

Summary
Highly effective vaccines elicit specific, robust, and durable adaptive immune responses. To advance informed vaccine design, it is critical that we understand the cellular dynamics underlying responses to different antigen formats. Here, we sought to understand how antigen-specific B and T cells were activated and participated in adaptive immune responses within the mucosal site. Using a human tonsil organoid model, we tracked the differentiation and kinetics of the adaptive immune response to influenza vaccine and virus modalities. Each antigen format elicited distinct B and T cell responses, including differences in their magnitude, diversity, phenotype, function, and breadth. These differences culminated in substantial changes in the corresponding antibody response. A major source of antigen format-related variability was the ability to recruit naive vs. memory B and T cells to the response. These findings have important implications for vaccine design and the generation of protective immune responses in the upper respiratory tract.
Keywords: adaptive immunity, organoids, influenza, vaccines, B cells, T cells, human immunology, antibodies, vaccine modalities, B/T cell repertoire
Graphical abstract
Highlights
-
•
Used tonsil organoids to compare influenza vaccine formats within an individual
-
•
Transcriptional fate of Ag-specific B cells is affected by vaccine format
-
•
Type I IFNs mediate some but not all of the vaccine-format-specific differences
-
•
Inactivated flu vaccine depends on pre-existing memory cells
Understanding the cellular dynamics underlying responses to different antigen formats is critical for rational vaccine design. Using a human immune organoid model, Kastenschmidt et al. evaluate the effect of influenza vaccine modalities on adaptive immunity and reveal key differences in the magnitude, breadth, and quality of B cell, T cell, and antibody responses.
Introduction
Vaccination is the most effective method of protection against influenza infection and influenza-related morbidity and mortality. However, vaccine effectiveness depends upon several factors, including the extent of antigenic match to circulating strains and host factors such as prior exposure and vaccination history. Influenza viruses also present a substantial pandemic risk due to their gene reassortment capabilities, which can lead to the emergence of novel strains against which most of the population has no pre-existing protective immunity. There is an urgent need to develop a universal flu vaccine that is robust, persistent, broadly cross-reactive, and effective in the majority of vaccinees.1 New strategies are under evaluation to develop broadly protective influenza vaccines, several of which show great promise.2,3,4,5 With the emergence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic and corresponding advances in vaccine development, there is also a renewed interest in utilizing intranasal vaccines to stimulate protective immunity against viruses in the upper respiratory tract.6,7
However, correlates of protection from mucosa-targeting vaccines have been difficult to quantify. The gold standard for 50% protection after intramuscular inactivated influenza vaccine (IIV) is a 1:40 hemagglutination inhibition (HAI) titer8 and seroconversion is often defined as a 4-fold increase in HAI titer.9 Identifying similar correlates of protection after immunization with an intranasal, live-attenuated influenza vaccine (LAIV) has had mixed results.10,11,12 LAIV has been shown to be more effective,11,13,14,15 as effective,16 or less effective14,17 than IIV in clinical studies in both young children11,13,15 and adults.14,16,17 A common explanation for these disparate outcomes is that high levels of antibodies at the mucosal surface may reduce LAIV efficacy; however, other reports suggest that pre-existing mucosal antibodies do not substantially contribute to a reduced response.18 Challenges persist in investigating the underlying mechanisms of these clinical observations due to limitations in sampling at the lymphoid tissues and in the respiratory tract.
To rationally design an influenza vaccine that stimulates protective mucosal responses, a better understanding of how antigen (Ag) modality affects adaptive immunity in the upper respiratory tract is needed. Since each individual’s influenza infection and vaccination history is unique, it has been difficult to address these questions in animal models or human clinical trials. Based on available clinical reports, we predicted that different Ag formats would have distinct effects on B and T cell activation, differentiation, diversity, and protection.
Here, we used a human tonsil organoid platform to elucidate the cellular dynamics of Ag-reactive B and T cells in primary human tonsils as they responded to IIV, LAIV, and influenza viruses. Tonsil organoids are a useful system for tracking human adaptive immunity and can recapitulate numerous aspects of the developing B and T cell response.19 Although in vitro systems have limitations, a major advantage of organoid platforms is the ability to test multiple conditions (including different compositions, doses, and time points) within the same donor. We used the tonsil organoid platform to investigate how host and Ag features altered the magnitude and quality of the B and T cell response on an individual basis. Importantly, we looked specifically at the mucosal and lymphoid tissue sites where influenza infection can occur and thus quantified relevant and local Ag-specific cells.
Results
Adult-derived tonsil organoids respond to influenza vaccine and virus antigens
In this study, tonsils were collected from otherwise healthy adults undergoing tonsillectomy (Table 1) and organoids were prepared using a 96-well high-throughput platform adapted from our previous method.19 On day 0, tonsil cells were stimulated with optimally titrated (Figure S1) doses of influenza Ags (IIV or LAIV from the 2019/20 influenza season, or A/California/07/2009 influenza virus [H1N1]) and cultured into organoids for 4–14 days (Figure 1A). A systems immunology approach was used to quantify various aspects of the adaptive immune response to define associated cellular signatures. B cell differentiation and antibody (Ab) secretion were measured on day 14 poststimulation; we found that IIV, LAIV, and H1N1 all successfully stimulated B cell responses in tonsil organoids (see Figures 1B, 1C, and S2A for sample gating). Germinal center (GC) and plasmablast B cells were induced by all influenza Ag modalities. However, the extent of activation and differentiation varied based on Ag format. LAIV induced more GC and plasmablast B cells than IIV or H1N1. H1N1-treated organoids stimulated pre-GC and GC B cell differentiation, but fewer plasmablasts formed than with LAIV (Figure 1C). In contrast, IIV elicited significantly fewer pre-GC, GC, and plasmablast B cells compared with live and live-attenuated viruses. Together these data show that immune organoids can respond to influenza antigens in various formats.
Table 1.
Donor demographics and experimental inclusion
| Donor number | Indication for surgery | Age | Sex | Self-reported race/ethnicity | Flow phenotype | Protein microarray | CCR6+PD1+ B cell phenotyping | scRNA- seq | B cell depletions | T cell depletions | Type I IFN supplementation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 002 | both | 20 | F | White, not Hispanic | X | X | X | – | – | X | – |
| 004 | both | 31 | M | White, Hispanic | X | X | – | X | – | – | – |
| 007 | hypertrophy | 18 | F | Native American | X | X | X | – | – | – | X |
| 010 | tonsillitis | 17 | F | White, not Hispanic | X | X | X | – | X | X | – |
| 011 | hypertrophy | 37 | M | White, Hispanic | X | X | X | – | X | – | – |
| 012 | both | 21 | M | White, not Hispanic | X | X | X | – | – | – | X |
| 013 | both | 23 | F | White, Hispanic | X | X | X | – | X | – | – |
| 014 | both | 28 | F | White, Hispanic | X | X | X | – | – | – | X |
| 015 | tonsillitis | 20 | F | White, not Hispanic | X | X | – | – | – | – | – |
| 016 | hypertrophy | 39 | M | White, not Hispanic | X | X | – | X | – | – | – |
| 017 | both | 32 | F | White, not Hispanic | X | X | – | X | X | – | – |
| 018 | both | 24 | F | White, not Hispanic | X | X | – | X | X | – | – |
| 019 | hypertrophy | 26 | F | White, not Hispanic | – | – | X | – | – | X | – |
| 021 | tonsillitis | 21 | F | White, not Hispanic | – | – | X | – | – | – | – |
| 022 | both | 22 | F | White, not Hispanic | – | – | X | – | – | – | – |
| 024 | hypertrophy | 13 | F | White, Hispanic | – | – | X | – | – | – | – |
| 025 | tonsillitis | 23 | F | White, not Hispanic | – | – | X | – | – | – | X |
| 026 | tonsillitis | 19 | M | White, not Hispanic | – | – | X | – | – | – | – |
| 027 | tonsillitis | 25 | F | White, not Hispanic | – | – | X | – | – | – | X |
| 028 | both | 34 | F | Asian | – | – | X | – | – | – | – |
| 029 | both | 26 | M | White, Hispanic | – | – | X | – | – | – | – |
| 030 | tonsillitis | 19 | F | Indian | – | – | X | – | – | – | – |
| 032 | tonsillitis | 17 | F | White, not Hispanic | – | – | X | – | – | – | X |
| 033 | both | 42 | F | Asian | – | – | X | – | – | – | – |
| 034 | tonsillitis | 19 | F | Black | – | – | X | – | – | X | X |
| 035 | hypertrophy | 35 | M | White, Hispanic | – | – | X | – | – | – | X |
| 037 | hypertrophy | 52 | M | White, not Hispanic | – | – | X | – | – | X | X |
| 038 | both | 20 | F | White, Hispanic | – | – | X | – | – | – | X |
| 041 | tonsillitis | 25 | F | White, not Hispanic/Mid East | – | – | X | – | – | – | X |
| 045 | hypertrophy | 33 | F | White, not Hispanic | – | – | – | – | – | X | – |
Figure 1.

Multiple influenza antigen modalities induce GC reactions and specific antibody secretion in human tonsil organoids
(A) Experimental design and workflow for tonsil organoids. Cells and supernatants were assessed using multiple readouts to define immune signatures. Created with Biorender.com.
(B and C) Representative flow cytometry staining (B) and summary data (C) for total B cell (CD45+CD19+CD3−) phenotypes from day 14 organoids stimulated with different influenza Ag modalities.
(D) Representative flow cytometry staining of HA+ (red) and nonspecific (gray) B cells from day 14 organoids. Numbers indicate frequency of HA+ B cells of a given phenotype out of total HA+ B cells.
(E) HA+ B cell frequencies.
(F) HA+ plasmablast frequencies.
(G) Quantification of HA-specific antibodies in day 14 organoid culture supernatants.
(H) Virus neutralization by day 14 organoid culture supernatants; neutralization quantification by area under the curve (AUC).
(I) Frequency of activated CXCR5+CD4 T cells and activated CD8 T cells on day 7. Activation was defined by dual expression of CD38 and HLA-DR. n = 12 donors (one experiment) for all data shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 using a Kruskal-Wallis test followed by paired Mann-Whitney U tests to compare groups. p values shown are for comparisons against the unstimulated control unless otherwise indicated by lines. Boxplots show the median, with hinges indicating the first and third quartiles and whiskers indicating the highest and lowest value within 1.5 times the interquartile range of the hinges. See also Tables 1 and S5 and Figures S1 and S2.
Influenza vaccines induce different quantities of Ag-specific B cells, plasmablasts, activated T cells, and neutralizing Abs
We next quantified the phenotypes of demonstrably Ag-specific B cells induced by each influenza stimulation. A/California 2009 hemagglutinin (HA)-specific B cells (HA+ B cells) were labeled as done previously,19,20,21 and their frequencies were quantified by flow cytometry (Figures 1D and S2A). Overall, LAIV stimulation produced more HA+ B cells (Figures 1D and 1E) and HA+ plasmablasts than either IIV or H1N1 (Figure 1F). Despite differences in total and HA+ B cell phenotypes between IIV and LAIV-stimulated organoids, both vaccines elicited detectable quantities of influenza-specific Abs against the four strain-matched HA proteins in the vaccine (Figure 1G). However, in line with HA+ B cell and plasmablast data, LAIV-stimulated cultures produced more neutralizing antibodies against A/California 2009 H1N1 virus (Figure 1H). On day 7, which we previously showed as the time point of peak T cell response,19 activated T follicular helper (TFH, CXCR5+ CD4) and activated CD8 T cells were significantly increased in response to LAIV and H1N1 stimulation, but not stimulation with IIV (Figures 1I and S2B). Altogether, a multifaceted adaptive immune response was elicited in tonsil organoids stimulated with influenza vaccines and viruses and the features of these responses were heavily dependent on Ag format.
LAIV induces greater influenza protein and strain breadth than IIV
Given the differences in Ab responses between IIV- and LAIV-stimulated organoids, we more deeply investigated their specificities and breadth using a high-throughput protein microarray22 composed of 169 influenza proteins (see STAR Methods), including HA and non-HA proteins spanning human (seasonal and pandemic) and zoonotic strains from 1918 to the present. Unbiased hierarchical clustering of the protein microarray data revealed shared patterns of Ab responses, defined by four main groups (Figure 2A). Samples strongly clustered by Ag modality rather than by donor. Group A, composed mostly of IIV-stimulated organoids, elicited a focused response against recently circulating (post-2009 H1N1) HA proteins and to nucleoprotein (NP) (Figure 2A). Group B consisted of IIV and H1N1-stimulated organoids with Abs narrowly specific for NP only. Group C, which contained mostly H1N1-stimulated organoids and a few vaccine-stimulated samples, showed broad cross-reactivity against numerous HA proteins (including pre- and post-2009 H1, H3, H5, and other nonseasonal HAs) and NP. Group D featured Ab specificities for influenza B HAs (in addition to several other HAs and NP) and were almost exclusively sourced from LAIV-stimulated organoids (Figure 2A).
Figure 2.

Influenza vaccine modality influences antibody magnitude, specificity, and breadth
(A) Heatmap of antibody-binding magnitude against influenza proteins on a high-throughput protein microarray. Culture supernatants are from day 14 organoids. Column dendrogram represents unbiased sample grouping based on similarity; top bar color represents the antigen stimulation. Rows represent individual proteins on the microarray and were manually arranged based on influenza strain origin and protein type.
(B) Summary antibody data from the protein microarray by protein type and virus source. Data represent median values. Each point is an individual donor. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 using a Kruskal-Wallis test followed by paired Mann-Whitney U tests to compare groups. p values shown are for comparisons against the unstimulated control unless otherwise indicated by lines. Boxplots show the median, with hinges indicating the first and third quartiles and whiskers indicating the highest and lowest value within 1.5 times the interquartile range of the hinges.
(C) Detection of cross-reactive Ab production from IIV- vs. LAIV-stimulated tonsil organoids. Each donor is a row. The numbers of protein antigens from H1N1 strains that circulated prior to each donor’s birth are shown to the right of the plot. Abs produced from the organoids were classified as either present or absent in the culture supernatants.
(D) Protein targets from Ab responses unique to LAIV-stimulated organoids. NA, neuraminidase; NS, nonstructural; NP, nucleoprotein. The number of strains uniquely targeted by Abs from LAIV stimulation are shown in doughnut centers.
(E) Organoid Ab responses to nonseasonal influenza strain proteins on the microarray. Ab presence or absence was classified and plotted as in (C). n = 12 donors for all analyses (1 experiment). See also Tables 1, S1, and S6.
To estimate Ab binding characteristics, we examined Ab quantities against all H1N1 HAs, H3N2 HAs, influenza B HAs, and neuraminidases (NAs). Compared with IIV and H1N1, LAIV induced more antibodies against H1N1 proteins (Figure 2B). HA responses following IIV and H1N1 stimulation showed a bimodal distribution, with some responders (mapping to group C) and some nonresponders (group B). The H3N2 HA response was also elevated in LAIV-stimulated organoids compared with most IIV-stimulated organoids. LAIV was the only stimulation to strongly induce influenza B-specific Abs (Figure 2B) and to consistently generate substantial quantities of NA-specific Abs.
Due to antigenic drift and shift, an important consideration in influenza vaccine design is the ability to produce broadly reactive Abs. Therefore, we investigated the extent to which influenza Ag modalities elicited cross-reactive Abs. We identified all H1N1-derived proteins on the microarray and, on a per-donor basis, selected only strains that circulated prior to each donor’s birth year. Thus, any Abs detected against these proteins were the result of cross-reactivity rather than direct exposure. Both IIV and LAIV induced cross-reactive H1N1 Abs. However, LAIV elicited cross-reactive Abs not detected with IIV (Figure 2C). These Abs were mostly directed against HA, and to a lesser extent NA, NP, and NS1 proteins (Figure 2D; Table S1). Generation of cross-reactive Abs against avian- and swine-origin strains is desirable from a pandemic preparedness perspective. Therefore, we extended our analysis to 75 zoonotic strains to which these donors had no previous exposure. For 11 of 12 donors tested, LAIV-stimulated organoids generated more heterosubtypic Abs than IIV (Figure 2E). Based on our combined Ab magnitude and diversity analysis, we conclude that LAIV stimulation elicits an increased magnitude and breadth of influenza-specific Abs compared with IIV.
Gene expression analysis reveals distinct B cell transcriptional profiles are driven by influenza Ag type
Given the distinct cellular and humoral responses to IIV, LAIV, and wild-type influenza virus, we next asked how different influenza Ags modulate the transcriptional signatures and B cell receptor (BCR) repertoires of total and HA+ B cells. We performed multimodal and kinetics analyses of Ag-specific and bystander B cells after influenza Ag stimulation and measured gene expression, targeted surface-protein expression, and BCRs at single-cell resolution from four donors. Integration of cells from all donors, time points, and stimulations revealed 11 distinct B cell subsets (see Figures 3A, 3B, and S3A; Table S2 for all marker genes), including Act. 1 (high metabolic activity, including PGK1 and LDHA), Act. 2 (antiviral signature), and a GC-entry cluster, which had intermediate IgD protein expression, PGK1 and LDHA expression, and some CD83, suggesting a transition toward a light zone (LZ) GC profile. A LZ B cell cluster was also identified (CD83 expression and BCL2A1, MIR155HG, which are indicative of CD40 signaling). Memory B cell subsets were also identified in organoid B cells, including classical memory (CD27+IgD−, with AIM2 and CD82 expression23), activated memory (IgDlo, mixed CCR6 protein expression,24,25 and an interferon (IFN) gene expression profile) (Figures 3A, 3B, and S3A), and a distinct population (FCRL4+ pre-mem B; expressing high FCRL4 and surface expression of PD-1 and CCR6) described by others as “atypical” in the periphery26 or as low-affinity memory precursors in human and mouse GCs25 (Figures 3A, 3B, and S3A). Two distinct plasmablast populations were identified, “PB1” and “PB2” (expressing JCHAIN and XBP1), both of which expressed high surface CD38 and CD27 expression. Finally, we identified a cluster of proliferative cells (MKI67+) that co-expressed markers for dark zone (DZ) B cells and plasmablasts (XBP1, JCHAIN), which we defined as “DZ-like” (Figures 3A, 3B, and S3A). The relative frequency of each B cell cluster was tracked over time and across stimulations (Figures 3C and S3B). In unstimulated controls, B cell profiles were relatively stable. Upon stimulation with LAIV or H1N1, many B cell clusters upregulated genes associated with activation, metabolic activity (GK1, LDHA), and response to type I IFNs (ISG15 and IFIT3). Similarly, cells with a GC-entry transcriptional profile were elevated with LAIV and H1N1, but not with IIV (Figure 3C).
Figure 3.

Influenza vaccine modalities elicit distinct antigen-specific B cell populations
(A) Combined uniform manifold approximation and projection (UMAP) of scRNA-seq data from immune organoid B cells. Cells from days 4, 7, 10, and 14 and all stimulation conditions are shown in aggregate. Clusters are represented by different colors and labels were manually annotated based on transcriptional and protein profiles. n = 4 donors (1 experiment).
(B) Bubble plot of select marker genes for each B cell cluster. Bubble size indicates the frequency of cells with gene expression and color represents level of RNA expression.
(C) B cell scRNA-seq cluster frequencies over time for each stimulation. Each donor is a point. n = 4 donors (1 experiment).
(D) Gene ontology analysis comparing PB1 and PB2 transcriptional profiles.
(E) Kinetics of HA+ B cell frequencies as determined by flow cytometry. n = 12 donors (1 experiment).
(F) Distribution of day 7 HA+ B cells (colored points) on the total B cell (light gray) UMAP. n = 4 donors (1 experiment).
(G) Transcriptional profiles (cluster identity) and kinetics of HA+ B cells. Frequencies shown were calculated out of total HA+ B cells. n = 4 donors (1 experiment).
(H) Representative flow cytometry staining (left) and quantification (right) of CCR6+PD-1+ HA+ pre-memory B cells on day 7 poststimulation. n = 23 donors (3 experiments combined). Paired Mann-Whitney U tests were used to compare groups; ∗∗p < 0.01
(I) Analysis of public data from Turner et al.20 UMAP (left) and quantification (right) of FCRL4+ memory B cells in fine needle aspirates (FNAs) of an adult vaccinated with IIV. Data were derived from a public scRNA-seq dataset (GSE148633). n = 1 donor analyzed before vaccination and on days 5, 12, 28, and 60 postimmunization. See also Tables 1 and S2 and Figure S3.
Transcriptional analyses also showed that IIV-elicited plasmablasts were mostly of the PB1 phenotype (a 3-fold increase compared with PB2) and their numbers peaked early (Figure 3C). H1N1-stimulated plasmablasts were also primarily of the PB1 profile but peaked later, on day 10. The PB2 phenotype was sustained with LAIV stimulation (Figure 3C). Developmental and functional differences between these two subsets were predicted by gene ontology analysis, with PB1 enriched for gene signatures typically associated with early differentiated plasmablasts (Figure 3D; Table S3). However, PB2 was enriched for pathways associated with protein processing, Ab secretion, and export (Figure 3D; Table S3), which are canonical functions of developmentally mature plasmablasts. Furthermore, we observed enrichment of genes involved in protein modification (including N-linked glycosylation), which was previously shown to alter B cell selection; the abundance of appropriately glycosylated Abs also correlates with protective responses in humans after vaccination.27 Overall, our organoid single-cell RNA sequencing (scRNA-seq) analysis indicates that the antigen format mediates the development of distinct B cell activation and differentiation trajectories.
Influenza Ag modalities elicit different HA+ B cell transcriptional programs
Although Ag modality had a strong influence on global B cell phenotypes, we also investigated its role in the differentiation and function of Ag-specific B cells. LAIV elicited more HA+ B cells than IIV or H1N1 virus at all time points measured (Figure 3E). We observed numerous changes in HA+ B cell transcriptional profiles based on Ag modality (Figures 3F, 3G, and S3D). As observed within the total B cell pool, HA+ B cells from H1N1-virus-stimulated organoids acquired an early activation phenotype (associated with IFN signaling) and entered an activated phenotype around day 7 but were delayed in plasmablast differentiation (Figure 3G). Further, HA+ plasmablasts from H1N1 stimulation were mostly of the less mature PB1 phenotype. In contrast, HA+ B cells from LAIV-stimulated organoids were more likely to acquire the mature PB2 transcriptional profile, with more than 30% of all HA+ B cells acquiring this phenotype on day 10. HA+ B cells from the LAIV-stimulated organoids were the largest contributor to the PB2 cluster (Figure S3E).
HA+ B cells were also activated following IIV stimulation, but their transcriptional signatures were distinct compared with other Ag formats (Figures 3B and 3G). IIV induced little IFN response compared with LAIV or H1N1 (Figures 3B and 3G). HA+ plasmablasts following IIV stimulation strongly preferred the PB1 phenotype (Figure S3E). Although low in number, some PB2 “mature” HA+ plasmablasts were detected later in the IIV response (Figure 3G). We also found a FCRL4+ pre-memory B cell population that was dominated by IIV-stimulated cells. Given their preference for entry or re-entry to the memory B cell pool,23 we further characterized their kinetics. These pre-mem B cells co-expressed PD-1 and CCR6, and their frequencies from flow cytometry were similar to the FCRL4+ pre-mem B cell cluster identified by scRNA-seq (Figures 3G and 3H). In an independent donor cohort, we validated that this population was increased in HA+ B cells from IIV-stimulated cultures compared with other Ag modalities and unstimulated controls (Figure 3H). Finally, we confirmed that the kinetics and proportions of FCRL4+ pre-mem B cells in IIV-stimulated organoids were similar to IIV immunization in vivo. A recent study by Turner et al. used scRNA-seq to measure GC responses in the draining lymph nodes of IIV-vaccinated volunteers over time.20 We independently analyzed these data and identified a cluster that was also marked by high FCRL4 expression within the “pre-memory” pool (Figure 3I). The kinetics of expansion and decline of this population were similar to our observations in IIV-stimulated organoids. Together these analyses define the cellular dynamics of influenza-specific B cells following LAIV and IIV vaccination and viral infection, which likely contribute to the observed differences in humoral responses.
Antigen modality controls naive vs. memory B cell activation and BCR diversity in HA+ B cells
Gene expression data indicated that IIV and LAIV might be recruiting HA+ B cells from distinct B cell subsets, since early responding cells from IIV-stimulated organoids were more memory-like and cells stimulated with LAIV originated from both naive and memory pools (Figures 3G and S3D). One would predict that HA+ B cells recruited from the naive pool would be of the IgM isotype and contain a small number of mutations in their CDR3 region, whereas reactivated memory cells would be class switched with more mutations. To test this, we grouped HA+ B cells into four broad functional categories, based on their transcriptional states: activated, memory, GC, or plasmablasts (Figure 4A) and assessed somatic hypermutation (SHM) and class switching in each of these groups. The early response (day 4) was dominated by IgM isotypes (Figure 4B), indicating that most HA+ cells come from a naive or IgM memory origin. On day 7, the preference for expansion of class switched (and likely memory-derived) HA+ B cells from IIV-stimulated organoids was prominent; both GC and plasmablast HA+ B cells had already class switched to IgG1 (Figure 4B). In contrast, isotype usage from HA+ B cells in LAIV- and H1N1-stimulated organoids at day 7 was variable; several unswitched HA+ B cells were detected in both memory and plasmablast pools. At the conclusion of the culture (day 14), isotype differences were largely resolved, as plasmablasts generated from both vaccine formats had predominantly class switched to IgG1. A substantial proportion of H1N1 virus-stimulated plasmablasts remained unswitched even on day 14.
Figure 4.

IIV and LAIV elicit distinct BCR repertoires and transcriptional profiles
(A) Kinetics of HA+ B cells with a given transcriptional profile in tonsil organoids. Phenotypes were defined based on scRNA-seq and manual annotation of cluster identities. Activated, Act. 1, Act. 2, and GC-entry clusters. Memory, MemB, Act. MemB, and FCRL4 Pre-MemB clusters. GC, LZ and DZ-like clusters. PB, PB1 and PB2 clusters.
(B) BCR isotype usage in HA+ memory, GC, and PB B cells at different time points.
(C) BCR SHM frequencies of HA+ B cell subsets following organoid stimulation with IIV, LAIV, or H1N1. Unpaired Mann-Whitney U tests were used to compare groups; ∗p < 0.05 and ∗∗∗∗p < 0.0001.
(D) V gene usage of HA+ plasmablasts following stimulation with different influenza antigens on day 7.
(E) Shannon index as a metric of BCR diversity for HA+ plasmablasts. Boxplots show the median, with hinges indicating the first and third quartiles and whiskers indicating the highest and lowest value within 1.5 times the interquartile range of the hinges.
(F) Representative data (1 of 4 donors) of the number and size of HA+ BCR clonal families. Each wedge represents a clonal family. n = 4 donors (1 experiment) for all data. See also Table 1 and Figure S3.
We next measured the rates of SHM within HA+ B cells over time (Figure 4C). SHM rates on day 4 were similar among all influenza stimulations, suggesting little affinity maturation early in the response, as expected. However, by day 7, HA+ B cells from IIV-stimulated organoids had more BCR mutations compared with LAIV or H1N1 stimulation, pointing to their memory origin. By day 14, LAIV-elicited HA+ plasmablasts had similar rates of SHM as IIV-stimulated plasmablasts, and both were higher than H1N1-stimulated organoids. We also assessed V gene usage of HA+ BCRs, as a recent study demonstrated that broadly neutralizing, HA stalk anchor-binding Abs preferentially use certain VH genes, including VH3-23, VH3-30, and VH3-48.25 Others have also identified numerous influenza-specific, broadly neutralizing Abs that utilize VH1-69.24,26,28 On day 7, HA+ BCRs from IIV-stimulated organoids were about 15% IGHV1-69 (Figure 4D); HA+ B cells from LAIV-induced organoids also used VH1-69 (∼5%) but had higher VH3-23 usage (∼18%) than any other stimulation (Figure 4D). These findings are consistent with our cross-reactivity and neutralization data and suggest that LAIV Abs can target numerous epitopes and explains their enhanced strain breadth.
Since the memory B cell pool is restricted in its repertoire diversity compared with naive B cells, we expected that a response dominated by memory cells would have reduced BCR diversity compared with a response that incorporates naive B cells. Therefore, we calculated the Shannon index29,30 as a metric of BCR diversity for each donor within the HA+ plasmablast population. On day 7, BCRs from HA+ plasmablasts induced by IIV or LAIV had a higher Shannon index than BCRs from H1N1-stimulated B cells, indicating increased magnitude and/or evenness in BCR diversity in vaccine-stimulated cells (Figure 4E). Diversity decreased on day 14 compared to day 7, indicating that IIV- and LAIV-stimulated organoids underwent selection. This was not the case for H1N1-stimulated organoids, where BCR diversity was largely unchanged between days 7 and 14, indicating that the delayed differentiation kinetics could also be associated with deficiencies in affinity maturation. To validate our diversity findings, we assessed the numbers and sizes of BCR clonal families in HA+ plasmablasts on days 7 and 14 (Figures 4F and S3F). Overall, IIV induced larger clonal families (>5 cells per clonal family) than either LAIV or H1N1 stimulation on day 7. This finding is consistent with the expansion of a pre-existing memory pool and with our SHM and class switching data. On day 14, HA+ B cells from IIV continued to be dominated by a handful of large clonal families. LAIV also induced larger clonal families by day 14 (Figures 4F and S3F). In conclusion, the kinetics of clonal families, combined with SHM as well as isotype usage data, point to early memory dominance by the IIV B cell response and more diversified B cell responses with LAIV stimulation.
The quality of CD4 T cell response is affected by influenza antigen modality
CD4 T cells play a critical role in selection during the GC response. Therefore, we investigated the contribution of T cells in functional responses to LAIV and IIV. To model the influence of influenza Ag on the phenotypes of diverse tonsillar T cell subsets, we profiled CD4 T cells from IIV-, LAIV-, and H1N1-stimulated organoids with scRNA-seq (Figure 1A). Integration and harmonization of gene expression profiles of 113,082 cells from four donors allowed us to dissect the heterogeneity of T cell identities in the organoids. Tonsil organoids were composed of naive (high SELL and IL7R), central memory (TCM, high PGK1, and LDHA), TFH/GC TFH (PDCD1 and CXCR5), regulatory (IL2RA and FOXP3), and effector memory T cell subsets—Th17 (CCR4, CCR6, and RORC)—as well as resident memory Th17 (AQP3 and ITGA4) CD4 T cells (Figures 5A and 5B; Table S2). Parallel profiling of protein readouts allowed for a more nuanced classification of closely related T cell subsets (Figure S4A) such as diverse clusters within activated (CD38+HLA-DR+) and Th17 (CCR4+CCR6+) clusters.
Figure 5.

T cells respond to influenza vaccine and virus antigens with different functional abilities
(A) UMAP (generated from scRNA-seq data) of sorted CD4 T cells from organoids collected on days 4, 7, 10, and 14 from n = 4 donors (1 experiment).
(B) Bubble plot of top differentially expressed genes used to identify T cell clusters. Bubble size indicates percentage of cells expressing the marker and color indicates magnitude of expression.
(C) Frequencies of key T cell clusters in organoids at different time points poststimulation with influenza antigen modalities (n = 4 donors, 1 experiment).
(D) Frequencies of GC TFH following organoid stimulation (n = 12 donors, 1 experiment) as quantified by flow cytometry.
(E) GC TFH frequencies at the peak of CD4 T cell activation (day 7) as quantified by flow cytometry (n = 5 donors, 1 experiment).
(F) Top gene ontology (GO) terms of genes upregulated in LAIV relative to IIV-stimulated organoids in TFH and GC TFH on day 7.
(G) Violin plot of candidate genes (mapping to “cytokine-mediated signaling pathway”) upregulated with LAIV relative to IIV stimulation in TFH and GC TFH on day 7.
(H) Cytokine production by GC TFH under different stimulation conditions on day 7 as quantified by intracellular staining (n = 12 donors, 1 experiment).
(I) Frequencies of Th1- (CXCR3+CCR4−CCR6−), Th2- (CXCR3−CCR4+CCR6−), and Th17-like (CXCR3−CCR4+CCR6+) GC TFH on day 7 as quantified by flow cytometry (n = 12 donors, 1 experiment). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 using a Kruskal-Wallis test followed by paired Mann-Whitney U tests to compare groups. p values shown are for comparisons against the unstimulated control unless otherwise indicated by lines.
(J) Bar graphs comparing Chao diversity metric within activated (OX40+) T cells, TFH, and GC TFH on day 7 poststimulation.
(K) Secreted cytokines and chemokines in organoid supernatants on day 4, measured using Luminex (n = 4 donors, 1 experiment). Boxplots show the median, with hinges indicating the first and third quartiles and whiskers indicating the highest and lowest value within 1.5 times the interquartile range of the hinges.
See also Tables 1, S2, and S3 and Figures S4–S6.
As expected, both LAIV and H1N1 were strong inducers of IFN-responsive genes within naive, TFH, Th17, and resident memory Th17-like CD4 T cells (Figures 5A, 5B, and S4B), albeit modest induction of these responses was observed with IIV at early time points (Figure S4B). While both LAIV and IIV stimulation resulted in comparable induction of early activated (CD134 high) and late activated/exhausted CD4 T cells (Figure 5C), only LAIV and H1N1 expanded proliferating CD4 T cells (high MKI67) (Figure S4B). While overall frequencies of GC TFH were comparable across conditions (Figures 5D and S5A), H1N1 and LAIV stimulated more GC TFH (CXCR5hiPD-1hi) frequencies than IIV (Figure 5E). Functional enrichment of differential gene signatures between LAIV and IIV within the follicular helper T cell compartment (GC TFH and TFH) revealed upregulation of pathways associated with antiviral and cytokine signaling (Figure 5F). These included genes associated with activation (CD40LG, TNFRSF4, and CD38), NF-κB signaling (TRAF1 and STAT1) but also TNF and CXCL13 (Figure 5G). Qualitative differences in GC TFH responses related to Ag modality were confirmed using intracellular cytokine and surface chemokine receptor staining (Figures S5B and S5C); both LAIV and H1N1 stimulated stronger polyfunctional and Th1 (Figures 5H and 5I) responses relative to IIV at the peak of the response. Frequencies of IL-4 and IL-17 expressing GC TFH did not vary with stimulation (Figure 5H), but we noted attenuation of Th2- and Th17-like programs within GC TFH following LAIV and H1N1 stimulation (Figure 5I). Furthermore, TCR analyses of key CD4 T cell subsets on day 7 (Figures 5J, S6A, and S6B) revealed no differences in clone sizes or diversity within early activated cells (OX40+) or TFH subsets between IIV and LAIV. However, stimulation with LAIV and to some extent H1N1 resulted in clonal expansion of GC TFH (Figure 5J), as evidenced by reduced repertoire diversity (Figures S6A and S6B). Finally, we analyzed the secreted factors from organoid cultures on day 4 and found an early increase in Th2-associated IL-13 exclusively in IIV-stimulated organoids but no differences in Th17-associated IL-22 (Figure 5K). Further, secreted CXCL13 remained unchanged with stimulation, but IL-21 was significantly lower in LAIV- and H1N1-stimulated cultures compared with IIV and unstimulated controls (Figure 5K). Overall, these findings indicate enhanced T cell proliferation and clonal expansion in virus-stimulated organoids and differences in key T cell cytokines and chemokines depending on Ag format.
Type I interferon supplementation enhances B cell response to IIV
In addition to elevated IFN-α protein in supernatants from LAIV-stimulated cultures, we observed gene expression signatures of type I IFN signaling in several T and B cell subsets preferentially following LAIV stimulation (Figure 5K). Since LAIV responses were stronger and broader compared with IIV (Figures 1 and 2), we hypothesized that enhanced IFN signaling might contribute to the elevated LAIV response. To test this, we measured responses to IIV in the presence or absence of type I IFN supplementation. Although exogenous type I IFN increased the proportion of HA+ B cells, HA+ plasmablasts (Figures 6A and 6B), and HA-specific Abs (Figure 6C), type I IFN supplementation alone in IIV cultures was insufficient to reach the same magnitude of response as LAIV-stimulated organoids. These data demonstrate that type I IFN secretion and signaling is a major contributor to differences in the IIV vs. LAIV response, but that other additional factors must also contribute to the increased LAIV response seen in immune organoids.
Figure 6.

Cellular and signaling mechanisms underlying differences in the quality and magnitude of the IIV vs. LAIV antibody response
(A–C) Type I interferon supplementation of immune organoids. Frequencies of HA+ B cells (A), HA+ plasmablasts (B), and influenza-specific antibodies (C) on day 7.
(D–F) Effects of naive or memory CD4 T cell depletion on organoid responses. Frequencies of total plasmablasts (D), HA+ B cells (E), and HA-specific Abs (F) on day 7.
(G–J) Effects of memory B cell depletion on organoid responses. Flow cytometry analysis of wild-type and memory B cell-depleted tonsil organoids on day 7 poststimulation (G). Antibody quantification (top) or fold-change (bottom) of specific antibodies on day 7 against hemagglutinin (H), neuraminidase (I), and nucleoprotein (J) in wild-type and memory B cell-depleted organoids. n = 6 donors combined from 2 experiments for all data shown. Boxplots show the median, with hinges indicating the first and third quartiles and whiskers indicating the highest and lowest value within 1.5 times the interquartile range of the hinges. ∗p < 0.05 and ∗∗p < 0.01 using a Kruskal-Wallis test followed by paired Mann-Whitney U tests to compare groups. p values shown are for comparisons against the unstimulated control unless otherwise indicated by lines. See also Tables 1 and S5.
CD4 T cells are required for IIV- but not LAIV-induced antibody responses
Given the interplay between B cells and CD4 T cell helpers, we assessed the relative contribution of CD4 T cells toward humoral responses induced by various influenza Ag formats. Tonsil cells were sorted to isolate naive (CD4+CD45RA+) and memory (CD4+CD45RA−) CD4 T cells and organoids were generated in the presence or absence of these helper CD4 T cell populations. On day 7, organoids lacking any CD4 T cells (total CD4 T depleted) had substantially fewer plasmablasts compared with wild-type controls, and this effect was independent of antigen stimulation (Figure 6D), consistent with our previous work.19 When comparing naive vs. memory CD4 T cell depletion, we observed a stronger detrimental effect on plasmablast differentiation when memory CD4 T cells were depleted in IIV cultures (Figure 6D). Further, CD4 T cells were found to be critical for HA+ B cell expansion in IIV-stimulated organoids, but depletion of these populations had little effect on LAIV- or H1N1-stimulated organoids (Figure 6E). These observations were consistent with the magnitude of the Ab response, where total and memory CD4 T cells depletion attenuated the generation of HA-specific Abs during IIV stimulation, but LAIV was able to induce similar Ab quantities even in the absence of CD4 T cells (Figure 6F). Together, these data show the critical importance of CD4 T cells, and particularly memory CD4 T cells, in eliciting a productive IIV-specific response.
B cell depletion reveals dependence on pre-existing memory for IIV but not LAIV responses
The multiomic analysis strategy in this study allowed us to define distinct cellular, Ab, and repertoire signatures associated with influenza vaccine modalities and live virus. Several pieces of data, including Ab cross-reactivity, transcriptional profiles, and BCR repertoire and isotype usage, suggested that the IIV response was dominated by pre-existing memory B cells. To directly test the role of memory B cells in response to influenza vaccine or virus stimulation, we depleted total memory B cells (CD27+CD38− and CD27−CD38−IgD− B cells) and prepared tonsil organoids as “memB depleted” or “wild-type” (memory-depleted and reconstituted with memory B cells at their original frequency) controls and evaluated cellular and Ab responses on day 7. Although depletion of pre-existing memory B cells reduced the frequency of total and HA+ plasmablasts regardless of Ag format, HA+ B cells were nearly absent from memory-depleted, IIV-stimulated organoids (Figure 6G). Furthermore, influenza-specific Abs were nearly undetectable in these cultures (Figures 6H–6J). However, organoids that were depleted of pre-existing memory and stimulated with LAIV still elicited an influenza-specific Ab response, as measured by cellular responses and Ab production, albeit attenuated compared with wild-type controls. Collectively, these findings demonstrate that the IIV response is fully dependent on pre-existing memory while both naive and memory B cells contribute to the LAIV response.
Discussion
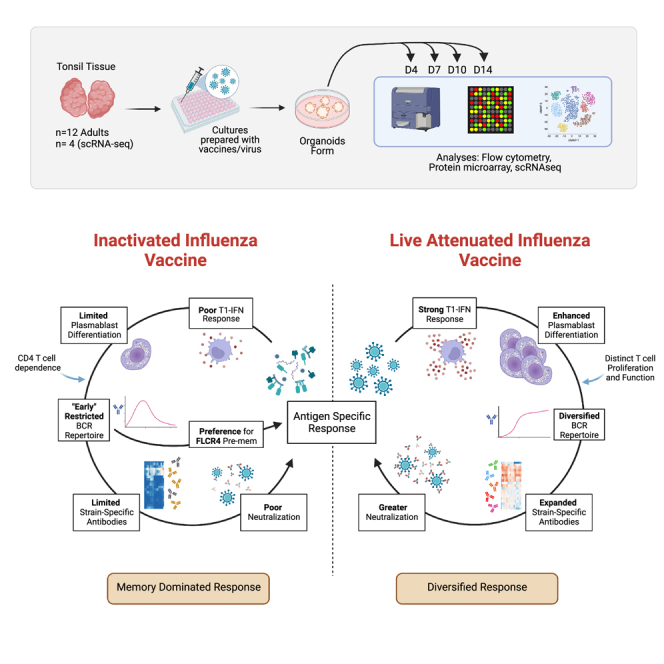
In this study, we used human tonsil organoids to identify key differences in adaptive immune responses to inactivated and live-attenuated influenza vaccines, both of which were distinct from the response to wild-type influenza A virus. LAIV stimulated more B cell differentiation, more HA+ B cells and HA+ plasmablasts, and a stronger neutralizing Ab response than the inactivated vaccine or wild-type virus. We also demonstrated that T cells were activated to varying degrees depending on the Ag format. The kinetics of these responses are in line with those of Ag-experienced individuals and consistent with the ages and demographics of our donors. LAIV stimulated a consistently strong and broadly cross-reactive Ab response compared with IIV. In particular, LAIV was the only Ag format to induce substantial quantities of NA-specific Abs, which are important for blocking viral egress; recent studies implicate a role for NA-specific Abs in protection.31,32 A major difference between IIV and LAIV is Ag composition; the inactivated vaccine manufacturing process focuses on HA content, and non-HA proteins and nucleic-acid content can vary dramatically between manufacturers and influenza seasons.33 Our transcriptional profiling data also point to a robust and early type I IFN response after LAIV stimulation, which may explain the increased magnitude and breadth of the response. Based on the findings presented here, we hypothesize that LAIV draws a larger pool of Ag-reactive B and T cells to enter the response and optimally activates them, leading to a diversified Ab response.
For respiratory infections such as influenza, the ideal vaccine ought to induce and sustain sterilizing mucosal immunity in the upper respiratory tract and lungs. We showed that the types and breadth of B and T cells recruited to the mucosal response was heavily dependent on the Ag format in pre-exposed individuals. This finding has critical implications for future intranasal vaccine development. We expect that different strategies could be implemented depending on the pathogen and the type of response to be induced. In individuals with protective pre-existing memory cells, boosting a narrowly focused memory pool near the site of infection might be favorable. Our data suggest that inactivated formulations might achieve this goal. In other cases where the pre-existing response is rapidly escaped by mutation (such as with influenza), enhanced diversification could promote protection. We propose that a LAIV for such a purpose is ideal.
A key finding from our study is that IIV responses in the tonsil are largely dependent on memory B cells. Several pieces of data suggested that the IIV response was dominated by and dependent on pre-existing memory B cells: (1) donors were divided by the type of Ab responses they made to IIV, (2) IIV did not generate any distinct cross-reactive Abs, and (3) IIV organoids had more class switching and increased SHM rates early in the response. These findings are in alignment and provide insights into clinical data from IIV immunization studies, which have shown that a single IIV immunization poorly stimulates Ab responses in naive subjects.34 An IIV efficacy meta-analysis in children under 2 years old (likely influenza naive) showed that protection after IIV immunization was no different than placebo.35 Since our studies focused on the tonsil, we expect that the bimodal distribution of responses observed to IIV stimulation in tonsil organoids can be explained by differences in influenza exposure history among the donors. We hypothesize that individuals with poor responses to IIV have not been recently exposed to influenza viruses, since IIV immunization is not sufficient to stimulate mucosal responses.36
Previous studies investigating early T cell correlates of influenza vaccine protection have used circulating TFH (cTFH) from blood as surrogates for their lymphoid tissue counterparts.37 In this study, we compared the early dynamics of lymphoid tissue-derived human GC T cells following IIV and LAIV stimulation. A key finding was the qualitative differences in GC TFH responses to different Ag modalities. In vivo human studies identified the emergence of activated cTFH at day 7 post-IIV administration,38,39 which strongly predict Ab titers at day 28.40,41 Here we show that within the same individual, LAIV was more stimulatory than IIV and induced a more potent polyfunctional Th1-like response in GC TFH. Previous studies have described Th1-associated upregulation of CXCR3 but Th17-associated downregulation of CCR6 in cTFH after IIV immunization.42 Our data suggest that both LAIV and, to a lesser extent, H1N1 preferentially skew GC TFH toward a Th1-like program and away from a Th2/Th17 program. cTFH have been shown to clonally expand in individuals following IIV stimulation.42 Here, we showed that LAIV stimulation preferentially selected larger GC TFH clones compared with both IIV and H1N1. While these adaptations correlated with higher frequencies of HA+ B cells and Abs on day 14, our CD4 T cell depletion data also showed that LAIV can elicit a response even without CD4 T cell help. Combined, these findings suggest that LAIV stimulates T and B cell responses through multiple independent and robust mechanisms.
A main observation from the scRNA-seq data was the appearance of several T and B cell clusters expressing IFN-stimulated genes (ISGs), predominantly from LAIV- and H1N1-stimulated organoids. IFN-ɑ secretion was also significantly higher with LAIV compared with IIV, and even H1N1, stimulation. This diminished type I IFN response in H1N1-stimulated organoids could be due to one of a multitude of viral evasion mechanisms43,44 and may explain the delayed development of the adaptive immune response in H1N1- versus LAIV-stimulated organoids. We also showed that the IIV response could be elevated by type I IFN supplementation, though not to levels of response equal to those found in LAIV-stimulated organoids. Our interpretation of these findings is that type I IFN secretion and signaling only partly explain the differences observed between IIV and LAIV responses. Future work will examine additional pathways that could be targeted to further enhance the magnitude and breadth of the IIV response.
Another important consideration is that the modes of immunization for IIV and LAIV are different in vivo. Although we did not replicate the natural route of administration, our study did address how Ag format affects the ability of Ag-specific cells to respond at the site of infection. Despite these differences, the transcriptional profiles of Ag-specific B cells within the draining lymph nodes of recently vaccinated IIV recipients20 show numerous similarities to the data we showed here. Like our tonsil organoid data, many IIV-immunized donors do not have a discernible GC response despite increases in peripherally circulating Abs, suggesting a reliance on pre-existing memory that bypasses the GC. A benefit from the organoid system is that multiple Ag formats and conditions can be tested within the same individual, which allows us to account for interindividual differences in a way that is not possible in human in vivo studies.
A main finding from our work is that when both vaccine antigens were delivered directly to immune cells, the magnitude of the LAIV response was greater than the IIV response. Our findings demonstrate that there are no intrinsic issues with LAIV immunogenicity. The infectious nature of LAIV may induce a stronger early innate response in general compared with an inactivated vaccine. Both were able to elicit influenza-specific responses in organoids, but our evaluation of cellular and Ab profiles highlights major differences in how B and T cells are activated by IIV and LAIV. Even in cases where the Ab response to both vaccines were of similar magnitude (5/12 donors for H1N1 antigens), there were notable differences in Ab breadth and B and T cell transcriptional profiles.
In conclusion, this study establishes human tonsil organoids as a robust system to interrogate adaptive immune responses to influenza Ags. Importantly, this platform allowed us to address the effect of Ag modality in human cells while controlling for the interindividual variability that is intrinsically present in vaccine clinical trials. We have established the cellular and Ab dynamics associated with distinct influenza vaccine modalities, which can be used in the future to profile in vivo immune responses during clinical trials. These characteristics are imperative for rational vaccine design, which further impacts human health by providing a better understanding of immune responses with the ultimate goal of universal flu vaccine development and pandemic avoidance.
Limitations of the study
One limitation of the in vitro organoid system is that peripheral factors impacting the response in vivo are not considered. Influenza virus-exposed adults likely have influenza-specific mucosal Abs; pre-existing Abs can play an important role in shaping Ag accessibility during recall responses.45,46 In vivo, Ab responses after IIV immunization tend to be short-lived (often less than 1 year), while Abs induced by infection are long-lived. Here we show that even when pre-existing Abs are eliminated as a factor in Ag accessibility, and indeed even when memory B cells are eliminated, IIV poorly stimulates naive B cell responses. This memory dependence might be even more substantial in vivo, where competitive factors additionally influence the response. It is also important to note that we were only able to specifically track A/California H1N1 HA+ B cells. This specificity likely represents only a fraction of the total Ag-specific response, since we did not examine HA-specific cells against H3N2 and influenza B, nor to other influenza proteins. These cells would fall into the HA− B cell pool, which we also quantified, but these specificities could not be separated from irrelevant B cells. This might explain why some of the strong differences we observed in the HA+ B cells were also detected in the HA− B cell pool, though to a lesser extent. Future work will more deeply explore the role of pre-existing memory and influenza vaccination vs. infection on the magnitude and breadth of the adaptive immune response to other HA and non-HA Ags.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human TruStain FcX | Biolegend | cat # 422302; RRID: AB_2818986 |
| Anti-Human CCR4 (L291H4) BV421 | Biolegend | cat # 359414; RRID: AB_2562435 |
| Anti-Human CCR6 (G034E3) BV650 | Biolegend | cat # 353426; RRID: AB_2563869 |
| Anti-Human CCR7 (G043H7) PE-Dazzle594 | Biolegend | cat # 353236; RRID: AB_2563641 |
| Anti-Human CD127 (A019D5) BV605 | Biolegend | cat # 351334; RRID: AB_2562022 |
| Anti-Human CD19 (HIB19) BV605 | Biolegend | cat # 302244; RRID: AB_2562015 |
| Anti-Human CD19 (HIB19) BV650 | Biolegend | cat # 302238; RRID: AB_2562097 |
| Anti-Human CD19 (HIB19) PE | Biolegend | cat # 302208; RRID: AB_314238 |
| Anti-Human CD25 (BC96) APC-Cy7 | Biolegend | cat # 302614; RRID: AB_314284 |
| Anti-Human CD27 (O323) PE-Cy7 | Biolegend | cat # 302838; RRID: AB_2561919 |
| Anti-Human CD3 (HIT3a) AF700 | Biolegend | cat # 300324; RRID: AB_493739 |
| Anti-Human CD3 (OKT3) BV605 | Biolegend | cat # 317322; RRID: AB_2561911 |
| Anti-Human CD3 (SK7) PerCp-Cy5.5 | Biolegend | cat # 344808; RRID: AB_10640736 |
| Anti-Human CD38 (HIT2) APC | Biolegend | cat # 303510; RRID: AB_314362 |
| Anti-Human CD38 (HIT2) FITC | Biolegend | cat # 303504; RRID: AB_314356 |
| Anti-Human CD38 (HIT2) PE-Dazzle594 | Biolegend | cat # 303538; RRID: AB_2564105 |
| Anti-Human CD4 (OKT4) BV650 | Biolegend | cat # 317436; RRID: AB_2563050 |
| Anti-Human CD4 (OKT4) FITC | Biolegend | cat # 317408; RRID: AB_571951 |
| Anti-Human CD4 (OKT4) PE-Cy5 | Biolegend | cat # 317412; RRID; AB_571957 |
| Anti-Human CD45 (HI30) AF700 | Biolegend | cat # 304024; RRID: AB_493761 |
| Anti-Human CD45RA (HI100) BV785 | Biolegend | cat # 304140; RRID: AB_2563816 |
| Anti-Human CD56 (5.1H11) PerCP-Cy5.5 | Biolegend | cat # 362506; RRID: AB_2563914 |
| Anti-Human CD8 (SK1) FITC | Biolegend | cat # 344704; RRID: AB_1877178 |
| Anti-Human CD8 (RPA-T8) PerCP-Cy5.5 | Biolegend | cat # 301032; RRID: AB_893422 |
| Anti-Human CXCR3 (G025H7) BV711 | Biolegend | cat # 353732; RRID: AB_2563533 |
| Anti-Human CXCR5 (J252D4) FITC | Biolegend | cat # 356914; RRID:AB_2561896 |
| Anti-Human CXCR5 (J252D4) PE | Biolegend | cat # 356904; RRID: AB_2561813 |
| Anti-Human CXCR5 (J252D4) PE-Cy7 | Biolegend | cat # 356924; RRID: AB_2562355 |
| Anti-Human HLA-DR (L243) Pac blue | Biolegend | cat # 307633; RRID: AB_1595444 |
| Anti-Human IFN-g (4S.B3) PE-Cy7 | Biolegend | cat # 502528; RRID: AB_2123323 |
| Anti-Human IgD (IA6-2) APC-Cy7 | Biolegend | cat # 348218; RRID: AB_11203722 |
| Anti-Human IL-10 (JES3-9D7) PE-Dazzle594 | Biolegend | cat # 501426; RRID: AB_2566744 |
| Anti-Human IL-17A (BL168) AF700 | Biolegend | cat # 512318; RRID: AB_2124868 |
| Anti-Human IL-4 (MP4-25D2) FITC | Biolegend | cat # 500806; RRID: AB_315125 |
| Anti-Human PD-1 (NAT105) APC | Biolegend | cat # 367406; RRID: AB_2566067 |
| Anti-Human PD-1 (EH12.2H7) Pac Blue | Biolegend | cat # 329916; RRID: AB_2283437 |
| Anti-Human PD1 (EH12.2H7) AF700 | Biolegend | cat # 329952; RRID: AB_2566364 |
| Streptavidin APC conjugate | Invitrogen | cat # 17-4317-82 |
| Streptavidin PE conjugate | Invitrogen | cat # 12-4317-87 |
| Anti-Human TCRgd (5A6.E9) PE | Invitrogen | cat # MHGD04; RRID: AB_10374518 |
| Anti-Human TNF-a (MAb11) Pac-Blue | Biolegend | cat # 502920; RRID: AB_528965 |
| TotalSeq C0071 Anti-Human CCR4 (L291H4) | Biolegend | cat # 359425; RRID: AB_2800988 |
| TotalSeq C0088 Anti-Human PD-1 (EH12.2H7) | Biolegend | cat # 329963; RRID: AB_2800862 |
| TotalSeq C0140 Anti-Human CXCR3 (G025H7) | Biolegend | cat # 353747; RRID: AB_2800949 |
| TotalSeq C0144 Anti-Human CCR6 (G034E3) | Biolegend | cat # 353440; RRID: AB_2810563 |
| TotalSeq C0144 Anti-Human CXCR5 (J252D4) | Biolegend | cat # 356939; RRID: AB_2800968 |
| TotalSeq C0154 Anti-Human CD27 (O323) | Biolegend | cat # 302853; RRID: AB_2800747 |
| TotalSeq C0159 Anti-Human HLA-DR (L243) | Biolegend | cat # 307663; RRID: AB_2800795 |
| TotalSeq C0251 Anti-Human Hashtag 1 (LNH-94; 2M2) | Biolegend | cat # 394661; RRID: AB_2801031 |
| TotalSeq C0252 Anti-Human Hashtag 2 (LNH-94; 2M2) | Biolegend | cat # 394663; RRID: AB_2801032 |
| TotalSeq C0253 Anti-Human Hashtag 3 (LNH-94; 2M2) | Biolegend | cat # 394665; RRID: AB_2801033 |
| TotalSeq C0254 Anti-Human Hashtag 4 (LNH-94; 2M2) | Biolegend | cat # 394667; RRID: AB_2801034 |
| TotalSeq C0255 Anti-Human Hashtag 5 (LNH-94; 2M2) | Biolegend | cat # 394669; RRID: AB_2801035 |
| TotalSeq C0256 Anti-Human Hashtag 6 (LNH-94; 2M2) | Biolegend | cat # 394671; RRID: AB_2820042 |
| TotalSeq C0257 Anti-Human Hashtag 7 (LNH-94; 2M2) | Biolegend | cat # 394673; RRID: AB_2820043 |
| TotalSeq C0258 Anti-Human Hashtag 8 (LNH-94; 2M2) | Biolegend | cat # 394675; RRID: AB_2820044 |
| TotalSeq C0384 Anti-Human IgD (IA6-2) | Biolegend | cat # 348245; RRID: AB_2810553 |
| TotalSeq C0389 Anti-Human CD38 (HIT2) | Biolegend | cat # 303543; RRID: AB_2800758 |
| anti-human IgG Qdot 800 (Custom) | Invitrogen | cat # C47091; RRID: AB_10556783 |
| horseradish peroxidase (HRP)-conjugated anti-mouse IgG | KPL | cat # 074-1802 |
| anti-NP mAbs (Millipore Cat. Nos. MAB 8257 and MAB 8258 | Millipore Sigma | cat # MAB8257F; RRID: AB_347730 |
| Anti-Influenza A Antibody, nucleoprotein, clone A1 | Millipore Sigma | cat # MAB8258; RRID: AB_95232 |
| monoclonal influenza IgG antibody (clone CR9114) | Creative Biolabs | cat # PABX-199 |
| Goat Anti-Human IgG+IgM+IgA H&L (HRP) | abcam | cat # ab102420; RRID: AB_10712551 |
| Bacterial and virus strains | ||
| A/California/07/2009 H1N1 virus | Wagar Lab | N/A |
| A/California/07/09 xX A/Puerto Rrico/8/1934 reassortant H1N1 virus | BEI | cat # NR-44004 |
| Biological samples | ||
| Healthy Human Tonsils | Universty of California, Irvine Medical Center | Table 1 |
| Fetal Bovine Serum, heat inactivated | R&D Systems | cat # S11550 |
| Bovine Serum Albumin (BSA) | Fisher Scientific | cat # BP9700100 |
| Bovine Serum Albumin (BSA) | Millipore Sigma | cat # A9085 |
| Chemicals, peptides, and recombinant proteins | ||
| Ham’s F12 medium | Gibco | cat # 31765035 |
| Normocin | InvivoGen | cat # ant-nr-1 |
| Penicillin-Streptomycin | Gibco | cat # 15140122 |
| Antibiotic-Antimycotic | Gibco | cat # 15240096 |
| Lymphoprep | Stemcell | cat # 7851 |
| DMSO | Sigma | cat # D4540-100ML |
| RPMI1640 with glutamax | Gibco | cat # 61870036 |
| Nonessential amino acids | Gibco | cat # 11140050 |
| Sodium pyruvate | Gibco | cat # 11360-070 |
| Insulin, selenium, transferrin supplement | Gibco | cat # 41400045 |
| Recombinant human BAFF | Biolegend | cat # 559602 |
| Influenza vaccine (LAIV FluMist® Quadrivalent) 2019-2020 | medImmune | cat # NDC 66017-306-01 |
| Influenza vaccine (IIV, Fluzone® Quadrivalent) 2019-2020 | Sanofi Pasteur | cat # NDC 49281-419-88 |
| PBS | Gibco | cat # 10010-023 |
| Sodium azide | Thermo Fisher | cat # 71448-16 |
| EDTA 0.5M, pH 8.0 | Invitrogen | cat # 15-575-020 |
| Sodium bicarbonate | Sigma | cat # S5761 |
| Sodium carbonate | Sigma | cat # 223530 |
| TPCK-treated trypsin | Worthington Biochemical | cat # NC9783694 |
| Eagle's Minimum Essential Medium (EMEM) | ATCC | cat # 30-2003 |
| Triton X-100 | Thermo Scientific | cat # 85111 |
| 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate (SureBlue) | Seracare | cat # 5120-007 |
| Tween-20 | Sigma | cat # P1379-250ML |
| Universal Type I IFN Protein | R and D Systems | cat # 11200-1 |
| Hemagglutinin A/California/07/2009 (H1N1) | Immune Technology | cat # IT-003-SW12ΔTMp |
| Hemagglutinin A/Brisbane/02/2018 (H1N1) | Immune Technology | cat # IT-003-0011ΔTMp |
| Hemagglutinin A/Kansas/14/2017 (H3N2) | Immune Technology | cat # IT-003-00436p |
| Hemagglutinin B/Phuket/3073/2013 | Immune Technology | cat # IT-003-B11ΔTMp |
| Hemagglutinin B/Colorado/06/2017 | Immune Technology | cat # IT-003-B21▵TMP |
| Influenza A H1N1 (A/California/04/2009) Neuraminidase | Sino Biologicals | cat # 11058-V08B |
| Influenza A H1N1 (A/California/07/2009) Nucleoprotein / NP Protein | Sino Biologicals | cat # 40205-V08B |
| Critical commercial assays | ||
| Cyto-Fast Fix/Perm buffer | Biolegend | cat # 426803 |
| Zombie Aqua™ Fixable Viability Kit | Biolegend | cat # 423101 |
| EZ-link micro NHS-PEG4-biotinylation kit | Thermo Scientific | cat # 21955 |
| immPACT AEC Substrate Kit, Peroxidase | Vector | cat # SK-4205 |
| ProcartaPlex Human Cytokine/Chemokine Panel 1A 34 plex | Invitrogen | cat # EPX340-12167-901 |
| Chromium Next GEM Single Cell 5’ Kit | 10x Genomics | cat # 1000263 |
| 5’ Feature Barcode Kit | 10x Genomics | cat # 1000256 |
| Chromium Single Cell Human BCR Amplification Kit, | 10x Genomics | cat # 1000253 |
| Chromium Single Cell Human TCR Amplification Kit | 10x Genomics | cat # 1000252 |
| Chromium Next GEM Chip K Single Cell Kit | 10x Genomics | cat # 1000286 |
| Deposited data | ||
| Raw and analyzed data | This paper | NCBI SRA: PRJNA980687 |
| Experimental models: Cell lines | ||
| Madin-Darby Canine Kidney (MDCK) cells | ATCC | cat # CCL-34 |
| Software and algorithms | ||
| GraphPad Prism | GraphPad Software | Version 8.0 |
| FlowJo | BD Bioscience | Version 10.8.1 |
| Seurat | R Package | Version 4.3 |
| Belysa software | Sigma | Version 1.2 |
| Cell Ranger Single Cell Software | 10x Genomics | Version 6.1.1 |
| Metascape | metascape.org | Version 3.5 |
| Immcantation toolbox | R/Python Package | Version 4.0 |
| Vegan | R Package | Version 2.6.2 |
| Immunarch | R Package | Version 0.7.0 |
| SoftMax Pro Software | Molecular Devices | Version 7.1 |
| Other | ||
| MultiScreen-IP Filter Plate, 0.45 μm (PVDF) | Millipore | cat # MAIPS4510 |
| Costar Assay plate 96 well, high-binding | Corning | cat # 3361 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lisa Wagar (lwagar@hs.uci.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Donors and ethics
Tonsils from 30 consented individuals undergoing surgery for obstructive sleep apnea/hypertrophy, recurrent tonsillitis, or both were collected in accordance with the University of California, Irvine Institutional Review Board (IRB). Ethics approval was granted by the University of California Irvine IRB (protocol #2020-6075) and all participants provided written informed consent. In the cohort used for this study, the participants were aged 13-52 years. Overall, tonsil tissue was healthy in appearance (Table 1).
Method details
Tissue processing and generation of immune organoids
Samples were processed as previously described.19 Briefly, whole tonsils were collected in saline after surgery and immersed in an antimicrobial bath of Ham’s F12 medium (Gibco) containing Normocin (InvivoGen), and penicillin-streptomycin (Gibco) for 30-60 min at 4°C for decontamination of the tissue. Tonsils were then briefly rinsed with PBS and mechanically dissociated; debris was removed using gradient centrifugation (Lymphoprep, Stemcell). Samples were cryopreserved in fetal bovine serum (FBS) with 10% DMSO and stored in nitrogen until use. To generate organoids, cryopreserved cells were thawed, enumerated, and then plated at a final density of 7.5 x106/ml (200 μl final volume in ultra-low attachment plates (Corning)). Organoid media was composed of RPMI1640 with glutamax, 10% FBS, 1x nonessential amino acids, 1x sodium pyruvate, 1x Antibiotic-Antimycotic (Gibco), 1x Normocin (InvivoGen), 1x insulin, selenium, transferrin supplement (Gibco), and 0.5 μg/ml recombinant human BAFF (Biolegend). Antigen (Ag) dose was titrated and optimal dosing was selected based on the ability to induce B cell activation and Ab production without affecting organoid viability (Figure S1). The following amounts of influenza antigens were added to immune organoids at culture setup: A/California/07/2009 H1N1 virus - 2.5 hemagglutination units (HAU) per culture; 2019/20 live attenuated influenza vaccine (LAIV, FluMist® Quadrivalent) - 1:2,000 final dilution; and 2019/20 inactivated influenza vaccine (IIV, Fluzone® Quadrivalent) - 1:10,000 final dilution. Cultures were incubated at 37°C, 5% CO2 with humidity and media was replenished every other day by exchanging 30% of the volume with fresh organoid media.
Flow cytometry surface staining
Immune organoids were harvested and cells were washed with FACS buffer (PBS + 0.1% BSA, 0.05% sodium azide, and 2 mM EDTA) to remove any residual antibodies or factors generated during culture. All samples were stained with Ab cocktails (Table S4) prepared in FACS buffer for 30 minutes on ice while protected from light. Data were collected using a Quanteon ACEA NovoCyte Quanteon (Agilent) flow cytometer. Cell sorting was performed using a BD FACSAria Fusion instrument.
Flow cytometry analysis and isolation of HA+ B cells
A/California/07/2009 (H1N1) hemagglutinin protein (Immune Technology) was biotinylated per manufacturer instructions using EZ-link micro NHS-PEG4-biotinylation kit (Thermo Scientific) and as described previously.20 Protein was incubated with 50 mmol excess biotin at room temperature (RT) for 45 minutes and buffer was exchanged using zeba spin desalting column to remove excess biotin. For staining of HA+ B cells, cells were harvested as described above and first incubated with biotinylated HA (2μg/ml final concentration) and Human TruStain FcX (Biolegend) for 30 minutes on ice. Cells were thoroughly washed with FACS buffer and stained with antibody cocktails (see Table S4) with streptavidin-PE and streptavidin-APC conjugates to detect antigen-specific cells.
Intracellular cytokine staining
Immune organoids were harvested, and cells were washed with FACS buffer. Samples were surface stained with antibody cocktails prepared in FACS buffer for 30 minutes on ice while protected from light. Cells were washed in FACS buffer and fixed with 1X Cyto-Fast Fix/Perm buffer (Biolegend) at RT for 20 minutes in the dark. Pellets were washed twice in 1X Cyto-Fast Perm Wash buffer and stained with a cocktail of intracellular antibodies (see Table S4) for an hour at RT in the dark. Cells were then washed once with 1X Cyto-Fast Perm Wash buffer, twice with FACS buffer, resuspended in FACS buffer, and analyzed on Quanteon ACEA NovoCyte Quanteon (Agilent) flow cytometer.
Antibody detection by ELISA
Influenza-specific antibodies were detected as previously described with the following modifications.19 High-binding assay plates (Corning) were coated overnight with influenza proteins (Table S5) at a final concentration of 2 μg/ml in 100mM sodium carbonate/bicarbonate ELISA coating buffer. Non-specific binding was blocked by incubating plates with 1% BSA (in PBS) for 2 hours at RT. Culture supernatants were diluted 1:20 or 1:80 in PBS and added to coated, blocked plates for 1 hour at RT. Horseradish peroxidase-conjugated anti-human secondary antibodies to IgM/IgG/IgA (Abcam) were used to detect bound antibodies. A monoclonal influenza IgG antibody (clone CR9114) was used as a standard to estimate HA-specific Ab concentration; for other Ab specificities, the optical density (A450) was reported as a semi-quantitative measure of Ab concentration.
ELISpot
HA-specific antibody-secreting cells (ASCs) were detected using an ELISpot protocol as previously described with modifications.19 Briefly, cultures that were either stimulated with influenza antigens for 7 days or left unstimulated were resuspended and enumerated, then plated on HA-coated (see Table S5) and blocked 96-well PVDF membrane plates (Millipore). Each sample was plated in duplicate and total live-cell counts ranged from 3.6 x 104 to 1.08 x 105 cells per well. Cells were incubated on these membranes, undisturbed for 5h at 37°C. Plates were then washed and treated with horseradish peroxidase-conjugated anti-IgG/IgA/IgM secondary antibody (Abcam). After incubation overnight at 4°C, plates were washed and developed with AEC substrate (Vector), washed 20 times with water, dried, and spots were enumerated. The frequency of ASCs out of total B cells was determined from B cell flow cytometry data analysis and the direct cell enumeration counts.
Microneutralization assays
Microneutralization assays (MN) in this study were performed with modifications from previously described protocols.47 Briefly, Madin-Darby canine kidney (MDCK) cells were maintained in Eagle’s minimum essential medium (EMEM; ATCC) containing penicillin/streptomycin (Gibco) and 10% heat-inactivated fetal calf serum (ATCC) and cultured at 37°C with 5% CO2 in a humid environment. Cells were sub-cultured when 80–85% confluency and only early passage cells were used for MN assays. One day prior to assay, MDCK cells were sub-cultured into flat-bottomed 96 well plates at 1.2 x 104 cells/well in 100μL. Organoid culture supernatants were diluted 1/5 in virus growth media (serum-free EMEM containing 0.6% BSA and 1 μg/mL N-p-Tosyl-l-phenylalanine chloromethyl ketone (TPCK)-treated trypsin (Worthington Biochemical)) then serially diluted (two-fold) in virus growth medium in a separate 96-well plate. A/California/07/09 x A/Puerto Rico/8/1934 reassortant H1N1 virus (BEI) was diluted to 50 TCID50 per 50 μL in virus growth media and then added to serially diluted supernatants and incubated for 1h at 37°C, 5% CO2. Wells containing only virus and growth media were also prepared to serve as control samples. Following incubation, media from cell monolayers were replaced with the serum-virus mixtures and were incubated for an additional 1h. Serum-virus mixtures were then replaced with 200 μL of virus growth media plus 2% FBS, and plates were incubated for 48 hours at 37°C. Cells were then fixed in 4% paraformaldehyde in PBS for 30 min, washed in PBS, and then permeabilized in 0.1% PBS/Triton X-100 at RT for 15 min. Cells were washed and blocked in a blocking buffer of 3% BSA (Sigma-Aldrich) in PBS for 1h at RT. Influenza virus nucleoprotein (NP) was detected using anti- influenza NP mAbs (Millipore Sigma) diluted 1/1000 dilution in blocking buffer, followed by horseradish peroxidase (HRP)-conjugated anti-mouse IgG (KPL) diluted to 1/3000 in blocking buffer. Plates were developed in 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate (SureBlue) and reactions were quenched using 0.18 M H2SO4. Assays were quantified in an ELISA plate reader at 450 nm using SoftMax Pro 7.1 software.
Protein microarray and antibody diversity analysis
The influenza protein specificities of antibodies in organoids culture supernatants were assessed using a high throughput protein microarray containing 169 influenza proteins (Table S6) that were generated for this study. Similar protein microarrays have been previously utilized to assess influenza-specific antibodies.22 Day 14 culture supernatants were diluted 1/5 in protein array blocking buffer (GVS) and incubated on arrays overnight at 4oC with gentle rocking. Arrays were washed with Tris-buffered saline with 0.05% Tween-20 (T-TBS) and were incubated for 1.5 hours with goat anti-human IgG Qdot 800 (Invitrogen; 1:400) in blocking buffer. Arrays were washed 3x in T-TBS, 3x in TBS, rinsed in water, and air dried. Images were acquired and intensities were quantified using an ArrayCAM imager and software (Grace Bio-Labs). Data were normalized using a composite of previously described methods.48,49 Briefly, control spots were normalized using a quantile-based normalization method. Then, the sum of the control spots was calculated. Finally, for each sample, a rescaling factor was calculated by dividing the sum of the normalized control spots by the sum of its control spots. The resulting factor was then multiplied by the reactivity of each spot. The specificities and cross-reactivity of influenza-specific antibodies were analyzed using R. To ensure a rigorous signal was detected for cross-reactive Abs, we established a minimum threshold for detection. fluorescence intensity > log10(3.5). We then assigned a positive/negative signal for each Ag and compared whether these cross-reactive Abs were present or absent after organoid stimulation with IIV or LAIV. Samples were classified as positive for IIV only, LAIV only, both IIV and LAIV (IIV and LAIV), or not induced (Neither). A dendrogram heatmap was generated using the R heatmap2 package. Other visualizations were generated using ggplot2.
Luminex
Secreted factors in culture supernatants were measured using ProcartaPlex Human Cytokine/Chemokine Panel 1A 34 plex (Invitrogen) with additional simplex analytes (CD40L, CD134, CXCL13). Diluted samples (1/2) were processed per manufacturer’s instructions and in duplicates in a 384-well plate format on the xMAP INTELLIFLEX System. Standard curves were fit using 5-parameter logistic regression on Belysa software (Millipore Sigma).
scRNA-seq
Ex vivo tonsil cells and immune organoids from days 4, 7, 10, and 14 were harvested and cells were washed with FACS buffer to remove any residual antibodies/factors generated during culture. Samples were stained with a cocktail containing fluorescent-labeled antibodies and HA proteins (Table S4), DNA oligo-tagged antibodies (Table S4), and sample-specific hashtag antibodies (BioLegend). Samples were washed with FACS buffer and sorted to enrich total CD4 T cells, HA+ B cells, and HA- B cells. Sample hashing with hashtag oligos (HTOs) allowed us to pool several stimulation conditions into one tube. Pooled samples were resuspended in ice-cold PBS with 0.04% BSA in a final concentration of 2400 cells/μl. Single-cell suspensions were then immediately loaded on a 10X Genomics Chromium Controller with a loading target of 30,000 cells. Libraries were generated using the Chromium Next Gem Single Cell 5’ Reagent Kit v2 (Dual Index) per the manufacturer’s instructions, with additional steps for amplification of TotalSeq/HTO barcodes and V(D)J libraries (10X Genomics). Quality and quantity of libraries were measured on tapestation, qubit, and Bioanalyzer, and sequenced on Illumina NovaSeq 6000 with a sequencing target of 30,000 reads per cell for gene expression libraries, and 5000 reads per cell for HTO, TCR, and BCR libraries.
5ʹ gene expression analysis
Raw reads from gene expression, TCR, BCR, and HTO libraries were aligned and quantified using Cell Ranger Single Cell Software Suite with Feature Barcode addition (version 6.1.1; 10X Genomics) against GRCh38 human reference genome (GRCh38-2020-A) using the STAR aligner (version 2.7.2a). Alignment was performed using feature and vdj option (vdj_GRCh38_alts_ensembl-5.0.0) in cellranger. Following alignment, hashing and cell surface features (Antibody Capture) from feature barcoding alignments were manually updated in cellranger-generated feature files. Sample assignment and doublet removal was performed using the HTODemux function. Droplets with poor quality RNA/ambient RNA (cells with fewer than 400 detected genes) and dying cells (more than 20% mitochondrial gene expression) were excluded during initial QC. Data normalization and variance stabilization were performed on the integrated object using NormalizeData and ScaleData functions in Seurat50 where regularized negative binomial regression corrected for differential effects of mitochondria and ribosomal gene expression. Dimension reduction was performed using the RunPCA function to obtain the first 30 principal components, of which the first 20 were used for clustering using Seurat’s RunUMAP function with a resolution of 0.8. Cell types were broadly assigned as CD3D expressing T cells and CD19 or MS4A1 expressing B cells, which were further subsetted for reclustering individually as described above. Batch variability between biological replicates was corrected using Harmony.51 A cluster of dying cells with low protein and RNA expression was removed from T cell clusters. The final description of UMAP for B and T cell compartments is provided in Figures 3A and 5A, respectively. Gene and protein markers for each cluster were identified using FindMarkers function. For gene expression, we used Wilcoxon Rank Sum tests for differential marker detection using a log2 fold change cutoff of at least 0.4 and clusters were identified using a known catalog of markers defined for T and B cells from human tonsil.52,53 A similar approach was followed for protein expression, following normalization of antibody capture using centered logratio (CLR) transformation. A combination of both positive and negative protein markers from each cluster was used to aid annotations. A list of final cluster-specific markers for T and B cells is provided in Table S2. Cluster-specific differential expression analysis across conditions was tested using the wilcoxon rank-sum test with default settings in Seurat. Only statistically significant genes (log2 fold change cutoff of 0.4; adjusted p-value < 0.05) were included in downstream analyses. Functional enrichment of candidate genes was done in Metascape (version 3.5). Module scores for specific gene sets were incorporated cluster-wise using the AddModuleScores function.
Analysis of in vivo vaccine response
In vivo responses to IIV were analyzed from the previously published dataset GSE148633.20 Briefly, we aggregated data from fine needle aspirates from one individual vaccinated with IIV (days 0, 5, 12, 28, and 60). Poor quality cells were filtered out (RNA features less than 400 or more than 4000, percentage mitochondria > 10%). Data normalization, variance stabilization, dimension reduction, and clustering were performed as described in the 5’ gene expression analysis section. Cluster-specific differential expression analysis over time was performed using the wilcoxon rank-sum test with default settings in Seurat.
BCR analysis
The BCR sequence data were processed using the Immcantation toolbox (version 4.0.0; https://immcantation.readthedocs.io/) using the IgBLAST and IMGT germline sequence databases, with default parameter values unless otherwise noted. The IgBLAST database was used to assign V(D)J gene annotations to the BCR FASTA files for each sample using the Change-O package,30 resulting in a matrix containing sequence alignment information for each sample. Cells were clustered using Scoper spectralClones function with vj method, which infers clones using junction region homology combined with mutation profiles of V and J segments.54 The quantity and sizes of B cell clonal families were calculated using the Alakazam30 countClones function. BCR mutation frequencies were then estimated using SHazaM30 observedMutations function with germline D mask. Shannon-Weiner diversity index for samples was calculated using R package Vegan 2.6.2. All BCR analysis was performed in python or R v.4.1.0 with visualization performed using base R and ggplot2.
TCR analysis
TCR sequence data were processed using Immunarch (https://immunarch.com/, version 0.7.0). Briefly, annotations generated by cellranger were filtered based on cells that passed all filters in the final UMAP. Analysis was performed on a cluster-, time-, and donor-specific manner. Data were first parsed on Immunarch using the repLoad function with a paired option, where only cells with one productive alpha and one productive beta chain were retained for downstream analyses. Clonality (size, top clones, rare clones, and clonal homeostasis) was examined using the repExplore function. Diversity estimates (D50 and Chao) were measured using the repDiversity function.
Quantification and statistical analysis
All statistical analyses were performed using R. Where multiple groups and multiple comparisons were made, a Kruskal-Wallis test was used to initially identify groups with significant differences; paired or unpaired Mann-Whitney or Wilcoxon signed-rank as appropriate were used to identify differences between groups of normally or non-normally distributed data respectively. In the main figures, for comparisons against unstimulated controls p values are placed above the relevant comparison group. p values that compare two stimulated conditions are identified by lines.
Acknowledgments
The authors thank the tonsillectomy patients for participating in this study, Dr. Jennifer Atwood and the UCI Institute for Immunology flow cytometry core for technical assistance, Drs. Edwards and Tifrea at UCI Medical Center pathology for sample coordination, Drs. Vamsee Mallajosyula and Ali Ellebedy for experimental and reagent advice, Dr. Dequina Nicholas for equipment access, Dr. Tim Yates for guidance on BCR repertoire analysis, and Dr. Michael Cahalan for critical feedback on the manuscript. This work is supported by funding from the Wellcome Leap HOPE Program (to L.E.W.) and the Bill and Melinda Gates Foundation (INV-038114, to L.E.W.). J.M.K. receives postdoctoral fellowship salary support in part from an F. Hoffman-La Roche sponsored research agreement. This work was made possible by the Genome Technology Access Center at the McDonnell Genome Institute at Washington University School of Medicine. The Center is partially supported by NCI Cancer Center Support grant P30 CA91842 to the Siteman Cancer Center. This work was also made possible through access to the Genomics Research and Technology Hub Shared Resource of the UCI Cancer Center Support Grant (P30CA-062203), the Single Cell Analysis Core shared resource (U54CA217378), the UCI Genomics-Bioinformatics Core of the Skin Biology Center (P30AR075047), and NIH shared instrumentation grants 1S10RR025496-01, 1S10OD010794-01, and 1S10OD021718-01. This publication is solely the responsibility of the authors and does not necessarily represent the official view of these resources or funding sources.
Author contributions
J.M.K., S.S., N.S., D.H.D., and L.E.W. conceived the study and guided it throughout. J.M.K., S.S., A.J., J.E.H.-D., R.d.A., Z.W.W., A.M.S., M.T.M., A.I.B., and E.L. performed assay development, experiments, and/or data analysis. J.M.K., S.S., and L.E.W. interpreted results and implications. G.A., Q.Z., D.T., J.B., and A.M.S. contributed to study recruitment, sample acquisition, and tissue processing. J.M.K., S.S., and L.E.W. prepared the manuscript, and all authors reviewed and approved it.
Declaration of interests
L.E.W. is the co-inventor of intellectual property assigned to Stanford University, “Systems and Methods to Model Adaptive Immune Responses,” which describes the immune organoid technology.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: July 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2023.06.019.
Supplemental information
Data and code availability
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Erbelding E.J., Post D.J., Stemmy E.J., Roberts P.C., Augustine A.D., Ferguson S., Paules C.I., Graham B.S., Fauci A.S. A universal influenza vaccine: the strategic plan for the National Institute of Allergy and Infectious Diseases. J. Infect. Dis. 2018;218:347–354. doi: 10.1093/infdis/jiy103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nachbagauer R., Feser J., Naficy A., Bernstein D.I., Guptill J., Walter E.B., Berlanda-Scorza F., Stadlbauer D., Wilson P.C., Aydillo T., et al. A chimeric hemagglutinin-based universal influenza virus vaccine approach induces broad and long-lasting immunity in a randomized, placebo-controlled phase I trial. Nat. Med. 2021;27:106–114. doi: 10.1038/s41591-020-1118-7. [DOI] [PubMed] [Google Scholar]
- 3.Krammer F., Fouchier R.A.M., Eichelberger M.C., Webby R.J., Shaw-Saliba K., Wan H., Wilson P.C., Compans R.W., Skountzou I., Monto A.S. NAction! How can neuraminidase-based immunity contribute to better influenza virus vaccines? mBio. 2018;9:e02332-17. doi: 10.1128/mBio.02332-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park J., Fong S., Schwartzman L.M., Sheng Z.-M., Freeman A., Matthews L., Xiao Y., Ramuta M.D., Batchenkova N.A., Qi L., et al. An inactivated multivalent influenza A virus vaccine is broadly protective in mice and ferrets. Sci Transl. Med. 2021;14:eabo2167. doi: 10.1126/scitranslmed.abo2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivachtchenko A.V., Ivanenkov Y.A., Mitkin O.D., Yamanushkin P.M., Bichko V.V., Shevkun N.A., Karapetian R.N., Leneva I.A., Borisova O.V., Veselov M.S. Novel oral anti-influenza drug candidate AV5080. J. Antimicrob. Chemother. 2014;69:1892–1902. doi: 10.1093/jac/dku074. [DOI] [PubMed] [Google Scholar]
- 6.Oh J.E., Song E., Moriyama M., Wong P., Zhang S., Jiang R., Strohmeier S., Kleinstein S.H., Krammer F., Iwasaki A. Intranasal priming induces local lung-resident B cell populations that secrete protective mucosal antiviral IgA. Sci. Immunol. 2021;6:eabj5129. doi: 10.1126/sciimmunol.abj5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H., Ren H., Zhang Y., Cao L., Xu W. Intranasal vaccination with a recombinant protein CTA1-DD-RBF protects mice against hRSV infection. Sci. Rep. 2021;11:18641. doi: 10.1038/s41598-021-97535-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hobson D., Curry R.L., Beare A.S., Ward-Gardner A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J. Hyg. (Lond.) 1972;70:767–777. doi: 10.1017/s0022172400022610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beyer W.E.P., Palache A.M., Lüchters G., Nauta J., Osterhaus A.D.M.E. Seroprotection rate, mean fold increase, seroconversion rate: which parameter adequately expresses seroresponse to influenza vaccination? Virus Res. 2004;103:125–132. doi: 10.1016/j.virusres.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 10.Wright P.F., Hoen A.G., Ilyushina N.A., Brown E.P., Ackerman M.E., Wieland-Alter W., Connor R.I., Jegaskanda S., Rosenberg-Hasson Y., Haynes B.C., et al. Correlates of immunity to influenza as determined by challenge of children with live, attenuated influenza vaccine. Open Forum Infect. Dis. 2016;3:ofw108. doi: 10.1093/ofid/ofw108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belshe R.B., Gruber W.C., Mendelman P.M., Mehta H.B., Mahmood K., Reisinger K., Treanor J., Zangwill K., Hayden F.G., Bernstein D.I., et al. Correlates of immune protection induced by live, attenuated, cold-adapted, trivalent, intranasal influenza virus vaccine. J. Infect. Dis. 2000;181:1133–1137. doi: 10.1086/315323. [DOI] [PubMed] [Google Scholar]
- 12.Cox R.J. Correlates of protection to influenza virus, where do we go from here? Hum. Vaccin. Immunother. 2013;9:405–408. doi: 10.4161/hv.22908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoft D.F., Babusis E., Worku S., Spencer C.T., Lottenbach K., Truscott S.M., Abate G., Sakala I.G., Edwards K.M., Creech C.B., et al. Live and inactivated influenza vaccines induce similar humoral responses, but only live vaccines induce diverse T-cell responses in young children. J. Infect. Dis. 2011;204:845–853. doi: 10.1093/infdis/jir436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X.S., Holmes T.H., Zhang C., Mahmood K., Kemble G.W., Lewis D.B., Dekker C.L., Greenberg H.B., Arvin A.M. Cellular immune responses in children and adults receiving inactivated or live attenuated influenza vaccines. J. Virol. 2006;80:11756–11766. doi: 10.1128/JVI.01460-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilyushina N.A., Haynes B.C., Hoen A.G., Khalenkov A.M., Housman M.L., Brown E.P., Ackerman M.E., Treanor J.J., Luke C.J., Subbarao K., et al. Live attenuated and inactivated influenza vaccines in children. J. Infect. Dis. 2015;211:352–360. doi: 10.1093/infdis/jiu458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Treanor J.J., Kotloff K., Betts R.F., Belshe R., Newman F., Iacuzio D., Wittes J., Bryant M. Evaluation of trivalent, live, cold-adapted (CAIV-T) and inactivated (TIV) influenza vaccines in prevention of virus infection and illness following challenge of adults with wild-type influenza A (H1N1), A (H3N2), and B viruses. Vaccine. 1999;18:899–906. doi: 10.1016/S0264-410X(99)00334-5. [DOI] [PubMed] [Google Scholar]
- 17.Monto A.S., Ohmit S.E., Petrie J.G., Johnson E., Truscon R., Teich E., Rotthoff J., Boulton M., Victor J.C. Comparative efficacy of inactivated and live attenuated influenza vaccines. N. Engl. J. Med. 2009;361:1260–1267. doi: 10.1056/NEJMoa0808652. [DOI] [PubMed] [Google Scholar]
- 18.Cole M.E., Kundu R., Abdulla A.F., Andrews N., Hoschler K., Southern J., Jackson D., Miller E., Zambon M., Turner P.J., et al. Pre-existing influenza-specific nasal IgA or nasal viral infection does not affect live attenuated influenza vaccine immunogenicity in children. Clin. Exp. Immunol. 2021;204:125–133. doi: 10.1111/cei.13564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagar L.E., Salahudeen A., Constantz C.M., Wendel B.S., Lyons M.M., Mallajosyula V., Jatt L.P., Adamska J.Z., Blum L.K., Gupta N., et al. Modeling human adaptive immune responses with tonsil organoids. Nat. Med. 2021;27:125–135. doi: 10.1038/s41591-020-01145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turner J.S., Zhou J.Q., Han J., Schmitz A.J., Rizk A.A., Alsoussi W.B., Lei T., Amor M., McIntire K.M., Meade P., et al. Human germinal centres engage memory and naive B cells after influenza vaccination. Nature. 2020;586:127–132. doi: 10.1038/s41586-020-2711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whittle J.R.R., Wheatley A.K., Wu L., Lingwood D., Kanekiyo M., Ma S.S., Narpala S.R., Yassine H.M., Frank G.M., Yewdell J.W., et al. Flow cytometry reveals that H5N1 vaccination elicits cross-reactive stem-directed antibodies from multiple Ig heavy-chain lineages. J. Virol. 2014;88:4047–4057. doi: 10.1128/JVI.03422-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakajima R., Supnet M., Jasinskas A., Jain A., Taghavian O., Obiero J., Milton D.K., Chen W.H., Grantham M., Webby R., et al. Protein microarray analysis of the specificity and cross-reactivity of influenza virus hemagglutinin-specific antibodies. mSphere. 2018;3:e00592-18. doi: 10.1128/mSphere.00592-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suan D., Kräutler N.J., Maag J.L.V., Butt D., Bourne K., Hermes J.R., Avery D.T., Young C., Statham A., Elliott M., et al. CCR6 defines memory B cell precursors in mouse and human germinal centers, revealing light-zone location and predominant low antigen affinity. Immunity. 2017;47:1142–1153.e4. doi: 10.1016/j.immuni.2017.11.022. [DOI] [PubMed] [Google Scholar]
- 24.Sui J., Hwang W.C., Perez S., Wei G., Aird D., Chen L.M., Santelli E., Stec B., Cadwell G., Ali M., et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guthmiller J.J., Han J., Utset H.A., Li L., Lan L.Y.-L., Henry C., Stamper C.T., McMahon M., O’Dell G., Fernández-Quintero M.L., et al. Broadly neutralizing antibodies target a haemagglutinin anchor epitope. Nature. 2022;602:314–320. doi: 10.1038/s41586-021-04356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pappas L., Foglierini M., Piccoli L., Kallewaard N.L., Turrini F., Silacci C., Fernandez-Rodriguez B., Agatic G., Giacchetto-Sasselli I., Pellicciotta G., et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature. 2014;516:418–422. doi: 10.1038/nature13764. [DOI] [PubMed] [Google Scholar]
- 27.Wang T.T., Maamary J., Tan G.S., Bournazos S., Davis C.W., Krammer F., Schlesinger S.J., Palese P., Ahmed R., Ravetch J.V. Anti-HA glycoforms drive B cell affinity selection and determine influenza vaccine efficacy. Cell. 2015;162:160–169. doi: 10.1016/j.cell.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekiert D.C., Bhabha G., Elsliger M.A., Friesen R.H.E., Jongeneelen M., Throsby M., Goudsmit J., Wilson I.A. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi X., Shao T., Huo F., Zheng C., Li W., Jiang Z. An analysis of abnormalities in the B cell receptor repertoire in patients with systemic sclerosis using high-throughput sequencing. PeerJ. 2020;8:e8370. doi: 10.7717/peerj.8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta N.T., Vander Heiden J.A., Uduman M., Gadala-Maria D., Yaari G., Kleinstein S.H. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics. 2015;31:3356–3358. doi: 10.1093/bioinformatics/btv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y.Q., Wohlbold T.J., Zheng N.Y., Huang M., Huang Y., Neu K.E., Lee J., Wan H., Rojas K.T., Kirkpatrick E., et al. Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell. 2018;173:417–429.e10. doi: 10.1016/j.cell.2018.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajendran M., Nachbagauer R., Ermler M.E., Bunduc P., Amanat F., Izikson R., Cox M., Palese P., Eichelberger M., Krammer F. Analysis of anti-influenza virus neuraminidase antibodies in children, adults, and the elderly by ELISA and enzyme inhibition: evidence for original antigenic Sin. mBio. 2017;8:e02281-16. doi: 10.1128/mBio.02281-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koroleva M., Batarse F., Moritzky S., Henry C., Chaves F., Wilson P., Krammer F., Richards K., Sant A.J. Heterologous viral protein interactions within licensed seasonal influenza virus vaccines. npj Vaccines. 2020;5:3. doi: 10.1038/s41541-019-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lerman S.J., Wright P.F., Patil K.D. Antibody decline in children following A/New Jersey/76 influenza virus immunization. J. Pediatr. 1980;96:271–274. doi: 10.1016/s0022-3476(80)80823-7. [DOI] [PubMed] [Google Scholar]
- 35.Jefferson T., Smith S., Demicheli V., Harnden A., Rivetti A., Di Pietrantonj C. Assessment of the efficacy and effectiveness of influenza vaccines in healthy children: systematic review. Lancet. 2005;365:773–780. doi: 10.1016/S0140-6736(05)17984-7. [DOI] [PubMed] [Google Scholar]
- 36.Krammer F. The human antibody response to influenza A virus infection and vaccination. Nat. Rev. Immunol. 2019;19:383–397. doi: 10.1038/s41577-019-0143-6. [DOI] [PubMed] [Google Scholar]
- 37.Ueno H. Tfh cell response in influenza vaccines in humans: what is visible and what is invisible. Curr. Opin. Immunol. 2019;59:9–14. doi: 10.1016/j.coi.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herati R.S., Reuter M.A., Dolfi D.V., Mansfield K.D., Aung H., Badwan O.Z., Kurupati R.K., Kannan S., Ertl H., Schmader K.E., et al. Circulating CXCR5+PD-1+ response predicts influenza vaccine antibody responses in young adults but not elderly adults. J. Immunol. 2014;193:3528–3537. doi: 10.4049/jimmunol.1302503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herati R.S., Muselman A., Vella L., Bengsch B., Parkhouse K., Del Alcazar D., Kotzin J., Doyle S.A., Tebas P., Hensley S.E., et al. Successive annual influenza vaccination induces a recurrent oligoclonotypic memory response in circulating T follicular helper cells. Sci. Immunol. 2017;2:eaag2152. doi: 10.1126/sciimmunol.aag2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koutsakos M., Wheatley A.K., Loh L., Clemens E.B., Sant S., Nüssing S., Fox A., Chung A.W., Laurie K.L., Hurt A.C., et al. Circulating TFH cells, serological memory, and tissue compartmentalization shape human influenza-specific B cell immunity. Sci. Transl. Med. 2018;10:eaan8405. doi: 10.1126/scitranslmed.aan8405. [DOI] [PubMed] [Google Scholar]
- 41.Bentebibel S.E., Lopez S., Obermoser G., Schmitt N., Mueller C., Harrod C., Flano E., Mejias A., Albrecht R.A., Blankenship D., et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci. Transl. Med. 2013;5:176ra32. doi: 10.1126/scitranslmed.3005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill D.L., Pierson W., Bolland D.J., Mkindi C., Carr E.J., Wang J., Houard S., Wingett S.W., Audran R., Wallin E.F., et al. The adjuvant GLA-SE promotes human Tfh cell expansion and emergence of public TCRβ clonotypes. J. Exp. Med. 2019;216:1857–1873. doi: 10.1084/jem.20190301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez-Sesma A., Marukian S., Ebersole B.J., Kaminski D., Park M.S., Yuen T., Sealfon S.C., García-Sastre A., Moran T.M. Influenza virus evades innate and adaptive immunity via the NS1 protein. J. Virol. 2006;80:6295–6304. doi: 10.1128/JVI.02381-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hale B.G., Albrecht R.A., García-Sastre A. Innate immune evasion strategies of influenza viruses. Future Microbiol. 2010;5:23–41. doi: 10.2217/fmb.09.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linderman S.L., Hensley S.E. Antibodies with “original antigenic sin” properties are valuable components of secondary immune responses to influenza viruses. PLoS Pathog. 2016;12:e1005806. doi: 10.1371/journal.ppat.1005806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dugan H.L., Guthmiller J.J., Arevalo P., Huang M., Chen Y.Q., Neu K.E., Henry C., Zheng N.Y., Lan L.Y.-L., Tepora M.E., et al. Preexisting immunity shapes distinct antibody landscapes after influenza virus infection and vaccination in humans. Sci. Transl. Med. 2020;12:eabd3601. doi: 10.1126/scitranslmed.abd3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gross F.L., Bai Y., Jefferson S., Holiday C., Levine M.Z. Measuring influenza neutralizing antibody responses to A(H3N2) viruses in human sera by microneutralization assays using MDCK-SIAT1 cells. J. Vis. Exp. 2017 doi: 10.3791/56448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duarte J., Serufuri J.-M., Mulder N., Blackburn J. In: Bioinformatics of Human Proteomics Translational Bioinformatics. Wang X., editor. Springer; 2013. Protein function microarrays: design, use and bioinformatic analysis in cancer biomarker discovery and quantitation; pp. 39–74. [DOI] [Google Scholar]
- 49.Bolstad B.M., Irizarry R.A., Astrand M., Speed T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinform. Oxf. Engl. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 50.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Massoni-Badosa R., Soler-Vila P., Aguilar-Fernández S., Nieto J.C., Elosua-Bayes M., Marchese D., Kulis M., Vilas-Zornoza A., Bühler M.M., Rashmi S., et al. 2022. An atlas of cells in the human tonsil. Preprint at bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King H.W., Orban N., Riches J.C., Clear A.J., Warnes G., Teichmann S.A., James L.K. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci. Immunol. 2021;6:eabe6291. doi: 10.1126/sciimmunol.abe6291. [DOI] [PubMed] [Google Scholar]
- 54.Nouri N., Kleinstein S.H. A spectral clustering-based method for identifying clones from high-throughput B cell repertoire sequencing data. Bioinformatics. 2018;34:i341–i349. doi: 10.1093/bioinformatics/bty235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.