

Abstract
Human African Trypanosomiasis (HAT), caused by Trypanosoma brucei, is one of the neglected tropical diseases with a continuing need for new medication. We here describe the discovery of 5-phenylpyrazolopyrimidinone analogs as a novel series of phenotypic antitrypanosomal agents. The most potent compound, 30 (NPD-2975), has an in vitro IC50 of 70 nM against T. b. brucei with no apparent toxicity against human MRC-5 lung fibroblasts. Showing good physicochemical properties, low toxicity potential, acceptable metabolic stability, and other pharmacokinetic features, 30 was further evaluated in an acute mouse model of T. b. brucei infection. After oral dosing at 50 mg/kg twice per day for five consecutive days, all infected mice were cured. Given its good drug-like properties and high in vivo antitrypanosomal potential, the 5-phenylpyrazolopyrimidinone analog 30 represents a promising lead for future drug development to treat HAT.
Introduction
Human African Trypanosomiasis (HAT), also known as African sleeping sickness, is a neglected parasitic disease (NPD).1 Although the detected cases have dropped from 25,000 in 1995 to less than 1000 in 2019, the parasite still represents a risk for 65 million people living in 36 African countries.2
The kinetoplastid parasite Trypanosoma brucei is the causative agent of HAT3 and is transmitted by tsetse flies (Glossina sp.). Two subspecies are responsible for the infection in human beings. One of the subspecies, T. b. gambiense, causes most human infections (95%) and occurs in Central and West Africa, which is also known as West African trypanosomiasis. While T. b. rhodesiense is responsible for infections (5%) in East and Southern Africa, known as East African trypanosomiasis.4 HAT develops in two clinical stages.3 In the first stage, the parasites multiply in the bloodstream and lymphatic system, causing non-specific symptoms such as headache, fever, and joint pain. Once the parasites penetrate the central nervous system, the disease reaches the second stage, leading to the most common symptom, namely a disturbed sleep pattern. If left untreated, the second stage of HAT will eventually result in a coma or death.2
There are only a few treatment options available to control both stages of trypanosomiasis.5−7 Thus far, all of them, except fexinidazole (Figure S1), suffer from difficult administration and moderate to severe side effects.8−10 As for many infectious diseases, drug resistance often develops, and the most recently approved fexinidazole shares cross-resistance with nifurtimox.11−13 Although the clinical trial of a promising new drug, acoziborole (Figure S1), is in progress, it is not easy to reach the goal of HAT elimination by 2030.14 Hence, a high urgency to explore novel antitrypanosomal agents remains.
Despite this urgency, only a limited number of programs are currently involved in research toward a new HAT treatment.15−18 Among them, phosphodiesterases (PDEs) are an important class of targets for HAT drug discovery. PDEs are enzymes that hydrolyze the second messengers cAMP and cGMP to their non-cyclic analogs, AMP and GMP. As these enzymes are found in both humans and parasites, some of them have been proven to be important targets for treating human diseases and potential targets for parasitic diseases.19−22 For instance, sildenafil was developed for the treatment of erectile dysfunction by targeting human PDE5.23 With abundant knowledge regarding human PDEs, we aimed to develop new antiparasitic treatments via a PDE target-based drug discovery strategy and a phenotypic screening strategy within the EU-funded consortium PDE4NPD (Phosphodiesterase Inhibitors for Neglected Parasitic Diseases).24 In 2007, T. brucei PDE B1 (TbrPDEB1)19 was validated as a target, and in 2018, Blaazer et al.25 reported optimization efforts toward TbrPDEB1-selective compounds targeting a parasitic-specific pocket (P-pocket26). Besides these target-based efforts, phenotypic screening/evaluation also played an important role. In 2015, BIPPO (1, Figure 1) analogs were reported as a novel class of potent anti-Plasmodium falciparum agents, potentially acting via their effect on cyclic nucleotide levels.27 Considering their antimalarial potency, low molecular weights, good drug-like properties (Table S1), and the fact that these molecules might act as parasite PDE inhibitors, a small additional series of BIPPO analogs (Table 1) was synthesized and phenotypically screened against a panel of protozoan parasites containing T. b. brucei, Trypanosoma cruzi, and Leishmania infantum. This study presents our efforts to characterize novel 5-phenylpyrazolopyrimidinone analogs as potent antitrypanosomal agents with in vivo efficacy.
Figure 1.

Design of 5-phenylpyrazolopyrimidinone analogs targeting T. b. brucei.
Table 1. Exploration of SAR in the R1 Position.
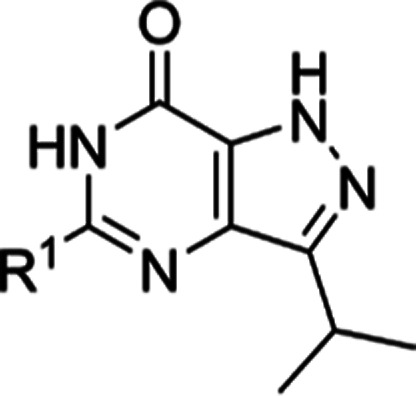
| code | R1 | MWa | cLogPa | PSAa | T. b. brucei pIC50b | MRC-5 pIC50b |
|---|---|---|---|---|---|---|
| 1 (BIPPO) | Bn | 268.3 | 2.2 | 70.1 | 4.5 ± 0.2 | <4.2 |
| 9 (NPD-2960) | 4-PyCH2 | 269.3 | 1.0 | 83.0 | <4.2 | <4.2 |
| 10 (NPD-0434) | C6H5OCH2 | 284.3 | 2.1 | 79.4 | <4.2 | <4.2 |
| 11 (NPD-3281) | C6H5(CH2)2 | 282.3 | 2.3 | 70.1 | <4.2 | <4.2 |
| 12 (NPD-3380) | Me | 192.2 | 0.7 | 70.1 | <4.2 | <4.2 |
| 13 (NPD-3645) | nBu | 234.3 | 2.1 | 70.1 | 5.0 ± 0.0 | <4.2 |
| 14 (NPD-3379) | iPr | 220.3 | 1.8 | 70.1 | 4.4 ± 0.1 | <4.2 |
| 15 (NPD-3200) | Ph | 254.3 | 2.3 | 70.1 | 6.6 ± 0.2 | <4.2 |
| 16 (NPD-3488) | 4-Py | 255.3 | 1.2 | 83.0 | 5.7 ± 0.0 | <4.2 |
| 17 (NPD-2973) | 4-thiazole | 261.3 | 1.2 | 83.0 | 4.9 ± 0.0 | <4.2 |
cLogP and PSA (polar surface area) are calculated using collaborative drug discovery (CDD).
Mean values of at least two independent experiments.
Results
Chemistry
The designed pyrazolopyrimidinone analogs were synthesized via the route shown in Scheme 1. The synthesis of 1, 4–8, 11, 12, and 15 was reported previously.27−30 This route starts with condensation and ring closure reactions to form the pyrazole ester intermediate 4. These two steps can be combined into a one-pot reaction. Following hydrolysis and nitration, intermediate 6 was obtained. The nitration reaction is a key step, and the rate of adding the reagent and the reaction temperature need to be carefully controlled.31 The first four (a–d) steps can be performed at a four hundred-gram scale without column purification. Following primary amide formation and reduction of the nitro group, the key intermediate 8 was formed. The final compounds can be obtained with two different methods from 8. Analogs 9–37 and 40–42 were prepared by amide coupling followed by ring closure reactions under basic conditions (Scheme 1A), whereas analogs 38 and 43–45 were obtained by ring closure reactions with the corresponding aldehydes and iodine (Scheme 1B) due to starting material availability and reactivity. Analog 39 was obtained following hydrolysis of 38, and [2 + 3] cycloaddition of 37 with NaN3 yielded tetrazole 46 with a decent yield. Due to tautomerism of the pyrazole ring in the structures, some carbon signals are too broad or invisible in the 1D NMR spectra, and earlier publications27,29 did not report complete chemical characterizations (especially 13C NMR signals) for the published analogs.25,27 Here, we report the results of 13C NMR combined with 2D NMR (HSQC and HMBC) or high-temperature NMR needed to obtain full characterization. For analog 30, additional efforts with salt formation to prevent tautomerism ended up with sharp 13C NMR signals; the further “1,n-ADEQUATE” experiment confirmed its structure (Figures S90–94).
Scheme 1. Synthesis of Pyrazolopyrimidinone Analogs.

Reagents and conditions: (a) 3-methylbutan-2-one, NaOEt, EtOH, 60 °C, 2 h; (b) N2H4·H2O, EtOH, reflux, 2 h, 54% over two steps; (c) NaOH, 1,4-dioxane/H2O 1:1 (v/v), 50 °C, 3 h, 62%; (d) conc. H2SO4, 65% HNO3, 60 °C, 3 h, 64%; (e) cat. DMF, (COCl)2, DCM, 0 °C, 1 h, then RT, 2 h; (f) 7 M NH3 in MeOH, 0 °C, 30 min, 69% over two steps; (g) 10% Pd/C, H2 (g), EtOH, 60 °C, 18 h, 93%; (h) RCOOH, TEA, bromo-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBroP), DCE, microwave irradiation (MW) 120 °C, 20 min; (i) KOtBu, iPrOH, MW 130 °C, 30 min, 4–90% over two steps; (j) NaN3, NH4Cl, DMF, MW 160 °C, 2 h, 69%; (k) RCHO, I2, DMF, 80 °C, 16 h, 23–47%; (l) LiOH, 1,4-dioxane/H2O 1:1 (v/v), reflux, 2 h, 75%.
In Vitro Evaluation of Anti-T. brucei Activity
In our PDE4NPD program, we initially synthesized a small library of 5-benzyl-3-isopropyl-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-one (BIPPO, 1, Figure 1) analogs as this scaffold was reported to inhibit PDEs from P. falciparum.27 Consequently, the pyrazolopyrimidinone scaffold was considered an interesting start to identifying antitrypanosomal compounds. The first small series focused on variations in the 5-position of the BIPPO scaffold (Table 1). The benzyl moiety was replaced with a number of aromatic and aliphatic substituents, and the linker length and flexibility between the pyrazolopyrimidinone moiety and the terminal aromatic substituent were varied to explore structure–activity relationships (SARs). The analogs were tested for activity against T. b. brucei, T. cruzi, L. infantum, and toxicity against the human MRC-5 cell line was determined to measure general toxicity and selectivity. Based on the results shown in Table S2, only T. b. brucei inhibition was observed; thereafter, we focused on modifications for improved antitrypanosomal activity.
As shown in Table 1, BIPPO (1) is only weakly active against T. b. brucei. A pyridine instead of a phenyl was introduced in 9 to increase solubility, but no potency improvement was observed. The same results were obtained with 10 and 11, in which, respectively, an oxygen atom or a methylene group was introduced into the linker. Pyrazolopyrimidinones with aliphatic substituents (12, 13, and 14) at R1 also exhibited low potency. The potency of analogs with different aromatic substituents varied considerably. When the aromatic rings were directly attached to the pyrazolopyrimidinone moiety, a drastic improvement in antiparasitic activity was observed. Analogs 15 and 16 were 125 and 16 times more potent than 1, respectively. However, 17 with a thiazole exhibited comparable activity to 1.
The phenyl-analog 15 had a pIC50 of 6.6 against T. b. brucei without noticeable toxicity against MRC-5 cells and was the most active compound in our first round of modifications.
Our second round of modifications (Table 2) logically were based on compound 15. Analogs with different substituents at the ortho, meta, and para positions were synthesized, and activities against T. b. brucei (Figure S2) and MRC-5 fibroblasts were determined. Analogs with an ortho substituent (18–21) showed comparable activity to 15, with the ortho-substituted methyl analog 21 being the most potent (pIC50 6.8). The analogs with an ortho-halogen substituent (18–20) were slightly less potent than 15. The ortho-methoxy-substitued 22 was clearly less effective, with a 25-fold reduction in potency. The meta-halogen analogs 23 and 24 proved to be significantly more potent than their ortho-analogs 18 and 19, with the meta-fluoro analog 23 showing a pIC50 of 7.0. Analog 25 with a meta-methyl group was equipotent to 15, while 26 with a meta-methoxy group was slightly less potent. In contrast, the introduction of meta-OH, −N(CH3)2, and −SO2CH3 (27–29) led to a more than 10-fold reduction in potency compared with 15.
Table 2. Exploration of SAR on the Phenyl Ring of 15.
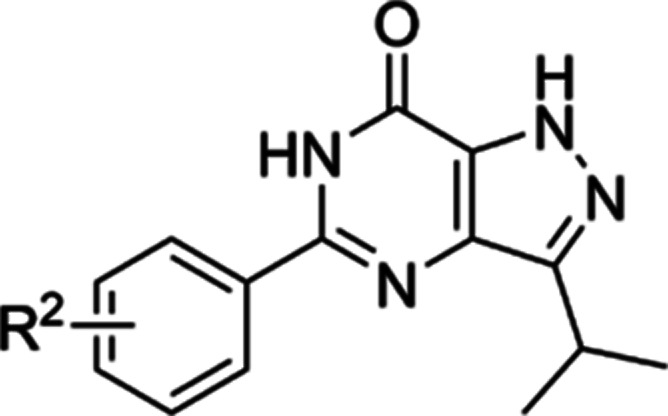
| code | R2 | MWa | cLogPa | PSAa | T. b. brucei pIC50b | MRC-5 pIC50b |
|---|---|---|---|---|---|---|
| 15 (NPD-3200) | H | 254.3 | 2.3 | 70.1 | 6.6 ± 0.2 | <4.2 |
| 18 (NPD-3199) | 2-F | 272.3 | 2.3 | 70.1 | 6.0 ± 0.1 | <4.2 |
| 19 (NPD-3538) | 2-Cl | 288.7 | 2.9 | 70.1 | 6.1 ± 0.2 | <4.2 |
| 20 (NPD-3539) | 2-Br | 333.2 | 2.9 | 70.1 | 6.2 ± 0.1 | <4.2 |
| 21 (NPD-3589) | 2-Me | 268.3 | 2.7 | 70.1 | 6.8 ± 0.2 | <4.2 |
| 22 (NPD-3590) | 2-OMe | 284.3 | 2.1 | 79.4 | 5.1 ± 0.1 | <4.2 |
| 23 (NPD-3202) | 3-F | 272.3 | 2.5 | 70.1 | 7.0 ± 0.1 | <4.2 |
| 24 (NPD-3591) | 3-Cl | 288.7 | 3.1 | 70.1 | 6.7 ± 0.3 | <4.2 |
| 25 (NPD-3382) | 3-Me | 268.3 | 2.8 | 70.1 | 6.6 ± 0.2 | <4.2 |
| 26 (NPD-3375) | 3-OMe | 284.3 | 2.4 | 79.4 | 6.3 ± 0.1 | <4.2 |
| 27 (NPD-2974) | 3-OH | 270.3 | 1.9 | 90.4 | 5.2 ± 0.0 | <4.2 |
| 28 (NPD-3381) | 3-N(CH3)2 | 297.4 | 2.5 | 73.4 | 5.1 ± 0.0 | <4.2 |
| 29 (NPD-3598) | 3-SO2CH3 | 332.4 | 1.2 | 104.3 | 4.5 ± 0.0 | <4.2 |
| 30 (NPD-2975) | 4-F | 272.3 | 2.5 | 70.1 | 7.2 ± 0.2 | <4.2 |
| 31 (NPD-3204) | 4-Cl | 288.7 | 3.1 | 70.1 | 7.0 ± 0.2 | <4.2 |
| 32 (NPD-2971) | 4-Br | 333.2 | 3.2 | 70.1 | 6.6 ± 0.3 | <4.2 |
| 33 (NPD-2972) | 4-OMe | 284.3 | 2.4 | 79.4 | 6.1 ± 0.1 | <4.2 |
cLogP and PSA (polar surface area) are calculated using CDD.
Mean values of at least two independent experiments.
For analogs with para-substituents, 30 with a para-F substituent was the most potent in this series with a pIC50 of 7.2 (Table 2). A comparable potency was observed for the para-Cl analog 31, but a four-fold reduction in potency was observed for the bromide 32. The para-methoxy group in 33 decreased the pIC50 to 6.1, which is three times less active than 15.
Based on the SAR results from Table 2, our third round of modifications (Table 3) focused on the para-position of the phenyl ring in 15 to further improve chemical diversity and physicochemical properties, such as polarity and solubility. Analogs with relatively small substituents 35–37 exhibited comparable potency with 15. Once bigger or polar substituents (38–46) were introduced, pIC50 values decreased dramatically (<5.0), which indicates that only a restricted set of substituents is accepted at the para-position of this phenyl ring.
Table 3. Exploration of SARs on the Para-position of the Phenyl Ring.
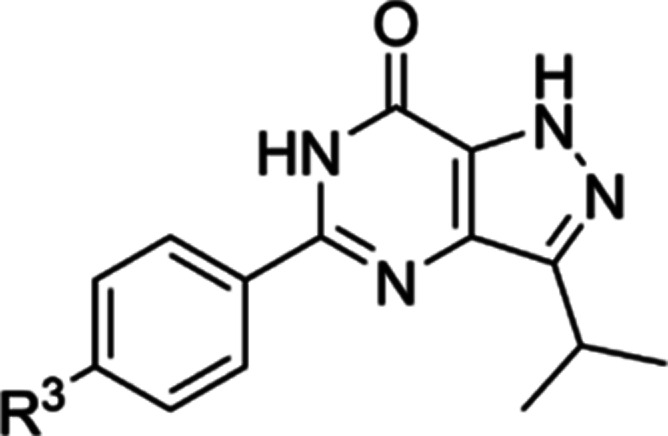
| code | R3 | MWa | cLogPa | PSAa | T. b. brucei pIC50b | MRC-5 pIC50b |
|---|---|---|---|---|---|---|
| 34 (NPD-3377) | -OiPr | 312.4 | 3.2 | 79.4 | <4.2 | <4.2 |
| 35 (NPD-3201) | -CF3 | 322.3 | 3.4 | 70.1 | 5.9 ± 0.1 | <4.2 |
| 36 (NPD-3597) | -OCF3 | 338.3 | 3.8 | 79.4 | 5.9 ± 0.2 | <4.2 |
| 37 (NPD-3203) | -CN | 279.3 | 2.1 | 93.9 | 6.6 ± 0.3 | <4.2 |
| 38 (NPD-3305) | -COOMe | 312.3 | 2.4 | 96.4 | <4.2 | <4.2 |
| 39 (NPD-3489) | -COOH | 298.3 | 2.1 | 107.4 | <4.2 | <4.2 |
| 40 (NPD-3371) | -CONH2 | 297.3 | 1.3 | 113.2 | <4.2 | <4.2 |
| 41 (NPD-3376) | -SO2CH3 | 332.4 | 1.2 | 104.3 | 4.3 ± 0.1 | <4.2 |
| 42 (NPD-3372) | -SO2NH2 | 297.3 | 1.0 | 113.2 | 4.4 ± 0.1 | <4.2 |
| 43 (NPD-3280) | -NHCOCH3 | 311.3 | 1.7 | 99.2 | <4.2 | <4.2 |
| 44 (NPD-3283) | -piperidine | 337.4 | 3.6 | 73.4 | <4.2 | <4.2 |
| 45 (NPD-3282) | -methylpiperazine | 352.4 | 2.1 | 76.6 | 4.7 ± 0.4 | 4.4 ± 0.1 |
| 46 (NPD-3490) | -tetrazole | 322.3 | 2.0 | 124.6 | <4.2 | <4.2 |
cLogP and PSA (polar surface area) are calculated using CDD.
Mean values of at least 2 independent experiments.
As 30 (NPD-2975) turned out to be the most interesting compound in this series in terms of in vitro potency and physicochemical properties (cLogP), this compound was selected for further antiparasitic profiling. Table 4 shows the activity of 30 against a panel of protozoan parasites, revealing nanomolar potency against T. b. brucei and no activity against T. cruzi, L. infantum, or P. falciparum. Moreover, no toxicity was observed against the human MRC-5 cell line or peritoneal mouse macrophages (PMM), endorsing its high selectivity.
Table 4. Potency of 30 against Other parasites.
| code | T. b. brucei pIC50 | T. cruzi pIC50 | L. infantum pIC50 | P. falciparum pIC50 | MRC-5 pIC50 | PMM pIC50 |
|---|---|---|---|---|---|---|
| 30 (NPD-2975) | 7.2 | <4.2 | <4.2 | <4.2 | <4.2 | <4.2 |
In Vitro Profiling of 30
Given its good potency and lack of toxicity, 30 was evaluated in detail in several in vitro assays. The target-based approach in the PDE4NPD consortium had shown that targeting both TbrPDEB1 and TbrPDEB2 with specific inhibitors25 will kill trypanosomes, thereby confirming earlier target validation work by Seebeck et al.19,32 As the scaffold of 30 was earlier identified to target PDE enzymes,2730 was tested against TbrPDEB1 but proved to be inactive (Table 5, Figure S3). Since both TbrPDEB1 and TbrPDEB2 need to be inhibited to kill trypanosomes,19 this observation immediately dismisses the PDE-hypothesis for the observed antiparasitic activity of 30.
Table 5. Detailed In Vitro Profiling of 30 (NPD-2975).
| assays | 30 |
|---|---|
| TbrPDEB1 inhibition | pKi < 5.0 |
| CYP450 IC50 | 1A2: 0.16 μM |
| 2C9: >10 μM | |
| 2C19: 0.42 μM | |
| 2D6: >10 μM | |
| 3A4: >10 μM | |
| hERG | >10 μM |
| mitochondrial toxicity | <10% toxicity at 10 μM |
| mini-ames | negative |
To identify potential safety issues, 30 was subsequently tested at 10 μM in the Eurofins Safety-47 panel, which includes 24 GPCRs, two nuclear hormone receptors, three transporters, eight ion channels, six non-kinase enzymes, and four kinase enzymes (Table S3). The activity of these targets was modulated <50% at 10 μM, with the exception of human PDE4D2, which was inhibited by 75% at 10 μM. Since compound 30 also shows some structural similarities with sildenafil, 30 was also tested against PDE5. At 10 μM 30 inhibited PDE5 by only 52%. Screening of 30 as inhibitors of a panel of CYP enzymes resulted in IC50 values of 0.16 and 0.42 μM against CYP1A2 and CYP2C19, respectively. Inhibition of CYP2C9, CYP2D6, and CYP3A4 by 30 was not observed in the tested concentration range. Finally, negative results from a mini-Ames test, hERG affinity, and a mitochondrial toxicity test further supported the drug-like quality of 30 (Table 5).
Metabolic Stability
Metabolic stability was assessed by incubating 30 and the control diclofenac with mouse, rat, and human liver microsomes in the absence or presence of uridine diphosphate glucuronic acid (UDPGA) to stimulate phase-II metabolism. The results indicated that 30 was metabolized the slowest by human microsomes and to a moderate extent in rodent microsomes (Figure 2). Phase-I metabolism in mouse microsomes resulted in 55% of the parent drug remaining after 30 min of incubation, which can be defined as acceptable metabolic stability.33 No significant difference was observed when metabolism by UGT enzymes was induced by the addition of UDPGA (phase-II metabolism), suggesting that phase-I metabolism is the main route of metabolism in 30.
Figure 2.

In vitro metabolic stability of 30 in liver microsomes. (A) Simulated phase-I metabolic stability of 30 in mouse, rat, and human liver microsomes with diclofenac as the reference. (B) Simulated phase-II metabolic stability of 30 in mouse, rat, and human liver microsomes with diclofenac as reference. Source data are provided in Table S4.
Based on its low cytotoxicity, potent in vitro activity (Table 2), good selectivity (Table 4), and acceptable metabolic stability in mouse microsomes (Figure 2), 30 was progressed to in vivo evaluation in mouse.
In Vivo Pharmacokinetics
The pharmacokinetic profiles of 30 were determined after either oral (PO) or intraperitoneal (IP) administration (Figure 3), and their blood concentrations were used to derive various pharmacokinetic parameters (Table 6). Both PO and IP administration quickly led to micromolar blood concentrations that exceeded the IC50 more than 50-fold (Table 6, Figure 3). For the subsequent evaluation of 30 in a mouse model of acute T. b. brucei infection, 50 mg/kg PO administration was used.
Figure 3.

Blood levels (0–24 h) of 30 in mice after a single IP dose (10 mg/kg) or PO dose (50 mg/kg). Results are expressed in mean blood concentration (μM) ± standard error of the mean.
Table 6. Pharmacokinetic Parameters of 30.
| compound | dosing | Tmax (h) | Cmax (μM) | T1/2 (h) | AUC0–6h (ng·h/mL) | Cl (mL/min) |
|---|---|---|---|---|---|---|
| 30 | 50 mg/kg PO | 1 | 5.25 | 3.46 | 6064.75 | 58.5 |
| 10 mg/kg IP | 13.18 | 1.06 | 3928.37 | 171 |
In Vivo Evaluation of 30
Next to suramin (10 mg/kg IP s.i.d. for 5 days) as a positive reference in a mouse model of acute T. b. brucei infection, treatment with 30 at 50 mg/kg twice daily PO for 5 consecutive days resulted in apparent full clearance of parasitemia (Figure 4). In contrast to the high parasitemia and early mortality in the vehicle-treated mice, all 30-treated animals in the highest dosing group were devoid of peripheral blood parasites throughout the 60 days post-infection (dpi) follow-up period. An additional SL RNA qPCR confirmed the absence or undetectable levels of parasites in peripheral blood. A clear dose-dependent in vivo efficacy was recorded, as all animals treated at 25 mg/kg b.i.d. for 5 days relapsed and succumbed within 11 dpi.
Figure 4.

In vivo evaluation of 30 in a stage-I mouse model of HAT. Survival rate (A) and parasitemia (B) of stage-I T. b. brucei-infected mice treated with vehicle (n = 3), suramin (n = 3) at 10 mg/kg or 30 (n = 3) at 50, 25, 12.5, and 6.25 mg/kg.
Discussion
We present a series of 5-phenylpyrazolopyrimidinone analogs with low nanomolar IC50 values and promising in vivo efficacy against T. b. brucei. Based on the published anti-P. falciparum inhibitor 1 (BIPPO), a library of 38 analogs was designed and phenotypically screened against T. b. brucei. The shortening of the linker between the phenyl group and the pyrazolopyrimidinone moiety drastically increased its potency, resulting in 15 with a pIC50 of 6.6. Further modification based on the structure of 15 with various substituents on its phenyl ring led to the discovery of 30 with an IC50 of 70 nM against T. b. brucei and no noticeable toxicity for a number of other protozoan parasites or human cell lines. Additional modifications based on the structure of 30 to improve solubility did not yield analogs with improved activity. Potentially, this might be a result of limited space or a lipophilic environment in the binding pocket of its target, which is currently unknown.
Follow-up analysis revealed that 30 did not inhibit the TbrPDEB1 enzyme, a validated target for HAT, and showed good selectivity over a range of targets (24 GPCRs, 2 nuclear hormone receptors, 3 transporters, 8 ion channels, 6 non-kinase enzymes, and 4 kinase enzymes). Also, 30 tested negative in a mini-Ames test and showed no hERG liability, two crucial criteria in drug discovery for a ‘lead’ compound. With respect to metabolism, 30 did not inhibit CYP2C9, CYP2D6, or CYP3A4 but did have moderate CYP1A2 and 2C19 liability and was metabolically stable in human and rodent liver microsomes, resulting in micromolar levels of 30 in mouse serum after PO and IP administration. In an acute mouse model of HAT, 30 yielded a 100% survival rate at 50 mg/kg b.i.d. for 5 days, making it an interesting lead for HAT treatment. Unfortunately, its mode of action is still unknown and is currently being investigated with a metabolomics approach34,35 and an RNAi method, as previously reported.36
Although the number of HAT patients is decreasing every year, many people living in sub-Saharan Africa are still at risk of infection. In the past few years, a number of publications25,33,37−44 indeed specifically focused on antitrypanosomal drug discovery. Compared to these published scaffolds, the pyrazolopyrimidinones (including 30) have a relatively low molecular weight, which is for most of the analogs lower than 300 Dalton. This is a very nice feature of this compound series and can be further exploited during lead optimization. Next to that, other physicochemical properties (such as cLogP, number of hydrogen bond donors/acceptors) of 30 fit with Lipinski’s rule of five,45 indicating good drug-like properties. Also, the selectivity and metabolic stability of 30 are remarkable. Last but not least, its in vivo pharmacokinetic features and in vivo efficacy are outstanding since a complete cure was obtained at 50 mg/kg b.i.d. PO for 5 days without relapse at 60 dpi.
Conclusions
To conclude, our pyrazolopyrimidinone analogs with phenyl substituents are novel, potent, and selective antitrypanosomal agents. Compound 30 with a para-fluorophenyl group exhibits an IC50 of 70 nM against T. b. brucei. Follow-up physiochemical feature analysis, metabolic stability, and pharmacokinetic testing revealed its excellent drug-like properties. Importantly, the absence of detectable parasite levels in peripheral blood following an oral dose of 50 mg/kg b.i.d. for 5 days in mice disclosed its promising in vivo potential and deserves further exploration for future drug development.
Experimental Section
In Vitro Evaluation
All compounds tested (1, 9–46) pass a publicly available pan-assay interference compound filter.46,47 The antiparasitic assays and the TbrPDEB1 enzyme assay were carried out as described in Blaazer et al.25 Briefly, phosphodiesterase activity assays were performed using the PDELight HTS cAMP phosphodiesterase Kit (Lonza, Walkersville, USA) at 25 °C in non-binding, low volume 384 wells plates (Corning, Kennebunk, ME, USA). PDE activity was measured in “stimulation buffer” (50 mM Hepes, 100 mM NaCl, 10 mM MgCl2, 0.5 mM EDTA, 0.05 mg/mL BSA, pH 7.5). Dose response curves were measured in triplo. The compounds were diluted in DMSO. Inhibitor dilutions (2.5 μL) were transferred to the 384 wells plate, 2.5 μL PDE in stimulation buffer was added and mixed, 5 μL cAMP (at 2 × km up to 20 μM) was added, and the assay was incubated for 20 min at 300 rpm. The reaction was terminated with 5 μL Lonza Stop Buffer supplemented with 10 μM NPD-0001. Luminescence was determined in a Victor3 luminometer. Antitrypanosomal assays were carried out with the T. brucei Squib 427 strain cultured in Hirumi’s modified Iscove’s medium 9 (HMI-9) supplemented with 10% fetal bovine serum at 37 °C in a 5% CO2 atmosphere. Parasites were seeded at a concentration of 1.5 × 104 parasites/well. Four-fold dilutions of the test compounds were added, with the highest in-test concentration of 64 μM. After 72 h of drug exposure, viability was determined by incubation with 10 μg/mL resazurin (Sigma-Aldrich, St. Louis, MO, USA) and fluorescence reading after 24 h. The percentage growth inhibition compared to untreated control wells was used to calculate the 50% inhibitory concentration (IC50). The CYP450 assays, hERG cardiotoxicity assay, mitochondrial toxicity assay, and data analysis were carried out as described in Moraes et al.48 The mini-Ames test (Wuxi49) and the Eurofins Safety47 panel (Eurofins50) screens were outsourced to CROs.
Metabolic Stability
The microsomal stability assay was carried out based on the BD Biosciences Guidelines for Use (TF000017 Rev1.0) with minor adaptations. Male mouse and pooled human liver microsomes (Corning) were purchased and stored at −80 °C until use. Both CYP450 and other NADPH-dependent enzymes (phase-I metabolism) and UGT enzymes (phase-II metabolism) were evaluated for NPD-2975 (30) with a working concentration of 5 μM. Diclofenac was used as a reference drug. Compound 30 was incubated with 0.5 mg/mL liver microsomes in potassium phosphate buffer, and reactions were initiated by the addition of 1 mM NADPH and 2 mM UDPGA cofactors for phase-I and phase-II metabolism, respectively. Samples were collected after 0, 15, 30, and 60 min. At these time points, 20 μL was withdrawn from the reaction mixture, and 80 μL cold acetonitrile (MeCN), containing the internal standard tolbutamide, was added to inactivate the enzymes and precipitate the proteins. The mixture was vortexed for 30 s and centrifuged at 4 °C for 5 min at 15,000 rpm. The supernatant was stored at −80 °C until analysis. The loss of parent compound was determined using liquid chromatography (UPLC) (Waters Aquity) coupled with tandem quadrupole mass spectrometry (MS2) (Waters Xevo), equipped with an electrospray ionization (ESI) interface, and operated in the multiple reaction monitoring (MRM) mode.
Pharmacokinetics
Analog 30 (NPD-2975) was evaluated for its pharmacokinetic properties after a single dose, either IP at 10 mg/kg or PO at 50 mg/kg. Blood drops from the animals were sampled before treatment and at 0.5, 1, 2, 4, 6, and 24 h after PO dosage; sampling after IP dosage was identical with an additional time point of 0.25 h. The blood drops were analyzed adopting the dry blood spot collection and analysis by LC–MS2. Briefly, blood samples were collected from the tail vein using capillary tubes and dropped (15 μL) on Whatman FTA DMPK cards (B). The spots were left to air dry at room temperature for at least 2 h. For analysis, a 6 mm disk was punched out and extracted in 75:25% MeCN/water containing the internal standard, tolbutamide. The brain tissue of the animals was collected on ice at autopsy 24 h post-treatment after perfusion. The tissue was immediately homogenized using a GentleMacs tissue homogenizer. The homogenates were then either immediately processed for analysis or stored at −80 °C. The tissue samples were subjected to protein precipitation by adding MeCN, followed by a centrifugation step at 4 °C for 5 min at 15,000 rpm. The supernatant was further diluted in 75:25% MeCN/water. The bio-analysis used liquid chromatography (UPLC) (Waters Aquity) coupled with tandem quadrupole mass spectrometry (MS2) (Waters Xevo), equipped with an ESI interface, and operated in the MRM mode. Standard curves in whole blood were made for calibration and validation. Standard pharmacokinetic parameters were determined using Topfit software.
Acute Mouse Model
Mice were allocated to groups of three and were infected by an IP injection with 104 trypomastigotes of T. b. brucei (suramin-sensitive Squib 427 strain). Compound 30 (NPD-2975) was formulated in PEG400 at 12.5 and 6.25 mg/mL, envisaging a maximal dosing volume of 100 μL/25 g live body weight. Next to including a vehicle control group with only PEG400, suramin was included as the reference drug and was injected IP s.i.d. for 5 consecutive days at 10 mg/kg. 30 was administered PO b.i.d. for 5 consecutive days at 25 or 50 mg/kg. The first treatment was given 30 min prior to the artificial infection. Drug efficacy was evaluated by microscopic determination of the parasitemia in tail vein blood samples at several time points until 63 dpi. An additional SL RNA qPCR assay was performed for all surviving animals to confirm parasitological cure. Animals were observed for the occurrence/presence of clinical or adverse effects during the course of the experiment. In cases of very severe clinical signs, either due to toxicity or clinical disease, animals were euthanized for animal welfare reasons. All animal experiments were conducted in compliance with institutional guidelines and following approval by the Ethical Committee of the University of Antwerp, Belgium [UA-ECD 2014–96].
Chemistry
General Information
All starting materials were obtained from commercial suppliers and used without purification. Anhydrous THF, DCM, and DMF were obtained by passing through an activated alumina column prior to use. All reactions were carried out under a nitrogen atmosphere, unless mentioned otherwise. TLC analyses were performed using Merck F254 aluminum-backed silica plates and visualized with 254 nm UV light. Flash column chromatography was executed using Biotage Isolera equipment. All HRMS spectra were recorded on a Bruker microTOF mass spectrometer using ESI in positive-ion mode. All NMR spectra were recorded on either a Bruker Avance 300, 400, 500, or 600 spectrometer at 25 °C, unless mentioned otherwise. The peak multiplicities are defined as follows: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; dd, doublet of doublets; dt, doublet of triplets; td, triplet of doublets; br, broad; m, multiplet, app, and apparent. The spectra were referenced to the internal solvent peak as follows: CDCl3 (δ = 7.26 ppm in 1H NMR, δ = 77.16 ppm in 13C NMR), DMSO-d6 (δ = 2.50 ppm in 1H NMR, δ = 39.52 ppm in 13C NMR). IUPAC names were adapted from ChemBioDraw Ultra 19.0. Purities were measured with the aid of analytical LC–MS using a Shimadzu LC-20AD liquid chromatography pump system with a Shimadzu SPDM20A diode array detector, with MS detection performed with a Shimadzu LCMS-2010EV mass spectrometer operating in the positive (or negative) ionization mode. The column used was an Xbridge (C18) 5 μm column (100 mm × 4.6 mm). The following solutions are used for the eluents. Solvent A: H2O/HCOOH 999:1, and solvent B: MeCN/HCOOH 999:1. The eluent program used is as follows: flow rate: 1.0 mL/min, start with 95% A in a linear gradient to 10% A over 4.5 min, hold 1.5 min at 10% A, in 0.5 min in a linear gradient to 95% A, hold 1.5 min at 95% A, and total run time: 8.0 min. Compound purities were calculated as the percentage peak area of the analyzed compound by UV detection at 254 nm. All final compounds (1, 9-46) are >95% pure by HPLC analysis (Figures S4–S142). Note: not all 13C signals are visible in the spectrum due to the rapid tautomerism of non-N-substituted pyrazoles. HSQC and HMBC were measured to assign 13C signals if needed.
Ethyl 3-Isopropyl-1H-pyrazole-5-carboxylate (4)
Diethyl oxalate 2 (20.0 mL, 147 mmol) and 3-methylbutan-2-one (15.8 mL, 150 mmol) were added to a mixture of NaOEt (14.0 g, 206 mmol) in EtOH (300 mL) at RT over 1 h. The reaction mixture was heated at 60 °C for 2 h, after which AcOH (47.7 mL, 294 mmol) and 64–65% N2H4 aqueous solution (9.10 g, 160 mmol) were added, and the mixture was stirred under reflux for 2 h. Water (200 mL) was added after the reaction mixture was evaporated under reduced pressure, and the mixture was extracted with EtOAc (3 × 150 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was subjected to silica gel column chromatography and eluted with EtOAc/cyclohexane 1:1 to give the title product 4 as a white solid (14.5 g, 54% for two steps). 1H NMR (300 MHz, CDCl3): δ 7.75 (br s, 1H), 6.63 (s, 1H), 4.37 (q, J = 7.1 Hz, 2H), 3.05 (hept, J = 7.7 Hz, 1H), 1.37 (t, J = 7.0 Hz, 3H), 1.30 (d, J = 6.8 Hz, 6H). 13C NMR (151 MHz, CDCl3): δ 161.5, 155.0, 140.6, 105.0, 61.2, 26.6, 22.5, 14.4. LC–MS: tR = 3.61 min, purity: >99%, not ionized. Spectral data agree with a previous report.29
3-Isopropyl-1H-pyrazole-5-carboxylic Acid (5)
Ester 4 (27.0 g, 148 mmol) and NaOH (14.8 g, 370 mmol) were dissolved in 1,4-dioxane (400 mL) and water (400 mL) and heated to 50 °C for 3 h. The reaction mixture was concentrated under reduced pressure and washed with EtOAc (3 × 200 mL); the pH was adjusted to 1 with the concentrated aqueous HCl solution, and the title product 5 was filtered as an off-white solid (14.2 g, 62%). 1H NMR (300 MHz, DMSO-d6): δ 6.47 (s, 1H), 2.94 (hept, J = 7.1 Hz, 1H), 1.21 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 163.3, 153.1, 141.6, 104.6, 26.1, 22.8. LC–MS: tR = 2.72 min, purity: >99%, m/z [M – H]−: 153. Spectral data agree with a previous report.29
3-Isopropyl-4-nitro-1H-pyrazole-5-carboxylic Acid (6)
Carboxylic acid 5 (14.2 g, 92.0 mmol) was added portion-wise to concentrated H2SO4 (74 mL, 1.36 mol) at RT with stirring. The reaction mixture was heated to 60 °C, and HNO3 (65%, 17.8 mL, 279 mmol) was added dropwise. The reaction was then stirred at 60 °C for 3 h, cooled to RT, and poured onto 200 g of ice with stirring. After 15 min, the white precipitate was isolated by filtration, washed with water (300 mL), and dried under reduced pressure to give the title product 6 as a white solid (11.7 g, 64%). 1H NMR (600 MHz, DMSO-d6): δ 3.49 (hept, J = 7.1, Hz, 1H), 1.29 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 162.8, 149.8, 129.4, 25.5, 21.2. LC–MS: tR = 2.70 min, purity: >99%, m/z [M – H]−: 198. Spectral data agree with a previous report.29
3-Isopropyl-4-nitro-1H-pyrazole-5-carboxamide (7)
Oxalyl chloride (16.6 mL, 189 mmol) was added dropwise to a suspension of carboxylic acid 6 (12.6 g, 63.1 mmol) in DCM (240 mL) containing DMF (300 μL, 3.87 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h, allowed to warm to RT, and stirred for another 2 h. The reaction mixture was added dropwise to a solution of 7 M NH3 in MeOH (54.1 mL, 379 mmol) at 0 °C, stirred for 0.5 h. The reaction mixture was concentrated in vacuo and purified by flash column chromatography on silica gel with a gradient elution of MeOH in DCM (0–10%) to yield the title compound 7 as a white solid (8.65 g, 69%). 1H NMR (300 MHz, DMSO-d6): δ 13.84 (br s, 1H), 7.99 (s, 1H), 7.71 (s, 1H), 3.53 (hept, J = 7.2 Hz, 1H), 1.28 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 162.2, 149.4, 143.5, 128.2, 25.2, 20.8. LC–MS: tR = 2.61 min, purity: >99%, m/z [M + H]+: 199. Spectral data agree with a previous report.29
4-Amino-3-isopropyl-1H-pyrazole-5-carboxamide (8)
Carboxamide 7 (5.70 g, 28.8 mmol) and 10% palladium on carbon (1.00 g, 0.940 mmol) in EtOH (90 mL) were stirred under H2 atmosphere at 60 °C for 18 h. The reaction mixture was filtered, and the solid was washed with MeOH (50 mL). The filtrate was concentrated in vacuo and purified by flash column chromatography on silica gel eluting with DCM/MeOH 9:1 to give the title product 8 as an off-white solid (4.50 g, 93%). 1H NMR (400 MHz, DMSO-d6, 373 K): δ 12.08 (br s, 1H), 6.81 (br s, 2H), 3.05–2.89 (m, 3H), 1.22 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 166.4, 133.4, 132.4, 128.0, 23.4, 21.4. LC–MS: tR = 2.11 min, purity: 99%, m/z [M + H]+: 169. Spectral data agree with a previous report.29
General Procedure for the Synthesis of Analogs 1, 9–46
Method A: 4-Amino-3-isopropyl-1H-pyrazole-5-carboxamide 8 (1.0 equiv) and the corresponding acid (1.0 equiv), PyBrop (1.1 equiv), and TEA (2.0 equiv) were combined in DCE and heated using microwave irradiation at 120 °C for 20 min. The reaction mixture was purified by column chromatography with an eluent of DCM and MeOH to get amide intermediates as off-white solids. Then, the amide intermediate was combined with KOtBu (2.0 equiv) in iPrOH (10 mL) and heated using microwave irradiation at 130 °C for 30 min. The reaction mixture was concentrated in vacuo and purified by column chromatography to get the final products.
Method B: 4-Amino-3-isopropyl-1H-pyrazole-5-carboxamide 8 (1.0 equiv) and the corresponding aldehyde (1.0 equiv), I2 (2.0 equiv) were combined in DMF and heated at 80 °C 16 h. The reaction was quenched with a saturated Na2S2O3 aqueous solution and extracted with EtOAc. The combined organic layers were washed with water, concentrated in vacuo, and purified by column chromatography to obtain the final products.
5-Benzyl-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 1 (NPD-0019)
Prepared from 8 (80 mg) via Method A as a white solid (87 mg, 68% for two steps). 1H NMR (500 MHz, DMSO-d6): δ 13.63 (br s, 1H), 12.18 (br s, 1H), 7.37–7.28 (m, 4H), 7.25–7.20 (m, 1H), 3.90 (s, 2H), 3.24 (hept, J = 6.8 Hz, 1H), 1.32 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 152.4 (HMBC), 150.3 (HMBC), 137.1, 128.7, 128.4, 126.6, 40.3, 25.8 (HSQC), 21.8. LC–MS: tR = 3.66 min, purity: >99%, m/z [M + H]+: 269; HR-MS: calcd for C15H16N4O [M + H]+, 269.1397; found, 269.1385. Spectral data agree with a previous report.27
3-Isopropyl-5-(pyridin-4-ylmethyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 9 (NPD-2960)
Prepared from 8 (80 mg) via Method A as a white solid (75 mg, 59% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 8.48 (d, J = 5.3 Hz, 2H), 7.32 (d, J = 5.6 Hz, 2H), 3.95 (s, 2H), 3.22 (hept, J = 6.9 Hz, 1H), 1.29 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6 + 1 drop of D2O): δ 150.6 (HMBC), 149.6, 145.9, 124.2, 39.5, 26.1 (HSQC), 21.8. LC–MS: tR = 2.26 min, purity: 98%, m/z [M + H]+: 270; HR-MS: calcd for C14H15N5O [M + H]+, 270.1349; found, 270.1341.
5-(Benzyloxy)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 10 (NPD-0434)
Prepared from 8 (197 mg) via Method A as a white solid (200 mg, 60% for two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.73 (br s, 1H), 12.40 (br s, 1H), 7.33–7.28 (m, 2H), 7.08–7.03 (m, 2H), 6.99–6.95 (m, 1H), 4.95 (s, 2H), 3.26 (app s, 1H), 1.33 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 157.9, 150.8 (HMBC), 148.9 (HMBC), 142.3 (HMBC), 129.5, 121.2, 114.8, 67.8, 26.2 (HSQC), 21.8. LC–MS: tR = 3.80 min, purity: >99%, m/z [M + H]+: 285; HR-MS: calcd for C15H16N4O2 [M + H]+, 285.1346; found, 285.1341.
3-Isopropyl-5-phenethyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 11 (NPD-3281)
Prepared from 8 (80 mg) via Method A as a white solid (96 mg, 71% for two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.52 (br s, 1H), 12.11 (br s, 1H), 7.29–7.25 (m, 4H), 7.20–7.16 (m, 1H), 3.26 (app s, 1H), 3.07–2.97 (m, 2H), 2.88 (app s, 2H), 1.34 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 154.3 (HMBC), 153.5 (HMBC), 150.8 (HMBC), 141.3, 137.5 (HMBC), 128.9, 128.7, 126.5, 36.2, 33.2, 26.6 (HSQC), 22.3. LC–MS: tR = 3.87 min, purity: >99%, m/z [M + H]+: 283; HR-MS: calcd for C16H18N4O [M + H]+, 283.1553; found, 283.1545. Spectral data agree with a previous report.27
3-Isopropyl-5-methyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 12 (NPD-3380)
Prepared from 8 (156 mg) via Method A as a white solid (0.16 g, 90% for two steps). 1H NMR (300 MHz, DMSO-d6): δ 13.52 (br s, 1H), 11.99 (br s, 1H), 3.23 (hept, J = 6.2 Hz, 1H), 2.31 (s, 3H), 1.32 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6): δ 151.1, 141.1 (HMBC), 26.4 (HSQC), 22.4, 21.5. LC–MS: tR = 2.46 min, purity: >99%, m/z [M + H]+: 193; HR-MS: calcd for C9H12N4O [M + H]+, 193.1084; found, 193.1090. Spectral data agree with a previous report.30
5-Butyl-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 13 (NPD-3645)
Prepared from 8 (72 mg) via Method A as a white solid (88 mg, 42% for two steps). 1H NMR (500 MHz, DMSO-d6): δ 13.52 (br s, 1H), 12.02 (br s, 1H), 3.29–3.17 (m, 1H), 2.61–2.53 (m, 2H), 1.65 (app p, J = 7.6 Hz, 2H), 1.37–1.28 (m, 8H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6): δ 154.5 (HMBC), 150.6 (HMBC), 141.4 (HMBC), 34.2, 29.7, 26.5, 22.3, 22.1, 14.2. LC–MS: tR = 3.58 min, purity: >99%, m/z [M + H]+: 235; HR-MS: calcd for C12H18N4O [M + H]+, 235.1553; found, 235.1562.
3,5-Diisopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 14 (NPD-3379)
Prepared from 8 (0.15 g) via Method A as a white solid (0.17 g, 87% for two steps). 1H NMR (300 MHz, DMSO-d6): δ 13.54 (br s, 1H), 11.88 (br s, 1H), 3.23 (hept, J = 6.8 Hz, 1H), 2.87 (hept, J = 7.8 Hz, 1H), 1.34 (d, J = 6.9 Hz, 6H), 1.22 (d, J = 6.8 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 158.1 (HMBC), 150.7 (HMBC), 32.8, 26.3 (HSQC), 21.8, 20.7. LC–MS: tR = 3.42 min, purity: >99%, m/z [M + H]+: 221; HR-MS: calcd for C11H16N4O [M + H]+, 221.1397; found, 221.1405.
3-Isopropyl-5-phenyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 15 (NPD-3200)
Prepared from 8 (0.15 g) via Method A as a white solid (73 mg, 32% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 8.04–7.98 (m, 2H), 7.55–7.48 (m, 3H), 3.33 (hept, J = 7.0 Hz, 1H), 1.37 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6 + 1 drop of D2O): δ 151.8 (HMBC), 150.4 (HMBC), 143.3 (HMBC), 133.6, 131.3, 129.3, 128.0, 26.6 (HSQC), 22.4. LC–MS: tR = 3.78 min, purity: >99%, m/z [M + H]+: 255; HR-MS: calcd for C14H14N4O [M + Na]+, 277.1060; found, 277.1070. Spectral data agree with a previous report.27
3-Isopropyl-5-(pyridin-4-yl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 16 (NPD-3488)
Prepared from 8 (86 mg) via Method A as a white solid (43 mg, 33% for two steps). 1H NMR (300 MHz, DMSO-d6 + 1 drop of D2O): δ 8.69 (d, J = 6.0 Hz, 2H), 7.96 (d, J = 6.0 Hz, 2H), 3.33 (app s, 1H), 1.36 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6 + 1 drop of D2O): δ 150.6, 148.1 (HMBC), 143.8 (HMBC), 140.7, 121.8, 26.8 (HSQC), 22.3. LC–MS: tR = 2.63 min, purity: >99%, m/z [M + H]+: 256; HR-MS: calcd for C13H13N5O [M + H]+, 256.1193; found, 256.1186.
3-Isopropyl-5-(thiazol-4-yl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 17 (NPD-2973)
Prepared from 8 (80 mg) via Method A as a white solid (67 mg, 54% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 9.26 (s, 1H), 8.50 (s, 1H), 3.33 (app s, 1H), 1.38 (d, J = 6.8 Hz, 6H). 13C NMR (126 MHz, DMSO-d6 + 1 drop of D2O): δ 155.4 (HSQC), 149.1, 149.0 (HMBC), 144.5, 135.8 (HMBC), 121.9, 25.8, 22.0. LC–MS: tR = 3.40 min, purity: >99%, m/z [M + H]+: 262; HR-MS: calcd for C11H11N5OS [M + H]+, 262.0757; found, 262.0756.
5-(2-Fluorophenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 18 (NPD-3199)
Prepared from 8 (80 mg) via Method A as a white solid (43 mg, 33% for two steps). 1H NMR (500 MHz, DMSO-d6): δ 13.82 (br s, 1H), 12.38 (br s, 1H), 7.71 (app t, J = 7.2 Hz, 1H), 7.62–7.54 (m, 1H), 7.40–7.31 (m, 2H), 3.32–3.26 (m, 1H), 1.35 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6): δ 159.5 (d, J = 249.6 Hz), 146.6, 132.3 (d, J = 8.3 Hz), 131.1 (d, J = 1.6 Hz), 124.6 (d, J = 3.3 Hz), 122.7 (d, J = 13.1 Hz), 116.1 (d, J = 21.4 Hz), 25.5, 21.9. LC–MS: tR = 3.66 min, purity: >99%, m/z [M + H]+: 273; HR-MS: calcd for C14H13FN4O [M + H]+, 273.1146; found, 273.1144.
5-(2-Chlorophenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 19 (NPD-3538)
Prepared from 8 (156 mg) via Method A as a white solid (0.15 g, 56% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 7.57–7.48 (m, 3H), 7.47–7.42 (m, 1H), 3.33–3.21 (m, 1H), 1.30 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6 + 1 drop of D2O): δ 152.5 (HMBC), 149.9 (HMBC), 144.3 (HMBC), 134.2, 132.5, 132.5, 131.8, 130.6, 128.2, 26.6 (HSQC), 22.6. LC–MS: tR = 3.66 min, purity: >99%, m/z [M + H]+: 289; HR-MS: calcd for C14H13ClN4O [M + H]+, 289.0851; found, 289.0850.
5-(2-Bromophenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 20 (NPD-3539)
Prepared from 8 (0.16 g) via Method A as a white solid (0.28 mg, 88% for two steps). 1H NMR (600 MHz, DMSO-d6 + 1 drop of D2O): δ 7.76 (dd, J = 8.0, 1.0 Hz, 1H), 7.59 (dd, J = 7.6, 1.7 Hz, 1H), 7.52 (app td, J = 7.5, 1.2 Hz, 1H), 7.46 (app td, J = 7.7, 1.8 Hz, 1H), 3.27 (hept, J = 6.9 Hz, 1H), 1.35 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6 + 1 drop of D2O): δ 150.4 (HMBC), 136.6, 133.1, 131.8, 131.6, 128.1, 122.0, 26.3, 22.3. LC–MS: tR = 3.69 min, purity: >99%, m/z [M + H]+: 333; HR-MS: calcd for C14H13BrN4O [M + H]+, 333.0346; found, 333.0333.
3-Isopropyl-5-(o-tolyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 21 (NPD-3589)
Prepared from 8 (0.16 g) via Method A as a white solid (0.11 g, 43% for two steps). 1H NMR (300 MHz, DMSO-d6): δ 13.76 (br s, 1H), 12.25 (br s, 1H), 7.49–7.43 (m, 1H), 7.42–7.37 (m, 1H), 7.36–7.27 (m, 2H), 3.30–3.23 (m, 1H), 2.36 (s, 3H), 1.36 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 151.0 (HMBC), 142.4 (HMBC), 136.4, 134.4, 130.6, 129.7, 129.4, 125.8, 26.3 (HSQC), 21.9, 19.7. LC–MS: tR = 3.79 min, purity: >99%, m/z [M + H]+: 269; HR-MS: calcd for C15H16N4O [M + H]+, 269.1397; found, 269.1405.
3-Isopropyl-5-(2-methoxyphenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 22 (NPD-3590)
Prepared from 8 (0.20 g) via Method A as a white solid (17 mg, 5% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 7.63 (d, J = 7.5 Hz, 1H), 7.50 (app t, J = 7.8 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 7.07 (app t, J = 7.4 Hz, 1H), 3.83 (s, 3H), 3.28 (app s, 1H), 1.34 (d, J = 6.8 Hz, 6H). 13C NMR (126 MHz, DMSO-d6 + 1 drop of D2O): δ 157.0, 150.9 (HMBC), 149.1 (HMBC), 142.2 (HMBC), 131.7, 130.5, 123.1, 120.5, 111.8, 55.8, 26.1 (HSQC), 21.9. LC–MS: tR = 3.96 min, purity: >99%, m/z [M + H]+: 285; HR-MS: calcd for C15H16N4O2 [M + H]+, 285.1346; found, 285.1333.
5-(3-Fluorophenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 23 (NPD-3202)
Prepared from 8 (0.16 g) via Method A as a white solid (95 mg, 37% for two steps). 1H NMR (300 MHz, CD3OD): δ 7.87 (d, J = 7.8 Hz, 1H), 7.84–7.78 (m, 1H), 7.63–7.52 (m, 1H), 7.32 (app td, J = 8.5, 2.5 Hz, 1H), 3.49 (app s, 1H), 1.50 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, CD3OD): δ 164.3 (d, J = 244.9 Hz), 153.7 (HMBC), 150.7 (HMBC), 145.1 (HMBC), 135.7 (d, J = 7.2 Hz), 131.8 (d, J = 8.3 Hz), 123.0 (app s), 118.6 (d, J = 21.6 Hz), 115.5 (d, J = 24.3 Hz), 28.1, 22.3. LC–MS: tR = 3.98 min, purity: 98%, m/z [M + H]+: 273; HR-MS: calcd for C14H13FN4O [M + Na]+, 295.0966; found, 295.0954.
5-(3-Chlorophenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 24 (NPD-3591)
Prepared from 8 (0.17 g) via Method A as a white solid (12 mg, 4% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 8.06 (s, 1H), 7.98 (d, J = 7.2 Hz, 1H), 7.62–7.52 (m, 2H), 3.34 (app s, 1H), 1.38 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, DMSO-d6 + 1 drop of D2O): δ 151.9 (HMBC), 149.1 (HMBC), 143.5 (HMBC), 135.3, 133.5, 130.6, 130.4, 127.3, 126.3, 26.4 (HSQC), 22.0. LC–MS: tR = 4.25 min, purity: 99%, m/z [M + H]+: 289; HR-MS: calcd for C14H13ClN4O [M + Na]+, 311.0670; found, 311.0676.
3-Isopropyl-5-(m-tolyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 25 (NPD-3382)
Prepared from 8 (0.15 g) via Method A as a white solid (0.15 g, 63% for two steps). 1H NMR (300 MHz, DMSO-d6 + 1 drop of D2O): δ 7.83–7.72 (m, 2H), 7.44–7.27 (m, 2H), 3.32 (app s, 1H), 2.36 (s, 3H), 1.35 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6 + 1 drop of D2O): δ 151.5 (HMBC), 149.7 (HMBC), 137.7, 133.1, 131.1, 128.4, 128.0, 124.7, 26.6, 21.9, 21.0. LC–MS: tR = 4.10 min, purity: >99%, m/z [M + H]+: 269; HR-MS: calcd for C15H16N4O [M + H]+, 269.1397; found, 269.1386.
3-Isopropyl-5-(3-methoxyphenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 26 (NPD-3375)
Prepared from 8 (0.18 g) via Method A as a white solid (0.10 g, 33% for two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.73 (br s, 1H), 12.40 (br s, 1H), 7.69 (s, 1H), 7.68–7.63 (m, 1H), 7.43 (app t, J = 8.0 Hz, 1H), 7.10 (dd, J = 8.1, 2.0 Hz, 1H), 3.86 (s, 3H), 3.33 (1H, confirmed with HSQC), 1.41 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 159.3, 149.3 (HMBC), 143.0 (HMBC), 134.4, 129.7, 119.8, 116.5, 112.5, 55.3, 26.2 (HSQC), 21.9. LC–MS: tR = 3.87 min, purity: >99%, m/z [M + H]+: 285; HR-MS: calcd for C15H16N4O2 [M + H]+, 285.1346; found, 285.1339.
5-(3-Hydroxyphenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 27 (NPD-2974)
Prepared from 8 (80 mg) via Method A as a white solid (53 mg, 41% for two steps). 1H NMR (500 MHz, DMSO-d6): δ 13.72 (br s, 1H), 12.31 (br s, 1H), 9.73 (s, 1H), 7.51–7.46 (m, 2H), 7.30 (app t, J = 8.0 Hz, 1H), 6.94 – 6.89 (m, 1H), 3.30 (app s, 1H), 1.39 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, DMSO-d6): δ 157.6, 154.4, 151.5, 150.1, 137.4, 134.7, 130.1, 126.1, 118.6, 118.0, 114.5, 26.6, 22.2. LC–MS: tR = 3.23 min, purity: >99%, m/z [M + H]+: 271; HR-MS: calcd for C14H14N4O2 [M + H]+, 271.1190; found, 271.1185.
5-(3-(Dimethylamino)phenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 28 (NPD-3381)
Prepared from 8 (0.15 g) via Method A as a white solid (0.12 g, 45% for two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.67 (br s, 1H), 12.33 (br s, 1H), 7.38 (d, J = 7.3 Hz, 2H), 7.30 (app t, J = 7.9 Hz, 1H), 6.86 (d, J = 8.7 Hz, 1H), 3.33 (1H, confirmed with D2O), 2.98 (s, 6H), 1.40 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 154.2, 151.0, 150.4, 142.3 (HMBC), 137.1, 129.1, 125.9, 115.4, 114.3, 111.1, 40.2, 26.3, 21.9. LC–MS: tR = 3.71 min, purity: >99%, m/z [M + H]+: 298; HR-MS: calcd for C16H19N5O [M + H]+, 298.1662; found, 298.1662.
3-Isopropyl-5-(3-(methylsulfonyl)phenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 29 (NPD-3598)
Prepared from 8 (0.13 g) via Method A as a white solid (61 mg, 24% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 8.54 (s, 1H), 8.37 (d, J = 7.8 Hz, 1H), 8.05 (d, J = 7.8 Hz, 1H), 7.80 (app t, J = 7.8 Hz, 1H), 3.35 (hept, J = 6.1 Hz, 1H), 3.27 (s, 3H), 1.38 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6 + 1 drop of D2O): δ 151.6 (HMBC), 148.8 (HMBC), 143.2 (HMBC), 141.4, 134.7, 132.9, 130.3, 129.1, 126.4, 43.8, 26.3 (HSQC), 22.2. LC–MS: tR = 3.29 min, purity: 95%, m/z [M + H]+: 333; HR-MS: calcd for C15H16N4O3S [M + H]+, 333.1016; found, 333.1012.
5-(4-Fluorophenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 30 (NPD-2975)
Prepared from 8 (80 mg) via Method A as a white solid (80 mg, 62% for two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.73 (br s, 1H), 12.38 (br s, 1H), 8.18–8.12 (m, 2H), 7.39–7.33 (m, 2H), 3.36–3.32 (m, 1H), 1.39 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 163.5 (d, J = 248.2 Hz), 151.4 (HMBC), 148.8 (HMBC), 130.0 (d, J = 8.8 Hz), 129.8 (d, J = 2.7 Hz), 115.5 (d, J = 22.1 Hz), 26.2 (HSQC), 21.9. LC–MS: tR = 3.89 min, purity: >99%, m/z [M + H]+: 273; HR-MS: calcd for C14H13FN4O [M + H]+, 273.1146; found, 273.1144; Anal. calcd for C14H13FN4O: C, 61.76; H, 4.81; N, 20.58; O, 5.88. Found: C, 61.92; H, 4.89; N, 20.5; O 6.52. More detailed 2D NMR analysis is attached in the Supporting Information.
5-(4-Chlorophenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 31 (NPD-3204)
Prepared from 8 (0.16 g) via Method A as a white solid (0.10 g, 37% for two steps). 1H NMR (300 MHz, CD3OD): δ 8.01 (d, J = 8.1 Hz, 2H), 7.55 (d, J = 8.2 Hz, 2H), 3.56–3.37 (m, 1H), 1.47 (d, J = 6.6 Hz, 6H). 13C NMR (126 MHz, CD3OD): δ 138.0, 133.5, 130.2, 130.0, 28.0, 22.3. LC–MS: tR = 4.32 min, purity: 98%, m/z [M + H]+: 289; HR-MS: calcd for C14H13ClN4O [M + H]+, 289.0851; found, 289.0839.
5-(4-Bromophenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 32 (NPD-2971)
Prepared from 8 (80 mg) via Method A as a white solid (86 mg, 54% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 8.00 (d, J = 8.7 Hz, 2H), 7.72 (d, J = 8.7 Hz, 2H), 3.32 (hept, J = 7.0 Hz, 1H), 1.38 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, DMSO-d6): δ 148.8, 132.5, 131.6, 129.6, 124.3, 29.1, 21.9. LC–MS: tR = 4.38 min, purity: >99%, m/z [M + H]+: 333; HR-MS: calcd for C14H13BrN4O [M + H]+, 333.0346; found, 333.0347.
3-Isopropyl-5-(4-methoxyphenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 33 (NPD-2972)
Prepared from 8 (80 mg) via Method A as a white solid (79 mg, 58% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 8.08 – 8.02 (m, 2H), 7.05 (d, J = 8.8 Hz, 2H), 3.82 (s, 3H), 3.31 (app s, 1H), 1.38 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, DMSO-d6): δ 161.2, 129.1, 125.4, 114.0, 55.4, 26.3, 21.9. LC–MS: tR = 3.86 min, purity: >99%, m/z [M + H]+: 285; HR-MS: calcd for C15H16N4O2 [M + H]+, 285.1346; found, 285.1343.
5-(4-Isopropoxyphenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 34 (NPD-3377)
Prepared from 8 (0.15 g) via Method A as a white solid (0.10 g, 36% for two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.68 (br s, 1H), 12.21 (br s, 1H), 8.05 (d, J = 8.8 Hz, 2H), 7.03 (d, J = 8.8 Hz, 2H), 4.73 (hept, J = 5.9 Hz, 1H), 3.31 (app s, 1H), 1.40 (d, J = 6.9 Hz, 6H), 1.30 (d, J = 6.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 159.4, 151.1 (HMBC), 149.3 (HMBC), 129.1, 125.0, 115.2, 69.4, 26.3 (HSQC), 21.9, 21.8. LC–MS: tR = 4.40 min, purity: >99%, m/z [M + H]+: 313; HR-MS: calcd for C17H20N4O2 [M + H]+, 313.1659; found, 313.1651.
3-Isopropyl-5-(4-(trifluoromethyl)phenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 35 (NPD-3201)
Prepared from 8 (0.16 g) via Method A as a white solid (0.13 g, 42% for two steps). 1H NMR (300 MHz, CD3OD): δ 8.16 (d, J = 6.9 Hz, 2H), 7.80 (d, J = 7.4 Hz, 2H), 3.52–3.36 (m, 1H), 1.43 (d, J = 5.7 Hz, 6H). 13C NMR (126 MHz, CD3OD): δ 154.1 (HMBC), 150.7 (HMBC), 145.5 (HMBC), 138.5, 133.3 (q, J = 32.6 Hz), 129.4, 126.7 (q, J = 3.6 Hz), 125.4 7 (q, J = 271.8 Hz), 28.1 (HSQC), 22.3. LC–MS: tR = 4.43 min, purity: >99%, m/z [M + H]+: 323; HR-MS: calcd for C15H13F3N4O [M + Na]+, 345.0934; found, 345.0920.
3-Isopropyl-5-(4-(trifluoromethoxy)phenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 36 (NPD-3597)
Prepared from 8 (0.20 g) via Method A as a white solid (60 mg, 15% for two steps). 1H NMR (500 MHz, DMSO-d6 + 1 drop of D2O): δ 8.16–8.09 (m, 2H), 7.49 (d, J = 8.1 Hz, 2H), 3.32 (app s, 1H), 1.36 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, DMSO-d6 + 1 drop of D2O): δ 151.3 (HMBC), 149.8, 132.6, 129.8, 123.1 (HMBC), 120.4 (q, J = 257.1 Hz), 26.4 (HSQC), 21.9. LC–MS: tR = 4.54 min, purity: >99%, m/z [M + H]+: 339; HR-MS: calcd for C15H13F3N4O2 [M + H]+, 339.1063; found, 339.1069.
4-(3-Isopropyl-7-oxo-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)benzonitrile 37 (NPD-3203)
Prepared from 8 (0.16 g) via Method A as a white solid (98 mg, 37% for two steps). 1H NMR (300 MHz, CD3OD): δ 8.20 (d, J = 8.4 Hz, 2H), 7.90 (d, J = 8.5 Hz, 2H), 3.46 (hept, J = 7.1 Hz, 1H), 1.48 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, CD3OD): δ 137.6, 132.2, 128.0, 117.8, 113.8, 20.9. LC–MS: tR = 3.69 min, purity: >99%, m/z [M + H]+: 280; HR-MS: calcd for C15H13N5O [M + H]+, 280.1193; found, 280.1182.
Methyl 4-(3-isopropyl-7-oxo-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)benzoate 38 (NPD-3305)
Prepared from 8 (0.15 g) via Method B as a white solid (95 mg, 34%). 1H NMR (600 MHz, DMSO-d6): δ 13.79 (s, 1H), 12.57 (s, 1H), 8.21 (d, J = 8.3 Hz, 2H), 8.08 (d, J = 8.3 Hz, 2H), 3.89 (s, 3H), 3.33 (1H, confirmed with HSQC), 1.39 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 165.7, 154.0, 151.3, 148.7, 137.2, 136.9, 131.1, 129.3, 127.9, 126.1, 52.4, 26.3, 21.9. LC–MS: tR = 3.83 min, purity: 97%, m/z [M + H]+: 313; HR-MS: calcd for C16H16N4O3 [M + H]+, 313.1295; found, 313.1284.
4-(3-Isopropyl-7-oxo-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)benzoic Acid 39 (NPD-3489)
Ester 38 (0.28 g, 0.89 mmol) and LiOH (37 mg, 0.89 mmol) were added to a mixture of 1,4-dioxane (10 mL) and water (10 mL) and refluxed for 2 h. The reaction mixture was concentrated under reduced pressure and washed with EtOAc (3 × 20 mL); the pH was adjusted to 1 with 1 M HCl aqueous solution, and the product was filtered as an off-white solid (0.20 g, 75%). 1H NMR (600 MHz, DMSO-d6): δ 13.80 (br s, 1H), 13.20 (br s, 1H), 12.47 (br s, 1H), 8.18 (d, J = 8.5 Hz, 2H), 8.05 (d, J = 8.5 Hz, 2H), 3.33 (1H, confirmed with HSQC), 1.40 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 166.8, 151.2 (HMBC), 148.7 (HMBC), 136.9, 132.3, 129.4, 127.7, 26.0 (HSQC), 21.9. LC–MS: tR = 3.16 min, purity: 99%, m/z [M + H]+: 299; HR-MS: calcd for C15H14N4O3 [M + H]+, 299.1139; found, 299.1101.
4-(3-Isopropyl-7-oxo-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)benzamide 40 (NPD-3371)
Prepared from 8 (0.15 g) via Method A as a white solid (49 mg, 18% for two steps). 1H NMR (300 MHz, DMSO-d6): δ 13.77 (br s, 1H), 12.45 (br s, 1H), 8.19–8.06 (m, 3H), 7.99 (d, J = 8.1 Hz, 2H), 7.51 (s, 1H), 3.33 (1H, confirmed with HSQC), 1.40 (d, J = 6.8 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 167.2, 151.3 (HMBC), 149.0 (HMBC), 142.9 (HMBC), 135.8, 135.5, 127.6, 127.3, 26.3 (HMBC), 21.9. LC–MS: tR = 2.76 min, purity: 97%, m/z [M + H]+: 298; HR-MS: calcd for C15H15N5O2 [M + Na]+, 320.1118; found, 320.1105.
3-Isopropyl-5-(4-(methylsulfonyl)phenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 41 (NPD-3376)
Prepared from 8 (0.15 g) via Method A as a white solid (90 mg, 30% for two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.82 (br s, 1H), 12.64 (br s, 1H), 8.30 (d, J = 8.5 Hz, 2H), 8.06 (d, J = 8.3 Hz, 2H), 3.33 (1H, confirmed with HSQC), 3.29 (s, 3H), 1.40 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 154.0 (HMBC), 151.4 (HMBC), 148.3 (HMBC), 143.6, 142.2, 137.7, 128.5, 127.1, 43.3, 26.3, 21.9. LC–MS: tR = 3.27 min, purity: 97%, m/z [M + H]+: 333; HR-MS: calcd for C15H16N4O3S [M + H]+, 333.1016; found, 333.1011.
4-(3-Isopropyl-7-oxo-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)benzenesulfonamide 42 (NPD-3372)
Prepared from 8 (0.15 g) via Method A as a white solid (56 mg, 19% for two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.82 (br s, 1H), 12.56 (br s, 1H), 8.23 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.3 Hz, 2H), 7.51 (s, 2H), 3.33 (1H, confirmed with HSQC), 1.41 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 151.3 (HMBC), 145.6, 136.1, 128.2, 125.8, 26.3 (HSQC), 21.9. LC–MS: tR = 2.96 min, purity: >99%, m/z [M + H]+: 334; HR-MS: calcd for C14H15N5O3S [M + H]+, 334.0969; found, 334.0953.
N-(4-(3-Isopropyl-7-oxo-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)phenyl)acetamide 43 (NPD-3280)
Prepared from 8 (0.15 g) via Method B as a white solid (0.13 g, 47%). 1H NMR (600 MHz, DMSO-d6): δ 13.66 (br s, 1H), 12.26 (br s, 1H), 10.18 (s, 1H), 8.03 (d, J = 8.7 Hz, 2H), 7.70 (d, J = 8.5 Hz, 2H), 3.37–3.34 (m, 1H), 2.08 (s, 3H), 1.39 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 168.7, 150.9 (HMBC), 149.4 (HMBC), 142.4 (HMBC), 141.4, 128.1, 127.4, 118.4, 27.3 (HSQC), 24.1, 21.9. LC–MS: tR = 3.06 min, purity: >99%, m/z [M + H]+: 312; HR-MS: calcd for C16H17N5O2 [M + H]+, 312.1455; found, 312.1443.
3-Isopropyl-5-(4-(piperidin-1-yl)phenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 44 (NPD-3283)
Prepared from 8 (0.15 g) via Method B as a white solid (70 mg, 23%). 1H NMR (300 MHz, DMSO-d6): δ 13.62 (br s, 1H), 12.03 (br s, 1H), 7.97 (d, J = 8.9 Hz, 2H), 7.00 (d, J = 9.0 Hz, 2H), 3.32–3.26 (m, 5H), 1.60 (app s, 6H), 1.39 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 152.5, 150.6 (HMBC), 141.8 (HMBC), 128.4, 121.5, 114.1, 48.3, 26.3 (HMBC), 24.9, 24.0, 21.9. LC–MS: tR = 4.34 min, purity: >99%, m/z [M + H]+: 338; HR-MS: calcd for C19H23N5O [M + H]+, 338.1975; found, 338.1964.
3-Isopropyl-5-(4-(4-methylpiperazin-1-yl)phenyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 45 (NPD-3282)
Prepared from 8 (0.15 g) via Method B as a white solid (0.11 g, 35%). 1H NMR (600 MHz, CDCl3): δ 10.78 (br s, 1H), 7.89 (d, J = 7.9 Hz, 2H), 6.87 (app s, 2H), 3.48 (hept, J = 6.6 Hz, 1H), 3.22 (app s, 4H), 2.53 (app s, 4H), 2.33 (s, 3H), 1.51 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, CDCl3): δ 155.8, 152.9, 152.2 (HMBC), 149.5, 139.0, 128.3, 122.7, 114.8, 54.8, 47.7, 46.2, 26.9, 22.0. LC–MS: tR = 2.53 min, purity: 98%, m/z [M + H]+: 353; HR-MS: calcd for C19H24N6O [M + H]+, 353.2084; found, 353.2078.
5-(4-(1H-Tetrazol-5-yl)phenyl)-3-isopropyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 46 (NPD-3490)
A mixture of 37 (0.15 g, 0.54 mmol), NaN3 (0.56 g, 8.6 mmol), and NH4Cl (0.46 g, 8.6 mmol) in DMF (5 mL) was heated using microwave irradiation at 160 °C for 2 h, after which the reaction mixture was dissolved in water (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine, concentrated in vacuo, and purified by flash column chromatography on silica gel with a gradient elution of EtOAc in cyclohexane (20%–50%) to give the title compound as a white solid (0.12 g, 69%). 1H NMR (600 MHz, DMSO-d6): δ 13.80 (br s, 1H), 12.57 (br s, 1H), 8.31 (d, J = 8.2 Hz, 2H), 8.19 (d, J = 8.0 Hz, 2H), 3.33 (1H, confirmed with HSQC), 1.42 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, DMSO-d6): δ 151.2 (HMBC), 148.7 (HMBC), 142.6 (HMBC), 135.4, 128.5, 127.1, 125.8 (HMBC), 26.3 (HMBC), 21.9. LC–MS: tR = 3.10 min, purity: >99%, m/z [M + H]+: 323; HR-MS: calcd for C15H14N8O [M + H]+, 323.1363; found, 323.1357.
Acknowledgments
Lars Binkhorst, Odessa Visser, and Shresta Giasi are thanked for their synthetic assistance. Hans Custers, Andrea van de Stolpe, Pim-Bart Feijens, and Mathias Sempels are thanked for their technical assistance. Elwin Janssen is thanked for his NMR support. This work was supported by the European Commission 7th Framework Program FP7-HEALTH-2013-INNOVATION-1 under project reference 602666 “Parasite-specific cyclic nucleotide phosphodiesterase inhibitors to target Neglected Parasitic Diseases” (PDE4NPD). YZ acknowledges the China Scholarship Council (CSC) for funding (Grant no. 201506220185). LMPH is a partner of the Excellence Centre ‘Infla-Med’ (www.uantwerpen.be/infla-med) and participates in COST Action CA21111.
Glossary
Abbreviations
- ADQUATE
adequate sensitivity double-quantum spectroscopy
- CDD
collaborative drug discovery
- dpi
days post infection
- HAT
human African Trypanosomiasis
- MRC-5
medical research council cell strain 5
- NPD
neglected parasitic disease
- PDE
phosphodiesterase
- PMM
peritoneal macrophage
- RT
room temperature
- UGT
uridine 5′-diphospho-glucuronosyltransferase
- WHO
World Health Organization.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00161.
Structures of approved HAT treatments; physicochemical properties of 1 (BIPPO); phenotypic activity of close BIPPO analogues against T. b. brucei, T. cruzi, and L. infantum; antitrypanosomal potency of 23, 30, and 31; anti-TbrPDEB1 activity of 30; safety profile of analogue 30 (NPD-2975) from Eurofins; in vitro metabolic stability of 30 using mouse, rat, and human S9 microsomal fractions; chemical characterization of final compounds; antitrypanosomal potency of final compounds; and LCMS, 1H NMR, 13C NMR, and 2D NMR spectroscopy data. (PDF)
Author Contributions
⊥ Y.Z., M.v.d.K., G.C. and R.L. contributed equally to this manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Kennedy P. G. E. Clinical Features, Diagnosis, and Treatment of Human African Trypanosomiasis (Sleeping Sickness). Lancet Neurol. 2013, 12, 186–194. 10.1016/s1474-4422(12)70296-x. [DOI] [PubMed] [Google Scholar]
- World Health Organization . Trypanosomiasis. https://www.who.int/health-topics/human-african-trypanosomiasis (accessed December 15, 2022).
- Büscher P.; Cecchi G.; Jamonneau V.; Priotto G. Human African Trypanosomiasis. Lancet 2017, 390, 2397–2409. 10.1016/s0140-6736(17)31510-6. [DOI] [PubMed] [Google Scholar]
- WHO . WHO HAT 2020. https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed 04 18, 2022).
- Lindner A. K.; Lejon V.; Chappuis F.; Seixas J.; Kazumba L.; Barrett M. P.; Mwamba E.; Erphas O.; Akl E. A.; Villanueva G.; Bergman H.; Simarro P.; Kadima Ebeja A.; Priotto G.; Franco J. R. New WHO Guidelines for Treatment of Gambiense Human African Trypanosomiasis Including Fexinidazole: Substantial Changes for Clinical Practice. Lancet Infect. Dis. 2020, 20, e38–e46. 10.1016/s1473-3099(19)30612-7. [DOI] [PubMed] [Google Scholar]
- De Koning H. P. The Drugs of Sleeping Sickness: their Mechanisms of Action and Resistance, and a Brief History. Trop. Med. Infect. Dis. 2020, 5, 14. 10.3390/tropicalmed5010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco J.; Scarone L.; Comini M. A. Drugs and Drug Resistance in African and American Trypanosomiasis. Annu. Rep. Med. Chem. 2018, 51, 97–133. 10.1016/BS.ARMC.2018.08.003. [DOI] [Google Scholar]
- Benaim G.; Lopez-Estrano C.; Docampo R.; Moreno S. N. J. A Calmodulin-Stimulated Ca2+ Pump in Plasma-Membrane Vesicles from Trypanosoma Brucei; Selective Inhibition by Pentamidine. Biochem. J. 1993, 296, 759–763. 10.1042/bj2960759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairlamb A. H.; Henderson G. B.; Cerami A. Trypanothione Is the Primary Target for Arsenical Drugs against African Trypanosomes. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 2607–2611. 10.1073/pnas.86.8.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann H.; Strassmann G. Suramin Inhibits the Stimulation of Acute Phase Plasma Protein Genes by IL-6-Type Cytokines in Rat Hepatoma Cells. J. Immunol. 1993, 151, 1456–1462. 10.4049/jimmunol.151.3.1456. [DOI] [PubMed] [Google Scholar]
- Vincent I. M.; Creek D.; Watson D. G.; Kamleh M. A.; Woods D. J.; Wong P. E.; Burchmore R. J. S.; Barrett M. P. A Molecular Mechanism for Eflornithine Resistance in African Trypanosomes. PLoS Pathog. 2010, 6, e1001204 10.1371/journal.ppat.1001204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S. R.; Taylor M. C.; Horn D.; Kelly J. M.; Cheeseman I. A Mechanism for Cross-Resistance to Nifurtimox and Benznidazole in Trypanosomes. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5022–5027. 10.1073/pnas.0711014105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Foth B. J.; Kelner A.; Sokolova A. Y.; Berriman M.; Fairlamb A. H. Nitroheterocyclic Drug Resistance Mechanisms in Trypanosoma Brucei. J. Antimicrob. Chemother. 2016, 71, 625–634. 10.1093/jac/dkv376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casulli A. New Global Targets for NTDs in the WHO Roadmap 2021–2030. PLoS Neglected Trop. Dis. 2021, 15, e0009373 10.1371/journal.pntd.0009373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rycker M.; Wyllie S.; Horn D.; Read K. D.; Gilbert I. H. Anti-Trypanosomatid Drug Discovery: Progress and Challenges. Nat. Rev. Microbiol. 2022, 21, 35–50. 10.1038/s41579-022-00777-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle A. S.; Khare S.; Kumar A. B.; Supek F.; Buchynskyy A.; Mathison C. J. N.; Chennamaneni N. K.; Pendem N.; Buckner F. S.; Gelb M. H.; Molteni V. Recent Developments in Drug Discovery for Leishmaniasis and Human African Trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamura F.; Rajesh R.; Catta-Preta C. M. C.; Moretti N. S.; Cestari I. The Current Drug Discovery Landscape for Trypanosomiasis and Leishmaniasis: Challenges and Strategies to Identify Drug Targets. Drug Dev. Res. 2022, 83, 225–252. 10.1002/ddr.21664. [DOI] [PubMed] [Google Scholar]
- Field M. C.; Horn D.; Fairlamb A. H.; Ferguson M. A. J.; Gray D. W.; Read K. D.; de Rycker M.; Torrie L. S.; Wyatt P. G.; Wyllie S.; Gilbert I. H. Anti-Trypanosomatid Drug Discovery: An Ongoing Challenge and a Continuing Need. Nat. Rev. Microbiol. 2017, 15, 217–231. 10.1038/nrmicro.2016.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer M.; Marti G.; Baresic M.; Kunz S.; Hemphill A.; Seebeck T. The Trypanosoma Brucei CAMP Phosphodiesterases TbrPDEBl and TbrPDEB2: Flagellar Enzymes That Are Essential for Parasite Virulence. FASEB J. 2007, 21, 720–731. 10.1096/fj.06-6818com. [DOI] [PubMed] [Google Scholar]
- Kunz S.; Balmer V.; Sterk G. J.; Pollastri M. P.; Leurs R.; Müller N.; Hemphill A.; Spycher C. The Single Cyclic Nucleotide-Specific Phosphodiesterase of the Intestinal Parasite Giardia Lamblia Represents a Potential Drug Target. PLoS Neglected Trop. Dis. 2017, 11, e0005891 10.1371/journal.pntd.0005891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Yan Z.; Geng J.; Kunz S.; Seebeck T.; Ke H. Crystal Structure of the Leishmania Major Phosphodiesterase LmjPDEB1 and Insight into the Design of the Parasite-Selective Inhibitors. Mol. Microbiol. 2007, 66, 1029–1038. 10.1111/j.1365-2958.2007.05976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long T.; Rojo-Arreola L.; Shi D.; El-Sakkary N.; Jarnagin K.; Rock F.; Meewan M.; Rascón A. A.; Lin L.; Cunningham K. A.; Lemieux G. A.; Podust L.; Abagyan R.; Ashrafi K.; McKerrow J. H.; Caffrey C. R. Phenotypic, Chemical and Functional Characterization of Cyclic Nucleotide Phosphodiesterase 4 (PDE4) as a Potential Anthelmintic Drug Target. PLoS Neglected Trop. Dis. 2017, 11, e0005680 10.1371/journal.pntd.0005680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boolell M.; Allen M. J.; Ballard S. A.; Gepi-Attee S.; Muirhead G. J.; Naylor A. M.; Osterloh I. H.; Gingell C. S. An Orally Active Type 5 Cyclic GMP-Specific Phosphodiesterase Inhibitor for the Treatment of Penile Erectile Dysfunction. Int. J. Impot. Res. 1996, 8, 47–52. [PubMed] [Google Scholar]
- Eruopean Commision . PDE4NPD, 2018https://cordis.europa.eu/project/id/602666. [Google Scholar]
- Blaazer A. R.; Singh A. K.; De Heuvel E.; Edink E.; Orrling K. M.; Veerman J. J. N.; Van Den Bergh T.; Jansen C.; Balasubramaniam E.; Mooij W. J.; Custers H.; Sijm M.; Tagoe D. N. A.; Kalejaiye T. D.; Munday J. C.; Tenor H.; Matheeussen A.; Wijtmans M.; Siderius M.; De Graaf C.; Maes L.; De Koning H. P.; Bailey D. S.; Sterk G. J.; De Esch I. J. P.; Brown D. G.; Leurs R. Targeting a Subpocket in Trypanosoma Brucei Phosphodiesterase B1 (TbrPDEB1) Enables the Structure-Based Discovery of Selective Inhibitors with Trypanocidal Activity. J. Med. Chem. 2018, 61, 3870–3888. 10.1021/acs.jmedchem.7b01670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen C.; Wang H.; Kooistra A. J.; De Graaf C.; Orrling K. M.; Tenor H.; Seebeck T.; Bailey D.; De Esch I. J. P.; Ke H.; Leurs R. Discovery of Novel Trypanosoma Brucei Phosphodiesterase B1 Inhibitors by Virtual Screening against the Unliganded TbrPDEB1 Crystal Structure. J. Med. Chem. 2013, 56, 2087–2096. 10.1021/jm3017877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard B. L.; Harvey K. L.; Stewart R. J.; Azevedo M. F.; Crabb B. S.; Jennings I. G.; Sanders P. R.; Manallack D. T.; Thompson P. E.; Tonkin C. J.; Gilson P. R. Identification of Potent Phosphodiesterase Inhibitors That Demonstrate Cyclic Nucleotide-Dependent Functions in Apicomplexan Parasites. ACS Chem. Biol. 2015, 10, 1145–1154. 10.1021/cb501004q. [DOI] [PubMed] [Google Scholar]
- Newkirk R.; Maenz D.; Classen H.. Oilseed processing. U.S. patent, 0,124,222A1, 2003, 1 (19), 1–4.
- DeNinno M. P.; Andrews M.; Bell A. S.; Chen Y.; Eller-Zarbo C.; Eshelby N.; Etienne J. B.; Moore D. E.; Palmer M. J.; Visser M. S.; Yu L. J.; Zavadoski W. J.; Michael Gibbs E. The Discovery of Potent, Selective, and Orally Bioavailable PDE9 Inhibitors as Potential Hypoglycemic Agents. Bioorg. Med. Chem. Lett. 2009, 19, 2537–2541. 10.1016/j.bmcl.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Moravcová D.; Havlicek L.; Krystof V.; Lenobel R.; Strnad M.. Pyrazolo[4,3-d]Pyrimidines, Processes for Their Preparation and Methods for Therapy. EP1348707A1, 2003.
- Dale D. J.; Dunn P. J.; Golightly C.; Hughes M. L.; Levett P. C.; Pearce A. K.; Searle P. M.; Ward G.; Wood A. S. The Chemical Development of the Commercial Route to Sildenafil: A Case History. Org. Process Res. Dev. 2000, 4, 17–22. 10.1021/op9900683. [DOI] [Google Scholar]
- de Koning H. P.; Gould M. K.; Sterk G. J.; Tenor H.; Kunz S.; Luginbuehl E.; Seebeck T. Pharmacological Validation of Trypanosoma Brucei Phosphodiesterases as Novel Drug Targets. J. Infect. Dis. 2012, 206, 229–237. 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulpia F.; Mabille D.; Campagnaro G. D.; Schumann G.; Maes L.; Roditi I.; Hofer A.; de Koning H. P.; Caljon G.; Van Calenbergh S. Combining Tubercidin and Cordycepin Scaffolds Results in Highly Active Candidates to Treat Late-Stage Sleeping Sickness. Nat. Commun. 2019, 10, 5564–5611. 10.1038/s41467-019-13522-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creek D. J.; Mazet M.; Achcar F.; Anderson J.; Kim D.-H.; Kamour R.; Morand P.; Millerioux Y.; Biran M.; Kerkhoven E. J.; Chokkathukalam A.; Weidt S. K.; Burgess K. E. V.; Breitling R.; Watson D. G.; Bringaud F.; Barrett M. P. Probing the Metabolic Network in Bloodstream-Form Trypanosoma Brucei Using Untargeted Metabolomics with Stable Isotope Labelled Glucose. PLoS Pathog. 2015, 11, e1004689 10.1371/journal.ppat.1004689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I. M.; Creek D. J.; Burgess K.; Woods D. J.; Burchmore R. J. S.; Barrett M. P. Untargeted Metabolomics Reveals a Lack Of Synergy between Nifurtimox and Eflornithine against Trypanosoma Brucei. PLoS Neglected Trop. Dis. 2012, 6, e1618 10.1371/journal.pntd.0001618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann Burkard G.; Jutzi P.; Roditi I. Genome-Wide RNAi Screens in Bloodstream Form Trypanosomes Identify Drug Transporters. Mol. Biochem. Parasitol. 2011, 175, 91–94. 10.1016/j.molbiopara.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Klug D. M.; Tschiegg L.; Diaz R.; Rojas-Barros D.; Perez-Moreno G.; Ceballos G.; García-Hernández R.; Martinez-Martinez M. S.; Manzano P.; Ruiz L. M.; Caffrey C. R.; Gamarro F.; Pacanowska D. G.; Ferrins L.; Navarro M.; Pollastri M. P. Hit-to-Lead Optimization of Benzoxazepinoindazoles As Human African Trypanosomiasis Therapeutics. J. Med. Chem. 2020, 63, 2527–2546. 10.1021/acs.jmedchem.9b01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulpia F.; Bouton J.; Campagnaro G. D.; Alfayez I. A.; Mabille D.; Maes L.; de Koning H. P.; Caljon G.; Van Calenbergh S.; Van Calenbergh S. C6–O-Alkylated 7-Deazainosine Nucleoside Analogues: Discovery of Potent and Selective Anti-Sleeping Sickness Agents. Eur. J. Med. Chem. 2020, 188, 112018. 10.1016/j.ejmech.2019.112018. [DOI] [PubMed] [Google Scholar]
- Pedron J.; Boudot C.; Brossas J.-Y.; Pinault E.; Bourgeade-Delmas S.; Sournia-Saquet A.; Boutet-Robinet E.; Destere A.; Tronnet A.; Bergé J.; Bonduelle C.; Deraeve C.; Pratviel G.; Stigliani J.-L.; Paris L.; Mazier D.; Corvaisier S.; Since M.; Malzert-Fréon A.; Wyllie S.; Milne R.; Fairlamb A. H.; Valentin A.; Courtioux B.; Verhaeghe P. New 8-Nitroquinolinone Derivative Displaying Submicromolar in Vitro Activities against Both Trypanosoma Brucei and Cruzi. ACS Med. Chem. Lett. 2020, 11, 464–472. 10.1021/acsmedchemlett.9b00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B.; Bernatchez J. A.; McCall L. I.; Calvet C. M.; Ackermann J.; Souza J. M.; Thomas D.; Silva E. M.; Bachovchin K. A.; Klug D. M.; Jalani H. B.; Bag S.; Buskes M. J.; Leed S. E.; Roncal N. E.; Penn E. C.; Erath J.; Rodriguez A.; Sciotti R. J.; Campbell R. F.; McKerrow J.; Siqueira-Neto J. L.; Ferrins L.; Pollastri M. P. Scaffold and Parasite Hopping: Discovery of New Protozoal Proliferation Inhibitors. ACS Med. Chem. Lett. 2020, 11, 249–257. 10.1021/acsmedchemlett.9b00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccoliti F.; Madia V. N.; Tudino V.; De Leo A.; Pescatori L.; Messore A.; De Vita D.; Scipione L.; Brun R.; Kaiser M.; Mäser P.; Calvet C. M.; Jennings G. K.; Podust L. M.; Pepe G.; Cirilli R.; Faggi C.; Di Marco A.; Battista M. R.; Summa V.; Costi R.; Di Santo R. Design, Synthesis, and Biological Evaluation of New 1-(Aryl-1 H-Pyrrolyl)(Phenyl)Methyl-1 H-Imidazole Derivatives as Antiprotozoal Agents. J. Med. Chem. 2019, 62, 1330–1347. 10.1021/acs.jmedchem.8b01464. [DOI] [PubMed] [Google Scholar]
- Bachovchin K. A.; Sharma A.; Bag S.; Klug D. M.; Schneider K. M.; Singh B.; Jalani H. B.; Buskes M. J.; Mehta N.; Tanghe S.; Momper J. D.; Sciotti R. J.; Rodriguez A.; Mensa-Wilmot K.; Pollastri M. P.; Ferrins L. Improvement of Aqueous Solubility of Lapatinib-Derived Analogues: Identification of a Quinolinimine Lead for Human African Trypanosomiasis Drug Development. J. Med. Chem. 2019, 62, 665–687. 10.1021/acs.jmedchem.8b01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodring J. L.; Bachovchin K. A.; Brady K. G.; Gallerstein M. F.; Erath J.; Tanghe S.; Leed S. E.; Rodriguez A.; Mensa-Wilmot K.; Sciotti R. J.; Pollastri M. P. Optimization of Physicochemical Properties for 4-Anilinoquinazoline Inhibitors of Trypanosome Proliferation. Eur. J. Med. Chem. 2017, 141, 446–459. 10.1016/j.ejmech.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodland A.; Thompson S.; Cleghorn L. A. T.; Norcross N.; De Rycker M.; Grimaldi R.; Hallyburton I.; Rao B.; Norval S.; Stojanovski L.; Brun R.; Kaiser M.; Frearson J. A.; Gray D. W.; Wyatt P. G.; Read K. D.; Gilbert I. H. Discovery of Inhibitors of Trypanosoma Brucei by Phenotypic Screening of a Focused Protein Kinase Library. ChemMedChem 2015, 10, 1809–1820. 10.1002/cmdc.201500300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. 10.1016/s0169-409x(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Xie X.-Q.PAINS Remover. https://www.cbligand.org/PAINS/ (accessed 01 24, 2023).
- Moraes C. B.; Witt G.; Kuzikov M.; Ellinger B.; Calogeropoulou T.; Prousis K. C.; Mangani S.; Di Pisa F.; Landi G.; Iacono L. D.; Pozzi C.; Freitas-Junior L. H.; dos Santos Pascoalino B.; Bertolacini C. P.; Behrens B.; Keminer O.; Leu J.; Wolf M.; Reinshagen J.; Cordeiro-da-Silva A.; Santarem N.; Venturelli A.; Wrigley S.; Karunakaran D.; Kebede B.; Pöhner I.; Müller W.; Panecka-Hofman J.; Wade R. C.; Fenske M.; Clos J.; Alunda J. M.; Corral M. J.; Uliassi E.; Bolognesi M. L.; Linciano P.; Quotadamo A.; Ferrari S.; Santucci M.; Borsari C.; Costi M. P.; Gul S. Accelerating Drug Discovery Efforts for Trypanosomatidic Infections Using an Integrated Transnational Academic Drug Discovery Platform. SLAS Discovery 2019, 24, 346–361. 10.1177/2472555218823171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetic Toxicology Screening Assays at Wuxi. https://labtesting.wuxiapptec.com/safety-assessment-services/genetic-in-vitro-toxicology/ (accessed 12 06, 2022).
- Safety47 Panel Dose Response . SAFETYscan Eurofins. https://www.eurofinsdiscoveryservices.com/catalogmanagement/viewItem/Safety47-Panel-Dose-Response-SAFETYscan-DiscoverX/87-1003DR (accessed 12 06, 2022).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.