

Significance
Protein trafficking and secretion through the endomembrane system is a defining feature of eukaryotes. While the secretory pathway is central to the unique biology and pathology of the obligate intracellular malaria parasite, tools for studying secreted protein function are limited. Knock-sideways is a powerful strategy to interrogate protein function by conditionally sequestering a protein away from its site of action; however, this approach is generally not applicable to secreted proteins. We developed a simple approach to conditionally sequester Plasmodium-secreted proteins in the ER by inducible C-terminal fusion with a KDEL ER-retrieval sequence that can be used for trafficking, topology, and loss-of-function studies. The knockER strategy is broadly applicable to functional dissection of proteins that traverse the eukaryotic secretory pathway.
Keywords: Plasmodium, ER, ClpB1, PTEX, knock-sideways
Abstract
Malaria parasites uniquely depend on protein secretion for their obligate intracellular lifestyle but approaches for dissecting Plasmodium-secreted protein functions are limited. We report knockER, a unique DiCre-mediated knock-sideways approach to sequester secreted proteins in the ER by inducible fusion with a KDEL ER-retrieval sequence. We show conditional ER sequestration of diverse proteins is not generally toxic, enabling loss-of-function studies. We employed knockER in multiple Plasmodium species to interrogate the trafficking, topology, and function of an assortment of proteins that traverse the secretory pathway to diverse compartments including the apicoplast (ClpB1), rhoptries (RON6), dense granules, and parasitophorous vacuole (EXP2, PTEX150, HSP101). Taking advantage of the unique ability to redistribute secreted proteins from their terminal destination to the ER, we reveal that vacuolar levels of the PTEX translocon component HSP101 but not PTEX150 are maintained in excess of what is required to sustain effector protein export into the erythrocyte. Intriguingly, vacuole depletion of HSP101 hypersensitized parasites to a destabilization tag that inhibits HSP101-PTEX complex formation but not to translational knockdown of the entire HSP101 pool, illustrating how redistribution of a target protein by knockER can be used to query function in a compartment-specific manner. Collectively, our results establish knockER as a unique tool for dissecting secreted protein function with subcompartmental resolution that should be widely amenable to genetically tractable eukaryotes.
Protein secretion is a fundamental biological process of all living cells. In eukaryotes, this is accomplished by a dedicated secretory pathway that begins with import into the Endoplasmic Reticulum (ER) for initial folding and posttranslational modification, followed by sorting in the Golgi and post-Golgi compartments. As an obligate intracellular parasite, secreted proteins play a critical role in the biology of the malaria parasite, Plasmodium. Through the secretory pathway, proteins are trafficked into both the plasma membrane and several organelles, including a relict plastid called the apicoplast that is critical for parasite metabolism (1, 2). During schizogony, another subset of proteins is packaged into several distinct secretory organelles that are subsequently discharged to facilitate host cell attachment and penetration (3). Invagination of the host membrane by the parasite during invasion generates an intracellular microenvironment called the parasitophorous vacuole (PV) which forms the interface for host–parasite interactions (4). Resident PV proteins secreted into the vacuole perform critical functions at this interface, including nutrient/waste exchange and protein export (5). The latter process enables parasites to deploy a battery of secreted effector proteins across the PV membrane (PVM) and into the host compartment, dramatically remodeling the erythrocyte to meet nutritional demands and avoid host defenses (6, 7). Remarkably, it is estimated that up to 10% of the parasite genome is devoted to encoding exported effectors, further highlighting the central importance of protein secretion to this parasitic lifestyle (8–12).
As with other eukaryotes, entry into the malaria parasite secretory pathway generally begins with cotranslational import of proteins containing a signal peptide or transmembrane domain(s) into the ER via the Sec61 complex, followed by sorting from the Golgi to internal compartments, the PV or beyond into the host (13). ER-resident proteins that escape the ER via bulk flow in COPII vesicles are returned to the ER in COPI vesicles by ERD2 surveillance, which binds a C-terminal retrieval sequence (“KDEL” and variants) in the cis Golgi and releases it in the ER in a pH-dependent manner (14–16). Similar to other eukaryotes, ER retention of secreted reporters or chimeric proteins can be achieved in Plasmodium spp. by a C-terminal KDEL or functional variants (17–22).
In this present work, we developed and characterized a unique DiCre-based knock-sideways tool called knockER that enables conditional fusion of KDEL to the C-terminus of secreted proteins, subjecting the target protein to ERD2 surveillance in the cis Golgi to cause its retrieval to the ER. Using this approach, we show successful conditional retrieval of five endogenous Plasmodium proteins (ClpB1, RON6, EXP2, PTEX150, and HSP101) that traffic through the ER to diverse compartments including the apicoplast, rhoptries, dense granules, and the PV. Importantly, conditional ER retrieval of both highly expressed reporters and endogenously tagged proteins does not generally impact fitness, indicating that knockER-mediated phenotypes are not the indirect result of an integrated stress response, making this system suitable for protein loss-of-function studies. Taking advantage of the unique ability to redistribute secreted proteins from their terminal destination to the ER, we reveal a surprising indifference to ER retrieval of HSP101 in P. falciparum, in contrast to the other PTEX components, indicating that HSP101 is maintained in excess in the PV. Interestingly, redistribution of HSP101 from the PV to the ER hypersensitized parasites to a posttranslational destabilization tag that disrupts HSP101 interaction with PTEX but not to translational knockdown, providing support for an HSP101 function in the early secretory pathway in addition to its role in protein translocation across the PVM in the assembled PTEX complex, as recently proposed (23–25). These results establish knockER as a unique tool to study eukaryotic secreted proteins.
Results
Conditional ER Retrieval of a Secreted Reporter Does Not Impact Parasite Fitness.
To facilitate conditional ER retrieval of a protein of interest, we designed a strategy based on the dimerizable Cre recombinase (DiCre) system (26, 27) where a gene of interest is C-terminally fused to a 3xFLAG tag and a stop codon flanked by loxP sequences imbedded in small introns (28). Activation of DiCre by rapamycin results in excision of the loxP-intervening sequence, removing the 3xFLAG tag and stop codon, and bringing into frame a 3xHA tag followed by the ER-retention sequence KDEL and a stop codon (Fig. 1A). While the ER plays a crucial role in quality control and is sensitive to added stress, several previous studies have exogenously expressed various KDEL or SDEL fusion proteins in P. falciparum and P. berghei without apparent impact on parasite fitness (19, 29–36). To directly ascertain whether ER retrieval of a highly expressed secreted protein impacted parasite fitness, we first appended the knockER system to an mNeonGreen (mNG) reporter and fused this to the exp2 5′ UTR and signal peptide, enabling expression from the strong exp2 promoter and entry into the ER followed by default secretion into the PV (37–39). This assembly was placed under control of the anhydrotetracycline (aTc)-responsive TetR-DOZI-aptamers system to prevent expression until aTc is present in the culture (Fig. 1A) (40, 41). To combine the Bxb1 integrase system with DiCre, we first introduced a DiCre expression cassette at the benign pfs47 locus (27) in the P. falciparum NF54attB strain (42) (SI Appendix, Fig. S1) and then integrated the mNG-knockER reporter construct at the attB site in the cg6 locus on chromosome 6 in this NF54attB-DiCre line.
Fig. 1.

Conditional ER retrieval of a secreted reporter does not impact parasite fitness. (A) Schematic of reporter expression cassette installed at the attB site of chromosome 6. An mNeonGreen reporter is fused to the signal peptide of EXP2 and expressed under control of the exp2 promoter. A 3xFLAG tag and stop codon are flanked by loxP-containing introns (triangles) followed by a 3xHA-KDEL, stop codon, and 10× aptamer array (10×-Apt). Aptamer interaction with TetR-DOZI (expressed from another cassette in the plasmid) prevents translation of the reporter, which is relieved in the presence of anhydrotetracycline (aTc) to enable expression. Activation of DiCre by rapamycin treatment excises the FLAG tag, bringing into frame the HA-KDEL tag. (B) Time course of excision following rapamycin treatment detected by PCR using primers P7/32. (C) Western blot of mNG reporter 24 h posttreatment with DMSO or rapamycin. Molecular weights after signal peptide cleavage are predicted to be 30.8 kDa for mNG-3xFLAG and 30.9 kDa for mNG-3xHA-KDEL. (D) Live microscopy of DMSO- or rapamycin-treated parasites 24 h posttreatment. (E) Quantification of percent internal mNG fluorescence 24 h and 168 h posttreatment with DMSO or rapamycin. Data are pooled from two independent experiments and bar indicates mean (****P < 0.0001; unpaired t test). (F) Representative growth of asynchronous parasites (n = 2 biological replicates) treated with DMSO or rapamycin. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). All cultures were maintained in media supplemented with 500 nM aTc. (Scale bar, 5 μm.)
Upon treatment with 10 nM rapamycin for 3 h, parasites efficiently excised the knockER cassette (kER) with essentially complete conversion of the population within one parasite developmental cycle (48 h) resulting in the expected tag switching from FLAG to HA (Fig. 1 B and C). Examination of DMSO vehicle–treated parasites by live microscopy showed a peripheral mNG signal, consistent with secretion to the PV (Fig. 1D). In contrast, following treatment with rapamycin the mNG signal was dramatically altered to an intracellular, perinuclear pattern that colocalized with ER Tracker, confirming retention in the ER (Fig. 1D). Quantification of the distribution of mNG fluorescence between the parasite periphery and interior showed substantial, stable relocalization following KDEL fusion, indicating robust retrieval by ERD2 (Fig. 1E, mean % internal mNG at 24 h: 48.78 ± 5.8% in DMSO vs. 76.87 ± 8.9% in Rap, and at 168 h: 42.54 ± 6.0% in DMSO vs. 84.66 ± 6.9% in Rap). Importantly, ER retention of mNG had no impact on parasite growth (Fig. 1F) showing that knockER can successfully retrieve a secreted reporter to the ER without impacting parasite fitness.
knockER Reveals Golgi-Dependent Trafficking and Essential Apicoplast Functionality of ClpB1.
Next, we applied knockER to an endogenous gene for conditional loss-of-function mutagenesis. We chose ClpB1, a poorly characterized, nuclear-encoded AAA+ class 1 Clp/HSP100 chaperone that is targeted to the apicoplast (43). As apicoplast ablation can be chemically rescued with isopentenyl pyrophosphate (IPP) (44), we reasoned that IPP supplementation would provide a simple validation that any loss of parasite fitness is a direct effect of sequestering ClpB1 away from its site of action and not an indirect result of ER stress. ClpB1 contains both a signal peptide for ER entry and a transit peptide for apicoplast targeting, and localization of ClpB1 to this organelle was previously confirmed by both microscopy and proteomic analyses (35, 43). Using CRISPR/Cas9 editing, we fused mNG to the C-terminus of ClpB1 followed by the kER cassette (Fig. 2A). As expected, IFA demonstrated colocalization with the apicoplast marker ACP1, confirming ClpB1 localization to this organelle (Fig. 2B). Treatment with rapamycin resulted in efficient excision (Fig. 2C) and switching to the 3xHA-KDEL tag produced a shift in migration by Western blot consistent with an increase in molecular weight of ~14 kDa in the rapamycin-treated samples, indicating that the N-terminal transit peptide (residues 24 to 152, 13.9 kDa) is no longer cleaved in the ClpB1-KDEL fusion (Fig. 2D). Since this maturation event takes place in the apicoplast (45), this suggested that ClpB1 was no longer being trafficked to this organelle. Indeed, live microscopy showed a dramatic relocalization of mNG from puncta to a perinuclear distribution that colocalized with ER Tracker, indicative of successful ER retrieval (Fig. 2E).
Fig. 2.

Lethal ER retention of the apicoplast chaperone ClpB1 is rescued by IPP. (A) Schematic showing strategy for appending kER to the C-terminus of clpB1. (B) IFA of PFA fixed parasites stained with mouse anti-Flag and rabbit anti-ACP1 antibodies. (C) PCR showing excision in ClpB1-kER parasites 24 h after rapamycin treatment using primers P7/8. (D) Western blot 24 h posttreatment with DMSO or rapamycin. Molecular weights after transit peptide cleavage are predicted to be 135 kDa for ClpB1-mNG-3xFLAG and 135.1 kDa for ClpB1-mNG-3xHA-KDEL. (+) represents the mature form of ClpB1 while the (*) represents ClpB1 with the transit peptide still attached. (E) Live microscopy of DMSO- or rapamycin-treated parasites 48 h posttreatment. (F) Representative growth of asynchronous parasites (n = 3 biological replicates) treated with DMSO or rapamycin, with or without supplementation with 200 µM IPP. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). (G) IFA of PFA fixed parasites stained with mouse anti-Flag or anti-HA and rabbit anti-ACP1 antibodies after three growth cycles where rapamycin-treated parasites were supplemented with 200 µM IPP. (Scale bars, 5 μm.)
While its functional importance has not been directly tested, ClpB1 is expected to be essential for blood-stage survival based on saturating insertional mutagenesis in P. falciparum and pooled knockout screens in P. berghei (46, 47). Indeed, ER retention of ClpB1 was lethal and resulted in delayed parasite death (Fig. 2F), consistent with an essential role in the apicoplast (48, 49). Moreover, supplementation with IPP fully rescued parasite growth, confirming the observed phenotype was apicoplast-specific and not due to a disruption of ER homeostasis following ClpB1 retention (Fig. 2F). Rapamycin-treated parasites supplemented with IPP showed dispersed, vesicular ACP1 signal consistent with plastid disruption, further indicating that ClpB1 is required for apicoplast maintenance (Fig. 2G). Taken together, these results show for the first time that ClpB1 is an indispensable component of the apicoplast machinery and illustrate that knockER can be used for protein loss-of-function studies in P. falciparum.
knockER Induces ER Retrieval of Rhoptry Proteins.
Having demonstrated that knockER is suitable to generate loss-of-function phenotypes by protein mislocalization to the ER, we next tested this knock-sideways approach on an expanded set of endogenous soluble and membrane proteins that traffic through the secretory pathway to diverse destinations, including secretory organelles and the PV/PVM. We began by attempting to retrieve RON6, a soluble rhoptry-neck protein that is injected into the PV during invasion (SI Appendix, Fig. S2 A and B). Attempts predating CRISPR technology to disrupt Pfron6 or truncate its Cysteine-rich C-terminus were unsuccessful (50). More recently, saturating transposon mutagenesis in P. falciparum recovered a single insertion within an intron (46) expected to generate a truncation at residue 713 of 950, leaving most of the protein intact (SI Appendix, Fig. S2A). Curiously, RON6 orthologs in rodent malaria parasites lack the cysteine-rich region which accounts for the C-terminal ~23% of P. falciparum RON6, roughly corresponding to the truncated region in the insertional mutant. As RON6 was not targeted in the PlasmoGEM dataset, its suspected essentiality remains uncertain (47). To directly test the importance of RON6, we generated RON6-kER parasites, which underwent efficient excision upon rapamycin treatment (SI Appendix, Fig. S2 C and D). Rapamycin treatment resulted in relocalization of RON6-mNG from the rhoptries to the ER and produced a minor but reproducible growth defect (SI Appendix, Fig. S2 D–F). These results suggest that RON6 may be dispensable in the blood stage, although we cannot exclude the possibility that small amounts of RON6-KDEL may escape ERD2 surveillance at levels sufficient to mediate a critical function. Additionally, the minor fitness defect that results from RON6-KDEL fusion could be the indirect result of modest ER stress induced by RON6 retention. Nonetheless, these findings clearly demonstrate that knockER efficiently retrieves secretory proteins synthesized during the brief period of merozoite formation at the terminal phase of intraerythrocytic development.
ER Retention of EXP2 Produces a Lethal Defect Independent to Loss of PVM Transport Function.
Having successfully retained several soluble proteins in the ER using knockER, we next tested conditional retrieval of a membrane protein. Solute permeation across the PVM is mediated by a nutrient-permeable channel (51, 52) whose identity was recently tied to EXP2, a PVM protein broadly conserved among vacuole-dwelling apicomplexans (53, 54). EXP2 contains a signal peptide followed by an N-terminal amphipathic helix and oligomerizes to form a heptameric pore in the PVM (25, 55, 56). It is currently unknown how pore formation is specifically constrained to the PVM to avoid perforating other membranes along the secretory pathway or the parasite plasma membrane. While not structurally homologous to the EXP2 pore, bacterial α-helical pore-forming toxins are functionally analogous and provide a possible model for EXP2 pore formation. These proteins are typically maintained in a monomeric state that shields a membrane-spanning amphipathic helix, enabling soluble trafficking until contact with components of a target membrane triggers conformational rearrangement, leading to membrane insertion and oligomerization (57).
If EXP2 traffics in a similar configuration, then its C-terminus will enter the secretory pathway lumen and be sensitive to ER-retrieval by KDEL fusion. Indeed, we endogenously tagged EXP2 with a version of the kER cassette lacking mNG and found that KDEL fusion disrupted EXP2 trafficking to the PV and resulted in ER retention, confirming that its C-terminus resides in the secretory pathway lumen (Fig. 3 A–D). Notably, ER retrieval of EXP2 appeared to cause more rapid parasite death than previously observed by conditional knockdown or knockout approaches (53, 58, 59) (Fig. 3E). To confirm this observation, we engineered parasites to enable DiCre-mediated conditional knockout of EXP2 (EXP2-cKO) to allow for matched induction kinetics to EXP2-kER (SI Appendix, Fig. S3 A–C). Following activation of DiCre in synchronized, ring-stage parasites, the EXP2 knockout progressed to the next cycle with equivalent new ring parasitemia to the DMSO control before death in the second cycle; in contrast, EXP2-kER parasites died immediately in the first cycle and did not form new rings (SI Appendix, Fig. S3D). To test whether parasite death was solely due to loss of EXP2 transport functions in the PV and not an indirect result of retaining EXP2 in the ER, we generated parasites expressing a second copy of EXP2-mNG-kER from the attB locus under the control of its own promoter, leaving the endogenous EXP2 locus unaltered (SI Appendix, Fig. S3 E–G). Retrieval of this second copy of EXP2 to the ER also resulted in parasite death, indicating that EXP2 retention in the ER is toxic independent of compromised transport functions at the PVM (Fig. 3 F and G). In contrast, ER retrieval of a second copy of EXP2 lacking the amphipathic helix did not cause parasite death (Fig. 3 H and I and SI Appendix, Fig. S3 E–G). As expected, prior to KDEL fusion, the ∆TM version of EXP2 localized to the PV and the digestive vacuole (due to endocytosis of PV proteins along with RBC cytosol). Surprisingly, most cells also showed EXP2ΔTM-mNG export into the RBC cytoplasm (arrow, Fig. 3H and SI Appendix, Fig. S3H). The basis for EXP2ΔTM-mNG export is unclear but may result from interactions with other PTEX components that somehow result in recognition as cargo in the absence of the amphipathic helix. The requirement for the amphipathic helix in EXP2-kER toxicity suggests that increasing EXP2 dwell time in the early secretory pathway may trigger an unfolded protein response or cause aberrant EXP2 pore formation in the ER membrane. Thus, this may provide a new tool for studying EXP2 pore formation but also indicates knockER may not be appropriate for loss-of-function studies of certain membrane proteins.
Fig. 3.

ER retention of EXP2 causes a lethal fitness defect independent of loss of PVM transport functions. (A) Schematic showing strategy for appending kER to the endogenous C-terminus of EXP2 without mNG. (B) PCR showing excision in EXP2-kER 24 h after treatment with rapamycin using primers P9/10. (C) Western blot of EXP2-kER parasites 24 h posttreatment with DMSO or rapamycin. Molecular weights after signal peptide cleavage are predicted to be 34.6 kDa for EXP2-3xFLAG and 34.7 kDa for EXP2-3xHA-KDEL. (D) IFA of EXP2-kER 24 h posttreatment with DMSO or rapamycin. (E) Growth of asynchronous parasites (n = 2 biological replicates) treated with DMSO or rapamycin. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). (F) Live microscopy of parasites expressing a full-length second copy of EXP2 with an mNG-kER fusion from the attB site under the control of the endogenous exp2 promoter (EXP2-FL-kERattB). Parasites were viewed 24 h after treatment with DMSO or rapamycin. (G) Representative growth of asynchronous EXP2-FL-kERattB parasites (n = 2 biological replicates) treated with DMSO or rapamycin. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). (H) Live microscopy of parasites expressing a second copy of EXP2 with mNG-kER fusion and lacking the amphipathic helix from the attB site (EXP2∆TM-kERattB). Parasites were viewed 24 h after treatment with DMSO or rapamycin. Two representative examples of unexcised parasites are presented, showing localization of the ∆TM version of EXP2 to the PV and host cell in infected RBCs (arrow). Digestive vacuole fluorescence was also observed as is typical for PV proteins. (I) Representative growth of asynchronous EXP2∆TM-kERattB parasites (n = 2 biological replicates) treated with DMSO or rapamycin. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). Second copy EXP2-kER lines were maintained in media supplemented with 500 nM aTc. (Scale bars, 5 μm.)
ER Retention of PTEX150 but not HSP101 Results in Loss of PTEX Function in P. falciparum.
In malaria parasites, the nutrient-permeable channel has been additionally functionalized by the flange-like adaptor PTEX150 which docks the AAA+ chaperone HSP101 onto EXP2 to form the Plasmodium Translocon of Exported proteins (PTEX), transforming EXP2 into a protein-conducting pore to translocate effector proteins across the PVM and into the erythrocyte (23–25, 53, 60). Interestingly, HSP101 shows a dual localization to both the PV and the ER as opposed to EXP2 and PTEX150, which localize exclusively to the PV and form a subcomplex independent of HSP101 (34, 39, 61, 62). We verified this unique ER localization of HSP101 by protease protection assays in parasite lines expressing fluorescently tagged versions of each PTEX core component, confirming an internal, perinuclear pool of HSP101 but not EXP2 or PTEX150 (SI Appendix, Fig. S4). This arrangement suggests that in addition to powering translocation in the assembled PTEX complex, HSP101 may also function upstream of the vacuole, possibly initiating cargo selection early in the secretory pathway (61). While several HSP101 conditional knockdown (24, 61) or inactivation (23) strategies have demonstrated an essential role in protein export, it is difficult to resolve distinct compartmental functions with these mutants given that they impact the total protein pool and have no subcompartmental resolution. In contrast, knockER can potentially resolve functions across distinct compartments by redistributing a target protein pool from its terminal destination to the ER.
In an attempt to separate HSP101 ER function from its role in the PV, we generated a kER fusion to the endogenous copy of HSP101, expecting that redistribution of HSP101 to the ER would be lethal but might provide insight into any ER localized function which could be preserved compared to the phenotype of HSP101 knockdown mutants. In parallel, we also generated a PTEX150-kER line, reasoning that since HSP101 and PTEX150 have similar expression timing and are exclusively involved in protein export but not nutrient uptake (53), PTEX150 would provide a control for knockER inactivation of PTEX activity independent of ER function. As expected, the PTEX150-KDEL fusion was efficiently retrieved to the ER, producing a lethal growth defect and accompanying block in protein export (Fig. 4 A–E), similar to glmS ribozyme-mediated knockdown of PTEX150 (24). Importantly, ER retention of a second copy of PTEX150-kER in parasites where the endogenous PTEX150 locus was unmodified did not impact growth, demonstrating that the phenotype observed upon ER retention of the endogenously tagged protein is a direct result of functional disruption of PTEX150 (SI Appendix, Fig. S5 A–D).
Fig. 4.
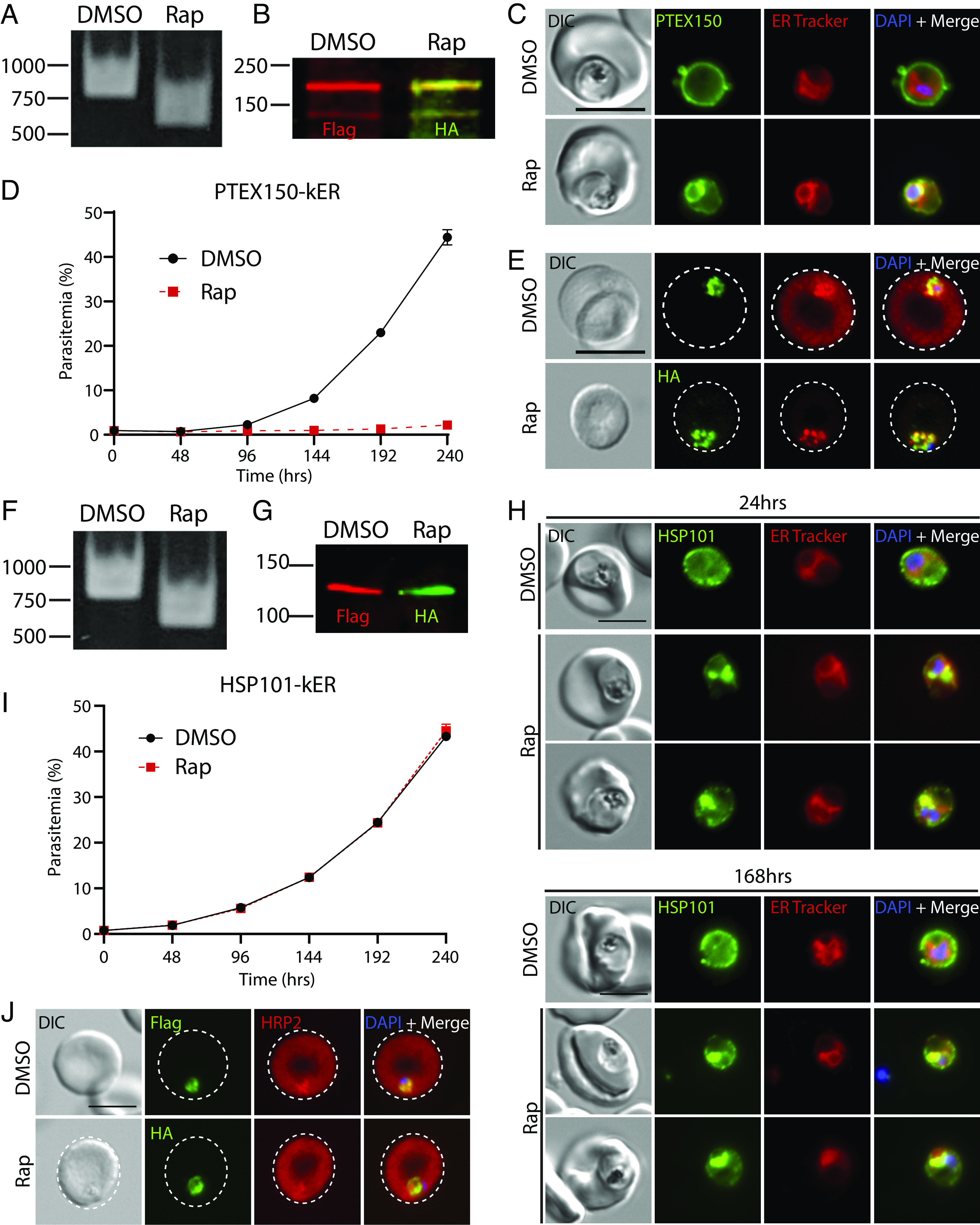
ER retention of PTEX150 but not HSP101 causes a lethal block in protein export in P. falciparum. (A) PCR showing excision in PTEX150-kER parasites 24 h after rapamycin treatment using primers P7/8. (B) Western blot of PTEX150-kER 24 h posttreatment with DMSO or rapamycin. Molecular weights after signal peptide cleavage are predicted to be 141 kDa for PTEX150-mNG-3xFLAG and 141.1 kDa for PTEX150-mNG-3xHA-KDEL. Note that PTEX150 is observed to migrate at higher molecular weight than predicted (60). (C) Live microscopy of PTEX150-kER 24 h posttreatment with DMSO or rapamycin. (D) Representative growth of asynchronous PTEX150-kER parasites (n = 3 biological replicates) treated with DMSO or rapamycin. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). (E) IFA of synchronized, ring-stage PTEX150-kER parasites 48 h posttreatment with DMSO or rapamycin and probed with anti-HRP2 and anti-FLAG or anti-HA antibodies. (F) PCR showing excision in HSP101-kER parasites 24 h after rapamycin treatment using primers P7/8. (G) Western blot of HSP101-kER parasites 14 d posttreatment with DMSO or rapamycin. Molecular weights after signal peptide cleavage are predicted to be 130.5 kDa for HSP101-mNG-3xFLAG and 130.6 kDa for HSP101-mNG-3xHA-KDEL. (H) Live microscopy of HSP101-kER 24 h and 168 h posttreatment with DMSO or rapamycin. (I) Representative growth of asynchronous HSP101-kER parasites (n = 3 biological replicates) treated with DMSO or rapamycin. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). (J) IFA of synchronized, ring-stage HSP101-kER parasites 48 h posttreatment with DMSO or rapamycin and probed with anti-HRP2 and anti-FLAG or anti-HA antibodies. (Scale bar, 5 μm.)
Unexpectedly, although rapamycin-induced HSP101-KDEL fusion increased ER localization similar to PTEX150 (Fig. 4 F–H), parasite growth was completely unaffected and protein export was not detectibly impacted (Fig. 4 I and J), sharply contrasting with the lethal export defect observed with HSP101 depletion or inactivation mutants (23, 24, 61). Indeed, HSP101-kER parasites could be cultured indefinitely following excision and displayed sustained ER retention of HSP101 (Fig. 4H and SI Appendix, Fig. S5 E and F, mean Rap internal HSP101: 58.82 ± 9.58% at 24 h vs. 54.82 ± 9.84% at 7 d). As an orthogonal approach to monitor protein redistribution by KDEL fusion, we released infected RBCs with saponin to permeabilize the PVM and quantified fluorescence via flow cytometry with or without proteinase K treatment. Again, we observed an increase in protected fluorescence following rapamycin treatment, consistent with redistribution of HSP101 to the ER (SI Appendix, Fig. S5G). Importantly, PCR and Western blot indicated that rapamycin-treated parasites had undergone complete excision with the preexcised FLAG-tagged version of HSP101 no longer detectible (Fig. 4 F and G). Furthermore, both the mNG-FLAG and mNG-HA-KDEL tagged versions of HSP101 migrated as a single band at the expected size for the full-length fusion proteins with no apparent breakdown products, ensuring that the mNG signal distribution observed by live microscopy reflected an intact HSP101 fusion (Fig. 4G and SI Appendix, Figs. S7B and S8B).
While a large fraction of PTEX150 and HSP101 was clearly ER-retained following knockER induction, some peripheral signal was still apparent after rapamycin treatment (Fig. 4 C and H). To determine whether the observed phenotypic differences between HSP101 and PTEX150 were due to a difference in sensitivity to ERD2 surveillance, we quantified the internal mNG fluorescence signal in HSP101- and PTEX150-kER parasites following KDEL fusion. In synchronized parasites, a similar increase in internal signal was observed in both lines at 24 h post Rap treatment (SI Appendix, Fig. S5E, mean Rap internal HSP101 58.82 ± 9.58% vs. 56.32 ± 18.56% for PTEX150). Since a comparison could not be made with PTEX150-kER parasites at 7 d due to parasite death by the second cycle following excision, we compared HSP101-kER parasites with second copy PTEX150-kERattB control parasites 7 d after excision and again observed a comparable internal signal redistribution (SI Appendix, Fig. S5F, mean Rap internal HSP101 54.82 ± 9.84% vs. 61.75 ± 16.27% for PTEX150 at 7 d). As major differences in ER retrieval cannot account for parasite insensitivity to HSP101-kER, these results indicate that vacuolar HSP101 levels are unexpectedly maintained in excess from what is required for PTEX function at the PVM.
ER Retention of P. berghei HSP101 Results in a Lethal Export Defect.
To determine if ER retention of HSP101 was similarly tolerated in other Plasmodium species, we tagged HSP101 and PTEX150 with kER in a P. berghei rodent malaria DiCre parasite. We also included a downstream cassette in this tagging plasmid for expression of a secreted mRuby under control of the constitutive Pbhsp70 promoter to label the PV (SI Appendix, Fig. S6A). Similar to P. falciparum knockER mutants, rapamycin treatment produced efficient excision and ER retrieval of PbPTEX150 and PbHSP101 (Fig. 5 A–C and F). Interestingly, PbHSP101-kER and PbPTEX150-kER parasites displayed greater internal retention than the corresponding P. falciparum knockER mutants (HSP101: mean % internal mNG 77.42 ± 7.72% in P. berghei vs. 58.82 ± 9.58% in P. falciparum; and PTEX150: mean % internal mNG 69.55 ± 10.86% in P. berghei vs. 56.32 ± 18.56% in P. falciparum), suggesting that KDEL-mediated ER retrieval is more stringent in rodent parasites (Fig. 5 D and G). To evaluate protein export, we generated versions of the PbPTEX150-kER and PbHSP101-kER mutants where the PV-mRuby reporter was replaced with a cassette for expression of the exported protein IBIS1 fused to mRuby (SI Appendix, Fig. S6A) (63). In contrast to P. falciparum, KDEL fusion to both PbPTEX150 and PbHSP101 produced a stark block in IBIS1 export (Fig. 5 I and J). To determine whether KDEL fusion to PbHSP101 impacted parasite fitness, parasites were induced ex vivo followed by overnight culture to allow for tag switching before equal numbers of DMSO- or rapamycin-treated parasites were IV injected into naïve mice. Strikingly, while DMSO controls became patent by day two postinjection, neither KDEL-fused PbPTEX150 nor PbHSP101 parasites appeared over the course of 10 d before the experiment was terminated (Fig. 5 E and H and SI Appendix, Fig. S6B).
Fig. 5.
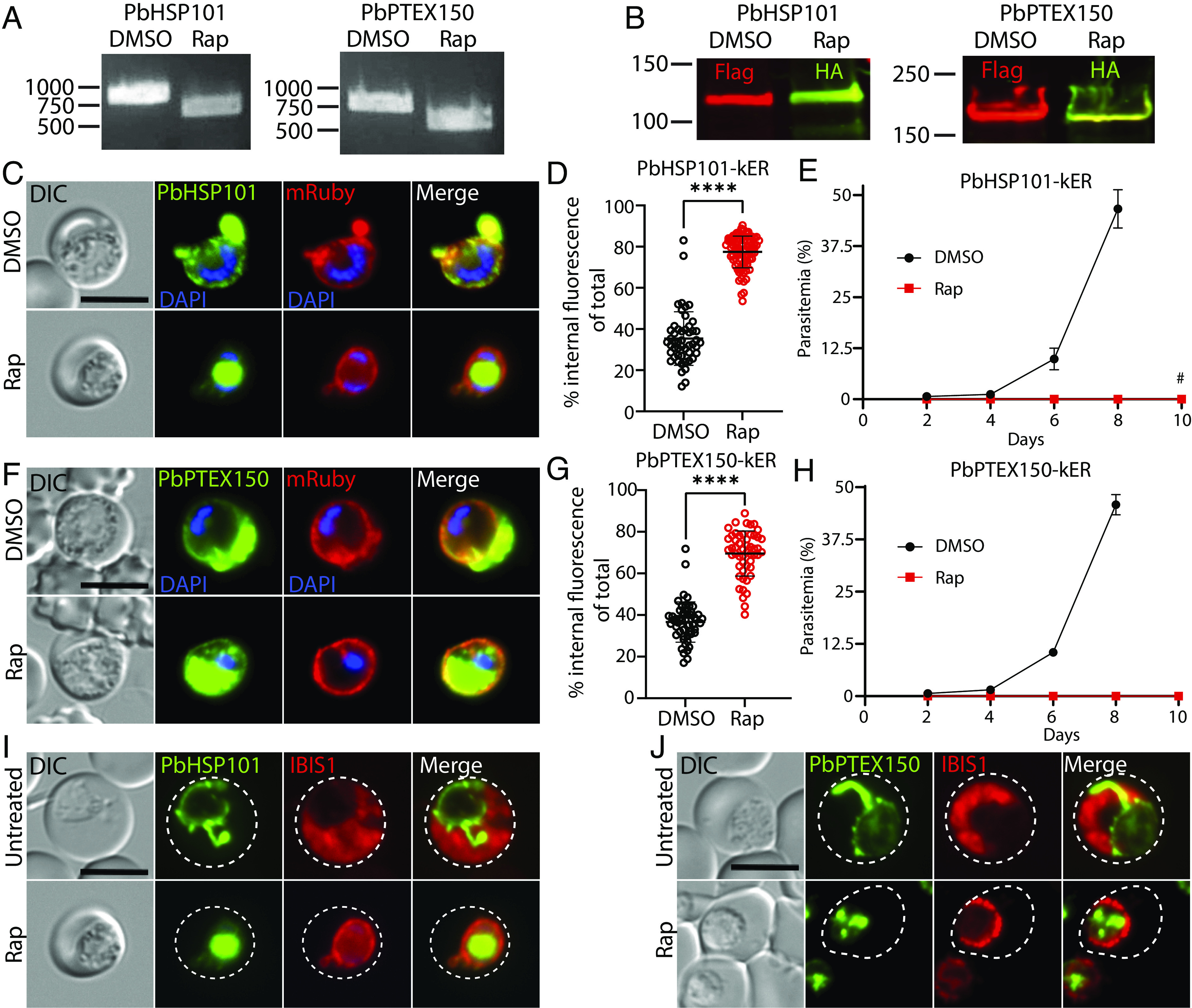
ER retention of HSP101 causes a lethal block in protein export in P. berghei. (A) PCR showing excision in PbPTEX150-kER and PbHSP101-kER P. berghei parasites treated with rapamycin and cultured 18 h ex vivo using primers P7/P60. (B) Western blot of PbPTEX150-kER and PbHSP101-kER parasites 24 h posttreatment with DMSO or rapamycin. Molecular weights after signal peptide cleavage are predicted to be 130.9 kDa for PbHSP101-mNG-3xFLAG, 131 kDa for PbHSP101-mNG-3xHA-KDEL, 130.5 kDa for PbPTEX150-mNG-3xFLAG, and 130.6 kDa for PbPTEX150-mNG-3xHA-KDEL. Note that PbPTEX150 is observed to migrate at higher molecular weight than predicted (64). (C) Live microscopy of PbHSP101-kER parasites 18 h posttreatment with DMSO or rapamycin. This transgenic line also contains a downstream cassette for expression of mRuby bearing a signal peptide for secretion into the PV (SI Appendix, Fig. S6A). (D) Quantification of percent internal mNG fluorescence in PbHSP101-kER 24 h posttreatment with DMSO or rapamycin. Data are pooled from two independent experiments and bar indicates mean (****P < 0.0001; unpaired t test). (E) Parasitemia of PbHSP101-kER infected mice inoculated by tail vein injection of parasites treated ex vivo with DMSO or rapamycin. Data are presented as means ± SD of three independent experiments (one mouse infected per treatment in each experiment). No parasites were observed in mice injected with rapamycin-treated parasites except that one mouse injected with rapamycin-treated PbHSP101-kER parasites became patent on day 10 (#). However, PCR showed these parasites had not undergone excision (SI Appendix, Fig. S6B). (F) Live microscopy of PbPTEX150-kER parasites as in C. (G) Quantification of percent internal mNG fluorescence in PbPTEX150-kER as in D. (H) Parasitemia of PbPTEX150-kER infected mice as in E. No parasites were observed in mice injected with rapamycin-treated parasites. (I and J) Live microscopy of PbHSP101-kER or PbPTEX150-kER parasites containing a downstream cassette for expression of an IBIS1-mRuby fusion that is exported into the RBC (SI Appendix, Fig. S6A). Parasites were imaged before or 24 h after treatment with rapamycin. Dashed lines indicate RBC boundaries traced from DIC images. Images are representative of n = 2 biological replicates. (Scale bars, 5 μm.)
The block in IBIS1 export suggests that PbHSP101-KDEL is sufficiently retained to produce a lethal export defect, in contrast with PfHSP101-KDEL; however, phenotypes can be masked by the rich resources available in culture (65, 66) and many exported effectors critical for survival in the vertebrate host are dispensable in vitro (12) where parasites do not have to contend with host defenses (67, 68). To distinguish whether the fitness defect incurred by ER retention of PbHSP101 resulted simply from parasite death or from the unique pressures imposed by the host, we evaluated parasite development of synchronized ring-stage cultures ex vivo. Importantly, both PbHSP101-kER and PbPTEX150-kER parasites also failed to develop into terminal schizonts in vitro following rapamycin treatment, indicating that the in vivo fitness defect is not merely the result of host defenses (SI Appendix, Fig. S6 C–E). Taken together, our data show that ER retention of HSP101-KDEL is more efficient in P. berghei, resulting in a lethal defect in protein export.
ER Retention of P. falciparum HSP101 Hypersensitizes Parasites to a Destabilization Tag that Inhibits HSP101-PTEX Complex Formation.
The lack of an observable phenotype after ER-retention of HSP101 in P. falciparum suggests that the level of retention does not reach a critical threshold required to impact HSP101 function at the PV. To determine the amount of HSP101 needed to support P. falciparum growth in vitro, we combined knockER with the TetR-DOZI-aptamers system to generate a HSP101-kER line that also allows for titration of HSP101 expression with aTc (Fig. 6A). Like the original HSP101-kER line, rapamycin-treated HSP101-kERTetR-DOZI parasites exhibited ER retention of HSP101 without impacting parasite growth or export of representative PEXEL and PNEP proteins (Fig. 6 B–D). However, HSP101 depletion following aTc washout resulted in a robust block in protein export and parasite death (Fig. 6D and SI Appendix, Fig. S7 A and B), consistent with previous HSP101 knockdown or inactivation studies (23, 24, 61). To determine the level of total HSP101 reduction sufficient to impact growth, we grew synchronized parasites at a range of aTc concentrations following DMSO or rapamycin treatment and measured mNG fluorescence via flow cytometry in parallel with parasite growth (Fig. 6 E and F). Interestingly, rapamycin-treated parasites grown at higher aTc concentrations displayed a small but reproducible increase in total HSP101, consistent with a reduction in HSP101 turnover by endocytosis from the PV to the digestive vacuole following redistribution to the ER (Fig. 6E). More importantly, we determined that a ~70% reduction in total HSP101 levels was necessary to impact parasite growth, showing that a relatively small amount of HSP101 is needed for survival (Fig. 6 E and F, 5 nM aTc), consistent with redistribution of HSP101 by knockER not meeting the threshold required to impact HSP101 PV function and parasite fitness in P. falciparum.
Fig. 6.
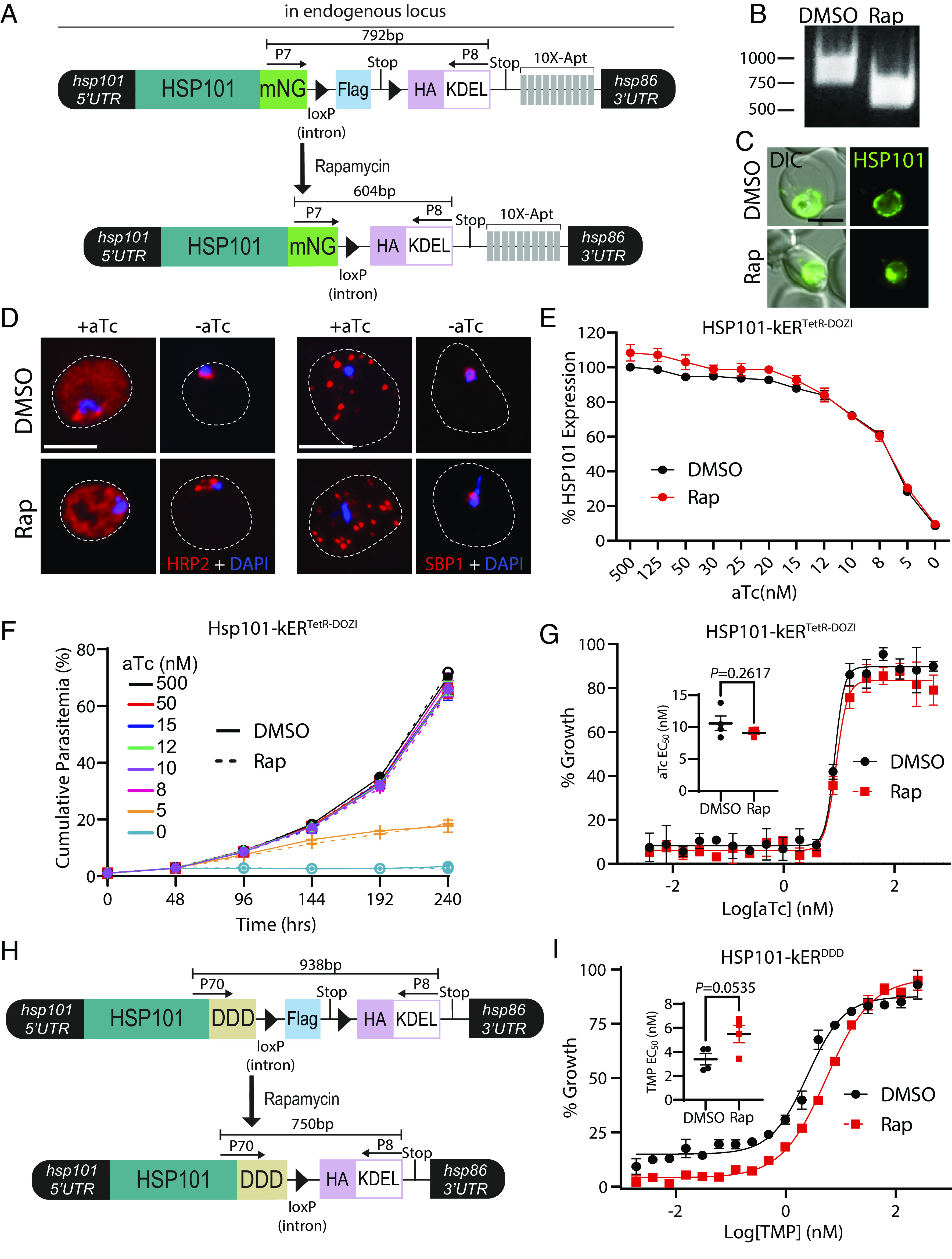
Depletion of HSP101 PV levels hypersensitizes parasites to posttranslational destabilization of the HSP101-PTEX complex but not HSP101 translational knockdown. (A) Schematic showing strategy for appending kER and the TetR-DOZI aptamers system to the endogenous C-terminus of HSP101. (B) PCR showing excision in HSP101-kERTetR-DOZI parasites 24 h after rapamycin treatment using primers P7/8. (C) Live microscopy of HSP101-kERTetR-DOZI parasites 168 h posttreatment with DMSO or rapamycin. (D) IFA detecting the soluble PEXEL protein HRP2 or the transmembrane PNEP SBP1 in synchronized, ring-stage HSP101-kERTetR-DOZI parasites grown for 48 h +/− aTc and treated with DMSO or rapamycin 1 cycle prior. Dashed lines indicate RBC boundaries traced from DIC images. (E) Quantification of HSP101 levels by flow cytometry measurement of mNeonGreen fluorescence in HSP101-kERTetR-DOZI parasites grown for 48 h at indicated aTc concentrations and treated with DMSO or rapamycin 2 cycles prior. MFI data were normalized to DMSO-treated parasites grown in 500 nM aTc (100%) and presented as mean ± SEM (n = 5 biological replicates). (F) Representative growth curves (n = 4 biological replicates) of HSP101-kERTetR-DOZI parasites grown at indicated aTc concentrations and treated with DMSO or rapamycin 2 cycles prior. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). (G) Representative EC50 assays (n = 4 biological replicates) with twofold dilutions of aTc starting at 500 nM. Parasites were treated with DMSO or rapamycin 72 h prior to adjusting aTc concentrations. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). EC50 graph Inset presented as mean ± SEM (n = 4 biological replicates): DMSO = 10.58 ± 1.16 nM and rapamycin = 9.11 ± 0.29 nM. (H) Schematic showing strategy for appending kER and the DDD system to the endogenous C-terminus of HSP101. (I) Representative EC50 assays (n = 4 biological replicates) with twofold dilutions of TMP starting at 250 nM. Parasites were treated with DMSO or rapamycin 72 h prior to adjusting TMP concentrations. Data are presented as means ± SD from one biological replicate (n = 3 technical replicates). EC50 graph Inset presented as mean ± SEM (n = 4 biological replicates): DMSO = 3.39 ± 0.48 nM and rapamycin 5.48 ± 0.73 nM. (Scale bars, 5 μm.)
If HSP101 functions exclusively in the PV, then knockER-mediated depletion of vacuolar HSP101 levels should hypersensitize parasites to HSP101 knockdown. However, dose–response to aTc was unaltered by rapamycin treatment (Fig. 6G) and the growth defect of KDEL-fused HSP101 parasites maintained at 5 nM aTc was indistinguishable from the DMSO control even over an extended 10-d growth assay (Fig. 6F), possibly suggesting that the PV-localized function of HSP101 may not fully account for its role in parasite fitness. To ensure depletion of HSP101 from the vacuole by knockER sensitizes parasites to compromised HSP101 PV function, we generated an HSP101-knockER line where mNG was replaced by a ligand-sensitive DHFR-based destabilization domain (DDD) (Fig. 6H) (23). In the absence of trimethoprim (TMP), the DDD blocks HSP101 interaction with EXP2/PTEX150 but not cargo binding, specifically targeting function of the assembled PTEX complex in the PV (23). In contrast to translational knockdown, HSP101-kERDDD parasites were hypersensitized to reduction of TMP levels following rapamycin treatment, indicating a synergistic effect when the reduction of HSP101 PV levels is combined with disruption of PTEX complex formation (Fig. 6I and SI Appendix, Fig. S7 C−E). Taken together, these results illustrate how knockER can be used to study protein function with subcompartmental resolution and may support a distinct HSP101 activity in the ER upstream of its PVM translocation function at the assembled PTEX complex.
Discussion
Several genetic tools are now available for protein loss-of-function studies in Plasmodium spp. (69), including knock-sideways for conditional sequestration of a target protein away from its normal site of function (70, 71). While this system is not currently amenable to parasite proteins that enter the secretory pathway, conditional control of secretory traffic has been achieved in P. falciparum by ligand-mediated alteration of the N-terminal transit peptide that controls apicoplast targeting, enabling redirection of plastid proteins into the default secretory pathway (72). Additionally, conditional ER release systems have been developed in model eukaryotes based on ER retrieval machinery (73). In contrast, knockER provides a simple, DiCre-mediated knock-sideways strategy that exploits the ERD2/KDEL receptor (KDELR) system for conditional ER retrieval, providing a generalizable approach for interrogating secreted proteins in genetically tractable eukaryotes.
In this study, we demonstrate that knockER simultaneously allows for trafficking and loss-of-function studies. The ability to determine whether a protein traffics from the ER through the cis Golgi is an important consideration for nuclear-encoded apicoplast proteins since Golgi-dependent and -independent trafficking routes have been reported (31, 33, 74). While these apicoplast trafficking studies were carried out with reporters and chimeras, knockER allows for monitoring cis Golgi transit of endogenous proteins. The sensitivity of ClpB1 to KDEL-mediated ER retrieval demonstrates a Golgi-dependent trafficking route to the apicoplast. Moreover, we show that this AAA+ chaperone is essential for apicoplast maintenance. ClpB1 is one of four Clp/HSP100 chaperones encoded by Plasmodium spp., three of which localize to the parasite plastid while the fourth HSP101 (previously known as ClpB2) localizes to the PV and ER (43). The other apicoplast-targeted members of this quartet include the nuclear-encoded ClpC, which mediates regulated proteolysis together with the serine protease ClpP (75, 76) and ClpM, now the only uncharacterized member of this group and one of the few proteins encoded on the apicoplast genome. Similar to the well-studied function of its closest orthologs (77), ClpB1 likely acts as a disaggregase that, together with ClpC/P and ClpM, forms a critical proteostasis system required to maintain and regulate the apicoplast proteome (78).
ER perturbations can trigger an integrated stress response leading to cell arrest or death (79, 80). Plasmodium possesses one of the widely conserved ER stress sensors, the PERK-eIF2α pathway which halts protein translation and flux through the secretory pathway (20, 81–84). Importantly, conditional ER retrieval of proteins produced from their endogenous locus or by heterologous expression was not generally toxic, enabling loss-of-function studies with knockER. The single notable exception was EXP2, which rapidly killed parasites when retained in the ER independent to loss of its PVM transport function, indicating that knockER experiments should be controlled for indirect effects. EXP2-KDEL toxicity was dependent on the amphipathic helix that forms the membrane-spanning region in the oligomeric EXP2 pore (25), suggesting that accumulation of EXP2 in the ER might trigger an unfolded protein response or lead to premature pore formation in the early secretory pathway. More work is required to determine whether this outcome is specific to EXP2, but the observation that parasites tolerate SDEL-mediated retention of several TM domain-containing exported proteins indicates that this is not a general problem for unnatural ER-retrieval of membrane proteins (85). Indirect fitness defects aside, retrieval of EXP2-KDEL demonstrates that its C-terminus enters the secretory pathway lumen, consistent with a soluble EXP2 trafficking state that precedes membrane integration at the PVM. Regardless of the basis for EXP2-kER toxicity, these experiments highlight that knockER can also be used to investigate membrane topology. While ER-resident type I membrane proteins utilize a different mechanism for retrieval as their C-termini face the cytosol of the cell (86, 87), ERD2/KDELR can retrieve membrane proteins whose C-termini face the lumen (88) and thus knockER should be able to distinguish the topology of single and multipass transmembrane domain-containing proteins.
A unique feature of knockER is the ability to redistribute secreted proteins from their terminal destination to the ER. We tagged both HSP101 and the translocon adaptor PTEX150 with knockER, reasoning that ER retrieval of either protein would produce a lethal export defect but that differential impacts on exported cargo trafficking might be exposed. Unexpectedly, ER retrieval of HSP101 had no impact on P. falciparum growth or export of representative PEXEL and PNEP proteins in vitro. Similar PV depletion of PTEX150 produced the anticipated lethal export block, revealing that HSP101 PV levels are uniquely maintained in excess. As a AAA+ ATPase, HSP101 is the only enzyme in the PTEX core complex and its activity may exceed what is required to support parasite fitness, at least during in vitro culture, as previously seen for the aspartic protease Plasmepsin V that licenses proteins for export in the ER (89, 90). Interestingly, parallel experiments in P. berghei resulted in greater retention and produced a lethal export defect for both PTEX150 and HSP101, showing that knockER is also suitable for conditional mutagenesis in this important model species and suggesting that retrieval may be more stringent in P. berghei, at least for some proteins. Notably, the strength of ERD2/KDELR binding between different C-terminal retrieval motifs varies by as much as 10-fold in higher eukaryotes (16). While KDEL-mediated retrieval was generally robust across the diverse set of reporters and endogenous proteins surveyed in P. falciparum and P. berghei, a range of functional variants have been validated or can be inferred in Plasmodium spp. (19) and other retrieval motifs may enable tuning the strength of retention by knockER.
Intriguingly, redistribution of HSP101 from the PV to the ER hypersensitized parasites to DDD-mediated HSP101 inactivation but not translational knockdown. While TetR-DOZI knockdown depletes the entire HSP101 pool, HSP101-DDD acts posttranslationally by disrupting complex formation with EXP2/PTEX150 but not cargo recognition (23), specifically targeting HSP101 function in PVM translocation. While these results do not enable strong conclusions about potential extra vacuolar activity of HSP101, they suggest that the PV-localized function may not fully account for HSP101’s contribution to parasite fitness, in line with a hypothesized role in exported cargo recognition and/or trafficking at the ER (61). Notably, IBIS1 was not retained in the ER with PbHSP101-KDEL but accumulated in the PV and was not exported into the RBC, indicating that HSP101 is not required to directly chaperone cargo from the ER to the PV. Future work dissecting the specific extra-vacuolar role(s) of HSP101 may resolve the long-standing question of how early events in this pathway that mark proteins for export, such as PEXEL processing during ER entry, are connected to cargo recognition and ultimate PVM translocation into the host cell at the assembled PTEX complex.
Materials and Methods
Parasite Maintenance.
P. falciparum NF54attB-DiCre and derivatives were cultured with deidentified, Institutional Review Board-exempt RBCs obtained from the American National Red Cross in RPMI 1,640 supplemented as described (39). The P. berghei marker-free HP DiCre line (91) and derivatives were maintained in Swiss Webster mice (Charles River). All experiments involving rodents were reviewed and approved by the Iowa State University Institutional Animal Care and Use Committee.
Genetic Modification of P. falciparum and P. berghei, knockER Induction and Phenotypic Analysis.
DiCre-mediated excision was induced with 10 nM rapamycin for 3 h and monitored by diagnostic PCR. Detailed methods for plasmid construction, generation of individual parasite strains, growth assays, microscopy, and Western blotting are provided in SI Appendix, Supplementary Methods section. Primer sequences are given in SI Appendix, Table S1.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by NIH grant HL133453 to (J.R.B.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. We thank J. McBride, D. Cavanaugh and EMRR for the EXP2 antibody and A. Waters for the HP DiCre P. berghei parasites.
Author contributions
M.A.F. and J.R.B. designed research; M.A.F., T.H., L.J.C., and J.R.B. performed research; M.A.F. and J.R.B. analyzed data; and M.A.F. and J.R.B. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.van Dooren G. G., Striepen B., The algal past and parasite present of the apicoplast. Annu. Rev. Microbiol. 67, 271–289 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Akuh O. A., Elahi R., Prigge S. T., Seeber F., The ferredoxin redox system - an essential electron distributing hub in the apicoplast of Apicomplexa. Trends Parasitol. 38, 868–881 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cova M. M., Lamarque M. H., Lebrun M., How apicomplexa parasites secrete and build their invasion machinery. Annu. Rev. Microbiol. 76, 619–640 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Matz J. M., Beck J. R., Blackman M. J., The parasitophorous vacuole of the blood-stage malaria parasite. Nat. Rev. Microbiol. 18, 379–391 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Garten M., Beck J. R., Structured to conquer: Transport across the Plasmodium parasitophorous vacuole. Curr. Opin. Microbiol. 63, 181–188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Koning-Ward T. F., Dixon M. W., Tilley L., Gilson P. R., Plasmodium species: Master renovators of their host cells. Nat. Rev. Microbiol. 14, 494–507 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Beck J. R., Ho C. M., Transport mechanisms at the malaria parasite-host cell interface. PLoS Pathog. 17, e1009394 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sargeant T. J., et al. , Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 7, R12 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boddey J. A., et al. , Role of plasmepsin V in export of diverse protein families from the Plasmodium falciparum exportome. Traffic 14, 532–550 (2013). [DOI] [PubMed] [Google Scholar]
- 10.van Ooij C., et al. , The malaria secretome: From algorithms to essential function in blood stage infection. PLoS Pathog. 4, e1000084 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulze J., et al. , The plasmodium falciparum exportome contains non-canonical PEXEL/HT proteins. Mol. Microbiol. 97, 301–314 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Jonsdottir T. K., Gabriela M., Crabb B. S., de Koning-Ward T. F., Gilson P. R., Defining the essential exportome of the malaria parasite. Trends Parasitol. 37, 664–675 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Florentin A., Cobb D. W., Kudyba H. M., Muralidharan V., Directing traffic: Chaperone-mediated protein transport in malaria parasites. Cell Microbiol. 22, e13215 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munro S., Pelham H. R., A C-terminal signal prevents secretion of luminal ER proteins. Cell 48, 899–907 (1987). [DOI] [PubMed] [Google Scholar]
- 15.Brauer P., et al. , Structural basis for pH-dependent retrieval of ER proteins from the Golgi by the KDEL receptor. Science 363, 1103–1107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerondopoulos A., et al. , A signal capture and proofreading mechanism for the KDEL-receptor explains selectivity and dynamic range in ER retrieval. Elife 10, e68380 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elmendorf H. G., Haldar K., Identification and localization of ERD2 in the malaria parasite Plasmodium falciparum: Separation from sites of sphingomyelin synthesis and implications for organization of the Golgi. EMBO J. 12, 4763–4773 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Greca N., Hibbs A. R., Riffkin C., Foley M., Tilley L., Identification of an endoplasmic reticulum-resident calcium-binding protein with multiple EF-hand motifs in asexual stages of Plasmodium falciparum. Mol. Biochem. Parasitol. 89, 283–293 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Kulzer S., Gehde N., Przyborski J. M., Return to sender: Use of Plasmodium ER retrieval sequences to study protein transport in the infected erythrocyte and predict putative ER protein families. Parasitol. Res. 104, 1535–1541 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Kudyba H. M., et al. , The endoplasmic reticulum chaperone PfGRP170 is essential for asexual development and is linked to stress response in malaria parasites. Cell Microbiol. 21, e13042 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cobb D. W., et al. , A redox-active crosslinker reveals an essential and inhibitable oxidative folding network in the endoplasmic reticulum of malaria parasites. PLoS Pathog. 17, e1009293 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fierro M. A., et al. , An endoplasmic reticulum CREC family protein regulates the egress proteolytic cascade in malaria parasites. mBio 11, e03078-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck J. R., Muralidharan V., Oksman A., Goldberg D. E., PTEX component HSP101 mediates export of diverse malaria effectors into host erythrocytes. Nature 511, 592–595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elsworth B., et al. , PTEX is an essential nexus for protein export in malaria parasites. Nature 511, 587–591 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Ho C. M., et al. , Malaria parasite translocon structure and mechanism of effector export. Nature 561, 70–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins C. R., et al. , Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol. Microbiol. 88, 687–701 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knuepfer E., Napiorkowska M., van Ooij C., Holder A. A., Generating conditional gene knockouts in Plasmodium - a toolkit to produce stable DiCre recombinase-expressing parasite lines using CRISPR/Cas9. Sci. Rep. 7, 3881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones M. L., et al. , A versatile strategy for rapid conditional genome engineering using loxP sites in a small synthetic intron in Plasmodium falciparum. Sci. Rep. 6, 21800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Przyborski J. M., et al. , Trafficking of STEVOR to the maurer’s clefts in Plasmodium falciparum-infected erythrocytes. EMBO J. 24, 2306–2317 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Dooren G. G., et al. , Development of the endoplasmic reticulum, mitochondrion and apicoplast during the asexual life cycle of Plasmodium falciparum. Mol. Microbiol. 57, 405–419 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Tonkin C. J., Struck N. S., Mullin K. A., Stimmler L. M., McFadden G. I., Evidence for Golgi-independent transport from the early secretory pathway to the plastid in malaria parasites. Mol. Microbiol. 61, 614–630 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Lee M. C., Moura P. A., Miller E. A., Fidock D. A., Plasmodium falciparum Sec24 marks transitional ER that exports a model cargo via a diacidic motif. Mol. Microbiol. 68, 1535–1546 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heiny S. R., Pautz S., Recker M., Przyborski J. M., Protein traffic to the Plasmodium falciparum apicoplast: Evidence for a sorting branch point at the golgi. Traffic 15, 1290–1304 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Matz J. M., et al. , The Plasmodium berghei translocon of exported proteins reveals spatiotemporal dynamics of tubular extensions. Sci. Rep. 5, 12532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boucher M. J., et al. , Integrative proteomics and bioinformatic prediction enable a high-confidence apicoplast proteome in malaria parasites. PLoS Biol. 16, e2005895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birnbaum J., et al. , A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 367, 51–59 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Waller R. F., Reed M. B., Cowman A. F., McFadden G. I., Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. EMBO J. 19, 1794–1802 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adisa A., et al. , The signal sequence of exported protein-1 directs the green fluorescent protein to the parasitophorous vacuole of transfected malaria parasites. J. Biol. Chem. 278, 6532–6542 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Nessel T., et al. , EXP1 is required for organisation of EXP2 in the intraerythrocytic malaria parasite vacuole. Cell Microbiol. 22, e13168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganesan S. M., Falla A., Goldfless S. J., Nasamu A. S., Niles J. C., Synthetic RNA-protein modules integrated with native translation mechanisms to control gene expression in malaria parasites. Nat. Commun. 7, 10727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajaram K., Liu H. B., Prigge S. T., Redesigned TetR-aptamer system to control gene expression in plasmodium falciparum. mSphere 5, e00457-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adjalley S. H., et al. , Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc. Natl. Acad. Sci. U.S.A. 108, E1214–E1223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El Bakkouri M., et al. , The Clp chaperones and proteases of the human malaria parasite Plasmodium falciparum. J. Mol. Biol. 404, 456–477 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Yeh E., DeRisi J. L., Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 9, e1001138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Dooren G. G., Su V., D’Ombrain M. C., McFadden G. I., Processing of an apicoplast leader sequence in Plasmodium falciparum and the identification of a putative leader cleavage enzyme. J. Biol. Chem. 277, 23612–23619 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Zhang M., et al. , Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360, eaap7847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bushell E., et al. , Functional profiling of a plasmodium genome reveals an abundance of essential genes. Cell 170, 260–272.e268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fichera M. E., Roos D. S., A plastid organelle as a drug target in apicomplexan parasites. Nature 390, 407–409 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Dahl E. L., Rosenthal P. J., Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 51, 3485–3490 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Proellocks N. I., et al. , Characterisation of PfRON6, a Plasmodium falciparum rhoptry neck protein with a novel cysteine-rich domain. Int. J. Parasitol. 39, 683–692 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Desai S. A., Krogstad D. J., McCleskey E. W., A nutrient-permeable channel on the intraerythrocytic malaria parasite. Nature 362, 643–646 (1993). [DOI] [PubMed] [Google Scholar]
- 52.Desai S. A., Rosenberg R. L., Pore size of the malaria parasite’s nutrient channel. Proc. Natl. Acad. Sci. U.S.A. 94, 2045–2049 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garten M., et al. , EXP2 is a nutrient-permeable channel in the vacuolar membrane of Plasmodium and is essential for protein export via PTEX. Nat. Microbiol. 3, 1090–1098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gold D. A., et al. , The toxoplasma dense granule proteins GRA17 and GRA23 mediate the movement of small molecules between the host and the parasitophorous vacuole. Cell Host Microbe 17, 642–652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanders P. R., et al. , The N-terminus of EXP2 forms the membrane-associated pore of the protein exporting translocon PTEX in Plasmodium falciparum. J. Biochem. 165, 239–248 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Hakamada K., Watanabe H., Kawano R., Noguchi K., Yohda M., Expression and characterization of the Plasmodium translocon of the exported proteins component EXP2. Biochem. Biophys. Res. Commun. 482, 700–705 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Dal Peraro M., van der Goot F. G., Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 14, 77–92 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Charnaud S. C., Kumarasingha R., Bullen H. E., Crabb B. S., Gilson P. R., Knockdown of the translocon protein EXP2 in Plasmodium falciparum reduces growth and protein export. PLoS One 13, e0204785 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mesen-Ramirez P., et al. , EXP1 is critical for nutrient uptake across the parasitophorous vacuole membrane of malaria parasites. PLoS Biol. 17, e3000473 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Koning-Ward T. F., et al. , A newly discovered protein export machine in malaria parasites. Nature 459, 945–949 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gabriela M., et al. , A revised mechanism for how Plasmodium falciparum recruits and exports proteins into its erythrocytic host cell. PLoS Pathog. 18, e1009977 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Charnaud S. C., et al. , Spatial organization of protein export in malaria parasite blood stages. Traffic 19, 605–623 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Ingmundson A., Nahar C., Brinkmann V., Lehmann M. J., Matuschewski K., The exported Plasmodium berghei protein IBIS1 delineates membranous structures in infected red blood cells. Mol. Microbiol. 83, 1229–1243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matthews K., et al. , The plasmodium translocon of exported proteins (PTEX) component thioredoxin-2 is important for maintaining normal blood-stage growth. Mol. Microbiol. 89, 1167–1186 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Nair S., et al. , Nutrient limitation magnifies fitness costs of antimalarial drug resistance mutations. Antimicrob. Agents Chemother. 66, e0152921 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gupta A., et al. , Complex nutrient channel phenotypes despite mendelian inheritance in a Plasmodium falciparum genetic cross. PLoS Pathog. 16, e1008363 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matz J. M., et al. , In vivo function of PTEX88 in malaria parasite sequestration and virulence. Eukaryot. Cell 14, 528–534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chisholm S. A., et al. , Contrasting inducible knockdown of the auxiliary PTEX component PTEX88 in P. falciparum and P. berghei unmasks a role in parasite virulence. PLoS One 11, e0149296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kudyba H. M., Cobb D. W., Vega-Rodriguez J., Muralidharan V., Some conditions apply: Systems for studying Plasmodium falciparum protein function. PLoS Pathog. 17, e1009442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Birnbaum J., et al. , A genetic system to study Plasmodium falciparum protein function. Nat. Methods 14, 450–456 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Hughes K. R., Waters A. P., Rapid inducible protein displacement in Plasmodiumin vivo and in vitro using knocksideways technology. Wellcome Open Res. 2, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roberts A. D., Nair S. C., Guerra A. J., Prigge S. T., Development of a conditional localization approach to control apicoplast protein trafficking in malaria parasites. Traffic 20, 571–582 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Praznik A., et al. , Regulation of protein secretion through chemical regulation of endoplasmic reticulum retention signal cleavage. Nat. Commun. 13, 1323 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prasad A., Mastud P., Patankar S., Dually localised proteins found in both the apicoplast and mitochondrion utilize the Golgi-dependent pathway for apicoplast targeting in Toxoplasma gondii. Biol. Cell 113, 58–78 (2021). [DOI] [PubMed] [Google Scholar]
- 75.Florentin A., et al. , PfClpC is an essential Clp chaperone required for plastid integrity and Clp protease stability in plasmodium falciparum. Cell Rep. 21, 1746–1756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Florentin A., Stephens D. R., Brooks C. F., Baptista R. P., Muralidharan V., Plastid biogenesis in malaria parasites requires the interactions and catalytic activity of the Clp proteolytic system. Proc. Natl. Acad. Sci. U.S.A. 117, 13719–13729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mogk A., Bukau B., Kampinga H. H., Cellular handling of protein aggregates by disaggregation machines. Mol. Cell 69, 214–226 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Bouchnak I., van Wijk K. J., Structure, function, and substrates of Clp AAA+ protease systems in cyanobacteria, plastids, and apicoplasts: A comparative analysis J. Biol. Chem. 296, 100338 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pakos-Zebrucka K., et al. , The integrated stress response. EMBO Rep. 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Costa-Mattioli M., Walter P., The integrated stress response: From mechanism to disease. Science 368, eaat5314 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chaubey S., Grover M., Tatu U., Endoplasmic reticulum stress triggers gametocytogenesis in the malaria parasite. J. Biol. Chem. 289, 16662–16674 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harbut M. B., et al. , Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design. Proc. Natl. Acad. Sci. U.S.A. 109, 21486–21491 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang M., et al. , PK4, a eukaryotic initiation factor 2alpha(eIF2alpha) kinase, is essential for the development of the erythrocytic cycle of Plasmodium. Proc. Natl. Acad. Sci. U.S.A. 109, 3956–3961 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang M., et al. , Inhibiting the plasmodium eIF2alpha kinase PK4 prevents artemisinin-induced latency. Cell Host Microbe 22, 766–776.e764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levray Y. S., et al. , Formation of ER-lumenal intermediates during export of Plasmodium proteins containing transmembrane-like hydrophobic sequences. PLoS Pathog. 19, e1011281 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gaynor E. C., te Heesen S., Graham T. R., Aebi M., Emr S. D., Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J. Cell Biol. 127, 653–665 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stornaiuolo M., et al. , KDEL and KKXX retrieval signals appended to the same reporter protein determine different trafficking between endoplasmic reticulum, intermediate compartment, and Golgi complex. Mol. Biol. Cell 14, 889–902 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tang B. L., Wong S. H., Low S. H., Hong W. J., Retention of a type-II surface-membrane protein in the endoplasmic-reticulum by the Lys-Asp-Glu-Leu sequence. J. Biol. Chem. 267, 7072–7076 (1992). [PubMed] [Google Scholar]
- 89.Sleebs B. E., et al. , Inhibition of plasmepsin V activity demonstrates its essential role in protein export, PfEMP1 display, and survival of malaria parasites. PLoS Biol. 12, e1001897 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Polino A. J., Nasamu A. S., Niles J. C., Goldberg D. E., Assessment of biological role and insight into druggability of the plasmodium falciparum protease plasmepsin V. ACS Infect. Dis. 6, 738–746 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kent R. S., et al. , Inducible developmental reprogramming redefines commitment to sexual development in the malaria parasite plasmodium berghei. Nat. Microbiol. 3, 1206–1213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.