

Abstract
Mitochondria are central regulators of healthspan and lifespan, yet the intricate choreography of multiple, tightly controlled steps regulating mitochondrial biogenesis remains poorly understood. Here, we uncover a pivotal role for specific elements of the 5′‐3′ mRNA degradation pathway in the regulation of mitochondrial abundance and function. We find that the mRNA degradation and the poly‐A tail deadenylase CCR4‐NOT complexes form distinct foci in somatic Caenorhabditis elegans cells that physically and functionally associate with mitochondria. Components of these two multi‐subunit complexes bind transcripts of nuclear‐encoded mitochondria‐targeted proteins to regulate mitochondrial biogenesis during ageing in an opposite manner. In addition, we show that balanced degradation and storage of mitochondria‐targeted protein mRNAs are critical for mitochondrial homeostasis, stress resistance and longevity. Our findings reveal a multifaceted role of mRNA metabolism in mitochondrial biogenesis and show that fine‐tuning of mRNA turnover and local translation control mitochondrial abundance and promote longevity in response to stress and during ageing.
Keywords: ageing, mitochondria, mRNA metabolism, protein synthesis, stress
Subject Categories: Metabolism, Organelles, Translation & Protein Quality
mRNA degradation pathway factors form local foci to regulate mitochondria‐targeted transcript content and mitochondrial function during systemic stress response and ageing in worms.
Introduction
Aberrant mitochondrial number and function contribute to premature ageing and increased susceptibility to disease. The rate at which a mitochondrial population is refreshed is determined by turnover, the balance between mitochondrial biogenesis and clearance (Palikaras et al, 2015a). Mitochondrial biogenesis spawns healthy mitochondria both under normal conditions and in response to stress. It is well established that mitochondrial biogenesis necessitates the selective translation of nuclear‐encoded, mitochondria‐targeted protein transcripts (MTPTs). Two regulators of MTPT transcription in the nematode Caenorhabditis elegans are the SKN‐1 (SKiNhead‐1) transcription factor, the nematode homologue of the mammalian NRF2/NFE2L2 (nuclear factor‐erythroid 2‐related factor 2; Palikaras et al, 2015b) and the AMP‐activated protein kinase, AAK‐2. The latter promotes MTPT transcription and activation of downstream pathways, especially under conditions of energy shortage (Hardie, 2011). Following their transcription, MTPTs are exported from the nucleus to the cytoplasm and then transferred to mitochondria, where they are anchored on the outer mitochondrial membrane (OMM) by the OMM proteins MDI (AKAP‐1, the C. elegans MDI homologue) and TOM20 (the nematode TOMM‐20) and are locally translated and imported into the organelles (Eliyahu et al, 2010; Gehrke et al, 2015; Zhang et al, 2016). These factors promote local translation of MTPTs, facilitated by OMM‐bound ribosomes or by free cytoplasmic ribosomes found in the mitochondrial vicinity as revealed by proximity‐specific ribosome profiling in yeast (Williams et al, 2014; Gold et al, 2017). However, the impact of their genetic inhibition on the organismal level has not been studied yet in multicellular eukaryotes. In addition, local translation of MTPTs is considered as a tightly regulated process (Das et al, 2021a, 2021b). Despite this, the mechanism by which mRNAs are selectively processed and locally translated remains poorly understood.
Processing bodies (P‐bodies) are highly conserved, non‐membranous cytoplasmic granules that consist of untranslated mRNAs and RNA‐binding proteins. Their formation and composition are dynamic exhibiting prominent changes during ageing and upon stress (Rieckher et al, 2018). P‐bodies control mRNA storage and 5′‐3′ exonucleolytic decay, while a subset of their bound mRNAs returns to translation when conditions become permissive (Aizer et al, 2014; Temme et al, 2014; Luo et al, 2018). Like yeast, flies and mammals, 5′‐3′ exonucleolytic decay in C. elegans also requires shortening of the poly‐A tail by the CCR4‐NOT deadenylase complex (Nousch et al, 2013). Notably, mRNA poly‐A tail lengths positively correlate with mRNA translation efficiency. Previous work indicated that P‐bodies enclose select transcripts that encode mostly regulatory proteins than housekeeping ones (Hubstenberger et al, 2017). Due to their ability to regulate the fate of these mRNAs, P‐bodies might act as translation regulatory hubs for select target transcripts. Intriguingly, the P‐body component Rck/p54/Dhh1, which is involved in translational repression, was found to localize to the vicinity of mitochondria in HUVEC and HeLa cells (Huang et al, 2011). Further studies are needed to provide mechanistic insight into this association. Moreover, the molecular basis of MTPT regulation and its physiological relevance is still elusive.
Here, we report that post‐transcriptional mechanisms in the cytoplasm, such as 5′‐3′ exonucleolytic decay, act in the vicinity of mitochondria to regulate MTPTs. Our findings reveal that components of the mRNA degradation and CCR4‐NOT complexes function as pivotal regulators of mitochondrial biogenesis to ultimately modulate stress resistance and longevity, by fine‐tuning the number and function of mitochondria, in response to environmental and genetic stimuli.
Results
The mRNA degradation and the CCR4‐NOT complexes physically and functionally associate with mitochondria in an age‐dependent manner
Mitochondrial biogenesis entails co‐translational import of locally expressed transcripts into mitochondria (Bykov et al, 2020). Such a mechanism requires targeting and processing of mRNAs prior to their translation. We hypothesized that post‐transcriptional regulation of MTPTs would also occur in the vicinity of the organelle, similar to their translation. To identify putative post‐transcriptional cytoplasmic regulators, we investigated the localization of P‐body components relative to mitochondria in vivo. We observed that the CCR4‐NOT complex component NTL‐2 and the decapping complex component EDC‐3 are localized in close proximity to mitochondria under physiological conditions (Fig 1A–D). In addition, we found that the vast majority of the NTL‐2 or EDC‐3 foci are located very close to mitochondria (0–1 μm distance), suggesting non‐random associations (Fig 1E). Moreover, we performed mitochondrial fractionation analysis and found that the CCR4‐NOT complex subunits NTL‐2 and CCF‐1 and the mRNA degradation complex subunits EDC‐3 and DCAP‐2 co‐precipitated with mitochondria, indicative of their physical association with the organelles (Fig 1F). In order to further verify that the complexes, rather than the single proteins, physically associate with mitochondria, we measured the distances that degradation and CCR4‐NOT complex components obtain from mitochondria. We found that DCAP‐1::DsRed foci were present in close proximity to mitochondria in 1‐day‐old animals (Appendix Fig S1). In addition, DCAP‐2::mCherry and CCF‐1::GFP foci were localized very close to mitochondria in vivo and also co‐fractionated with them in 1‐day‐old animals (Appendix Figs S2 and S3). Taken together, these results demonstrate that mRNA degradation and CCR4‐NOT complex components physically associate with mitochondria.
Figure 1. mRNA degradation and CCR4‐NOT complex components physically and functionally associate with mitochondria.
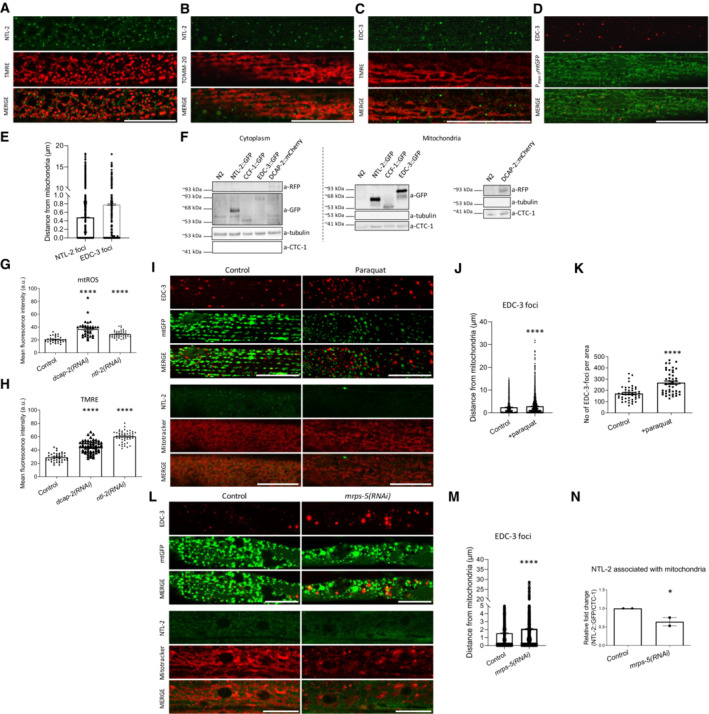
- NTL‐2 foci localize in close proximity to mitochondria in young adult animals; hypodermis is imaged (green: NTL‐2, red: TMRE (tetramethylrhodamine, ethyl ester, perchlorate), a mitochondrial membrane potential‐dependent dye) (n = 3 independent experiments).
- Localization of NTL‐2 foci relative to mitochondria in young adult animals; body wall muscle cells are imaged (green: NTL‐2, red: TOMM‐20, an outer mitochondrial membrane (OMM) protein marker of mitochondria) (n = 3 independent experiments).
- EDC‐3 foci localize in close proximity to mitochondria in young adult animals; body wall muscle cells are imaged (green: EDC‐3, red: TMRE) under control conditions (n = 3 independent experiments).
- Localization of EDC‐3 foci relative to mitochondria in young adult animals; body wall muscle cells are imaged (red: EDC‐3, green: mitochondrial matrix targeted by GFP) (n = 3 independent experiments).
- Quantification of NTL‐2 and EDC‐3 foci (shown in dots) with depicted distances from mitochondria is shown (n = 3 independent experiments) based on experiments presented in images (A–D).
- Immunoblot analysis of the cytoplasmic and mitochondria‐containing fractions obtained from whole animal extracts, showing that NTL‐2, CCF‐1, EDC‐3 and DCAP‐2 are localized in the cytoplasm and co‐precipitate with mitochondria (n = 3 independent experiments).
- Mitochondrial ROS production is elevated in animals subjected to either dcap‐2 or ntl‐2 RNAi, as evidenced by staining with Mitotracker Red CM‐H2X ROS (n = 3 independent experiments with at least 113 animals/experiment; ****P < 0.0001; one‐way analysis of variance (ANOVA)).
- Mitochondrial membrane potential (Δψ) is increased in animals subjected to either dcap‐2 or ntl‐2 RNAi as evidenced by TMRE staining (n = 3 independent experiments with at least 144 animals/experiment; ****P < 0.0001; one‐way analysis of variance (ANOVA)).
- EDC‐3 and NTL‐2 foci lose their specific localization close to mitochondria upon paraquat treatment of transgenic animals expressing mitochondria‐targeted GFP; top: EDC‐3 foci, bottom: NTL‐2 foci (n = 3 independent experiments).
- Measurement of the distances between EDC‐3 foci (shown in dots) and mitochondria under paraquat treatment as compared with their control counterparts (n = 3 independent experiments with at least 30 animals/experiment; ****P < 0.0001; two‐tailed unpaired t‐test).
- EDC‐3 foci are increased upon paraquat treatment (n = 3 Independent experiments with at least 30 animals/experiment; ****P < 0.0001; two‐tailed unpaired t‐test).
- EDC‐3 and NTL‐2 foci lose their specific localization in the vicinity of mitochondria upon genetic inhibition of mrps‐5; top: EDC‐3 foci, bottom: NTL‐2 foci (n = 3 independent experiments).
- Measurement of the distances between EDC‐3 foci (shown in dots) and mitochondria upon genetic inhibition of mrps‐5 as compared to control (n = 3 independent experiments with at least 30 animals/experiment; ****P < 0.0001; two‐tailed unpaired t‐test).
- Quantification of NTL‐2 protein bound on mitochondria under control conditions and upon genetic inhibition of mrps‐5 (western blot shown in Fig 5E, n = 2 independent experiments; *P < 0.05; one‐way analysis of variance (ANOVA) followed by Dunnett's test).
Data information: Images were acquired using an ×63 objective lens. Scale bars, 20 μm. Error bars denote SEM.
We then sought to investigate the potential functional significance of NTL‐2 or EDC‐3 associations with mitochondria. To this end, we tested whether these associations change during ageing, which is known to be accompanied by alterations in mitochondrial quality and function (Sun et al, 2016). Indeed, as expected, mitochondrial network integrity was highly perturbed in aged animals (Fig EV1A and E, compare left and right middle panels). We also found that the abundance of NTL‐2 foci was markedly reduced in unstressed, aged animals and inevitably their contacts with mitochondria are lost as animals age, as verified also by immunoblotting of NTL‐2 in isolated mitochondria (Fig EV1A–D). Despite the fact that EDC‐3 foci remained or even increased in aged worms, their association with mitochondria was significantly abolished as compared with their young counterparts (Fig EV1E–H). Similar changes during ageing were observed in a strain that co‐expresses NTL‐2 and EDC‐3 (Appendix Fig S4). To exclude the possibility that loss of contacts between the remaining EDC‐3 foci and mitochondria is simply a consequence of mitochondrial network deterioration that accompanies normal ageing, we mimicked these mitochondrial alterations in young adult animals by genetically inhibiting atp‐3, which encodes the nematode homologue of mammalian mitochondrial ATP5O (ATP synthase peripheral stalk subunit OSCP). Although atp‐3 genetic inhibition in young adult animals caused severe mitochondrial fragmentation and alterations in mitochondrial morphology, it did not markedly alter the associations between EDC‐3 foci and mitochondria, while it enhanced further the associations of NTL‐2 foci with mitochondria (Fig EV1I–M). Similarly, knockdown of cyc‐1, which encodes a putative subunit of mitochondrial respiratory complex III, also disrupted the mitochondrial network and enhanced the associations of EDC‐3 and NTL‐2 foci with mitochondria, as evidenced by measuring the distance of EDC‐3::GFP or NTL‐2::GFP foci from mitochondria in 1‐day‐old animals subjected to RNAi against cyc‐1 (Appendix Fig S5). Thus, the disruption of EDC‐3–mitochondria contacts appears to be directly linked to ageing. Together, these findings corroborate that the associations of mitochondria with the foci examined are not random but rather functional, and display severe age‐related changes. In line with this, the contacts of DCAP‐1, DCAP‐2 and CCF‐1 foci with mitochondria were significantly reduced in 7‐day‐old animals compared to 1‐day‐old worms, as shown by both in vivo and in vitro experiments (Appendix Figs S1–S3).
Figure EV1. The association of CCR4‐NOT and mRNA degradation complex components with mitochondria is age‐dependent.
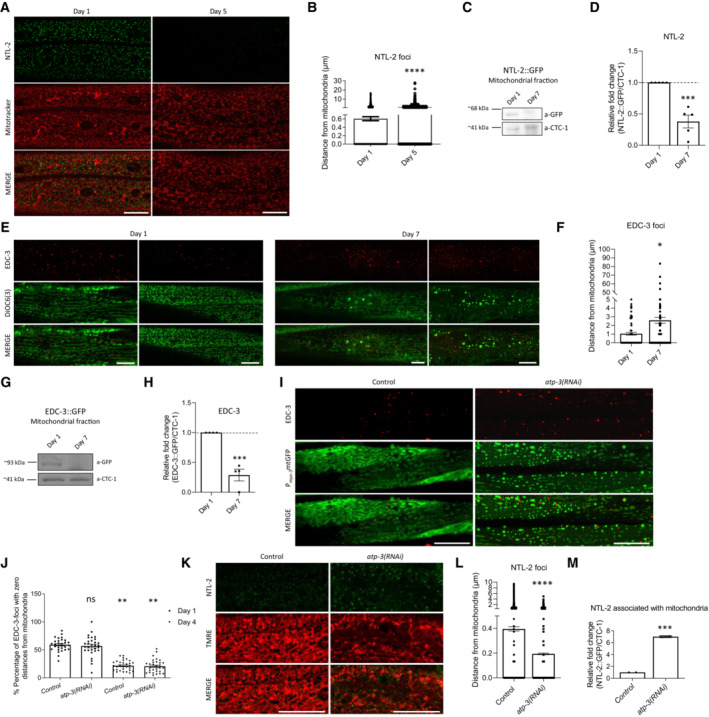
- Representative images showing the localization of NTL‐2 foci relative to mitochondria in young versus old animals (green: NTL‐2, red: Mitotracker Deep Red FM, a mitochondrial‐specific dye; n = 2 independent experiments).
- Quantification of the distances, the NTL‐2 foci obtain from mitochondria in young versus older animals (n = 3 independent experiments with at least 40 animals/experiment; ****P < 0.0001; two‐tailed unpaired t‐test).
- Representative image from immunoblot analysis showing that NTL‐2–mitochondria association decreases during ageing.
- Respective quantification (n = 5 independent experiments, ***P < 0.001; two‐tailed unpaired t‐test).
- Representative images showing the localization of EDC‐3 foci relative to mitochondria in young (left) versus old animals (right) (red: EDC‐3, green: DiOC6(3)) ((3,3′‐dihexyloxacarbocyanine Iodide), a mitochondrial‐specific dye; n = 2 independent experiments; image of day 1 adults in left is reused in Fig EV2K as conditions shown in (E) and Fig EV2K are part of the same experimental setup).
- Quantification of the distances EDC‐3 foci obtain from mitochondria in young versus older animals (n = 2 independent experiments with at least 40 animals/experiment; *P < 0.05; two‐tailed unpaired t‐test).
- Representative image from immunoblot analysis showing that EDC‐3 is less associated with mitochondria in older animals compared to younger ones.
- Respective quantification (n = 4 independent experiments; ***P < 0.001; two‐tailed unpaired t‐test).
- Representative images showing the localization of EDC‐3 foci relative to mitochondria in young adult animals upon genetic inhibition of atp‐3 (red: EDC‐3, green: mitochondrial matrix targeted by GFP; n = 2 independent experiments).
- Quantification of the percentage of EDC‐3 foci with zero distance from mitochondria under control conditions and following genetic inhibition of atp‐3 at days 1 and 4 of adulthood (n = 2 independent experiments with at least 40 animals/experiment; **P < 0.01; one‐way analysis of variance (ANOVA)).
- Representative images showing the localization of NTL‐2 foci relative to mitochondria in control conditions and upon genetic inhibition of atp‐3 (green: NTL‐2, red: TMRE, a mitochondrial membrane potential‐dependent dye; n = 2 independent experiments).
- Quantification of the distances of NTL‐2 foci from mitochondria in control conditions and upon genetic inhibition of atp‐3 (n = 2 independent experiments with at least 30 animals/experiment; **P < 0.01; two‐tailed unpaired t‐test).
- Quantification of the NTL‐2 protein bound to mitochondria under control conditions and upon genetic inhibition of atp‐3 (immunoblot analysis is shown in Fig 5E, n = 2 independent experiments; **P < 0.01; unpaired two‐tailed t‐test).
Data information: Images were acquired using an ×63 objective lens. Scale bars, 20 μm. Error bars denote SEM.
To further confirm that the association of mitochondria with the CCR4‐NOT and the mRNA degradation regulatory proteins is affected during ageing, we examined their contacts in long‐ and short‐lived animals. We found that the associations of NTL‐2 and CCF‐1 foci with mitochondria are increased in long‐lived daf‐2(RNAi) animals at day 1 of adulthood, whereas they are decreased in 1‐day‐old short‐lived nhr‐49(RNAi) worms compared to age‐matched controls (Fig EV2A–F). By contrast, the associations of DCAP‐1, DCAP‐2 and EDC‐3 with mitochondria are decreased in both daf‐2(RNAi) and nhr‐49(RNAi) animals (Fig EV2G–N). Similar results for NTL‐2 and EDC‐3 association with mitochondria in long‐ and short‐lived animals were obtained with a strain that co‐expresses both components (Appendix Fig S6). These findings further support the age‐dependent role of the CCR4‐NOT and the mRNA degradation components near mitochondria.
Figure EV2. Association of the CCR4‐NOT and the mRNA degradation complex components with mitochondria is tightly and differentially regulated in long‐ and short‐lived animals.
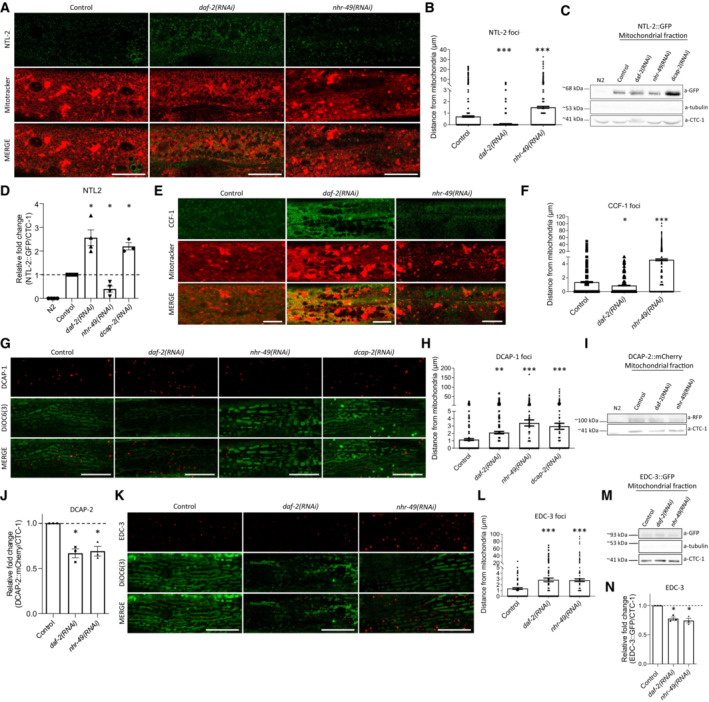
- Representative images showing the localization of NTL‐2 foci relative to mitochondria upon genetic inhibition of either daf‐2 known to extend lifespan or nhr‐49 known to shorten lifespan (green: NTL‐2, red: Mitotracker Deep Red FM, a mitochondria‐specific dye).
- Quantification of the distances NTL‐2 foci obtain from mitochondria upon genetic inhibition of daf‐2 or nhr‐49 (n = 3 independent experiments with at least 45 animals/experiment; ***P < 0.001; one‐way analysis of variance (ANOVA)).
- Immunoblot analysis in isolated mitochondria of 1‐day‐old animals showing the protein levels of NTL‐2 present in the mitochondrial isolate derived from animals under control conditions and upon the indicated genetic inhibitions.
- Quantification of western blot presented in B (n = at least 3 independent experiments; *P < 0.05; Welch's one‐way analysis of variance (ANOVA) followed by Dunnett's T3 multiple‐comparisons test).
- Representative images showing the localization of CCF‐1 foci relative to mitochondria upon genetic inhibition of daf‐2 or nhr‐49 in day 1 adults; image in control is reused in Appendix Fig S3A as conditions shown in Appendix Fig S3A and (E) are part of the same experimental setup (green: CCF‐1, red: Mitotracker Deep Red FM, a mitochondrial‐specific dye).
- Quantification of the distances CCF‐1 foci obtain from mitochondria upon genetic inhibition of daf‐2 or nhr‐49 (n = 3 independent experiments with at least 45 animals/experiment; *P < 0.05, ***P < 0.001; one‐way analysis of variance (ANOVA)).
- Representative images showing the localization of DCAP‐1 foci relative to mitochondria upon genetic inhibition of daf‐2, nhr‐49 or dcap‐2 (red: DCAP‐1, green: DiOC6(3), a mitochondria‐specific dye).
- Quantification of the distances DCAP‐1 foci obtain from mitochondria upon genetic inhibition of daf‐2, nhr‐49 or dcap‐2 (n = 3 independent experiments with at least 45 animals/experiment; **P < 0.01, ***P < 0.001; one‐way analysis of variance (ANOVA)).
- Immunoblot analysis in isolated mitochondria of 1‐day‐old animals showing the protein levels of DCAP‐2 present in the mitochondrial isolate in control conditions and upon the indicated genetic inhibitions.
- Respective quantifications (n = at least 3 independent experiments; *P < 0.05; Welch's one‐way analysis of variance (ANOVA) followed by Dunnett's T3 multiple‐comparisons test).
- Representative images showing the localization of EDC‐3 foci relative to mitochondria upon genetic inhibition of daf‐2 or nhr‐49 in day 1 adults (red: EDC‐3, green: DiOC6(3), a mitochondria‐specific dye; image in control is reused in Fig EV1E as conditions shown in Figs EV1E and (K) are part of the same experimental setup).
- Quantification of the distances EDC‐3 foci (shown in dots) obtain from mitochondria upon genetic inhibition of daf‐2 or nhr‐49 (n = 3 independent experiments with at least 45 animals/experiment; ***P < 0.001; one‐way analysis of variance (ANOVA)).
- Immunoblot analysis in isolated mitochondria of 1‐day‐old animals showing the protein levels of EDC‐3 present on mitochondria in control conditions and upon the indicated genetic inhibitions.
- Respective quantification (n = 3 independent experiments; *P < 0.05; Welch's one‐way analysis of variance (ANOVA) followed by Dunnett's T3 multiple‐comparisons test).
Data information: Images were acquired using a ×63 objective lens. Scale bars, 20 μm. Error bars denote SEM.
To verify the functional features of the associations between mRNA degradation/CCR4‐NOT components and mitochondria, we tested whether perturbation of either the degradation or the CCR4‐NOT complex influences mitochondrial function. Indeed, we observed that both dcap‐2 and ntl‐2 knockdown increased total mitochondrial ROS (mtROS) levels and mitochondrial membrane potential (Fig 1G and H). We also tested the effects of other genes involved in mRNA degradation and storage. Similar to dcap‐2, knockdown of either edc‐3 or xrn‐1 increased mtROS levels (Appendix Fig S7), mitochondrial membrane potential (Appendix Fig S8), ATP production (Appendix Fig S9) as well as basal and maximal oxygen consumption rates (Appendix Fig S10), indicative of a healthier mitochondrial bioenergetics status compared to control (Maglioni et al, 2019). By contrast, knockdown of genes encoding proteins belonging to the CCR4‐NOT complex (ccf‐1, let‐711) caused an increase in mtROS production (Appendix Fig S7) and mitochondrial membrane potential (Appendix Fig S8), but a decrease in mitochondrial ATP levels (Appendix Fig S9). To ensure that a direct functional association between mitochondria and the degradation/CCR4‐NOT complex components exists, we impaired mitochondrial function expecting that the localization pattern, the formation of the two types of foci or both phenotypes, would change. Consistently, perturbation of mitochondrial function either pharmacologically, by treatment with the oxidative stress inducer paraquat, or genetically, by knocking down mrps‐5 that encodes a mitochondrial ribosomal subunit, extensively and oppositely affected the abundance of the two types of foci (Fig 1I, K and L). Notably, EDC‐3 foci became more abundant, although their associations with mitochondria were weaker when mitochondrial function was impaired by paraquat treatment or mrps‐5 knockdown (Fig 1I top, J, L top and M). By contrast, formation of NTL‐2 foci was almost abolished following mitochondrial dysfunction, and thus their association with mitochondria was reduced compared to control (Fig 1I, L bottom and N), indicating that mitochondrial perturbations can affect the dynamic nature of both the EDC‐3 and NTL‐2 foci. It is important to note, however, that not all mitochondrial perturbations are expected to affect the abundance of mRNA degradation and CCR4‐NOT foci, as well as their associations with mitochondria in the same manner. For example, we observed that genetic inhibition of electron transport chain genes, such as atp‐3 and cyc‐1, did not alter or even strengthened the associations of EDC‐3 and NTL‐2 foci with mitochondria, as previously shown. Collectively, these findings support the notion that components of the mRNA degradation and the CCR4‐NOT complex form physical and functional contacts with mitochondria that change with age.
mRNA degradation and CCR4‐NOT complex components oppositely regulate mitochondrial biogenesis and abundance by functioning in discrete foci
To gain deeper insight into the functional association of mitochondria with the mRNA degradation and the CCR4‐NOT complex, we examined whether depletion of their components can affect mitochondrial abundance and network integrity. We observed that genetic inhibition of dcap‐2 caused mitochondrial network disruption and mitochondrial fragmentation (Fig 2A and B). Moreover, genetic inhibition of ntl‐2 affected mitochondrial network integrity and triggered mitochondrial globularization and swelling (Fig 2A).
Figure 2. Components of the CCR4‐NOT and the degradation complexes differentially control mitochondrial abundance.
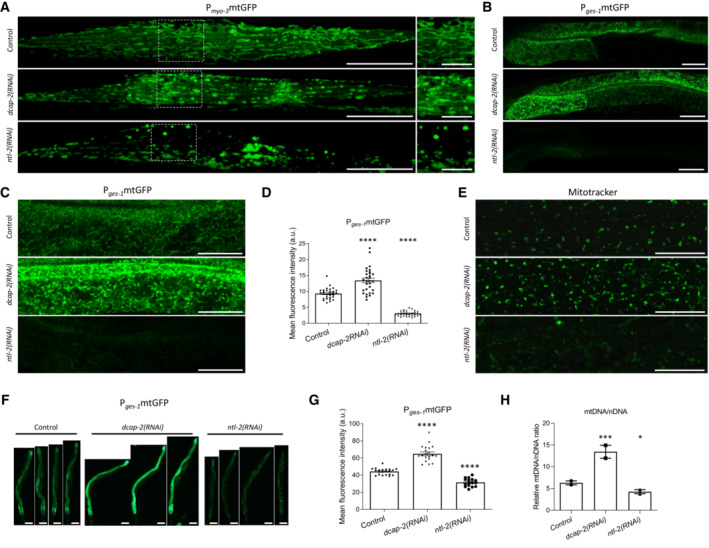
-
AConfocal images of young adult animals expressing mitochondria‐targeted GFP in body wall muscle cells upon dcap‐2 or ntl‐2 RNAi treatment (right panels indicate higher‐magnification images of inlays in left panels; right panel scale bar, 10 μm). Images were acquired using an ×40 objective lens (n = 3 independent experiments).
-
B, CConfocal images of young adult animals that express mitochondria‐targeted GFP in the intestine upon dcap‐2 or ntl‐2 knockdown. Images were acquired using an ×40 objective lens (n = 3 independent experiments).
-
DQuantification of intestinal mitochondrial mass upon knockdown of either dcap‐2 or ntl‐2, in animals shown in (B) and (C) (n = 3 independent experiments with at least 30 animals/experiment; ****P < 0.0001; one‐way analysis of variance (ANOVA)).
-
EConfocal images of the hypodermis in young adult animals stained with Mitotracker Green. Images were acquired using an ×40 objective lens (n = 3 independent experiments).
-
FRepresentative images showing the effect of dcap‐2 and ntl‐2 genetic inhibition on the intestinal cell mitochondrial content in 7‐day‐old nematodes (n = 3 independent experiments; images were acquired using an ×5 objective lens).
-
GQuantification of the mean GFP fluorescence intensity in the matrix of intestinal mitochondria as shown in (F) (n = 3 independent experiments with at least 57 animals/experiment; ****P < 0.0001; one‐way analysis of variance (ANOVA)).
-
HQuantification of the mtDNA/nDNA ratio under control conditions and upon genetic inhibition of either dcap‐2 or ntl‐2 (n = 2 independent experiments; ***P < 0.001, *P < 0.05; one‐way analysis of variance (ANOVA)).
Data information: Scale bars, 20 μm. Error bars denote SEM.
We next tested whether and how these interventions influence mitochondrial content. We found that genetic inhibition of dcap‐2 increases mitochondrial abundance in several tissues, in sharp contrast to ntl‐2 genetic inhibition that decreases it (Fig 2A–D). The effect of these treatments on mitochondrial abundance was sustained or even propagated when wild‐type animals were chronically subjected to RNAi against these genes (Fig 2E–G). More importantly, these features characterize the mRNA degradation and CCR4‐NOT complexes and not only the individual components DCAP‐2 and NTL‐2. Indeed, knockdown of other genes encoding mRNA degradation proteins (edc‐3, xrn‐1) also increased intestinal mitochondrial content in 1‐day old animals, similar to dcap‐2 knockdown (Appendix Fig S11). By contrast, knockdown of genes encoding components of the CCR4‐NOT complex (ccf‐1, let‐711) decreased mitochondrial abundance in the intestine of 1‐day‐old animals compared to controls, similar to ntl‐2 deficiency (Appendix Fig S11). Consistently, we found that dcap‐2 genetic inhibition significantly increased the mtDNA/nDNA ratio in contrast to ntl‐2 genetic inhibition, further corroborating our previous findings (Fig 2H). Considering the opposing effects of dcap‐2 and ntl‐2 genetic inhibition on mitochondrial mass and their impact on mitochondrial functionality, we conclude that perturbation of the mRNA degradation complex increases the functional mitochondrial population, whereas perturbation of the CCR4‐NOT complex leads to fewer and aberrantly functioning mitochondria compared to control conditions.
Mitochondrial biogenesis and mitophagy are the two opposing cellular processes that co‐ordinately regulate mitochondrial content in response to various intracellular or environmental stimuli (Palikaras et al, 2015b). To unravel the mechanism that governs the opposing effects of the degradation and CCR4‐NOT complexes on mitochondrial abundance, we first examined the involvement of mitophagy, using two different systems to monitor the process in vivo. We observed that genetic inhibition of either ntl‐2 or dcap‐2 does not affect mitophagy under normal conditions and instead blocks mitophagy upon paraquat exposure (Fig EV3A–C). Collectively, our findings exclude the possibility that mitophagy directly mediates the opposing effects of the degradation and CCR4‐NOT complexes on mitochondrial abundance.
Figure EV3. Perturbation of CCR4‐NOT and mRNA degradation complex components blocks mitophagy induction.
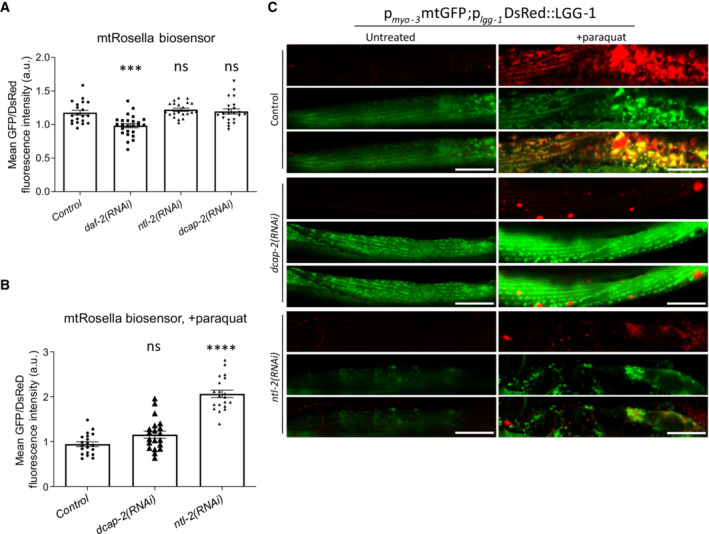
-
A, BTransgenic animals expressing the mitochondria‐targeted Rosella (mtRosella) biosensor in body wall muscles were used to assess mitophagy (A) upon genetic inhibition of either ntl‐2 or dcap‐2 under control conditions; daf‐2 RNAi is used as a positive control (n = 3 independent experiments with at least 88 animals/experiment; ***P < 0.001; one‐way analysis of variance (ANOVA)) and (B) after paraquat treatment (n = 2 independent experiments with at least 30 animals/experiment; ****P < 0.0001; one‐way analysis of variance (ANOVA)). Mitophagy induction is signified by the reduction in the ratio between pH‐sensitive GFP to pH‐insensitive DsRed.
-
CRepresentative images showing mitophagy events under control conditions and following paraquat treatment in animals expressing a mitochondria‐targeted GFP, together with the autophagosomal marker LGG‐1 fused with DsRed in body wall muscle cells and subjected to either dcap‐2 or ntl‐2 RNAi (green: mitochondria, red: autophagosomes; yellow: mitophagy events; n = 2 independent experiments).
Data information: Images were acquired using an ×40 objective lens. Scale bars, 20 μm. Error bars denote SEM.
We also considered whether the degradation and the CCR4‐NOT complex components modulate mitochondrial abundance by differentially influencing mitochondrial biogenesis. To test this hypothesis, we focused on SKN‐1 and AAK‐2, two key regulators of mitochondrial biogenesis (Reznick et al, 2007; Palikaras et al, 2015b). We found that genetic inhibition of dcap‐2 significantly increased the protein levels and the transcriptional activity of SKN‐1, as evidenced by the expression of its target gene gst‐4, while genetic inhibition of ntl‐2 had the opposite effects (Fig 3A–C, Appendix Fig S12A). This induction was dependent on SKN‐1 activity since it was abrogated by skn‐1 knockdown (Appendix Fig S12B and C). Similar effects were observed under genetic or pharmacological interventions known to activate SKN‐1 (Fig 3C and D, respectively, and Appendix Fig S12A). Notably, neither dcap‐2 nor ntl‐2 genetic inhibition affected skn‐1 transcription (Fig 3E). These results indicate that DCAP‐2 and NTL‐2 oppositely regulate SKN‐1 levels and activity at a post‐transcriptional level. To ensure that these effects reflect the impact of the mRNA degradation and the CCR4‐NOT complex, we tested additional genes encoding components of the two complexes. Similar to dcap‐2 genetic inhibition, knockdown of either edc‐3 or xrn‐1 also increased SKN‐1 levels and the transcriptional activity of gst‐4 gene promoter (Fig 3A and B, Appendix Fig S13). By contrast, knockdown of ntl‐2, ccf‐1 or let‐711 reduced SKN‐1 levels and gst‐4 transcriptional activity (Fig 3A and B, Appendix Fig S13).
Figure 3. DCAP‐2 and NTL‐2 modulate mitochondrial abundance by oppositely regulating SKN‐1 and AAK‐2 at the post‐transcriptional level.
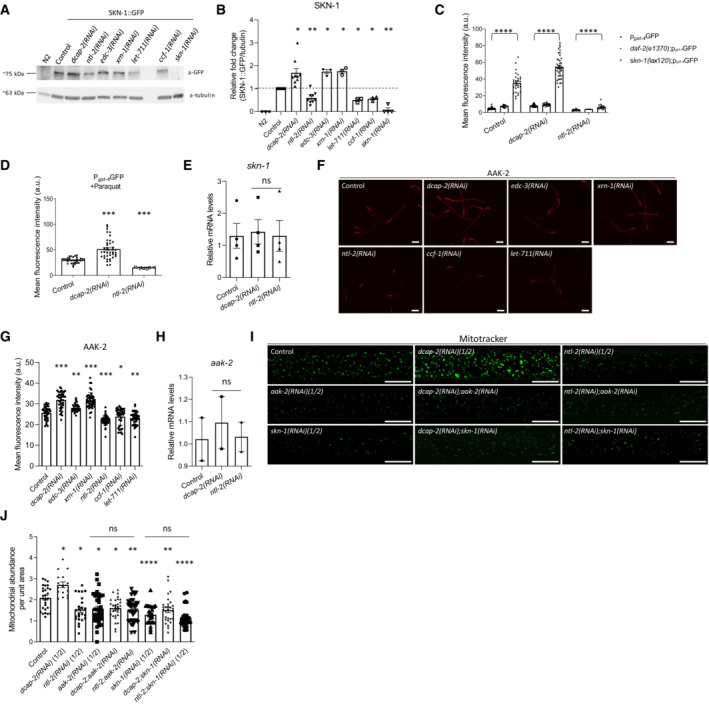
- Immunoblot analysis of SKN‐1 protein levels upon the indicated genetic inhibitions.
- Quantification showing the normalized SKN‐1 protein levels (n = at least 3 independent experiments; *P < 0.05, **P < 0.01; Welch's one‐way analysis of variance (ANOVA) followed by Dunnett's T3 multiple‐comparisons test).
- Measurement of the promoter activity of the SKN‐1 target gene gst‐4 upon genetic inhibition of either dcap‐2 or ntl‐2 in wild‐type background, daf‐2 mutant background and in a mutant background (skn‐1(lax120)) where SKN‐1 is constitutively active (n = 3 independent experiments with at least 256 animals/experiment; ****P < 0.0001; two‐way analysis of variance (ANOVA), followed by Turkey's multiple‐comparison test).
- Measurement of the promoter activity of the SKN‐1 target gene gst‐4 upon genetic inhibition of either dcap‐2 or ntl‐2 in wild‐type animals following paraquat administration (n = 3 independent experiments with at least 96 animals/experiment; ***P < 0.001; one‐way analysis of variance (ANOVA)).
- Relative expression of skn‐1 mRNA in wild‐type, DCAP‐2‐ and NTL‐2‐depleted animals (n = 4 independent experiments, P(dcap‐2(RNAi)) = 0.98, P(ntl‐2(RNAi)) > 0.99; one‐way analysis of variance (ANOVA)).
- Representative images showing AAK‐2 protein levels in young adult animals fed bacteria expressing either control or the indicated RNAi construct (n = 3 independent experiments; images were acquired using an ×4 objective lens), Scale bars, 40 μm.
- Quantification of AAK‐2 protein levels in animals shown in F under the indicated treatments (n = 3 independent experiments with at least 300 animals/experiment; ***P < 0.001, **P < 0.01, *P < 0.05; one‐way analysis of variance (ANOVA)).
- Relative expression of aak‐2 mRNA in wild‐type, DCAP‐2‐ and NTL‐2‐depleted animals (n = 2 independent experiments; P(dcap‐2(RNAi)) = 0.85, P(ntl‐2(RNAi)) > 0.99; one‐way analysis of variance (ANOVA)).
- Representative images showing the effect of the indicated genetic inhibitions on the mitochondrial mass of young adult animals stained with Mitotracker Green. Images were acquired using an ×40 objective lens (n = 3 independent experiments; scale bars, 20 μm).
- Quantification of the mitochondrial mass in the animals shown in (I) under the indicated treatments (n = 3 independent experiments with at least 255 animals/experiment; *P < 0.05, **P < 0.01, ****P < 0.0001; one‐way analysis of variance (ANOVA)).
Data information: Error bars denote SEM.
Next, we tested whether dcap‐2 and ntl‐2 influence AAK‐2 levels. We found that dcap‐2 genetic inhibition increased AAK‐2 abundance in contrast to ntl‐2 downregulation, which significantly decreased it (Fig 3F and G). In line with these findings, knockdown of either edc‐3 or xrn‐1 also increased AAK‐2 levels, whereas ccf‐1 or let‐711 knockdown reduced AAK‐2 abundance (Fig 3F and G). Similar to skn‐1, aak‐2 mRNA levels were not affected by these treatments, indicating that aak‐2 expression is regulated post‐transcriptionally under these conditions (Fig 3H).
To determine whether dcap‐2 and ntl‐2 directly regulate mitochondrial content through SKN‐1 and AΑK‐2, we measured mitochondrial abundance in animals subjected to RNAi against skn‐1 or aak‐2 combined with dcap‐2 or ntl‐2. We found that the increased mitochondrial mass of dcap‐2 RNAi‐treated animals was decreased to levels similar to the ones caused by either skn‐1 or aak‐2 genetic inhibition, while the lower mitochondrial mass of ntl‐2‐depleted animals was not further decreased by the combined depletion of either skn‐1 or aak‐2 with ntl‐2 (Fig 3I and J). Together, these findings indicate that DCAP‐2 and NTL‐2 oppositely regulate mitochondrial abundance acting through SKN‐1 and AAK‐2.
To gain insight into the different phenotypes observed upon knockdown of the mRNA degradation and the CCR4‐NOT complex components, we generated transgenic animals that co‐express EDC‐3::DsRed and NTL‐2::GFP fusion proteins for monitoring their interaction and alterations under physiological or stress conditions in vivo. We found that these components form distinct foci based on Pearsons correlation coefficient, rather than functioning in the same pathway within P‐bodies (Fig 4A and B). To verify the distinct localization pattern of the CCR4‐NOT and the mRNA degradation complex components, we generated animals that co‐express the following translational reporters: CCF‐1::GFP with DCAP‐2::mCherry, NTL‐2::GFP with DCAP‐2::mCherry and EDC‐3::GFP with DCAP‐2::mCherry. We found that DCAP‐2 foci do not co‐localize with CCF‐1 or NTL‐2 foci, while they extensively co‐localize with EDC‐3 foci under control conditions (Appendix Fig S14). Although localization of the CCR4‐NOT and the degradation complex components in distinct foci has never been reported in vivo, recent evidence from in vitro analyses supports this idea (Ozgur et al, 2015; Youn et al, 2018). Considering the functional features of their constituents, we will refer hereafter to the mRNA degradation components (DCAP‐1/DCAP‐2/EDC‐3 and XRN‐1)‐containing foci as degradation bodies and to the CCR4‐NOT (CCF‐1, NTL‐2 and LET‐711)‐containing ones as storage bodies (Fig 4C).
Figure 4. Components of the CCR4‐NOT and the mRNA degradation complexes show discrete localization patterns, are oppositely expressed during ageing, and oppositely influence global translation rates.
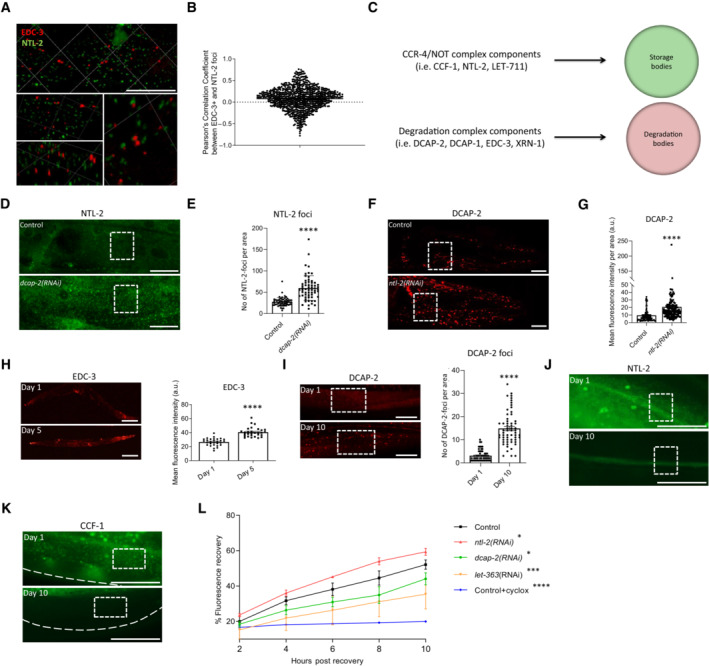
- 3D representation of the subcellular localization of EDC‐3‐ (red) and NTL‐2‐ (green)‐labelled foci (lower panels are higher magnification and rotation images of the top panel image). Images were acquired using an ×63 objective lens (n = 3 independent experiments).
- Diagram showing Pearson's correlation coefficient values after measuring the correlation between EDC‐3 and NTL‐2 foci.
- Schematic representation of the components of storage and degradation bodies referred to in our study.
- Representative images showing an increase in NTL‐2 foci upon dcap‐2 genetic inhibition (n = 3 independent experiments).
- Quantification of NTL‐2 foci upon dcap‐2 genetic inhibition (n = 3 independent experiments with at least 40 animals/experiment; ****P < 0.0001; two‐tailed unpaired t‐test).
- Representative images showing an increase in the signal of DCAP‐2 foci upon ntl‐2 genetic inhibition. Images were acquired using an ×40 objective lens (n = 3 independent experiments).
- Respective quantification (n = 3 independent experiments with at least 80 animals/experiment; ****P < 0.0001; two‐tailed unpaired t‐test).
- (left) Representative images depicting EDC‐3 expression in whole animals during ageing and (right) quantification of EDC‐3 levels during ageing. Images were acquired using ×5 objective lens (n = 3 independent experiments with at least 47 animals/experiment; ****P < 0.0001; two‐tailed unpaired t‐test).
- (left) Representative images showing DCAP‐2 expression levels in young versus old animals. Images were acquired using an ×40 objective lens and (right) quantification (n = 3 independent experiments with at least 20 animals/experiment; ****P < 0.0001; two‐tailed unpaired t‐test).
- Representative images of NTL‐2 expression levels in young (top) versus old (bottom) animals showing that NTL‐2 foci are barely detectable by day 10 of adulthood.
- CCF‐1 expression levels in young (top) versus old (bottom) animals showing that CCF‐1 foci are barely detectable by day 10 of adulthood (n = 3 independent experiments; images were acquired using an ×40 objective lens).
- Quantification of the percentage of fluorescence recovery after photobleaching (FRAP) to measure the rates of de novo protein synthesis upon genetic inhibition of either ntl‐2 or dcap‐2 in 6‐day‐old adult animals; the protein synthesis inhibitor cycloheximide (500 μM) and let‐363 genetic inhibition were used as controls. Measurements started after animals have recovered for 2 h post photobleaching (n = 3 independent experiments with at least 50 animals/experiment).
Data information: Scale bars, 20 μm, boxes in white‐dashed lines include representative areas of interest for comparison. Error bars denote SEM.
To establish that the two types of bodies are distinct, we tested whether they are formed independently of each other. In fact, perturbation of degradation bodies through dcap‐2 knockdown caused an increase in NTL‐2 foci and vice versa, perturbation of storage bodies through ntl‐2 knockdown increased the abundance of DCAP‐2 foci (Fig 4D–G). Consistently, knockdown of the mRNA storage‐related genes ccf‐1 and let‐711 increased DCAP‐2 levels (Appendix Fig S15A and B). Likewise, knockdown of edc‐3 or xrn‐1 increased NTL‐2 levels (Appendix Fig S15C and D). These results demonstrate that the two types of bodies form antagonistically of each other and in a rather interdependent manner. It is also noteworthy that degradation body components accumulate with age (Fig 4H and I), in contrast to storage body components (Fig 4J and K), further corroborating that the two types of bodies are distinct entities.
We then hypothesized that storage and degradation bodies acquire discrete functional properties, differentially influencing essential cellular processes. Recent findings elucidated a mechanism through which P‐bodies modulate global protein synthesis in the soma (Rieckher et al, 2018). To test whether the two types of bodies oppositely influence protein synthesis rates in vivo, we performed fluorescence recovery after photobleaching (FRAP) analysis (Kourtis & Tavernarakis, 2017) and found that knockdown of dcap‐2 reduced global protein synthesis rates, similarly to let‐363 genetic inhibition, while ntl‐2 knockdown had the opposite effect (Fig 4L). We confirmed this finding by measuring total protein levels in wild‐type animals subjected to RNAi against either dcap‐2 or ntl‐2. We found that DCAP‐2‐depleted animals have lower total protein content at day 1 of adulthood, while NTL‐2‐depleted animals have higher total protein levels compared to age‐matched wild‐type worms (Appendix Fig S16). Such alterations in protein synthesis may, to some extent, be linked with the decapping and deadenylation activities of the DCAP‐2 and NTL‐2 enzymes respectively. Together, these results indicate that the CCR4‐NOT and the degradation complex are parts of distinct foci which form interdependently, mutually antagonize each other and oppositely control bulk protein synthesis.
Storage and degradation bodies co‐regulate MTPT fate near mitochondria
Current evidence supports that MTPTs are transcripts that are mostly locally translated. Our findings suggest that storage and degradation bodies oppositely affect mitochondrial biogenesis possibly by modulating cytoplasmic translation in the vicinity of the organelles. To further test this notion, we sought to identify MTPTs bound to storage body components that are expected to form more stable associations with their target mRNAs compared to degradation body components which probably associate with mRNAs transiently to degrade them. To test whether MTPTs are bound by storage body components in vivo, we performed RNA immunoprecipitation, using whole protein extracts from NTL‐2::GFP animals, followed by quantitative real‐time PCR (qRT–PCR) analysis. We found that NTL‐2 binds MTPTs, including atp‐5, atp‐1, f46b6.6, mrpl‐13, nuo‐5, t20h4.5 and skn‐1 (Fig 5A). Enrichment of these transcripts in the NTL‐2::GFP immunoprecipitated sample is specific since mRNAs encoding nuclear proteins (FIB‐1, NPP‐22), endoplasmic reticulum (SPCS‐1) or cytoplasmic (RHI‐1) proteins have not been detected (Appendix Fig S17A). Binding of MTPTs to NTL‐2 was impaired upon mitochondrial stress such as CCCP treatment and during ageing (Appendix Fig S17B). By contrast, knockdown of dcap‐2 enhanced the association between MTPTs and NTL‐2 (Appendix Fig S17B). This result is further strengthened by our finding that dcap‐2 genetic inhibition enhances the associations of storage bodies with mitochondria, while it disrupts associations of degradation bodies with the organelles (Appendix Fig S18, Figs EV2C and D, and 2G and H). Taken together, these findings indicate that storage bodies specifically bind MTPTs and modulate their abundance near mitochondria, in cooperation with degradation bodies.
Figure 5. Storage body components bind MTPTs and associate with mitochondria in a local translation‐dependent manner.
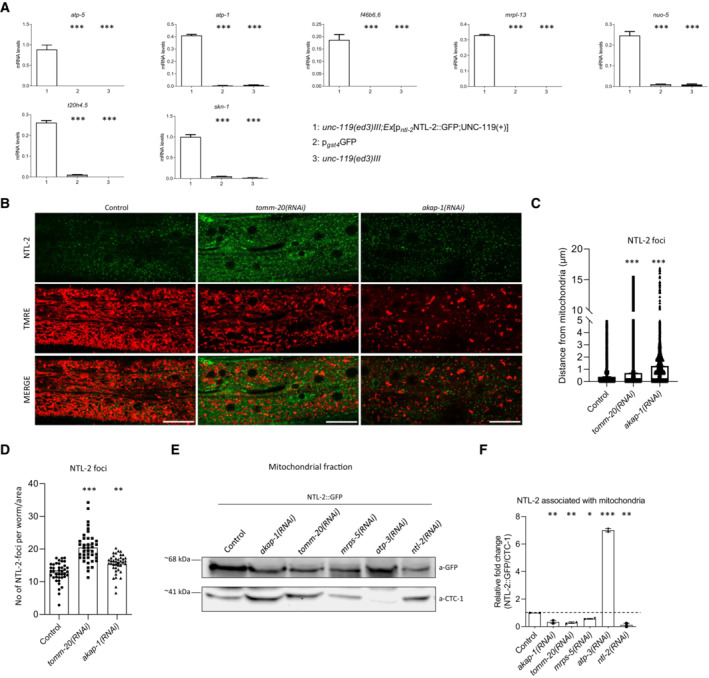
- Analysis of expression of select MTPTs by RT–qPCR following RNA immunoprecipitation (RIP) in anti‐GFP isolates from NTL‐2::GFP transgenic animals, unc‐119(ed3)III (no GFP expression) and p gst‐4 GFP (used as an additional negative control) counterparts (n = 2 independent experiments, ***P < 0.001; one‐way analysis of variance (ANOVA)).
- Representative images showing the localization of NTL‐2/storage bodies relative to mitochondria upon genetic inhibition of either tomm‐20 or akap‐1 (green: NTL‐2, red: TMRE, a mitochondrial membrane potential‐dependent dye; n = 3 independent experiments). Scale bars, 20 μm. Images were acquired using an ×63 objective lens.
- Quantification of the distances NTL‐2/storage bodies acquire from mitochondria upon genetic inhibition of either tomm‐20 or akap‐1 (n = 3 independent experiments with at least 45 animals/experiment; ***P < 0.001; one‐way analysis of variance (ANOVA)).
- Quantification of the number of NTL‐2/storage bodies upon genetic inhibition of either tomm‐20 or akap‐1 (n = 3 independent experiments with at least 45 animals/experiment; **P < 0.01, ***P < 0.001; one‐way analysis of variance (ANOVA)).
- Immunoblot analysis in mitochondria isolates of whole animal extracts showing the protein levels of NTL‐2 contained in the isolate in control conditions and upon genetic inhibition of akap‐1, tomm‐20, mrps‐5 and atp‐3; genetic inhibition of ntl‐2 is used as a control; NTL‐2 protein is detected by an anti‐GFP antibody in transgenic animals that contain the NTL‐2::GFP protein fusion (n = 2 independent experiments).
- Quantification of NTL‐2 protein bound to mitochondria under control conditions and upon genetic inhibitions shown in (E); CTC‐1 is used as a loading control for mitochondria (n = 2 independent experiments; *P < 0.05, **P < 0.01, ***P < 0.001; one‐way analysis of variance (ANOVA) followed by Dunnett's test).
Data information: Error bars denote SEM.
We next investigated whether storage bodies have a role in local translation of MTPTs. We found that knockdown of either ntl‐2 or let‐711 increases the levels of the mitochondrial local translation inducer TOMM‐20, as indicated by the increased signal of transgenic animals expressing TOMM‐20::RFP or TOMM::20mKate2 fusion (Appendix Fig S19). Moreover, knockdown of ntl‐2 increased the protein levels of MTPTs such as ATP‐1, F46B6.6, MRPL‐13 and T20h4.5 (Appendix Fig S20). This increase was abrogated by depletion of either tomm‐20 or akap‐1, implicating mRNA storage components in localized translation of MTPTs near the OMM. Then, we examined whether tomm‐20 or akap‐1 knockdown affects storage bodies–mitochondria associations. While these interventions did not decrease the formation of storage bodies, they impaired their associations with mitochondria as shown by in vivo experiments, as well as by immunoblot detection of NTL‐2 in isolated mitochondria (Fig 5B–F).
Balanced mRNA storage and degradation promotes stress resistance and longevity
Mitochondrial homeostasis is of paramount importance for stress resistance and longevity. Based on our data indicating that storage and degradation bodies acquire opposing roles in coordinating mitochondrial biogenesis, we hypothesized that they would oppositely affect stress responses and organismal ageing as well. To test this hypothesis, we exposed animals to specific stressors that lead to mitochondrial perturbation, such as heat stress, CCCP and paraquat, and assessed stress resistance (Castello et al, 2007; Labbadia et al, 2017; Miyazono et al, 2018). We found that DCAP‐2‐depleted animals were more resistant to all three stressors compared to wild‐type worms (Fig 6A–C, Tables [Link], [Link]). Likewise, depletion of either EDC‐3 or XRN‐1 increased heat stress resistance compared to controls (Fig EV4A, Table EV1). By contrast, NTL‐2 depletion compromised stress responses during adulthood in both wild‐type animals and daf‐2 mutants, known to be resistant to oxidative stress (Honda & Honda, 1999) (Fig 6A–D, Tables [Link], [Link]). Similarly, depletion of either CCF‐1 or LET‐711 rendered animals susceptible to stress (Fig EV4B and C, Table EV1). Notably, ntl‐2 knockdown suppressed the enhanced resistance of EDC‐3‐ or DCAP‐2‐deficient animals to heat stress and paraquat treatment (Fig EV4C and D, Tables EV1 and EV2). Together, these findings indicate that the integrity of storage and degradation bodies as well as their relative coordination is intimately related to the ability of the organism to cope with the detrimental effects elicited by stressors that interfere with mitochondrial function.
Figure 6. Storage and degradation body components oppositely influence stress resistance and longevity.
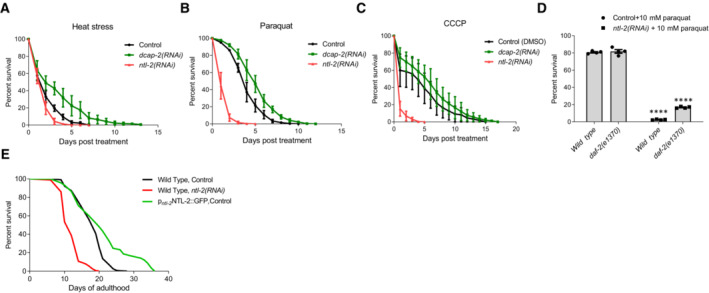
- Per cent survival of wild‐type, DCAP‐2‐ and NTL‐2‐depleted animals subjected to heat stress for 5 h at 37°C and then counted every 24 h (n = 4 independent experiments with at least 294 animals/experiment).
- Per cent survival of wild‐type, DCAP‐2‐ and NTL‐2‐depleted animals following paraquat (8 mM) administration and counted every 24 h (n = 4 independent experiments with at least 144 animals/experiment).
- Per cent survival of wild‐type, DCAP‐2‐ and NTL‐2‐depleted animals counted every 24 h post‐CCCP (15 μM) treatment (n = 4 independent experiments with at least 116 animals/experiment).
- Per cent survival of wild‐type and daf‐2(e1370)‐mutant animals subjected to ntl‐2 knockdown 24 h post‐paraquat (10 mM) administration (n = 4 independent experiments with at least 150 animals/experiment; ****P < 0.0001, two‐way analysis of variance (ANOVA)).
- Knockdown of ntl‐2 shortens the lifespan of wild‐type animals, while NTL‐2 overexpression extends lifespan.
Data information: Error bars denote SEM. Lifespan assays were performed at 20°C; detailed data are given in Tables [Link], [Link].
Figure EV4. Balance between storage and degradation body components is indispensable for animal stress resistance.
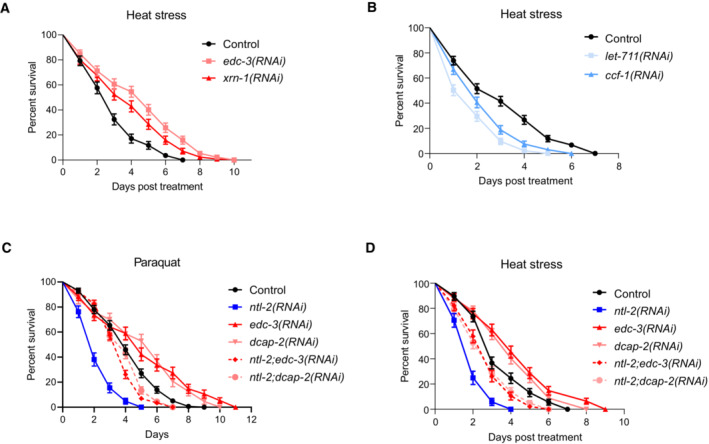
- Per cent survival of wild‐type, EDC‐3‐ and XRN‐1‐depleted animals exposed to heat stress for 5 h at 37°C and then counted every 24 h (n = 3 independent experiments with at least 369 animals/experiment).
- Per cent survival of wild‐type, LET‐711‐ and CCF‐1‐depleted animals exposed to heat stress performed for 5 h at 37°C counted every 24 h (n = 3 independent experiments with at least 429 animals/experiment).
- Per cent survival of wild‐type animals fed with bacteria expressing either control or the indicated RNAi construct diluted 1:1 following paraquat (8 mM) administration and counted every 24 h (n = 3 independent experiments with at least 603 animals/experiment).
- Per cent survival of wild‐type animals fed with bacteria expressing either control or the indicated RNAi construct diluted 1:1 and subjected to heat stress for 5 h at 37°C counted every 24 h (n = 3 independent experiments with at least 603 animals/experiment).
Data information: Error bars denote SEM. Stress assays were performed at 20°C; detailed data are given in Tables [Link], [Link].
We also tested whether changes in storage and degradation body components influence lifespan. Perturbation of mRNA storage, through ntl‐2 genetic inhibition, dramatically reduced the lifespan of wild‐type animals in contrast to its overexpression (Fig 6E, Table EV4). In addition, knockdown of other storage body genes, such as ccf‐1 and let‐711 in wild‐type animals, also shortened lifespan (Appendix Fig S21A, Table EV4), while their overexpression prolonged lifespan (Appendix Fig S21B and C, Table EV4). By contrast, and in line with previous reports (Rieckher et al, 2018), downregulation of specific degradation body components, including edc‐3, dcap‐2 and xrn‐1, extended lifespan, while their overexpression compromised life expectancy (Appendix Fig S21A, D and E, Table EV4).
We next examined whether a balance between mRNA storage and degradation is required to modulate ageing and found that depletion of degradation body components further extended the lifespan of animals overexpressing storage body components (Appendix Fig S21B and C, Table EV4), while genetic inhibition of storage body components decreased the lifespan of animals overexpressing degradation body components (Appendix Fig S21D and E, Table EV4). Moreover, deficiency in storage body components compromised longevity mediated by deficiency of degradation body components (Appendix Fig S22A–E, Table EV4). Notably, ntl‐2 was required for the longevity of long‐lived mutants such as daf‐2, age‐1 and akt‐1 (Fig EV5A–C, Table EV4). In addition to its detrimental effects on longevity, ntl‐2 genetic inhibition also markedly affected healthspan, manifested as decreased locomotor activity in middle‐aged animals, while dcap‐2 genetic inhibition enhanced healthspan (Movies [Link], [Link]).
Figure EV5. Genetic inhibition of ntl‐2 shortens the lifespan of long‐lived insulin/IGF‐1 pathway mutants and differentially impacts longevity of animals with compromised mitochondrial function.
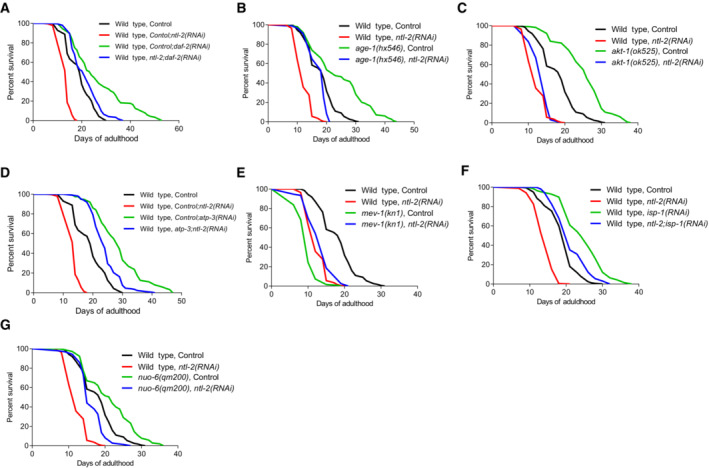
- Knockdown of ntl‐2 shortens the lifespan of daf‐2(RNAi) long‐lived animals.
- Knockdown of ntl‐2 shortens the lifespan of age‐1(hx546) mutants.
- Knockdown of ntl‐2 shortens the lifespan of akt‐1(ok525) mutants.
- Knockdown of atp‐3 rescues the short lifespan of NTL‐2‐depleted animals.
- mev‐1(kn1) mutation is beneficial for the lifespan of NTL‐2‐depleted animals.
- Knockdown of isp‐1 rescues the short lifespan of NTL‐2‐depleted animals.
- nuo‐6(qm200) mutation is beneficial for the lifespan of NTL‐2‐depleted animals.
Data information: Lifespan assays were performed at 20°C; detailed data are given in Table EV4.
To investigate whether local translation in the mitochondrial vicinity interfaces mechanistically with NTL‐2‐mediated processes to co‐regulate lifespan, we tested the effect of akap‐1 knockdown on wild‐type, ntl‐2 overexpressing and ntl‐2‐depleted animals (Fig 7A–C, Table EV4). Genetic inhibition of akap‐1 extended the lifespan of wild‐type animals, similar to tomm‐20 genetic inhibition, indicating that perturbation of local translation events may be beneficial for lifespan (Appendix Fig S23, Table EV4). Furthermore, akap‐1 knockdown rescued the short lifespan of ntl‐2 heterozygote mutants but did not further extend the lifespan of long‐lived ntl‐2‐overexpressing worms, implying that the two components function in the same genetic pathway (Fig 7A and B, Table EV4). Intriguingly, akap‐1 RNAi could ameliorate the lifespan‐shortening effect associated with NTL‐2 deficiency even when administered diluted (Fig 7C, Table EV4). Together, these findings suggest that the detrimental effects of NTL‐2 depletion on lifespan are probably due to increased local translation rates or aberrant translation of target MTPTs on mitochondria, and this effect is ameliorated upon akap‐1 knockdown.
Figure 7. The detrimental effects of ntl‐2 genetic inhibition on lifespan are ameliorated upon local translation perturbation.
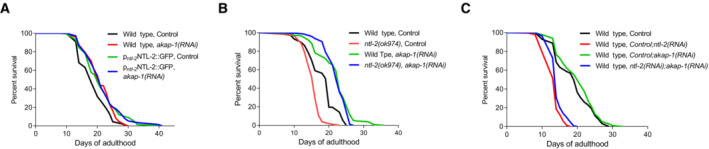
- Knockdown of akap‐1 extends the lifespan of wild‐type animals, while it does not further extend the lifespan of NTL‐2‐overexpressing animals.
- Knockdown of akap‐1 reverses the lifespan shortening of ntl‐2‐mutant worms.
- Knockdown of akap‐1 ameliorates the lifespan shortening caused by ntl‐2 genetic inhibition (double RNAi administered as 1:1 mixture).
Data information: Lifespan assays were performed at 20°C; detailed lifespan data are given in Table EV4.
To determine whether overexpression/aberrant translation of select MTPTs mediates the adverse effects of NTL‐2 depletion on longevity, we assayed the lifespan of several MTPT mutants upon ntl‐2 knockdown. Notably, we found that both nuo‐6 and mev‐1 mutations ameliorated the short lifespan of NTL‐2‐deficient animals (Fig EV5D and E, Table EV4). Consistent with this, genetic inhibition of isp‐1 and atp‐3 reversed the short lifespan of NTL‐2‐deficient worms (Fig EV5F and G, Table EV4). Combined, these results support an NTL‐2‐dependent regulation of local translation of specific MTPTs that interfaces with mechanisms mediating stress resistance and longevity in C. elegans.
Discussion
Our findings unravel a pivotal and surprising role for select components of the 5′‐3′ mRNA degradation pathway in controlling mitochondrial biogenesis, eventually contributing to mitochondrial homeostasis during ageing. We show in vivo that components of the mRNA degradation and the CCR4‐NOT complex are located in distinct foci, the degradation and storage bodies respectively. The two types of bodies form physical and functional associations with mitochondria, which are attenuated during ageing. Unexpectedly, components of the two types of bodies oppositely impact mitochondrial biogenesis by differentially regulating SKN‐1 and AAK‐2 expression and activity at a post‐transcriptional level.
Our results also reveal that the two types of bodies differentially influence global protein synthesis (Fig 4L) in a manner that is consistent with the enzymatic activities of their constituents. In fact, reduced protein synthesis rates upon DCAP‐2 depletion can be attributed to the accumulation of deadenylated mRNAs, which are not decapped and degraded and may therefore be inefficiently translated and/or interfere with the translation of other mRNAs. By contrast, increased protein synthesis rates upon NTL‐2 depletion can, to some extent, be due to inhibition of deadenylation, leading to mRNA stabilization and increased translation rates (Collart, 2016). Delineating the complex cross‐talk between the mechanisms that regulate mRNA metabolism and mRNA translation regulatory mechanisms, as well as the molecular players involved are forthcoming challenges.
Previous studies have shown that reduced global protein synthesis increases lifespan and stress resistance in diverse model organisms, including C. elegans, yet the precise mechanism that mediates longevity remains unknown (Hansen et al, 2007; Syntichaki et al, 2007).
Our results indicate that increased abundance of NTL‐2/storage bodies upon depletion of mRNA degradation components, such as DCAP‐2, correlates with a drop in global translation rates in somatic cells, enhanced associations of storage bodies with mitochondria and binding on MTPTs, as well as increased stress resistance and lifespan extension. Conversely, depletion of NTL‐2 increases the abundance of DCAP‐2/degradation bodies and global mRNA translation, reducing survival and stress resistance, in accordance with findings in human cell lines (Ito et al, 2011). These findings suggest that a balance between storage and degradation bodies locally may act as a regulatory mechanism that adjusts local translation events to the cell needs even at a single transcript level. In line with this idea, it has been proposed that spatiotemporal associations of mRNAs with RNA‐binding proteins can specifically determine mRNA fate, that is, storage, degradation or translation (Re et al, 2016). Therefore, even adjacent mRNAs could be differently regulated, enabling cells to maintain a proper number of functional mitochondria to achieve allostasis during ageing and upon stress (Picard et al, 2018).
Previous studies in yeast have shown that P‐body components can bind mitochondrial transcripts and regulate their expression in response to stress (Wang et al, 2018). In line with this, our findings indicate that storage bodies bind MTPTs and regulate their local translation in the vicinity of the organelles, supporting the evolutionary conservation of the molecular mechanism by which mRNA fate near mitochondria is influenced, and influences translation, in turn. In addition, accumulating evidence suggests that a quick and efficient response to stress requires rapid translation of select transcripts, as shown in neuronal axons during synaptic plasticity (Sutton & Schuman, 2006; Besse & Ephrussi, 2008; Rangaraju et al, 2019). Under these conditions, storage bodies offer this advantage to neighbouring mitochondria as inferred from the increased susceptibility of NTL‐2‐deficient animals to mitochondrial stress.
Our findings also indicate that storage bodies associate stronger with mitochondria in the long‐lived DAF‐2‐depleted animals, in contrast to the short‐lived NHR‐49‐depleted ones, in which mitochondria–storage body association is decreased. Regarding degradation bodies, we find that their associations with mitochondria are decreased both in long‐ and short‐lived animals. In long‐lived animals, decreased association of degradation components with mitochondria linked to an enhanced association of storage bodies with the organelles may represent a state of balance between mRNA storage and degradation which contributes to the maintenance of cellular and organismal homeostasis. Accordingly, in short‐lived animals, the reduced associations of the mRNA degradation and the storage components with mitochondria reflect a state in which both mRNA storage and degradation near mitochondria are impaired, possibly contributing to their decreased survival and compromised stress responses. Importantly, this finding may explain the shortened lifespan of daf‐2 mutants upon ntl‐2 genetic inhibition and also adds to previous findings showing that knockdown of the degradation body component edc‐3 further extends the lifespan of daf‐2 mutants (Rieckher et al, 2018). Deciphering the complete mRNA cargo of storage bodies and investigating whether and how it changes during ageing and in response to stress or whether it is affected by the subcellular localization of these bodies in diverse cell types and genetic backgrounds are worth considering in the future.
Our work implicates, for the first time, components of the mRNA degradation and the CCR4‐NOT complex in the regulation of mitochondrial function and abundance during ageing, suggesting the importance of their balance for cellular and organismal homeostasis. Notably, impaired mitochondrial biogenesis is associated with common neurodegenerative disorders, heart failure, acute kidney injury (AKI) and type 2 diabetes, among others (Funk & Schnellmann, 2012; Joseph & Hood, 2014; Pisano et al, 2016; Golpich et al, 2017; Gureev et al, 2019). Identification of novel players in mitochondrial biogenesis that could potentially be exploited for therapeutic interventions to fight mitochondrial‐associated diseases is a forthcoming challenge for future medicine. Given their tight evolutionary conservation, components of storage and degradation bodies might be promising candidate targets for possible translational applications.
Materials and Methods
Reagents and Tools table
| Chemicals | ||
|---|---|---|
| Reagent or resource | Source | Identifier |
| Mitotracker Green FM | Molecular Probes, Invitrogen | M7514 |
| TMRE | Molecular Probes, Invitrogen | T669 |
| Mitotracker Red CM‐H2X ROS | Molecular Probes, Invitrogen | M7513 |
| Cycloheximide | Sigma Aldrich | C7698 |
| Mitotracker Deep Red FM | Molecular Probes, Invitrogen | M22426 |
| Paraquat | Sigma Aldrich | 856177 |
| CCCP | Sigma‐Aldrich | 857815 |
| Magnetic Beads | Merc, Millipore | LSKMAGAG02 |
| Levamisole | Sigma‐Aldrich | 196142 |
| RNAse inhibitor | Roche | 3335399001 |
| Nanobeads 100 nm | Polysciences | 6401015 |
| DMSO | AppliChem | A3672,0250 |
| Glycoblue | ThermoFisher Scientific | AM9515 |
| CIP enzyme | NEB | M0290 |
| Completemini Proteinase inhibitor coctail | Roche | 11836153001 |
| Protector RNase Inhibitor | Roche | 3335399001 |
| Zirconium Oxide Beads 0.5 mm | Next Advance | |
| DiO6(3) | Invitrogen | D275 |
| BioTracker ATP‐Red live cell dye | Merck | SCT045 |
| Antibodies | ||
| Type | Source | Identifier |
| Anti‐GFP | Minotech | ‐ |
| Anti‐MTCOI | Abcam | ab14705 |
| Anti‐alpha tubulin | DSHB | AA4.3 (concentrate) |
| JLA20 (anti‐actin) | DHSB | ‐ |
| Anti‐ATP5A | Abcam | ab14748 |
| Anti‐MRPL13 | ThermoFisher Scientific | PA5‐51007 |
| Anti‐NDUFS8 | abbexa | abx026882 |
| Anti‐RFP | Minotech | ‐ |
| Experimental models | ||
| Organism/Strain | Source | |
| C. elegans: Strain N2: Wild Type | Caenorhabditis Genetics Center | |
| C. elegans: Strain SJ4103: N2;Is[p myo‐3 mtGFP] | Caenorhabditis Genetics Center | |
| C. elegans: Strain SJ4143: N2;Is[p ges‐1 mtGFP] | Caenorhabditis Genetics Center | |
| C. elegans: Strain CL2166: N2;Is[p gst‐4 GFP] | Caenorhabditis Genetics Center | |
| C. elegans: Strain OP168: unc‐119(ed3) III;[skn‐1::TY1::EGFP::3xFLAG;unc‐119(+)] | Caenorhabditis Genetics Center | |
| C. elegans: Strain AGD383: uthIs202 [aak‐2 (intron 1)::aak‐2(aa1‐aa321)::Tomato::unc‐54 3′UTR;pRF4] | Caenorhabditis Genetics Center | |
| C. elegans: Strain CB1370: daf‐2(e1370)III | Caenorhabditis Genetics Center | |
| C. elegans: Strain MQ887: isp‐1(qm150) | Caenorhabditis Genetics Center | |
| C. elegans: Strain TJ1052: age‐1(hx546) | Caenorhabditis Genetics Center | |
| C. elegans: Strain RB759: akt‐1(ok525) | Caenorhabditis Genetics Center | |
| C. elegans: Strain MQ133: nuo‐6(qm200) | Caenorhabditis Genetics Center | |
| C. elegans: Strain TK22: mev‐1(kn1)III | Caenorhabditis Genetics Center | |
| C. elegans: Strain IR1284: N2;Is[p myo‐3 mtGFP];Ex011[p lgg‐1 DsRed::LGG‐1] | Palikaras et al (2015b) | |
| C. elegans: Strain SPC167: skn‐1(lax120)IV;P gst‐4 GFP | Caenorhabditis Genetics Center | |
| C. elegans: daf‐2(e1370);Is[p gst‐4 GFP] | This paper | |
| C. elegans: N2;EX[p edc‐3 EDC‐3::DsRed;pRF4] | Rieckher et al (2018) | |
| C. elegans: unc‐119 (ed3)III;EX[p ntl‐2 NTL‐2::GFP;unc‐119(+)] | This paper | |
| C. elegans: unc‐119 (ed3)III;Ex[p edc‐3 EDC‐3::DsRed;p ntl‐2 NTL‐2::GFP;unc‐119(+)] | This paper | |
| C. elegans: unc‐119 (ed3)III;EX[p dcap‐2 DCAP‐2::mcherry;unc‐119(+)] | This paper | |
| C. elegans: unc‐119 (ed3)III;Ex[p ccf‐1 CCF‐1::GFP;unc‐119(+)] | This paper | |
| C. elegans: N2;Ex[p ife‐2 GFP;pRF4] | Syntichaki et al (2007) | |
| C. elegans: unc‐119 (ed3)III;Ex[p ntl‐2 NTL‐2::GFP;unc‐119(+)];syEx1155 [p myo‐3 TOMM‐20::mRFP::3xMyc;unc‐119(+)] | This paper | |
| C. elegans: SJ4103; N2;Ex[p edc‐3 EDC‐3::DsRed; pRF4] | This paper | |
| C. elegans: unc‐119 (ed3)III;Ex[p edc‐3 EDC‐3::GFP;unc‐119(+)] | This paper | |
| C. elegans: unc‐119 (ed3)III;Ex003[p myo‐3 TOMM‐20::Rosella;unc‐119(+)] | This paper | |
| C. elegans: Strain PS6187: syEx1155[p myo‐3 TOMM‐20::mRFP::3xMyc;unc‐119(+)] | Caenorhabditis Genetics Center | |
| C. elegans: Strain JJ1850: unc‐119(ed3)III; Is[his‐72(1kb 5′UTR)::his‐72::SRPVAT::GFP::his72 (1KB 3′UTR)] | Caenorhabditis Genetics Center | |
| C. elegans: Strain VC682: ntl‐2(ok974)/mIn1 [mIs14 dpy‐10(e128)]II | Caenorhabditis Genetics Center | |
| C. elegans: Strain N2;Ex[ pdcap‐1 DCAP‐1::DsRed;pRF4] | Rieckher et al (2018) | |
| C. elegans: Strain SJZ328: foxSi75[eft‐3p::tomm‐20::mKate2::HA::tbb‐2 3′UTR]I | Caenorhabditis Genetics Center | |
| C. elegans: Strain unc‐119(ed3)III;Ex[p ccf‐1 CCF‐1::GFP;p dcap‐2 DCAP‐2::mCherry;unc‐119(+)] | This paper | |
| C. elegans: Strain N2;Ex[p ntl‐2 NTL‐2::GFP;p dcap‐2 DCAP‐2::mCherry;pRF4] | This paper | |
| C. elegans: Strain unc‐119(ed3)III;Ex[p edc‐3 EDC‐3::GFP;p dcap‐2 DCAP‐2::mCherry;unc‐119(+)] | This paper | |
| C. elegans: Strain unc‐119(ed3)III;Ex[p f46b6.6 F46B6.6::GFP;unc‐119(+)] | This paper | |
| Oligonucleotides | ||
| Name | Sequence | |
| Primer: ntl‐2 promoter Forward | ACACGACGGATCATTTCACGAG | |
| Primer: ntl‐2 promoter Reverse | GGATCCCTGAAAGAAAATCGATTT | |
| Primer: ntl‐2 coding Forward | GGATCCATTTATGAGTAGAACGTAGCCAT | |
| Primer: ntl‐2 coding Reverese | ACCGGTGGGTTTGGAGTAGCTCG | |
| Primer: dcap‐2 promoter Forward | ACATTGCACTATACCCCTCTTATTGC | |
| Primer: dcap‐2 promoter Reverse | ACGAGAGATTCAAGCAAGTGGTGTC | |
| Primer: dcap‐2 coding part 1 Forward | GCGGCCGCATGCAGCAACA | |
| Primer: dcap‐2 coding part 1 Reverse | TCTAGACGTTTCAGCGAGTAATGAAACTTTTG | |
| Primer: dcap‐2 coding part 2 Forward | TCTAGAGATAGTGAAGCCCTTTCTCTTTT | |
| Primer: dcap‐2 coding part 2 Reverse | CCCGGGTGGTAATTGTGGTC | |
| Primer: ccf‐1 promoter Forward | TTCGTGTTTTGAAGAATTATCTTGTAAAATGAG | |
| Primer: ccf‐1 promoter Reverse | AAGCTTCTAGAATTTTCGTTTTAGAGTGAACG | |
| Primer: ccf‐1 coding Forward | TTCCAGATATCATAAAAATGGCTTCTAGTAGC | |
| Primer: ccf‐1 coding Reverse | ACCGGTGGGGCTTGTTGTGGAACT | |
| Primer: f46b6.6 promoter Forward | AGTTATCATTACAGTTTCGCTGGTTTTATATATGAAAC | |
| Primer: f46b6.6 promoter Reverse | GCTAGCCTGGAACTTAATTATGCAACGAT | |
| Primer: f46b6.6 coding Forward | ATGTCCAAAACGATGATTCTACAGTTGTTG | |
| Primer: f46b6.6 coding Reverse | ACCGGTAAATCCTGGTGGATACCAGT | |
| Primer: ntl‐2 RNAi Forward | ATGTTAGCAGACGACCATCAAGTCG | |
| Primer: ntl‐2 RNAi Reverse | TTAGTTTGGAGTAGCTCGCGCC | |
| Primer: atp‐3 RNAi Forward | AGAACAAGCTCGACCAGATTTC | |
| Primer: atp‐3 RNAi Reverse | GGGCATCCTTGTATTTCTTGAC | |
| Primer: mrps‐5 RNAi Forward | ATGGCATCACTTTTGCCATTTGTC | |
| Primer: mrps‐5 RNAi Reverse | ACCGGTCTTTTTGGGAACCACATGCGA | |
| Primer: tomm‐20 RNAi Forward | ATGTCGGACACAATTCTTGGTTTCAAC | |
| Primer: tomm‐20 RNAi Reverse | CTCCAAGTCGTCGGTGTCATCGA | |
| Primer: akap‐1 RNAi Forward | ACCGGTGAACATTTTCTCACTTACTG | |
| Primer: akap‐1 RNAi Reverse | TTTGCGACGAGAATGATGGTC | |
| Primer: xrn‐1 RNAi Forward | GATGAATTCACAAGTGGAAAAAGCTCTGAC | |
| Primer: xrn‐1 RNAi Reverse | GATGAATTCGATGAAGCCGTCGGA | |
| Primer: nhr‐49 RNAi Forward | CATCTGAATCACATCCACGATC | |
| Primer: nhr‐49 RNAi Reverse | ATTATTCTGCTCACTGTTCAAAATG | |
| Primer: Mito1 Forward | GTTTATGCTGCTGTAGCGTG | |
| Primer: Mito1 Reverse | CTGTTAAAGCAAGTGGACGAG | |
| Primer: ama‐1 Forward | TGGAACTCTGGAGTCACACC | |
| Primer: ama‐1 Reverse | CATCCTCCTTCATTGAACGG | |
| Primer: skn‐1 Forward | TCCACCAGGATCTCCATTCG | |
| Primer: skn‐1 Reverse | CTCCATAGCACATCAATCAAGTCG | |
| Primer: aak‐2 Forward | CAGAGCGCATTGCAGCGTC | |
| Primer: aak‐2 Reverse | GTTGAGCACCTTCCACTCCATGTC | |
| Primer: atp‐5 Forward | ACTGGTCGAAGCTCGCCGAG | |
| Primer: atp‐5 Reverse | ACTCGGCTGGAACCTCTCCG | |
| Primer: f46b6.6 Forward | GTTTGGAAGCAAATGAATCCTTCAGA | |
| Primer: f46b6.6 Reverse | CTGCAAAATCGAAACATCGTCAAGT | |
| Primer: h28o16.1 Forward | ACCTTGAGGAGACCGGAAAGGTT | |
| Primer: h28o16.1 Reverse | TTCCGAAGACGACAACTCCGA | |
| Primer: mrpl‐13 Forward | GTGATTGATGCGAATCAACAAGAC | |
| Primer: mrpl‐13 Reverse | CATATTGAACTTATAAATTGTGTGTTTCCA | |
| Primer: mrps‐5 Forward | AGAAAATTCTCGAAGAACGAGATACTGGA | |
| Primer: mrps‐5 Reverse | ACAATAAGTTTCAAAATCATCGAAATTCACA | |
| Primer: nuo‐5 Forward | CGGCTCCACCGAAAAAGGTC | |
| Primer: nuo‐5 Reverse | TCACCGACTTCTCCACCTCTACCA | |
| Primer: spcs‐1 Forward | GTCGCCGAGAGAACTTACCAGGTAA | |
| Primer: spcs‐1 Reverse | ACAATCGGGTTCTTTCTGAAGAGGA | |
| Primer: t20h4.5 Forward | GACCACTCAGTTCAAGATTCAGAGG | |
| Primer: t20h4.5 Reverse | ATAATCCACAATAGATGCACTTGGTC | |
| Primer: pmp‐3 Forward | ATGATAAATCAGCGTCCCGAC | |
| Primer: pmp‐3 Reverse | TTGCAACGAGAGCAACTGAAC | |
| Primer: fib‐1 Forward | TCGTTGGACCAGAAGGAATCGTT | |
| Primer: fib‐1 Reverse | AAAGATGACATCGACCATTCCGA | |
| Primer: npp‐22 Forward | ACTCAATTTTTCGCATATCCAAGTGAAT | |
| Primer: npp‐22 Reverse | ACCATGCTGAGAATCCAGAAACCAT | |
| Primer: rhi‐1 Forward | GAGCTTCTCAATGCTGACAAGGAGG | |
| Primer: rhi‐1 Reverse | GGAACCTTCTTTGATGGAAACAGAC | |
Methods and Protocols
Strains and transgenic lines
We followed standard procedures for C. elegans maintenance, crosses and other genetic manipulations (Brenner, 1974). Nematode rearing temperature was kept at 20°C unless noted otherwise. The following strains, available at CGC, were used for this study: N2: Wild‐type Bristol isolate, SJ4103: N2;Is[p myo‐3 mtGFP] to examine mitochondrial mass and mitochondrial changes in body wall muscle cells, SJ4143: N2;Is[p ges‐1 mtGFP] to analyse mitochondria in matrix of intestinal cells, OP178: wgIs178[skn‐1::TY1::EGFP::3xFLAG;unc‐119(+)], CL2166: N2;Is[p gst‐4 GFP] to measure SKN‐1 protein levels, and PS6187: syEx1155 [p myo‐3 TOMM‐20::mRFP::3xMyc;unc‐119(+)] to monitor mitochondrial changes and TOMM‐20 protein levels in body wall muscle cells. To measure the ubiquitous expression of TOMM‐20, we used SJZ328: foxSi75[eft‐3p::tomm‐20::mKate2::HA::tbb‐2 3′ UTR]I. To investigate the localization and abundance of degradation body components, we used the N2;Ex[p edc‐3 EDC‐3::DsRed;pRF4] (Rieckher et al, 2018), N2;Ex[ pdcap‐1 DCAP‐1::DsRed;pRF4] (Rieckher et al, 2018) and the unc‐119(ed3)III;Ex[p dcap‐2 DCAP‐2::mCherry;unc‐119(+)] (generated for this study) transgenic animals. To investigate the localization and abundance of storage body components, we used the unc‐119(ed3)III;Ex[p ntl‐2 NTL‐2::GFP;unc‐119(+)] and the unc‐119(ed3)III;Ex[p ccf‐1 CCF‐1::GFP;unc‐119(+)] transgenic animals both generated for this study. To investigate the relative positions of the degradation and the CCR4‐NOT complex components, we used the unc‐119(ed3)III;Ex[p edc‐3 EDC‐3::DsRed;p ntl‐2 NTL‐2::GFP;unc‐119(+)], unc‐119(ed3)III;Ex[p ccf‐1 CCF‐1::GFP;p dcap‐2 DCAP‐2::mCherry;unc‐119(+)] and N2;Ex[p ntl‐2 NTL‐2::GFP;p dcap‐2 DCAP‐2::mCherry;pRF4] generated for this study. To investigate the relative positions of the degradation complex components, we used unc‐119(ed3)III;Ex[p edc‐3 EDC‐3::GFP;p dcap‐2 DCAP‐2::mCherry;unc‐119(+)] generated for this study. To monitor global protein synthesis levels by FRAP, we used the Ex[p ife‐2 IFE‐2::GFP;pRF4] transgenic animals (Syntichaki et al, 2007). To study the localization of degradation bodies in relation to mitochondria in body wall muscle cells, we generated the following strains, in addition to the strains mentioned above: N2;Is[p myo‐3 mtGFP];Ex[p edc‐3 EDC‐3::DsRed;pRF4] and unc‐119(ed3)III;Ex[p edc‐3 EDC‐3::GFP;unc‐119(+)]. To study the relative positions of storage bodies and mitochondria in body wall muscle cells, we established additionally to before‐mentioned strains: unc‐119(ed3)III;Ex[p ntl‐2 NTL‐2::GFP;unc‐119(+)];syEx1155[p myo‐3 TOMM‐20::mRFP::3xMyc;unc‐119(+)]. To monitor mitophagy, we used the following strains and transgenic animals: IR1284: N2;Is[p myo‐3 mtGFP];Ex011[p lgg‐1 DsRed::LGG‐1] (Palikaras et al, 2015b) and N2; Ex[p myo‐3 TOMM‐20:: Rosella; unc‐119(+)]. To measure gst‐4 expression levels in an inducing genetic background, we used the daf‐2(e1370);Is[p gst‐4 GFP] strain generated by cross and the SPC167: P gst‐4 GFP;skn‐1(lax120) which is available in CGC. To measure AAK‐2 abundance, we used AGD383: uthIs202[aak‐2 (intron 1)::aak‐2(aa1‐aa321)::Tomato::unc‐54 3′UTR;pRF4] available from CGC. To measure F46B6.6/MTIF2 abundance, we used unc‐119(ed3)III;Ex[p f46b6.6 F46B6.6::GFP;unc‐119(+)] generated for this study. To test the lifespan of ntl‐2‐depleted animals, we used VC682: ntl‐2(ok974)/mIn1 [mIs14 dpy‐10(e128)]II available from CGC. To test possible unspecific mRNA binding on the GFP protein for the RIP experiment, we used CL2166: N2;Is[p gst‐4 GFP] and unc‐119(ed3)III animals. For lifespan experiments, we additionally used CB1370: daf‐2(e1370)III, TJ1052: age‐1(hx546)II, RB759: akt‐1(ok525)V, MQ133: nuo‐6(qm200)I, TK22: mev‐1(kn1)III and VC682: ntl‐2(ok974)/mln1[mls14 dpy‐10(e128)]II, available at CGC.
Molecular cloning
To generate the p ntl‐2 NTL‐2::GFP reporter construct, we amplified the coding region of the gene from C. elegans genomic DNA using the ntl‐2‐coding Forward primer (which contains the BamHI restriction enzyme sequence) and ntl‐2‐coding Reverse primer (which contains the AgeI restriction enzyme sequence). This amplified region was inserted in the TOPO‐pCRII vector (Invitrogen). In parallel, a ~2,000 bp region upstream of ntl‐2 gene‐coding sequence was amplified using the primers ntl‐2 promoter Forward and ntl‐2 promoter Reverse which was also inserted in the TOPO‐pCRII vector. Subsequently, ntl‐2 coding region was extracted from TOPO‐pCRII vector with BamHI/AgeI and inserted in frame at the amino (N) terminus of GFP, in the pPD95.77 plasmid vector. This genetic cassette was linearized using BamHI restriction enzyme and dephosphorylated using CIP. The promoter region was extracted from TOPO‐pCRII vector with BamHI and inserted into the dephosphorylated genetic cassette described previously. The construct was checked for correct promoter orientation with several diagnostic restriction digestions.
To generate the p edc‐3 EDC‐3::GFP reporter construct, we isolated the p edc‐3 EDC‐3 fragment from the pPD95.77 plasmid vector containing DsRed (Rieckher et al, 2018) and subcloned it into the pPD95.77 plasmid vector containing GFP.
To generate the p dcap‐2 DCAP‐2::mcherry reporter construct, we amplified the coding region of the gene from C. elegans genomic DNA in two steps owing to the big size of the sequence (~7,000 bp) using the primers dcap‐2 coding part 1 Forward (which contains the NotI restriction enzyme sequence) and dcap‐2 coding part 1 Reverse (which contains the XbaI restriction enzyme sequence) and dcap‐2 coding part 2 Forward (which contains the XbaI restriction enzyme sequence) and dcap‐2 coding part 2 Reverse (which contains the XmaI restriction enzyme sequence). Both amplified regions were separately inserted in the TOPO‐pCRII vector. In parallel, a ~1,700 bp region of the operon promoter was amplified using the primers dcap‐2 promoter Forward and dcap‐2 promoter Reverse and also inserted in the TOPO‐pCRII vector. In the last genetic cassette, which contains the operon promoter in the TOPO‐pCRII vector, we ligated the first fragment of dcap‐2 coding region cut by NotI/XbaI. Then, the TOPO‐pCRII vector containing the operon promoter and dcap‐2 fragment 1 was cut with XbaI and dephosphorylated by CIP. Subsequently, dcap‐2 coding region fragment 2 was inserted and the correct orientation of this fragment insertion was verified. Next, the operon promoter and the full dcap‐2 coding sequence were extracted from TOPO‐pCRII vector as one fragment in a single step with BamHI/XmaI and ligated in frame at the amino (N) terminus of mCherry, in the pPD95.77 plasmid vector which was cut with BamHI/AgeI taking advantage of the AgeI compatibility to XmaI.
To generate the p ccf‐1 CCF‐1::GFP reporter construct, we amplified the coding region of the gene from C. elegans genomic DNA using the primers ccf‐1 coding Forward and ccf‐1 coding Reverse (which contains the AgeI restriction enzyme sequence). To amplify the operon promoter in which ccf‐1 belongs, we used the primers ccf‐1 promoter Forward and ccf‐1 promoter Reverse. Both amplified regions were separately inserted in the TOPO‐pCRII vector. Next, ccf‐1 coding region was extracted from TOPO‐pCRII vector and inserted in frame at the amino (N) terminus of GFP, in the pPD95.77 plasmid vector. Then, the pPD95.77‐containing the ccf‐1 coding region‐ genetic cassette was linearized with HindIII and dephoshorylated by CIP. ccf‐1 promoter region was extracted from the TOPO‐pCRII vector with HindIII digest and ligated to the linearized vector. The correct orientation of the promoter was verified with additional diagnostic digests.
To generate the p f46b6.61 F46B6.6::GFP reporter construct, we amplified the coding region of the gene from C. elegans genomic DNA using the primers f46b6.6 coding Forward and f46b6.6 coding Reverse (which contains the AgeI restriction enzyme sequence). To amplify the operon promoter in which f46b6.6 belongs, we used the primers promoter Forward and f46b6.6 promoter Reverse. Both amplified regions were separately inserted in the TOPO‐pCRII vector. Next, f46b6.6‐coding region was extracted from TOPO‐pCRII vector and inserted in frame at the amino (N) terminus of GFP, in the pPD95.77 plasmid vector. Following, the pPD95.77‐containing the ccf‐1 coding region‐ genetic cassette was linearized. f46b6.6 promoter region was extracted from the TOPO‐pCRII vector and ligated to the linearized vector. The correct orientation of the promoter was verified with additional diagnostic digests.
To generate the RNAi constructs, gene‐specific fragments of interest were obtained by PCR amplification directly from the C. elegans genomic DNA using appropriate primer sets. The PCR‐generated fragments were initially inserted into the TOPO‐pCRII vector and then sub‐cloned into the pL4440 plasmid vector and the resulting constructs were transformed into HT115 (DE3) E. coli bacteria. For ntl‐2 RNAi, we amplified a 1,548 bp region by using the primers ntl‐2 RNAi Forward and ntl‐2 RNAi Reverse. For atp‐3 RNAi, we amplified a 694 bp region by using the primers atp‐3 RNAi Forward and atp‐3 RNAi Reverse. For mrps‐5 RNAi, a 2,150 bp region was amplified by using the primers mrps‐5 RNAi Forward and mrps‐5 RNAi Reverse. For tomm‐20 RNAi, a 984 bp region was amplified by using the primers tomm‐20 RNAi Forward and tomm‐20 RNAi Reverse. For akap‐1 RNAi, a 1,997 bp region was amplified by using the primers akap‐1 RNAi Forward and akap‐1 RNAi Reverse. For xrn‐1 RNAi, a 998 bp region was amplified by using the primers xrn‐1 RNAi Forward (containing the EcoRI restriction enzyme) and xrn‐1 RNAi Reverse (containing the EcoRI restriction enzyme). For nhr‐49 RNAi, a 1,465 bp region was amplified by using the primers nhr‐49 RNAi Forward and nhr‐49 RNAi Reverse. For cyc‐1 RNAi, let‐711 RNAi and ccf‐1 RNAi, the Ahringer C. elegans RNAi feeding library was used. For edc‐3 RNAi, we used the same construct as in Rieckher et al, 2018.
mRNA quantification
Total RNA was extracted using TRIzol reagent. For cDNA synthesis, mRNA was reverse transcribed using the iScriptTM cDNA Synthesis Kit (BioRad). Quantitative PCR was performed with the Eva Green qPCR Kit (Biotium). All kits were used according to the manufacturer's instructions. To quantify skn‐1 mRNA levels, we used the primers skn‐1 Forward and skn‐1 Reverse. To quantify aak‐2 mRNA levels, we used the primers aak‐2 Forward and aak‐2 Reverse. To quantify atp‐5 mRNA levels, we used the primers atp‐5 Forward and atp‐5 Reverse. To quantify f46b6.6 mRNA levels we used the primers f46b6.6 Forward and f46b6.6 Reverse. To quantify h28o16.1 mRNA levels, we used the primers h28o16.1 Forward and h28o16.1 Reverse. To quantify mrpl‐13 mRNA levels, we used the primers mrpl‐13 Forward and mrpl‐13 Reverse. To quantify mrps‐5 mRNA levels, we used the primers mrps‐5 Forward and mrps‐5 Reverse. To quantify nuo‐5 mRNA levels, we used the primers nuo‐5 Forward and nuo‐5 Reverse. To quantify spcs‐1 mRNA levels, we used the primers spcs‐1 Forward and spcs‐1 Reverse. To quantify t20h4.5 mRNA levels, we used the primers t20h4.5 Forward and t20h4.5 Reverse. Results were normalized to the expression levels of the housekeeping gene, pmp‐3, quantified by using the primers pmp‐3 Forward and pmp‐3 Reverse. To quantify fib‐1 mRNA levels, we used the primers fib‐1 Forward and fib‐1 Reverse. To quantify npp‐22 mRNA levels, we used the primers npp‐22 Forward and npp‐22 Reverse. To quantify rhi‐1 mRNA levels, we used the primers rhi‐1 Forward and rhi‐1 Reverse. Quantitative PCR was performed using a Bio‐Rad CFX96 Real‐Time PCR system.
Mitochondrial DNA quantification
Total RNA was extracted using TRIzol reagent. For cDNA synthesis, mRNA was reverse transcribed using the iScriptTM cDNA Synthesis Kit (BioRad). Quantitative PCR was performed in triplicate using a BioRad CFX96 Real‐Time PCR system (BioRad). To quantify mtDNA levels, we used the primers Mito1 Forward and Mito1 Reverse. The results were normalized to genomic DNA, quantifying the ama‐1 gene using the primers: ama‐1 Forward and ama‐1 Reverse. Quantitative PCR was performed using a Bio‐Rad CFX96 Real‐Time PCR system.
Fluorescent recovery after photobleaching (FRAP) analysis
To determine the role of mRNA degradation and storage body components in protein synthesis control, we performed FRAP analysis as previously described (Kourtis & Tavernarakis, 2017). Specifically, we measured global translation rates using the N2;Ex[p ife‐2 GFP;pRF4] strain which expresses the GFP protein under the ife‐2 gene promoter which is ubiquitously expressed in somatic tissues. We performed the assay on the agar plates, seeded with bacteria without the use of any anaesthetics (step B1a described in the aforementioned study). We bleached whole animals for 6 min at the 10× lens using the Zeiss AxioImager Z2 epifluorescence microscope. Time zero is the time point following photobleaching. Fluorescent recovery of each animal was measured every 1 h for at least 6 h. For each and every measurement one photo per animal was acquired and this was used to measure the mean fluorescence at each time point. Cycloheximide (Sigma Aldrich, C7698), which is an inhibitor of global protein synthesis, was used as a control in a final concentration of 500 μg/ml. At least 20 animals were bleached in each experiment and each condition tested. Animals with signs of damage after photobleaching were excluded from the final analysis. Fluorescence intensity quantification was performed using Image J.
Degradation and storage body monitoring
Quantification of storage and degradation body components was performed either tissue specifically or in whole animals. In the first case, we acquired images using the Zeiss LSM 710 confocal microscope and LEICA TCS SP8 Laser scanning confocal microscope unit. We acquired thin, single confocal slices and subsequently measured the number of particles using the Volocity software. In the second case, we used the Zeiss AxioImager Z2 epifluorescence microscope and the EVOS Cell Imaging Systems and acquired images of whole animals using a 5× or a 4× lens. Then, we quantified the mean fluorescence intensity of each animal using the Image J software. The co‐localization analysis of storage and degradation bodies was performed by acquiring 2 μm slices in Zeiss LSM 710 confocal microscope and LEICA TCS SP8 Laser scanning confocal microscope unit using the 63× lens. The 2 μm thickness of the stack is small enough to enable us to capture several foci in more than one slice. Co‐localization analysis and Pearson's correlation coefficient were measured with the Volocity software. The distances of storage/degradation bodies from mitochondria were acquired in the Zeiss LSM 710 confocal microscope and LEICA TCS SP8 Laser scanning confocal microscope unit using a 63× lens. Single slices were acquired and distances between each foci from adjacent mitochondria were measured with the Volocity software. For confocal microscopy experiments, we immobilized animals on 5% agarose pads with Nanobeads 100 nm (Polysciences, 6401015) in M9 to avoid the use of anaesthetics. For epifluorescence microscopy, we anesthetized animals using levamisole 20 mM (Sigma Aldrich, 196142).
Measurement of the distances between foci and mitochondria
Using the Volocity 6.3 software, we designed a different protocol for each type of foci protocol using the “Measurements” tab; first, the tool “find objects” so that the two different populations (the foci and mitochondria) are discriminated by the software. Next, we used the option “Clip toROIs” so that only areas of interest are measured and areas containing unspecific fluorescent signal (i.e. intestinal autofluorescence) are excluded from our analysis. Last, we selected the tool “measure distances” and then the option to measure distances from the edge of one object to the edge of the other. Last, the protocol ran and we got an automated calculation of the distance each foci acquires from mitochondria in μm.
Mitochondrial imaging
To monitor the mitochondrial network formation, we acquired single slices in the 40× lens of a confocal microscope using the SJ4103 strain which expresses GFP in the matrix of body wall muscle cell mitochondria. To monitor the relative positions of mitochondria and storage/degradation bodies, we either used the SJ4103 strain (crossed with the N2;Ex[p edc‐3 EDC‐3::DsRed;pRF4] strain) or stained mitochondria with specific dyes. In these experiments, we acquired images of single slices using a 63× lens of the confocal microscope. To measure mitochondrial mass, we used several approaches: (1) we measured the mean fluorescence intensity in whole animals expressing GFP in the matrix of intestinal mitochondria with the strain SJ4143. Given that in principle the intestine is the most abundant tissue in C. elegans, measuring mitochondrial mass in this tissue would be representative of the global mitochondrial mass. Mean fluorescence intensity was quantified using the Image J software. (2) Using the same strain we acquired single confocal slices of defined size and measured fluorescence intensity in a certain number of slices using the Volocity software. (3) We used the same approach as in “2” but this time we stained animals with Mitotracker Green and measured fluorescence in the hypodermis as Mitotracker staining stains more efficiently in that tissue.
Stress assays
Heat stress
We performed a heat shock assay to measure thermotolerance of dcap‐2(RNAi)‐ and ntl‐2(RNAi)‐treated animals versus control. We bleached animals and put the isolated eggs on plates seeded with HT115 bacteria expressing the desired RNAi. We transferred animals on fresh seeded plates every 2 days and at day 5 of adulthood we performed the heat stress assay. We first incubated seeded plates without worms at 37°C for 1.5 h to pre‐warm them. Afterwards, we placed the animals on the pre‐warmed plates (we placed around 40 animals on each plate and used at least three plates per condition in each experiment). Next, we placed the plates containing the animals in the incubator at 37°C for 5 h. Afterwards, we put the animals in a 20°C incubator and let them recover for at least 12 h. The next day (after 12 h), we scored for deaths and we continued scoring every 24 h until all animals died. Four independent experiments were performed.
Paraquat stress
For the oxidative stress resistance assay, we placed 5‐day‐old adult animals on paraquat‐containing plates (we added paraquat (Sigma Aldrich, 856177) to the top of pre‐seeded plates) at a final concentration of 8 mM. We placed 40 animals per plate and at least three plates for every condition tested. Every 3 days we placed animals on fresh plates and measured death events every day until all animals died. Four independent experiments were performed.
CCCP stress
For the CCCP stress resistance assay, we diluted CCCP (Sigma Aldrich, 857815) of an initial concentration of 49 mM in M9 and placed it on the top of pre‐seeded plates at a final concentration of 15 μΜ. Given that CCCP is dissolved in DMSO (AppliChem, A3672,0250), we used DMSO‐containing plates in the same concentration (15 μM) as a control. Forty 5‐day‐old adult animals were placed in each plate and at least three plates were used per condition tested. Death events were monitored every day until all animals died. Four independent experiments were performed.
Western blotting
Sample preparation: Synchronous animal populations were collected and washed in M9 buffer. After washing, two volumes of homogenization buffer (20 mM Tris, pH 7,4, 20 mM NaCl and 1 mM MgCl2) plus complete mini proteinase inhibitor cocktail (Roche) in a final concentration 1× was used. Following which one volume of beads (0.5 mm zirconium oxide beads) was added and samples were placed in the Bullet Blender Homogenizer (Model BT24M, Next Advance, Troy, NY, USA) for 3 min at Speed 10. After homogenization 6×, Laemmli sample buffer with b‐mercaptoethanol was supplemented in a final concentration of 1×. Samples were boiled for 5 min at 95°C. Protein samples were analysed by Tricine‐SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred on nitrocellulose membrane. Boiled samples are stored at −20°C or directly run in a gel. For protein quantification, the following antibodies were used: anti‐GFP (enzyQUEST), anti‐MTCOI (Abcam, ab14705), anti‐alpha tubulin (DSHB, AA4.3 (concentrate)), anti‐ATP5A (ab14748), anti‐MRPL13 (ThermoFisher Scientific # PA5‐51007), anti‐NDUFS8 (abbexa, Catalogue No: abx026882), JLA20 (anti‐actin) (DHSB) and anti‐RFP (enzyQUEST).
Total protein quantification
Synchronous animal populations were collected and washed in M9 buffer. After washing, two volumes of homogenization buffer (20 mM Tris, pH 7.4, 20 mM NaCl and 1 mM MgCl2) plus complete mini proteinase inhibitor cocktail (Roche) in a final concentration of 1× was used. Following which one volume of beads (0.5 mm zirconium oxide beads) was added and samples were placed in the Bullet Blender Homogenizer (Model BT24M, Next Advance, Troy, NY, USA) for 3 min at Speed 10. Total protein concentration in whole‐worm lysates was measured in a NanoDrop Microvolume Spectophotometer.
Mitochondria isolation
Mitochondria isolation was performed as described previously (Palikaras et al, 2015b).
RΝΑ immunoprecipitation
To identify possible MTPT targets bound to storage bodies, we performed RNA immunoprecipitation as follows: first, we incubated in three different 1.5 ml Eppendorf tubes (one for the NTL‐2::GFP sample, one for the gst‐4p::GFP control sample and one for the unc‐119(ed3)III sample used as no GFP control) 40 μl of magnetic beads (Merc, Millipore, LSKMAGAG02) with a‐GFP diluted in PBST overnight at 4°C rotating. Next, we used a magnetic stand and washed the beads bound with a‐GFP twice in wash buffer (50 mM Hepes, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, proteinase inhibitor and 1/100 PMSF). We removed wash buffer and added the whole‐worm protein extracts (diluted in 50 mM Hepes, 2 M NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, proteinase inhibitor and 1/100 PMSF, 0.1% NP‐40 and 1/1000 Protector RNAse Inhibitor (Roche, 3335399001)) in the antibody‐bound magnetic beads pellet. We incubated 5 h in the cold room rotating. After 5 h, the Eppendorf tubes were placed on a magnetic stand and supernatant was removed. Beads were washed twice with wash buffer plus NP‐40 0, 1%. Wash buffer was removed and 2× Laemmle sample buffer containing b‐mercaptoethanol was added and boiled at 95°C form 5 min. Eppendorf tubes were placed on a magnetic stand and supernatant was collected in new tubes. We then added 1 ml TRIzol to the isolated supernatant and continued with the RNA extraction protocol. Glycoblue (Invitrogen, ThermoFisher Scientific, AM9515) was added before isopropanol. Whole‐animal protein extracts were prepared by gridding worms that were flash‐freezed in liquid nitrogen (worm pellets were diluted in 50 mM Hepes, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% GLYCEROL, 1 mM DTT, proteinase inhibitor and 1/100 PMSF, 1% NP‐40). When a fine powder was produced, we stopped gridding and added the powder in a 15 ml Eppendorf and let it melt in ice. We centrifuged at 9,300 g at 4°C and kept supernatant for the rest of the protocol described above.
Lifespan assays
Lifespan assays were performed at 20°C unless noted otherwise. Synchronous animal populations were generated by hypochlorite treatment of gravid adults to obtain tightly synchronized embryos that were allowed to develop into adulthood under appropriate, defined conditions. When they reach the L4 stage, 20–25 worms were placed on NGM plates containing 2 mM IPTG and seeded with HT115(DE3) bacteria transformed with either the pL4440 vector (empty vector) as a control or the test RNAi construct. At least a total of 150–200 animals were tested per condition in each experiment. The day of egg collection and initiation of RNAi is t = 0. Animals were transferred to fresh plates every 2 days thereafter and examined every day for touch‐provoked movement and pharyngeal pumping, until death. Worms that died owing to internally hatched eggs, an extruded gonad or desiccation due to crawling on the edge of the plates were censored and incorporated as such into the dataset. Each survival assay was repeated at least twice, and figures represent typical assays. Survival curves were created using the product‐limit method of Kaplan and Meier. The log‐rank (Mantel–Cox) test was used to evaluate differences between survivals and determine P‐values. We used the Prism software package (GraphPad Prism 8 Software) for statistical analysis and to determine lifespan values. Full lifespan statistics are summarized in Table EV4.
Movement tracking
Mobility capacity of 15‐day‐old animals was recorded while they were on NGM plates seeded with bacteria transformed with either the PL4440 vector (empty vector) or the test RNAi construct. To ensure that all subjects are alive, animals were examined for touch‐provoked movement and pharyngeal pumping before the videos were captured.
Mitochondrial staining
For staining of the animals with Mitotracker Green FM (Molecular Probes, Invitrogen, M7514), Mitotracker Red CM‐H2X ROS (Molecular Probes, Invitrogen, M7513), Mitotracker Deep Red FM (Molecular Probes, Invitrogen, M22426), DiO6(3) (Invitrogen, Catalogue number: D275), BioTracker ATP‐Red live‐cell dye (Merck, SCT045) and TMRE (Molecular Probes, Invitrogen, T669), we diluted the stain in M9 and added it to the top of pre‐seeded plates in a final concentration of 0.15 μM. We let the worms feed overnight and the next day we measured parameters of interest as described previously.
Measurement of basal and maximal respiration
Assessment of OCR under different treatments was performed strictly at day 1 of adulthood using a Seahorse XFe96 Analyzer (Agilent Technologies Inc., USA) as previously described (Koopman et al, 2016). Briefly, approximately 25 adults per well were transferred in a Seahorse analyser microplate (Utility plate). CCCP (20 μΜ from 20 mM stock) and sodium azide (40 mM from 400 mM stock) were injected at the indicative time points to assess different aspects of animals' respiration. The first measurements of OCR were excluded from analysis and only the time points 3–5 before CCCP injection were considered for basal OCR calculation. Time points 7–9 between CCCP and sodium azide (NaN3) injections were considered for maximal OCR calculation. After the measurement, the number of worms per well was estimated and used to normalize oxygen consumption values for each time point.
Microparticle bombardment
The Biolistic PDS‐1000/He particle delivery system (BioRad) was used for biolistic transformation. For generating transgenic strains, we linearized 10–15 μg plasmid DNA, which was bombarded onto unc‐119(ed3)III L4/adult hermaphrodites. Animals were grown on NGM plates seeded with Na22 bacteria and the rest of the procedure was done according to Isik and Berezikov (2013).
Quantification and statistical analysis
Quantification was performed using the Image J software (NIH, http://imagej.nih.gov/ij/) and the Volocity 6.3 software as specified in the Figure legends. Statistical analyses were carried out using the Prism software package (GraphPad Software Inc., San Diego, USA) and the Microsoft Office 2010 Excel software package (Microsoft Corporation, Redmond, WA, USA). The statistical tests applied for each experiment are specified in the Figure legends. For all experiments, at least three independent experiments were performed unless otherwise noted. The brightness and contrast features for part of the images have been processed in PowerPoint. In the latter case, images of the same experimental setup were equally handled.
Author contributions
Ioanna Daskalaki: Conceptualization; resources; formal analysis; validation; investigation; visualization; methodology; writing – original draft. Maria Markaki: Conceptualization; formal analysis; supervision; investigation; methodology; writing – original draft; writing – review and editing. Ilias Gkikas: Formal analysis; validation; investigation; visualization; methodology. Nektarios Tavernarakis: Conceptualization; resources; formal analysis; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Movie EV1
Movie EV2
Movie EV3
PDF+
Acknowledgements
We thank Angela Pasparaki for valuable support with confocal microscopy and Konstantinos Palikaras for providing us the daf‐2(e1370);Is[p gst‐4 GFP] and the unc‐119(ed3);Ex[ punc‐119 mtRosella;unc‐119(+)] strain. We also thank DikaiaTsagkari and Kostas Kounakis for providing us with the nhr‐49 RNAi construct. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by the European Union Horizon 2020 FETOPEN, project “Dynamic”, under the grant agreement “GA‐863203”, the European Research Council, under the grant agreement “ERC‐GA695190‐MANNA”, the National Research Infrastructure Program “BIOIMAGING‐GR” (MIS5002755), which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020), and the European Commission Research Executive Agency Excellence Hub “CHAngeing” (GA‐101087071) to NT.
The EMBO Journal (2023) 42: e112446
Data availability
This study includes no data deposited in external repositories.
References
- Aizer A, Kalo A, Kafri P, Shraga A, Ben‐Yishay R, Jacob A, Kinor N, Shav‐Tal Y (2014) Quantifying mRNA targeting to P‐bodies in living human cells reveals their dual role in mRNA decay and storage. J Cell Sci 127: 4443–4456 [DOI] [PubMed] [Google Scholar]
- Besse F, Ephrussi A (2008) Translational control of localized mRNAs: restricting protein synthesis in space and time. Nat Rev Mol Cell Biol 9: 971–980 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykov YS, Rapaport D, Herrmann JM, Schuldiner M (2020) Cytosolic events in the biogenesis of mitochondrial proteins. Trends Biochem Sci 45: 650–667 [DOI] [PubMed] [Google Scholar]
- Castello PR, Drechsel DA, Patel M (2007) Mitochondria are a major source of paraquat‐induced reactive oxygen species production in the brain. J Biol Chem 282: 14186–14193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart MA (2016) The CCR4‐NOT complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip Rev RNA 7: 438–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Vera M, Gandin V, Singer RH, Tutucci E (2021a) Author correction: intracellular mRNA transport and localized translation. Nat Rev Mol Cell Biol 22: 505 [DOI] [PubMed] [Google Scholar]
- Das S, Vera M, Gandin V, Singer RH, Tutucci E (2021b) Intracellular mRNA transport and localized translation. Nat Rev Mol Cell Biol 22: 483–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliyahu E, Pnueli L, Melamed D, Scherrer T, Gerber AP, Pines O, Rapaport D, Arava Y (2010) Tom20 mediates localization of mRNAs to mitochondria in a translation‐dependent manner. Mol Cell Biol 30: 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk JA, Schnellmann RG (2012) Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol 302: F853–F864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke S, Wu ZH, Klinkenberg M, Sun YP, Auburger G, Guo S, Lu BW (2015) PINK1 and parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metab 21: 95–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold VA, Chroscicki P, Bragoszewski P, Chacinska A (2017) Visualization of cytosolic ribosomes on the surface of mitochondria by electron cryo‐tomography. EMBO Rep 18: 1786–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A (2017) Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther 23: 5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureev AP, Shaforostova EA, Popov VN (2019) Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC‐1alpha signaling pathways. Front Genet 10: 435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C (2007) Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans . Aging Cell 6: 95–110 [DOI] [PubMed] [Google Scholar]
- Hardie DG (2011) AMP‐activated protein kinase‐an energy sensor that regulates all aspects of cell function. Genes Dev 25: 1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y, Honda S (1999) The daf‐2 gene network for longevity regulates oxidative stress resistance and Mn‐superoxide dismutase gene expression in Caenorhabditis elegans . FASEB J 13: 1385–1393 [PubMed] [Google Scholar]
- Huang L, Mollet S, Souquere S, Le Roy F, Ernoult‐Lange M, Pierron G, Dautry F, Weil D (2011) Mitochondria associate with P‐bodies and modulate microRNA‐mediated RNA interference. J Biol Chem 286: 24219–24230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubstenberger A, Courel M, Benard M, Souquere S, Ernoult‐Lange M, Chouaib R, Yi Z, Morlot JB, Munier A, Fradet M et al (2017) P‐body purification reveals the condensation of repressed mRNA regulons. Mol Cell 68: 144–157 [DOI] [PubMed] [Google Scholar]
- Isik M, Berezikov E (2013) Biolistic transformation of Caenorhabditis elegans . Methods Mol Biol 940: 77–86 [DOI] [PubMed] [Google Scholar]
- Ito K, Inoue T, Yokoyama K, Morita M, Suzuki T, Yamamoto T (2011) CNOT2 depletion disrupts and inhibits the CCR4‐NOT deadenylase complex and induces apoptotic cell death. Genes Cells 16: 368–379 [DOI] [PubMed] [Google Scholar]
- Joseph AM, Hood DA (2014) Relationships between exercise, mitochondrial biogenesis and type 2 diabetes. Med Sport Sci 60: 48–61 [DOI] [PubMed] [Google Scholar]
- Koopman M, Michels H, Dancy BM, Kamble R, Mouchiroud L, Auwerx J, Nollen EAA, Houtkooper RH (2016) A screening-based platform for the assessment of cellular respiration in Caenorhabditis elegans . Nature Protoc 11: 1798–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N, Tavernarakis N (2017) Protein synthesis rate assessment by fluorescence recovery after photobleaching (FRAP). Bio Protoc 7: e2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Brielmann RM, Neto MF, Lin YF, Haynes CM, Morimoto RI (2017) Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep 21: 1481–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Na ZK, Slavoff SA (2018) P‐bodies: composition, properties, and functions. Biochemistry 57: 2424–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglioni S, Mello DF, Schiavi A, Meyer JN, Ventura N (2019) Mitochondrial bioenergetic changes during development as an indicator of C. elegans health‐span. Aging 11: 6535–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono Y, Hirashima S, Ishihara N, Kusukawa J, Nakamura KI, Ohta K (2018) Uncoupled mitochondria quickly shorten along their long axis to form indented spheroids, instead of rings, in a fission‐independent manner. Sci Rep 8: 350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nousch M, Techritz N, Hampel D, Millonigg S, Eckmann CR (2013) The CCR4‐NOT deadenylase complex constitutes the main poly(A) removal activity in C. elegans . J Cell Sci 126: 4274–4285 [DOI] [PubMed] [Google Scholar]
- Ozgur S, Basquin J, Kamenska A, Filipowicz W, Standart N, Conti E (2015) Structure of a human 4E‐T/DDX6/CNOT1 complex reveals the different interplay of DDX6‐binding proteins with the CCR4‐NOT complex. Cell Rep 13: 703–711 [DOI] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N (2015a) Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ 22: 1399–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N (2015b) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans . Nature 521: 525–U241 [DOI] [PubMed] [Google Scholar]
- Picard M, McEwen BS, Epel ES, Sandi C (2018) An energetic view of stress: focus on mitochondria. Front Neuroendocrinol 49: 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisano A, Cerbelli B, Perli E, Pelullo M, Bargelli V, Preziuso C, Mancini M, He L, Bates MG, Lucena JR et al (2016) Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end‐stage ischemic heart failure. Cardiovasc Pathol 25: 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Lauterbach M, Schuman EM (2019) Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell 176: 73–84 [DOI] [PubMed] [Google Scholar]
- Re A, Waldron L, Quattrone A (2016) Control of gene expression by RNA binding protein action on alternative translation initiation sites. PLoS Comput Biol 12: e1005198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA et al (2007) Aging‐associated reductions in AMP‐activated protein kinase activity and mitochondrial biogenesis. Cell Metab 5: 151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieckher M, Markaki M, Princz A, Schumacher B, Tavernarakis N (2018) Maintenance of proteostasis by P body‐mediated regulation of eIF4E availability during aging in Caenorhabditis elegans . Cell Rep 25: 199–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T (2016) The mitochondrial basis of aging. Mol Cell 61: 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM (2006) Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127: 49–58 [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Troulinaki K, Tavernarakis N (2007) eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans . Nature 445: 922–926 [DOI] [PubMed] [Google Scholar]
- Temme C, Simonelig M, Wahle E (2014) Deadenylation of mRNA by the CCR4‐NOT complex in Drosophila: molecular and developmental aspects. Front Genet 5: 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Schmich F, Srivatsa S, Weidner J, Beerenwinkel N, Spang A (2018) Correction: context‐dependent deposition and regulation of mRNAs in P‐bodies. Elife 7: e41300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CC, Jan CH, Weissman JS (2014) Targeting and plasticity of mitochondrial proteins revealed by proximity‐specific ribosome profiling. Science 346: 748–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JY, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, Bagci H, Rathod B, MacLeod G, Eng SWM et al (2018) High‐density proximity mapping reveals the subcellular organization of mRNA‐associated granules and bodies. Mol Cell 69: 517–532 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen Y, Gucek M, Xu H (2016) The mitochondrial outer membrane protein MDI promotes local protein synthesis and mtDNA replication. EMBO J 35: 1045–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]