

Abstract
Background—
Cyclic nucleotides play critical roles in cardiovascular biology and disease. Phosphodiesterase 10A (PDE10A) is able to hydrolyze both cAMP and cGMP. PDE10A expression is induced in various human tumor cell lines and PDE10A inhibition suppresses tumor cell growth. Chemotherapy drug such as doxorubicin (DOX) is widely used in chemotherapy. However, cardiotoxicity of DOX remains to be a serious clinical complication. In the current study, we aim to determine the role of PDE10A and the effect of PDE10A inhibition on cancer growth and cardiotoxicity induced by DOX.
Methods—
We used global PDE10A knockout (PDE10A-KO) mice and PDE10A inhibitor TP-10 to block PDE10A function. DOX-induced cardiotoxicity was evaluated in C57Bl/6J mice and nude mice with implanted ovarian cancer xenografts. Isolated adult mouse cardiomyocytes (CMs) and a human ovarian cancer cell line were used for in vitro functional and mechanistic studies.
Results—
We found that PDE10A deficiency or inhibition alleviated DOX-induced myocardial atrophy, apoptosis, and dysfunction in C57Bl/6J mice. RNA-seq study revealed a number of PDE10A-regulated signaling pathways involved in DOX-induced cardiotoxicity. PDE10A inhibition increased the death, decreased the proliferation, and potentiated the effect of DOX on various human cancer cells. Importantly, in nude mice with implanted ovarian cancer xenografts, PDE10A inhibition attenuated tumor growth while protecting DOX-induced cardiotoxicity. In isolated CMs, PDE10A contributed to DOX-induced CM death via increasing Top2β expression, mitochondrial dysfunction, and DNA damage by antagonizing cGMP/PKG signaling. PDE10A contributed to CM atrophy via potentiating FoxO3 signaling via both cAMP/PKA and cGMP/PKG dependent signaling.
Conclusions—
Taken together, our study elucidates a novel role for PDE10A in cardiotoxicity induced by DOX and cancer growth. Given that PDE10A has been already proven to be a safe drug target, PDE10A inhibition may represent a novel therapeutic strategy in cancer therapy, with effects preventing DOX-induced cardiotoxicity and simultaneously antagonizing cancer growth.
Keywords: Cardio-Oncology, Cardiotoxicity, Heart Failure, Cyclic nucleotide, phosphodiesterase, doxorubicin, cardiotoxicity, ovarian cancer
Graphical Abstract
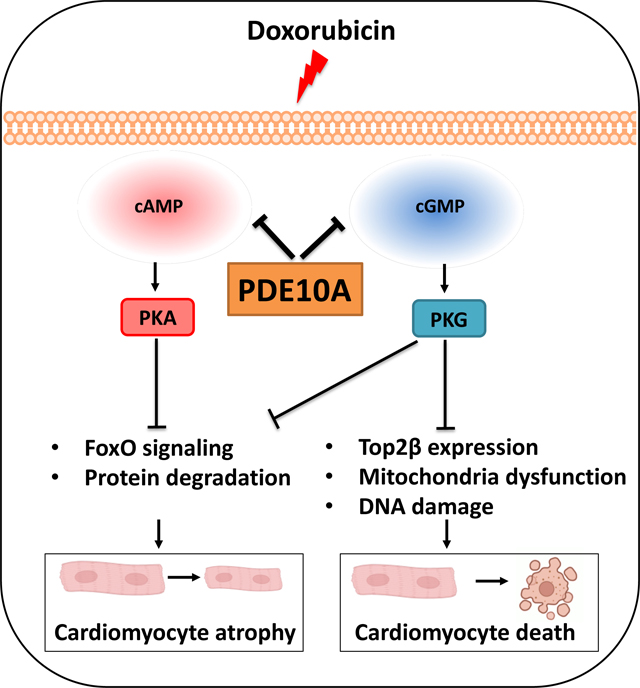
Introduction
Cardiovascular disease is the second leading cause of morbidity and mortality in cancer patients after recurrent malignancy 1. This recognition has stimulated the communication between oncology and cardiovascular communities and boosted a rapid development of cardio-oncology (or onco-cardiology). The goal of cardio-oncology is to improve patient outcomes through the detection, monitoring and treatment of cardiotoxicity occurring as a side effect of cancers and chemotherapies.
Chemotherapeutic drugs, such as anthracycline doxorubicin (DOX), are effective against a variety of cancers including breast, prostate and ovarian cancers. Despite being widely used in clinics, cardiotoxicity remains a serious problem for patients with cancer and cancer survivors. Acute cardiotoxicity occurs within days of treatment, which is characterized by depression of myocardial contractility 2. Chronic cardiotoxicity is associated with cardiomyopathy and heart failure 2. DOX-induced cardiotoxicity is associated with increased protein degradation and downregulation of proteins involved in sarcomere, contractile apparatus, mitochondria, and sarcoplasmic reticulum, therefore causing cardiac atrophy and cardiac dysfunction. DOX also promotes reactive oxygen species (ROS) production and DNA damage, leading to cardiomyocyte (CM) death. Similar effects and mechanisms may also be shared by other types of cancer therapeutic agents. Despite the growing interest in assessing the cardiac effects and understanding the mechanisms, there is no standard effective therapy for the prevention of cardiotoxicity induced by chemotherapy.
Cyclic nucleotides, cAMP and cGMP, regulate numerous biological processes in the heart, including inotropic/chronotropic functions, metabolism, cell growth/survival and structural remodeling 3, 4. cAMP/cGMP degradation is catalyzed by a large family of phosphodiesterases (PDEs) with more than 100 variants derived from 21 genes and grouped into 11 broad families (PDE1-PDE11) 5. Among 11 PDE families, PDE1–5, and PDE8–10 have been reported in the heart 6. Each regulates unique cyclic nucleotide signaling pathways and play distinct roles in different cardiac cells and cardiac diseases. PDE10A is able to hydrolyze both cAMP and cGMP 7–9. Its high expression in medium spiny neurons of the human striatum led to the development of several PDE10A inhibitors with the intent to treat various psychiatric/neurodegenerative disorders 10, 11. Importantly, several PDE10A inhibitors have been previously tested in humans for the treatment of schizophrenia and Huntington’s disease and successfully passed phase I (safety) clinical trials. Although PDE10A expression is normally low in peripheral tissues, PDE10A can be induced in a number of peripheral tissues during various pathological conditions. For example, we have recently reported the induction and function of PDE10A in cardiac myocytes and fibroblasts of diseased hearts with pathological cardiac remodeling and heart failure induced by chronic pressure overload and neurohormonal activation 12. PDE10A is also induced in various cancers including colorectal and lung cancer, and involved in cancer growth 13, 14, which leads to a proposed clinical trial for using PDE10A inhibitors against cancers (NCT03786484). However, the role of PDE10A in cardiac cell viability remains unknown. Given the relationship of PDE10A with the heart and cancers, in the current study, we aim to explore the role of PDE10A in tumor growth and cardiotoxicity with chemotherapy. Using genetic and pharmacological approaches, we demonstrated the protective effect of PDE10A inactivation against DOX-induced cardiomyopathy and dysfunction while suppressing cancer growth, e.g. human ovarian tumor grafts. We further reported a critical role and potential mechanisms by which PDE10A promotes CM death and atrophy. Collectively, these results provide proof-of-concept that targeting PDE10A can mitigate cardiovascular complications in cancer therapies.
Materials and Methods
Data Availability
Detailed experimental methods and Major Resource Table are provided in the Supplemental Material. Data will be made available upon reasonable request, by contacting the corresponding author. Raw and processed RNA-seq are available in the NCBI GEO database (GSE224157). The graphic abstract was generated using BioRender.com.
Animal models
All animal procedures were performed in accordance with the National Institutes of Health (NIH) and University of Rochester institutional guidelines. For experiments with genetically modified mice, age/sex/genetic background matched mice were randomly separated into indicated groups. For the acute DOX-induced cardiac dysfunction model, Male or female mice received pretreatment of TP-10 (3.2mg/kg/day) or the same volume of vehicle (10% DMSO in 40% β-cyclodextrin) via subcutaneous injection before DOX treatment for 2 days via intra-peritoneal injection and then continuously received the same treatment for another 5 days. TP-10 was dissolved in a vehicle consisting of 10% DMSO and 40% β-cyclodextrin. After pretreatment, mice received DOX treatment (15mg/kg) in one bolus via intra-peritoneal injection for 7 days. For the chronic DOX-induced cardiac dysfunction model, Male or female C57BL/6J PDE10A-WT or PDE10A-KO mice at the age of 12 weeks were treated with DOX (4 mg/kg) on days 0, 7, and 14. For the ovarian tumor xenograft model, A2780 cells (5 × 106 cells) with expression of luciferase were mixed with 50 μL matrigel matrices. The mixture of cells was then injected into flanks of female BALB/cAnNCr-nu/nu mice at the age of 6 weeks. 10 days post-inoculation of the tumor cells, the animals were randomly divided into four groups and received saline (intra-peritoneal injection), DOX (1.5mg/kg/day, intra-peritoneal injection), vehicle (10% DMSO in 40% β-cyclodextrin v/v, subcutaneous injection), TP-10 succinate (4.5 mg/kg/day, dissolved in a vehicle consisting of 10% DMSO and 40% β-cyclodextrin, subcutaneous injection), or DOX plus TP-10 daily for 2 weeks. Tumor sizes were measured twice weekly and tumor volume was calculated by ab2/2 where “a” and “b” are the long and short axes of tumor. Cardiac function was evaluated via non-invasive echocardiography one day before cell injection, one day before drug treatment, and at 1 and 2 weeks after drug treatment.
Adult mouse CM isolation and culture
Adult mouse CMs were isolated by enzymatic dissociation using collagenase type II (Worthington) in a Langendorff perfusion system as previously described 15–17. CMs were cultured in the presence of blebbistatin (a myosin II inhibitor) to block myocyte contraction and to extend their survival during the culture as described 15, 17, 18.
Statistics
The exact sample size in each group is listed in Supplementary Table S5-6 or mentioned in corresponding figure legends. All data are presented as mean ± SEM or median with interquartile range (IQR) based on data distribution. We used the Shapiro-Wilk test and Brown-Forsythe test to help determine the assumptions of normality and variance equality, respectively. Proper corrections were applied to control the inflation of type I error in multiple post-hoc comparisons, and the corresponding adjusted p-values were reported. All parametric and non-parametric tests were two-sided with a significance level set at 5%. Linear mixed effect models were used to test the differences of cell surface area, cell death (as measured by trypan blue staining and LDH cytotoxicity assay), and quantification of fluorescence intensity among groups, with the cell providing animal (mouse) accounted as random effect 19. The model was fitted using the lmerTest packages in R 4.2.2. (https://www.R-project.org/). All the other statistical analyses were conducted using GraphPad Prism 8.0. Detailed statistical methods were described in figure legends.
Results
PDE10A deficiency or inhibition alleviates acute DOX-induced cardiotoxicity in mice.
We observed an upregulation of PDE10A mRNA (Figure 1A) and PDE10A protein levels (Figure 1B) in mouse hearts after DOX treatment. We further evaluated the association between PDE10A levels and DOX treatment using previous published RNA-sequencing (RNA-Seq) data from human embryonic stem cell (ESC)-derived cardiomyocytes (CMs) 20, human induced pluripotent stem cell (iPSC)-derived CMs 21, and neonatal rat ventricular myocytes (NRVMs). We found that PDE10A mRNA levels significantly changed upon DOX stimulation (Supplemental Table S1). These results imply the association between PDE10A and DOX-induced cardiotoxicity. We then evaluated the effects of PDE10A deficiency or PDE10A inhibitor TP-10 on DOX-induced cardiomyopathy and cardiac dysfunction. Using TP-10 in both PDE10A wildtype (PDE10A-WT) and PDE10A knockout (PDE10A-KO) mice allows us not only to evaluate the pharmacological effect of TP-10 but also to validate the specificity of TP-10 on targeting PDE10A. An acute DOX model was first used, in which male or female C57BL/6J PDE10A-WT or -KO mice at the age of 12 weeks were treated with vehicle or PDE10A inhibitor TP-10 subcutaneously 2 days prior to a single dose of saline or DOX, and continued for 1 week (Figure 1C). Consistent with clinical evidence of weight loss in DOX-treated patients, we observed that DOX induced a significant decrease in body weight in mice with both genotypes (Supplemental Figure S1A–B). As anticipated, in PDE10A-WT hearts, DOX treatment induced cardiac atrophy, as evidenced by significant reductions in global heart size (Figure 1D, and Supplemental Figure S1C), and ratio of ventricular weight and tibia length (HW/TL) (Figure 1E). These DOX-induced changes in PDE10A-WT hearts were markedly attenuated by PDE10A inhibitor TP-10 treatment or in PDE10A-KO hearts (Figure 1D–E, and Supplemental Figure S1C). Echocardiography was performed before and 1 week after DOX administration to monitor the progression of cardiac structural and functional changes (Figure 1F–G, Supplemental Figure S1D–F, and Supplemental Table S2). There was a significant reduction of cardiac function in PDE10A-WT mice after DOX treatment as assessed by fraction shortening and ejection fraction, which was significantly improved by TP-10 or in PDE10A-KO mice (Figure 1F–G, Supplemental Figure S1D–F, and Supplemental Table S2). In addition, cardiac injury markers such as plasma concentrations of cardiac troponin I changed in a manner consistent with observed functional impairments after DOX stimulation (Figure 1H), which was alleviated by TP-10 and PDE10A deficiency (Figure 1H). Histological analysis of myocardial surface areas (CSA) further showed CM atrophy induced by DOX in PDE10A-WT mice, which was significantly attenuated by TP-10 or in PDE10A-KO mice (Figure 1I–J). Next, we examined myocyte apoptosis by TUNEL staining in the heart sections. We found that myocardial apoptotic cell number was markedly increased by DOX treatment in PDE10A-WT mice (Figure 1K–L). PDE10A deficiency or TP-10 protected from DOX-induced myocardial apoptosis (Figure 1K–L). Additionally, we observed that acute DOX-induced cardiac dysfunction and atrophy is more severe in male mice than in female mice (Supplemental Figure S2A–T). In conclusion, these results indicate that PDE10A inhibition or deficiency significantly suppressed DOX-induced cardiac atrophy and dysfunction in both male and female mice. Importantly, TP-10 had no further effect on PDE10A-KO mice, suggesting the effect of TP-10 is through PDE10A inhibition.
Figure 1: PDE10A inhibition or deficiency alleviates doxorubicin-induced cardiac dysfunction in vivo.

Male or female C57BL/6J PDE10A-WT or PDE10A-KO mice at the age of 12 weeks were treated with vehicle or PDE10A inhibitor TP-10 (3.2 mg/kg/day) subcutaneously 2 days prior to a single dose of saline or DOX (15mg/kg), and continued for 1 week. (A) Pde10a mRNA levels assessed by qPCR in the hearts of PDE10A-WT mice after saline or DOX treatment as indicated, normalized to Gapdh. n = 5 for PDE10A-WT/Veh; n = 10 for PDE10A-WT/DOX. (B) PDE10A protein levels measured by western blotting in mouse tissues from PDE10A-WT mice treated with saline or DOX, bar graph shows the average of n=4 WT/Veh; n=5 WT/DOX. (C) Experiment timeline. (D) Representative images of hematoxylin and eosin staining in mouse hearts after drug treatment. Scale bars: 1000 μm. (E) Quantification of heart weight/tibia length. (F-G) Echocardiography was performed 1 week after DOX administration to monitor the progression of cardiac structural and functional changes. (F) Representative M-mode echocardiographic images of each study group at 1 week point. (G) Percent fractional shortening at 1 week point. (H) Plasma troponin I levels in each treatment group. (I) Representative images of wheat germ agglutinin (WGA)-fluorescein isothiocyanate-staining in mouse hearts, showing cardiomyocyte (CM) cross-sectional area (CSA). Scale bars: 50 μm. (J) Quantitative data of CM atrophy assessed by CSA, n = 6–15 hearts per group with 300–800 CMs analyzed per heart. (K) Representative image of TUNEL staining of heart sections. White arrows indicate TUNEL-positive CM. Scale bars = 50 μm. (L) Quantification of TUNEL staining, n = 6–12 hearts per group with 10 random fields analyzed per heart. Data were represented as mean ± SEM (A, E, H, and L) or median with IQR (B, G and J). Statistics: Welch’s t test in A, Mann-Whitney test in B, two-way ANOVA with Holm-Sidak post-hoc test for 3 comparisons in E and H, Kruskal-Wallis’s test with Dunn’s corrections for 3 comparisons in G, mixed effect model with Sidak corrections for 3 comparisons in J, and Welch ANOVA with Dunnett’s T3 corrections for 3 comparisons in L. Animal numbers: E, G-H, and J, n = 3–19.
PDE10A deficiency alleviates chronic DOX-induced cardiotoxicity and dysfunction in mice.
Next, we explored the role of PDE10A in chronic DOX-induced cardiotoxicity. Male or female C57BL/6J PDE10A-WT or PDE10A-KO mice at the age of 12 weeks were treated with DOX on days 0, 7, and 14 (Figure 2A). Consistent with the acute DOX model, we observed that chronic DOX treatment induced a significant decrease in body weight in both genotypes (Supplemental Figure S3A–B). We also observed a progressive worsening of multiple hallmark features of cardiac atrophy and dysfunction, including global heart size (Figure 2B, and Supplemental Figure S3C), HW/TL (Figure 2C), fractional shortening (%FS) (Figure 2D–E, and Supplemental Table S3), ejection fraction (%EF) (Supplemental Figure S3D, and Supplemental Table S3), plasma concentrations of cardiac troponin I (Figure 2F), CM atrophy (Figure 2G–H), CM apoptosis (Figure 2I), and cardiac fibrosis (Figure 2J–K). All these DOX-induced cardiac changes were significantly reduced in PDE10A-KO mice (Figure 2B–K, Supplemental Figure S3C–D, and Supplemental Table S3). We also observed that chronic DOX-induced cardiac dysfunction and atrophy is more severe in male mice than in female mice (Supplemental Figure S4A–R). Taken together, these results indicate that PDE10A deficiency significantly inhibits DOX-induced cardiotoxicity and dysfunction.
Figure 2: PDE10A deficiency alleviates DOX-induced cardiotoxicity and dysfunction in mice.
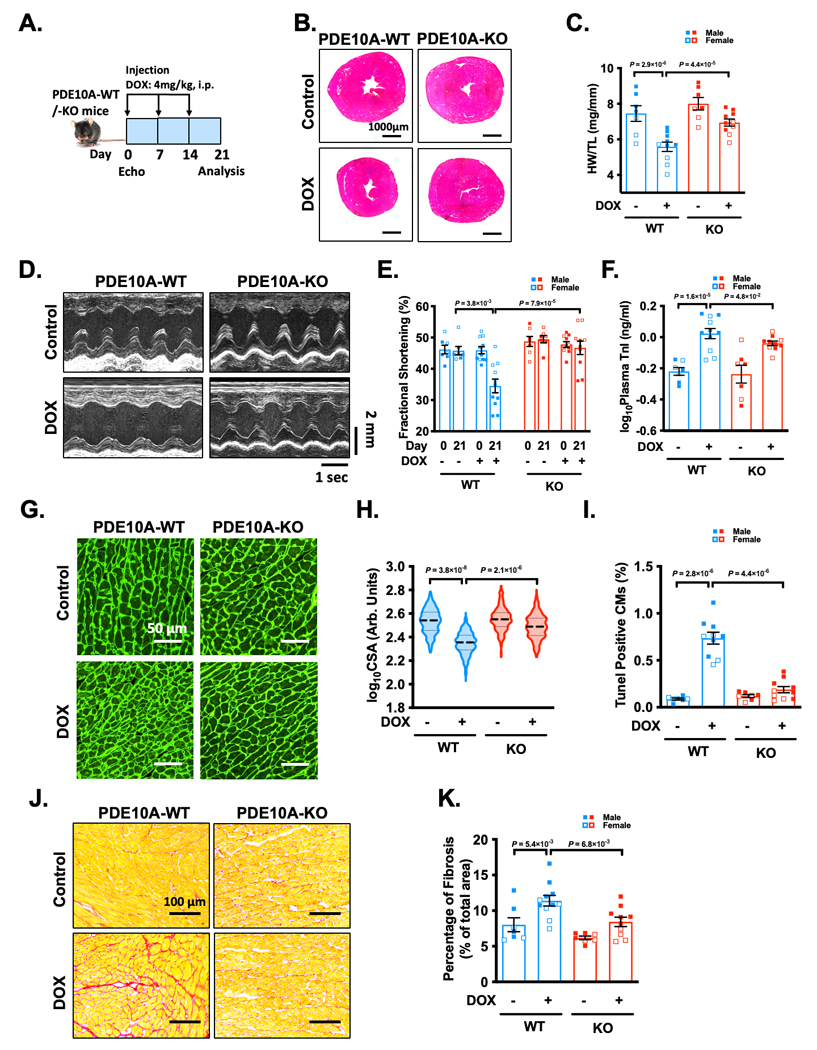
Male or female C57BL/6J PDE10A-WT or PDE10A-KO mice at the age of 12 weeks were treated with DOX (4 mg/kg) on days 0, 7, and 14. (A) Experiment timeline. (B) Representative images of hematoxylin and eosin staining in mouse hearts after drug treatment. Scale bars: 1000 μm. (C) Quantification of heart weight/tibia length. (D-E) Echocardiography was performed prior to the DOX administration as well as 1 week after the last dose of DOX administration. (D) Representative M-mode echocardiographic images of each study group at 21 day point. (E) Percent fractional shortening at 0 or 21 day point. (F) Plasma troponin I levels in each treatment group. (G) Representative images of wheat germ agglutinin (WGA)-fluorescein isothiocyanate-staining in mouse hearts, showing cardiomyocyte (CM) cross-sectional area (CSA). Scale bars: 50 μm. (H) Quantitative data of CM atrophy assessed by CSA, n = 7–10 hearts per group with 300–500 CMs analyzed per heart. (I) Quantification of TUNEL staining, n = 7–10 hearts per group with 10 random fields analyzed per heart. (J) Representative images of heart sections stained with picrosirius red. Red staining shows fibrotic areas. Scale bars: 100 μm. (K) Quantification of total fibrosis. Data were represented as mean ± SEM (C, F, I, and K) or median with IQR (E and H). Statistics: two-way ANOVA with Holm-Sidak post-hoc test for 2 comparisons in C, F and K, Kruskal-Wallis’s test with Dunn’s corrections for 2 comparisons in E, mixed effect model with Sidak corrections for 2 comparisons in H, and Welch ANOVA with Dunnett’s T3 corrections for 2 comparisons in I. Animal numbers: C, E, F, H-I, and K, n = 7–11.
RNA-sequencing identifies transcriptomes that are regulated by DOX and PDE10A.
To support our functional observations of PDE10A at the molecular level, we performed whole genome transcriptome profiling via RNA-seq with heart samples from both PDE10A-WT and PDE10A-KO mice treated with or without DOX. Principle component analysis (PCA) demonstrated distinct patterns of transcriptomes in four groups, including PDE10A-WT/Vehicle (WT/Veh), PDE10A-WT/DOX (WT/DOX), PDE10A-KO/Vehicle (KO/Veh), and PDE10A-KO/DOX (KO/DOX) (Supplemental Figure S5A). We found that biological replicates clustered together among all groups, while each experimental group demonstrated a unique transcriptome profile. Interestingly, the clusters of KO/Veh and KO/DOX are much closer than the clusters of WT/Veh and WT/DOX, indicating the changes from DOX on cardiac transcriptomes were diminished in PDE10A-KO mice (Supplemental Figure S5A).
The comparisons of various groups revealed a total 240 genes that were changed by DOX in the WT group (145 upregulated and 95 downregulated) but reversed in the KO/DOX group, suggesting that these genes are altered by PDE10A (Supplemental Figure S5B). Gene set enrichment analysis (GSEA) revealed that oxidative phosphorylation signaling (P adj. < 0.05) was one of top downregulated gene sets in WT/DOX group versus WT/Veh group, which was not seen between KO/DOX and KO/Veh groups (Supplemental Figure S5C). Abnormal regulation of the oxidative phosphorylation signaling by DOX has been implicated in myocardium damage 22. In addition, we also observed that forkhead box O (FoxO) signaling (P adj. < 0.05) and cell death signaling pathway (P adj. < 0.05) were among the top upregulated gene sets in WT/DOX group versus WT/Veh group but not in KO groups (Supplemental Figure S5C). It has been known that FoxO-mediated activation of ubiquitin/proteasome system (UPS) and/or autophagy play important roles in protein degradation and lead to cardiac atrophy 23. Cell death is involved in DOX-induced cardiomyopathy 24. Therefore, these observations are in line with our findings that PDE10A KO attenuated CM atrophy, CM death, and myocardial damage induced by DOX.
PDE10A inhibition enhances DOX-induced ovarian cancer cell death/apoptosis and proliferation.
Previous studies have revealed a role of PDE10A inhibition in suppressing various cancer cell growth such as colon and lung cancer cell growth 13, 14, 25. We found that DOX and TP-10 increased PDE10A expression in ovarian cancer cell line A2780 (Figure 3A) and decreased PDE10A expression in ovarian cancer cell line OCC1 (Supplemental Figure S6A). We also detected PDE10A expression in human colon cancer cell line HCT116 and two human prostate cancer cell lines C4–2 and PC-3 (Supplemental Figure S6B). We thus determined the effects of DOX, TP-10 or both on the growth and survival of human ovarian cancer. As anticipated, DOX decreased A2780 and OCC1 cell viability in a dose-dependent manner as assessed by Cell Counting Kit-8 (CCK8) assay (Figure 3B, and Supplemental Figure S6C). Similar to DOX, TP-10 also decreased A2780 and OCC1 cell viability in a dose-dependent manner (Figure 3C, and Supplemental Figure S6D). Interestingly, cotreatment with TP-10 resulted in an additive effect on DOX-induced reduction of cell viability (Figure 3D–E, and Supplemental Figure S6E–F). We found similar anti-cancer effect of DOX and TP-10 treatment in HCT116 (Supplemental Figure S6G), C4–2 (Supplemental Figure S6H), and PC-3 (Supplemental Figure S6I). Cell death assay with NucGreen staining consistently showed that DOX or TP-10 individually induced A2780 death and DOX plus TP-10 elicited an additive effect (Figure 3F–G). DOX is known to stimulate DNA damage and cell death, we therefore examined DNA damage by measuring phosphorylation of H2AX at Ser139 (p-H2AX) in the cell nucleus. We found that DOX induced DNA damage in A2780 cells, which was further enhanced by TP-10 treatment (Figure 3H–I). Moreover, we examined cell apoptosis by using Annexin-V-FITC and propidium iodide (PI) staining followed by flow cytometry analysis. We found that DOX or TP-10 induced A2780 cell apoptosis (Figure 3J–K). Cotreatment with TP-10 and DOX had a significantly greater effect on apoptosis compared to DOX or TP-10 alone (Figure 3J–K). Next, we determined the effect of DOX and TP-10 on A2780 cell proliferation. A2780 cells were treated with DOX, TP-10, or the combination of DOX and TP-10. A2780 cell growth was evaluated by different approaches. We found that DOX or TP-10 alone suppressed A2780 cell proliferation as accessed by decreased [3H]-thymidine incorporation (Figure 3L), and decreased expression of nuclear cell proliferation antigen Ki-67 (Supplemental Figure S6J–K). The combinational treatment with DOX and TP-10 had greater effects on A2780 proliferation compared to individual ones (Figure 3L, and Supplemental Figure S6J–K). Taken together, these results support a critical role of PDE10A inhibition in promoting death/apoptosis and suppressing proliferation as well as potentiating the effects of DOX on cancer cells.
Figure 3: PDE10A inhibition enhanced DOX-induced ovarian cancer cell death/apoptosis and growth.

(A) Human ovarian cancer cell line A2780 were treated with vehicle, 300 nM TP-10, 1 μM DOX or combination of TP-10 and DOX as indicated for 6 hours. qPCR analysis of Pde10a, normalized to Gapdh, n = 5 replicates for each group. (B-E) Human ovarian cancer cell line A2780 were treated with vehicle or increasing dose of DOX (B), vehicle or increasing dose of TP-10 (C), 300 nM TP-10 plus vehicle or increasing dose of DOX (D), vehicle, 300 nM TP-10, 1 μM of DOX or combination of 300 nM TP-10 and 1 μM of DOX (E) as indicated for 24 hours. Cell viability was measured by cell counting kit-8 (CCK8) assay, n = 3 replicates for each group. (F-I) Human ovarian cancer cell line A2780 were treated with vehicle, 3 μM TP-10, 1 μM of DOX or combination of 3 μM TP-10 and 1 μM of DOX as indicated for 24 hours. (F) Representative images of cells stained with Dapi (total cells) and NucGreen (dead cells), scale bars: 100 μm. (G) Bars represent the percentage of NucGreen positive cells over the total cells. n = 10 random fields from 3 independent experiments for each group. (H) Representative images of immunostaining of p-H2AX, A2780 cells were fixed, and immunostained for p-H2AX, and counterstained for nuclei with DAPI. Scale bars: 50 μm. (I) Quantification of p-H2AX fluorescence intensity, n = 200–300 cells for each group. (J) A2780 cells were stained with annexin V-FITC and PI, the apoptotic cells were analyzed by a dot-plot using a flow cytometer. Q1, Annexin V-FITC−/PI+, necrosis; Q2, Annexin V-FITC+/PI+, late apoptosis; Q3, Annexin V-FITC+/PI−, early apoptosis; Q4, living cell population. (K) Quantification of the percentage of apoptotic cells (Annexin V+/PI− and Annexin V+/PI+), n = 6 replicates from 3 independent experiments for each group. (L) DNA synthesis measured via [3H]-thymidine incorporation in A2780 cells. n = 9–11 replicates from 3 independent experiments. Data were represented as mean ± SEM (A-D, G, K and L) or median with IQR (I). Statistics: Welch ANOVA with Dunnett’s T3 corrections for 3 comparisons in A and G, two-way ANOVA in D, one-way ANOVA with Holm-Sidak post-hoc test for 3 comparisons in E, K and L, Kruskal-Wallis’s test with Dunn’s corrections for 3 comparisons in I.
PDE10A inhibition attenuates ovarian xenograft tumor growth and simultaneously protects against DOX-induced cardiotoxicity in nude mice.
Given the cancer cell killing effect while the cardiac protective effect of PDE10A inactivation, we sought to further evaluate the effects of DOX, TP-10, or both on cancer xenografts and cardiac properties in nude mice in vivo, using human ovarian cancer cell line as an example. As illustrated in Figure 4A, A2780 cells with expression of luciferase were injected into flanks of nude mice. 10 days post-inoculation of the tumor cells, tumors became visible and measurable. Meanwhile, the animals received the injection of saline, DOX, TP-10, or DOX plus TP-10 daily for 2 weeks. The cardiac function was assessed by echocardiography prior to the tumor cell injection and drug treatment as well as once a week during the drug treatment period. Tumor development was monitored by luciferin signal and tumor volumes were measured every three days. At the end, tumors on both left and right flanks were isolated and weighted individually. Mouse hearts were harvested for further evaluation of morphological and histological changes. Consistent with what we found in A2780 cell line, DOX and TP-10 treatments induced an upregulation of PDE10A expression in A2780 xenografts (Figure 4B). We found that DOX or TP-10 alone significantly attenuated tumor growth, as shown by the images of luciferin (Figure 4C), tumor volume progression over 2 weeks (Figure 4D), as well as the final tumor weight (Figure 4E–F). Interestingly, TP-10 and DOX cotreatment enhanced the efficacy of DOX to antagonize tumor growth (Figure 4E–F).
Figure 4: PDE10A inhibition attenuates ovarian xenograft tumor growth in nude mice.

A2780 cells with expression of luciferase were injected into flanks of female nude mice at the age of 6 weeks. 10 days post-inoculation of the tumor cells, the animals received the injection of saline, DOX (1.5mg/kg/day), TP-10 (succinate form, 4.5 mg/kg/day) (please note that we increased the mass of TP-10 succinate because its higher molecular weight (MW: 1301) compared to the free base form (MW: 1003)), or DOX plus TP-10 daily for 2 weeks. (A) Experiment timeline. (B) PDE10A protein levels measured by western blotting in A2780 xenografts from nude mice treated with vehicle, DOX, TP-10, or DOX plus TP-10, bar graph shows the average of n = 6 in each group. (C) Representative bioluminescence imaging after transplantation of A2780 cells. Mice were imaged at indicated days. (D) Line graphs showing the inhibitory effects of DOX, TP-10, or both on the growth of xenograft tumors. Tumor growth of A2780 xenografts was determined by caliper measurement over 2 weeks, n = 10–15 tumors per treatment group. (E) Photographs of the excised tumors at the end of the study (2 weeks after drug treatment). (F) Weights of excised tumors were determined at the end of study (2 weeks after drug treatment), n = 8–12 tumors per treatment group. Data were represented as mean ± SEM. Statistics: Welch ANOVA with Dunnett’s T3 corrections for 3 comparisons in A and for 4 comparisons in F, repeated measures ANOVA with Holm-Sidak post-hoc test for 5 comparisons at day 13 in D.
Cardiac function was monitored by echocardiography throughout the study (Figure 5A and Supplemental Table S4). We found that the cardiac functions of control mice progressively declined likely due to tumor growth, as assessed by fractional shortening (%FS) (Figure 5B and Supplemental Table S4) and ejection fraction (%EF) (Supplemental Figure S7A, and Supplemental Table S4). This is consistent with previous findings that cancer itself is associated with cardiac dysfunction 26. Cardiac function was drastically reduced by the DOX treatment, which was reversed by TP-10 (Figure 5B, Supplemental Figure S7A, and Supplemental Table S4). In addition, plasma concentrations of cardiac troponin I significantly increased after DOX stimulation, which was alleviated by TP-10 treatment (Figure 5C). We also found that DOX treatment induced cardiac atrophy, as evidenced by a significant decrease in the global heart size (Figure 5D, and Supplemental Figure S7B), the ratio of HW/TL (Figure 5E), and the myocardial CSA (Figure 5F–G). We did not observe an apparent effect of TP-10 on cardiac fibrosis due to no significant fibrosis induced by DOX treatment in this model (data not shown). CM apoptosis assessed by the TUNEL staining was significantly increased in hearts treated with DOX compared to vehicle (Figure 5H). Importantly, DOX-induced cardiac atrophy (Figure 5D–G) and apoptosis (Figure 5H) were significantly attenuated by TP-10 treatment. Taken together, these findings suggest that PDE10A inhibition is able to attenuate ovarian xenograft tumor growth, while protecting against DOX-induced cardiotoxicity in nude mice.
Figure 5: PDE10A inhibition protects doxorubicin-induced cardiotoxicity in nude mice.
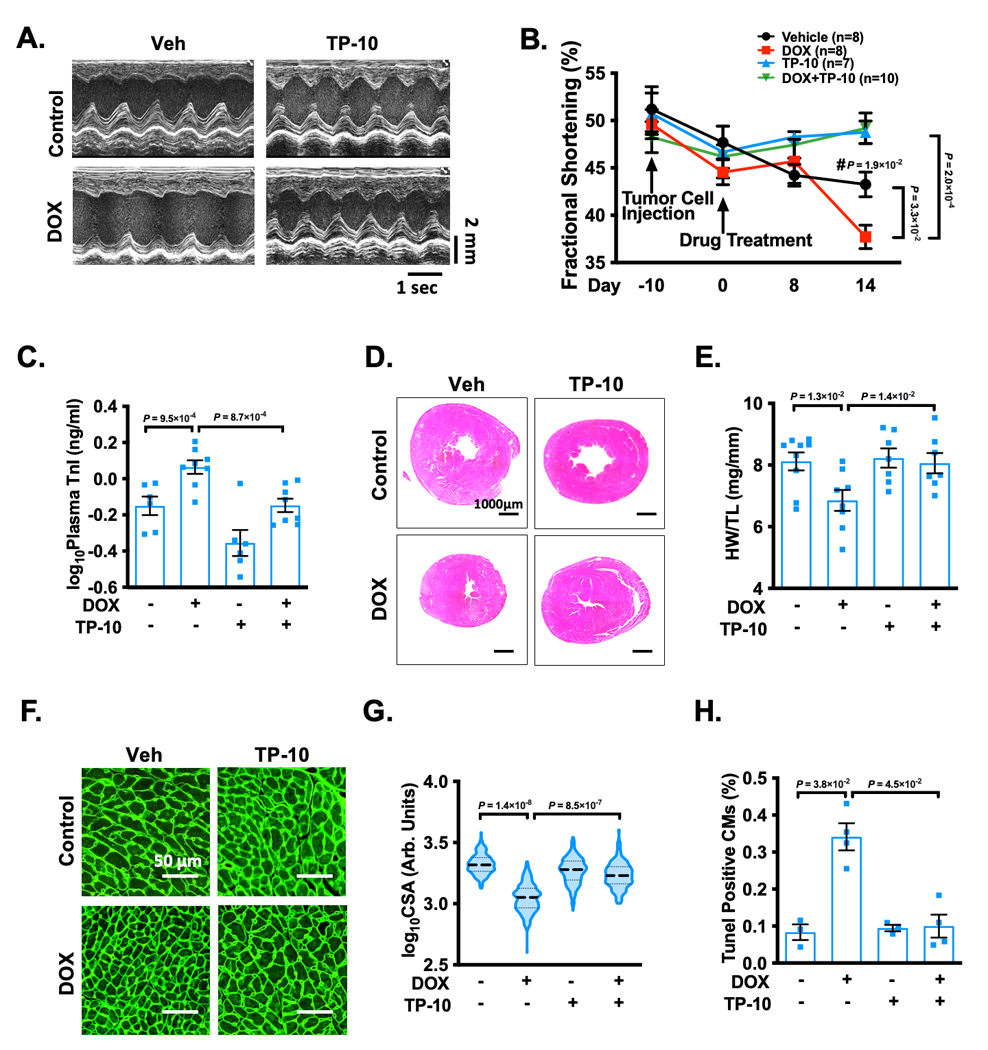
A2780 cells with expression of luciferase were injected into flanks of female nude mice at the age of 6 weeks. 10 days post-inoculation of the tumor cells, the animals received the injection of saline, DOX (1.5mg/kg), TP-10 (succinate form, 4.5 mg/kg/day), or DOX plus TP-10 daily for 2 weeks. Cardiac function was monitored by echocardiography throughout the study. (A) Representative M-mode echocardiographic images of each study group at 2 week point after drug treatment. (B) Progressive percent fractional shortening. (C) Plasma troponin I levels in each treatment group. (D) Representative images of hematoxylin and eosin staining in mouse hearts after treatment. Scale bars: 1000 μm. (E) Quantification of heart weight/tibia length. (F) Representative images of wheat germ agglutinin (WGA)-fluorescein isothiocyanate-staining in mouse hearts, showing cardiomyocyte (CM) cross-sectional area (CSA). Scale bars: 50 μm. (G) Quantitative data of CM hypertrophy assessed by CSA, n = 3–6 hearts per group with 300–800 CMs analyzed per heart. (H) Quantification of TUNEL staining, n = 3–6 hearts per group with 10 random fields analyzed per heart. Data were represented as mean ± SEM (B, C, and E) or median with IQR (G, H). Statistics: repeated measures ANOVA with Holm-Sidak post-hoc test for 2 comparisons at day 14, and 1 comparison between Vehicle/day 14 vs. Vehicle/day −10 (#) in B; one-way ANOVA with Holm-Sidak post-hoc test for 2 comparisons in C and E; mixed effect model with Sidak corrections for 2 comparisons in G; Kruskal-Wallis’s test with Dunn’s corrections for 2 comparisons in H. Animal numbers: n = 3–10.
PDE10A inhibition or deficiency alleviates DOX-induced CM death and atrophy in vitro.
We examined the effect of DOX on PDE10A expression at the 6 h time point, which was determined through our preliminary time course studies. We found DOX increased PDE10A expression in cultured adult mouse CMs (Figure 6A), suggesting a direct action of CM PDE10A in response to DOX. Next, we examined the effect of PDE10A inactivation on DOX-induced CM death using cultured adult mouse CMs in vitro. Female and male CMs were pretreated with TP-10. CM death was induced with DOX and cell death was evaluated by various methods. We found that DOX-induced cell death was alleviated by both PDE10A inhibitor TP-10, as assessed by trypan blue staining (Figure 6B–C, and Supplemental Figure S8A–C) and lactate dehydrogenase (LDH) cytotoxicity assay (Figure 6D–E, and Supplemental Figure S8D–E). We also examined the effects of DOX on isolated CMs from PDE10A-WT or PDE10A-KO mice. Consistently, we found that DOX-induced CM death was alleviated in PDE10A-KO CMs (Figure 6F). We also examined the effects of TP-10 on DOX-induced DNA damage in CM by assessing the p-H2AX level (Figure 6G). As anticipated, both TP-10 (Figure 6G–H, and Supplemental Figure S8F) and PDE10A deficiency (Figure 6G, 6I, and Supplemental Figure S8F) significantly inhibited DOX-induced increase of p-H2AX signal. Next, we examined the effect of PDE10A inactivation on CM atrophy using both female and male CMs. We found that DOX-induced CM atrophy, as indicated by decreases in CM CSA, was largely attenuated by TP-10 or PDE10A deficiency (Figure 6J–K, and Supplemental Figure S8G–H). Collectively, these results strongly suggest that PDE10A plays a critical causative role in DOX-induced CM death and atrophy. Since there is no sex difference regarding the effect of DOX and TP-10 on CM death and atrophy, and basal cell death in male CMs (40%) is higher than that in female CMs (20%). We mainly used female CMs for experiments throughout this study unless otherwise noted.
Figure 6: PDE10A inhibition or deficiency alleviates doxorubicin-induced cardiomyocyte death and atrophy in vitro.
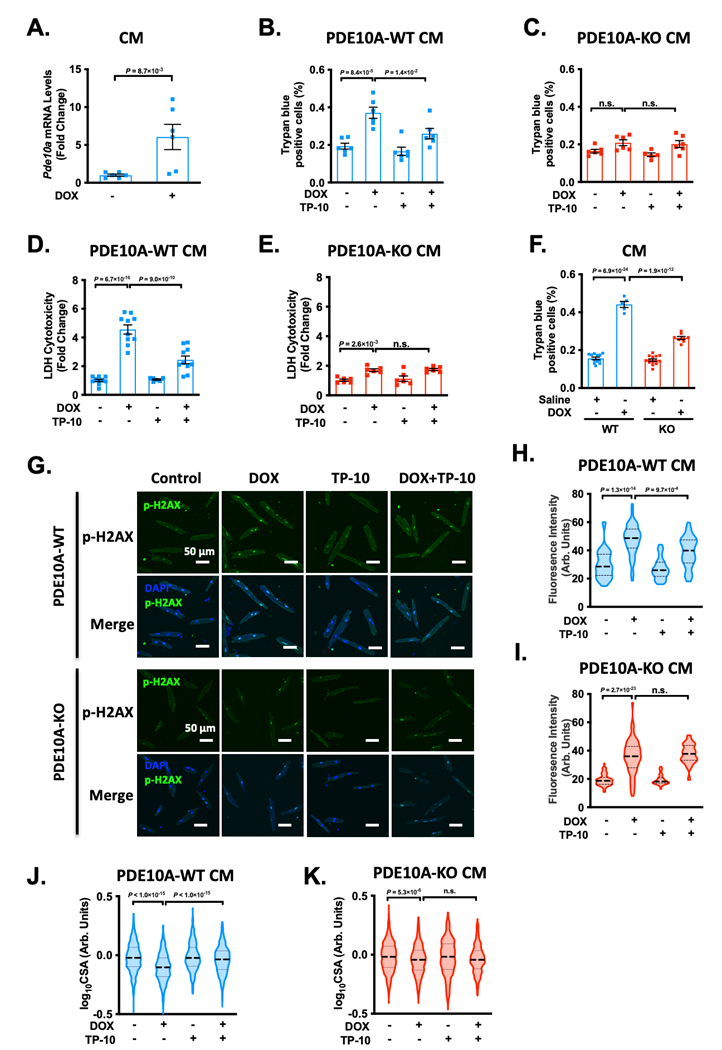
(A) Cardiomyocytes (CMs) isolated from PDE10A-WT mice were stimulated with or without DOX for 6 hours. qPCR analysis of Pde10a, normalized to Gapdh, n = 6 replicates for each group. (B-K) CMs isolated from PDE10A-WT or KO mice were stimulated with 10 μM DOX in the presence of 300 nM TP-10 or vehicle for 24 hours. Cell death was measured in PDE10A-WT CMs (B) and PDE10A-KO CMs (C) by trypan blue staining, 20 random fields were assessed per treatment group, n = 6 replicates from 3 mice for each group. (D-E) Lactate dehydrogenase (LDH) cytotoxicity was quantified in PDE10A-WT (D) and PDE10A-KO (E) CMs, n = 6–10 replicates from 4 mice for each group. (F) PDE10A-WT or PDE10A-KO mice were treated with DOX (25 mg/kg by intraperitoneal injection (i.p.)) for 16 hours and CMs were isolated and cultured for 24 hours. Cell death was measured in by trypan blue staining, 20 random fields were assessed per treatment group, n = 5–14 replicates from 3 mice. (G) Representative images of immunostaining of p-H2AX, showing the effect of PDE10A inhibitor TP-10 on DOX stimulated mouse CM DNA damage. Mouse CMs were fixed, and immunostained for p-H2AX, and counterstained for nuclei with DAPI. Scale bars: 50 μm. (H-I) Quantification of p-H2AX fluorescence intensity in PDE10A-WT (H) or PDE10A-KO (I) CM, n = 50–80 CMs from 3 isolations. (J-K) Cell surface areas (CSAs) were quantified from CMs isolated from PDE10A-WT (J) and PDE10A-KO (K) mice. CSAs were averaged from n = 500–1500 myocytes from 3 isolations per group. Data were represented as mean ± SEM (B-F) or median with IQR (A, H-K). Statistics: Mann-Whitney test in A; mixed effect model with Sidak corrections for 2 comparisons in B-F, and H-K. n.s.: no significance difference.
PDE10A contributes to CM death and mitochondrial dysfunction dependent on cGMP signaling.
Inhibiting PDE10A by TP-10 can increase both cAMP and cGMP levels in CMs 12. However, the links between PDE10A functions and cAMP or cGMP signaling remain unknown. The canonical cAMP and cGMP effectors include protein kinase A (PKA), exchange protein activated by cAMP (Epac1/2), and cGMP-dependent protein kinase (PKG). We first aimed to determine the role of cAMP signaling in PDE10A-mediated regulation of CM death induced by DOX. We used a selective PKA inhibitor PKI and an Epac1/2 inhibitor ESI-09. We found that treatment with either PKI or ESI-09 had no significant effect on the protective effects of PDE10A inhibition or PDE10A deficiency against CM death, as measured by trypan blue staining and LDH cytotoxicity assay (Supplemental Figure S9A–H). These results suggest that PDE10A regulation of CM death is independent of cAMP/PKA or cAMP/Epac signaling. We next determined the contribution of PKG signaling to PDE10A-mediated regulation of CM death. We found that the effects of TP-10 or PDE10A deficiency on suppressing DOX-induced CM death were reversed by different strategies of PKG inactivation, including PKG inhibitors Rp-8-Br-PET-cGMPS and DT-2 (Figure 7A–B, and Supplemental Figure S9I–J), as well as knocking down of PKG1 via lentivirus-mediated expression of two different PKG1 shRNAs (Figure 7C–D, and Supplemental Figure S9K–M). Consistently, PKG activator 8-pCPT-cGMP decreased DOX-induced CM death (Figure 7E, and Supplemental Figure S9N). Together, these observations suggest a critical role of cGMP/PKG signaling in PDE10A inhibition/deficiency mediated protection against CM death.
Figure 7: Role of cGMP signaling in PDE10A-mediated regulation of cardiomyocyte death, Top2β expression, DNA damage, and mitochondria dysfunction.

(A-B) Role of PKG in cardiomyocyte (CM) death induced by DOX. PDE10A-WT CMs (A) and PDE10A-KO CMs (B) were treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) in the presence of vehicle or 2 PKG inhibitors Rp-8-Br-PET-cGMPs (10 μM) and DT-2 (1 μM), and stimulated with DOX (10 μM) for 24 hours. Cell death was measured by trypan blue staining. (C-D) PDE10A-WT CMs (C) and PDE10A-KO CMs (D) were transduced with lentivirus expressing scramble shRNA, and 2 different PKG1shRNAs, and treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) and stimulated with DOX (10 μM) for 24 hours. Cell death was measured by trypan blue staining. (E) PDE10A-WT CMs were treated with PKG activator 8-pCPT-cGMP (10 μM) and stimulated with DOX (10 μM) for 24 hours. Cell death was measured by trypan blue staining. (F-G) qPCR analysis of Top2b expression. PDE10A-WT CMs (F) and PDE10A-KO CMs (G) were treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) in the presence of vehicle or 2 PKG inhibitors Rp-8-Br-PET-cGMPs (10 μM) and DT-2 (1 μM), and stimulated with DOX (10 μM) for 6 hours, normalized to Gapdh. n = 6 replicates. (H) Top2b mRNA expression was assessed by qPCR in the hearts of PDE10A-WT or PDE10A-KO mice after saline or DOX (15mg/kg, one bolus) treatment for 1 week as indicated, normalized to Gapdh. n = 3–5 hearts. (I-J) PDE10A-WT CMs were treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) in the presence of vehicle or 2 PKG inhibitors Rp-8-Br-PET-cGMPs (10 μM) and DT-2 (1 μM), and stimulated with DOX (10 μM) for 24 hours. (I) Representative images of immunostaining of p-H2AX in PDE10A-WT CMs. CMs were fixed, and immunostained for p-H2AX, and counterstained for nuclei with DAPI. Scale bars: 50 μm. (J) Quantification of p-H2AX fluorescence intensity in PDE10A-WT CMs, n = 50–100 CMs from 3 isolations. (K) Representative images of PDE10A-WT CM loaded with TMRE. The scale bar is 50 μm. (L-M) Quantification of TMRE fluorescence intensity. (L) PDE10A-WT CMs were treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) in the presence of vehicle or 2 PKG inhibitors Rp-8-Br-PET-cGMPs (10 μM) and DT-2 (1 μM), and stimulated with DOX (10 μM) for 24 hours. (M) PDE10A-WT CMs were transduced with lentivirus expressing scramble shRNA, and 2 different PKG1shRNAs, and treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) and stimulated with DOX (10 μM) for 24 hours. (N) Representative images of PDE10A-WT CM loaded with DHE. The scale bar is 50 μm. (O-P) Quantification of DHE fluorescence intensity. (O) PDE10A-WT CMs were treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) in the presence of vehicle or 2 PKG inhibitors Rp-8-Br-PET-cGMPs (10 μM) and DT-2 (1 μM), and stimulated with DOX (10 μM) for 24 hours. (P) PDE10A-WT CMs were transduced with lentivirus expressing scramble shRNA, and 2 different PKG1shRNAs, and treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) and stimulated with DOX (10 μM) for 24 hours. Data were represented as mean ± SEM (A-G) or median with IQR (H, J, L-M, and O-P). Statistics: mixed effect model with Sidak corrections for 2–4 comparisons as indicated in A-E, J, L-M, and O-P; Welch ANOVA with Dunnett’s T3 corrections for 3–4 comparisons as indicated in F-G; Kruskal-Wallis’s test with Dunn’s corrections for 2 comparisons in H. n.s.: no significance difference. For trypan blue staining, 20 random fields were assessed per treatment group, n = 6–22 replicates from 3–5 mice for each group. For TMRE and DHE staining, 8–10 random fields were assessed per treatment group, n = 20–60 cells from 3 mice for each group.
We next determined the role and mechanistic action of cGMP/PKG signaling in antagonizing DOX-induced CM death. CMs express topoisomerase 2β (Top2β), which is an enzyme that can control and alter the topologic states of DNA during replication and transcription 27. DOX intercalates into DNA and forms complexes with Top2β that blocks the function of Top2β and induces DNA damage, leading to mitochondria injury and cell death 27, 28. Since PDE10A is important in DNA damage (Figure 6G–I), we thus determined the role of PDE10A/cGMP/PKG signaling in the regulation of Top2β, DNA damage, and mitochondria function. We found that DOX increased Top2β expression in PDE10A-WT CMs (Figure 7F), which was markedly down-regulated by both PDE10A inhibitor TP-10 (Figure 7F) or PDE10A deficiency (Figure 7G). Consistently, Top2β expression was also upregulated in mouse hearts treated with DOX, and downregulated by PDE10A deficiency (Figure 7H). Interestingly, inhibition of PKG by Rp-8-Br-PET-cGMPs or DT-2 significantly reversed the effect of PDE10A inhibition or deficiency on Top2β expression (Figure 7F–G). Consistently, inhibition of PKG by Rp-8-Br-PET-cGMPs or DT-2 abolished the effect of PDE10A inhibition or deficiency on CM DNA damage by assessing p-H2AX level (Figure 7I–J, and Supplemental Figure S10A–C).
Our RNA-seq and GSEA analysis results revealed that PDE10A may regulate pathways pertinent to oxidative phosphorylation signaling (Supplemental Figure S5C), a key signaling pathway involved in mitochondrial function regulation. Given the role of PDE10A and Top2β in mitochondria function, we tested if PDE10A/cGMP/PKG would have functional effects on mitochondria. Using tetramethyl-rhodamine (TMRE) and dihydroethidium (DHE) staining, we measured changes of mitochondria membrane potential (Figure 7K–M, and Supplemental Figure S11) and ROS generation (Figure 7N–P, and Supplemental Figure S12), respectively, in both PDE10A-WT and PDE10A-KO CMs. As anticipated, DOX treatment decreased mitochondrial membrane potential (Figure 7K–M) and increased ROS generation (Figure 7N–P) in PDE10A-WT CMs, as indicated by decreased TMRE intensity and increase DHE intensity, respectively. PDE10A inhibitor TP-10 and PDE10A deficiency attenuated the loss of mitochondrial membrane potential (Figure 7K–M, and Supplemental Figure S11) and increased ROS generation induced by DOX (Figure 7N–P, and Supplemental Figure S12). Interestingly, inhibiting PKG by Rp-8-Br-PET-cGMPs or DT-2, or knocking down PKG1 with 2 different PKG1 shRNAs blocked the effect of PDE10A inactivation on mitochondrial functions (Figure 7L–M, 7O–P, and Supplemental Figure S11–12). To confirm the effect of PDE10A deficiency on DOX-mediated mitochondria dysfunction in vivo, we examined the expression of several key components in the electron transport chain (such as Ndufs1, Sdha, and Atp5b) and mitochondria biogenesis (such as Ppargc1a, Ppargc1b, and Tfam). We observed upregulation of these genes in hearts from DOX-treated PDE10A-WT mice but not from DOX-treated PDE10A-KO mice (Supplemental Figure S13A–B). Finally, we determined the effects of DOX and TP-10 on PKG activity in CMs. Since cGMP binding to allosteric sites of the PKG1 regulatory domain is a key step in PKG1 activation 29, we therefore detected cGMP and PKG1 binding by proximity ligation assay (PLA) (Supplemental Figure S14A). Compared with control, DOX decreased PLA signals (red dots). The PLA signals further increased by TP-10 treatment, indicating increased PKG activation upon TP10 treatment (Supplemental Figure S14B–D). Together, these results indicate that PDE10A-regulated cGMP/PKG signaling plays an inhibitory role in DOX-induced Top2β expression, DNA damage, and mitochondria dysfunction in CMs.
PDE10A contributes to CM atrophy dependent on both cAMP and cGMP signaling.
Similarly, we determined the roles of cAMP and cGMP signaling in PDE10A regulation of DOX-induced CM atrophy. Different from CM death, PKA inhibitor PKI blocked the protective effect of PDE10A inhibition (Figure 8A) or PDE10A deficiency (Figure 8B) against CM atrophy. There are a number of PKA regulatory subunits belonging to type I (RI) and type II (RII) subunits. PKA RIa and PKA RIIa are two major isoforms expressed in the heart. To determine the PKA regulatory subunit involved in DOX-induced CM atrophy, we knocked down PKA RIa (Prkar1a) or PKA RIIa (Prkar2a) via selective lentivirus-mediated expression of shRNAs in CM, respectively. We found that PKA RIIa was specifically involved in PDE10A inhibition/deficiency mediated CM atrophy (Supplemental Figure S15A). Therefore, to determine PKA activity, we detected cAMP and PKA RII binding by PLA (Supplemental Figure S15B). We found that DOX decreased PLA signals (red dots) while TP-10 increased PLA signals in CMs, indicating increased PKA activation upon TP-10 treatment (Supplemental Figure S15C–E). To determine the role Epac in CM atrophy, we used Epac1/2 inhibitor ESI09. We found that ESI09 had no detectable influence on the protective effect of PDE10A inhibition or deficiency against CM atrophy (Supplemental Figure S15F–G). These results suggest that the role of PDE10A inactivation against CM atrophy is dependent on PKA but not Epac signaling.
Figure 8: Role of cAMP and cGMP signaling in PDE10A-mediated regulation of cardiomyocyte atrophy.
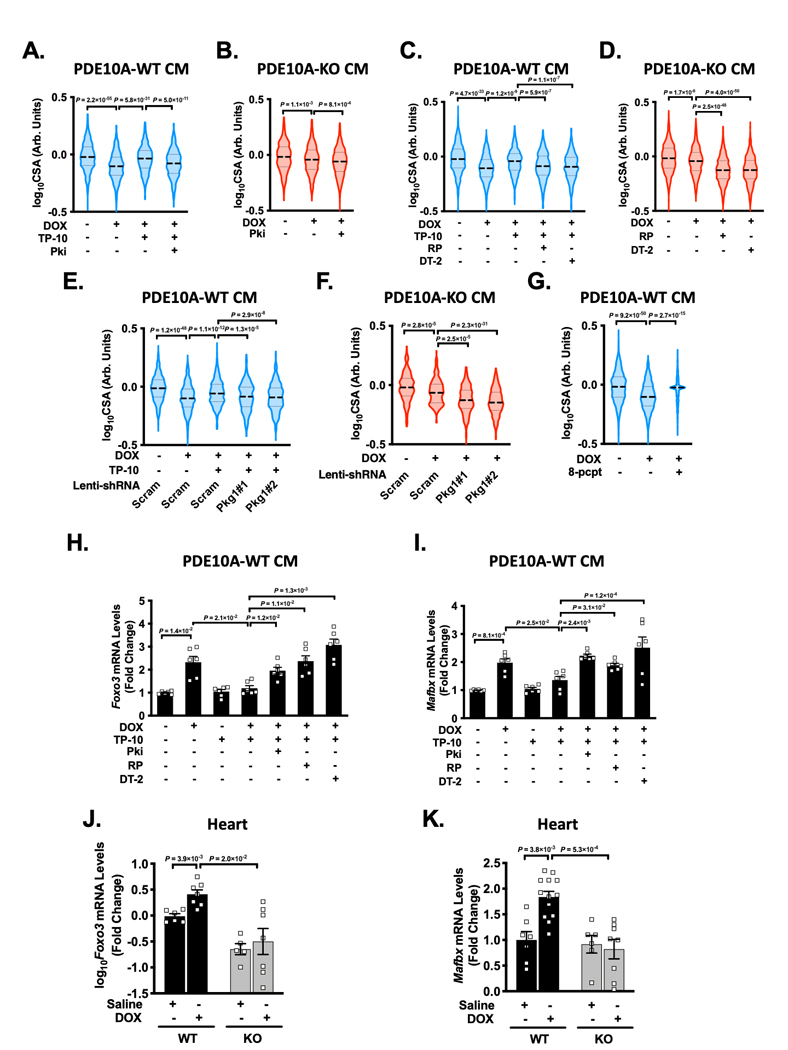
(A-B) Role of PKA in cardiomyocyte (CM) atrophy induced by DOX. Quantified data of cell surface area of PDE10A-WT CMs (A) and PDE10A-KO CMs (B) treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) in the presence of vehicle or PKA inhibitor PKI (5 μM) and stimulated with DOX (10 μM) for 24 hours. (C-G) Role of PKG in CM atrophy induced by DOX. Quantified data of cell surface area of PDE10A-WT CMs (C) and PDE10A-KO CMs (D) treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) in the presence of vehicle or 2 PKG inhibitors Rp-8-Br-PET-cGMPs (10 μM) and DT-2 (1 μM), and stimulated with DOX (10 μM) for 24 hours. (E-F) Quantified data of cell surface area of PDE10A-WT CMs (E) and PDE10A-KO CMs (F) transduced with lentivirus expressing scramble shRNA, and 2 different PKG1shRNAs, and treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) and stimulated with DOX (10 μM) for 24 hours. (G) Quantified data of cell surface area of CM PDE10A-WT CMs treated with PKG activator 8-pCPT-cGMP (10 μM) and stimulated with DOX (10 μM) for 24 hours. (H-I) PDE10A-WT CMs were treated with or without vehicle or the PDE10A inhibitor TP-10 (300 nM) in the presence of vehicle, PKA inhibitor PKI (5 μM) or 2 PKG inhibitors Rp-8-Br-PET-cGMPs (10 μM) and DT-2 (1 μM), and stimulated with DOX (10 μM) for 6 hours. qPCR analysis of cardiac atrophic genes Foxo3 (H) and Mafbx (I), normalized to Gapdh, n = 6 replicates for each group. (J-K) Cardiac atrophic genes Foxo3 (J) and Mafbx (K) were assessed by qPCR in the hearts of PDE10A-WT or PDE10A-KO mice after saline or DOX (15mg/kg, one bolus) treatment for 1 week as indicated, normalized to Gapdh. n = 3–10 hearts. Data were represented as mean ± SEM (H-J) or median with IQR (A-G, K). Statistics: mixed effect model with Sidak corrections for 2–4 comparisons as indicated in A-G; Welch ANOVA with Dunnett’s T3 corrections for 2–4 comparisons in H-J. Kruskal-Wallis’s test with Dunn’s corrections for 2 comparisons in K. For quantification of cell surface area, n > 1000 CMs from 3–4 isolations for each group.
Next, we evaluated the contribution of cGMP/PKG on PDE10A-mediated regulation of CM atrophy. Interestingly, inhibition of PKG by Rp-8-Br-PET-cGMPs or DT-2 also significantly blocked the effect of PDE10A inhibition or deficiency on CM atrophy (Figure 8C–D). Similarly, knocking down PKG1 via lentivirus-mediated expression of two different PKG1 shRNAs abolished the protective effect of PDE10A inhibition or deficiency (Figure 8E–F). Consistently, PKG activation by 8-pCPT-cGMP significantly alleviated DOX-induced CM atrophy (Figure 8G). These results suggest cGMP/PKG signaling also contributes to the anti-atrophic effect of PDE10A inactivation.
Our RNA-seq results revealed that PDE10A may regulate FoxO signaling pathway (Supplemental Figure S5C). FoxO family of transcription factors are important for the expression of protein ligases involved in atrophy, such as MAFbx 30. Thus, we further determined the role of PDE10A regulated cAMP/PKA and cGMP/PKG signaling in the expression of the molecules involved in atrophy. We found that Foxo3 and Mafbx expression was significantly induced by DOX and reduced by both TP-10 (Figure 8H–I) and PDE10A deficiency (Supplemental Figure S15H–I). The inhibition of PKA by PKI as well as inhibition of PKG by Rp-8-Br-PET-cGMPs and DT-2 reversed the effect of PDE10A inactivation on Foxo3 and Mafbx expression, consistent with their effects on CM CSA (Figure 8H–I, and Supplemental Figure S15H–I). We also found upregulation of Foxo3 and Mafbx in hearts from DOX-treated PDE10A-WT mice but not in hearts from DOX-treated PDE10A-KO mice (Figure 8J–K). Together these results strongly suggest that PDE10A plays a critical causative role in DOX-induced CM atrophy via regulating FoxO signaling in a manner dependent on both cAMP/PKA and cGMP/PKG signaling.
Discussion
Cardiotoxicity is one of the most serious adverse effects of chemotherapy. The most exciting novel finding of the present study is that PDE10A inhibition is able to significantly suppress ovarian cancer graft growth and potentiate the chemotherapeutic efficacy of DOX on ovarian cancer, while concurrently ameliorate DOX-induced cardiac toxicity and dysfunction. In addition to anthracycline DOX, various other popular chemotherapy agents, such as monoclonal antibodies and tyrosine kinase inhibitors, can also cause significant risk of cardiovascular complications 31. Thus, the role of PDE10A in the cardiotoxicity induced by other cancer therapy agents also warrants to be comprehensively investigated in the future. PDE10A has been proven to be a safe drug target in a number of clinical trials 12. Thus, findings from our study provide proof-of-concept that the PDE10A inhibitors may represent as a novel class of anticancer drugs when used alone or in combination with chemotherapy drugs.
A previous study has identified PDE10A as one of 11 prognostic biomarkers that robustly and accurately predicts overall survival in patients with ovarian cancer 32. PDE10A has also been reported in a number of other types of cancers. For example, PDE10A expression is induced in human lung cancers 14 and colon cancers 13, 25, 33. Some studies have reported that PDE10A knockdown or inhibition effectively attenuates the growth of these cancer cells in vitro in a cGMP/PKG-dependent manner, likely through inhibiting the β-catenin signaling 13, 14, 25, 33. While a recent study has found that PDE10A inhibition blocked colon cancer cell growth independent of cGMP signaling 34. During the preparation of this manuscript, a new published study reported that PDE10A inhibition decreases ovarian cancer growth and induces apoptosis, which supports our observation in ovarian cancer cells and in nude mice xenograft 35. In addition, a PDE10A single nucleotide polymorphism (SNP) variant in a noncoding exon is associated with the risk of tobacco-induced developing non-small cell lung cancer (NSCLC) 36. A Pro360Ala mutation of PDE10A has been shown to play a potential oncogenic role in NSCLC 37. PDE10A sequence variations are higher in prostate cancer patients than those in the normal population 38. Whole-exome sequencing identified a PDE10A mutation in peritoneal metastatic gastric adenocarcinoma 39. In the current study, we showed that PDE10A inhibition not only suppresses the survival of ovarian cancer, but suppresses the survival of various other cancers including colon cancer and prostate cancer. These lines of evidence together indicate that PDE10A is important for multiple different cancers and suggest that PDE10A inhibition may be a promising anti-cancer approach.
DOX exhibits both chronic and acute cardiotoxicity 2. In this study, we demonstrated the protective effect of PDE10A inactivation against DOX-induced myocardial death, cardiac atrophy and dysfunction in both acute and chronic DOX models (Figure 1 and 2). Chronic effects of DOX in the heart show the development of cardiac fibrosis 40. We also found that cardiac fibrosis induced by chronic DOX treatment was attenuated by PDE10A inhibition, which is consistent with our previous finding that PDE10A plays a critical role in cardiac fibroblast activation and proliferation as well as in cardiac fibrosis 12. We are aware of that the protective cardiac effects resulted from systemic PDE10 inhibition or global PDE10A deficiency may be also contributed by other cells in the heart (such as endothelial cells or immune cells) and/or other non-cardiac tissues. The contribution of immune cells appears to be limited since we observed similar cardiac protective effects of TP-10 between C57BL/6J and nude mice with inhibited immune system.
Besides PDE10A, there are several other PDEs reported to link to cancers and DOX-induced cardiotoxicity. For example, PDE5A (a cGMP-specific hydrolyzing PDE) upregulation has been found in variety of human cancers 41, and PDE5 inhibitors antagonize the proliferation, mortality and/or invasion of cancer cells in vitro or in vivo or sensitize chemotherapeutic agents to cancer cells 41, 42. PDE5 inhibitors attenuate CM apoptosis and/or protects from DOX-induced cardiotoxicity and dysfunction in mice 6. One study with nude mice also showed that sildenafil increases chemotherapeutic efficacy of DOX in prostate cancer and concomitantly provides a cardioprotective effect 43. In addition, PDE1C has been implicated in cancer cells 44, 45. We have recently reported that PDE1C activation promotes CM death 17. PDE1C inhibition/deficiency attenuates DOX-induced CM death in vitro as well as cardiac toxicity and dysfunction in vivo 17. Although PDE1C, PDE5A, or PDE10A all play important roles in CM survival in the presence of DOX in mice, their mechanistic actions could be different. PDE1C inhibition/deficiency protects CM death by facilitating protective A2R/cAMP/PKA signaling 6, 46. PDE5A inhibition prevents CM death, likely dependent upon nitric oxide (NO)-cGMP/PKG signaling 6. The results from our current study suggest that the protective effect of PDE10A deficiency/inhibition against CM death is dependent on cGMP/PKG signaling but not cAMP/PKA or cAMP/Epac signaling. In CMs, cGMP could be produced from two sources: via the soluble guanylyl cyclase (sGC) upon NO stimulation or particular GC (pGC) upon natriuretic peptide (NP) stimulation. PDE5A and PDE10A have different subcellular localization in CMs: PDE5A has been shown to complex with α-actinin at the Z-disk in CMs 47 and PDE10A is primarily localized at the plasma membrane 12, suggesting that PDE5A and 10A may regulate different sources of cGMP in CMs. The role of NO/sGC or NP/pGC in PDE10A-mediated regulation of CM survival remains to be determined. Nevertheless, these differences in mechanistic actions suggest an interesting option for simultaneously targeting more than one PDE to achieve potential additive or synergistic beneficial effects. Indeed, combined inhibition of PDE5 and PDE10 or dual knockdown of PDE5A and PDE10A suppresses colon cancer cell growth to a greater extent than inhibition/knockdown of individual ones 33. Therefore, the cardioprotective effects from combined PDE inhibitor treatments also deserve to be evaluated.
Our current study demonstrates a critical role of PDE10A in CM death induced by DOX both in vitro and in vivo. Our results suggest a critical role of cGMP/PKG signaling activation via PDE10A deficiency/inhibition in suppressing DOX-induced Top2β upregulation, DNA damage, mitochondria dysfunction, and CM death (Figure 7). It is possible that cGMP/PKG regulates mitochondria DNA transcription/post-translational modification. The molecular mechanisms underlying these processes need to be further investigated. The protective effect of cGMP/PKG signaling in CMs have been reported in a number of previous studies. For example, activating cGMP/PKG signaling by NO donor SNAP, cGMP analog 8-pCPT-cGMP, or expressing active PKG1 attenuates hypoxia/reoxygenation (H/R)-induced neonatal or adult rat CM apoptosis in vitro 48, 49. Inhibiting PDE5 by sildenafil attenuates DOX-induced adult mouse CM apoptosis in vitro and in vivo 50. However, several other studies appear to have inconsistent findings. For example, NO donors, cell-permeant cGMP analogs, sGC activators, or ANP/CNP have been shown to induce apoptosis under basal conditions in both adult or neonatal rat CMs 51–54. It has been reported that NO-induced CM apoptosis is not dependent on cGMP production and cGMP analog does not have effects on CM viability 55. These discrepancies may be due to different types or doses of reagents used to activate the cGMP/PKG signaling. For example, cGMP analogs such 8-Br-cGMP and 8-pCPT-cGMP may have different effects on CM survival at different doses. This is because high doses of cGMP analogs is able to activate PKA signaling 56. ANP at a low dose (≈10−9 M) and high dose (≈10−6 M) shows different effects on CM apoptosis 57. NO- and NP-induced cGMP/PKG signaling are localized in different cellular compartments, which may differentially regulate CM viability. In addition, it appears that cGMP signaling exhibits distinct effects under basal and stimulated conditions. CM apoptosis induced by NO or cGMP under the basal state is not seen in CMs stimulated with hypoxia 52. Therefore, the role of cGMP/PKG signaling in CM viability deserves to be further investigated in manners dependent on specific contexts.
It is also interesting to find that PDE10A inhibition elicits opposite effects on CMs and cancer cells: suppressing CM death induced by DOX whereas promoting cancer cell death. Indeed, findings from a number of previous studies have suggested that cGMP can exert distinct or even opposing functions in different cell types via regulating different downstream signaling pathways 29, 58. For example, activation of cGMP/PKG signaling leads to CM and neuron cell protection against apoptosis through regulating the Bcl-2 family member 49, 59. Oppositely, elevating intracellular cGMP levels induces the apoptosis of endothelial 60 or smooth muscle cells 61. In colon cancer cells, activation of cGMP/PKG signaling suppresses cell proliferation and induces cell apoptosis via mediating MAPK/ERK signaling 62. In particular, previous studies have also reported that inhibition of PDE10A suppresses colon cancer cell growth and promotes colon cancer cell death in a cGMP-PKG-dependent manner, through inhibiting β-catenin signaling 13, 14, 25. While PDE10A inhibition exerts anti-ovarian cancer effect via mediating both cAMP/PKA and cGMP/PKG signaling, and subsequently leads to inhibition of β-catenin signaling and decreased activation of the MAPK and AKT pathways 35.
In the current study, we found that PDE10A contributes to CM atrophy induced by DOX. Previously, we also showed that PDE10A contributes to CM pathological hypertrophy induced by chronic pressure overload and/or neurohormonal stimulation, but not physiological hypertrophy 12. It is of great interest that PDE10A regulates both CM atrophy and hypertrophy. CM atrophy is due to over-activation of UPS and/or autophagy, and subsequent increase of protein ligase expression and protein degradation 30, 63, which can be mediated by transcription factors such as FoxO 30. Indeed, we found that FoxO signaling pathway is one of the most significantly enriched terms among the genes altered by DOX and regulated by PDE10A (Supplemental Figure S5C). DOX treatment significantly promoted the expression of FoxO3 and the muscle-specific ligase MAFbx in CMs, which was significantly suppressed by PDE10A inhibition and deficiency (Figure 8H–I, and Supplemental Figure S15H–I). Different from CM atrophy, CM hypertrophy is due to the activation of the fetal gene program that accelerates global protein synthesis in CMs 64. Transcription factors such as MEF2 and NFAT are important for CM fetal gene expression and CM hypertrophy 65. It is possible that multiple discrete PDE10A-coupled signaling pathways existing in CMs regulate different sets of transcription factor expression and contribute to different biological functions. For example, we found that PDE10A-mediated regulation of CM atrophy and FoxO signaling is dependent on both cAMP/PKA and cGMP/PKG signaling pathways, while PDE10A-mediated regulation of CM viability is dependent on the cGMP/PKG signaling pathway. The signaling pathway and molecular mechanism involved in PDE10A regulation of CM hypertrophy remains unknown and is under investigation in our lab.
Supplementary Material
What Is Known?
Chemotherapeutic drugs such as doxorubicin (DOX) are widely used in cancer treatment. However, side effects such as DOX cardiotoxicity remain as a serious clinical complication.
Phosphodiesterase 10A (PDE10A) is able to hydrolyze both cAMP and cGMP. PDE10A expression is induced in various human tumor cell lines and PDE10A inhibition suppresses tumor cell growth.
PDE10A is upregulated in human and mouse failing hearts. PDE10A deficiency/inhibition protects against cardiac hypertrophy and heart failure induced by chronic pressure overload or neurohormonal overstimulation.
What New Information Does This Article Contribute?
In C57Bl/6J mice, PDE10A inactivation alleviates DOX-induced cardiotoxicity including myocardial atrophy, apoptosis, and dysfunction. In nude mice with implanted human ovarian cancer xenografts, PDE10A inhibition attenuates tumor growth while protecting against DOX-induced cardiotoxicity in mice.
In cancer cells, PDE10A inhibition increases the death, decreases the proliferation, and potentiates the effect of DOX on various human cancer cell lines.
In cardiomyocytes (CMs), PDE10A contributes to DOX-induced CM death via increasing Top2β expression, mitochondrial dysfunction, and DNA damage by antagonizing cGMP/PKG signaling. PDE10A contributes to CM atrophy via potentiating FoxO3 signaling via both cAMP/PKA and cGMP/PKG dependent signaling.
The cardiotoxicity associated with anthracyclines (e.g., doxorubicin – DOX) continues to be a serious clinical problem. Herein, we report a critical role and potential mechanisms by which PDE10A promotes cardiomyocyte death and atrophy. PDE10A inactivation alleviates DOX-induced cardiotoxicity in C57Bl/6J mice, including myocardial atrophy, apoptosis, and dysfunction. In cancer cells, PDE10A inhibition increases the death, decreases the proliferation, and potentiates the effect of DOX in various human cancer-cell lines. In nude mice with implanted human ovarian cancer xenografts, PDE10A inhibition attenuates tumor growth while protecting against DOX-induced cardiotoxicity. In CMs, PDE10A/cGMP/PKG signaling regulates Top2β expression, DNA damage, mitochondria dysfunction, therefore contributes to CM viability. Both PDE10A/cAMP/PKA and PDE10A/cGMP/PKG signaling contribute to CM atrophy via regulating the FoxO3 signaling. Findings from this study provide proof-of-concept that the PDE10A inactivation may represent a novel therapeutic strategy in combatting chemotoxicity.
Acknowledgments
The authors would like to thank Pfizer Inc. for sharing TP-10 and TP-10 succinate, and the Genomics Research Center for performing RNA-sequencing analysis.
Sources of Funding
This work was financially supported by National Institute of Health HL134910 and HL154318 (to C.Y.).
Non-standard Abbreviations and Acronyms
- PDE
Phosphodiesterase
- DOX
Doxorubicin
- KO
Knockout
- CM
Cardiomyocytes
- ROS
Reactive oxygen species
- ESC
Embryonic stem cell
- iPSC
Human induced pluripotent stem cell
- NRVM
Neonatal rat ventricular myocytes
- WT
Wildtype
- CSA
Cell surface area
- FS
Fractional shortening
- EF
Ejection fraction
- PCA
Principle component analysis
- GSEA
Gene set enrichment analysis
- FoxO
Forkhead box O
- UPS
Ubiquitin proteasome system
- CCK8
Cell counting kit-8
- PI
Propidium iodide
- LDH
Lactate dehydrogenase
- PKA
Protein kinase A
- PKG
cGMP-dependent protein kinase
- Epac
Exchange protein activated by cAMP
- Top2β
Topoisomerase 2β
- TMRE
Tetramethyl rhodamine
- DHE
Dihydroethidium
- PLA
Proximity ligation assay
- SNP
Single nucleotide polymorphism
- NSCLC
non-small cell lung cancer
- NO
Nitric oxide
- GC
Guanylyl cyclase
- NP
Natriuretic peptide
- H/R
Hypoxia/reoxygenation
Footnotes
Disclosures
None
References
- 1.Henson KE, Reulen RC, Winter DL, Bright CJ, Fidler MM, Frobisher C, Guha J, Wong KF, Kelly J, Edgar AB, McCabe MG, Whelan J, Cutter DJ, Darby SC and Hawkins MM. Cardiac Mortality Among 200 000 Five-Year Survivors of Cancer Diagnosed at 15 to 39 Years of Age: The Teenage and Young Adult Cancer Survivor Study. Circulation. 2016;134:1519–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ and Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52:1213–25. [DOI] [PubMed] [Google Scholar]
- 3.Guellich A, Mehel H and Fischmeister R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflugers Arch. 2014;466:1163–75. [DOI] [PubMed] [Google Scholar]
- 4.Rainer PP and Kass DA. Old dog, new tricks: novel cardiac targets and stress regulation by protein kinase G. Cardiovasc Res. 2016;111:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bender AT and Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. [DOI] [PubMed] [Google Scholar]
- 6.Chen S and Yan C. An update of cyclic nucleotide phosphodiesterase as a target for cardiac diseases. Expert Opin Drug Discov. 2020:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujishige K, Kotera J, Michibata H, Yuasa K, Takebayashi S, Okumura K and Omori K. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J Biol Chem. 1999;274:18438–45. [DOI] [PubMed] [Google Scholar]
- 8.Loughney K, Snyder PB, Uher L, Rosman GJ, Ferguson K and Florio VA. Isolation and characterization of PDE10A, a novel human 3’, 5’-cyclic nucleotide phosphodiesterase. Gene. 1999;234:109–17. [DOI] [PubMed] [Google Scholar]
- 9.Soderling SH, Bayuga SJ and Beavo JA. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci U S A. 1999;96:7071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grauer SM, Pulito VL, Navarra RL, Kelly MP, Kelley C, Graf R, Langen B, Logue S, Brennan J, Jiang L, Charych E, Egerland U, Liu F, Marquis KL, Malamas M, Hage T, Comery TA and Brandon NJ. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther. 2009;331:574–90. [DOI] [PubMed] [Google Scholar]
- 11.Beaumont V, Zhong S, Lin H, Xu W, Bradaia A, Steidl E, Gleyzes M, Wadel K, Buisson B, Padovan-Neto FE, Chakroborty S, Ward KM, Harms JF, Beltran J, Kwan M, Ghavami A, Haggkvist J, Toth M, Halldin C, Varrone A, Schaab C, Dybowski JN, Elschenbroich S, Lehtimaki K, Heikkinen T, Park L, Rosinski J, Mrzljak L, Lavery D, West AR, Schmidt CJ, Zaleska MM and Munoz-Sanjuan I. Phosphodiesterase 10A Inhibition Improves Cortico-Basal Ganglia Function in Huntington’s Disease Models. Neuron. 2016;92:1220–1237. [DOI] [PubMed] [Google Scholar]
- 12.Chen S, Zhang Y, Lighthouse JK, Mickelsen DM, Wu J, Yao P, Small EM and Yan C. A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction. Circulation. 2020;141:217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee K, Lindsey AS, Li N, Gary B, Andrews J, Keeton AB and Piazza GA. beta-catenin nuclear translocation in colorectal cancer cells is suppressed by PDE10A inhibition, cGMP elevation, and activation of PKG. Oncotarget. 2016;7:5353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu B, Lindsey A, Li N, Lee K, Ramirez-Alcantara V, Canzoneri JC, Fajardo A, Madeira da Silva L, Thomas M, Piazza JT, Yet L, Eberhardt BT, Gurpinar E, Otali D, Grizzle W, Valiyaveettil J, Chen X, Keeton AB and Piazza GA. Phosphodiesterase 10A is overexpressed in lung tumor cells and inhibitors selectively suppress growth by blocking beta-catenin and MAPK signaling. Oncotarget. 2017;8:69264–69280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, Small EM, Abe J and Yan C. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016;113:E7116-E7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen S, Wang Z, Xu B, Mi X, Sun W, Quan N, Wang L, Chen X, Liu Q, Zheng Y, Leng J and Li J. The Modulation of Cardiac Contractile Function by the Pharmacological and Toxicological Effects of Urocortin2. Toxicol Sci. 2015;148:581–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Knight W, Chen S, Mohan A and Yan C. Multiprotein Complex With TRPC (Transient Receptor Potential-Canonical) Channel, PDE1C (Phosphodiesterase 1C), and A2R (Adenosine A2 Receptor) Plays a Critical Role in Regulating Cardiomyocyte cAMP and Survival. Circulation. 2018;138:1988–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kabaeva Z, Zhao M and Michele DE. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am J Physiol Heart Circ Physiol. 2008;294:H1667–74. [DOI] [PubMed] [Google Scholar]
- 19.Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, Lyon AR, Harding SE and MacLeod KT. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res. 2017;113:1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maillet A, Tan K, Chai X, Sadananda SN, Mehta A, Ooi J, Hayden MR, Pouladi MA, Ghosh S, Shim W and Brunham LR. Modeling Doxorubicin-Induced Cardiotoxicity in Human Pluripotent Stem Cell Derived-Cardiomyocytes. Sci Rep. 2016;6:25333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kattih B, Shirvani A, Klement P, Garrido AM, Gabdoulline R, Liebich A, Brandes M, Chaturvedi A, Seeger T, Thol F, Gohring G, Schlegelberger B, Geffers R, John D, Bavendiek U, Bauersachs J, Ganser A, Heineke J and Heuser M. IDH1/2 mutations in acute myeloid leukemia patients and risk of coronary artery disease and cardiac dysfunction-a retrospective propensity score analysis. Leukemia. 2021;35:1301–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallace KB, Sardao VA and Oliveira PJ. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ Res. 2020;126:926–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ronnebaum SM and Patterson C. The FoxO family in cardiac function and dysfunction. Annu Rev Physiol. 2010;72:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christidi E and Brunham LR. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021;12:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li N, Lee K, Xi Y, Zhu B, Gary BD, Ramirez-Alcantara V, Gurpinar E, Canzoneri JC, Fajardo A, Sigler S, Piazza JT, Chen X, Andrews J, Thomas M, Lu W, Li Y, Laan DJ, Moyer MP, Russo S, Eberhardt BT, Yet L, Keeton AB, Grizzle WE and Piazza GA. Phosphodiesterase 10A: a novel target for selective inhibition of colon tumor cell growth and beta-catenin-dependent TCF transcriptional activity. Oncogene. 2015;34:1499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertero E, Ameri P and Maack C. Bidirectional Relationship Between Cancer and Heart Failure: Old and New Issues in Cardio-oncology. Card Fail Rev. 2019;5:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF and Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–42. [DOI] [PubMed] [Google Scholar]
- 28.Sawyer DB. Anthracyclines and heart failure. N Engl J Med. 2013;368:1154–6. [DOI] [PubMed] [Google Scholar]
- 29.Francis SH, Busch JL and Corbin JD. cGMP-Dependent Protein Kinases and cGMP Phosphodiesterases in Nitric Oxide and cGMP Action. Molecular Physiology. 2010;62:525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferdous A, Battiprolu PK, Ni YG, Rothermel BA and Hill JA. FoxO, autophagy, and cardiac remodeling. J Cardiovasc Transl Res. 2010;3:355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han X, Zhou Y and Liu W. Precision cardio-oncology: understanding the cardiotoxicity of cancer therapy. NPJ Precis Oncol. 2017;1:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, Yang SM, Jin YL, Xiong GW, Wang P, Snijders AM, Mao JH, Zhang XW and Hang B. A Robust Gene Expression Prognostic Signature for Overall Survival in High-Grade Serous Ovarian Cancer. J Oncol. 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li N, Chen X, Zhu B, Ramirez-Alcantara V, Canzoneri JC, Lee K, Sigler S, Gary B, Li Y, Zhang W, Moyer MP, Salter EA, Wierzbicki A, Keeton AB and Piazza GA. Suppression of beta-catenin/TCF transcriptional activity and colon tumor cell growth by dual inhibition of PDE5 and 10. Oncotarget. 2015;6:27403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou Y, Wren A, Mylarapu N, Browning K, Islam BN, Wang R, Vega KJ and Browning D. Inhibition of colon cancer cell growth by phosphodiesterase inhibitors is independent of cGMP signaling. J Pharmacol Exp Ther. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borneman RM, Gavin E, Musiyenko A, Richter W, Lee KJ, Crossman DK, Andrews JF, Wilhite AM, McClellan S, Aragon I, Ward AB, Chen X, Keeton AB, Berry K, Piazza GA, Scalici JM and da Silva LM. Phosphodiesterase 10A (PDE10A) as a novel target to suppress beta-catenin and RAS signaling in epithelial ovarian cancer. J Ovarian Res. 2022;15:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fusco JP, Pita G, Pajares MJ, Andueza MP, Patino-Garcia A, de-Torres JP, Gurpide A, Zulueta J, Alonso R, Alvarez N, Pio R, Melero I, Sanmamed MF, Rodriguez Ruiz M, Gil-Bazo I, Lopez-Picazo JM, Casanova C, Baz Davila R, Agudo A, Lozano MD, Gonzalez A, Sala N, Ardanaz E, Benitez J, Montuenga L, Gonzalez-Neira A and Perez-Gracia JL. Genomic characterization of individuals presenting extreme phenotypes of high and low risk to develop tobacco-induced lung cancer. Cancer Med. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Q, Cheng F, Song H, Lu W, Zhao J, An X, Liu M, Chen G, Zhao Z and Zhang J. Proteome-Scale Investigation of Protein Allosteric Regulation Perturbed by Somatic Mutations in 7,000 Cancer Genomes. Am J Hum Genet. 2017;100:5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Alexandre RB, Horvath AD, Szarek E, Manning AD, Leal LF, Kardauke F, Epstein JA, Carraro DM, Soares FA, Apanasovich TV, Stratakis CA and Faucz FR. Phosphodiesterase sequence variants may predispose to prostate cancer. Endocr Relat Cancer. 2015;22:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu H, Li F, Zhu Y, Li T, Huang H, Lin T, Hu Y, Qi X, Yu J and Li G. Whole-exome sequencing to identify somatic mutations in peritoneal metastatic gastric adenocarcinoma: A preliminary study. Oncotarget. 2016;7:43894–43906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, McDermott BJ and Grieve DJ. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010;70:9287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barone I, Giordano C, Bonofiglio D, Ando S and Catalano S. Phosphodiesterase type 5 and cancers: progress and challenges. Oncotarget. 2017;8:99179–99202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savai R, Pullamsetti SS, Banat GA, Weissmann N, Ghofrani HA, Grimminger F and Schermuly RT. Targeting cancer with phosphodiesterase inhibitors. Expert Opin Investig Drugs. 2010;19:117–31. [DOI] [PubMed] [Google Scholar]
- 43.Das A, Durrant D, Mitchell C, Mayton E, Hoke NN, Salloum FN, Park MA, Qureshi I, Lee R, Dent P and Kukreja RC. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc Natl Acad Sci U S A. 2010;107:18202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowther FB, Wei W, Dawson TP, Ashton K, Singh A, Madiesse-Timchou MP, Thomas DG, Darling JL and Warr T. Cyclic nucleotide phosphodiesterase-1C (PDE1C) drives cell proliferation, migration and invasion in glioblastoma multiforme cells in vitro. Mol Carcinog. 2016;55:268–79. [DOI] [PubMed] [Google Scholar]
- 45.Shimizu K, Murata T, Watanabe Y, Sato C, Morita H and Tagawa T. Characterization of phosphodiesterase 1 in human malignant melanoma cell lines. Anticancer Res. 2009;29:1119–22. [PubMed] [Google Scholar]
- 46.Chen S, Knight WE and Yan C. Roles of PDE1 in Pathological Cardiac Remodeling and Dysfunction. J Cardiovasc Dev Dis. 2018;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ and Kass DA. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96:100–9. [DOI] [PubMed] [Google Scholar]
- 48.Fiedler B, Feil R, Hofmann F, Willenbockel C, Drexler H, Smolenski A, Lohmann SM and Wollert KC. cGMP-dependent protein kinase type I inhibits TAB1-p38 mitogen-activated protein kinase apoptosis signaling in cardiac myocytes. J Biol Chem. 2006;281:32831–40. [DOI] [PubMed] [Google Scholar]
- 49.Das A, Xi L and Kukreja RC. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008;283:29572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fisher PW, Salloum F, Das A, Hyder H and Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111:1601–10. [DOI] [PubMed] [Google Scholar]
- 51.Shimojo T, Hiroe M, Ishiyama S, Ito H, Nishikawa T and Marumo F. Nitric oxide induces apoptotic death of cardiomyocytes via a cyclic-GMP-dependent pathway. Exp Cell Res. 1999;247:38–47. [DOI] [PubMed] [Google Scholar]
- 52.Taimor G, Hofstaetter B and Piper HM. Apoptosis induction by nitric oxide in adult cardiomyocytes via cGMP-signaling and its impairment after simulated ischemia. Cardiovasc Res. 2000;45:588–94. [DOI] [PubMed] [Google Scholar]
- 53.Wu CF, Bishopric NH and Pratt RE. Atrial natriuretic peptide induces apoptosis in neonatal rat cardiac myocytes. J Biol Chem. 1997;272:14860–6. [DOI] [PubMed] [Google Scholar]
- 54.Han B, Fixler R, Beeri R, Wang Y, Bachrach U and Hasin Y. The opposing effects of endothelin-1 and C-type natriuretic peptide on apoptosis of neonatal rat cardiac myocytes. Eur J Pharmacol. 2003;474:15–20. [DOI] [PubMed] [Google Scholar]
- 55.Arstall MA, Sawyer DB, Fukazawa R and Kelly RA. Cytokine-mediated apoptosis in cardiac myocytes: the role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ Res. 1999;85:829–40. [DOI] [PubMed] [Google Scholar]
- 56.Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA and Butt E. Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods. 2008;5:277–8. [DOI] [PubMed] [Google Scholar]
- 57.Kato T, Muraski J, Chen Y, Tsujita Y, Wall J, Glembotski CC, Schaefer E, Beckerle M and Sussman MA. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J Clin Invest. 2005;115:2716–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pilz RB and Casteel DE. Regulation of gene expression by cyclic GMP. Circ Res. 2003;93:1034–46. [DOI] [PubMed] [Google Scholar]
- 59.Johlfs MG and Fiscus RR. Protein kinase G type-Ialpha phosphorylates the apoptosis-regulating protein Bad at serine 155 and protects against apoptosis in N1E-115 cells. Neurochem Int. 2010;56:546–53. [DOI] [PubMed] [Google Scholar]
- 60.Suenobu N, Shichiri M, Iwashina M, Marumo F and Hirata Y. Natriuretic peptides and nitric oxide induce endothelial apoptosis via a cGMP-dependent mechanism. Arterioscler Thromb Vasc Biol. 1999;19:140–6. [DOI] [PubMed] [Google Scholar]
- 61.Fukuo K, Hata S, Suhara T, Nakahashi T, Shinto Y, Tsujimoto Y, Morimoto S and Ogihara T. Nitric oxide induces upregulation of Fas and apoptosis in vascular smooth muscle. Hypertension. 1996;27:823–6. [DOI] [PubMed] [Google Scholar]
- 62.Soh JW, Mao Y, Liu L, Thompson WJ, Pamukcu R and Weinstein IB. Protein kinase G activates the JNK1 pathway via phosphorylation of MEKK1. J Biol Chem. 2001;276:16406–10. [DOI] [PubMed] [Google Scholar]
- 63.Kavazis AN, Smuder AJ and Powers SK. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J Appl Physiol (1985). 2014;117:223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hannan RD, Jenkins A, Jenkins AK and Brandenburger Y. Cardiac hypertrophy: a matter of translation. Clin Exp Pharmacol Physiol. 2003;30:517–27. [DOI] [PubMed] [Google Scholar]
- 65.Akazawa H and Komuro I. Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res. 2003;92:1079–88. [DOI] [PubMed] [Google Scholar]
- 66.Bolger AM, Lohse M and Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liao Y, Smyth GK and Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 69.Love MI, Huber W and Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marini F and Binder H. Development of Applications for Interactive and Reproducible Research: a Case Study. Genomics and Computational Biology. 2016;3:e39. [Google Scholar]
- 71.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu G, Wang LG, Han Y and He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kanehisa M and Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Detailed experimental methods and Major Resource Table are provided in the Supplemental Material. Data will be made available upon reasonable request, by contacting the corresponding author. Raw and processed RNA-seq are available in the NCBI GEO database (GSE224157). The graphic abstract was generated using BioRender.com.