

SUMMARY
DNA damage-activated signaling pathways are critical for coordinating multiple cellular processes, which must be tightly regulated to maintain genome stability. To provide comprehensive and unbiased perspective of DDR signaling pathways, we performed 30 fluorescence-activated cell sorting–based genome-wide CRISPR screens in human cell lines with antibodies recognizing distinct endogenous DNA damage–signaling proteins to identify critical regulators involved in DNA damage response (DDR). We discovered that proteasome-mediated processing is an early and prerequisite event for cells to trigger camptothecin- and etoposide-induced DDR signaling. Furthermore, we identified PRMT1 and PRMT5 as modulators that regulate ATM protein level. Moreover, we discovered that GNB1L is a key regulator of DDR signaling via its role as a co-chaperone specifically regulating PIKK proteins. Collectively, these screens offer a rich resource for further investigation of DDR, which may provide insight into strategies of targeting these DDR pathways to improve therapeutic outcomes.
Graphical Abstract
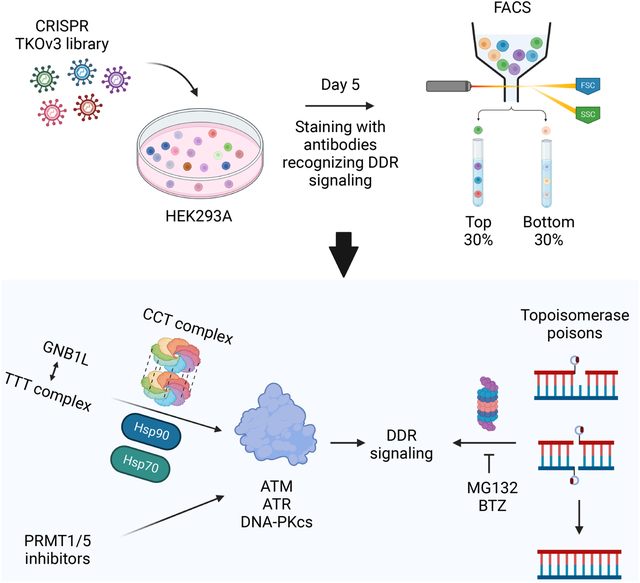
eTOC Blurb (In brief)
Huang et al. performed FACS-based CRISPR screens with antibodies recognizing DDR signaling proteins and identified regulators involved in DDR signaling pathways, which demonstrate the feasibility and power of FACS-based screens for the studies of signal transduction pathways.
INTRODUCTION
DNA, the carrier of genetic information, must be precisely maintained and passed to the next generation. However, cells inevitably encounter many threats to the integrity of their genetic information. These threats, such as exposure to DNA-damaging agents and endogenous and/or induced replication stress, can lead to DNA lesions that must be detected and repaired immediately. Therefore, timely sensing of DNA damage and further transducing these signals to downstream effectors to initiate DNA repair, cell-cycle checkpoint control, or other downstream pathways is of great importance for genome maintenance. These processes together are termed DNA damage response (DDR), which include DDR signaling, DNA repair, and cell-cycle regulation that work together to cope with DNA lesions1–4.
Cells have evolved specific repair mechanisms for many types of DNA lesions. For example, base excision repair pathway works on small chemical alterations of DNA bases, nucleotide excision repair removes damaged oligonucleotide-containing bases, and mismatch repair directly replaces misincorporated DNA bases with correct ones. Double strand breaks, the most deleterious DNA lesions, can be repaired by homologous recombination and non-homologous end joining in an error-free and error-prone fashion, respectively4–6.
Besides these different DNA repair pathways, cells have also evolved pathways to sense DNA damage and transduce these signals to downstream effectors and/or specific repair pathways. Just like the employment of various DNA repair pathways, cells also use different sensors to initiate DDR signaling pathways. In particular, the MRE11/RAD50/NBS1 (MRN) and KU70/80 complexes bind to double strand breaks to recruit ATM and DNA-dependent protein kinase catalytic subunit (DNA-PKcs), respectively, whereas single strand breaks at stalled replication forks are coated by replication protein A (RPA), which recruits ATR1–3. Once these DNA damage sensors and the cirtical transducers, namely, the phosphatidylinositol 3-kinase–like protein kinase (PIKK) family members, consisting of ATM, ATR, and DNA-PKcs, are recruited to sites of DNA damage, these PIKKs are activated and spread the DNA damage signals to downstream effectors by phosphorylating large numbers of substrates and further stimulating cellular response to cope with DNA damage7. Because these PIKKs play critical roles in DDR, inhibitors of PIKKs have been developed, which are in clinical trials8–10. Therefore, comprehensive understanding of DDR signaling network will not only help us appreciate the intricacy of these DDR signaling pathways but also may provide therapeutic opportunities for the application of these inhibitors in clinical settings.
Genome-wide CRISPR/Cas9 screening, a powerful and unbiased approach, has been widely used to uncover genetic interactions and expand knowledge in many scientific disciplines, including DDR. The CRISPR screens performed in the DDR field have led to the discovery of novel factors such as shieldin as well as new vulnerabilities to anticancer agents, such as RNASEH2 loss to ATR and poly(ADP-ribose) polymerase inhibitors11–13. A recent study also illustrated a genetic map of DDR in human cells14, which further demonstrates the power of CRISPR screens.
Besides genetic screens, fluorescence-activated cell sorting (FACS)–based CRISPR/Cas9 screens using reporter cell lines can be used to study specific signaling pathways with the goal of identifying key regulators involved in these signaling pathways. For example, KIRREL and KMT2A were identified as new factors involved in the Hippo and Wnt signaling pathways, respectively, via CRISPR screens with a Hippo GTII-dRFP reporter and endogenous Wnt β-catenin reporter15,16. However, these are potential artifacts associated with the use of engineered cell lines15. Alternatively, genome-wide small interfering RNA screens with antibodies such as those against γH2AX and RAD51 have been conducted to identify regulators and pathways contributing to these signals17,18. Unfortunately, the off-target effect of small interfering RNAs and high cost of these microscopy-based analyses limited the application of the small interfering RNA screens. Here, we reason that FACS-based CRISPR screens with antibodies specifically recognizing endogenous signals can be employed as a promising strategy for uncovering the regulatory network of any signaling pathway including DDR.
As described above, DDR signaling is critically important for sensing DNA lesions and transducing the signals to downstream effectors to facilitate DNA repair and initiate cell-cycle checkpoint control. DDR signaling is mostly regulated through posttranslational modifications (PTMs) of proteins, which can be examined by antibodies specifically recognizing the modified proteins. Thus, we carried out FACS-based genome-wide CRISPR screens with six antibodies recognizing different endogenous DDR signals in untreated cells and cells treated with four distinct types of DNA-damaging agents. These six antibodies and five conditions produced a total of 30 genome-wide screens that facilitate our understanding of the sophisticated DDR network. These studies not only demonstrated a positive role for the proteasome-mediated pathway in regulating camptothecin (CPT)- and etoposide (ETO)-induced DDR signaling but also identified PRMT1 and PRMT5 as regulators of ATM. Moreover, we identified and validated GNB1L as a specific regulator of PIKKs, probably by functioning as a co-chaperone for PIKKs. These studies not only provide broad insight into the DDR signaling network but also demonstrate the feasibility and power of FACS-based screens applicable to many other signal transduction pathways that are important for human health.
RESULTS
Workflow of FACS-based CRISPR screens
Before conducting FACS-based CRISPR screens with antibodies recognizing DDR signaling proteins, we first determined whether the available antibodies could be used for FACS. We examined eight antibodies specifically recognizing γH2AX (S139), pKAP1 (S824), pRPA2 (S4/S8), pRPA2 (S33), pATM (S1981), pChk2 (T68), pATR (S1989), and pChk1 (S345) in the absence or presence of CPT exposure to determine whether CPT-induced DNA damage signals can be detected by FACS (Figure S1). We observed marked or noticeable changes, exhibiting as shift by FACS analyses, when using the antibodies recognizing γH2AX, pKAP1, pRPA2 (S4/S8), pRPA2 (S33), pATM, and pChk2 but not those recognizing pATR or pChk1 in CPT-treated samples when compared to those in untreated control samples. We reasoned that these antibodies can be used to detect DDR signaling via FACS. Thus, we conduct a total of 30 FACS-based genome-wide screens using these six antibodies under five experimental conditions: no treatment or treatment with ionizing radiation (IR), CPT, ETO, or hydroxyurea (HU).
As described previously19, we conducted CRISPR screens by infecting HEK293A cells with the TKOv3 lentiviral library and selection with puromycin, we then cultured sgRNA-infected cells for 5 days. On day 5, we exposed cells to different treatments, which was followed by fixation, immunostaining, and sorting (Figure 1A). We collected the top 30% cell populations (those with the highest signals) and bottom 30% cell populations (those with the lowest signals) for subsequent next-generation sequencing. We conducted bioinformatic analysis by comparing the enrichment of sgRNAs in the top 30% and bottom 30% cell populations using DrugZ analysis20 to identify both positive and negative regulators of DDR signals (Figures 1A and S2). Of note, the hits with negative NormZ values were positive regulators whose depletion led to reduced signals. On the other hand, hits with positive NormZ values were negative regulators, defects of which resulted in increased signals. These data provided a comprehensive view of the regulatory network controlling DDR signaling pathways (Tables S1 and S2).
Figure 1. Workflow for FACS-based CRISPR screens.
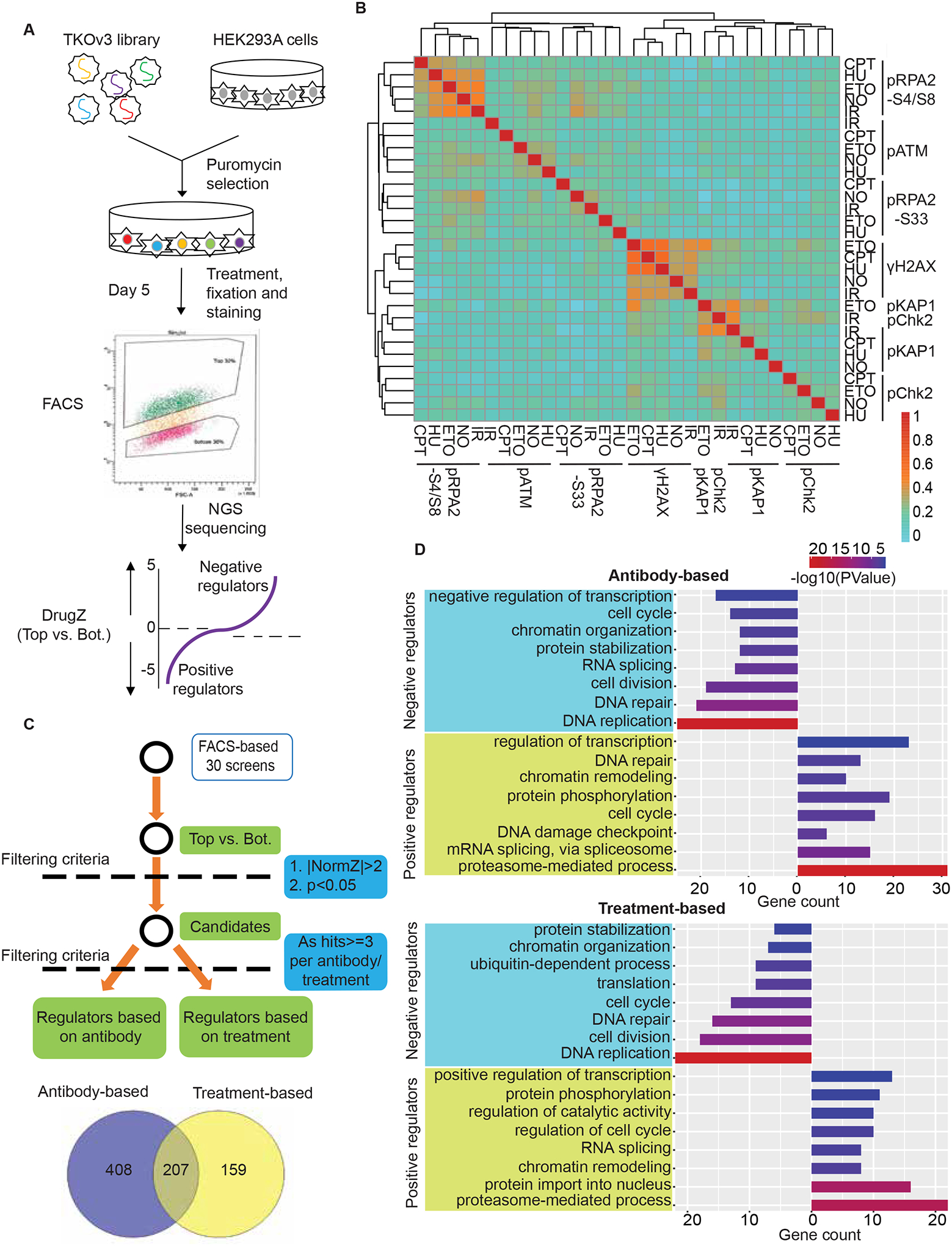
(A) Workflow of the FACS-based CRISPR screening strategy.
(B) Heat map shows the correlation of 30 screens based on NormZ scores. Also see Table S1.
(C) The criteria for analyzing positive and negative regulators of DDR signaling.
(D) Representative GO terms enriched in antibody-based analyses (top panel) and treatment-based analyses (bottom panel) of negative/positive regulators of DDR signaling. The GO term list is provided in Table S3.
The correlation among the 30 screens based on NormZ scores is shown as a heat map in Figure 1B. We performed each screen with two replicates. Screens performed with the same antibody regardless of treatment are clustered together in the heat map. Next, we employed the following criteria for identifying both potential positive and negative regulators of DDR signals: candidates with NormZ values either less than −2 or greater than 2 as well as p values less than 0.05 (Figure 1C). Furthermore, we applied an additional threshold to these candidates, which must be detected as hits three or more times, because we focused on uncovering common regulators of DDR signaling pathways instead of regulators that only function in one or two specific contexts in this study. Common regulators are identified based on the antibody (i.e., DDR signaling indicated by a specific antibody) or treatment, consisting of 615 and 366 hits, respectively. Specifically, we identified 207 overlapping hits as well as 159 and 408 hits exclusively in treatment- and antibody-based analysis, respectively (Figure 1C).
Notably, the IR-treated screen with the anti-pChk2 antibody clustered very tightly with that with the anti-pKAP1 antibody (Figure 1B), which could be explained by that ATM is the major kinase activated following IR exposure and is responsible for both pChk2 and pKAP1. KAP1, also known as TRIM28 or TIF1β, was identified as a transcriptional co-repressor due to its interaction with a large KRAB-containing zinc finger protein (KRAB-ZFP) transcription factor family21–24. Different from its canonical function in transcriptional control, the phosphorylated KAP1 (S824) is known to participate in DNA repair via contributing to ATM-mediated chromatin relaxation25. DrugZ analysis showed that TRIM28 was the top hit in the IR-treated pChk2 screen (Figure S3A). Moreover, hits identified in the IR-treated pChk2 and pKAP1 screens exhibited considerable overlap (Figure S3B), raising the possibility that the antibody against pChk2 recognizes both pChk2 and pKAP1. We confirmed this via Western blotting (Figure S3C). These results suggested that FACS-based CRISPR screening is a promising strategy for antibody validation, especially for antibodies that require high specificity. Indeed, researchers at Thermo Fisher Scientific have proposed this to accelerate neurobiology research. Ideally, the target gene should be one of the top hits.
To gain further insight into the negative and positive regulatory networks of DDR signaling pathways, we performed functional term enrichment analysis using both antibody- and treatment-based hits (Figure 1D; Table S3). Positive regulators were enriched for Gene Ontology (GO) terms directly associated with DDR signaling, such as DNA damage checkpoint (GO:0000077), DNA repair (GO:0006281), cell cycle (GO:0007049), and protein phosphorylation (GO:0006468). Moreover, chromatin remodeling (GO:0006338) appeared in this analysis. Although chromatin remodeling is not directly associated with DDR signaling, this process is well known to be highly relevant to DNA damage signaling, as decondensation of chromatin around DNA lesions is required to facilitate DNA repair, and chromatin needs to be condensed once DNA lesions are repaired26. Other cellular processes not directly associated with DDR signaling are transcription-related pathways such as mRNA splicing (GO:0000398) and DNA-templated regulation of transcription (GO:0006355). The identification of these processes is consistent with current knowledge that R-loop formation may result in DDR signaling27, but of course these processes may also participate in DDR signaling via other mechanisms. Functional terms such as cell cycle, DNA repair, and RNA splicing are also highly enriched in hits of negative regulators. Notably, DNA replication (GO:0006260) is specifically and most significantly enriched as a negative regulator of DDR signaling, suggesting that defects in DNA replication result in replication stress, which further enhance DDR signaling28. Intriguingly, the proteasome-mediated, ubiquitin-dependent protein catabolic process is significantly enriched in GO terms as the pathway that positively regulates DDR signaling (Figure 1D), implying that proteasome inhibition leads to impaired DDR signaling.
The regulatory network of DDR signaling
To obtain a global view of the regulatory network contributing to DDR signaling, we used the 615 and 366 hits identified via antibody- and treatment-based analysis for further evaluation. Since we were initially aiming to identify regulators of DDR signaling, we first analyzed the data in antibody-based perspective (Figures 2A–2C), but we found that these data could also be analyzed in the perspective of treatment type (Figures S4A–S4C), which could uncover proteins specifically involved in DDR signaling in response to certain DNA damaging agent-induced lesions. For example, we identified several well-known proteins involved in resolving the CPT-induced TOP1 cleavage complex and ETO-induced TOP2 cleavage complex in our screens regardless of the antibodies used but these hits exhibited exquisitely restricted profiles (Figures S4B and S5A–S5D). We identified TOP1 and TDP1 as positive and negative regulators in CPT-treated screens respectively, in most if not all the CPT-treated screens. Similarly, we identified TOP2A, TDP2, and ZNF451 among the top hits in the ETO-treated screens. These findings are consistent with the current working models of the removal and repair of the TOP1 and TOP2 cleavage complexes. For example, TDP2 and ZNF451 were uncovered as negative regulators in the ETO-treated screens, which corresponds to their known functions, i.e. TDP2 hydrolase has a unique role in removal of the TOP2 cleavage complex, which is further enhanced by the SUMO ligase ZNF45129,30. Moreover, ABCC1 is a multidrug resistance-associated protein, which mediates resistance to etoposide and daunorubicin31–33. Consistently, ABCC1 was identified as a specific negative regulator in response to ETO treatment (Figures S4B and S5A). In addition, we identified RECQL5 as a negative regulator in both CPT- and ETO-treated screens, which was more obvious in CPT-treated screens (Figures S4B, S5A and S5D). RECQL5 has not been fully studied, although it belongs to RecQ family of helicases which consists of RECQL1, BLM, WRN, RECQL4 and RECQL5,. Unlike BLM, WRN and RECQL4, which are associated with Bloom Syndrome, Werner Syndrome and Rothmund- Thompson Syndrome respectively, RECQL5 and RECQL1 remain to be explored for their correlations with specific diseases. Nevertheless, it is known that RECQL5 participates in DSB repair, regulation of DNA replication stress as well as resolution of conflicts between transcription and DNA replication34–36, and therefore may be involved in DDR. Determining precisely how RECQL5 negatively modulates the response to these topoisomerase inhibitors requires further investigation. Thus, our data suggest that FACS-based screens can be used effectively for studying any stimuli or treatment, in this case, treatment with TOP1 or TOP2 poisons.
Figure 2. The regulatory network of DDR signaling revealed by antibody-based analyses.
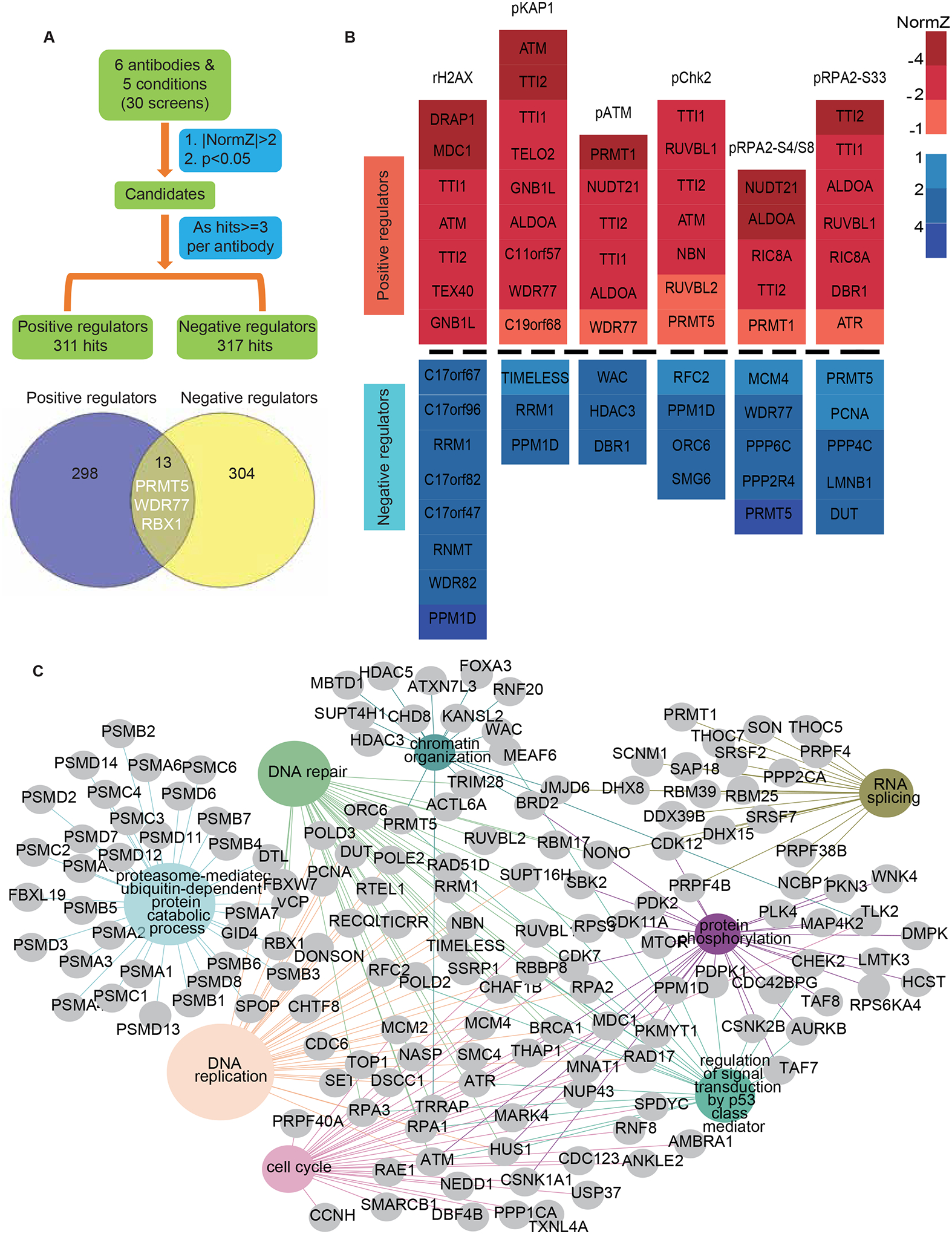
(A) The criteria for analyzing positive and negative regulators of DDR signaling based on antibody.
(B) Heatmap plots show the normZ values of hub candidates. Red and blue colors represent positive and negative regulators, respectively. NormZ values were grouped by their absolute values labelled by different colors.
(C) Representation of the Reactome pathway network for the proteins in A. The pathways highlighted in color were significantly enriched. The GO term list is provided in Table S4.
Comparing to the hits exclusively enriched in treatment-based analysis, which includes TDP1, TDP2, ZNF451 and ABCC1, the antibody-based analysis also identified TOP1 and TOP2A, which are known to regulate replication and transcription30 therefore possibly regulating some DDR signals. Besides the same hits uncovered between these two analyses, the antibody-based analysis also identified specific regulators contributing to DDR signaling regardless of the treatment used. For example, MDC1 was identified as a specific positive regulator contributing to γH2AX, which is consistent with its known function (Figure 2B). Moreover, PRMT5 and WDR77 were uncovered as specific positive regulators to signals of pATM and pKAP1 as well as negative regulators to that of pRPA2(S4/8) and pRPA2(S33) (Figure 2B), which were later validated with PRMT5 inhibitor.
Many of the common hits that were identified in both antibody- and treatment- based perspective are involved in DNA damage checkpoints, DNA repair, DNA replication, the cell cycle, p53 class mediator-regulated signal transduction, proteasome-mediated process, protein phosphorylation, RNA splicing, and chromatin organization (Figures 2C and S4C; Table S4). As expected, we identified well-known genes involved in DNA damage checkpoints as well as detection of DNA damage, such as ATM, ATR, RPA1, RPA2, RPA3, HUS1, RAD17, CHK2, MRE11A, RAD50, and NBN. Several well-established DNA repair pathways were also enriched, including homologous recombination, non-homologous end joining, nucleotide excision repair, and base excision repair. As described above, DNA replication was enriched as a negative regulator of DDR signaling. Additionally, we identified pre-replicative complex assembly, DNA replication initiation, and DNA replication regulation as the core subnetwork together with DNA replication.
Several genes that are unassigned and/or have functions outside of their known functional networks may deserve further investigation. These genes include C11orf57, C21orf59, C19orf68, C17orf67, ALDOA, NUDT21, and GNB1L. For example, NUDT21 was 1 of the 5 most frequent hits in our screens (Figure S6A). Moreover, NUDT21 was the outlier in the screens with the anti-pRPA2 (S4/S8) antibody (Figure S6B). These data imply that NUDT21 affects DDR signaling globally. A recent report identified the RNA-processing factor Nudt21 as a novel regulator of cell fate through chromatin signaling37, but its role in DDR remains unclear, which warrants additional inquiry. ALODA, which encodes a fructose-bisphosphate aldolase A38, was identified as another common positive regulator (Figure S6A). We therefore examined whether Aldolase (ALDOA) knockdown would broadly reduce DDR signaling. We employed two different short hairpin RNAs (shRNAs) targeting ALDOA, which significantly reduced endogenous ALDOA protein level in HEK293A cells (Figure S6C). Indeed, ALDOA knockdown markedly reduced CPT-, ETO- or HU-induced γH2AX, pRPA2-S4/S8, and pRPA2-S33 but not RPA2 protein level (Figure S6C). In addition, we validated these results in U2OS cells (Figure S6D). Since ALDOA is a key metabolic enzyme, we speculate that ALDOA regulates DDR signaling indirectly.
Furthermore, we validated several other hits with unknown functions such as C21orf59 and C11orf57, which were identified as specific positive regulators of DDR signaling in our screens. C11orf57 was identified as a specific positive regulator of pKAP1 signals (Figure S7A). However, the commercial C11orf57 antibody only recognized exogenously expressed proteins, but not endogenous C11orf57. We chose three sgRNAs that could decrease the exogenously expressed C11orf57 by co-expression of C11orf57 with its sgRNAs in HEK293A cells (Figure S7B). We showed that cells infected with the indicated sgRNAs of C11orf57 led to decreased CPT-induced pKAP1 when compared with those in control cells (Figure S7C). However, the lack of commercial antibody that recognizes endogenous C11orf57 limited further investigation of C11orf57.
In addition, C21orf59 was identified as a positive regulator in the screens with antibodies recognizing endogenous pRPA2S33 (Figure S7D). We chose three shRNAs (shRNA2, shRNA4 and shRNA6), which reduced endogenous C21orf59 protein level (Figure S7E). As shown in Figure S7F, knockdown C21orf59 with three different shRNAs impaired CPT-induced pRPA2-S33. Consistent with the flow cytometry results, knockdown C21orf59 impaired CPT-induced pRPA2S33 as well as pChk1 and pKAP1, but only modestly affected expression levels of RPA2 and DDR kinases (Figure S8A). Intriguingly, knockdown C21orf59 also impaired the basal pRPA2S33 level (Figure S8A). Therefore, we further tested whether C21orf59 could regulate chromatin-associated RPA2 level. Consistent with CPT-induced increasing level of pRPA2S33, we observed that chromatin-bound RPA2 increased after CPT treatment comparing to those in control cells (Figure S8B). Notably, C21orf59 knockdown markedly impaired chromatin-associated RPA2 in the absence or presence of CPT treatment, but only modestly affected the soluble and whole cell levels of RPA2 (Figure S8B), implying that C21orf59 may affect DNA replication. We then performed immuno-staining with EdU incorporation to detect S phase cells. The results showed that C21orf59 depletion reduced the percentage of EdU positive cells when compared to that in control cells (Figure S8C). Next, we examined the localization of C21orf59, which may provide additional insight into its effect on DNA replication. Unfortunately, the commercial antibody we used for Western blotting could not recognize endogenous C21orf59 by immunostaining. We thus performed chromatin fractionation assay to examine chromatin-bound C21orf59 in the absence or presence of CPT treatment. As shown in Figure S8D, the amount of chromatin-bound RPA2 increased after CPT treatment (1 μM and 5 μM) when compared to that in DMSO-treated cells. However, we could not detect chromatin-associated C21orf59 with or without CPT treatment, suggesting that C21orf59 is not recruited or recruited at a very low level to DNA damage sites. To determine whether C21orf59 knockdown would sensitize cells to different genotoxic agents, we performed CellTiter-Glo assay by employing two different shRNAs targeting C21orf59 in HEK293A cells and showed that C21orf59 knockdown cells only exhibited modest sensitivity to HU (Figure S8E). Since C21orf59 is a common essential gene according to depmap portal, we speculated that C21orf59 may indirectly regulate DDR signaling via its effect on DNA replication and/or cell survival.
To visualize genes enriched in the network, we zoomed in on the network for some clearly defined submodules, especially the pathways that are not directly associated with DDR signaling (Figures 2C and S4C). For example, PRMT5 and PRMT1, both belonging to the protein arginine methyltransferase family39, were enriched in chromatin organization and RNA splicing, respectively. Although their roles in DDR pathways were implicated in previous studies, their functions in DDR signaling have yet to be fully elucidated.
Proteasome inhibition reduces both CPT- and ETO-induced DDR signaling
The GO analysis (Figure 1D) and network analysis (Figure 2C) described above strongly suggested that the proteasome-mediated process is positively involved in DDR signaling. Intrigued by these results, we examined whether suppressing proteasome activity reduces DDR signaling. Treatment of HEK293A cells with MG132, a commonly used proteasome inhibitor, for 1 h prior to DNA damaging treatments markedly abolished both CPT- and ETO-induced DDR signaling but had a mild or no effect on IR- and HU-triggered DNA damage signals (Figure 3A). A recent report indicated that cells pretreated with different proteasome inhibitors displayed drastically decreased ETO-induced γH2AX40. We observed not only clear reduction in γH2AX and pKAP1 but also a modest decrease in pRPA2 (S33) signals, which were induced by exposing cells to CPT or ETO (Figure 3A). Additionally, we observed the suppressive effect of proteasome inhibition on CPT- and ETO-induced DDR signaling in both HeLa and U2OS cells (Figure S9A). Moreover, CPT or ETO treatment increased chromatin-bound topoisomerases when cells were preincubated with MG132 (Figure S9B), indicating that chromatin-bound topoisomerases are stabilized upon treatment with proteasome inhibitors.
Figure 3. Proteasome inhibition reduces both CPT- and ETO-induced DDR signaling.
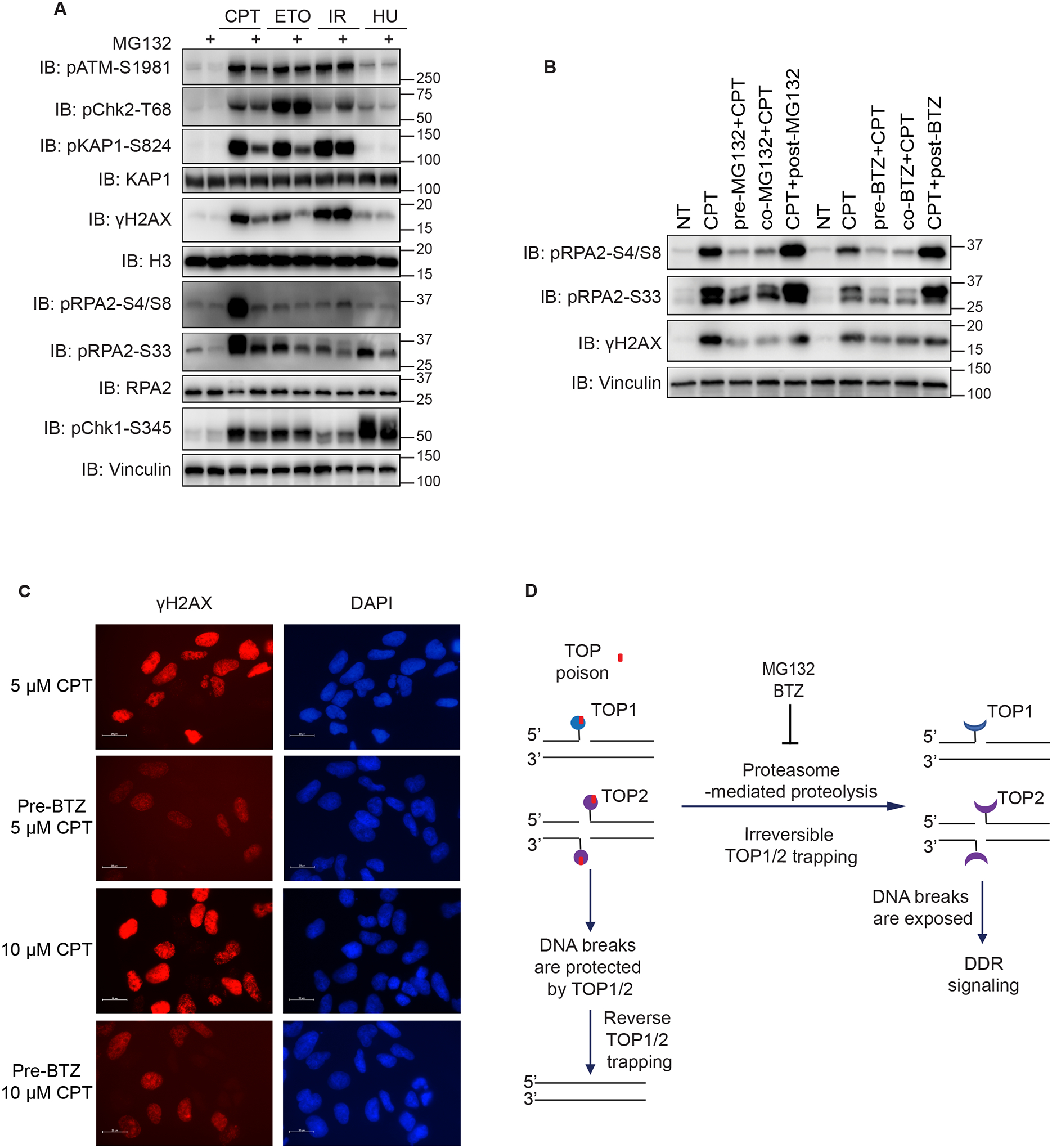
(A) Immunoblots of DDR signaling proteins from HEK293A cells. Cells were treated with 10 μM MG132 for 1 h or subjected to mock treatment and then treated with CPT (1 μM, 1 h), ETO (10 μM, 1 h), IR (10 Gy, allowed recovery for 1 h), or HU (10 mM, 1 h). Cells were then directly lysed by SDS loading buffer for Western blotting. Anti-Vinculin, anti-KAP1, anti-RPA2 and anti-H3 are the loading controls for Western blotting.
(B) Immunoblots of indicated proteins from HEK293A cells treated with either MG132 or BTZ and CPT. Specifically, cells were treated with 1 μM CPT for 1 h together with 1 h of pretreatment, co-treatment, and posttreatment with 10 μM BTZ or MG132.
(C) Immunofluorescent staining of γH2AX in HEK293A cells. Cells were treated with 10 μM BTZ for 1 h and then 5 μM or 10 μM CPT for 1 h. Representative images of γH2AX were shown (scale bar 20 μm).
(D) Schematic of the working hypothesis of proteasome-mediated proteolysis and DDR signaling.
We further validated our results using another proteasome inhibitor, bortezomib (BTZ). Pretreatment with BTZ efficiently suppressed CPT-induced γH2AX, pRPA2 (S4/S8), and pRPA2 (S33) signals (Figure 3B). Also, consistent with Western blot results, immunostaining demonstrated that preincubation with BTZ impaired CPT-induced γH2AX (Figure 3C). Of note, prior or simultaneous incubation of MG132 or BTZ with CPT markedly reduced γH2AX, pRPA2 (S4/S8), and pRPA2 (S33) signals to a similar extent. However, posttreatment with proteasome inhibitors could not prevent CPT-induced DDR signaling (Figure 3B). These data suggested that a proteasome-mediated process occurs very early in the activation of CPT-induced DDR signaling and implied that proteasome-mediated processing of TOP1 cleavage complex is irreversible, which could destruct ubiquitinated TOP141.
In summary, we validated our screening data and confirmed that proteasome inhibition impaired CPT- and ETO-induced DDR signaling. Accordingly, we proposed that proteasome processing, e.g. possibly proteolysis, of the TOP1 or TOP2 cleavage complex are likely early and irreversible events that are critical for the activation of DNA damage signaling following treatment with TOP1/2 poisons. When proteasome processing is inhibited, intact TOP1/2 can protect DNA breaks from being recognized and avoid TOP poison-induced DDR (Figure 3D). Moreover, the protease Spartan is known to degrade DNA-protein crosslinks42. Therefore, Spartan may function similarly as proteasome in the removal of chromatin-bound TOP1/2, which warrants further investigation. In addition, MRN complex also contributes to the removal of chromatin-associated TOP2, which acts directly on DNA to remove TOP2-DNA complex when TOP2 fails to complete catalysis43, raising the possibility that MRN complex functions independently of proteasome for the removal of TOP2 cleavage complex. Notably, proteasome inhibition prior to CPT and ETO treatment exhibited some obvious variations on DDR signaling as reflected by different DDR antibodies (Figure 3A). It is possible that MG132 treatment may block the phosphorylation of a subset of ATM substrates. Alternatively, MG132 treatment may differentially influence ATM or DNA-PKcs activation, which could have overlapping roles in these downstream phosphorylation events. Further investigation of these differences is needed to determine precisely how TOP cleavage complexes are processed and activate different DNA damage-responsive pathways, which are likely to be regulated in a cell cycle–dependent and – independent manner, e.g. the effect of proteasome inhibition in suppressing CPT-induced γH2AX may not be cell cycle-regulated, but mainly observed in S phase cells (Figure S9C).
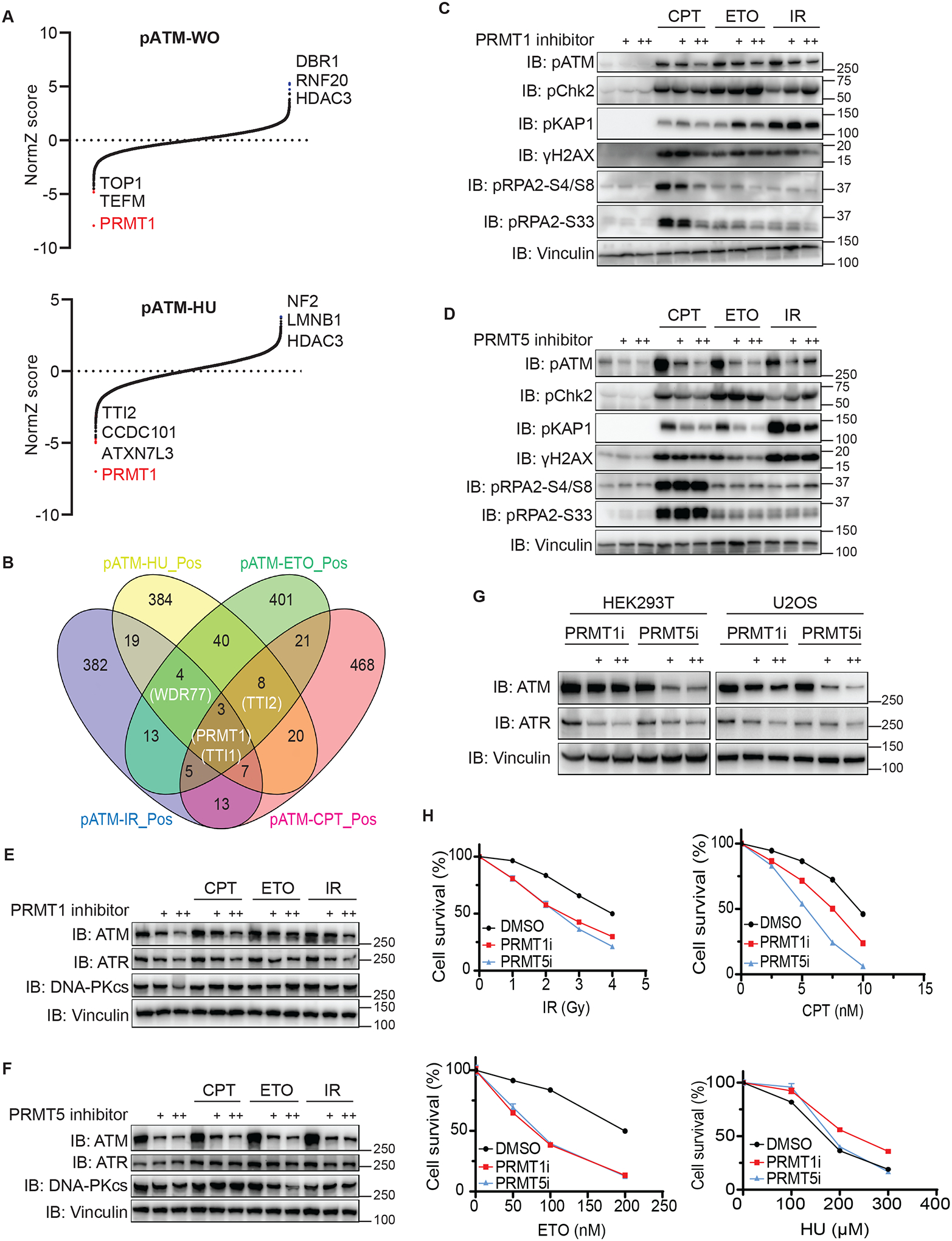
PRMT1 and PRMT5 regulate ATM protein levels
As shown in Figure S6A, we identified PRMT1, ATM, TTI1, and TTI2 as common positive regulators of DDR signaling in our screens. ATM is well known to be one of the key kinases contributing to DDR signaling44. In addition, the TTT complex, consisting of TTI1, TTI2, and TELO2, has an established role in regulating PIKK protein stability45–47. Therefore, we further investigated the ill-defined role of PRMT1 in regulating DDR signaling. Notably, PRMT1 was the most obvious outlier in the screens conducted using the anti-pATM antibody (Figure 4A). We further performed Venn diagram analysis of the screening data obtained using the anti-pATM antibody. Besides PRMT1, TTI1, and TTI2, we identified WDR77 as a common positive regulator of pATM (Figure 4B). WDR77, also known as MEP50, forms a stable and tight heterooctameric complex with PRMT5, which is required for PRMT5 activity. Moreover, PRMT5/WDR77 complex requires different partners to recognize and methylate corresponding substrates, therefore regulating gene transcription, chromatin remodeling and RNA splicing48. On the other hand, PRMT1 can function largely by itself49. PRMT1 and PRMT5 are type ǀ and type ǁ protein arginine methyltransferases that generate asymmetric dimethylarginine and symmetric dimethylarginine, respectively39. Considering that both WDR77 and PRMT5 were high-confidence hits in our screens, we chose PRMT1 and PRMT5 as potential positive regulators of DDR signaling for further validation.
Figure 4. PRMT1 and PRMT5 regulate ATM protein levels.
(A) Results of DrugZ analysis in FACS-based screens performed with an antibody recognizing endogenous pATM.
(B) Venn diagram of the overlap of hits in screens performed with the pATM antibody. Courtesy: Oliveros, J.C. (2007–2015) Venny. An interactive tool for comparing lists with Venn’s diagrams. Publicly available at http://bioinfogp.cnb.csic.es/tools/venny/index. html.
(C) Immunoblots of DDR signaling proteins in HEK293A cells treated with 1 μM or 10 μM PRMT1 inhibitor EPZ019997 for 3 days, which was followed by DNA-damaging treatment.
(D) Immunoblots of DDR signaling proteins in HEK293A cells treated with 1 μM or 10 μM PRMT5 inhibitor EPZ015666 for 3 days, which was followed by DNA-damaging treatment.
(E) Immunoblots of the indicated proteins in the samples in C.
(F) Immunoblots of the indicated proteins in the samples in D.
(G) Immunoblots of the indicated proteins prepared from HEK293T and U2OS cells. Cells were treated with the PRMT1 or PRMT5 inhibitor for 3 days.
(H) Treatment of PRMT1 or PRMT5 inhibitor sensitized cells to DNA damaging agent. HEK293A cell survival was determined with the use of CellTiter-Glo luminescence assays. Data are represented as mean ± SEM. n= 6 biological independent replicates.
To examine the roles of PRMT1 and PRMT5 in regulating DDR signaling, we employed inhibitors of PRMT1 and PRMT5 in validation experiments50,51. Cells treated with either PRMT1 or PRMT5 inhibitor exhibited cell proliferation defects at day 3 (Figure S10A). Of note, the effects of the PRMT1 and PRMT5 inhibitors on damage-induced DDR signals were quite different. Furthermore, inhibition of PRMT1 and PRMT5 impaired DNA-damaging agent–induced pATM levels to different extents, with the PRMT5 inhibitor exhibiting a more profound effect than the PRMT1 inhibitor (Figures 4C, 4D, S10B, and S10C). In contrast with the PRMT1 inhibitor, which impaired most CPT-induced DDR signals, the PRMT5 inhibitor specifically reduced CPT-triggered pATM and pKAP1 signals and had no effect on other DDR signals, such as pChk2 levels (Figures 4C and 4D), which may be due to the activation of DNA-PKcs in the absence of ATM activity52. These data validated our screening results, suggesting that PRMT1 and PRMT5 have overlapping and distinct functions in regulating DDR signaling pathways.
We next investigated precisely how PRMT1 and PRMT5 affect DNA damage-induced signals. Given that PRMT1 and PRMT5 are known to be involved in transcriptional regulation, we speculated that inhibiting PRMT1 or PRMT5 may affect ATM protein levels. Of note, PRMT1 inhibition led to downregulation of ATM protein as well as mild downregulation of ATR protein (Figure 4E). Moreover, PRMT5 inhibition caused marked downregulation of ATM but not ATR (Figure 4F). However, ATM mRNA level decreased in PRMT1 inhibitor-treated cells but was unaffected in PRMT5 inhibitor-treated cells (Figure S10D), suggesting that PRMT1 or PRMT5 may affect different biological processes contributing to the regulation of ATM protein expression. In addition, we observed that the DNA-PKcs protein level was not affected by the PRMT1 or PRMT5 inhibitor (Figures 4E and 4F). We next expanded treatment with these inhibitors to HEK293T and U2OS cells, which produced results similar to those observed in HEK293A cells (Figure 4G). To further validate the effects of PRMT1 and PRMT5 on ATM protein levels, we knocked down PRMT1 and PRMT5 with two independent shRNAs in HEK293A and U2OS cells. As expected, knockdown of PRMT1 or PRMT5 in both cell lines affected ATM protein levels (Figures S11A and S11B). The reduction of ATM protein levels in these knockdown experiments was relatively mild when compared to that observed following the inhibitor treatment, probably due to inadequate knockdown efficiency.
Ataxia-telangiectasia patients carrying ATM mutations have deficiencies in DDR53. These patients are characterized by extreme radiation sensitivity, cancer predisposition, and progressive neurodegeneration. Given that inhibition of PRMT1 or PRMT5 caused obvious defects in ATM protein levels, we suspected that both PRMT1 and PRMT5 inhibitors could be used as radiation sensitizers. Indeed, we found that both PRMT1 inhibitor- and PRMT5 inhibitor–treated HEK293A cells were hypersensitive to IR (Figure 4H). Moreover, we found that PRMT1 and PRMT5 inhibitors also sensitized cells to CPT and ETO (Figure 4H), which is probably due to that both PRMT1 and PRMT5 could affect the expression of several DNA repair genes54–58. For example, it was noted that treatment with PRMT1 inhibitor led to decreased BRCA1, BRCA2 and RAD51, which could be upregulated by PRMT5 in response to DNA damage56,58. In addition, we showed that HEK293T cells treated with PRMT1 or PRMT5 inhibitor were hypersensitive to IR, CPT and ETO (Figure S11C). However, U2OS cells treated with PRMT1 or PRMT5 inhibitor only showed clear hypersensitivity to HU (Figure S11C), suggesting that the genetic context of different cell lines may affect their overall sensitivity to different DNA damaging agents.
Taken together, these results suggested that PRMT1 and PRMT5 regulate ATM protein levels and that treatments with inhibitors of PRMT1 or PRMT5 sensitize cells to DNA-damaging agent, raising the possibility of combining PRMT1 or PRMT5 inhibitor with radiation therapy and/or chemotherapy for cancer treatment.
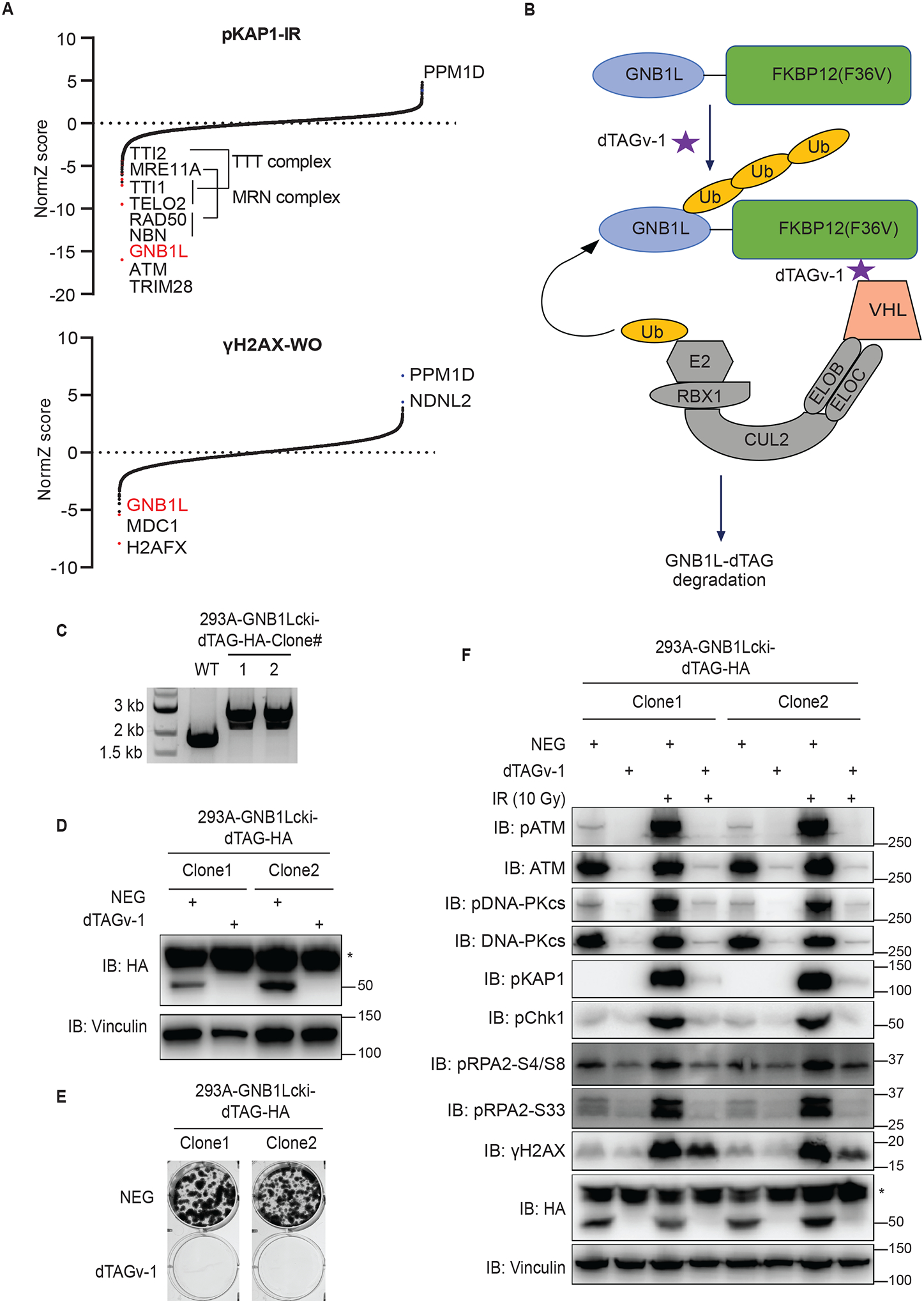
GNB1L is a critical regulator of DDR signaling
Our screens identified GNB1L59, a high-confidence hit, as a positive regulator of DDR signaling (Figures 5A and S12A), which was unassigned in functional network analysis (Figure 2A). In fact, GNB1L was a top hit in multiple screens presented in this study. For example, GNB1L, ATM, and the TTT complex were the top positive regulators of pKAP1 in the presence of IR, CPT, or ETO (Figures 5A and S12A). ATM is known to directly phosphorylate KAP1, whereas the TTT complex may affect damage-induced pKAP1 by stabilizing ATM and other PIKK proteins45,60. Moreover, the MRE11/RAD50/NBS1 complex members were also high-confidence hits in pKAP1 screening following IR exposure (Figure 5A). Additionally, the high-confidence hits in the screens for γH2AX revealed GNB1L, ATM, and MDC1 (Figures 5A and S12A). Given the known functions of these proteins and complexes in DDR signaling, we thus focused on uncovering how GNB1L, a protein with a hitherto unknown function, participates in DDR.
Figure 5. GNB1L depletion significantly reduces both basal and IR-induced DDR signaling.
(A) Results of DrugZ analysis in FACS-based screens performed with antibodies recognizing endogenous pKAP1 or γH2AX.
(B) Schematic of the dTAG system using VHL-recruiting dTAGv-1 to promote the degradation of GNB1L-dTAG protein.
(C) PCR validation of GNB1Lcki-dTAG clones of HEK293A cells.
(D) Western blot validation of the GNB1Lcki-dTAG cell lines.
(E) Clonogenic survival of GNB1L-dTAG cells in the presence of dTAGv-1 or dTAGv-1-NEG (NEG).
(F) Immunoblots of DDR signaling proteins in GNB1L-dTAG cells. Cells were treated with dTAGv-1 or dTAGv-1-NEG for 3 days and followed by exposure to IR (10 Gy, allowed recovery for 1 h) or mock treatment.
GNB1L is located on human chromosome 22q11, deletion of which causes 22q11 deletion syndromes, including DiGeorge and velocardiofacial syndromes, and GNB1L is also associated with schizophrenia59,61,62. However, nothing has been known about GNB1L protein function. Loss of Gnb1l function led to lethality during early embryogenesis in a previous study63. This result, together with data available in the depmap portal (https://depmap.org/portal/gene/GNB1L?tab=overview), suggests that GNB1L is essential for cell proliferation. We thus employed the degradation tag (dTAG; FKBP12F36V) system to investigate the function of GNB1L. This system takes advantage of the endogenous E3 ligase complex (e.g. VHL E3 ubiquitin ligase complex) and therefore does not require expression of any exogenous proteins (Figure 5B)64,65. We identified two different clones (clones 1 and 2) with homozygous FKBP12F36V (dTAG)-HA C-terminus knock-in at the GNB1L locus using polymerase chain reaction (PCR) (Figure 5C), which were further validated using Western blotting, as the GNB1L-dTAG-HA protein decreased to markedly lower levels in the presence of dTAGv-1 than that in cells treated with dTAG-NEG (Figure 5D). As expected, both clones exhibited striking defects in cell proliferation in the presence of dTAGv-1, but cells treated with dTAG-NEG proliferated normally (Figure 5E), suggesting that GNB1L is critical for cell survival.
Taking advantage of these powerful GNB1L-dTAG cells, we next examined whether depletion of GNB1L affects DDR signaling. Notably, depletion of GNB1L diminished all IR-induced DDR signaling in both clones (Figure 5F). Moreover, depletion of GNB1L also significantly reduced basal DDR signaling as well as ATM and DNA-PKcs protein levels (Figure 5F). Taken together, we validated our screening data and showed that depletion of GNB1L strikingly impaired both basal and IR-induced DDR signaling. These data strongly suggested that GNB1L is a major regulator of DDR signaling.
GNB1L associates with chaperones and may act as a co-chaperone to regulate PIKKs
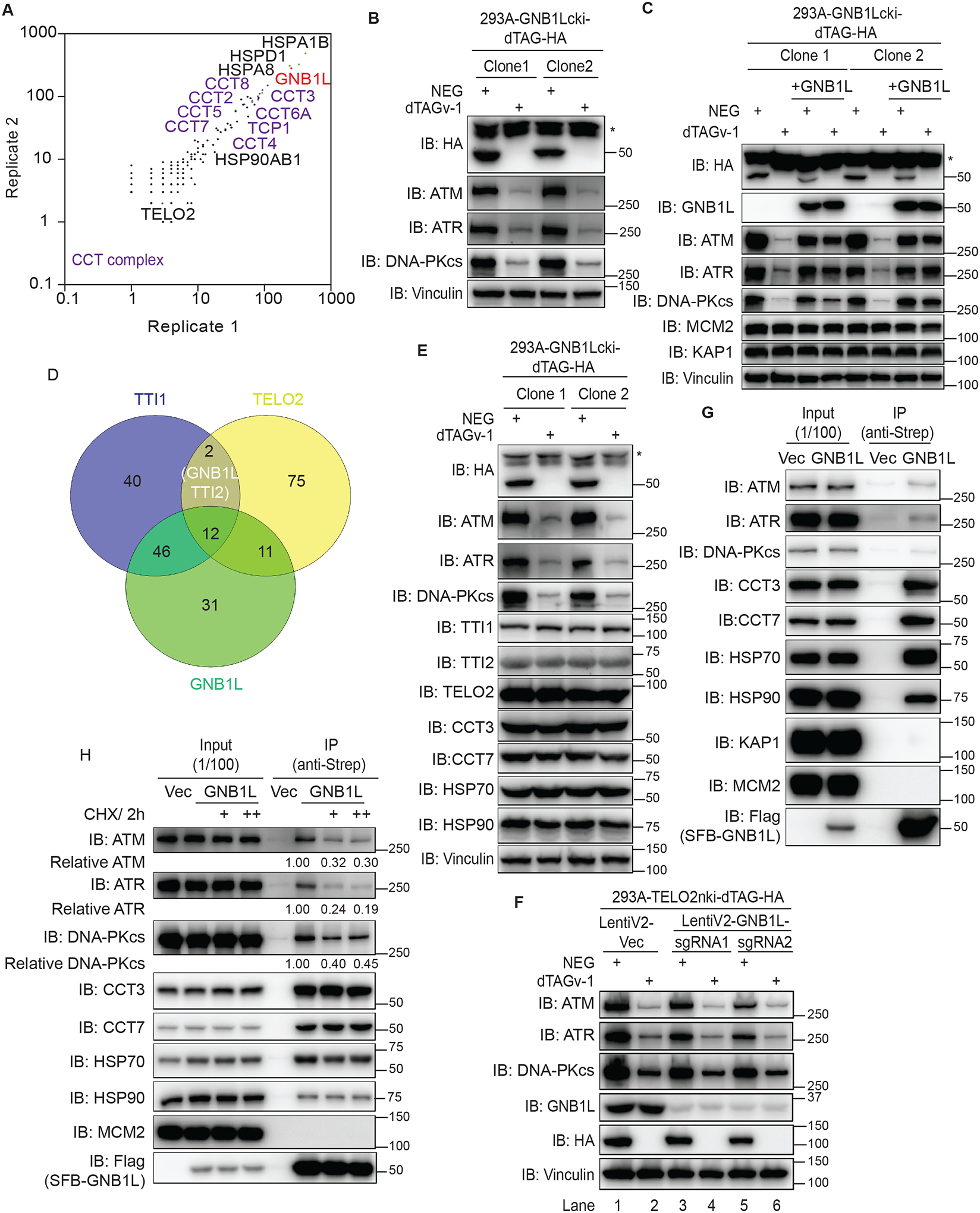
We next explored the mechanisms underlying impaired DDR signaling due to GNB1L depletion. We generated HEK293T cell line with stable expression of SFB-GNB1L, which was localized mainly in cytosol and to a lesser extent in the nucleus (Figure S12B). Using mass spectrometry, we identified GNB1L-associated proteins in HEK293T cells via tandem affinity purification of SFB-GNB1L–associated proteins. Proteins highly associated with GNB1L were chaperones, which include heat shock protein 70 (HSP70), HSP90, and all components of the chaperonincontaining tailless complex polypeptide 1 (CCT) complex (Figure 6A). The CCT complex is an essential eukaryotic chaperone complex that protects a variety of client proteins during their folding process to prevent the toxic effects of misfolding and aggregation of normal proteins66–69. For example, CCT complex is required for the folding of actin and tubulin, which allows them to be further assembled into microfilaments and microtubules70,71. As described above, depletion of GNB1L led to striking defects in both basal and IR-induced DDR signals as well as ATM and DNA-PKcs protein levels. Thus, we speculated that GNB1L functions with chaperones to regulate PIKK proteins. We tested this hypothesis using two different GNB1L-dTAG cell line clones, which both displayed significant reduction of ATM, ATR, and DNA-PKcs protein levels in the presence of dTAGv-1 for 3 days (Figure 6B). Of note, these GNB1L-dTAG cells treated with dTAGv-1 were still proliferating and exhibited mild proliferation defects at day 3 (Figure S12C), thus excluding the possibility that reduction of these PIKKs is due to cell death. However, although treatment with dTAGv-1 caused almost immediate reduction of GNB1L protein levels (Figure S12D), treatment with dTAGv-1 for 1 day had a mild effect on PIKK proteins; it had a greater effect after a longer treatment period (Figure S12E). These results demonstrated that GNB1L may only influence newly synthesized PIKK proteins, an effect that can accumulate in a time-dependent manner when GNB1L is depleted.
Figure 6. GNB1L associates with the CCT complex and regulates PIKK proteins.
(A) The profile of the GNB1L interactome in HEK293T cells identified using mass spectrometry.
(B) Immunoblots of the indicated proteins in lysates prepared from GNB1L-dTAG cells. The cells were treated with dTAGv-1 or dTAGv-1-NEG for 3 days.
(C) Immunoblots of the indicated proteins prepared from GNB1L-dTAG cells and the reconstituted GNB1L cell lines generated from GNB1L-dTAG cells. Cells were treated with dTAGv-1 or dTAGv-1-NEG for 3 days.
(D) Venn diagram showing the overlapping co-dependency genes for TTI1, TELO2, and GNB1L. The co-dependency gene lists for TTI1, TELO2, and GNB1L are from depmap.
(E) Immunoblots of the indicated proteins in the samples in B.
(F) HEK293A-TELO2-dTAG cells were treated with 1 μM NEG or dTAGv-1 and further infected with control virus (vector) or virus expressing LentiV2-GNB1L-sgRNA1/2. Cells were lysed directly after 3 days and immunoblotted for the indicated proteins.
(G) Co-immunoprecipitation assay identification of the interaction of GNB1L with endogenous PIKKs, CCT3, CCT7, HSP70, and HSP90.
(H) Co-immunoprecipitation assay identification of the interaction of GNB1L with endogenous PIKKs, CCT3, or HSP90 in the presence of CHX (100 μg/ml or 200 μg/ml) for 2 h in HEK293T cells as indicated. The quantification was performed with Image J.
To confirm these results, we reconstituted GNB1L expression in the two GNB1L-dTAG clones described above. As shown in Figure 6C and Figure S12F, both reduction of PIKK protein levels and cell proliferation defects in dTAGv-1–treated cells were restored by GNB1L expression. Moreover, depletion of GNB1L had no effect on either MCM2 or KAP1 protein levels, implying that GNB1L specifically modulates some client proteins, such as PIKKs (Figure 6C). To determine whether GNB1L depletion specifically affects PIKKs instead of other DDR factors, we carried out label-free quantitative proteomics analysis by comparing the proteomes in NEG-treated versus in dTAGv-1-treated GNB1L-dTAG cells. As shown in Figure S13A and Table S5, GNB1L depletion specifically reduced PIKK kinases and ATRIP rather than other DDR factors. Obviously, PIKK kinases, ATRIP and mTOR are major downregulated substrates affected by GNB1L depletion. Meanwhile, we also performed RNA-seq to rule out the possibility that GNB1L regulates PIKK protein levels via transcriptional regulation. The differentially expressed genes (DEGs) as well as the GO analysis confirmed that depleting GNB1L had no effect on PIKK mRNA levels (Figures S13B and Figure S13C; Table S6). These data suggested that GNB1L specifically regulates PIKK protein levels instead of other DDR factors.
Notably, data from the depmap portal demonstrate highly overlapping co-dependency hits between GNB1L and the TTT complex (Figure 6D). In addition, Asa1, the homologous gene of GNB1L in yeast, has been linked to TTT complex by high-resolution phenotypic profiling72. Because the TELO2/TTI1/TTI2 complex is known to be a PIKK-specific co-chaperone73, these data strongly suggest that GNB1L works together with or has an overlapping function with the TTT complex. To elucidate the potential genetic interaction between GNB1L and TTT complex, we generated HEK293A-TELO2nki-dTAG cells and validated two different clones, both of which displayed the striking reduction in TELO2 protein level and defects in cell proliferation when cells were treated with dTAGv-1 (Figures S14A and S14B). Consistent with the published data45,73, depletion of TELO2 drastically decreased PIKK protein levels in a time-dependent manner which was similar with the effect of GNB1L on PIKKs (Figure S14C). Given that GNB1L consists of several WD (trp-asp) repeats that may facilitate the formation of multiprotein complexes, we tested whether GNB1L modulates PIKK proteins through the TTT complex. We found that cells with GNB1L depletion did not display any reduction in the expression of TTI1, TTI2 and TELO2 (Figure 6E), excluding the possibility that GNB1L affects TTT complex formation and thereby indirectly affects PIKK proteins. Similarly, TELO2 depletion had no effect on GNB1L protein level (Figure S14C). Additionally, cells depleted of TELO2 led to decreased PIKK protein levels to the same or similar degree as cells depleted of both TELO2 and GNB1L (Figure 6F; Lane 2, 4 and 6). Moreover, additional depletion of TTI1 or TELO2 in combination with GNB1L depletion did not further impair PIKK protein levels (Figure S14D; Lane 4, 6, 8 and 10). These data suggest that there is no additional effect when depleting both GNB1L and TTT complex, and thus raising the possibility that GNB1L and TTT complex may function together to regulate PIKK protein levels.
Considering the strong interaction of GNB1L with the CCT complex, HSP70, and HSP90, we also examined whether GNB1L ablation affects CCT complex formation and/or HSP70/HSP90 protein levels. As shown in Figure 6E and Table S5, depletion of GNB1L had no effect on CCT3, CCT7, HSP70, or HSP90 protein levels, indicating that GNB1L likely functions as a co-chaperone rather than directly affecting chaperones to regulate PIKK protein stability. To test our hypothesis, we then examined the interaction between GNB1L and PIKKs as well as chaperones using co-immunoprecipitation (Figure 6G). We observed interaction of GNB1L with endogenous ATM, ATR, and DNA-PKcs. We also detected interaction of GNB1L with chaperones, including CCT3, CCT7, HSP70, and HSP90. KAP1 and MCM2, which we included as negative control, did not associate with GNB1L (Figure 6G). These findings suggested that GNB1L modulates PIKKs via several mechanisms, for example, as a co-chaperone for the CCT complex, HSPs, and/or the TTT complex.
As described above, our data demonstrated that GNB1L may function with chaperones and only affect newly synthesized PIKK proteins. To determine whether GNB1L modulates PIKK maturation, we treated SFB-GNB1L–expressing cells with cycloheximide (CHX) to prevent protein translation. As shown in Figure 6H, the association of GNB1L with ATM, ATR, and DNA-PKcs decreased following CHX exposure. However, the interaction of GNB1L with CCT3, CCT7 and HSP90 was not affected, but its association with HSP70 was mildly decreased. MCM2 was included as the negative control in these experiments. Because short treatment with CHX mainly affects newly synthesized proteins, we thus speculated that GNB1L functions as a co-chaperone to regulate newly translated PIKK proteins and thereby modulating DDR signaling. Moreover, inhibiting either HSP70 or HSP90 mildly impaired the interaction between GNB1L and PIKKs (Figure S14E), which implies that the effect of GNB1L on PIKKs is at least partially dependent on HSP70 and/or HSP90.
To better understand how GNB1L functions in the cell, we mapped the domains of GNB1L which would be important for its functions in cell proliferation, regulating PIKK protein levels and/or interacting with other proteins. We generated GNB1L truncation mutants depleting one of the seven potential WD repeats within GNB1L. Depleting any WD repeat of GNB1L could mimic the effect of depleting GNBL1 on PIKK protein levels and cell proliferation (Figures S14F and S14G), which probably indicate that GNB1L is a small protein therefore depleting any WD repeat would affect its conformation and/or functions. In addition, all the truncation mutants exhibited impaired interaction with both PIKKs and TELO2. However, these GNB1L truncation mutants exhibited different defects in associating with CCT7 and/or HSP90 (Figure S14H), which need to be further investigated. Moreover, GNB1L depletion led to accumulation of DSBs (Figures S15A and S15B) and chromosomal fragments (Figures S15C and S15D). Taken together, our data suggest that GNB1L is a key regulator of DDR signaling by specifically regulating PIKK protein levels and thus plays a critical role in maintaining genomic stability.
DISCUSSION
DDR signals transduced by PIKKs are critical for coordinating DNA replication, DNA repair, cell-cycle checkpoints, and other cellular processes and protecting cells from exogenous and endogenous DNA damage. Over the past 2 decades, researchers have extensively studied DDR signaling pathways. However, new factors involved in the modulation of these pathways are still being identified, indicating the existence of additional regulators of these critical DDR pathways. In the present study, we carried out 30 unbiased FACS-based genome-wide screens to provide a comprehensive view of the regulatory network involved in DDR signaling. The data obtained from these systematic screens not only validate many known regulators of DDR signaling, but also uncover previously understudied and uncharacterized modulators as well as pathways involved in DDR signaling.
The success of our FACS-based screens with antibodies recognizing endogenous proteins and signals highlights the potential of this type of screening in studies of many other signaling pathways, since such screens do not require engineered reporter cell lines and therefore genuinely assess endogenous signaling events and can be conducted in any available cell lines. Moreover, our data also demonstrated that FACS-based screens performed with different DNA-damaging agents exhibit unique profiles, suggesting that this type of screening can be used to dissect proteins and pathways specifically involved in one process but not in related processes. Furthermore, FACS-based screening is a promising method for antibody validation, especially for critical antibodies used in clinical investigations that require high specificity and accuracy.
One of the major differences between FACS-based screens and synthetic lethality-based screens is the duration of the experiments. Synthetic lethality-based CRISPR screens are usually required to maintain cell proliferation for 21 days, whereas FACS-based screens only take a few days after lentiviral infection and puromycin selection. Therefore, the FACS-based screens allow for the identification of essential genes that are eliminated or ignored in synthetic lethality-based CRISPR screens. For example, our screens identified several essential genes, including MRE11, RAD50, NBS1, TTI1, TTI2, TELO2, PRMT1, PRMT5, and GNB1L, involved in DDR signaling. Therefore, identifying uncharacterized essential genes in FACS-based screens is possible.
In this study, we characterized a previously unknown protein, GNB1L. Of note, analysis of The Cancer Genome Atlas data on GNB1L mRNA levels in tumor and normal tissue samples as well as their correlations with patient survival implied that GNB1L expression associates with cancer progression (Figures S16A and S16B). Particularly, GNB1L mRNA levels are consistently higher in tumor samples than in normal tissue samples among 31 cancer types. Moreover, high levels of GNB1L expression correlate with poor survival for some cancer types, including ACC, BRCA, CHOL, and LIHC. Whether high levels of GNB1L expression in tumors facilitate the maturation and stabilization of PIKKs, which promote cell survival as well as resistance to DNA damage, remains to be determined. Furthermore, the CCT complex, the strong binding partner of GNB1L, is a potential chemotherapeutic target69,74,75. These data raise the significance of developing inhibitors specifically targeting GNB1L. Uncovering GNB1L’s structure and the molecular details of its association with the CCT complex and HSP70/90 will be helpful in this regard.
Limitations of the study
The present study highlights the potential of FACS-based CRISPR screens using antibodies recognizing endogenous signaling molecules to investigate the regulation of signal transduction pathways. However, not all antibodies are suitable for FACS-based screens. In our study, we did not include antibodies recognizing pATR (S1989) or pChk1 (S317/S345), as these antibodies did not have high specificity for our application (i.e. increased signal following treatment with CPT). In addition, we only performed our studies with HEK293A cells using four different treatment conditions. Future studies with additional cell lines and treatments will undoubtedly enable us to further expand the DDR network. Moreover, we mainly focused on the identification of common DDR regulators in this study, which most likely regulate DDR signaling broadly via affecting PIKKs. Other specific DDR regulators, e.g. C11orf57 and C21orf59, may be involved in the control of a subset of the DDR signaling pathway in response to a particular type of DNA damage. We did not highlight or study these types of regulators in this study. Additionally, although we validated the effect of ALDOA, C11orf57 and C21orf59 on DDR signaling, much more mechanistic work is required to fully understand how these regulators contribute to DDR signaling.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Junjie Chen (JChen8@mdanderson.org).
Materials availability
Requests for cell lines and plasmids generated in this study should be directed to the lead contact.
Data and code availability
The datasets are publicly available. The mass spectrometry data (whole-proteome profiling and GNB1L-associated protein identification) have been deposited to the MassIVE data sets with the dataset identifier MSV000092198. Raw RNA-sequencing data have been deposited to NCBI under GEO: GSE235200. All FACS-based genome-wide CRISPR screens next-generation sequencing raw data and processed data are provided in Tables S1 and S2. Raw immunoblot images are available at Mendeley Data with the DOI 10.17632/x7d248vz6f.1 and are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Study Participant Details
Cell lines
HEK293A, HEK293T, U2OS and HeLa cells were purchased from the ATCC and cultured in Dulbecco’s modified Eagle’s medium (Corning) with 10% fetal bovine serum (Sigma). HEK293A-GNB1L-CKI-dTAG and HEK293A-TELO2-NKI-dTAG cell lines were generated via co-transfection of GNB1L C-terminus knock-in sgRNA or TELO2 N-terminus knock-in sgRNA together with a donor vector containing dTAG that includes a linker, FKBPF36V-2HA and a P2A self-cleavage site, and blasticidin flanked by about 1 kb of homology arms in a PUC19 backbone as reported previously76. After selection with 10 μg/ml blasticidin for 5 days, the surviving cells were seeded in 96-well plates. Positive clones of dTAG knock-in were screened using genomic PCR and further validated using Western blotting.
METHOD DETAILS
Constructs and cloning
GNB1L sgRNA (AGGATCAGCGGATCAGCCTC) and TELO2 sgRNA (CCCAGAT CTGTCCTGCA GGA) were cloned into pX330 (Addgene: 42230;77) for knock-in of C-terminal and N-terminal dTAG tag at the endogenous GNB1L and TELO2 locus respectively. The DNA fragments’ dTAG for CKI and NKI was amplified via PCR from pCRIS-PITChv2-dTAG-blasticidin (Addgene: 91795) and pCRIS-PITChv2-blasticidin-dTAG (Addgene: 91792) respectively. A donor vector for GNB1L/TELO2 knock-in was generated using Gibson assembly of the 5’ homolog arm, dTAG, and 3’ homolog arm into a PUC19 vector. The human GNB1L open reading frame was subcloned into a modified pLEX_307 SFB vector using Gateway recombination cloning technology.
FACS-based CRISPR/Cas9 screens
As described previously19, we conducted CRISPR screens using the TKOv3 human wholegenome library, which included 70,948 single guide RNAs (sgRNAs) targeting 18,053 genes. HEK293T cells were co-transfected with the TKOv3 library, the packaging vector psPAX2, and the envelope vector pMD2.G using the X-tremeGENE HP DNA transfection reagent (Sigma-Aldrich; cat. no. 6366546001) for generation of TKOv3 library lentivirus. The virus-containing media were collected 24 h after transfection and used to infect HEK293A cells at a low multiplicity of infection (<0.3) or frozen at −80°C. Twenty-four hours after infection, cells were then selected using 2 μg/ml puromycin for 2 days. The selected populations were passaged and then prepared for treatment on day 5. Each screen was performed with two replicates, with 100 million cells per replicate exposed to treatment (IR: 10 Gy, repair 1 h; ETO: 10 μM for 1 h; CPT: 0.5 μM for 3 h; HU: 5 mM for 3 h) and collected in a 15-ml tube, which was followed by precold 70% ethanol fixation.
All the following procedures were performed using a 15-ml tube filled with buffer on a rotating mixer for immunostaining. After fixation, HEK293A cells were permeabilized with 0.5% Triton X-100 (in phosphate-buffered saline [PBS]) for 15 min. The samples were then blocked with 3% bovine serum albumin (in PBS) for 30 min at room temperature. Next, cells were incubated with indicated antibodies (1:1000 diluted in 3% bovine serum albumin) at 4°C overnight and then incubated with a secondary antibody (1:500 diluted in 3% bovine serum albumin) for 45 min at room temperature. Finally, cells were resuspended in PBS for flow cytometry selection (the top 30% with the highest signals and bottom 30% with the lowest signals cell populations, respectively). Genomic DNA was extracted from flow cytometryselected cells using a QIAamp Blood Maxi Kit (QIAGEN) and was further amplified and labeled with barcodes via PCR. The amplicons were sequenced. Raw sequencing reads were aligned to TKOv3 library using MAGeCK78 and raw counts for each sgRNA were generated (Table S2). As described in the paper20, the DrugZ software was used to evaluate the differential gRNA representation between the high-signal groups and the low-signal groups, which generated Z-score (Drug-Z score) for each gene. Then, genes were ranked according to their Z-scores (Figure S2 and Table S1).
Western blot analysis
HEK293A, HEK293A-TELO2nki-dTAG or HEK293A-GNB1Lcki-dTAG cells were seeded in a six-well plate with EPZ015666, EPZ019997, or ligands (dTAGv-1-NEG and dTAGv-1) for 3 days. This was followed by mock treatment or treatment with indicated DNA-damaging agents. Cells were then directly lysed using a sodium dodecyl sulfate gel-loading buffer and boiled for further analysis. Samples were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed using immunoblotting with indicated antibodies.
Immunofluorescent staining
Immunofluorescent staining was performed as described previously79. Briefly, HEK293A or HEK293T cells were seeded on cover glasses overnight. Next, cells were pretreated with BTZ for 1 h and then treated with CPT and fixed in 3% paraformaldehyde for 15 min. After fixation, cells were permeabilized with 0.5% Triton X-100 (in PBS) for 15 min and blocked with 3% bovine serum albumin in PBS for 30 min. Cells were further incubated with antibodies against γH2AX or Flag overnight and then incubated with a secondary antibody the next day. The cells were later counterstained with DAPI, and images were acquired using a Leica microscope.
CellTiter-Glo cell viability assay
For short-term cell survival assays, HEK293A cells were seeded in 96-well plates (800 cells/well) with 1 μM EPZ015666 or EPZ019997. Once cells attached to the plates, the cells were subjected to DNA-damaging treatment (IR, CPT, ETO, or HU). Four days later, cell viability was evaluated with the use of CellTiter-Glo luminescence assay (Promega).
Colony formation assay
For colony formation assay, GNB1L-dTAG 293A cells were seeded with 1 μM dTAGv-1-NEG or dTAGv-1 in six-well plates in triplicate (200 cells/well). Eight days later, cells were stained with crystal violet solution (Sigma-Aldrich).
Co-immunoprecipitation and mass spectrometry
HEK293T cells were co-transfected with pLenti-SFB (S protein-Flag-Streptavidin–binding peptide)-GNB1L, the packaging vector psPAX2, and the envelope vector pMD2.G using the X-tremeGENE transfection reagent for generation of SFB-GNB1L–expressing lentivirus. HEK293T cells were then infected with lentivirus and selected with 2 μg/ml puromycin for 2 days. The selected cells were harvested and lysed with NETN buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 0.5% Nonidet P-40, protease inhibitor cocktail) at 4°C for 30 min, which was followed by centrifugation at 15,000 rpm for 30 min. The supernatant was incubated with streptavidin beads for 2 h at 4°C, and the beads were washed four times with NETN buffer and eluted with NETN buffer containing 2 mg/ml biotin and protease inhibitor cocktail. The elution was incubated with S-protein beads for 2 h, and the S bead–associated proteins were eluted and analyzed using mass spectrometry.
To detect protein-protein interactions, HEK293T cells were transfected with SFBGNB1L. Forty-eight hours later, cells were harvested, lysed with NETN buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 0.5% Nonidet P-40, protease inhibitor cocktail) for 30 min at 4°C, and centrifuged. The supernatant was incubated with streptavidin beads and washed four times with NETN buffer. Next, the beads were treated with sodium dodecyl sulfate gel-loading buffer and boiled for further analysis. Samples were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed using immunoblotting with indicated antibodies.
Metaphase spread
HEK293A-GNB1L-dTAG cells were treated with 1 μM NEG or dTAGv-1 for 3 days, then synchronized with 100 ng/mL Nocodazole for 16 hours and harvested. The harvested cells were subject to hypotonization with 0.075 M KCl for 15 min at 37 °C and fixed by fresh methanol: glacial acetic acid (3:1) solution. A single drop of fixed cells was released vertically onto the slide from ~20 cm height. Slides were subject to Giemsa staining after air dry. Briefly, Gurrs stain (R66) Giemsa (Life Tech, 10092–013) was added to the slide for 5 min, which later was rinsed with Gurrs 6.8 buffer (Life Tech, 10092–013) twice, and then mounted with nail polish (VWR, 100491–940). Photos were captured by Olympus BX43F (40x Lens /0.65) and CellSens Standard software.
Neutral Comet assay
The neutral comet assay was conducted with CometAssay kit (4250–050-K; R&D systems) following the instructions as described previously52. Briefly, HEK293A-GNB1L-dTAG cells were treated with 1 μM NEG or dTAGv-1 for 3 days, followed by DMSO or ETO (10 μM for 1 h) treatment. Cells were collected and washed with PBS once, then diluted to the concentration of 1 × 105 cells/mL in PBS. We then resuspended 20 μL cells with 200 μL of pre-warmed LMAgarose and immediately spread 50 μL mixture onto CometSlide, followed by placing the slides flat at 4 °C for 15 min. Samples were lysed by immersing slides in lysis solution for 1 h at 4 °C. After being removed from the lysis solution, slides were washed and immersed in 1X neutral electrophoresis buffer (50 nM Tris base and 150 mM sodium acetate) for 30 min and then subjected to electrophoresis at 25 V for 25 min in 1X neutral electrophoresis buffer. Then slides were washed and dried overnight, stained with SYBR-gold for imaging. Images were obtained using a Nikon 90i microscope at ×10 magnification. Collected images were analyzed using OpenComet80, and the olive tail movements are shown.
Label-free quantitative proteomics analysis
HEK293A-GNB1L-dTAG cells were treated with 1 μM NEG or dTAGv-1 for 3 days. The samples were then prepared as previously described81. Briefly, cells were lysed with 8 M urea and subjected to reduction, alkylation, and Trypsin digestion sequentially. After desalination with Sep-Pak column, tryptic peptides were subjected to fractionation. The eluent was combined into 10 fractions and then analyzed in a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific) in data-dependent mode. The proteingroup.txt file was imported into Perseus software (version 1.6.7.0)82, followed by filtering proteins annotated with “reverse,” “potential contamination,” and “only identified by site.” Proteins with at least two unique peptides and 70% valid value in total samples were kept. The significantly differential proteins were set as a permutation-based false discovery rate of <0.05 and S0 = 0.1.
RT-qPCR
HEK293A-GNB1L-dTAG cells were treated with 1 μM NEG or dTAGv-1 for 3 days followed by RNA extraction with kit (Qiagen; 217004) and reverse-transcribed cDNA was synthesized with iScript cDNA synthesis kit (Biorad; 1708891). The qPCR reactions were run in an ABI Q6 RTPCR instrument. Levels of ATM mRNA were detected by the SYBR green (Thermo Fisher Scientific; A25776) and normalized by β-actin mRNA. The primers used in this study were as follows: (β-actin forward: CACCATTGGCAATGAGCGGTTC; β-actin reverse: AGGTCTTTGCGGATGTCCACGT; ATM_Primer1 forward: ATAGATTGTGTAGGTTCC GATGG; ATM_Primer1 reverse: CATCTTGTCTCAGGTCATCACG83; ATM_Primer2 forward: CC AGCTGTGCAGCGAACAAT; ATM_Primer2 reverse: TCTAAGCACGTTTCTGCTAACCAGT84.
RNA-seq and data analysis
HEK293A-GNB1L-dTAG cells were treated with 1 μM NEG or dTAGv-1 for 3 days followed by RNA extraction with kit (Qiagen; 217004). We prepared 3 replicates for each group. All the following mRNA sequencing processes until the generation of FASTQ files were completed by Cancer Genomics Center at the University of Texas Health Science Center at Houston. After removal of low-quality reads, we used clean reads of samples for preprocessing and aligning to human GRCh38 genome using hisat2 (version 2.2.1)85. Unique mapped reads were obtained and annotated to the latest GTF file downloaded from GENCODE (https://www.gencodegenes.org/) by HTseq (version 0.9.1)86. The edgeR package in R 4.0.5 was used to identify differentially expressed genes with cutoff (p-value <0.01 and fold change >2)87.
TCGA analysis
The gene expression data of tumor patients and adjacent normal samples and the clinical data of matched patients were obtained from The Cancer Genome Atlas (TCGA) data portal (https://TCGAData.nci.nih.gov/TCGA/). For differential expressed gene (DEG) analysis of mRNA, we used the limma package in R 4.0.5, and genes with a p-value <0.05 and fold change >1.5 were selected to be differentially expressed. FPKM of GNB1L was showed in boxplot by the ggplot2 package in R 4.0.5. For Kaplan-Meier (K-M) analysis and Cox regression analysis, tumor patients were divided into a low group (below the median expression of GNB1L) and a high group (above the median expression of GNB1L). The high and low groups were stratified and visualized using K-M survival curves and analyzed for statistical significance using the log-rank test. Cox regression analysis and K-M curves with the log-rank test were conducted by the glmnet and survival packages in R 4.0.5.
Gene Set Enrichment analysis
GO terms of differential expressed genes (DEGs) were enriched using DAVID (the database for annotation, visualization, and integrated discovery, https://david.ncifcrf.gov/)88, which is an online tool for functional annotation and enrichment analysis to reveal biological pathways related to the given gene lists. The visualization of representative biological pathways was performed by the ggplot2 package in R4.0.5. The network was generated using the Cytoscape plug-in ClueGO89.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphpad Prism version 9 was used for all statistical analysis. Statistical tests used and sample sizes are described in figure legends.
Supplementary Material
Table S1. FACS-based CRISPR screens NormZ results, related to Figures 1A and S2. NormZ values calculated with DrugZ for each CRISPR screen reported in this work.
Table S2. FACS-based CRISPR screens raw counts, related to Figures 1A and S2. Raw counts for each sgRNA of 30 screens reported in this work.
Table S3. Gene Ontology analysis of positive and negative regulators, related to Figure 1D. Here are listed all the gene ontology terms for the 311 and 317 hits respectively identified as positive and negative regulators based on the antibody as well as 205 and 163 hits respectively identified as positive and negative regulators based on the treatment.
Table S4. Gene Ontology analysis of screen hits for network, related to Figures 2C and S4C. Here are listed all the gene ontology terms for the 615 and 366 hits that respectively identified based on the antibody and treatment.
Table S5. Proteome profiling in the absence of GNB1L, related to Figures 6C and S13A. List of proteins with significantly changes by the log2 ratio of protein abundance in the GNB1L depletion versus control cells.
Table S6. RNA-seq analysis in the absence of GNB1L, related to Figures 6C, S13B and S13C. Here are listed all the gene ontology terms for differentially expressed genes with cutoff (p-value <0.01 and fold change >2).
Table S7. SgRNA and shRNA target sequences, related to the STAR Methods.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-pKAP1-S824 | Cell Signaling Technology | Cat#4127S, RRID: AB_2209906 |
| Anti-pATM-S1981 | Cell Signaling Technology | Cat#4526L, RRID: AB_2062663 |
| Anti-pChk2-T68 | Cell Signaling Technology | Cat#2661S, RRID: AB_331479 |
| Anti-PRMT1 | Cell Signaling Technology | Cat#2449S, RRID: AB_2237696 |
| Anti-PRMT5 | Cell Signaling Technology | Cat#79998S, RRID: AB_2799945 |
| Anti-Aldolase/ALDOA | Cell Signaling Technology | Cat#3188S, RRID: AB_2226674 |
| Anti-MCM2 | Cell Signaling Technology | Cat#4007S, RRID: AB_2142134 |
| Anti-HA | Cell Signaling Technology | Cat#2999S, RRID: AB_1264166 |
| Anti-HSP70 | Cell Signaling Technology | Cat#4872S, RRID: AB_2279841 |
| Anti-HSP90 | Cell Signaling Technology | Cat#4877S, RRID: AB_2233307 |
| Anti-pChk1-S345 | Cell Signaling Technology | Cat#2348S, RRID: AB_331212 |
| Anti-ATM | Cell Signaling Technology | Cat#2873S, RRID: AB_2062659 |
| Anti-ATR | Cell Signaling Technology | Cat#2790S, RRID: AB_2227860 |
| Anti-pRPA2-S4/S8 | Bethyl | Cat#A300-245A, RRID: AB_210547 |
| Anti-pRPA2-S33 | Bethyl | Cat#A300-246A, RRID: AB_2180847 |
| Anti-TOP1 | Bethyl | Cat#A302-589A, RRID: AB_2034865 |
| Anti-TTI1 | Bethyl | Cat#A303-451A, RRID: AB_10953982 |
| Anti-TTI2 | Bethyl | Cat#A303-476A, RRID: AB_10948973 |
| Anti-KAP1 | Bethyl | Cat#A300-274A, RRID: AB_185559 |
| Anti-DNA-PKcs | Abcam | Cat#ab70250, RRID: AB_1209452 |
| Anti-DNA | Abcam | Cat#ab27156, RRID: AB_470907 |
| Anti-RPA2 | Abcam | Cat#ab2175, RRID: AB_302873 |
| Anti-pDNA-PKcs-S2056 | Abcam | Cat#ab18192, RRID: AB_869495 |
| Anti-Flag | Sigma-Aldrich | Cat#F3165, RRID: AB_259529 |
| Anti-Vinculin | Sigma-Aldrich | Cat#V9264, RRID: AB_10603627 |
| Anti-GNB1L | Sigma-Aldrich | Cat#HPA034627, RRID: AB_10600662 |
| Anti-TELO2 | Proteintech | Cat#15975-1-AP, RRID: AB_2203337 |
| Anti-CCT3 | Proteintech | Cat#10571-1-AP, RRID: AB_2073658 |
| Anti-CCT7 | Proteintech | Cat#15994-1-AP, RRID: AB_2073903 |
| Anti-C11orf57 | Proteintech | Cat#21181-1-AP, RRID: AB_2878825 |
| Anti-C21orf59 | Proteintech | Cat#21461-1-AP, RRID: AB_2878863 |
| Anti-TOP1cc | Millipore | Cat#MABE1084, RRID: AB_2756354 |
| Anti-TOP2α | Millipore | Cat#MAB4197, RRID: AB_2205862 |
| Anti-C21orf59 | GeneTex | Cat#89274-274 |
| Anti-TOP2β | BD Biosciences | Cat#611492, RRID: AB_398952 |
| Anti-γH2AX | Millipore | Cat#05-636-I, RRID: AB_2755003 |
| Anti-γH2AX | BioLegend | Cat#613402, RRID: AB_315795 |
| Alexa Fluor 488–conjugated goat anti-mouse IgG (H+L) | Thermo Fisher Scientific | Cat#A32723, RRID: AB_2633275 |
| FITC–conjugated goat antirabbit IgG (H+L) | Thermo Fisher Scientific | Cat#F-2765, RRID: AB_2536525 |
| Chemicals, peptides, and recombinant proteins | ||
| PRMT5 inhibitor EPZ015666 | Selleck Chemicals | S7748; CAS: 1616391-65-1 |
| PRMT1 inhibitor EPZ019997 | Selleck Chemicals | S8858; CAS: 2227587-26-8 |
| HSP70 inhibitor VER155008 | Selleck Chemicals | S7751; CAS: 1134156-31-2 |
| HSP90 inhibitor 17-AAG | Selleck Chemicals | S1141; CAS: 75747-14-7 |
| Cisplatin | Selleck Chemicals | S1166; CAS: 15663-27-1 |
| Camptothecin (CPT) | Selleck Chemicals | S1288; CAS: 7689-03-4 |
| dTAGv-1-NEG | Tocris Bioscience | Cat#6915 |
| dTAGv-1 | Tocris Bioscience | Cat#6914 |
| Etoposide | Sigma-Aldrich | E1383; CAS: 33419-42-0 |
| Hydroxyurea (HU) | Sigma-Aldrich | H8627; CAS: 127-07-1 |
| MG132 | Sigma-Aldrich | 474790; CAS: 133407-82-6 |
| Cycloheximide (CHX) | Sigma-Aldrich | C4859; CAS: 66-81-9 |
| Bortezomib (BTZ) | Sigma-Aldrich | 504314; CAS: 179324-69-7 |
| Streptavidin-Sepharose beads | GE-Healthcare | 17-5113-01 |
| Critical commercial assays | ||
| CometAssay kit | R&D systems | Cat:4250-050-K |
| Deposited data | ||
| RNA-seq data | This study | GEO: GSE235200 |
| Whole-proteome profiling and GNB1L-associated proteins | This study | MassIVE: MSV000092198 |
| CRISPR screen data | This study | Tables S1 and S2 |
| Immunoblot data | This study | Mendeley, DOI: 10.17632/x7d248vz6f.1 |
| Experimental models: Cell lines | ||
| HEK293A | ATCC | N/A |
| HEK293T | ATCC | N/A |
| U2OS | ATCC | N/A |
| HeLa | ATCC | N/A |
| HEK29A-GNB1L-dTAG | This paper | N/A |
| HEK29A-TELO2-dTAG | This paper | N/A |
| Oligonucleotides | ||
| GNB1L sgRNA: AGGATCAGCGGATCAGCCTC | This paper | N/A |
| TELO2 sgRNA: CCCAGATCTGTCCTGCAGGA | This paper | N/A |
| β-actin forward: CACCATTGGCAATGAGCGGTTC | Wang et al., 2022 | N/A |
| β-actin reverse: AGGTCTTTGCGGATGTCCACGT | Wang et al., 2022 | N/A |
| ATM_Primer1 forward: ATAGATTGTGTAGGTTCCGATGG | Lee et al., 2011 | N/A |
| ATM_Primer1 reverse: CATCTTGTCTCAGGTCATCACG | Lee et al., 2011 | N/A |
| ATM_Primer2 forward: CCAGCTGTGCAGCGAACAAT | Rondeau et al., 2015 | N/A |
| ATM_Primer2 reverse: TCTAAGCACGTTTCTGCTAACCAGT | Rondeau et al., 2015 | N/A |
| See Table S7 for more sgRNA and shRNA target sequences | ||
| Recombinant DNA | ||
| pX330 | Cong et al., 2013 | Addgene#42230 |
| pCRIS-PITChv2-dTAG-blasticidin | Nabet et al., 2018 | Addgene#91795 |
| pCRIS-PITChv2-blasticidin-dTAG | Nabet et al., 2018 | Addgene#91792 |
| SFB-GNB1L | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | Version 9.0.0 |
| FlowJo | BD Biosciences | Version 10.8.1 |
| Adobe Illustrator | Adobe | 2022 |
| OpenComet | Gyori et al., 2014 | https://cometbio.org/index.html |
| MaxQuant | Tyanova et al., 2016 | N/A |
| MAGeCK | Li et al., 2014 | N/A |
| DrugZ | Colic et al., 2019 | N/A |
| R version 4.0.5 | N/A | https://www.r-project.org/ |
| Perl version 5.32.1 | N/A | https://www.perl.org/ |
| GO (DAVID) | Huang et al., 2009 | https://david-d.ncifcrf.gov/ |
| HISAT2 | Kim et al., 2019 | https://daehwankimlab.github.io/hisat2/manual/ |
| edgeR | Robinson et al., 2010 | N/A |
| HTSeq | Anders et al., 2015 | N/A |
| Cytoscape 3.5.1 | Shannon et al., 2003 | https://cytoscape.org/ |
| Other | ||
Highlights.
FACS-based CRISPR screens define positive and negative regulators of DDR signaling
Proteasome inhibition reduces DDR signaling
PRMT1 or PRMT5 inhibitor sensitizes cells to DNA damaging agents
GNB1L is a critical regulator of DDR signaling via specifically regulating PIKK proteins
ACKNOWLEDGMENTS
We thank all the members of the Chen laboratory for their help and constructive discussions. We thank The University of Texas MD Anderson Cancer Center Science Park Next-Generation Sequencing Facility for their help with CRISPR library next-generation sequencing (supported by the NIH/NCI under award number P30CA016672). We thank the technical support from Cancer Genomics Center at the University of Texas Health Science Center at Houston for their help with RNA-seq (CPRIT RP180734). The graphical abstract was created with BioRender.com. This work was supported in part by MD Anderson institutional funds and the Pamela and Wayne Garrison Distinguished Chair in Cancer Research. J.C. received support from the Cancer Prevention & Research Institute of Texas (RP160667 and RP180813) and NIH/NCI (CA193124, CA210929, CA216911, CA216437, CA274234, and CA275712). T.H. received support from NIH (R35GM130119). T.H. is also an Andrew Sabin Family Fellow and a Cancer Prevention & Research Institute of Texas Scholar in Cancer Research. Editorial support was provided by Don Norwood of Editing Services, Research Medical Library at MD Anderson.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Marechal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanz MC, Dibitetto D, and Smolka MB (2019). DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J 38, e101801. 10.15252/embj.2019101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl T, and Barnes DE (2000). Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65, 127–133. 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 5.Ghosal G, and Chen J (2013). DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res 2, 107–129. 10.3978/j.issn.2218-676X.2013.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caldecott KW (2008). Single-strand break repair and genetic disease. Nat Rev Genet 9, 619–631. 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 7.Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 66, 801–817. 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Brown JS, O’Carrigan B, Jackson SP, and Yap TA (2017). Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov 7, 20–37. 10.1158/2159-8290.CD-16-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cleary JM, Aguirre AJ, Shapiro GI, and D’Andrea AD (2020). Biomarker-Guided Development of DNA Repair Inhibitors. Mol Cell. 10.1016/j.molcel.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lavin MF, and Yeo AJ (2020). Clinical potential of ATM inhibitors. Mutat Res 821, 111695. 10.1016/j.mrfmmm.2020.111695. [DOI] [PubMed] [Google Scholar]
- 11.Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. 10.1038/s41586-018-0340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, Muir M, Fluteau A, Aregger M, McEwan A, et al. (2018). CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289. 10.1038/s41586-018-0291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, Chen Z, Srivastava M, McLaughlin ME, Navone NM, et al. (2019). Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 38, 2451–2463. 10.1038/s41388-018-0606-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olivieri M, Cho T, Alvarez-Quilon A, Li K, Schellenberg MJ, Zimmermann M, Hustedt N, Rossi SE, Adam S, Melo H, et al. (2020). A Genetic Map of the Response to DNA Damage in Human Cells. Cell 182, 481–496 e421. 10.1016/j.cell.2020.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan C, Mahara S, Sun C, Doan A, Chua HK, Xu D, Bian J, Li Y, Zhu D, Sooraj D, et al. (2021). Genome-scale CRISPR-Cas9 screen of Wnt/beta-catenin signaling identifies therapeutic targets for colorectal cancer. Sci Adv 7. 10.1126/sciadv.abf2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C, Feng X, Su D, Chen Z, Wang S, Tang M, Huang M, Nie L, Zhang H, Li S, et al. (2022). Integrated screens uncover a cell surface tumor suppressor gene KIRREL involved in Hippo pathway. Proc Natl Acad Sci U S A 119, e2121779119. 10.1073/pnas.2121779119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herr P, Lundin C, Evers B, Ebner D, Bauerschmidt C, Kingham G, Palmai-Pallag T, Mortusewicz O, Frings O, Sonnhammer E, and Helleday T (2015). A genome-wide IR-induced RAD51 foci RNAi screen identifies CDC73 involved in chromatin remodeling for DNA repair. Cell Discov 1, 15034. 10.1038/celldisc.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. (2009). A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35, 228–239. 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang M, Feng X, Su D, Wang G, Wang C, Tang M, Paulucci-Holthauzen A, Hart T, and Chen J (2020). Genome-wide CRISPR screen uncovers a synergistic effect of combining Haspin and Aurora kinase B inhibition. Oncogene 39, 4312–4322. 10.1038/s41388-020-1296-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colic M, Wang G, Zimmermann M, Mascall K, McLaughlin M, Bertolet L, Lenoir WF, Moffat J, Angers S, Durocher D, and Hart T (2019). Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med 11, 52. 10.1186/s13073019-0665-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Douarin B, Nielsen AL, Garnier JM, Ichinose H, Jeanmougin F, Losson R, and Chambon P (1996). A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J 15, 6701–6715. [PMC free article] [PubMed] [Google Scholar]
- 22.Moosmann P, Georgiev O, Le Douarin B, Bourquin JP, and Schaffner W (1996). Transcriptional repression by RING finger protein TIF1 beta that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res 24, 4859–4867. 10.1093/nar/24.24.4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SS, Chen YM, O’Leary E, Witzgall R, Vidal M, and Bonventre JV (1996). A novel member of the RING finger family, KRIP-1, associates with the KRAB-A transcriptional repressor domain of zinc finger proteins. Proc Natl Acad Sci U S A 93, 15299–15304. 10.1073/pnas.93.26.15299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, and Rauscher FJ 3rd (1996). KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10, 2067–2078. 10.1101/gad.10.16.2067. [DOI] [PubMed] [Google Scholar]
- 25.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, BekkerJensen S, Bartek J, and Shiloh Y (2006). Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol 8, 870–876. 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 26.Price BD, and D’Andrea AD (2013). Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354. 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crossley MP, Bocek M, and Cimprich KA (2019). R-Loops as Cellular Regulators and Genomic Threats. Mol Cell 73, 398–411. 10.1016/j.molcel.2019.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hills SA, and Diffley JF (2014). DNA replication and oncogene-induced replicative stress. Curr Biol 24, R435–444. 10.1016/j.cub.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 29.Schellenberg MJ, Lieberman JA, Herrero-Ruiz A, Butler LR, Williams JG, Munoz-Cabello AM, Mueller GA, London RE, Cortes-Ledesma F, and Williams RS (2017). ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA-protein cross-links. Science 357, 1412–1416. 10.1126/science.aam6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pommier Y, Sun Y, Huang SN, and Nitiss JL (2016). Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17, 703–721. 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole SP (2014). Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol 54, 95–117. 10.1146/annurevpharmtox-011613-135959. [DOI] [PubMed] [Google Scholar]
- 32.Hanssen KM, Haber M, and Fletcher JI (2021). Targeting multidrug resistance-associated protein 1 (MRP1)-expressing cancers: Beyond pharmacological inhibition. Drug Resist Updat 59, 100795. 10.1016/j.drup.2021.100795. [DOI] [PubMed] [Google Scholar]
- 33.Yin JY, Huang Q, Yang Y, Zhang JT, Zhong MZ, Zhou HH, and Liu ZQ (2009). Characterization and analyses of multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphisms in Chinese population. Pharmacogenet Genomics 19, 206–216. 10.1097/FPC.0b013e328323f680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popuri V, Tadokoro T, Croteau DL, and Bohr VA (2013). Human RECQL5: guarding the crossroads of DNA replication and transcription and providing backup capability. Crit Rev Biochem Mol Biol 48, 289–299. 10.3109/10409238.2013.792770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saponaro M, Kantidakis T, Mitter R, Kelly GP, Heron M, Williams H, Soding J, Stewart A, and Svejstrup JQ (2014). RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157, 1037–1049. 10.1016/j.cell.2014.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamadeh Z, and Lansdorp P (2020). RECQL5 at the Intersection of Replication and Transcription. Front Cell Dev Biol 8, 324. 10.3389/fcell.2020.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brumbaugh J, Di Stefano B, Wang X, Borkent M, Forouzmand E, Clowers KJ, Ji F, Schwarz BA, Kalocsay M, Elledge SJ, et al. (2018). Nudt21 Controls Cell Fate by Connecting Alternative Polyadenylation to Chromatin Signaling. Cell 172, 106–120 e121. 10.1016/j.cell.2017.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akman HO, Raghavan A, and Craigen WJ (2011). Animal models of glycogen storage disorders. Prog Mol Biol Transl Sci 100, 369–388. 10.1016/B978-0-12-384878-9.00009-1. [DOI] [PubMed] [Google Scholar]
- 39.Jarrold J, and Davies CC (2019). PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol Med 25, 993–1009. 10.1016/j.molmed.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Sciascia N, Wu W, Zong D, Sun Y, Wong N, John S, Wangsa D, Ried T, Bunting SF, Pommier Y, and Nussenzweig A (2020). Suppressing proteasome mediated processing of topoisomerase II DNA-protein complexes preserves genome integrity. Elife 9. 10.7554/eLife.53447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desai SD, Liu LF, Vazquez-Abad D, and D’Arpa P (1997). Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J Biol Chem 272, 24159–24164. 10.1074/jbc.272.39.24159. [DOI] [PubMed] [Google Scholar]
- 42.Morocz M, Zsigmond E, Toth R, Enyedi MZ, Pinter L, and Haracska L (2017). DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA. Nucleic Acids Res 45, 3172–3188. 10.1093/nar/gkw1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsutsui K, et al. (2016). Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol Cell 64, 1010. 10.1016/j.molcel.2016.11.028. [DOI] [PubMed] [Google Scholar]
- 44.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 45.Takai H, Wang RC, Takai KK, Yang H, and de Lange T (2007). Tel2 regulates the stability of PI3K-related protein kinases. Cell 131, 1248–1259. 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 46.Takai H, Xie Y, de Lange T, and Pavletich NP (2010). Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev 24, 2019–2030. 10.1101/gad.1956410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurov KE, Cotta-Ramusino C, and Elledge SJ (2010). A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev 24, 1939–1950. 10.1101/gad.1934210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulvaney KM, Blomquist C, Acharya N, Li R, Ranaghan MJ, O’Keefe M, Rodriguez DJ, Young MJ, Kesar D, Pal D, et al. (2021). Molecular basis for substrate recruitment to the PRMT5 methylosome. Mol Cell 81, 3481–3495 e3487. 10.1016/j.molcel.2021.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim H, and Ronai ZA (2020). PRMT5 function and targeting in cancer. Cell Stress 4, 199–215. 10.15698/cst2020.08.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 11, 432–437. 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- 51.Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, Rioux N, Lingaraj T, Ribich SA, Pappalardi MB, et al. (2019). Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 36, 100–114 e125. 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 52.Zhang H, Xiong Y, Su D, Wang C, Srivastava M, Tang M, Feng X, Huang M, Chen Z, and Chen J (2022). TDP1-independent pathways in the process and repair of TOP1-induced DNA damage. Nat Commun 13, 4240. 10.1038/s41467-022-31801-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teive HA, Moro A, Moscovich M, Arruda WO, Munhoz RP, Raskin S, and Ashizawa T (2015). Ataxia-telangiectasia - A historical review and a proposal for a new designation: ATM syndrome. J Neurol Sci 355, 3–6. 10.1016/j.jns.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamard PJ, Santiago GE, Liu F, Karl DL, Martinez C, Man N, Mookhtiar AK, Duffort S, Greenblatt S, Verdun RE, and Nimer SD (2018). PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep 24, 2643–2657. 10.1016/j.celrep.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan DQ, Li Y, Yang C, Li J, Tan SH, Chin DWL, Nakamura-Ishizu A, Yang H, and Suda T (2019). PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep 26, 2316–2328 e2316. 10.1016/j.celrep.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Owens JL, Beketova E, Liu S, Tinsley SL, Asberry AM, Deng X, Huang J, Li C, Wan J, and Hu CD (2020). PRMT5 Cooperates with pICln to Function as a Master Epigenetic Activator of DNA Double-Strand Break Repair Genes. iScience 23, 100750. 10.1016/j.isci.2019.100750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hwang JW, Kim SN, Myung N, Song D, Han G, Bae GU, Bedford MT, and Kim YK (2020). PRMT5 promotes DNA repair through methylation of 53BP1 and is regulated by Src-mediated phosphorylation. Commun Biol 3, 428. 10.1038/s42003-020-01157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giuliani V, Miller MA, Liu CY, Hartono SR, Class CA, Bristow CA, Suzuki E, Sanz LA, Gao G, Gay JP, et al. (2021). PRMT1-dependent regulation of RNA metabolism and DNA damage response sustains pancreatic ductal adenocarcinoma. Nat Commun 12, 4626. 10.1038/s41467-021-24798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gong L, Liu M, Jen J, and Yeh ET (2000). GNB1L, a gene deleted in the critical region for DiGeorge syndrome on 22q11, encodes a G-protein beta-subunit-like polypeptide. Biochim Biophys Acta 1494, 185–188. 10.1016/s0167-4781(00)00189-5. [DOI] [PubMed] [Google Scholar]
- 60.White DE, Negorev D, Peng H, Ivanov AV, Maul GG, and Rauscher FJ 3rd (2006). KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res 66, 11594–11599. 10.1158/0008-5472.CAN-06-4138. [DOI] [PubMed] [Google Scholar]
- 61.Williams NM, Glaser B, Norton N, Williams H, Pierce T, Moskvina V, Monks S, Del Favero J, Goossens D, Rujescu D, et al. (2008). Strong evidence that GNB1L is associated with schizophrenia. Hum Mol Genet 17, 555–566. 10.1093/hmg/ddm330. [DOI] [PubMed] [Google Scholar]
- 62.Ishiguro H, Koga M, Horiuchi Y, Noguchi E, Morikawa M, Suzuki Y, Arai M, Niizato K, Iritani S, Itokawa M, et al. (2010). Supportive evidence for reduced expression of GNB1L in schizophrenia. Schizophr Bull 36, 756–765. 10.1093/schbul/sbn160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi CH, Oghalai J, Curran S, et al. (2006). Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci U S A 103, 7729–7734. 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol 14, 431–441. 10.1038/s41589-0180021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nabet B, Ferguson FM, Seong BKA, Kuljanin M, Leggett AL, Mohardt ML, Robichaud A, Conway AS, Buckley DL, Mancias JD, et al. (2020). Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat Commun 11, 4687. 10.1038/s41467-020-18377-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yebenes H, Mesa P, Munoz IG, Montoya G, and Valpuesta JM (2011). Chaperonins: two rings for folding. Trends Biochem Sci 36, 424–432. 10.1016/j.tibs.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 67.Grantham J (2020). The Molecular Chaperone CCT/TRiC: An Essential Component of Proteostasis and a Potential Modulator of Protein Aggregation. Front Genet 11, 172. 10.3389/fgene.2020.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kubota H, Hynes G, and Willison K (1995). The chaperonin containing t-complex polypeptide 1 (TCP-1). Multisubunit machinery assisting in protein folding and assembly in the eukaryotic cytosol. Eur J Biochem 230, 3–16. 10.1111/j.1432-1033.1995.tb20527.x. [DOI] [PubMed] [Google Scholar]
- 69.Vallin J, and Grantham J (2019). The role of the molecular chaperone CCT in protein folding and mediation of cytoskeleton-associated processes: implications for cancer cell biology. Cell Stress Chaperones 24, 17–27. 10.1007/s12192-018-0949-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Llorca O, McCormack EA, Hynes G, Grantham J, Cordell J, Carrascosa JL, Willison KR, Fernandez JJ, and Valpuesta JM (1999). Eukaryotic type II chaperonin CCT interacts with actin through specific subunits. Nature 402, 693–696. 10.1038/45294. [DOI] [PubMed] [Google Scholar]
- 71.Llorca O, Martin-Benito J, Ritco-Vonsovici M, Grantham J, Hynes GM, Willison KR, Carrascosa JL, and Valpuesta JM (2000). Eukaryotic chaperonin CCT stabilizes actin and tubulin folding intermediates in open quasi-native conformations. EMBO J 19, 5971–5979. 10.1093/emboj/19.22.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stirling PC, Bloom MS, Solanki-Patil T, Smith S, Sipahimalani P, Li Z, Kofoed M, Ben-Aroya S, Myung K, and Hieter P (2011). The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet 7, e1002057. 10.1371/journal.pgen.1002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goto GH, Ogi H, Biswas H, Ghosh A, Tanaka S, and Sugimoto K (2017). Two separate pathways regulate protein stability of ATM/ATR-related protein kinases Mec1 and Tel1 in budding yeast. PLoS Genet 13, e1006873. 10.1371/journal.pgen.1006873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin YF, Lee YF, and Liang PH (2012). Targeting beta-tubulin:CCT-beta complexes incurs Hsp90- and VCP-related protein degradation and induces ER stress-associated apoptosis by triggering capacitative Ca2+ entry, mitochondrial perturbation and caspase overactivation. Cell Death Dis 3, e434. 10.1038/cddis.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin YF, Tsai WP, Liu HG, and Liang PH (2009). Intracellular betatubulin/chaperonin containing TCP1-beta complex serves as a novel chemotherapeutic target against drug-resistant tumors. Cancer Res 69, 6879–6888. 10.1158/0008-5472.CAN-08-4700. [DOI] [PubMed] [Google Scholar]
- 76.Tang M, Feng X, Pei G, Srivastava M, Wang C, Chen Z, Li S, Zhang H, Zhao Z, Li X, and Chen J (2020). FOXK1 Participates in DNA Damage Response by Controlling 53BP1 Function. Cell Rep 32, 108018. 10.1016/j.celrep.2020.108018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. 10.1186/s13059014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang M, Zhou B, Gong J, Xing L, Ma X, Wang F, Wu W, Shen H, Sun C, Zhu X, et al. (2018). RNA-splicing factor SART3 regulates translesion DNA synthesis. Nucleic Acids Res 46, 4560–4574. 10.1093/nar/gky220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol 2, 457–465. 10.1016/j.redox.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nie L, Wang C, Liu X, Teng H, Li S, Huang M, Feng X, Pei G, Hang Q, Zhao Z, et al. (2022). USP7 substrates identified by proteomics analysis reveal the specificity of USP7. Genes Dev 36, 1016–1030. 10.1101/gad.349848.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 83.Lee KW, Tsai YS, Chiang FY, Huang JL, Ho KY, Yang YH, Kuo WR, Chen MK, and Lin CS (2011). Lower ataxia telangiectasia mutated (ATM) mRNA expression is correlated with poor outcome of laryngeal and pharyngeal cancer patients. Ann Oncol 22, 1088–1093. 10.1093/annonc/mdq569. [DOI] [PubMed] [Google Scholar]
- 84.Rondeau S, Vacher S, De Koning L, Briaux A, Schnitzler A, Chemlali W, Callens C, Lidereau R, and Bieche I (2015). ATM has a major role in the double-strand break repair pathway dysregulation in sporadic breast carcinomas and is an independent prognostic marker at both mRNA and protein levels. Br J Cancer 112, 1059–1066. 10.1038/bjc.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 89.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. FACS-based CRISPR screens NormZ results, related to Figures 1A and S2. NormZ values calculated with DrugZ for each CRISPR screen reported in this work.
Table S2. FACS-based CRISPR screens raw counts, related to Figures 1A and S2. Raw counts for each sgRNA of 30 screens reported in this work.
Table S3. Gene Ontology analysis of positive and negative regulators, related to Figure 1D. Here are listed all the gene ontology terms for the 311 and 317 hits respectively identified as positive and negative regulators based on the antibody as well as 205 and 163 hits respectively identified as positive and negative regulators based on the treatment.
Table S4. Gene Ontology analysis of screen hits for network, related to Figures 2C and S4C. Here are listed all the gene ontology terms for the 615 and 366 hits that respectively identified based on the antibody and treatment.
Table S5. Proteome profiling in the absence of GNB1L, related to Figures 6C and S13A. List of proteins with significantly changes by the log2 ratio of protein abundance in the GNB1L depletion versus control cells.
Table S6. RNA-seq analysis in the absence of GNB1L, related to Figures 6C, S13B and S13C. Here are listed all the gene ontology terms for differentially expressed genes with cutoff (p-value <0.01 and fold change >2).
Table S7. SgRNA and shRNA target sequences, related to the STAR Methods.
Data Availability Statement
The datasets are publicly available. The mass spectrometry data (whole-proteome profiling and GNB1L-associated protein identification) have been deposited to the MassIVE data sets with the dataset identifier MSV000092198. Raw RNA-sequencing data have been deposited to NCBI under GEO: GSE235200. All FACS-based genome-wide CRISPR screens next-generation sequencing raw data and processed data are provided in Tables S1 and S2. Raw immunoblot images are available at Mendeley Data with the DOI 10.17632/x7d248vz6f.1 and are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.