

Abstract
The global initiatives on sustainable and green energy resources as well as large methane reserves have encouraged more research to convert methane to hydrogen. Catalytic decomposition of methane (CDM) is one optimistic route to generate clean hydrogen and value-added carbon without the emission of harmful greenhouse gases, typically known as blue hydrogen. This Review begins with an attempt to understand fundamentals of a CDM process in terms of thermodynamics and the prerequisite characteristics of the catalyst materials. In-depth understanding of rate-determining steps of the heterogeneous catalytic reaction taking place over the catalyst surfaces is crucial for the development of novel catalysts and process conditions for a successful CDM process. The design of state-of-the-art catalysts through both computational and experimental optimizations is the need of hour, as it largely governs the economy of the process. Recent mono- and bimetallic supported and unsupported materials used in CDM process have been highlighted and classified based on their performances under specific reaction conditions, with an understanding of their advantages and limitations. Metal oxides and zeolites have shown interesting performance as support materials for Fe- and Ni-based catalysts, especially in the presence of promoters, by developing strong metal–support interactions or by enhancing the carbon diffusion rates. Carbonaceous catalysts exhibit lower conversions without metal active species and largely result in the formation of amorphous carbon. However, the stability of carbon catalysts is better than that of metal oxides at higher temperatures, and the overall performance depends on the operating conditions, catalyst properties, and reactor configurations. Although efforts to summarize the state-of-art have been reported in literature, they lack systematic analysis on the development of stable and commercially appealing CDM technology. In this work, carbon catalysts are seen as promising futuristic pathways for sustained H2 production and high yields of value-added carbon nanomaterials. The influence of the carbon source, particle size, surface area, and active sites on the activity of carbon materials as catalysts and support templates has been demonstrated. Additionally, the catalyst deactivation process has been discussed, and different regeneration techniques have been evaluated. Recent studies on theoretical models towards better performance have been summarized, and future prospects for novel CDM catalyst development have been recommended.
1. Introduction
Global energy consumption is rapidly increasing, causing an increase in CO2 emissions in the atmosphere, which in turn contributes to global warming.1−3 As climate change continues to take its toll, the development of clean and renewable energy sources is imperative to alleviate high usage of fossil fuels.4 Hydrogen, having a mass calorific value of ∼148 MJ/kg, is envisioned as a fuel of the future that can serve as an alternative sustainable energy carrier.5,6 Besides, it is a critical feedstock to various manufacturing industries, such as oil refineries, ammonia production, fuel-cell electric vehicles, aircrafts, and methanol production.4 The main pathways for H2 production are from fossil fuels (hydrocarbons), water (electro- or photolysis and thermochemical), or photosynthetic microorganisms (biological) (Table 1). However, low efficiencies and high production costs of H2 generation from water and biological routes make these technologies still in their early stages of development, with a large scope of further research needed.7,8 Globally, about 48% of hydrogen is produced from natural gas (steam methane reforming), 30% is produced from oil refining, 18% is produced from coal gasification, and only 4% is produced from water electrolysis. A major portion of H2 generated through steam methane reforming (SMR) is attributed to the availability of vast reserves of methane (a major component of natural gas), its easy handling, and the high H/C content.9
Table 1. Comparative Summary of Various Hydrogen Production Technologies.
| hydrogen production technologies | reactions involved | advantages | challenges |
|---|---|---|---|
| source: water | |||
| electrolysis | 2H2O → O2 + 4H+ + 4e–; ΔH = 285 kJ/mol, ΔG = 237 kJ/mol | • carbon-free H2 generation | • low efficiency of electrolyzer over a range of operational conditions |
| • possibility of using renewable/nuclear feedstock | • high capital and production costs | ||
| • low operational life of electrodes | |||
| • in early stages of development | |||
| photochemical | 2H2O → O2 + 4H+ + 4e– ; ΔH = 285 kJ/mol, ΔG = 237 kJ/mol | • ability to generate negligible or no greenhouse gas emissions | • only developed at lab-scale |
| • no intake of fossil fuels | • low efficiency and stability of photoelectrodes/photocatalysts due to high reaction impedance | ||
| • low operating temperatures | • high risk of photocorrosion of active materials | ||
| • spontaneous and rapid back reactions | |||
| • overwhelming production costs | |||
| thermochemical | 2H2O → O2 + 4H+ + 4e– ; ΔH = 285 kJ/mol, ΔG = 237 kJ/mol | • potentially negligible or no greenhouse gas emissions | • highly energy intensive with requirement of temperature up to 2000 °C |
| • follows a closed loop cycle consuming only water and producing only H2 and O2 | • low efficiency of thermochemical reactors and materials | ||
| • chemicals can be reused within a cycle | • high cost of solar concentrating mirrors | ||
| • developed at lab scale only | |||
| source: organic matter | |||
| biomass gasification | C6H12O6 + O2 + H2O → CO + CO2 + H2 + other byproducts (taking glucose molecules only) | • high efficiency and rapid process | • emission of COx and other components such as H2S, NH3 and tar |
| • availability of abundant and cheap raw materials | • requirement of gas separation and COx removal processes | ||
| • low cost syngas production | • high reactor and feedstock costs | ||
| • quality of H2 produced is poor | |||
| • demonstrated at lab scale only | |||
| photobiological | 2H2O → O2 + 2H2 ; ΔH = 285 kJ/mol, ΔG = 237 kJ/mol | • can be used in a wide range of water conditions | • in early stages of development |
| • the process is self-sustaining | • low efficiency and sustainability of microorganisms | ||
| • designing robust reactor configurations that can use sunlight effectively and produce H2 | |||
| source: fossil fuels (natural gas/methane) | |||
| steam methane reforming (SMR) | CH4+ H2O ↔ CO + 3H2; ΔH298K = 206 kJ/mol | • low-cost H2 production technology | • emission of greenhouse gases in to atmosphere |
| • high H/C ratio | • requirement of downstream separation and purification processes | ||
| • high efficiency | • a complex system of reactions including water-gas shift reaction and pressure-swing adsorption reactions | ||
| dry reforming of methane (DRM) | CH4 + CO2 ↔ 2CO + 2H2; ΔH = 247 kJ/mol | • utilizes CO2 to produce H2 | • more energy intensive than SMR |
| • can be used for the Fischer–Tropsch process | • high equipment cost | ||
| • complex downstream processes for H2 purification | |||
| • emission of greenhouse gases | |||
| partial oxidation of methane (POM) | CH4 + 1/2O2 ⇆ CO + 2H2; ΔH = −36 kJ/mol | • high efficiency and selectivity | • high emissions of COx and possibility of emissions of NOx |
| • very short residence time | • formation of soot byproduct | ||
| • requirement of pure O2 | |||
| catalytic decomposition of methane (CDM) | CH4 → C(s) + 2H2; ΔH298K = 75.4 kJmol | • simple and one-step process | • in early stages of development |
| • no COx or NOx emissions | • low stability of catalysts, requiring effective regeneration techniques | ||
| • produces high quality H2 | • unreacted methane in out stream | ||
| • produces solid carbon as a byproduct, which is easier to separate | |||
| • solid carbon in the form of value-added nanocarbons is generated | |||
Additionally, research is constantly evolving in producing H2 through dry reforming of methane (DRM) and partial oxidation of methane (POM). However, the product contains CO, CO2, and unconverted methane, demanding further downstream processes involving water–gas shift reaction, catalytic converters to convert CO to CO2, and subsequent separations to meet environmental regulations. As an alternative, catalytic decomposition of methane (CDM) provides an interesting route as it does not produce hazardous greenhouse gases, is moderately endothermic, and the byproduct is solid carbon. The H2 produced is pure, and the process does not require any separation/purification downstream processes.10,11
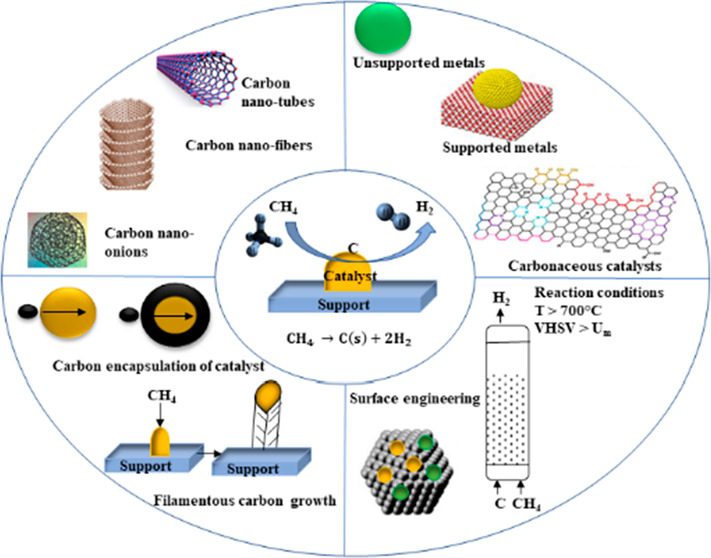
Despite various advantages of CDM over SMR process, it suffers from catalyst instability, raising economic concerns over catalyst reactivation, residual methane in the outlet, and low-grade nanocarbon byproducts. The desirable qualities of CDM catalysts are their high conversion, thermochemical stability, good carbon capacity, capability to withstand attrition, and light weight. To date, various transition metals (Ni, Co, Fe, and Cu), metal oxides (NiO, CuO, and FeO), noble metals (Pt, Pd, Rh, and Au), and carbon and their composites have been widely studied for reducing the activation energy barrier in the methane decomposition reaction for more sustainable and economic CDM process. Metal catalysts give a very high initial conversion but undergo fast deactivation due to the encapsulation by deposited carbon or deposition of coke over catalyst pores and internal cavities. On the other hand, carbonaceous catalysts are envisioned as advantageous due to their high-temperature resistance, fuel flexibility, insensitivity to sulfur poisoning, better availability at commercial scale, better durability than a metal catalyst, formation of high-value carbon, low cost, high and tunable surface area, and porosity.12−14
The majority of literature on CDM focuses on the types of catalysts and reactors used for this reaction.9,15,16 Often the published literature addresses different types of mono- and bimetallic catalysts on various templates, reporting the conversion and the types of carbon formed.17−19 This Review, however, begins with discussing the reaction mechanism of CDM, followed by a summary of catalysts widely studied by researchers, with a specific focus on porous carbonaceous catalysts and templates. Recent research results have been used to provide an insight into the catalytic capabilities of both commercial carbons (activated carbon, carbon black, carbon nanotubes, and metal-doped carbons) and noncommercial carbons (mesoporous carbons) toward the CDM reaction.22−20 Carbon materials have proven advantageous as catalysts in energy conversion and storage technologies, such as electrochemical and photoelectrochemical conversions, conversions through biological routes, and pyrolysis of natural gas.20−171 The use of carbon-based materials as catalysts has a long history, particularly to CDM in the recent past. Recently, various fascinating results in terms of the stability of catalyst and the yield of carbon nanomaterials have been reported in literature using carbon-based catalysts.21−172 This Review thus lays emphasis on reaction conditions, sustained catalyst activity, product composition, and the nature of carbon formed over the carbonaceous materials. Compilation of the literature indicates the use of various carbon materials, such as activated carbon (AC), carbon black (CB), graphite, carbon nanotubes (CNTs), and ordered mesoporous carbons (OMCs) as potential catalysts for CDM. Biomass-derived AC has also shown promising results in terms of activity and stability, and it could be a cheap and renewable source, potentially impacting the economics at a commercial scale. The Review also demonstrates a microscopic understanding of catalyst regeneration, providing a futuristic perspective for guiding research in hydrogen production through CDM. Additionally, recent studies in theoretical research done in this field have been summarized to attain an understanding of the current challenges and future requirements.
2. Catalytic Decomposition of Methane (CDM)
A methane molecule (CH4) consists of four identical sp3-hybridized C–H bonds and thus is a highly stable molecule at room-temperature and pressure, having bond-energy of 435 kJ/mol. The noncatalytic thermal decomposition of methane is highly endothermic (ΔH298K = 75.6 kJ/mol) and occurs at temperatures of or above 900 °C, where energy is required for C–H bond breaking.24 Methane degradation follows Le Chatelier’s principle, where low pressure and high temperature navigate the forward reaction. The influence of reaction temperature and pressure on the equilibrium conversion of methane can be seen in Figure 1(a).171 CDM or nonoxidative pyrolysis of methane, on the other hand, is a moderately endothermic reaction with comparatively lower energy demand. The activation energy of a CH4 molecule on the catalyst surface determines the extent of interaction between the active metal and the catalyst surface.173,26 A suitable catalyst reduces the activation energy and increases the rate of reaction. Figure 1(b) gives an estimation of the decrease in activation energy of the methane decomposition reaction on a Ni-based catalyst.172
Figure 1.

(a) Influence of the reaction parameters on the equilibrium conversion of methane (temperature and pressure). Reprinted with copyright permission from ref (171). Copyright Elsevier 2021. (b) Effect of using a catalyst in the CDM reaction. Reprinted with copyright permission from ref (172). Copyright Elsevier 2017. (c) Illustration of a gas–solid heterogeneous reaction mechanism for the formation of carbon nanomaterials (CNMs). Reprinted with copyright permission from ref (30). Copyright Elsevier 2021. Copyright 2020 Science Press and Dalian Institute of Chemical Physics, Chinese Academy of Sciences.
CDM is a heterogeneous catalytic reaction, which is a reversible two-phase gas–solid reaction. Methane molecules from the gas phase get adsorbed on the catalyst surface at interface 1 (Figure 1(c)), followed by the CH4 cracking.30 Adsorbed C atoms react with the metal catalyst to form a metastable metal-carbide phase, which under specific reaction conditions decompose, liberating C atoms to the metal subsurface. The “metallic phase” is a driving force for diffusion and precipitation of C atoms through the rear-end of the catalyst, forming various nanostructured filamentous carbons. At interface 2, partial exfoliation of carbonaceous nanomaterials (CNMs) takes place due to the atmospheric impact of produced hydrogen at higher temperatures due to the removal of any O-containing gases or simply water vapors. The equilibrium constant of reaction, Keq, depends on the type and nature of the catalyst used, and can be written as26
where Cs is the carbon solubility, P represents partial pressure of CH4 and H2, and the diffusion of filamentous carbon is determined by Keq. The temperature required for reasonable methane conversion is reported to be ∼500–700 °C for Ni-based catalysts, 700–900 °C for Fe-based catalysts, and 850–950 °C for carbon-based catalysts.15 This process generates solid carbons as byproducts on the catalyst surface, such as amorphous carbon or graphitic carbon, which have different nanostructures depending on the catalyst used and process conditions.27−29 The catalytic effect of different forms of amorphous carbons is also proven by several studies; however, the activity largely depends on the form of carbon produced. Factors, such as the design of the catalyst, the size of the active metal and the corresponding metal–support interaction (MSI), the type and concentration of the promoter, and other reaction conditions determine the quality and yield of graphitic nanocarbons generated. Generally, graphitic filamentous nanocarbons in the form of CNTs, carbon nanofibers (CNFs), and carbon nano-onions (CNOs) are produced by metallic catalysts over metal-oxide supports, while amorphous carbons are generated by undoped carbonaceous catalysts. Ideally, the produced carbon would be autocatalytic, but the literature shows that the deposited carbon is not as active as the original carbon catalyst and eventually degrades the overall catalyst performance.
Production of the solid carbon may not have enough avenues for consumption if the formation tends to be amorphous carbon, as 1/3 of the mass of methane would be converted to carbon. It is highly favorable if the formed carbon is graphitic, as it has high value and tremendous emerging applications. The nanostructured carbon is of high importance and commercial value due to its high demand in electrical devices, hydrogen storage materials, pharmaceuticals, nanosensors, water-purification, and metal-extraction industries.
2.1. CDM Reaction Mechanism
The general reaction mechanism of CDM is similar for all types of catalysts and supports; however, a comprehensive mechanism has yet to be fully elucidated. The initiation of the CDM process takes place by chemisorption of a CH4 molecule on the catalyst surface, followed by progressive C–H bond cleavage, leading to elemental carbon and hydrogen. The elemental hydrogen aggregates to form H2 molecules, followed by H2 desorption. The process of carbon formation is well-known to be influenced by the presence of hydrogen as well, since hydrogen tends to saturate the unsaturates, thereby controlling the rate of formation of carbon. The elemental carbon either accumulates and encapsulates the catalyst surface, resulting in catalyst decay, or diffuses over the catalyst rear-face from the leading face due to the difference in concentration. This nucleation of carbon with a high carbon-to-hydrogen ratio results in several polyaromatic structured carbons, amorphous and graphitic carbons, and nanostructured carbons, such as filaments, rods, and tubes.31 The catalyst stability, type of carbon formed, and overall performance depend upon the reaction conditions, support properties, promoter, and synthesis methods. The reaction kinetics for hydrogen formation is highly governed by the reaction temperature and pressure. The order of reaction and the activation energy for each transition state are, however, still challenging to determine due to complex characteristics of the process.32 Addition of support to the active-metal catalysts enhances all the parameters deciding the catalytic efficiency, such as the surface electronic state, crystal size, metal dispersibility, composition, specific surface area, and structural properties. The formation of a metal–promoter alloy allows the separation of metal active sites from each other, which in turn enhances the dispersibility and rate of carbon diffusion and hence the stability of catalyst. This causes the interaction of deposited carbon with the metal to decrease, thereby minimizing the encapsulation of carbon, and promotes the growth of filamentous carbon as well as the chemisorption of methane. However, an optimal metal loading is critical to balance the metal agglomeration and number of active metal sites. Apart from these factors, the catalyst synthesis method also plays a vital part in the overall activity. To date, CDM catalysts have been synthesized via wet impregnation, sol–gel, incipient wetness, fusion, and coprecipitation methods, resulting in different MSIs, porosities, and active metal dispersibility. In general, the sol–gel method results in a strong MSI and high metal reducibility, while the wet impregnation method produces catalysts with a weak MSI and thus less durability.
2.1.1. Dissociative Methane Adsorption
The mechanism of CDM reaction over metallic and carbonaceous catalysts widely follows the dissociative methane adsorption and can be described in five stages.31
-
1.First, CH4 molecule gets chemisorbed over the catalyst surface and the C atoms chemisorb over the surface of catalyst by donating nonbinding electron pairs to unfilled d orbitals of active metal to form catalyst–C σ bonds. At the same time, the first C–H bond-breaking also takes place (Figure 2(a)).

Here (g) and (a) denote the species in gaseous and adsorbed states, respectively. The activation energy of the first C–H bond (∼0.003 eV) is lesser than that of progressive C–H bonds (∼0.2–0.3 eV) (Figure 2(b)). This means that the chemisorption of CH4 and its cracking can be treated simultaneously. This is the rate-determining step of the whole reaction, as it requires the highest amount of activation energy compared to the other C–H bonds.
-
2.The first reaction is then followed by a number of progressive C–H bond-breaking steps.
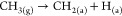
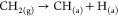
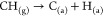
-
3.Emission of H2 gas after the formation of H2 molecules, resulting from the gathering of adsorbed H atoms.
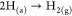
-
4.Atomic carbon either accumulates as the encapsulated carbon or diffuses through the tail-end as a result of the concentration gradient. Upon reaching saturation, carbon gets deposited on the catalyst rear-end.
where (c) denotes the crystalline phase of carbon.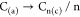
-
5.
Depending upon the growth of carbon nuclei, a variety of carbon nanostructures grow in the catalyst rear-side. The final structure and morphology of deposited carbon depend on the catalyst’s original properties, such as the nature, structure, and activity of the catalyst.
Figure 2.

(a) Schematic of the CDM reaction via dissociative methane adsorption. (b) Relative energies of CH4 decomposition on the Ni–Al2O3 (001) catalyst surface. Reprinted with copyright permission from ref (31). Copyright Elsevier 2017.
The reaction on a carbonaceous catalyst begins with the interaction of CH4 molecules with chemically reactive high-energy surfaces (HES) or other energetic abnormalities of carbon crystallites for C–H bond-breaking and to form new hexagonal layers of C–C at the periphery of carbon crystallites.33−36 However, a complete understanding of the process mechanism at the atomic level has yet to be achieved. It is reported that two simultaneous events take place for new carbon phase build-up, viz., carbon nucleation (Ea = 317 kJ/mol) and carbon crystallites growth (Ea = ∼227 kJ/mol).33 Carbon nucleation is proportional to the carbon surface area or HES concentration. The activation energies indicate that the growth of carbon crystallites is faster than that of nucleation. In the carbonaceous catalysts, this results in the production of a pseudo-ordered carbon (also known as turbostratic carbon), which ultimately leads to a decrease in the active surface area and the concentration of HES, thereby reducing the catalytic activity. Thus, based on the difference in crystallinity and surface morphologies, the difference in the catalytic activities over different carbon catalysts could be explained.
2.1.2. Nondissociative Methane Adsorption
Although the dissociative adsorption mechanism is widely accepted for methane decomposition, the initial disintegration of CH4 over catalysts is still debatable. Grabke, in 1965, proposed the concept of nondissociative adsorption of methane for some catalysts, such as γ-iron.34 Under the concept of a nondissociative mechanism, methane adsorption takes place first, followed by C–H bond breaking (Figure 3). Methane decomposition on (110) surfaces of Pt and Ir has been observed to follow this mechanism of adsorption.31
Figure 3.

Schematic diagram of a nondissociative model of methane adsorption. Reprinted with copyright permission from ref (31). Copyright Elsevier 2017.
Such a mechanism takes place under a low-energy incident beam on an Ir (110) surface where only a portion of CH4 molecules (<25%) are cracked while a major portion remained only in the adsorbed state. The following equation shows the initiation step of nondissociative methane adsorption on carbon. This first step is then followed by all the same steps as shown in dissociative adsorption, and the first C–H breaking step is considered the rate-determining step.
Various studies have been carried out to investigate the C–H bond activation in a methane molecule on several catalysts. It has been observed that C–H bond activation can take place by either a radical or polar mechanism. In the radical mechanism, C–H bond cleavage is homolytic, forming free-radical intermediates. The H radicals form the surface hydroxyl groups in the presence of O-containing species, and the methyl radical is released as gas.37−40 On the other hand, the C–H bond breaking is heterolytic in the polar mechanism and forms the hydroxyl groups with surface-bound O species present as surface hydration along with the methyl metallic species.41,42 The possibility of a radical mechanism of C–H bond cleavage is likely favored through oxidative C–H activation on metal oxides. The mechanism of C–H cleavage through nonoxidative activation has yet to be explored and offers complexity, as both Lewis acid and base sites are involved in the reaction simultaneously. Cholewinski et al. studied methane activation on γ-Al2O3 and developed a relationship between its physical characteristics and the catalytic activity using first-principles calculations.40 Various other studies were conducted by researchers to gain clear insight into the C–H bond activation.43−47 The majority of studies reflect the initial methane adsorption (C–H bond breaking) as the rate-determining step that depends on the structural aspects of the catalyst, such as type and adjacent coordination states of the metal, and the effect of promoters and supports. However, in a study by Snoeck et al., the adsorption of methane over Ni catalyst was observed to follow a dissociation mechanism, with the desorption of H from the surface of catalyst forming the rate-determining step.43 In a model developed by Alstrup et al., methane adsorption was seen to take place through the radical mechanism where the dissociative adsorption of methane was the rate-determining step.44 However, the C–H activation mechanism cannot be completely generalized but varies according to the reaction conditions.
2.2. Material Selection and Criteria
With respect to the material development, various mono- and bimetallic as well as carbon-based catalysts have been studied for the CDM process. Transition metals, such as Fe, Co, and Ni, have been widely studied owing to their partially filled 3d orbitals. These partially filled 3d orbitals promote C-H breaking by transferring electrons to the unoccupied antibonding orbitals of hydrocarbon molecule. Unsupported undoped metal catalysts show conversions of 20–40% at lower temperatures (500–650 °C), while the conversion may go higher to 85% at higher temperatures (700–900 °C) upon loading with noble or rare-earth metals. However, in all unsupported catalysts, sustenance of conversion and stability are detrimental parameters. The use of a support for improving the catalytic activity of metals was first proposed in 1935.28 Supported metal catalysts exhibit better pore-size distributions and specific surface areas, with an initial conversion ranging from 70% to 85% at temperatures between ∼600 and 800 °C. It is reported that Ni supported on various types of zeolites, such as USY, SiO2, HY, and SBA-15, showed variation in overall performances in terms of both catalytic conversion and stability (Figure 4(a)).45 The order of conversion was observed to be HY ∼ SiO2 > USY > SBA-15, while carbon yield (whiskers of nanofilaments) followed the order of HY > USY > SiO2 > SBA-15. Such an observation was attributed to a decrease in the size of Ni and the acidity of the support material.
Figure 4.

Methane conversion over (a) 30% Ni on various zeolites at 550 °C45 and (b) 20% Co on various supports at 500 °C.46 Panel a was reprinted with copyright permissions from ref (45). Copyright Elsevier 2007. Panel b was reprinted with copyright permissions from (46). Copyright American Chemical Society 2004.
Metal oxides are widely studied supports, which include MgO, Al2O3, SiO2, and TiO2. Takenaka et al. studied Co on various metal oxide-based supports, such as Al2O3, MgO, SiO2, and TiO2. The catalytic activity and stability followed the order of Co/Al2O3 > Co/MgO > Co/TiO2 > Co/SiO2 (Figure 4(b)). Deposition of Co over different supports resulted in various sizes of metal crystallites, where the smaller crystallites corresponding to Co/Al2O3 exhibited the highest conversion.46 Support composition, type, and structure are other critical parameters that establish the overall activity of catalysts. In the Ni/MgO catalyst, the presence of MgO nanoflakes having a particle size of ∼22 nm increased its overall activity, with a H2 yield of ∼50% and stability for 6 h. Carbon produced was mainly multiwalled carbon nanotubes (MWCNTs) and a few layers of graphene sheets, with no significant encapsulating carbon.48 Supports with mesoporous structures have shown better performance compared to those with micropores. Rastegarpanah et al. synthesized a nickel-loaded mesoporous MgO catalyst using the “one-pot” evaporation-induced self-assembly method and reported an improved conversion of 65%. Other factors that are responsible for influencing the catalytic performance are the electronic state, morphological state, dispersibility, pore structure, synthesis procedure, and catalyst composition.49,50
The development of alloy or bimetallic catalysts of Ni, Fe, and Co is believed to overcome issues of instability and low performance as is the case of pure metallic catalysts.51,52 It has been observed that a bimetallic system exhibits better conversion and higher stability, which could be attributed to the modifications in structural and electronic properties during the formation of alloys.50 In such a system, a stable metal oxide network is formed which prevents catalytic agglomeration and reduces the deactivation.53 Copper, having fully filled 3d orbitals, is often used as a promoter with other active metals. The addition of Cu to Ni or Fe during heat treatment is reported to highly enhance their performance and longevity. It has also been observed that adding Cu as a promoter with Ni, Fe, and Co could reduce carbon encapsulation and increase the formation of single or multiwalled carbon nanotubes (CNTs).54Table 2 shows a comparison of indicative catalytic performances of various catalysts reported in the literature.
Table 2. Summary of Recently Studied Catalytic Performances of Unsupported, Metal-Supported, Carbon-Based, and Carbon-Supported Catalysts.
| catalyst | initial CH4 conversion (%) | reaction temperature (°C) | initial H2 yield mmol/gcat·min | stability (hours) | ref |
|---|---|---|---|---|---|
| unsupported metal catalysts | |||||
| Ni | 25 | 550 | 2.0 | 2 | (54) |
| NiO | 575 | 11.7 | 2.5 | (55) | |
| Fe | 75 | 900 | 2.75 | 10 | (56) |
| Ni–Cu | 85 | 750 | 8 | 5 | (54) |
| 10% Ni–1% Pd | 57 | 600 | 5.8 | (57) | |
| f-Fe65–Al3.7 | 70 | 750 | 4.1 | 6.6 | (58) |
| NiCuLa | 79 | 700 | 22 | (59) | |
| NiMgAlMo | 36 | 650 | 55 | 0.5 | (60) |
| metal/support catalysts | |||||
| 50% Ni–10% Cu/SiO2 | 83 | 750 | 8.3 | (61) | |
| 50% Fe/Al2O3 | 45 | 700 | 3 | (62) | |
| 60% Ni/Al2O3 | 60 | 700 | 6 | (63) | |
| 9% Co/Al2O3 | 35 | 700 | 7 | (64) | |
| 40% Fe/MgO | 40 | 700 | 3 | (62) | |
| 50% Co/MgO | 700 | 7 | (64) | ||
| 55% Ni/MgO | 75 | 675 | 3.8 | 2.6 | (48) |
| 50% Fe/TiO2 | 17 | 700 | 3 | (64) | |
| 55% Ni/2MgO–Al2O3 | 55 | 600 | <6 | (65) | |
| 9% Ni–1% Co/Al2O3–TiO2 | 72 | 650 | 1 | (66) | |
| Co3–xAlxO4 | 44 | 500 | 3.3 | (67) | |
| Fe/CeO2 | 54 | 800 | 2.2 | 6 | (68) |
| Fe/WO3–ZrO2 | 92 | 800 | 1.8 | 4 | (69) |
| Ni–Fe/SiO2 | 59 | 650 | 2.8 | 1 | (70) |
| Ni–Fe/MgO | 64 | 700 | 2.6 | 3 | (71) |
| Ni/CeO2 | 46 | 550 | 0.08 | 4 | (72) |
| carbonaceous catalysts | |||||
| carbon black | 85 | 1120 | 2.5 | (73) | |
| activated carbon | 67 | 900 | >10 | (74) | |
| carbon black | 52 | 900 | >10 | (74) | |
| mesoporous carbon | 45 | 900 | >10 | (74) | |
| carbon nanofiber | 15 | 900 | >10 | (74) | |
| carbon black | 12 | 900 | 0.41 | 2 | (75) |
| metal/carbon catalysts | |||||
| 30% Fe/AC | 18 | 800 | 2.5 | >10 | (76) |
| K2CO3/C | 89 | 850 | 0.6 | 10 | (77) |
| 10% Ni/C (coal char) | 80 | 850 | 10 | 10 | (78) |
| 8% Ni/C | 55 | 850 | 1.3 | 10 | (79) |
| 10% Fe/AC | 29 | 850 | 1.1 | 8.3 | (76) |
| Fe–Al2O3/AC | 35 | 850 | 5 | (80) | |
| Ru/AC | 21 | 800 | 0.26 | 60 | (81) |
| Ru/activated biochar | 51 | 800 | 0.23 | 60 | (81) |
Metals loaded on Al2O3 and TiO2 have lower stability than those loaded on SiO2 at approximately the same temperature. Moreover, metals over CeO2 and ZrO2 are required to operate at higher temperatures for higher CH4 conversions. Although some metal-based catalysts have shown promising methane conversions and H2 yields, high cost, and low stability still remain a challenge for upscaling of the CDM process. As such, researchers have shifted their focus to carbon-based materials for use as catalysts and supports, owing to their low cost and better thermal and chemical resistance than their metallic counterparts. Although initial CH4 conversion in the case of carbon-based catalysts is lower (20–60%), they exhibit appreciably better stability compared to metal-based catalysts. Various structural modifications to carbonaceous catalysts have been reported to enhance high-energy active sites, which include metal/nonmetal doping to increase the density of functional groups and to modify the surface chemistry and electronic structure.82−84 Literature shows that metals impregnated on carbon supports show better stability and long-term conversion efficiency (Table 2); however, the temperature requirements for such catalysts are high (from 700 to 900 °C).
3. Carbon Catalysts
To date, the state-of-art development of a novel catalyst to withstand long operational hours and generate a high H2 yield has yet to be accomplished. The commercial appeal of this process lies in the continuous regeneration of catalysts where constant separation of solid carbon from the catalyst would be required, posing major hurdles to CDM commercialization. As such, carbon catalysts seem to avoid the necessity of a solid carbon/catalyst separation step. Different forms of carbon catalysts classified based on crystallinity and order are reported in the literature. The higher the order, the lower the concentration of high-energy sites (HES) and hence the activity. In the case of disordered carbons, such as amorphous carbons, the high-energy centers are significant due to the edges and corners of crystallites resulting in discontinuities and the irregular arrays of carbon bonds, which create free valences. Figure 5 shows the classification of carbon-based catalysts based on their degrees of order.85−87
Figure 5.

Classification of carbon-based catalysts based on degrees of order.
The surface density of high-energy sites increases with a decrease in the crystallite size and an increase in the active surface area. High-energy sites also get affected inversely by the degree of order or graphitization.88,89 Activated carbon (AC) and carbon black (CB) are highly active in an attempt to satisfy their valency and stabilize energies compared to the ordered carbons. During the CDM reaction, methane reacts with highly energetic edges and corners of carbon crystallites. This results in the dissociation of C–H bonds in the CH4 molecule and facilitates the formation of new C–C bonds in a hexagon layer of carbon. The variation in the rate of methane decomposition on various carbon materials could thus be determined by the difference in their crystallinity, surface morphology, surface area, and particle size.
AC, CB, mesoporous carbon, coal char, graphene, carbon nanotubes (CNTs), fullerenes, and ordered mesoporous carbon (OMC) have been extensively studied for CDM processes.90 However, AC, CB, and ordered mesoporous carbons (CMK, carbon mesostructured by KAIST) have attracted researchers the most due to their better activity and tunability. Their high initial conversion is related to the low threshold temperatures, which in turn are due to the high density of graphene defects. AC has the advantages of availability at an industrial scale and at lower cost. Although metal-based catalysts have higher deactivation-rates than that of ACs, the AC eventually deactivates over a longer period.91 In contrast, CB, due to a lack of porosity and lower susceptibility to pore blockage, has better stability but with lower initial activity. Typically, carbon has catalytic activity in the following order: amorphous > turbostratic > graphitic.32 AC, which is inherently an amorphous carbon, has an irregular order of carbon bonds with surface defects and dislocations, which increases the number of high-energy active sites and ultimately gives better catalytic functioning. However, the catalytic properties and stability of ACs highly depend on their source and method of preparation. Carbon materials have also been used as ideal supports for improving metal dispersion and tuning the morphology of metal catalysts.93 From the literature, it could be observed that the initial conversion on carbon catalysts for methane decomposition at 850 °C ranges between 25% and 35%.94−97 Ni supported on carbon has been extensively studied, with the highest initial conversion corresponding to AC and the lowest for CB. In a study on Ni/C/B2O3, results of CDM reaction at 850 °C showed that around 90% of initial methane conversion could be achieved. Additionally, the spent catalyst was successfully regenerated for 15 cycles using the technique of gasification in the presence of CO2.91 Chen and co-workers studied various loadings of Ni0 on CB (nanosized) and observed a better initial activity of the catalyst as compared to the CB only. However, the catalyst suffered significant deactivation due to the sintering effect at high temperature.92 Zhang et al. prepared a hierarchically porous carbon and used it as a support material for the Ni catalyst. Apart from showing an improved conversion from 27% to 61%, the catalyst was stable over a long duration for >10 h. Such a drastic enhancement in performance was attributed to the introduction of mesopores and macropores in the support network.93 Iron supported on carbon has also been widely studied for CDM, mainly with the aim to obtain highly graphitized CNTs. Wang et al. investigated various loadings of Fe on AC for a temperature range of 700–900 °C and observed that the highest methane conversion was ∼58% for a 30 wt % Fe/AC at 800 °C.94 In a study conducted by Rahul et al., carbon obtained from cellulosic biochar was used as an encapsulating support for iron nanoparticles. The catalyst performance varied from 68.3% to 83% and then to 95% at the reaction temperatures of 700, 750, and 800 °C, respectively.87 Some of the well-studied metal–carbon catalysts are summarized in Table 3, depicting conversions with respect to the reaction conditions.
Table 3. Overview of Recent Studies Conducted on Various Metal-Impregnated Carbonaceous Catalysts for the CDM Reactiona.
| no. | catalyst | preparation method | metal loading (wt. ratio %) | SBET (m2/g) | operating parameters | CH4 conversion (mol %) | carbon produced | ref |
|---|---|---|---|---|---|---|---|---|
| 1 | Ni/AC (coal) | impregnation | 6.7 | 0.2 g of catalyst | 13% (t = 0) | web-like filamentous carbon (tube size = 50 nm) | (98) | |
| T = 1023 K | 5% (t = 2h) | |||||||
| 50 mL/min CH4 | ||||||||
| 2 | Ni/AC (coconut shells) | impregnation | 30 | 565.12 (before) | 29% (t = 0) | highly degenerated soot and carbon fibers | (99) | |
| 179.0 (after) | 75% (t = 4h) | |||||||
| 3 | Ni/RC (shenhua coal) | in situ preparation with carbon | 10 | 589 | T = 850 °C | 15% (t = 0) | some filamentous carbon formed | (100) |
| 25% (t = 4 h) | ||||||||
| 4 | Ni/AC (coconut shells) | impregnation | 30 | 1185 (before) | VHSV= 1.62 L/h·gcatT = 850 °C | 10% (t = 15 min) | carbon nanofibers (diameter 100–200 nm and length 5–8 μm) surrounded by sooty carbon | (101) |
| 565.12 (after) | 30% (t = 4h) | |||||||
| 5 | Ni/CB (petroleum base) | impregnation | 30 | 120 (before) | 5% (t = 15 min) | |||
| 65.1(after) | 20% (t = 4 h) | |||||||
| 6 | Ni–Co/AC (saimengte coal) | in situ during activation | 20 (Ni) | 326 | 75% (t = 0) | spherical encapsulated carbon with some amount of fibers | (102) | |
| 20 (Co) | 34% (t = 5 h) | |||||||
| 7 | Ni–Fe/AC (saimengte coal) | 20 (Ni) | 68 | 20% (t = 0) | relatively higher amount of carbon fibers than spherical | |||
| 20 (Fe) | 75% (t = 5h) | |||||||
| 8 | Ni/CC (blue coke) | selective steam gasification | 6.87 | 265 (before) | VHSV = 12 L/h·gcat | 50% (t = 0) | fibrous carbon | (103) |
| 41 (after) | T = 850 °C. | 70% (t = 10h) | ||||||
| 9 | Ni/C (pine saw dust) | selective steam gasification (t = 30 min) | 57.31 | 16.48 | 70% (t = 0) | fibrous carbon | (104) | |
| 90% (t = 6 h) | ||||||||
| 10 | Ni–K/CC | selective steam gasification with residual K2CO3 (used as catalyst) | VHSV= 12 L/h·gcatT = 850 °C | 60% (t = 0) | encapsulated carbon | (105) | ||
| 25% (t = 2h) | ||||||||
| 11 | Ni/CC | selective steam gasification without residual K2CO3 | 17.66 | 128 (before) | 55% (t = 0) | carbon fibers | ||
| 25 (after) | 90% (t = 10h) | |||||||
| 12 | Ni–sucrose/C | selective steam gasification | 35.87 | 26.4 | VHSV = 24 L/h·gcat | ≈90% (t = 10h) | filamentous carbon (fibers + some encapsulation structures) | (106) |
| T = 850 °C | ||||||||
| 13 | Ni/3DOMC | steam-assisted crystallization | 47.4 | 884 | 0.336 g of catalyst | 10% (t = 0) | graphitic carbon | (107) |
| VHSV = 12.5 L/h·gcat | 50% (t = 10h) | |||||||
| T = 850 °C. | 48% (t = 4h) | |||||||
| 14 | Ni–Cu/CNT | polyol reduction method | 78(Ni):22(Cu) | 0.05 g of catalyst | 55% (t = 0) | branched CNFs | (108) | |
| Total flow rate = 20 mL/min | 50% (t = 14 h) | |||||||
| T = 850 °C. | 20% (t = 26h) | |||||||
| 15 | Fe–Al/AC (coconut shell) | impregnation | 22.16(Fe) | 492 | 0.1 g catalyst, | 37% (t = 0) | encapsulated carbon | (109) |
| VHSV = 15 L/h·gcat | 17% (t = 2.5h) | |||||||
| T = 850 °C. | ||||||||
| 16 | Fe/AC (Shenmu coal) | in situ during carbonization-activation | 23 | 762 | VHSV = 15 L/h·gcat | 25% (t = 0) | carbon fibers along with encapsulating carbon structures | (110) |
| T = 850 °C. | 14% (t = 6 h) | |||||||
| 17 | Pd/AC | impregnation | 10 | 751.56 (before) | VHSV = 1.62 L/h·gcat | 37.5% (t = 0) | carbon nanofibers (diameter 40–250 nm; 9 μm length) | (111) |
| 245.17 (after) | T = 1123 K | 52.5% (t = 4h) | ||||||
| 18 | Pt/AC (ash wood biomass) | impregnation | 20.5 | 919 | 0.05 g of catalyst | 11.3% (t = 0) | thin layer carbon deposition | (112) |
| 0.6 L/h methane | 1% (t = 1.5 h) | |||||||
| T = 850 °C | ||||||||
| 19 | Pd/AC (ash wood biomass) | impregnation | 19.42 | 864 | chaotically arranged filaments of different lengths (20–200 nm) | |||
| 20 | Ca/AC | impregnation | 15 | 1838 | 0.2 g of catalyst | 21% (t = 0) | hyaline carbon surface deposits with some carbon fibers. | (113) |
| VHSV = 15 L/h·gcat | 8% (t = 6 h) | |||||||
| T = 850 °C. | ||||||||
| 21 | Ru/AC | impregnation | 3.16 | 690 | WHSV = 0.1 h–1 | 20% (t = 60h) | CNT | (114) |
| T = 800 °C. |
Abbreviations are as follows: SBET = BET surface area, T = temperature, VHSV = vapor hour space velocity, t = time, AC = activated carbon, CC = coal char, 3DOMC= three-dimensional ordered mesoporous carbon, CNT = carbon nanotube, and CB = carbon black
Ordered mesoporous carbons (OMCs) are other promising candidates for catalytic applications and high stability owing to their high surface area and well-connected network of pores (Table S1). Mesoporous carbons are light in weight and can react well with large molecules by providing rapid diffusion pathways. A high degree of uniformity of mesopores in a three-dimensional periodic pore structure is observed in such porous materials. The synthesis of OMCs using templates where the resultant materials replicate the structural characteristics of the template promises success in obtaining the porous carbons. An OMC of type CMK-1 (carbon mesostructured by KAIST) was first synthesized by Ryoo et al. using a MCM-48 template.95 The prepared CMK-1 exhibited a cubic structure and a large total pore-volume (0.9–1.2 cm3/g). OMC of type CMK-2 that showed retention of the cubic structure of the template SBA-15 was again synthesized by Ryoo et al.96 Serrano et al. reported the use of CMK-3 and CMK-5 synthesized by a nanoreplication technique using SBA-15 as a template and furfuryl alcohol as the carbon source. CMK-5 showed superior performance, producing more than 20 g of solid carbon per gram of catalyst, having the morphology of graphene sheets (3–4 nm thick). The H2 production was enhanced from 0.9 mol/gcat for CB-bp (carbon black, Black Pearls 2000) to 1.8 mol/gcat for CMK-5. This was attributed to the fact that accumulating carbon was observed to grow on the exterior of catalysts, whereas the active sites continued to remain accessible to the reactant molecules for a longer time.97 Although utilization of the OMCs for CMD is hindered due to the intricate and complicated synthesis process, the results seem to be promising, especially in terms of catalyst stability. Until 2010, CB-bp was believed to show the highest initial activity among carbon-based catalysts. However, Serrano et al. reported that CMK-3 and CMK-5 possessed threshold temperatures (defined as the temperature at which H2 production is of the order of 0.1 mmol/g) of 744 and 753 K, respectively, compared to 778 K for CB-bp, proving that CMK-3 exhibited an even higher initial activity than CB-bp. Hence, OMCs are expected to gain increasing attention in the coming years owing to their favorable properties in promoting greater applications in the field of catalytic methane decomposition.
Although carbon catalysts could serve to be an economical option, due to their low activation energy they demand high operating temperatures in excess of 800 °C. However, it should be remembered that the CDM reactions are thermodynamically limited and favor high temperatures for high conversion. This limitation can be overcome by the metal-loaded carbon catalyst that is effective even at low temperatures (Table 3). The design of catalysts with optimal metal loading and characteristics is vital to ensuring high conversion and product quality. In general, unsupported carbonaceous catalysts yield amorphous carbon, while suitable metal–carbons favor the deposition of highly graphitic carbons.
3.1. Factors Influencing the Catalytic Properties of Carbon Catalysts
Multiple experimental investigations have been carried out on CH4 cracking over carbon-based catalysts to determine the relationship between catalytic activities and the reaction parameters.115,116 Some of the important factors are discussed below.
3.1.1. Carbon Source
Carbon derived from biomass has shown promising results and ensures the employment of waste materials at the same time. In a study conducted by Kim et al., AC originating from coal was tested for the CDM reaction at 850 °C in a fixed-bed reactor, and it was observed to have a linear correlation between coke deposition and catalytic deactivation. This was attributed to pore blocking by deposited carbon crystallites, making the accessibility of active sites lower over time.118 Another study was conducted by Al-Hassani et al. on activated carbons having sources of palm shells and hardwood (both activated via steam activation process). It was reported that AC (hardwood) resulted in longer catalytic activity compared to the AC from palm shells; however, the initial conversions were almost same for both sources at all temperatures between 820 and 940 °C (Figure 6(a and b)).116 The difference in stabilities was ascribed to the pore size distribution, specifically the proportion of mesopores over micropores. Mahmoudi et al. reported a high initial yield followed by a sharp decline in activity over AC (olive stones) (Figure 6(c)). This was again attributed to the blockage of micropores by the deposited coke (Figure 6 (d)).117
Figure 6.

(a and b) Methane decomposition over AC from two sources: ACPS:AC from palm shells and NORIT:commercial AC from hardwood at different temperatures. Reprinted with copyright permission from ref (116). Copyright Elsevier 2010. (c) Methane decomposition over activated carbon from olive stones and (d) N2 sorption isotherms at −196 °C. Reprinted with copyright permission from ref (117). Copyright Elsevier 2017.
Glassy carbon, a brittle, nongraphitizable polymeric carbon that has a negligible porosity, showed higher initial catalytic activity than ACs but relatively lower stability, with quick deactivation within 10 min.118 Lee et al. investigated different types of carbon blacks in a vertical fixed-bed reactor and observed stable activity at all temperatures between 850 and 1015 °C despite the deposition of coke.119 Some of the methane decomposition studies on carbon-based catalysts as a function of carbon source are listed in Table S2.
Investigations of carbon catalysts obtained from various sources make it necessary for experimentalists to consider textural and other surface properties in terms of surface area and pore size distribution associated with the sources of origin. There is a direct relation between the concentration of mesopores to micropores and the catalyst stability.120,121
3.1.2. Carbon Surface Area
Various studies have investigated the relationship between carbon catalyst kinetics and surface areas and depicted no particular trend or straightforward relation.122Figure 7(a) shows a dependence of the initial methane decomposition rate on the surface area of the carbon catalyst. Methane decomposition is heterogeneous in character over carbon materials; however, the characteristics deviate for ACs having very high surface areas. On the other hand, there is no particular relation between the activity and BET surface areas of catalysts (Figure 7(b)). This implies that the entire catalyst surface area is not active for the CDM reaction but instead only a part of the surface.
Figure 7.

Initial CDM rates of conversion on ACs (a) with respect to the carbon surface area and (b) versus the BET surface area of fresh ACs. Figures have been reprinted with copyright permissions from refs (32) and (119). Copyright Elsevier 2005 and 2004.
The surface chemistry of carbon, such as defects, is the deciding factor that governs the catalytic activity, while physical properties such as the BET surface area and pore volume govern the catalyst stability.90 The observation implies that the entire carbon surface is not active for methane decomposition but instead only a portion of it. Mostly, high surface area arises from the porosity of micropores rather than meso- or macropores, and hence knowing the surface area in terms of the proportion of micropores to mesopores could serve as a better indicator to assess the potential of a catalyst.
3.1.3. Particle Size
The influence of the size of catalyst particles on the overall mass-transfer kinetics of methane diffusion has reportedly been significant. As particle size increases, the length of the diffusion path would increase, increasing the diffusional resistance that controls the overall reaction rate. Figure 8(a) shows the dependence of CH4 conversion on the particle size of activated carbon. Kim et al. reported methane decomposition over commercial AC in a fluidized-bed reactor at 850 °C.118 The methane conversion increased with a decrease in the particle size, which is due to the higher surface area/particle volume and fluidization efficiency that enhanced the residence time between solid/fluid phases. However, in a fluidized-bed reactor, the catalyst particle size limits the range of the fluidization velocity. Larger catalyst particles require higher velocities to achieve the minimum fluidization for successfully suspending the denser particles. At a higher minimum fluidization velocity, a significant mass-flow rate of CH4 enables higher production of H2. However, for the same velocity, smaller catalyst particles will have lower CH4 conversion because of the lower minimum fluidization-velocity due to the formation of a bubble phase that would increase mass transfer resistance, resulting in lower conversion.
Figure 8.

Dependence of the CH4 conversion of ACs on (a) particle size and (b) different temperatures. (c) Influence of catalyst particle size on turnover costs. Figures have been reprinted with the copyright permissions from refs (119) and (120). Copyright Elsevier 2004 and 2022.
Lumbers et al. proposed a model that established the relation between catalyst particle size and catalyst turnover cost, which is defined as the total cost of the used catalyst per ton of H2 generated (Figure 8(c)). It was observed that larger particles could result in higher H2 production rates, thereby decreasing the overall turnover costs. However, a maximum size of 150 μm was noticed to be the threshold for smooth fluidization behavior. It was observed that for smooth fluidization, particles should remain within the range of the Geldart A classification, and the temperature should not exceed the sintering temperature of the catalyst.120
Particle size also determines the stability of carbon by alleviating pore-mouth blocking by deposited carbon. Carbon deposition takes place and forms coke or, at the outer shell of the carbon catalyst, encapsulates the pores. The inner surface for the smaller particles can thus be utilized to a larger extent compared with that in the larger particles. Thus, an optimum catalyst size needs to be large enough to provide a significant H2 yield and small enough to cause less fouling.123
3.1.4. Reaction Temperature
According to Le Chatelier’s principle, reaction temperature has a significant influence on the rate of methane decomposition because the reaction is endothermic in nature. An increase in temperature is thermodynamically bound to increase the conversion, as the equilibrium would shift to the left. However, with an increase in temperature, the rate of reaction increases, which is expected to increase the rate of carbon formation and hence deactivation (Figure 8(b)). There is always a stable temperature range for every catalyst, and raising the temperature beyond that range could cause catalyst sintering and ultimately catalyst deactivation.124,125 A study conducted on Ni loaded on biomorphic carbon (formed during the degradation of vine shoots in a reducing or inert environment at high temperatures and rates of heating) showed that the production, type, and quality of carbon material deposited over the catalyst highly depends upon the reaction temperature. At temperatures <850 °C, the carbon deposited was mainly carbon nanofibers (bamboo-type), while at higher temperatures it was graphite. The highest quality graphene was achieved at temperatures above 950 °C. It was explained by the high rate of carbon nucleation, which leads to the formation of numerous nucleation points at the surface of the metallic nanoparticles; this allows for multiple routes for the dissolved carbon to escape and ultimately the formation of graphitic materials. Regardless of the type of AC, the activation energy was estimated to be between 194 and 200 kJ/mol, which is less than the C–H bond energy in a CH4 molecule (∼440 kJ/mol).27 Ammendola et al. determined the rates of CH4 conversion and H2 yield in a fluidized-bed reactor and reported the rate to be of first order. The intrinsic kinetics of the reaction and carbon deposition were established by taking the dependence of CH4 conversion on the Damkohler number, where the activation energy was estimated to be 1.55 × 105 kJ/mol.122 In another study, Lumbers et al. developed a relationship between the frequency factor and the activation energy. Using the Arrhenius equation, the activation energy was estimated to be ∼90.3 kJ/mol.120 The CDM reaction rate is the summation of carbon crystallite growth and carbon nuclei formation, and activation energies for both processes are estimated to be ∼227 and ∼316 kJ/mol, respectively.10 Thus, for carbon-based catalysts, the activation energy is near that required for the growth of carbon crystallite, which forms the rate-determining step for CDM. Wang et al. demonstrated the effect of temperature (750–900 °C) on 30% Fe-doped ACs. A conversion of 58% was reported at 800 °C, while an extremely low conversion at 900 °C was observed due to the sintering effect.76 Henao et al. evaluated the effect of reaction temperature (650–950 °C) for Co–Cu/CDC catalysts on the yield and growth of CNTs. The maximum productivity of CNTS was recorded at 0.33 g/h·gcat with 28.6% CH4 conversion and 14.3% H2 at 800 °C. However, at a temperature above 800 °C, the carbon catalyst deactivated due to the sintering of the catalyst, which inhibited further methane diffusion through catalyst pores.123 Hence, it is necessary to obtain an optimized reaction temperature to get the best possible performance of carbon catalysts without losing their surface activity due to the sintering effect.
3.1.5. Space Velocity
Reaction space velocity refers to the ratio of total feed flow rate to the catalyst bed volume. A lower gas velocity in a fluidized-bed does not fluidize the bed due to low throughput per unit volume to accomplish the desired reaction. On the other hand, a large gas velocity reduces the contact time between the reactant species and as such decreases the residence time. Thus, an optimum velocity is required, taking the hydrogen yield and methane conversion into consideration. In the case of fixed bed reactors, increasing the flow rate at constant catalyst weight decreases the methane conversion and H2 yield due to the large handling capacity per unit catalyst weight. Zhang et al. reported the influence of volumetric hourly space velocity (VHSV) on CH4 cracking over 10% Ni/carbon catalysts. They observed that the conversion decreased as the VHSV increased from 15 to 30 l/gcat,·h and showed a negligible conversion at an even higher VHSV of 60 l/h·gcat.124 Pudukudy et al. demonstrated the effect of space velocity on hydrogen and carbon yields for Ni/TiO2, reporting a maximum H2 yield of 56% at a space-velocity of 9000 mL/h·gcat, while a further drop in H2 yield to 42% at a space-velocity of 12000 mL/h·gcat.125 Alves Silva et al. observed an increase in conversion over iron-based catalysts with an increase in the weight hourly space velocity (WHSV) up to 2 L/h·gcat, and the catalyst started deactivating when WHSV was increased to 6 L/h·gcat.126 This could be attributed to the fragmentation of particles at higher space velocities.127
3.1.6. Metal-Loaded Carbon Catalysts
Loading of metal in a carbon catalyst highly increases its activity by creating surface defects and high-energy active sites. Surface-defects, vacancies, and low-coordination sites strongly influence the surface properties of carbon catalysts. Free valences and other high-energy points, such as crystallite edges, are typically strong active sites of carbon catalysts. Carbon materials exhibit flexibility in surface area and porosity tuning, thereby facilitating better metal dispersibility. Due to the reducing nature of carbon catalysts at higher temperatures, metal oxides can undergo in situ reduction. Most commonly used metals are Fe, Ni, Co, and Cu. Wang et al. investigated the influence of Fe loading (5–30%) on carbon catalysts derived from Shenmu coal. They reported that the activity of Fe-loaded carbon catalysts increased with an increase in the Fe loading. At Fe proportion of 10 wt %, the conversion, as well as H2 yield, increased for the first 30 min but decreased in the next 2 h, which was attributed to the pore blockage by carbon formation.76 Zhang et al. analyzed the effect of cerium loading (5–20%) on 5–10% Ni/C catalysts synthesized by selective steam gasification of coal char (CC) and reported an increase in conversion from 65% to 90% with the addition of 20 wt % Ce to 10% Ni/C. Although virgin cerium supported on a carbon catalyst has poor activity, its addition to Ni/C catalysts was seen to improve the activity of the whole catalyst.128 Similarly, the effect of 1–20% Co and 1–20% Fe on Ni/AC catalyst derived from Saimengte coal was studied by Wang et al. It was found that bimetallic catalysts Ni–Co/AC and Ni–Fe/AC offered better stability and activity for CH4 conversion as compared to Ni/AC at 850 °C.129
Such increased activity of metal-loaded carbon catalysts is attributed to the increased concentration of active sites and the modification of the electronic state. There has to be an optimized loading of the metal to incorporate in the carbon matrix, as it has a significant influence on reducing the specific surface area and total pore volume. For low loading amounts of metals, the metal/carbon composites have better dispersibility and a tendency to reduce at lower temperatures.130
3.1.7. Surface Functional Groups
The presence of oxygenated surface groups, such as carboxylic, phenolic, lactonic, and carbonylic groups, is known to significantly affect the chemical and catalytic activities of carbon catalysts.131,132 Surface functional groups on ACs are located at the edges represented by the noncarbon heteroatoms such as nitrogen, phosphorus, hydrogen, and oxygen (Figure 9).30 The acidic nature of ACs is formed through bonding with these heteroatoms, which come from oxygen functional groups and can enhance the catalytic performance of carbon in the initial stage by either directly reacting with methane or by generating new reacting sites by releasing COx. Several concentrated acids such as HNO3, HCl, ammonium persulfate, and H2SO4 have been used as oxidizing agents to enhance the oxygen functional groups.133,134 3D metal–organic frameworks (MOFs), carboxylated carbon, nitrogenized porous carbon derived from biomass, and graphydine have also been reported to show good performance for CDM.
Figure 9.
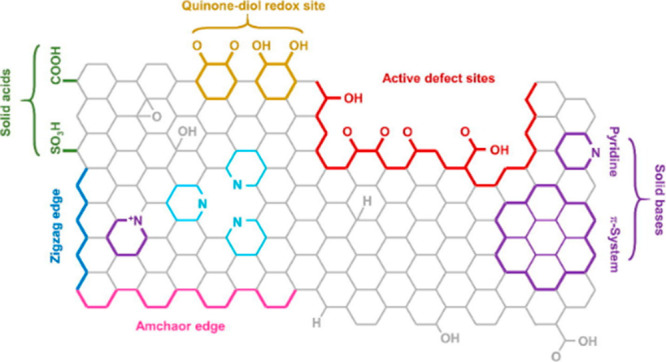
Illustration of functionalized active centers on the surface of a carbon catalyst due to the introduction of acidic groups. Figure is reprinted with copyright permission from ref (30). Copyright Elsevier 2021.
The adsorption capacity of Ni2+ on AC was reported to increase with the increase in the surface acidity of the carbon template.135 Bai et al. reported an increase in the total pore volume and surface area for AC treated with acids such as HCl, HF, and HNO3 as compared with the untreated ACs. All acid-treated ACs depicted a higher initial CH4 conversion as compared to untreated AC.136 Wang et al. reported that pretreating ACs with HCl and HF resulted in increased surface area, which is in good agreement with the findings of Bai et al.; however, excessive removal of the mineral content from the catalysts led to the collapse of the surface area and pore volume.137
3.1.8. Composition of Feedstock
In a few studies, researchers have attempted to analyze the effect of introducing impurities and varying the proportion of methane to impurities in the feedstock on the conversion capacity. Such an analysis is necessary to lay the foundation for the scale-up of CDM to the industrial level where feedstock is replaceable with natural gas. Results obtained from varying compositions of methane and other components of natural gas, such as other lower hydrocarbons, H2S, and N2 in the feedstock will aid in understanding the effect of other components of natural gas on the conversion and H2 yield as well as on the deactivation of the catalyst.
In a study on co-feeding methane with 20% ethylene, the authors presented higher H2 yield and stability on carbon-based catalysts.138 With the addition of acetylene to methane as well, authors reported an increase in the H2 concentration in the outlet stream, with higher longevity of the catalyst (carbon black) at higher temperatures >850 °C.139 Similarly, in another work reported by Rechnia et al., the addition of ethanol in feedstock was observed to enhance the H2 yield and catalyst stability over activated carbon.140 Other studies conducted on the addition of ethane, propane, propylene, benzene, and nitrogen in the inlet steam over carbon-based catalysts have shown similar results.141 Such an influence of feedstock impurities on the catalyst performance is attributed to the introduction of various O-containing functional groups and surface defects (discontinuities and dislocations) in the carbon lattices, which in turn increase the number of high-energy sites. Moreover, the characteristics of carbon deposits depend not only upon the type of catalyst but also on its surface chemistry.142 It has been observed that the carbon deposited over impurities in feedstock has better autocatalytic properties than that deposited in the pure methane feed and deposits in the form of filaments rather than encapsulation. Additionally, carbon catalysts are observed to catalyze the reaction effectively in the presence of H2S (a common component of natural gas) owing to their resistance to S-poisoning. However, this calls for the incorporation of additional components for further purification of H2 when operated on an industrial scale due to the generation of COx in the outlet stream.
4. Carbon Deposition
Current technologies for restoring catalyst activity, such as steam oxidation and air combustion to convert the deposited C to CO2 or CO, act against the environmentally benign advantage of the CDM process for producing clean hydrogen.143 Minimizing the carbon formation on the active sites is challenging in any high-temperature catalytic hydrocarbon conversion. However, this can be partially addressed by converting a major proportion of deposited carbon to high-value carbon. Carbon formation in a CDM reaction is a multistage process that initiates with the CH4 adsorption, and then various dehydrogenation steps conclude the formation of a conically ordered graphitic filamentous carbon over suitable catalysts and under the required reaction conditions. Attempts to understand the thermodynamics and driving force for carbon filament growth are widely reported in the literature.144 The gradients in temperature and/or concentration across the catalyst are believed to drive the carbon diffusion process through the catalyst.145 After the dehydrogenation step, carbon atoms diffuse from the surface through the catalyst (gaseous/metallic interlayer) and precipitate toward the rear-end (metallic/support interlayer). For the carbon filament formation, the isolated carbon atoms on the surface of the catalyst dissolve interstitially into the catalyst particles at the catalyst–gas interface and diffuse to the other end, which is the catalyst–support interface. A selvedge area having a high concentration of carbon is formed over the catalyst surface due to the segregation behavior of carbon in metal, and the concentration of carbon decreases from the selvedge to the bulk and to the rear-end over various atomic layers. The relationship between the surface carbon and the concentration of carbon in bulk, i.e., interstitially dissolved carbon in the catalyst, has been widely described using the Langmuir–McLean isotherm.146−148 Diffusion of carbon continues until the carbon solution of the catalyst (interstitially dissolved carbon in catalyst) is supersaturated, after which the nucleation of filamentous carbon begins as a result of carbon filament precipitation at the catalyst/support end. The pile-up of graphite layers detaches a metal particle from the support, and the metal remains at the tip of the filament due to the strong interaction with methane (Figure 10(a)). However, in some exceptions where the interactions between phases are too strong, filamentous carbon formation takes place from the metal–gas interface due to the inability of the stacked graphite layers to push the metal particle up, as is shown in Figure 10(b). As filamentous carbon begins to grow, the encapsulating carbon is also formed, which deactivates the catalyst and ultimately decreases the cracking rate. The following are the stages of filamentous carbon formation mechanism in a CDM process:145,149,150
dissolution of carbon in the catalyst to form carbide as an intermediate
decomposition of carbide
carbon or graphitic nucleation
precipitation of carbon at the interface of the metal and support
separation of metal particle from the support
Figure 10.

Schematic of filamentous carbon formation during a CDM reaction over a Ni catalyst: (a) tip growth and (b) base growth. The figure has been reprinted with copyright permission from ref (145). Copyright Elsevier 1997.
Due to their unique electronic and mechanical properties, CNTs and CNFs are considered highly important in nanoengineering.151,152 The structures and morphologies of these carbon nanomaterials are highly dependent on reaction temperature, feed composition, and pressure (Table S3). In an investigation on the formation of carbon nanomaterials, it was observed that the conversion to carbon nanotubes (CNTs) was low on monometallic catalysts when compared with bimetallic counterparts based on Co and Cu. However, with the insertion of Fe to Cu, graphitization and crystallinity of CNTs was observed to highly increase.153 In another study, the effect of the composition of support on the quality of CNTs was studied. It was noticed that with the aid of proper carbon diffusion through Co, and the subsequent dispersion through the support (ZrxMg1–xO), single-walled or multiwalled CNTs could be obtained with various diameters.154 The formation of carbon nanofibers (CNFs) has been widely studied on Ni-based catalysts. Ashok and co-workers observed that CNFs of varying diameters and lengths could be obtained by varying the ratio of Ni to support. It was pointed out that the tip and the diameter of CNFs were equal in dimension, suggesting that Ni particles were responsible for this observation.155 Carbon nano-onions (CNOs) can be obtained by chemical vapor deposition or pyrolysis. CNOs have been mostly observed over Fe-based catalysts at reaction temperatures beyond 700 °C. Specific understanding of CNO formation still remains ambiguous; however, some studies suggest CNO formation is due to the rearrangement of Fe atoms when the carbon deposition surpasses the carbon solubility.156
5. Carbon Deactivation and Regeneration
Catalyst deactivation is a major challenge in the continuous generation of hydrogen by CDM. The deposited carbon may diffuse through the catalyst and produce carbon filaments and other nanostructures. Excessive carbon deposition on the active sites results in coking, and carbon deposited can thus indicate either the extent of the reaction by the formation of carbon nanostructures inside the catalyst or the deactivation of the catalyst by the formation of encapsulating carbon. This, in turn, would be responsible for the decreased active surface area of the catalyst as the reaction proceeds with time.
The reaction order of carbon-based CDM has been reported to be 0.5 ± 0.1 by a number of studies, and thus the rate-equation can be written as157
In a study, the equations describing the empirical decay of the surface areas (S) for activated carbon (AC) and carbon black (CB) were given by35
where t denotes the time of reaction. On the other hand, carbon catalyst deactivation due to coking has been given by Voorhies equation157
where CC denotes the moles of deposited carbon on the surface of catalyst, t is time on stream, and k and n denote the fouling parameters, established experimentally. Since the Voorhies equation only reflects the quantity of carbon deposition rather than the activity factor (AF), there is a need to develop models forming the correlation between the activity factor (or surface area) and the amount of coke for a better understanding of catalyst deactivation. The activity of a catalyst depends on an equilibrium between the carbon diffusion rate and the carbon production rate in the catalyst pores. For excessive carbon deposition, the reaction rate becomes faster than the diffusion and hence the equilibrium is disturbed, leading to the deactivation of the catalyst. Therefore, a balance is sought between the reaction rate and the deactivation rate, which can be explored by recognizing the optimum reaction conditions with the help of a well-developed reaction-rate model. Over the years, researchers have made attempts to optimize the concentration of active centers by incorporating foreign particles (heteroatoms or surface functional groups), modifying the support, improving metal–oxide interactions, and altering synthesis strategies to tune the metal particle size, morphology, and defect states.119,158 However, for a carbon-based heterogeneous catalytic reaction, the development of a reaction model for the prediction of reaction kinetics is quite challenging, as the catalyst itself is evolving. Several models have been developed and reported in the literature to predict the CH4 conversion, H2 conversion, and deactivation rate (Table 4).
Table 4. List of Some Noteworthy Models Developed on the Deactivation of the Carbon-Based Catalyst during the CDM Reaction.
| models | catalysts | modeled parameters | refs |
|---|---|---|---|
| a 3D coupled mathematical model for microwave-assisted thermocatalytic decomposition of methane | activated carbon | temperature distribution and concentration profiles for CH4 and H2 in the catalyst bed | (159) |
| global rate model for a kinetic study in a solar-driven thermogravimetric reactor | activated carbon and carbon black | methane cracking rate and reaction deactivation rate | (160) |
| two-phase and three-phase models for a bubbling fluidized bed and one-parameter and multiparameter models for a turbulent fluidized bed reactor (FBR) | activated carbon | reactor geometry for H2 and C formation for scale-up of FBR | (161) |
| artificial neural network modeling based on Bayesian regularization and Levenberg–Marquardt-trained multilayer perceptron | carbon particles | studying the influence of reaction parameters on the catalyst synthesis and H2 generation | (162) |
| full factorial and ANOVA-based quadratic model | activated carbon | influence of reaction partial pressure, temperature, and catalyst weight on the initial rate of reaction | (163) |
| Dsmoke and plug-flow reactor model | carbon black | investigating gas emission with respect to the residence time | (164) |
| a mathematical model for a continually stirred tank model | graphite | H2 production rate and corresponding turnover cost based on reaction conditions | (120) |
| quadratic RCH4 model as a function of the time of decay and the reaction temperature | activated carbon | catalyst deactivation concerning textural properties, relative time, and reaction temperature | (163) |
A successful model should take the carbon deposition rate and reactivation into account besides the CH4 decomposition rate. The idea is to consider all of the competitive processes and predict the rates of CH4 decomposition and deactivation so that proper reactivation techniques can be applied for catalyst regeneration. Figure 11 illustrates a schematic that could be used as a benchmark for developing models to predict rates of CH4 decomposition, deactivation, and reactivation.
Figure 11.

A schematic of the CDM flow diagram for developing models for the CDM reactions.
Thermocatalytic decomposition of methane, deactivation of the catalyst due to carbon deposition, and reactivation of the catalyst external surface are three main competitive phenomena that need to be considered for a successful application of this process. The fluidized-bed operating conditions, such as gas velocity, differential pressure, and size of the catalyst, could be used as input parameters apart from other reaction conditions. Fluidized-bed reactors have a higher heat and mass transfer rate because of the constant mixing. The temperature of the bed may be maintained uniformly, facilitating the removal of solids and regeneration on a continual basis in order to continue stable conversion for a long period of time. However, there are only a few limited mathematical models focusing on both catalyst deactivation and regeneration for estimating the internal and external catalyst active surface areas. Models developed so far are simply based on the power law to determine the reaction rates. There is a need for further development of theoretical and artificial network modeling intelligence for a better understanding of the morphological and other textural-related parameters of active catalysts during catalytic methane cracking. Theoretical modeling is also required for the development of further novel materials considering the constraints of existing materials for CDM and also of being compatible with the reactor and process scale-up. The number of constraints for developing a model needs to be reduced for bringing the model closer to real systems. In the heterogeneous catalytic reactions, heat transfer processes are quite complex, and the determination of the gas temperature that in turn controls the extent of reaction is challenging. Thus, theoretical studies taking into consideration the reactor gas distribution to establish the reaction conversion rate are necessary.
For the recovery of catalyst activity, researchers have followed separation and regeneration (Table S4). Most of the accumulated carbons are removed by the physical separation method after each CDM cycle, which roughly restores the catalyst activity to a greater extent.165 One promising method of physical separation employed in industries is a physical shaking process. Carbon depositions (graphitic filamentous) grown over the catalyst are dropped to the bottom of the reactor by a rigorous shaking or rolling of the reactor, and from there they are collected for extraction. To reduce the blocking of reactor by the collected carbon, the use of molten metal, such as lead and copper, has been employed due to the density difference facilitating the accumulation of carbon above the liquid surface that can be easily skimmed off. However, carbon deposited inside the catalyst pores cannot be removed by simple physical separation processes, leading to the complete destruction of the catalyst with each CDM cycle. Therefore, to ensure the longevity of the catalyst, a regeneration process is required to eliminate the carbon deposited inside the catalyst pores.166 The initial CDM activity and carbon deposition may depend on the characteristic of the nanostructure; however, deposited carbon blocks the catalyst surface very quickly, with the deposit itself acting as a catalyst. As the process of methane decomposition proceeds, any further incremental layer of the deposit is less active than the preceding layer, thereby reducing the autocatalytic capability of the whole catalyst. The regeneration of catalysts using H2O or CO2 oxidation or gasification has been employed by many researchers to remove carbon and uncover the underlying layer. The vacancies and radical sites become exposed and account for an increase in the catalytic activity. However, such recovery is quite short-lived, since uniform oxidation of the deposited carbon layer is highly challenging without causing significant oxidation or removal of the original catalyst surface. Additionally, it is difficult to obtain uniform surface regression, as the surface during the regeneration process is not uniform. Thus, the extent of catalyst regeneration greatly depends upon the structural similarities between the original catalyst and the deposited carbon layers.13 Disordered carbon deposits having radicals as active sites are advantageous in terms of the overall catalytic activity of the spent catalyst. Ethanol co-feeding may increase stability but results in CO2 formation.
Currently, rapid catalytic deactivation is a deterrent step in taking the CDM process toward commercialization. A continuously circulating fluidized-bed system with a provision of separate cracking and regeneration modules could be envisaged as a potential solution. As the reaction proceeds, the catalyst would move through the reactor, where carbon filaments deposited are constantly removed in the regeneration unit, thereby exposing a fresh metal surface for the reaction. Besides, the development of high-conversion and durable catalysts that can withstand attrition is of crucial importance.
6. Challenges and Future Recommendations
One of the most serious challenges in the catalytic decomposition of methane is catalytic deactivation due to the deposition of unwanted carbon produced, leading to a significant reduction in performance. The need is to formulate catalysts by keeping the kinetic factors under consideration, where catalysts are optimized for cracking reactions and are nonselective for amorphous and encapsulating carbon. It is imperative to develop a catalyst that maximizes the conversion, hydrogen yield, and stability and minimizes formation of amorphous and encapsulating carbon.167,168 Scale-up and commercial adoption of the process largely depend on catalysts that are cost-effective, offer high methane conversion, are stable for long duration, offer a high carbon yield to catalyst ratio, and produce a high proportion of graphitic carbon. A better understanding of the mechanism of conversion, carbon formation, kinetics of decomposition, and the effect of vital process parameters, such as reaction temperature, pressure, flow rates, and feed composition, is important.169,170 Efforts to design suitable reactor configurations that facilitate continuous regeneration and withdrawal for a solid carbon product are mandatory for practical applications. Taking these factors into consideration, the following are some of the noteworthy futuristic directions that demand further research inputs:
-
1.
Synthesis of thermodynamically stable and high-quality crystals at a lower synthesis temperature in a one-step process.
-
2.
Recently, perovskites with structural formula ABO3 have shown better conversion with good selectivity for hydrogen evolution and the ability to suppress carbon deposition compared to Ni-based catalysts. Efforts should be directed toward optimizing such materials (such as through ionic substitution) and minimizing the dependency on noble metals.
-
3.
Layered double hydroxides (LDHs) are emerging catalyst materials, as they can be structurally, morphologically, and electronically tuned. These 2D layered materials exhibit ease-of-control in terms of elemental composition and nanostructures. Utilizing bimetallic systems based on LDH catalysts could also be an optimistic approach.
-
4.
As the activity of carbon material is low, demanding high temperatures of the order of 800–1100 °C to decompose methane (activation energy of ∼240 kJ/mol),91 investigations on the synergistic effects of metal and carbon catalysts are much needed. Further, as the catalytic activity highly depends on chemical composition and porosity, the design and control of the surface chemistry and pore network are highly favorable.
-
5.
The effect of process parameters and the design of a catalyst on the structure and morphology of the product carbon nanomaterials must be well understood, as it decides the overall economics of the process.
-
6.
Major concerns involving reactor design, such as reactor clogging, periodic removal of deposited carbon and catalyst, and large back pressure need to be well understood.
-
7.
Although various theories have been postulated, the exact reaction mechanism remains uncertain and unclear. Therefore, besides simulation techniques, experimental investigations, especially those using operando techniques, are highly recommended.
-
8.
For the coproduction of CNMs, their yield and quality directly depend on the catalyst design in terms of metal loading and the metal–support interaction, which in turn govern the quasi-stable life of metal carbide. In this regard, Fe catalysts are advantageous compared to Ni and hence must be explored for a better understanding.
7. Conclusions
Thermocatalytic methane decomposition is an interesting route to enable the generation of COx-free and sustainable hydrogen. Various metal-based and carbon-based catalysts have been studied for methane cracking, though this process is far from being used in practical applications owing to high catalyst turnover costs. However, as the demand for COx-free hydrogen rises, research in this field is advancing worldwide. The aim of this Review was to provide a critical analysis of catalysts and the mechanism of the CDM reaction. It could be determined that noble metals are uneconomical, iron-based catalysts show better conversion at higher temperatures, and Ni-based catalysts show poor catalytic conversion at higher temperatures. Various carbon-based materials, especially activated carbon and carbon black, have shown sustained methane conversions and the formation of value-added nanocarbons. The challenge of achieving higher CH4 conversions on carbon-based catalysts has been addressed to a greater extent by doping with transition metal catalysts. Impregnating bimetallic catalysts further provides tolerance to deactivation and enhances the catalytic performance by structural reconstruction and modification of the electronic properties. In addition to filamentous carbon, other valuable forms of carbon, such as bamboo and onion-shaped nanocarbons, have been observed to form under specific reaction conditions. Although sustained efforts are being made to develop a novel catalyst to increase the conversion and to reduce the fouling rate, adoption to commercial scales largely depends on the ability to continuously regenerate the catalyst and sustain the catalyst activity for a longer duration. A continuous system based on a closed-loop of a fluidized-bed reactor and a regeneration system with deposition particles circulating in a fluidized state could be employed. In the fluidized conditions, a constant flow of catalyst particles in the reactor is ensured by replacing the spent catalysts with fresh ones. This promotes the continual removal of deposited carbon and allows the homogeneous distribution of temperature in the reactor. Further theoretical studies are required to further realize the morphological and other important properties of catalysts toward their performances and kinetics. Optimum reaction conditions need to be devised, such as the extent of methane gas dilution in the gas feedline and other reaction parameters.
Acknowledgments
The authors express their gratitude to Khalifa University, Abu Dhabi, UAE, for providing financial support and other technical facilities for this and related research. The present work was supported by the Joint Research Program between Khalifa University and King Fahad University of Petroleum and Minerals 8474000337.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c01936.
Additional tables listing overview summaries of recent studies on ordered mesoporous carbons, conversions rates of carbon catalysts based on carbon source, filamentous carbons deposited in the CDM reaction, and catalyst regeneration techniques (PDF)
Author Contributions
∇ These authors contributed equally. I.R.H.: conceptualization, writing–original draft, editing, reviewing, formal analysis, and data curation. A.A.: writing–sections 3.1.4, 3.1.5, and 3.1.6. H.M.C.: writing–ordered mesoporous carbons. A.A.S.: Funding acquisition and project administration. C.S.: writing–review and editing. M.M.H.: review and proof-reading.
The authors declare no competing financial interest.
Supplementary Material
References
- Pudukudy M.; Yaakob Z. Methane decomposition over Ni, Co and Fe based monometallic catalysts supported on sol gel derived SiO2 microflakes. Chemical Engineering Journal. 2015, 262, 1009–1021. 10.1016/j.cej.2014.10.077. [DOI] [Google Scholar]
- Neeli S. T.; Ramsurn H. Synthesis and formation mechanism of iron nanoparticles in graphitized carbon matrix using biochar from biomass model compounds as a support. Carbon. 2018, 134, 480–490. 10.1016/j.carbon.2018.03.079. [DOI] [Google Scholar]
- Wang I. W.; Kutteri D. A.; Gao B.; Tian H.; Hu J. Methane pyrolysis for carbon nanotubes and COx -free H2 over transition-metal catalysts. Energy and Fuels. 2019, 33, 197–205. 10.1021/acs.energyfuels.8b03502. [DOI] [Google Scholar]
- Hamdani I. R.; Bhaskarwar A. N. Cu2O nanowires based p-n homojunction photocathode for improved current density and hydrogen generation through solar-water splitting. Int. J. Hydrogen Energy. 2021, 46, 28064–28077. 10.1016/j.ijhydene.2021.06.067. [DOI] [Google Scholar]
- Hamdani I. R.; Bhaskarwar A. N. Tuning of the structural, morphological, optoelectronic and interfacial properties of electrodeposited Cu2O towards solar water-splitting by varying the deposition pH. Solar Energy Materials and Solar Cells. 2022, 240, 111719. 10.1016/j.solmat.2022.111719. [DOI] [Google Scholar]
- Hamdani I. R.; Bhaskarwar A. N. Recent progress in material selection and device designs for photoelectrochemical water-splitting. Renewable and Sustainable Energy Reviews. 2021, 138, 110503. 10.1016/j.rser.2020.110503. [DOI] [Google Scholar]
- Spath P.L.; Mann M.K.. Life cycle assessment of hydrogen production via natural gas steam reforming; NREL/TP-570-27637; National Renewable Energy Laboratory: Golden, CO, 2000. 10.2172/764485. [DOI]
- Marbán G.; Valdés-Solís T. Towards the hydrogen economy?. Int. J. Hydrogen Energy. 2007, 32, 1625–1637. 10.1016/j.ijhydene.2006.12.017. [DOI] [Google Scholar]
- Iulianelli A.; Liguori S.; Wilcox J.; Basile A. Advances on methane steam reforming to produce hydrogen through membrane reactors technology: A review. Catal. Rev. Sci. Eng. 2016, 58, 1–35. 10.1080/01614940.2015.1099882. [DOI] [Google Scholar]
- Muradov N. Catalysis of methane decomposition over elemental carbon. Catal. Commun. 2001, 2, 89–94. 10.1016/S1566-7367(01)00013-9. [DOI] [Google Scholar]
- Ahmed S.; Aitani A.; Rahman F.; Al-Dawood A.; Al-Muhaish F. Decomposition of hydrocarbons to hydrogen and carbon. Appl. Catal. A Gen. 2009, 359, 1–24. 10.1016/j.apcata.2009.02.038. [DOI] [Google Scholar]
- Qian J. X.; Chen T. W.; Enakonda L. R.; Liu D. B.; Mignani G.; Basset J.-M.; Zhou L. Methane decomposition to produce COx-free hydrogen and nano-carbon over metal catalysts: A review. Int. J. Hydrogen Energy. 2020, 45, 7981–8001. 10.1016/j.ijhydene.2020.01.052. [DOI] [Google Scholar]
- Ashik U. P. M.; Wan Daud W. M. A.; Hayashi J. A review on methane transformation to hydrogen and nanocarbon: Relevance of catalyst characteristics and experimental parameters on yield. Renewable Sustainable Energy Rev. 2017, 76, 743–767. 10.1016/j.rser.2017.03.088. [DOI] [Google Scholar]
- Muhammad A. F. S.; Awad A.; Saidur R.; Masiran N.; Salam A.; Abdullah B. Recent advances in cleaner hydrogen productions via thermo-catalytic decomposition of methane: Admixture with hydrocarbon. Int. J. Hydrogen Energy. 2018, 43, 18713–18734. 10.1016/j.ijhydene.2018.08.091. [DOI] [Google Scholar]
- Gamal A.; Eid K.; El-Naas M. H.; Kumar D.; Kumar A. Catalytic methane decomposition to carbon nanostructures and COx-free hydrogen: A mini-review. Nanomaterials 2021, 11, 1226. 10.3390/nano11051226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Wal R.; Makiesse Nkiawete M. Carbons as catalysts in thermo-catalytic hydrocarbon decomposition: A Review. C J. Carbon Res. 2020, 6, 23. 10.3390/c6020023. [DOI] [Google Scholar]
- Naikoo G.A.; Arshad F.; Hassan I.U.; Tabook M.A.; Pedram M.Z.; Mustaqeem M.; Tabassum H.; Ahmed W.; Rezakazemi M. Thermocatalytic hydrogen production through decomposition of methane-A Review. Front. Chem. 2021, 9, 736801. 10.3389/fchem.2021.736801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. J.; Yang Y. L.; Zou X. X.; Shi X. L.; Chen Z. G. Flexible hollow TiO2@CMS/carbon-fiber van der Waals heterostructures for simulated-solar light photocatalysis and photoelectrocatalysis. J. Mater. Sci. Technol. 2022, 98, 143–150. 10.1016/j.jmst.2021.05.009. [DOI] [Google Scholar]
- Fan Z.; Weng W.; Zhou J.; Gu D.; Xiao W. Catalytic decomposition of methane to produce hydrogen: A review. Journal of Energy Chemistry. 2021, 58, 415–430. 10.1016/j.jechem.2020.10.049. [DOI] [Google Scholar]
- Schwarz H. Chemistry with methane: Concepts rather than recipes. Angewandte Chemie - International Edition. 2011, 50, 10096–10115. 10.1002/anie.201006424. [DOI] [PubMed] [Google Scholar]
- Hooshmand Zaferani S.; Sams M. W.; Shi X. L.; Mehrabian N.; Ghomashchi R.; Chen Z. G. Applications of thermoelectric generators to improve catalytic-assisted hydrogen production efficiency: future directions. Energy and Fuels. 2022, 36, 8096–8106. 10.1021/acs.energyfuels.2c01754. [DOI] [Google Scholar]
- Leal Pérez B. J.; Medrano Jiménez J. A.; Bhardwaj R.; Goetheer E.; Van Sint Annaland M.; Gallucci F. Methane pyrolysis in a molten gallium bubble column reactor for sustainable hydrogen production: Proof of concept & techno-economic assessment. Int. J. Hydrogen Energy. 2021, 46, 4917–4935. 10.1016/j.ijhydene.2020.11.079. [DOI] [Google Scholar]
- Ashik U. P. M.; Wan Daud W. M. A.; Abbas H. F. Methane decomposition kinetics and reaction rate over Ni/SiO2 nanocatalyst produced through co-precipitation cum modified StÖber method. Int. J. Hydrogen Energy. 2017, 42, 938–952. 10.1016/j.ijhydene.2016.09.025. [DOI] [Google Scholar]
- Raza J.; Khoja A. H.; Anwar M.; Saleem F.; Naqvi S. R.; Liaquat R.; Hassan M.; Javaid R.; Qazi U. Y.; Lumbers B. Methane decomposition of hydrogen production: A comprehensive review on catalyst selection and reactor systems. Renewable Sustainable Energy Rev. 2022, 168, 112774. 10.1016/j.rser.2022.112774. [DOI] [Google Scholar]
- Ahmad K.; Polychronopoulou K.; Abi Jaoude M. CH4 valorisation reactions: A comparative thermodynamic analysis and their limitations. Fuel. 2022, 320, 123877. 10.1016/j.fuel.2022.123877. [DOI] [Google Scholar]
- Li Y.; Li D.; Wang G. Methane decomposition to COx-free hydrogen and nano-carbon material on group 8–10 base metal catalysts: A review. Catal. Today 2011, 162, 1–48. 10.1016/j.cattod.2010.12.042. [DOI] [Google Scholar]
- Azuara M.; Latorre N.; Villacampa J.I.; Sebastian V.; Cazaña F.; Romeo E.; Monzón A. Use of Ni catalysts supported on biomorphic carbon derived from lignocellulosic biomass residues in the decomposition of methane. Front. Energy Res. 2019, 7, 34. 10.3389/fenrg.2019.00034. [DOI] [Google Scholar]
- Li Y.; Li D.; Wang G. Methane decomposition to COx-free hydrogen and nano-carbon material on group 8–10 base metal catalysts: A review. Catal. Today. 2011, 162, 1–48. 10.1016/j.cattod.2010.12.042. [DOI] [Google Scholar]
- Dufour A.; Celzard A.; Ouartassi B.; Broust F.; Fierro V.; Zoulalian A. Effect of micropores diffusion on kinetics of CH4 decomposition over a wood-derived carbon catalyst. Appl. Catal. A Gen. 2009, 360, 120–125. 10.1016/j.apcata.2009.02.033. [DOI] [Google Scholar]
- Fan Z.; Weng W.; Zhou J.; Gu D.; Xiao W. Catalytic decomposition of methane to produce hydrogen: A review. Journal of Energy Chemistry. 2021, 58, 415–430. 10.1016/j.jechem.2020.10.049. [DOI] [Google Scholar]
- Tong S.; Miao B.; Zhang L.; Chan S.H. Decarbonizing natural gas: a review of catalytic decomposition and carbon formation mechanisms. Energies 2022, 15, 2573. 10.3390/en15072573. [DOI] [Google Scholar]; Salam M. A.; Abdullah B. Catalysis mechanism of Pd-promoted γ-alumina in the thermal decomposition of methane to hydrogen: A density functional theory study. Mater. Chem. Phys. 2017, 188, 18–23. 10.1016/j.matchemphys.2016.12.022. [DOI] [Google Scholar]
- Muradov N.; Smith F.; T-Raissi A. Catalytic activity of carbons for methane decomposition reaction. Catal. Today 2005, 102-103, 225–233. 10.1016/j.cattod.2005.02.018. [DOI] [Google Scholar]
- Muradov N.Thermocatalytic CO2-Free production of hydrogen from hydrocarbon fuel cells; Florida Solar Energy Center: Cocoa, FL, 2002.
- Grabke H. J. Evidence on the surface concentration of carbon on gamma iron from the kinetics of the carburization in CH4-H. Metallurgical Transactions. 1970, 1, 2972–2975. 10.1007/BF03037847. [DOI] [Google Scholar]
- Tait S. L.; Dohnálek Z.; Campbell C. T.; Kay B. D. Methane adsorption and dissociation and oxygen adsorption and reaction with CO on Pd nanoparticles on MgO (100) and on Pd (111). Surf. Sci. 2005, 591, 90–107. 10.1016/j.susc.2005.06.024. [DOI] [Google Scholar]
- Gee A. T.; Hayden B. E.; Mormiche C.; Kleyn A. W.; Riedmüller B. The dynamics of the dissociative adsorption of methane on Pt (533). J. Chem. Phys. 2003, 118, 3334–3341. 10.1063/1.1538184. [DOI] [Google Scholar]
- Schwarz H. Chemistry with methane: Concepts rather than recipes. Angewandte Chemie - International Edition. 2011, 50, 10096–10115. 10.1002/anie.201006424. [DOI] [PubMed] [Google Scholar]
- Antony A.; Asthagiri A.; Weaver J.F. Pathways and kinetics of methane and ethane C-H bond cleavage on PdO (101). J. Chem. Phys. 2013, 139, 104702. 10.1063/1.4819909. [DOI] [PubMed] [Google Scholar]
- Weaver J. F.; Hakanoglu C.; Antony A.; Asthagiri A. Alkane activation on crystalline metal oxide surfaces. Chem. Soc. Rev. 2014, 43, 7536–7547. 10.1039/C3CS60420A. [DOI] [PubMed] [Google Scholar]
- Cholewinski M. C.; Dixit M.; Mpourmpakis G. Computational study of methane activation on γ-Al2O3. ACS Omega. 2018, 3, 18242–18250. 10.1021/acsomega.8b02554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljama H.; Nørskov J. K.; Abild-Pedersen F. Theoretical insights into methane C-H bond activation on alkaline metal oxides. J. Phys. Chem. C 2017, 121, 16440–16446. 10.1021/acs.jpcc.7b05838. [DOI] [Google Scholar]
- Krcha M. D.; Mayernick A. D.; Janik M. J. Periodic trends of oxygen vacancy formation and C-H bond activation over transition metal-doped CeO2 (111) surfaces. J. Catal. 2012, 293, 103–115. 10.1016/j.jcat.2012.06.010. [DOI] [Google Scholar]
- Snoeck J. W.; Froment G. F.; Fowles M. Kinetic study of the carbon filament formation by methane cracking on a nickel catalyst. J. Catal. 1997, 169, 250–262. 10.1006/jcat.1997.1635. [DOI] [Google Scholar]
- Alstrup I.; Chorkendorff I.; Ullmann S. Dissociative chemisorption of CH4 on Ni (100) with preadsorbed oxygen. Surf. Sci. 1990, 234, 79–86. 10.1016/0039-6028(90)90667-W. [DOI] [Google Scholar]
- Ashok J.; Naveen Kumar S.; Venugopal A.; Durga Kumari V.; Subrahmanyam M. COX-free H2 production via catalytic decomposition of CH4 over Ni supported on zeolite catalysts. J. Power Sources. 2007, 164, 809–814. 10.1016/j.jpowsour.2006.11.029. [DOI] [Google Scholar]
- Takenaka S.; Ishida M.; Serizawa M.; Tanabe E.; Otsuka K. Formation of carbon nanofibers and carbon nanotubes through methane decomposition over supported cobalt catalysts. J. Phys. Chem. B 2004, 108, 11464–11472. 10.1021/jp048827t. [DOI] [Google Scholar]
- Pudukudy M.; Yaakob Z.; Akmal Z. S. Direct decomposition of methane over Pd promoted Ni/SBA-15 catalysts. Appl. Surf. Sci. 2015, 353, 127–136. 10.1016/j.apsusc.2015.06.073. [DOI] [Google Scholar]
- Rastegarpanah A.; Rezaei M.; Meshkani F.; Zhang K.; Zhao X.; Pei W.; Liu Y.; Deng J.; Arandiyan H.; Dai H. Influence of group VIB metals on activity of the Ni/MgO catalysts for methane decomposition. Appl. Catal., B 2019, 248, 515–525. 10.1016/j.apcatb.2019.01.067. [DOI] [Google Scholar]
- Gao B.; Wang I. W.; Ren L.; Hu J. Catalytic methane decomposition over bimetallic transition metals supported on composite aerogel. Energy and Fuels. 2019, 33, 9099–9106. 10.1021/acs.energyfuels.9b01723. [DOI] [Google Scholar]
- Huang L. N.; Li D.; Tian D.; Jiang L.; Li Z.; Wang H.; Li K. Optimization of Ni-based catalysts for dry reforming of methane via alloy design: a review. Energy and Fuels. 2022, 36, 5102–5151. 10.1021/acs.energyfuels.2c00523. [DOI] [Google Scholar]
- Ashik U. P. M.; Wan Daud W. M. A.; Abbas H. F. Production of greenhouse gas free hydrogen by thermocatalytic decomposition of methane - A review. Renewable and Sustainable Energy Reviews. 2015, 44, 221–256. 10.1016/j.rser.2014.12.025. [DOI] [Google Scholar]
- Cunha A. F.; Órfão J. J. M.; Figueiredo J. L. Methane decomposition on Ni-Cu alloyed Raney-type catalysts. Int. J. Hydrogen Energy. 2009, 34, 4763–4772. 10.1016/j.ijhydene.2009.03.040. [DOI] [Google Scholar]
- Donat F.; Kierzkowska A.; Müller C. R. Chemical looping partial oxidation of methane: reducing carbon deposition through alloying. Energy and Fuels. 2022, 36, 9780–9784. 10.1021/acs.energyfuels.2c01345. [DOI] [Google Scholar]
- Lua A. C.; Wang H. Y. Decomposition of methane over unsupported porous nickel and alloy catalyst. Appl. Catal., B 2013, 132–133, 469–478. 10.1016/j.apcatb.2012.12.014. [DOI] [Google Scholar]
- Shen Y.; Lua A. C. Sol-gel synthesis of titanium oxide supported nickel catalysts for hydrogen and carbon production by methane decomposition. J. Power Sources. 2015, 280, 467–475. 10.1016/j.jpowsour.2015.01.057. [DOI] [Google Scholar]
- Qian J. X.; Liu D.; Basset J. M.; Zhou L. Methane decomposition to produce hydrogen and carbon nanomaterials over costless, iron-containing catalysts. J. Cleaner Prod. 2021, 320, 128879. 10.1016/j.jclepro.2021.128879. [DOI] [Google Scholar]
- Wang I. W.; Dagle R. A.; Khan T. S.; Lopez-Ruiz J. A.; Kovarik L.; Jiang Y.; Xu M.; Wang Y.; Jiang C.; Davidson S. D.; Tavadze P.; Li L.; Hu J. Catalytic decomposition of methane into hydrogen and high-value carbons: combined experimental and DFT computational study. Catal. Sci. Technol. 2021, 11, 4911–4921. 10.1039/D1CY00287B. [DOI] [Google Scholar]
- Zhou L.; Enakonda L. R.; Harb M.; Saih Y.; Aguilar-Tapia A.; Ould-Chikh S.; Hazemann J.; Li J.; Wei N.; Gary D.; Del-Gallo P.; Basset J. M. Fe catalysts for methane decomposition to produce hydrogen and carbon nano materials. Appl. Catal., B 2017, 208, 44–59. 10.1016/j.apcatb.2017.02.052. [DOI] [Google Scholar]
- Figueiredo J. L.; Órfão J. J. M.; Cunha A. F. Hydrogen production via methane decomposition on Raney-type catalysts. Int. J. Hydrogen Energy. 2010, 35, 9795–9800. 10.1016/j.ijhydene.2009.12.071. [DOI] [Google Scholar]
- Liang W.; Yan H.; Feng X.; Chen C.; Lin D.; Liu J.; Chen X.; Liu Y.; Yang C.; Shan H. NiMgAlMo catalyst derived from a guest-host MoO42- mediated layered double hydroxide: High performance for the methane decomposition reaction. Appl. Catal. A Gen. 2020, 597, 117551. 10.1016/j.apcata.2020.117551. [DOI] [Google Scholar]
- Saraswat S. K.; Pant K.K. Synthesis of hydrogen and carbon nanotubes over copper promoted Ni/SiO2 catalyst by thermocatalytic decomposition of methane. J. Nat. Gas Sci. Eng. 2013, 13, 52–59. 10.1016/j.jngse.2013.04.001. [DOI] [Google Scholar]
- Ibrahim A. A.; Al-Fatesh A. S.; Khan W. U.; Soliman M. A.; al Otaibi R. L.; Fakeeha A. H. Influence of support type and metal loading in methane decomposition over iron catalyst for hydrogen production. Journal of the Chinese Chemical Society. 2015, 62, 592–599. 10.1002/jccs.201500052. [DOI] [Google Scholar]
- Bayat N.; Rezaei M.; Meshkani F. COx-free hydrogen and carbon nanofibers production by methane decomposition over nickel-alumina catalysts. Korean Journal of Chemical Engineering. 2016, 33, 490–499. 10.1007/s11814-015-0183-y. [DOI] [Google Scholar]
- Awadallah A. E.; Aboul-Enein A. A.; Yonis M. M.; Aboul-Gheit A. K. Effect of structural promoters on the catalytic performance of cobalt-based catalysts during natural gas decomposition to hydrogen and carbon nanotubes. Fullerenes Nanotubes and Carbon Nanostructures. 2016, 24, 181–189. 10.1080/1536383X.2015.1132206. [DOI] [Google Scholar]
- Rastegarpanah A.; Meshkani F.; Rezaei M. COx-free hydrogen and carbon nanofibers production by thermocatalytic decomposition of methane over mesoporous MgO·Al2O3 nanopowder-supported nickel catalysts. Fuel Process. Technol. 2017, 167, 250–262. 10.1016/j.fuproc.2017.07.010. [DOI] [Google Scholar]
- Gao B.; Wang I. W.; Ren L.; Hu J. Catalytic methane decomposition over bimetallic transition metals supported on composite aerogel. Energy and Fuels. 2019, 33, 9099–9106. 10.1021/acs.energyfuels.9b01723. [DOI] [Google Scholar]
- Calgaro C. O.; Perez-Lopez O. W. Decomposition of methane over Co3–xAlxO4 (x = 0–2) coprecipitated catalysts: The role of Co phases in the activity and stability. Int. J. Hydrogen Energy. 2017, 42, 29756–29772. 10.1016/j.ijhydene.2017.10.082. [DOI] [Google Scholar]
- Pudukudy M.; Yaakob Z.; Jia Q.; Takriff M. S. Catalytic decomposition of methane over rare earth metal (Ce and La) oxides supported iron catalysts. Appl. Surf. Sci. 2019, 467–468, 236–248. 10.1016/j.apsusc.2018.10.122. [DOI] [Google Scholar]
- al -Fatesh A. S.; Kasim S. O.; Ibrahim A. A.; Al-Awadi A. S.; Abasaeed A. E.; Fakeeha A. H.; Awadallah A. E. Catalytic methane decomposition over ZrO2 supported iron catalysts: Effect of WO3 and La2O3 addition on catalytic activity and stability. Renew Energy. 2020, 155, 969–978. 10.1016/j.renene.2020.04.038. [DOI] [Google Scholar]
- Ayillath Kutteri D.; Wang I. W.; Samanta A.; Li L.; Hu J. Methane decomposition to tip and base grown carbon nanotubes and COx-free H2 over mono- and bimetallic 3d transition metal catalysts. Catal. Sci. Technol. 2018, 8, 858–869. 10.1039/C7CY01927K. [DOI] [Google Scholar]
- Al-Fatesh A.-S.; Barama S.; Ibrahim A.-A.; Barama A.; Khan W.-U.; Fakeeha A. Study of methane decomposition on Fe/MgO-based catalyst modified by Ni, Co, and Mn additives. Chem. Eng. Commun. 2017, 204, 739–749. 10.1080/00986445.2017.1311254. [DOI] [Google Scholar]
- Zhang C.; Zhang W.; Drewett N. E.; Wang X.; Yoo S. J.; Wang H.; Deng T.; Kim J. G.; Chen H.; Huang K.; Feng S.; Zheng W. Integrating catalysis of methane decomposition and electrocatalytic hydrogen evolution with Ni/CeO2 for improved hydrogen production efficiency. ChemSusChem. 2019, 12, 1000–1010. 10.1002/cssc.201802618. [DOI] [PubMed] [Google Scholar]
- Lee S. C.; Seo J.; Han G. Y. Hydrogen production by catalytic decomposition of methane over carbon black catalyst at high temperatures. Korean J. Chem. Eng. 2013, 30, 1716–1721. 10.1007/s11814-013-0107-7. [DOI] [Google Scholar]
- Nishii H.; Miyamoto D.; Umeda Y.; Hamaguchi H.; Suzuki M.; Tanimoto T.; Harigai T.; Takikawa H.; Suda Y. Catalytic activity of several carbons with different structures for methane decomposition and by-produced carbons. Appl. Surf. Sci. 2019, 473, 291–297. 10.1016/j.apsusc.2018.12.073. [DOI] [Google Scholar]
- Lee S. Y.; Ryu B. H.; Han G. Y.; Lee T. J.; Yoon K. J. Catalytic characteristics of specialty carbon blacks in decomposition of methane for hydrogen production. Carbon. 2008, 46, 1978–1986. 10.1016/j.carbon.2008.08.008. [DOI] [Google Scholar]
- Wang J.; Jin L.; Li Y.; Hu H. Preparation of Fe-doped carbon catalyst for methane decomposition to hydrogen. Ind. Eng. Chem. Res. 2017, 56, 11021–11027. 10.1021/acs.iecr.7b02394. [DOI] [Google Scholar]
- Zhang J.; Qi M.; Zhang G.; Hu H.; Xie L.; Ma X. Co-production of hydrogen and fibrous carbons by methane decomposition using K2CO3/carbon hybrid as the catalyst. Int. J. Hydrogen Energy. 2017, 42, 11047–11052. 10.1016/j.ijhydene.2017.03.113. [DOI] [Google Scholar]
- Zhang J.; Xie W.; Li X.; Hao Q.; Chen H.; Ma X. Methane decomposition over Ni/carbon catalysts prepared by selective gasification of coal char. Energy Convers Manag. 2018, 177, 330–338. 10.1016/j.enconman.2018.09.075. [DOI] [Google Scholar]
- Zhang J.; Xie W.; Li X.; Hao Q.; Chen H.; Ma X. In situ generation of nickel/carbon catalysts by partial gasification of coal char and application for methane decomposition. Int. J. Hydrogen Energy. 2019, 44, 2633–2644. 10.1016/j.ijhydene.2018.12.005. [DOI] [Google Scholar]
- Jin L.; Si H.; Zhang J.; Lin P.; Hu Z.; Qiu B.; Hu H. Preparation of activated carbon supported Fe-Al2O3 catalyst and its application for hydrogen production by catalytic methane decomposition. Int. J. Hydrogen Energy. 2013, 38, 10373–10380. 10.1016/j.ijhydene.2013.06.023. [DOI] [Google Scholar]
- Harun K.; Adhikari S.; Jahromi H. Hydrogen production: via thermocatalytic decomposition of methane using carbon-based catalysts. RSC Adv. 2020, 10, 40882–40893. 10.1039/D0RA07440C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Y.; Yang Y.; Shi X. L.; Feng L.; Hou X.; Ye X.; Zhang L.; Suo G.; Chen J.; Chen Z. G. Bi2O3/BiVO4@graphene oxide van der Waals heterostructures with enhanced photocatalytic activity toward oxygen generation. J. Colloid Interface Sci. 2021, 593, 196–203. 10.1016/j.jcis.2021.02.079. [DOI] [PubMed] [Google Scholar]
- Shilapuram V.; Ozalp N. Hydrogen production by carbon-catalyzed methane decomposition via thermogravimetry. J. Energy Resour. Technol. 2017, 139, 012005. 10.1115/1.4035145. [DOI] [Google Scholar]
- Yang L.; Xin C.; Xuan G.; Liu S.; Liu F.; Ren Y. Protrusions induction by carbon black on surface of activated carbon to enhance its catalytic activity. Fuel. 2022, 324, 124378. 10.1016/j.fuel.2022.124378. [DOI] [Google Scholar]
- Dunker A. M.; Kumar S.; Mulawa P. A. Production of hydrogen by thermal decomposition of methane in a fluidized-bed reactor - Effects of catalyst, temperature, and residence time. Int. J. Hydrogen Energy. 2006, 31, 473–484. 10.1016/j.ijhydene.2005.04.023. [DOI] [Google Scholar]
- Zou X.; Yang Y.; Chen H.; Shi X. L.; Suo G.; Ye X.; Zhang L.; Hou X.; Feng L.; Chen Z. G. Tuning wall thickness of TiO2 microtubes for an enhanced photocatalytic activity with thickness-dependent charge separation efficiency. J. Colloid Interface Sci. 2020, 579, 463–469. 10.1016/j.jcis.2020.06.081. [DOI] [PubMed] [Google Scholar]
- Kundu R.; Ramasubramanian V.; Neeli S. T.; Ramsurn H. Catalytic pyrolysis of methane to hydrogen over carbon (from cellulose biochar) encapsulated iron nanoparticles. Energy and Fuels. 2021, 35, 13523–13533. 10.1021/acs.energyfuels.1c01620. [DOI] [Google Scholar]
- Muradov N. Z.; Veziroǧlu T. N. From hydrocarbon to hydrogen-carbon to hydrogen economy. Int. J. Hydrogen Energy. 2005, 30, 225–237. 10.1016/j.ijhydene.2004.03.033. [DOI] [Google Scholar]
- Hadian M.; Buist K. A.; Bos A. N. R.; Kuipers J. A. M. Single catalyst particle growth modeling in thermocatalytic decomposition of methane. Chemical Engineering Journal. 2021, 421, 129759. 10.1016/j.cej.2021.129759. [DOI] [Google Scholar]
- Bai Z.; Chen H.; Li W.; Li B. Hydrogen production by methane decomposition over coal char. Int. J. Hydrogen Energy. 2006, 31, 899–905. 10.1016/j.ijhydene.2005.08.001. [DOI] [Google Scholar]
- Kang D.; Lee J. W. Enhanced methane decomposition over nickel-carbon-B2O3 core-shell catalysts derived from carbon dioxide. Appl. Catal., B 2016, 186, 41–55. 10.1016/j.apcatb.2015.12.045. [DOI] [Google Scholar]
- Chen J.; He M.; Wang G.; Li Y.; Zhu Z. J. Production of hydrogen from methane decomposition using nanosized carbon black as catalyst in a fluidized-bed reactor. Int. J. Hydrogen Energy. 2009, 34, 9730–9736. 10.1016/j.ijhydene.2009.10.030. [DOI] [Google Scholar]
- Zhang J.; Jin L.; Li Y.; Si H.; Qiu B.; Hu H. Hierarchical porous carbon catalyst for simultaneous preparation of hydrogen and fibrous carbon by catalytic methane decomposition. Int. J. Hydrogen Energy. 2013, 38, 8732–8740. 10.1016/j.ijhydene.2013.05.012. [DOI] [Google Scholar]
- Wang J.; Jin L.; Li Y.; Hu H. Preparation of Fe-doped carbon catalyst for methane decomposition to hydrogen. Ind. Eng. Chem. Res. 2017, 56, 11021–11027. 10.1021/acs.iecr.7b02394. [DOI] [Google Scholar]
- Ryoo R.; Joo S. H.; Jun S. Synthesis of highly ordered carbon molecular sieves via template-mediated structural transformation. J. Phys. Chem. B 1999, 103, 7743–7746. 10.1021/jp991673a. [DOI] [Google Scholar]
- Ryoo R.; Joo S. H.; Kruk M.; Jaroniec M. Ordered mesoporous carbons. Adv. Mater. 2001, 13, 677–681. . [DOI] [Google Scholar]
- Serrano D. P.; Botas J.Á.; Pizarro P.; Guil-López R.; Gómez G. Ordered mesoporous carbons as highly active catalysts for hydrogen production by CH4 decomposition. Chemical Communications. 2008, 6585–6587. 10.1039/b811800k. [DOI] [PubMed] [Google Scholar]
- Bai Z.; Chen H.; Li B.; Li W. Methane decomposition over Ni loaded activated carbon for hydrogen production and the formation of filamentous carbon. Int. J. Hydrogen Energy. 2007, 32, 32–37. 10.1016/j.ijhydene.2006.06.030. [DOI] [Google Scholar]
- Sarada Prasad J.; Dhand V.; Himabindu V.; Anjaneyulu Y. Production of hydrogen and carbon nanofibers through the decomposition of methane over activated carbon supported Ni catalysts. Int. J. Hydrogen Energy. 2011, 36, 11702–11711. 10.1016/j.ijhydene.2011.05.176. [DOI] [Google Scholar]
- Zhang J.; Jin L.; Li Y.; Hu H. Ni doped carbons for hydrogen production by catalytic methane decomposition. Int. J. Hydrogen Energy. 2013, 38, 3937–3947. 10.1016/j.ijhydene.2013.01.105. [DOI] [Google Scholar]
- Srilatha K.; Viditha V.; Srinivasulu D.; Ramakrishna S. U. B.; Himabindu V. Hydrogen production using thermocatalytic decomposition of methane on Ni30/activated carbon and Ni30/carbon black. Environmental Science and Pollution Research. 2016, 23, 9303–9311. 10.1007/s11356-015-5112-4. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang Y.; Zhao S.; Zhu J.; Jin L.; Hu H. Preparation of bimetallic catalysts Ni-Co and Ni-Fe supported on activated carbon for methane decomposition. Carbon Resources Conversion. 2020, 3, 190–197. 10.1016/j.crcon.2020.12.002. [DOI] [Google Scholar]
- Zhang J.; Xie W.; Li X.; Hao Q.; Chen H.; Ma X. Methane decomposition over Ni/carbon catalysts prepared by selective gasification of coal char. Energy Convers Manag. 2018, 177, 330–338. 10.1016/j.enconman.2018.09.075. [DOI] [Google Scholar]
- Zhang J.; Li X.; Xie W.; Hao Q.; Chen H.; Ma X. Handy synthesis of robust Ni/carbon catalysts for methane decomposition by selective gasification of pine sawdust. Int. J. Hydrogen Energy. 2018, 43, 19414–19419. 10.1016/j.ijhydene.2018.08.207. [DOI] [Google Scholar]
- Zhang J.; Xie W.; Li X.; Hao Q.; Chen H.; Ma X. In situ generation of nickel/carbon catalysts by partial gasification of coal char and application for methane decomposition. Int. J. Hydrogen Energy. 2019, 44, 2633–2644. 10.1016/j.ijhydene.2018.12.005. [DOI] [Google Scholar]
- Zhang J.; Ren M.; Li X.; Hao Q.; Chen H.; Ma X. Ni-based catalysts prepared for CO2 reforming and decomposition of methane. Energy Convers. Manag. 2020, 205, 112419. 10.1016/j.enconman.2019.112419. [DOI] [Google Scholar]
- Yang X.; Yang E.; Hu B.; Yan J.; Shangguan F.; Hao Q.; Chen H.; Zhang J.; Ma X. Nanofabrication of Ni-incorporated three-dimensional ordered mesoporous carbon for catalytic methane decomposition. J. Environ. Chem. Eng. 2022, 10, 107451. 10.1016/j.jece.2022.107451. [DOI] [Google Scholar]
- Shen Y.; Lua A. C. Synthesis of Ni and Ni-Cu supported on carbon nanotubes for hydrogen and carbon production by catalytic decomposition of methane. Appl. Catal., B 2015, 164, 61–69. 10.1016/j.apcatb.2014.08.038. [DOI] [Google Scholar]
- Jin L.; Si H.; Zhang J.; Lin P.; Hu Z.; Qiu B.; Hu H. Preparation of activated carbon supported Fe-Al2O3 catalyst and its application for hydrogen production by catalytic methane decomposition. Int. J. Hydrogen Energy. 2013, 38, 10373–10380. 10.1016/j.ijhydene.2013.06.023. [DOI] [Google Scholar]
- Wang J.; Jin L.; Li Y.; Hu H. Preparation of Fe-doped carbon catalyst for methane decomposition to hydrogen. Ind. Eng. Chem. Res. 2017, 56, 11021–11027. 10.1021/acs.iecr.7b02394. [DOI] [Google Scholar]
- Prasad J. S.; Dhand V.; Himabindu V.; Anjaneyulu Y.; Jain P. K.; Padya B. Production of hydrogen and carbon nanofibers through the decomposition of methane over activated carbon supported Pd catalysts. Int. J. Hydrogen Energy. 2010, 35, 10977–10983. 10.1016/j.ijhydene.2010.07.021. [DOI] [Google Scholar]
- Szymańska M.; Malaika A.; Rechnia P.; Miklaszewska A.; Kozłowski M. Metal/activated carbon systems as catalysts of methane decomposition reaction. Catal. Today. 2015, 249, 94–102. 10.1016/j.cattod.2014.11.025. [DOI] [Google Scholar]
- Wang J.; Jin L.; Zhou Y.; Li Y.; Hu H. Effect of Ca(NO3)2 addition in coal on properties of activated carbon for methane decomposition to hydrogen. Fuel Process. Technol. 2018, 176, 85–90. 10.1016/j.fuproc.2018.03.012. [DOI] [Google Scholar]
- Harun K.; Adhikari S.; Jahromi H. Hydrogen production: via thermocatalytic decomposition of methane using carbon-based catalysts. RSC Adv. 2020, 10, 40882–40893. 10.1039/D0RA07440C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradov N. Hydrogen via methane decomposition: an application for decarbonization of fossil fuels. Int. J. Hydrogen Energy 2001, 26, 1165–1175. 10.1016/S0360-3199(01)00073-8. [DOI] [Google Scholar]
- Al-Hassani A. A.; Abbas H. F.; Daud W. M. A. W. Hydrogen production via decomposition of methane over activated carbons as catalysts: Full factorial design. Int. J. Hydrogen Energy. 2014, 39, 7004–7014. 10.1016/j.ijhydene.2014.02.075. [DOI] [Google Scholar]
- Mahmoudi M.; Dentzer J.; Gadiou R.; Ouederni A. Evaluation of activated carbons based on olive stones as catalysts during hydrogen production by thermocatalytic decomposition of methane. Int. J. Hydrogen Energy. 2017, 42, 8712–8720. 10.1016/j.ijhydene.2016.11.177. [DOI] [Google Scholar]
- Kim M. H.; Lee E. K.; Jun J. H.; Han G. Y.; Kong S. J.; Lee B. K.; Lee T.-J.; Yoon K. J. Hydrogen production by catalytic decomposition of methane over activated carbons: deactivation study. Korean J. Chem. Eng. 2003, 20, 835–839. 10.1007/BF02697284. [DOI] [Google Scholar]
- Kim M. H.; Lee E. K.; Jun J. H.; Kong S. J.; Han G. Y.; Lee B. K.; Lee T. J.; Yoon K. J. Hydrogen production by catalytic decomposition of methane over activated carbons: Kinetic study. Int. J. Hydrogen Energy. 2004, 29, 187–193. 10.1016/S0360-3199(03)00111-3. [DOI] [Google Scholar]
- Lumbers B.; Agar D. W.; Gebel J.; Platte F. Mathematical modelling and simulation of the thermo-catalytic decomposition of methane for economically improved hydrogen production. Int. J. Hydrogen Energy. 2022, 47, 4265–4283. 10.1016/j.ijhydene.2021.11.057. [DOI] [Google Scholar]
- Bai Z.; Chen H.; Li B.; Li W. Catalytic decomposition of methane over activated carbon. J. Anal Appl. Pyrolysis. 2005, 73, 335–341. 10.1016/j.jaap.2005.03.004. [DOI] [Google Scholar]
- Ammendola P.; Chirone R.; Ruoppolo G.; Russo G.; Solimene R. Some issues in modelling methane catalytic decomposition in fluidized bed reactors. Int. J. Hydrogen Energy. 2008, 33, 2679–2694. 10.1016/j.ijhydene.2008.03.033. [DOI] [Google Scholar]
- Henao W.; Cazaña F.; Tarifa P.; Romeo E.; Latorre N.; Sebastian V.; Delgado J.J.; Monzón A. Selective synthesis of carbon nanotubes by catalytic decomposition of methane using Co-Cu/cellulose derived carbon catalysts: A comprehensive kinetic study. Chem. Eng. J. 2021, 404, 126103. 10.1016/j.cej.2020.126103. [DOI] [Google Scholar]
- Zhang J.; Jin L.; Li Y.; Hu H. Ni doped carbons for hydrogen production by catalytic methane decomposition. Int. J. Hydrogen Energy. 2013, 38, 3937–3947. 10.1016/j.ijhydene.2013.01.105. [DOI] [Google Scholar]
- Pudukudy M.; Yaakob Z.; Kadier A.; Takriff M. S.; Hassan N. S. M. One-pot sol–gel synthesis of Ni/TiO2 catalysts for methane decomposition into COx free hydrogen and multiwalled carbon nanotubes. Int. J. Hydrogen Energy. 2017, 42, 16495–16513. 10.1016/j.ijhydene.2017.04.223. [DOI] [Google Scholar]
- Alves Silva J.; Oliveira Santos J. B.; Torres D.; Pinilla J. L.; Suelves I. Natural Fe-based catalysts for the production of hydrogen and carbon nanomaterials via methane decomposition. Int. J. Hydrogen Energy. 2021, 46, 35137–35148. 10.1016/j.ijhydene.2021.08.065. [DOI] [Google Scholar]
- Pinilla J. L.; Utrilla R.; Karn R. K.; Suelves I.; Lázaro M. J.; Moliner R.; García A. B.; Rouzaud J. N. High temperature iron-based catalysts for hydrogen and nanostructured carbon production by methane decomposition. Int. J. Hydrogen Energy. 2011, 36, 7832–7843. 10.1016/j.ijhydene.2011.01.184. [DOI] [Google Scholar]
- Zhang J.; Xie W.; Li X.; Hao Q.; Chen H.; Ma X. Methane decomposition over Ni/carbon catalysts prepared by selective gasification of coal char. Energy Convers Manag. 2018, 177, 330–338. 10.1016/j.enconman.2018.09.075. [DOI] [Google Scholar]
- Wang Y.; Zhang Y.; Zhao S.; Zhu J.; Jin L.; Hu H. Preparation of bimetallic catalysts Ni-Co and Ni-Fe supported on activated carbon for methane decomposition. Carbon Resources Conversion. 2020, 3, 190–197. 10.1016/j.crcon.2020.12.002. [DOI] [Google Scholar]
- Ashik U. P. M.; Abbas H. F.; Abnisa F.; Kudo S.; Hayashi J.; Daud W. M. A. W. Methane decomposition with a minimal catalyst: An optimization study with response surface methodology over Ni/SiO2 nanocatalyst. Int. J. Hydrogen Energy. 2020, 45, 14383–14395. 10.1016/j.ijhydene.2020.03.164. [DOI] [Google Scholar]
- Strelko V.; Malik D. J.; Streat M. Characterisation of the surface of oxidised carbon adsorbents. Carbon. 2002, 40, 95–104. 10.1016/S0008-6223(01)00082-3. [DOI] [Google Scholar]
- Sangsiri P.; Laosiripojana N.; Laosiripojana W.; Daorattanachai P. Activity of a sulfonated carbon-based catalyst derived from organosolv lignin toward esterification of stearic acid under near-critical alcohol conditions. ACS Omega. 2022, 7, 40025–40033. 10.1021/acsomega.2c04693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafeeyan M. S.; Daud W. M. A. W.; Houshmand A.; Shamiri A. A review on surface modification of activated carbon for carbon dioxide adsorption. J. Anal Appl. Pyrolysis. 2010, 89, 143–151. 10.1016/j.jaap.2010.07.006. [DOI] [Google Scholar]
- Pittman C. U.; He G.-R.; Wu B.; Gardner S. D. Chemical modification of carbon fiber surfaces by nitric acid oxidation followed by reaction with tetraethylenepentamine. Carbon. 1997, 35, 317–331. 10.1016/S0008-6223(97)89608-X. [DOI] [Google Scholar]
- Wang S.; Lu G.Q. Effects of acidic treatments on the pore and surface properties of Ni catalyst supported on activated carbon. Carbon 1998, 36, 283–292. 10.1016/S0008-6223(97)00187-5. [DOI] [Google Scholar]
- Bai Z.; Li W.; Bai J.; Li B.; Chen H. The effects of textural properties and surface chemistry of activated carbon on its catalytic performance in methane decomposition for hydrogen production. Energy Sources, Part A: Recovery, Utilization and Environmental Effects. 2012, 34, 1145–1153. 10.1080/15567031003663174. [DOI] [Google Scholar]
- Wang Y.; Yang H.; Jin L.; Li Y.; Hu H.; Ding H.; Bai X. Effect of mineral in coal on preparation of activated carbon for methane decomposition to hydrogen. Fuel 2019, 258, 116138. 10.1016/j.fuel.2019.116138. [DOI] [Google Scholar]
- Malaika A.; Kozłowski M. Influence of ethylene on carbon-catalysed decomposition of methane. Int. J. Hydrogen Energy. 2009, 34, 2600–2605. 10.1016/j.ijhydene.2009.01.052. [DOI] [Google Scholar]
- Pinilla J. L.; Suelves I.; Lázaro M. J.; Moliner R. Influence on hydrogen production of the minor components of natural gas during its decomposition using carbonaceous catalysts. J. Power Sources. 2009, 192, 100–106. 10.1016/j.jpowsour.2008.12.074. [DOI] [Google Scholar]
- Rechnia P.; Malaika A.; Najder-Kozdrowska L.; Kozłowski M. The effect of ethanol on carbon-catalysed decomposition of methane. Int. J. Hydrogen Energy. 2012, 37, 7512–7520. 10.1016/j.ijhydene.2012.02.014. [DOI] [Google Scholar]
- Wei L.; Tan Y. S.; Han Y. Z.; Zhao J. T.; Wu J.; Zhang D. Hydrogen production by methane cracking over different coal chars. Fuel. 2011, 90, 3473–3479. 10.1016/j.fuel.2011.06.053. [DOI] [Google Scholar]
- Zhang J.; Li X.; Chen H.; Qi M.; Zhang G.; Hu H.; Ma X. Hydrogen production by catalytic methane decomposition: Carbon materials as catalysts or catalyst supports. Int. J. Hydrogen Energy. 2017, 42, 19755–19775. 10.1016/j.ijhydene.2017.06.197. [DOI] [Google Scholar]
- Zhang J.; Li X.; Chen H.; Qi M.; Zhang G.; Hu H.; Ma X. Hydrogen production by catalytic methane decomposition: Carbon materials as catalysts or catalyst supports. Int. J. Hydrogen Energy. 2017, 42, 19755–19775. 10.1016/j.ijhydene.2017.06.197. [DOI] [Google Scholar]
- Tong S.; Miao B.; Zhang L.; Chan S.H. Decarbonizing natural gas: a review of catalytic decomposition and carbon formation mechanisms. Energies 2022, 15, 2573. 10.3390/en15072573. [DOI] [Google Scholar]
- Snoeck J. W.; Froment G. F.; Fowles M. Filamentous carbon formation and gasification: thermodynamics, driving force, nucleation, and steady-state growth. J. Catal. 1997, 169, 240–249. 10.1006/jcat.1997.1634. [DOI] [Google Scholar]
- Chen Q.; Lua A.C. Kinetic reaction and deactivation studies on thermocatalytic decomposition of methane by electroless nickel plating catalyst. Chem. Eng. J. 2020, 389, 124366. 10.1016/j.cej.2020.124366. [DOI] [Google Scholar]
- Zhu X.; Huo P.; Zhang Y. P.; Cheng D. G.; Liu C. J. Structure and reactivity of plasma treated Ni/Al2O3 catalyst for CO2 reforming of methane. Appl. Catal., B 2008, 81, 132–140. 10.1016/j.apcatb.2007.11.042. [DOI] [Google Scholar]
- Frusteri F.; Italiano G.; Espro C.; Cannilla C.; Bonura G. H2 production by methane decomposition: catalytic and technological aspects. Int. J. Hydrogen Energy. 2012, 37, 16367–16374. 10.1016/j.ijhydene.2012.02.192. [DOI] [Google Scholar]
- Italiano G.; Delia A.; Espro C.; Bonura G.; Frusteri F. Methane decomposition over Co thin layer supported catalysts to produce hydrogen for fuel cell. Int. J. Hydrogen Energy. 2010, 35, 11568–11575. 10.1016/j.ijhydene.2010.05.012. [DOI] [Google Scholar]
- Snoeck J. W.; Froment G. F.; Fowles M. Kinetic study of the carbon filament formation by methane cracking on a nickel catalyst. J. Catal. 1997, 169, 250–262. 10.1006/jcat.1997.1635. [DOI] [Google Scholar]
- Suelves I.; Pinilla J. L.; Lázaro M. J.; Moliner R. Carbonaceous materials as catalysts for decomposition of methane. Chemical Engineering Journal. 2008, 140, 432–438. 10.1016/j.cej.2007.11.014. [DOI] [Google Scholar]
- Yu J.; Lucas J.; Strezov V.; Wall T. Coal and carbon nanotube production. Fuel. 2003, 82, 2025–2032. 10.1016/S0016-2361(03)00189-3. [DOI] [Google Scholar]
- Dasireddy V. D. B. C.; Likozar B. Activation and decomposition of methane over cobalt-, copper-, and iron-based heterogeneous catalysts for COx-free hydrogen and multiwalled carbon nanotube production. Energy Technology. 2017, 5, 1344–1355. 10.1002/ente.201600633. [DOI] [Google Scholar]
- Awadallah A. E.; Aboul-Enein A. A.; Mahmoud A. H.; Abd El Rehim S. S.; El-Ziaty A. K.; Aboul-Gheit A. K. ZrxMg1-xO supported cobalt catalysts for methane decomposition into COx-free hydrogen and carbon nanotubes. Int. J. Green Energy. 2018, 15, 568–576. 10.1080/15435075.2018.1510407. [DOI] [Google Scholar]
- Ashok J.; Reddy P. S.; Raju G.; Subrahmanyam M.; Venugopal A. Catalytic decomposition of methane to hydrogen and carbon nanofibers over Ni-Cu-SiO2 catalysts. Energy and Fuels. 2009, 23, 5–13. 10.1021/ef8003976. [DOI] [Google Scholar]
- Ibrahim A. A.; Fakeeha A. H.; Al-Fatesh A. S.; Abasaeed A. E.; Khan W. U. Methane decomposition over iron catalyst for hydrogen production. Int. J. Hydrogen Energy. 2015, 40, 7593–7600. 10.1016/j.ijhydene.2014.10.058. [DOI] [Google Scholar]
- Butt J. B.Reaction kinetics and reactor design, 2nd ed.; Marcel Dekker Inc.: New York, NY, 1999. [Google Scholar]
- Latorre N.; Romeo E.; Villacampa J. I.; Cazaña F.; Royo C.; Monzón A. Kinetics of carbon nanotubes growth on a Ni-Mg-Al catalyst by CCVD of methane: Influence of catalyst deactivation. Catal. Today 2010, 154, 217–223. 10.1016/j.cattod.2010.03.065. [DOI] [Google Scholar]
- Gadkari S.; Fidalgo B.; Gu S. Numerical analysis of microwave assisted thermocatalytic decomposition of methane. Int. J. Hydrogen Energy. 2017, 42, 4061–4068. 10.1016/j.ijhydene.2016.09.126. [DOI] [Google Scholar]
- Abanades S.; Kimura H.; Otsuka H. Kinetic investigation of carbon-catalyzed methane decomposition in a thermogravimetric solar reactor. Int. J. Hydrogen Energy. 2015, 40, 10744–10755. 10.1016/j.ijhydene.2015.07.023. [DOI] [Google Scholar]
- Muradov N.; Chen Z.; Smith F. Fossil hydrogen with reduced CO2 emission: Modeling thermocatalytic decomposition of methane in a fluidized bed of carbon particles. Int. J. Hydrogen Energy. 2005, 30, 1149–1158. 10.1016/j.ijhydene.2005.04.005. [DOI] [Google Scholar]
- Alsaffar M. A.; Ghany M. A. R. A.; Ali J. M.; Ayodele B. V.; Mustapa S. I. Artificial neural network modeling of thermo-catalytic methane decomposition for hydrogen production. Top Catal. 2021, 64, 456–464. 10.1007/s11244-020-01409-6. [DOI] [Google Scholar]
- Abbas H. F.; Baker I. F. Thermocatalytic decomposition of methane using activated carbon: Studying the influence of process parameters using factorial design. Int. J. Hydrogen Energy. 2011, 36, 8985–8993. 10.1016/j.ijhydene.2011.05.005. [DOI] [Google Scholar]
- Rodat S.; Abanades S.; Coulié J.; Flamant G. Kinetic modelling of methane decomposition in a tubular solar reactor. Chemical Engineering Journal. 2009, 146, 120–127. 10.1016/j.cej.2008.09.008. [DOI] [Google Scholar]
- Palmer C.; Upham D. C.; Smart S.; Gordon M. J.; Metiu H.; McFarland E. W. Dry reforming of methane catalysed by molten metal alloys. Nat. Catal. 2020, 3, 83–89. 10.1038/s41929-019-0416-2. [DOI] [Google Scholar]
- Postels S.; Abánades A.; von der Assen N.; Rathnam R. K.; Stückrad S.; Bardow A. Life cycle assessment of hydrogen production by thermal cracking of methane based on liquid-metal technology. Int. J. Hydrogen Energy. 2016, 41, 23204–23212. 10.1016/j.ijhydene.2016.09.167. [DOI] [Google Scholar]
- Khazaal M. H.; Staniforth J. Z.; Alfatlawi Z. A.; Ormerod R. M.; Darton R. J. Enhanced methane reforming activity of a hydrothermally synthesized codoped perovskite catalyst. Energy and Fuels. 2018, 32, 12826–12832. 10.1021/acs.energyfuels.8b02848. [DOI] [Google Scholar]
- Zhu J.; Li H.; Zhong L.; Xiao P.; Xu X.; Yang X.; Zhao Z.; Li J. Perovskite oxides: Preparation, characterizations, and applications in heterogeneous catalysis. ACS Catal. 2014, 4, 2917–2940. 10.1021/cs500606g. [DOI] [Google Scholar]
- Wang P.; Zhang X.; Shi R.; Zhao J.; Yuan Z.; Zhang T. Light-driven hydrogen production from steam methane reforming via bimetallic PdNi catalysts derived from layered double hydroxide nanosheets. Energy and Fuels. 2022, 36, 11627–11635. 10.1021/acs.energyfuels.2c01349. [DOI] [Google Scholar]
- Zhang J.; Jin L.; Liu S.; Xun Y.; Hu H. Mesoporous carbon prepared from direct coal liquefaction residue for methane decomposition. Carbon. 2012, 50, 952–959. 10.1016/j.carbon.2011.09.058. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.