At least 3 animals were analyzed for each group.
Funding
This work was supported by the National Institutes of Health: diversity supplement to R35GM122549 (TTT); T32NS096050, diversity supplement to R01NS090029 and F31NS106755 (EDG); Larry L. Hillblom Foundation fellowship (CMA), University Fellowship (KMB); American Heart Association pre-doctoral fellowship (XY), R01DK114008 (NFB); R01DK124769, R01DK106404 and R01DK060540 (CV) and R01NS090029, R35GM122549 and R35GM148416 (TC).
References
- 1.Nishimura Y., Kasahara K., Shiromizu T., Watanabe M. & Inagaki M. Primary Cilia as Signaling Hubs in Health and Disease. Adv. Sci. 6, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hearn T. et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat. Genet. 31, 79–83 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Hearn T. et al. Subcellular Localization of ALMS1 Supports Involvement of Centrosome and Basal Body Dysfunction in the Pathogenesis of Obesity, Insulin Resistance, and Type 2 Diabetes. Electrophoresis 54, 1581–1587 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Knorz V. J. et al. Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol. Biol. Cell 21, 3617–3629 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mykytyn K. & Sheffield V. C. Establishing a connection between cilia and Bardet-Biedl syndrome. Trends Mol. Med. 10, 106–109 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Berbari N. F., Lewis J. S., Bishop G. A., Askwith C. C. & Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc. Natl. Acad. Sci. U. S. A. 105, 4242–4246 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin H. et al. The conserved bardet-biedl syndrome proteins assemble a coat that traffics membrane proteins to Cilia. Cell 141, 1208–1219 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nachury M. V. et al. A Core Complex of BBS Proteins Cooperates with the GTPase Rab8 to Promote Ciliary Membrane Biogenesis. Cell 129, 1201–1213 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Liew G. M. et al. The intraflagellar transport protein ift27 promotes bbsome exit from cilia through the gtpase ARL6/BBS3. Dev. Cell 31, 265–278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engle S. E., Bansal R., Antonellis P. J. & Berbari N. F. Cilia signaling and obesity. Semin. Cell Dev. Biol. 110, 43–50 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davenport J. R. et al. Disruption of Intraflagellar Transport in Adult Mice Leads to Obesity and Slow-Onset Cystic Kidney Disease. Curr. Biol. 17, 1586–1594 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y. et al. Melanocortin 4 receptor signals at the neuronal primary cilium to control food intake and body weight. J. Clin. Invest. 131, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hughes J. W. et al. Primary cilia control glucose homeostasis via islet paracrine interactions. Proc. Natl. Acad. Sci. U. S. A. 117, 8912–8923 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaisse C., Reiter J. F. & Berbari N. F. Cilia and obesity. Cold Spring Harb. Perspect. Biol. 9, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larkins C. E., Gonzalez Aviles G. D., East M. P., Kahn R. A. & Caspary T. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol. Biol. Cell 22, 4694–4703 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Z. et al. A genetic screen in zebrafish indentifies cilia genes as a principal cause of cystic kidney. Development 131, 4085–4093 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Caspary T., Larkins C. E. & Anderson K. V. The Graded Response to Sonic Hedgehog Depends on Cilia Architecture. Dev. Cell 12, 767–778 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Kahn R. A., East M. P. & Francis J. W. ARF-Like (ARL) Proteins, in: Wittinghofer A. (Ed.), Ras Superfamily Small G Proteins: Biology and Mechanisms 2. Springer International Publishing, (2014). doi: 10.1007/978-3-319-07761-1 [DOI] [Google Scholar]
- 19.Zhou C., Cunningham L., Marcus A. I., Li Y. & Kahn R. A. Arl2 and Arl3 Regulate Different Microtubule-dependent Processes. Mol. Biol. Cell 17, 2476–2487 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gotthardt K. et al. A G-protein activation cascade from Arl13B to Arl3 and implications for ciliary targeting of lipidated proteins. Elife 4, 1–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q. et al. GTP-binding of ARL-3 is activated by ARL-13 as a GEF and stabilized by UNC-119. Sci. Rep. 6, 1–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humbert M. C. et al. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. U. S. A. 109, 19691–19696 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujisawa S. et al. ARL3 and ARL13B GTPases participate in distinct steps of INPP5E targeting to the ciliary membrane. Biol. Open 10, 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu H., Fujisawa S., Nozaki S., Katoh Y. & Nakayama K. Interaction of INPP5E with ARL13B is essential for its ciliary membrane retention but dispensable for its ciliary entry. Biol. Open 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chávez M. et al. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Dev. Cell 34, 338–350 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Gonzalo F. R. et al. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev. Cell 34, 400–409 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bielas S. L. et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 41, 1032–1036 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cantagrel V. et al. Mutations in the Cilia Gene ARL13B Lead to the Classical Form of Joubert Syndrome. Am. J. Hum. Genet. 83, 170–179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas S. et al. Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. Eur. J. Hum. Genet. 23, 621–627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaheen R. et al. Characterizing the morbid genome of ciliopathies. Genome Biol. 17, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bachmann-Gagescu R. et al. Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 52, 514–522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivanova A. A. et al. Biochemical characterization of purified mammalian ARL13B protein indicates that it is an atypical GTPase and ARL3 guanine nucleotide exchange factor (GEF). J. Biol. Chem. 292, 11091–11108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacoby M. et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 41, 1027–1031 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Xu W. et al. The joubert syndrome protein Inpp5e controls ciliogenesis by regulating phosphoinositides at the apical membrane. J. Am. Soc. Nephrol. 28, 118–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gigante E. D., Taylor M. R., Ivanova A. A., Kahn R. A. & Caspary T. Arl13b regulates sonic hedgehog signaling from outside primary cilia. Elife 9, 1–23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suciu S. K., Long A. B. & Caspary T. Smoothened and ARL13B are critical in mouse for superior cerebellar peduncle targeting. Genetics 218, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Sciver R. E., Long A. B., Katz H. G., Gigante E. D. & Caspary T. Ciliary ARL13B inhibits developmental kidney cystogenesis in mouse. Dev. Biol. 500, 1–9 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosario W. et al. The brain-to-pancreatic islet neuronal map reveals differential glucose regulation from distinct hypothalamic regions. Diabetes 65, 2711–2723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coleman D. L. & Eicher E. M. Fat (fat) and tubby (tub): Two autosomal recessive mutations causing obesity syndromes in the mouse. J. Hered. 81, 424–427 (1990). [DOI] [PubMed] [Google Scholar]
- 40.Kleyn P. W. et al. Identification and Characterization of the Mouse Obesity Gene. Cell 85, 281–290 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Noben-trauth K., Naggert J. K., Northt M. A. & Nishina P. M. A candidate gene for the mouse mutation tubby. 380, 534–538 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Han S. et al. TULP3 is required for localization of membrane-associated proteins ARL13B and INPP5E to primary cilia. Biochem. Biophys. Res. Commun. 509, 227–234 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Palicharla V. R. et al. Interactions between TULP3 tubby domain and ARL13B amphipathic helix promote lipidated protein transport to cilia. Mol. Biol. Cell 34, ar18 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukhopadhyay S. & Jackson P. K. The tubby family proteins. Genome Biol. 12, 1–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delous M. et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 39, 875–881 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Arts H. H. et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 39, 882–888 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Stratigopoulos G. et al. Hypomorphism for RPGRIP1L, a ciliary gene vicinal to the fto locus, causes increased adiposity in mice. Cell Metab. 19, 767–779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stratigopoulos G. et al. Hypomorphism of Fto and Rpgrip1l causes obesity in mice. J. Clin. Invest. 126, 1897–1910 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L. et al. Ciliary gene RPGRIP1L is required for hypothalamic arcuate neuron development. JCI Insight 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiegering A. et al. Rpgrip1l controls ciliary gating by ensuring the proper amount of Cep290 at the vertebrate transition zone. Mol. Biol. Cell 32, 675–689 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mariani L. E. et al. Arl13b regulates Shh signaling from both inside and outside the cilium. Mol. Biol. Cell 27, 3780–3790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alkanderi S. et al. ARL3 Mutations Cause Joubert Syndrome by Disrupting Ciliary Protein Composition. Am. J. Hum. Genet. 103, 612–620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai J. et al. Loss of ARL13 impedes BBSome-dependent cargo export from Chlamydomonas cilia. J. Cell Biol. 221, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cevik S. et al. Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J. Cell Biol. 188, 953–969 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mina A. I. et al. CalR: A Web-Based Analysis Tool for Indirect Calorimetry Experiments. Cell Metab. 28, 656–666.e1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Antonellis P. J., Engle S. E., Brewer K. M. & Berbari N. F. The hedgehog signaling pathway is expressed in the adult mouse hypothalamus and modulated by fasting. eNeuro 8, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Engle S. E. et al. A CreER mouse to study melanin concentrating hormone signaling in the developing brain. Genesis 56, 1–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]