

Abstract
Mosaic chromosomal alterations (mCAs) detected in white blood cells represent a type of clonal hematopoiesis (CH) that is understudied compared to CH-related somatic mutations. A few recent studies indicated their potential link with non-hematological cancers, especially lung cancer. In this study, we investigated the association between mCAs and lung cancer using the high-density genotyping data from the OncoArray study of INTEGRAL-ILCCO, the largest single genetic study of lung cancer with 18,221 lung cancer cases and 14,825 cancer-free controls. We identified a comprehensive list of autosomal mCAs, ChrX mCAs, and mosaic ChrY (mChrY) losses from these samples. Autosomal mCAs were detected in 4.3% of subjects, in addition to ChrX mCAs in 3.6% of females and mChrY losses in 9.6% of males. Multivariable logistic regression analysis indicated that the presence of autosomal mCAs in white blood cells was associated with an increased lung cancer risk after adjusting for key confounding factors including age, sex, smoking status, and race. This association was mainly driven by a specific type of mCAs: copy-neutral loss of heterozygosity (CN-LOH) on autosomal chromosomes. The association between autosome CN-LOH and increased risk of lung cancer was further confirmed in two major histological subtypes, lung adenocarcinoma and squamous cell carcinoma. Additionally, we observed a significant increase of ChrX mCAs and mChrY losses in smokers compared to non-smokers, and racial differences in certain types of mCA events. Our study established a link between mCAs in white blood cells and increased risk of lung cancer.
Keywords: Mosaic chromosomal alterations, Clonal hematopoiesis, Lung cancer risk
Introduction
In humans, hematopoietic stem cells reside in bone marrow, maintaining the ability to divide and differentiate into all types of blood cells. With increasing age, irreparable somatic mutations may occur and accumulate in a small fraction of hematopoietic stem cells1,2. Some of these mutations confer proliferative or survival advantages and lead to clonal expansion of the host cells in blood, a phenomenon called clonal hemopoiesis (CH). While most CH studies have focused on the detection of point mutations and short insertion/deletions (indels), mosaic chromosomal alterations (mCAs) have also been identified2,3. mCAs are somatic alterations including large chromosomal gains, losses and copy-neutral losses of heterozygosity that can be detected in a fraction of peripheral leukocytes.
Recently, two large-scale studies have been performed to identify mCAs from genotyping data of blood-derived DNA using the United Kingdom Biobank (UKBB)4 and BioBank Japan (BBJ)5, respectively. These studies revealed that the accumulation of mCAs is a feature of aging with a detection rate of 2–8% in subjects younger than 50 but a rapid increase afterward4,5. Particularly, in the BBJ cohort more than 35% of subjects with age ≥ 90 have mCAs5. Smokers are more likely to carry mCAs than non-smokers with matched age. In addition, the incidence of mCA in males is significantly higher than in females after adjusting for age and smoking status5. In both UKBB and BBJ studies, a significantly higher all-cause mortality rate has been observed for individuals with mCAs4,5. Importantly, it has been reported that blood cell mCAs are associated with a variety of human diseases, such as cardiovascular diseases6, autism spectrum disorder7 and infectious diseases8, and have been found to be associated with hematological cancers9,10. Individuals with detected mCAs had a ten times higher risk of developing hematological cancers compared to those without mCAs11. Moreover, mCAs involving larger genomic regions tend to be associated with an earlier onset and a higher rate of mortality of patients with hematological malignancy12.
The association of CH with selected non-hematological cancers has also been reported in previous publications13,14. However, most of these studies focused on point mutations and short indels without considering mCA events. The UKBB and BBJ cohorts come from a general population with a relatively small number of cancers at the time of study, which provided limited information for investigating the association between mCAs and specific cancer types. Interestingly, in a multicancer study, genotyping data from 13 cancer genome-wide association datasets were integrated for identifying mCAs in 31,717 cancer cases (including 31,259 non-hematologic cases from over 14 different cancer types) and 26,136 cancer-free controls10. This study found that mCAs were more frequently detected in subjects with non-hematologic cancers than in controls. When stratified based on cancer types, a significant association was observed in lung cancer (OR=1.56, 95% CI =1.18–2.08, p=0.002). In addition, mosaic loss of chromosome Y (mChrY loss) has been reported to be associated with increased lung cancer risk and prognosis15,16. These studies suggested a potential association between mCAs and lung cancer. To further verify this association, a more careful investigation using a large lung cancer cohort is required.
INTEGRAL (Integrative Analysis of Lung Cancer Etiology and Risk)-ILCCO (International Lung Cancer Consortium) is the largest single genetic study of lung cancer17. We focused on a major sub-cohort from the OncoArray Consortium Lung Study18,19, which provides high-density blood genotyping data for 33,046 subjects, including 18,221 lung cancer cases and 14,825 non-cancer controls. Moreover, the data provide high-quality demographic and clinical variables including age, sex, race, smoking status, and histological subtypes, allowing us to investigate the association between mCAs and lung cancer while considering the effect of these confounders.
Methods and Materials
The OncoArray data from the INTEGRAL-ILCCO cohort
The OncoArray study is a major part of the INTEGRAL-ILCCO cohort, which provides high-quality genotyping array data and clinical information for a total of 33,046 subjects, 18,221 lung cancer cases and 14,825 controls without lung cancer diagnosis. All blood samples are from this cohort were obtained after lung cancer diagnosis but before any treatments. The genotyping data were generated by using the Infinium OncoArray-500K BeadChip (Illumina, San Diego, CA) platform, which contains a total of 533,631 customized SNPs for studying cancer genetics19. The clinical information includes age, sex, race, smoking status, and lung cancer histological subtype. The OncoArray study has been approved by the institutional review board of all sites accruing participants.
Genotyping using the Oncotype platform
Genotyping and data processing were described by the previous studies17–19. Briefly, for the SNP array genotype data, DNA extracted from peripheral white blood cells was genotyped using the OncoArray microarray. We converted all the genotyping intensity files to VCF files with a BCFtools plugin gtc2vcf (https://github.com/freeseek/gtc2vcf). Samples with abnormal heterozygosity rate, sex discordance, <95% completion rates, and unexpected relatedness (identity-by-state > 10%) were discarded.
Identification of autosomal mCAs
We followed the methods of Loh et al.4,12 to identify mosaic chromosomal alterations from high-density SNP genotyping array data. Unphased VCF files were firstly split by chromosomes, then we phased each single-chromosome VCF file by SHAPEIT420 with default parameters. The phased output and unphased ChrY data were then concatenated into a single VCF file. We applied a MOsaic CHromosomal Alterations (MoChA) caller to detect mCAs with either B Allele Frequency (BAF) and Log R Ratio (LRR) or allelic depth (AD), with default parameters4,12. The highly polymorphic MHC (chr6:27486711–33448264) and KIR (chr19:54574747–55504099) regions were excluded from mCA calling. We then applied a series of filters to exclude potential constitutional duplications (e.g., germline chromosome alterations) and low quality mCA calls (Fig. S1). Constitutional duplications have expected deviations in allelic balance (|ΔBAF|) = 1/6, with corresponding LRR ≈ 0.3612. In order to exclude possible constitutional duplications, for mCA events of length > 10 Mb, we excluded events with LRR > 0.35 or with LRR within [0.2, 0.35] and |ΔBAF| > 0.16; for mCA events of length < 10 Mb, we excluded events with LRR > 0.2 or with LRR within [0.1, 0.2] and |ΔBAF| > 0.1. MoChA used a hidden Markov model (HMM) to detect mCAs either based on LRR and BAF or phased BAF (pBAF). LOD scores were used as the measurement of calling quality for model based on LRR and BAF (lod_lrr_baf) or for model based on pBAF (lod_baf_phase). To exclude low-quality mCA calls, we required either lod_lrr_baf or lod_baf_phase to be larger than 10 for mCA events of length > 2 Mb. For mCA events < 2 Mb we required lod_baf_phase > 30 and lod_lrr_baf > 10. In addition, a high-frequency reversion was found in Chr17q2121, which could cause intensively low heterozygosity and induce false calling results. Thus, we removed the mCA events overlapped with Chr17 42–47Mb.
Identification of ChrX mCAs and mChrY losses
The mCAs associated with ChrX were also identified by MoChA. We only identified mCAs in female subjects because MoChA can only call mCAs on diploid homologous chromosome regions. In principle, we can apply MoChA and use the intensities of SNPs located in the pseudo-autosomal regions on sex chromosome (PAR1 and PAR2) to identify ChrX and ChrY mCAs in male subjects. However, the OncoArray genotyping platform contains only a small number of variants (28 SNPs in PAR1 and 1 SNP in PAR2) in the two PARs, which limited the ability of MoChA for phase inference and ChrX/ChrY mCA detection in our male subjects.
Previous studies have reported frequent mosaic loss of ChrY in males, which has been associated with lung cancer15,16. We therefore identified mChrY losses in our male subjects by using the method proposed in previous studies22–24. Briefly, the LRR on non-PAR regions of ChrY was calculated and those with ChrY LRR lower than −0.15 were identified as mChrY loss according to the references24,25.
Determination of whole-chromosome and arm-level mCAs
We manually inspected the distribution of mCAs on chromosome arms. In autosomes, the vast majority of mosaic gain events were whole-chromosomal, while loss and CN-LOH might only occur at one arm of the chromosome. Thus, we divided autosomal mCAs into five categories: gain (+), loss on short arm (p-) and long arm (q-), CN-LOH on short arm (p=) and long arm (q=). Mosaic ChrX gains, losses and CN-LOHs were not divided into chromosome arm level categories, because most of ChrX mCAs covered nearly the whole chromosome. Altogether, this classification resulted in 103 types of mCA at the whole-chromosome or arm level. We tested the significance of co-occurrence between two mCA events by using the Fisher’s exact test. Co-occurred mCA pairs in at least three subjects with an FDR<0.05 were highlighted in the co-occurrence graph.
Multivariable regression model for determine the association of clinical variables with mCAs
To determine the association between clinical variables and mCAs, we constructed a multivariable logistic regression model as the following:
| (Model I) |
In the model, the response variable mCA is set as binary with 1 indicating the presence of mCAs in a subject, and 0 otherwise. In the independent variables, Age is represented as a continuous variable; Sex is set 1 for males and 0 for females; Smoking is set to 1 for current/ex-smokers and 0 for never-smokers; Race is a categorical variable with White as the baseline; and LungCancer is set to 1 for lung cancer cases and 0 for controls. The model was separately applied to the 3 mCA types: autosomal mCA, ChrX mCA, and ChrY loss. Of note, only female subjects were used for ChrX mCA analysis and male subjects for mChrY loss analysis, with the “Sex” variable removed from the model. The autosomal and ChrX mCAs were further divided into 3 subtypes: gain, loss, and CN-LOH.
Multivariable regression model for determine the contribution of mCAs to lung cancer risk
To quantify the contribution of mCAs to the risk of lung cancer while adjusting for key confounding variable, we constructed the following model:
| (Model II) |
The variables were defined in the same way as Model I. In the primary analysis, the model was applied to all lung cancer cases and non-cancer controls. In stratified analysis, the model was applied to three major lung cancer histological subtypes, LUAD, LUSC and SCLC. For each subtype, all non-cancer controls were included in the model for estimating coefficient and significance.
Genetic variants associated with mCA phenotypes
Prior to GWAS analysis, genotype imputation was performed for all subjects in our cohort by using 32,470 reference samples from the Haplotype Reference Consortium (HRC)26. Low quality variants and subjects were then filtered out following the method described in Byun et al.27. To minimize the bias from genetic structure, we only include White/Caucasian subjects in the association analyses. Rare variants with minor allele frequency (MAF) ≤ 1% were excluded from the analysis. For each variant, we separately performed the Hardy-Weinberg equilibrium (HWE) test in lung cancer cases and controls. The variants that significantly deviated from HWE (p-value < 5e-8, Chi-square test) in either lung cancer cases or controls were then excluded. We applied a logistic regression model to identify genetic variants associated with each category of mCA events. Present of mCAs (with/without mCA) in each subject was regarded as the dependent variable and genotype of each SNP as independent variables. Sex, age, lung cancer status, smoking and the first three principal components were included in the model as covariates. We calculated the correlation between mCA status and each SNP by the “glm” option of plink 2.028. To improve the statistical power, we required the sample size for each genotype ≥ 3 and the total sample size ≥ 30. The cutoff of p-value was set to 5e-818.
Results
Systematic identification of mCAs from the OncoArray data
The OncoArray dataset from the INTEGRAL-ILCCO cohort contains blood-derived genotyping array data for a total of 33,046 subjects, including 18,221 lung cancer cases and 14,825 cancer-free controls (Table 1)19. We applied the MoChA method4,12 to identify mCAs presenting on autosomal chromosomes in all subjects and ChrX in female subjects. MoChA harnesses chromosome phase information to combine nearby SNPs and can confidently identify mCAs presenting even in a small fraction of blood cells (cell fraction ≥1%)12. For male subjects, MoChA relies on variants in the pseudoautosomal regions (PAR1 and PAR2) of sex chromosomes16. However, the OncoArray genotyping platform has only a limited number of SNPs (n=29) in these regions. Therefore, we restricted ChrX-specific mCA detection to female subjects. Nevertheless, frequent mosaic loss of ChrY (mChrY loss) in male blood cells has been reported16,24,29–31, and found to be associated with an increased risk of lung cancer15,32. As such, we determined the mChrY loss events in our male subjects by using an established method from previous studies22–24.
Table 1. Characteristics of the OncoArray subjects.
For Age, the mean age and the standard deviation (in the parenthesis) are listed. For other variables, the number and percentage of subject are listed. LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; SCLC: small cell lung cancer.
| Phenotype | Variable | Lung Cancer | Control |
|---|---|---|---|
| Age | 64.9 (±10.0) | 62.1 (±10.4) | |
| Sex | Male | 11180 (61%) | 8915 (60%) |
| Female | 7041 (39%) | 5910 (40%) | |
| Race | White | 12896 (90%) | 10733 (86%) |
| Asian | 608 (4.30%) | 819 (6.60%) | |
| Black | 346 (2.40%) | 576 (4.60%) | |
| Other | 436 (3.10%) | 341 (2.70%) | |
| Smoking | Smoker | 15967 (89%) | 9754 (67%) |
| Non-smoker | 1984 (11%) | 4773 (33%) | |
| Cancer Subtype | LUAD | 6852 (38%) | |
| LUSC | 4408 (24%) | ||
| SCLC | 1648 (9%) | ||
| Other | 6960 (38%) | ||
| Total | 18221 | 14825 |
Distribution of mCAs in the human genome
From the OncoArray subjects, we identified a total of 1,808 autosomal mCAs presenting in ≥1% of blood cells. Out of these mCAs, 310 (17.1%), 586 (32.4%), and 763 (42.2%) were confidently categorized as gain, loss, and copy-number neutral loss of heterozygosity (CN-LOH), respectively. The remaining 149 mCAs (8%) were categorized as “undetermined”, because their copy number cannot be explicitly determined. Most of these mCAs present in a small fraction of blood cells with a median cell fraction of 5.6% (Fig. S1). Interestingly, mCAs were not evenly distributed across the genome with Chr11, Chr20, and Chr9 having the largest number of mCAs (Fig. 1A). These 1,808 autosomal mCAs were identified from 1,411 subjects, accounting for about 4.2% of the 33,046 subjects from our cohort. In the 12,951 female subjects, we identified 512 ChrX mCAs involving 397 subjects, which included 181 gain, 143 loss, 123 CN-LOH, and 65 undetermined events (Fig. 1A). Of note, 3.1% of female subjects harbor at least one mCA on ChrX, which is much higher than the detected mCA rate on all individual autosomal chromosomes.
Figure 1. The distribution of mCA events across the human genome.
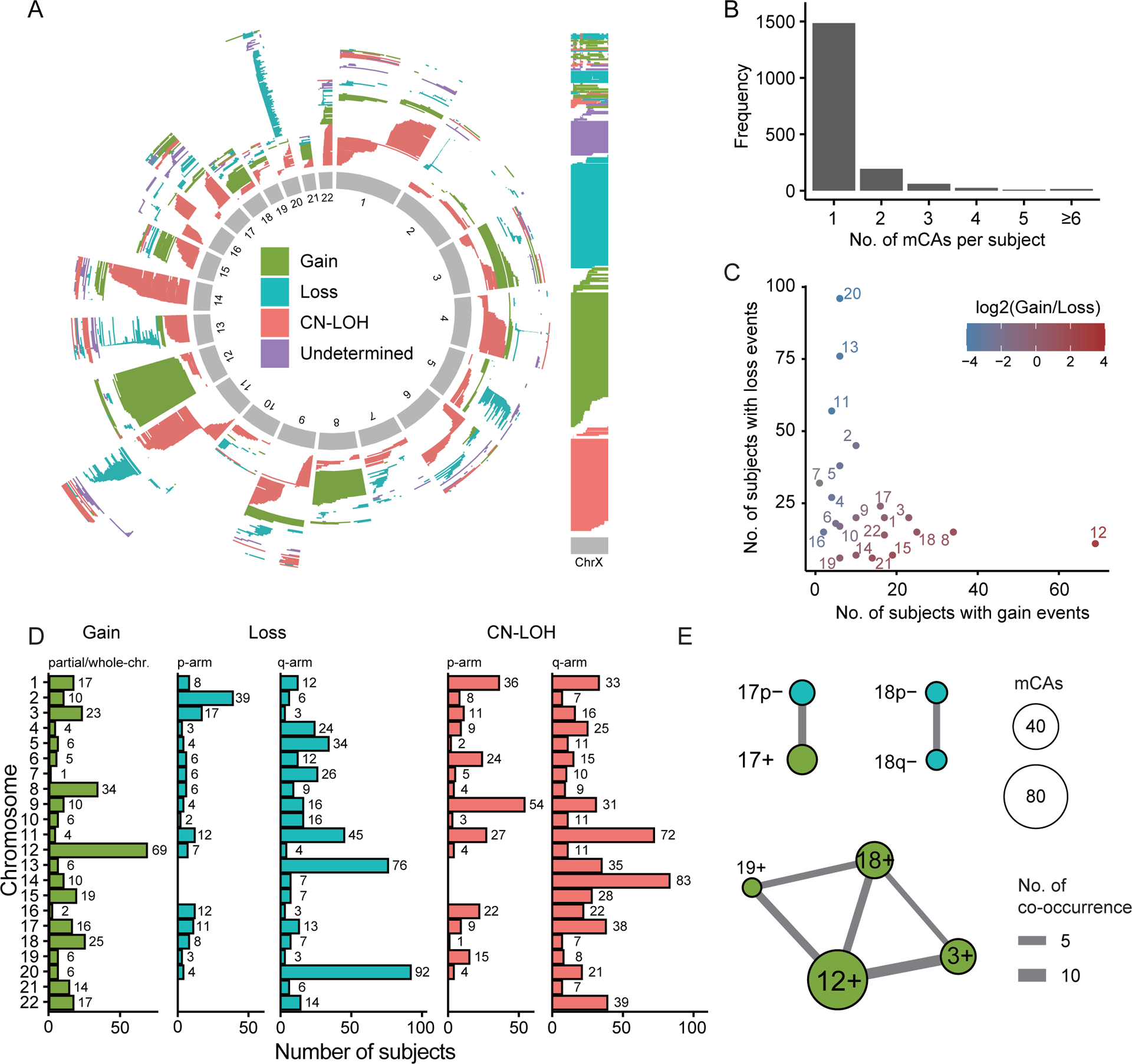
(A) Distribution of mCA events on each autosome and chromosome X. Each mCA event is shown as a line with indicated start and end positions on the corresponding chromosome. (B) Distribution of the number of autosomal and ChrX mCA events detected in each subject. As shown, most subjects have only one mCA event. (C) For each chromosome, the number of subjects with mCA gain (X-axis) and loss (Y-axis) events are counted and shown as a scatterplot. Each dot represents a chromosome. (D) The number of chromosome arm-level mCA events for each chromosome. Mosaic loss and CN-LOH events are further mapped into the long (q-arm) and short arms (p-arm). Most mosaic gain events involve the whole chromosome. (E) The co-occurrence graph for arm-level mCAs. Each edge connects two arm-level mCAs that are significantly co-occurred across subjects (FDR<0.05). Of note, p+/− and q+/− indicate the presence of mCA gain/loss event on the short and long arm, rather than gain/loss of the whole arm.
In the 1,786 mCA-positive subjects, the majority (n=1482, 83%) have only a single autosomal/ChrX mCA event, but a small fraction of subjects presented multiple mCAs (Fig. 1B). Most of the mCAs involved a broad genomic region with a median size of 19.5M bases. We compared the mosaic gain and loss events associated with each autosomal chromosome and found a negative correlation between them (ρ=−0.44, Spearman correlation). This indicated that most chromosomes or arms tended to have either gain or loss events (Fig. 1C). Consistent with the UKBB cohort12 , Chr12 has the largest number of mosaic gain events, while Chr13 and Chr20 were most enriched in mosaic loss events (Fig. 1C).
Most of autosomal mosaic gain events were whole-chromosome events. In contrast, most of the autosomal loss and CN-LOH mCAs involved only certain region of a chromosome. As such, we mapped the loss and CN-LOH mCA events to specific chromosome arms and denoted them as p/q- (loss) or p/q= (CN-LOH). At the arm-level, Chr12q is enriched for mosaic gain events; Chr13q and Chr20q are enriched for mosaic loss events; while Chr11q, Chr14q and Chr9p are enriched for mosaic CN-LOH events (Fig. 1D). At the chromosome/arm level, a small number of subjects (n=155, 9%) harbored multiple mCA events, in which we identified a few mCA pairs with significantly more co-occurrences than what expected by chance (Fig. 1E). Consistent with previous reported results from the UKBB cohort12, we found a cluster of mosaic gain events on Chr12, Chr3, Chr18 and Chr19 tend to present together. In addition, we found another two pairs of co-occurrences i) mosaic loss of Chr17 short arm (17p-) and mosaic gain of Chr17 (17+), and ii) mosaic loss of Chr18 long (18q-) and short (18p-) arms (Fig. 1E). Their occurrence has also been observed in the UKBB cohort, but did not reach the significant threshold12.
The detection rate of mCAs in blood cells is continuously increased with age
Accumulation of mCAs has been found to be a feature of aging4,5. We built a multivariable logistic regression model (Model I, refer to the Methods) to investigate how the presence of mCAs was affected by different subject features including age. Specifically, we investigated autosomal and ChrX mCAs, which were further divided into 3 subtypes (gain, loss, and CN-LOH), as well as mChrY losses. For all mCA types and subtypes, we observed a significant association with age; the probability of a subject being mCA-positive is significantly increased with age (Supplementary Table S1). As shown in Fig. 2A, the fraction of subjects with autosomal mCAs (in both males and females), ChrX mCAs (in females), and mChrY loss (in males) are continuously increasing with age. It is notable that mChrY loss showed a faster increase than the other mCA types: it was detected in less than 5% of males younger than 60 but in ~16% of males older than 80. We then divided all subjects into a young group (<65) and an old group (≥65), and observed a significantly higher fraction of mCA-positive samples in the old group for all mCA types (Fig. 2B). Our models also identified a sex difference: males are more likely to have autosomal mCA gains and losses compared to females (Fig. 2C).
Figure 2. Association of mCAs with age and sex.
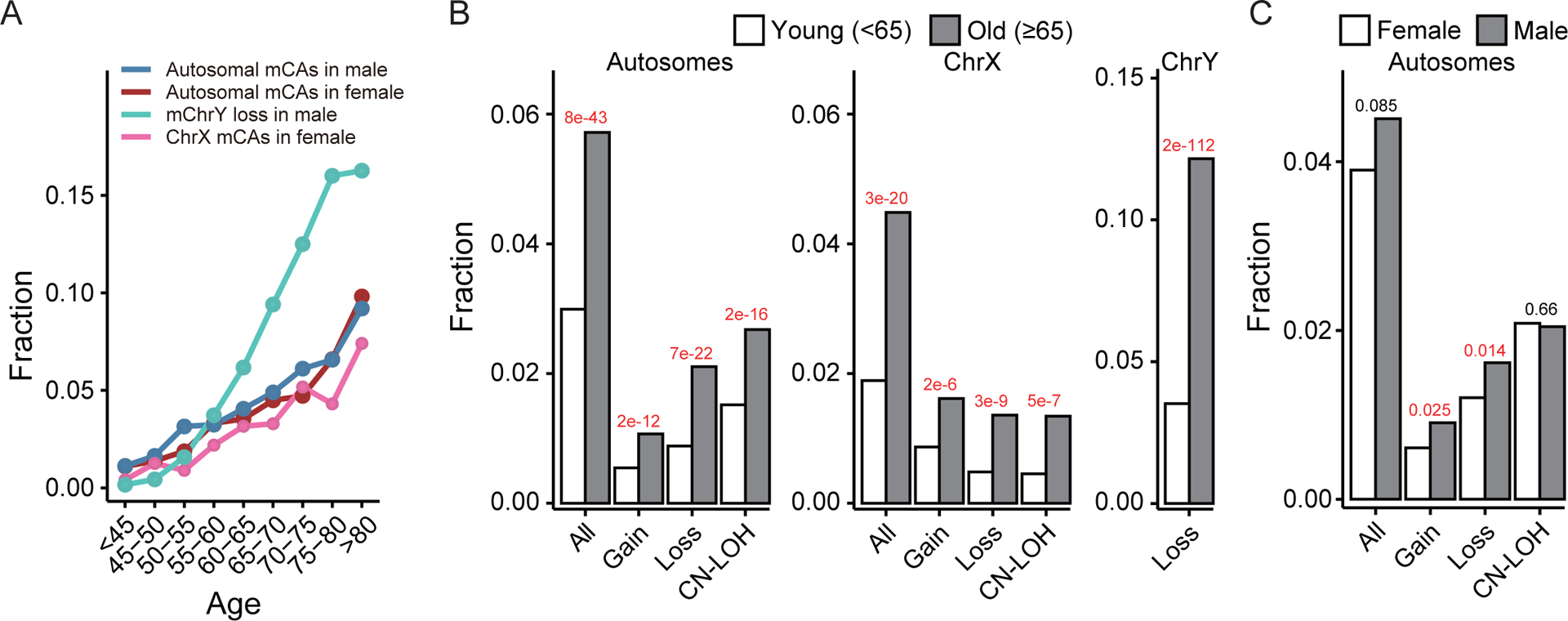
(A) Fraction of subjects with autosomal mCAs, ChrX mCAs or mChrY loss in each age group. The frequency of all types of mCAs increases with age in both males and females. (B) Comparisons of mCA rate between young (age<65) and old (age≥65) subjects. (C) Comparisons of autosomal mCA rate between males and females. Males tend to have a higher rate of autosomal gains and losses than females.
Significant increase of autosomal CN-LOH in patients with lung cancer
Model I indicated that lung cancer cases were more likely to accumulate autosomal mCAs in their blood cells compared to non-cancer controls (Table 2). As shown in Fig. 3A, in both lung cancer cases and controls the fraction of subjects with detected autosome mCAs continuously increase with age; but cases showed an increase starting 5–10 years earlier than controls. This suggests that lung cancer cases accumulate mCAs at earlier ages. In other words, the accumulation of autosomal mCAs with age is associated with increased lung cancer risk.
Table 2. The associations between different types of mCA and lung cancer while adjusting for age, sex, race, and smoking status.
Results are based on the logistic regression Model II. Significant associations were highlighted in bold. ALL, LUAD, LUSC, SCLC indicate all lung cancer, lung adenocarcinoma, squamous cell carcinoma, and small cell lung cancer cases, respectively. mCA freq.: percentage of samples with mCA events in lung cancer cases/controls.
| mCA | All | LUAD | LUSC | SCLC | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr. | Type | mCA freq. | OR (95%CI) | P-value | mCA freq. | OR (95%CI) | P-value | mCA freq. | OR (95%CI) | P-value | mCA freq. | OR (95%CI) | P-value |
| Autosome | All | 5.16%/3.47% | 1.33 (1.17–1.52) | 1e-5 | 5.29%/3.47% | 1.39 (1.18–1.63) | 7.4e-5 | 5.31%/3.47% | 1.35 (1.12–1.62) | 0.0018 | 4.32%/3.47% | 1.14 (0.84–1.54) | >0.1 |
| Gain | 0.89%/0.71% | 1.11 (0.83–1.48) | >0.1 | 0.75%/0.71% | 0.96 (0.65–1.41) | >0.1 | 0.95%/0.71% | 1.17 (0.76–1.78) | >0.1 | 0.41%/0.71% | 0.54 (0.22–1.36) | >0.1 | |
| Loss | 1.75%/1.21% | 1.27 (1.02–1.57) | 0.03 | 1.73%/1.21% | 1.25 (0.95–1.64) | >0.1 | 1.82%/1.21% | 1.25 (0.92–1.71) | >0.1 | 1.41%/1.21% | 1.04 (0.62–1.75) | >0.1 | |
| CN-LOH | 2.56%/1.59% | 1.44 (1.20–1.73) | 1e-4 | 2.71%/1.59% | 1.54 (1.22–1.93) | 2.1e-4 | 2.51%/1.59% | 1.41 (1.08–1.84) | 0.012 | 2.57%/1.59% | 1.48 (1–2.2) | 0.05 | |
| ChrX | All | 3.45%/2.81% | 0.97 (0.76–1.23) | >0.1 | 3.91%/2.81% | 1.18 (0.89–1.57) | >0.1 | 3.82%/2.81% | 0.97 (0.63–1.49) | >0.1 | 2.03%/2.81% | 0.59 (0.28–1.24) | >0.1 |
| Gain | 1.38%/1.14% | 0.99 (0.68–1.45) | >0.1 | 1.53%/1.14% | 1.17 (0.76–1.83) | >0.1 | 1.27%/1.14% | 0.86 (0.42–1.74) | >0.1 | 1.27%/1.14% | 0.88 (0.34–2.27) | >0.1 | |
| Loss | 1.04%/0.78% | 0.92 (0.59–1.43) | >0.1 | 1.27%/0.78% | 1.23 (0.74–2.02) | >0.1 | 1.27%/0.78% | 0.95 (0.45–1.99) | >0.1 | 0.51%/0.78% | 0.47 (0.11–2) | >0.1 | |
| CN-LOH | 1.00%/0.76% | 1.11 (0.70–1.74) | >0.1 | 1.23%/0.76% | 1.47 (0.88–2.46) | >0.1 | 1.15%/0.76% | 1.34 (0.60–2.98) | >0.1 | 0%/0.76% | 1.1e-06 (NA) | >0.1 | |
| ChrY | Loss | 8.95%/6.92% | 1.02 (0.91–1.16) | >0.1 | 9.62%/6.92% | 1.12 (0.95–1.31) | >0.1 | 9.18%/6.92% | 1.01 (0.86–1.19) | >0.1 | 7.53%/6.92% | 0.89 (0.67–1.18) | >0.1 |
Figure 3. The presence of mCAs is associated with increased risk of lung cancer.
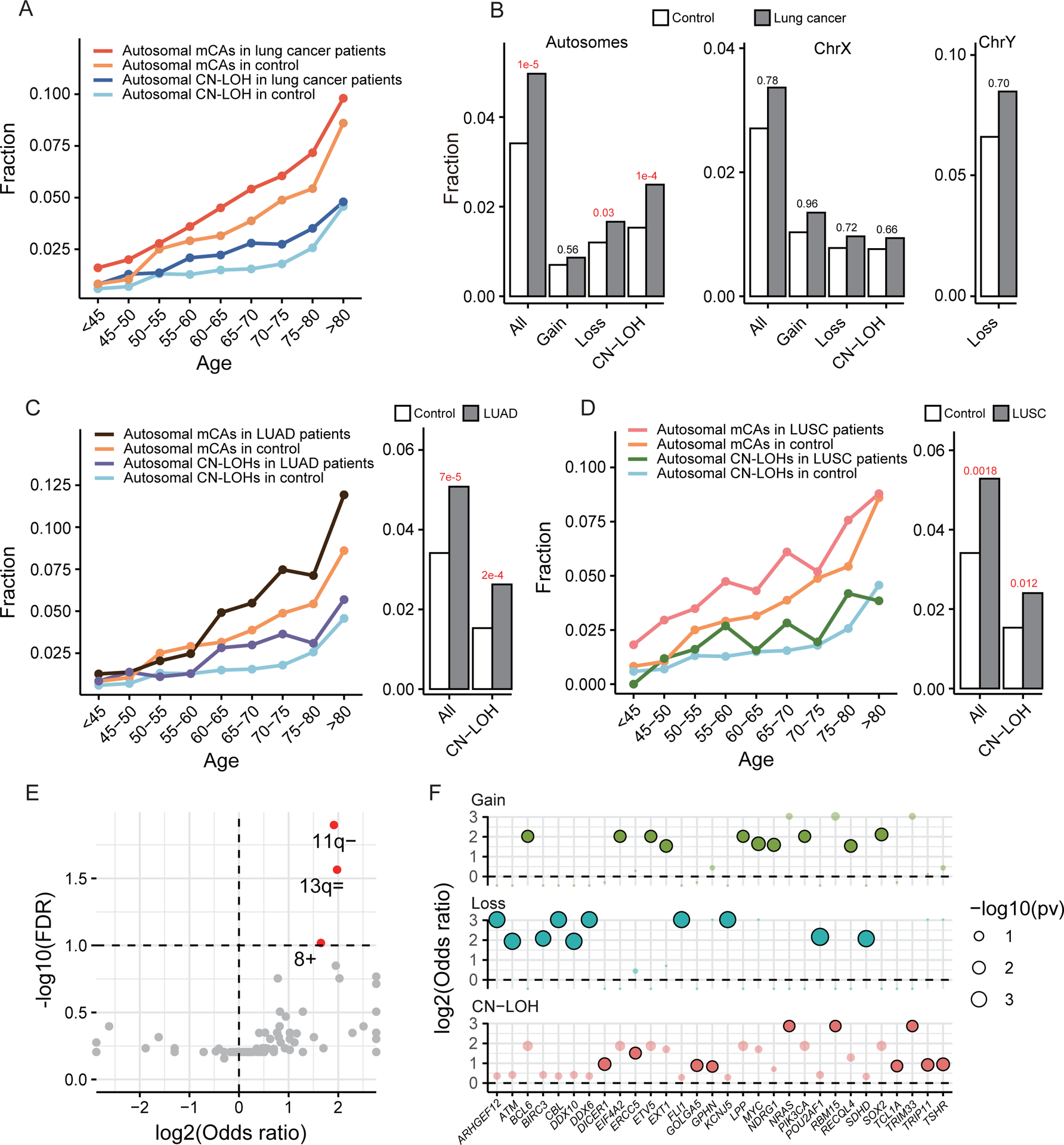
(A) Distribution of overall autosomal mCAs and CN-LOHs across age in lung cancer cases and controls. (B) Lung cancer cases show a significantly higher rate of autosomal mCAs, especially CN-LOHs and losses. (C-D) Distribution of overall autosomal mCAs and CN-LOHs across age in two major lung cancer subtypes, lung adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC). (E) Arm-level autosomal mCAs enriched in lung cancer cases. Significantly enriched mCAs were marked in red. (F) The top 10 most enriched cancer genes in each type of mCAs. Significantly enriched genes were highlighted in a deeper color.
To determine the contribution of mCA events to lung cancer risk while adjusting for major confounding variables (e.g., age, smoking status, etc.), we built another logistic regression model using lung cancer status as the response variable (Model II, see Methods). Our model indicated that the presence of autosomal mCA events increased the risk of lung cancer by 33% (odds ratio OR=1.33, p=1e-5) after adjusting for age, sex, race, and smoking status (Table 2). More specifically, mosaic autosomal loss and CN-LOH is associated with a 27% (p=0.03) and 44% (p=1e-4) increased risk of lung cancer, respectively, while mosaic autosomal gain is not significantly associated (Table 2 and Fig. 3B). In contrast, neither ChrX mCAs nor mChrY losses are significantly associated with lung cancer risk (Table 2 and Supplementary Table S2) after adjusting for increases associated with aging. Furthermore, we examined the three major lung cancer histological types: lung adenocarcinoma (LUAD), squamous cell carcinoma (LUSC), and small cell lung cancer (SCLC). Our results confirmed the association between autosomal mCAs and lung cancer risk in LUAD and LUSC (Table 2) and indicated that the association was mainly driven by mosaic autosomal CN-LOH events. As shown, the presence of autosomal CN-LOH events is associated with 54% increased risks of LUAD and 41% increased risks of LUSC (Fig. 3C–D, Table 2). While we did not identify significant associations between SCLC and mCAs potentially due to smaller sample sizes, the mosaic autosome CN-LOH events also showed weak correlation with SCLC (p=0.05, Table 2).
We compared the occurrences of chromosome/arm level mCAs between the lung cancer and the control groups. Some of the mCAs were more likely to present in the cancer group, including the mosaic loss of Chr11q (11q-), CN-LOH of Chr13q (13q=), and gain of Chr8 (8+) (Fig. 3E). Interestingly, no mCAs were enriched in the controls. Chr11q hosts several tumor suppressor genes including ATM and CBL33,34,35,36 and its frequent deletion has been reported in various cancers37. By enumerating genes in each detected mCA region, we identified genes that were more frequently covered by mCAs in lung cancer cases than in controls (Supplementary Table S3). We found many cancer-related genes curated by COSMIC (the Catalogue Of Somatic Mutations In Cancer)38 are over-represented in lung cancer mCA regions. Among the top ten cancer related genes enriched in the mCA regions in lung cancer versus controls, we found suppressor genes such as ARHGEF1239, DDX1040 and ATM were more likely lost in cancer; while, oncogenes such as BCL641, LPP42 and MYC were more likely to be gained in cancer. The oncogene NRAS was more likely CN-LOH in lung cancer cases (Fig. 3F).
Smokers have a higher rate of ChrX mCAs and mChrY loss
In addition to age and lung cancer status, other clinical factors were also found to be associated with the presence of mCAs in blood cells (Supplementary Table S1). Specifically, we found that smoking females are 42% more likely to harbor ChrX mCAs in their blood cells than non-smoking females (p=0.01), which was mainly driven by mChrX loss (OR=2.26, p=0.005). In males, smokers had a significantly higher fraction of mChrY loss (OR=2.27, p=1e-12) compared with non-smokers (Supplementary Table S1). Similar results were obtained when the analysis was restricted to non-cancer controls (Supplementary Table S4). The age-dependent increase of ChrX mCA and mChrY loss for smokers and non-smokers was demonstrated in Fig. 4A. As shown, the fraction of smokers with ChrX mCA and mChrY loss increased with age at a faster rate than non-smokers, especially for mChrY loss. The higher mCA rate of smokers was also shown in Fig. 4B with a significant difference observed for mChrY and mChrX loss. When smokers were further divided into current- and ex-smokers and compared with never-smokers, similar results were observed: the rate of mChrX and mChrY loss were significantly higher in both current-smokers and ex-smokers than in non-smokers (Supplementary Table S5). Interestingly, while we did not observe any correlation between overall smoking status and autosomal mCAs, current smokers tend to have more autosomal mCAs than ex-smokers (OR=1.16, p=0.043, Supplementary Table S5). A similar trend was also observed in mChrY losses (OR=1.69, p=2.6e-14, Supplementary Table S5), but not found in ChrX mCAs. These results suggested autosomes and ChrY may be more vulnerable to recent smoking harms.
Figure 4. Association of mCAs with smoking status and racial disparity in mCAs.
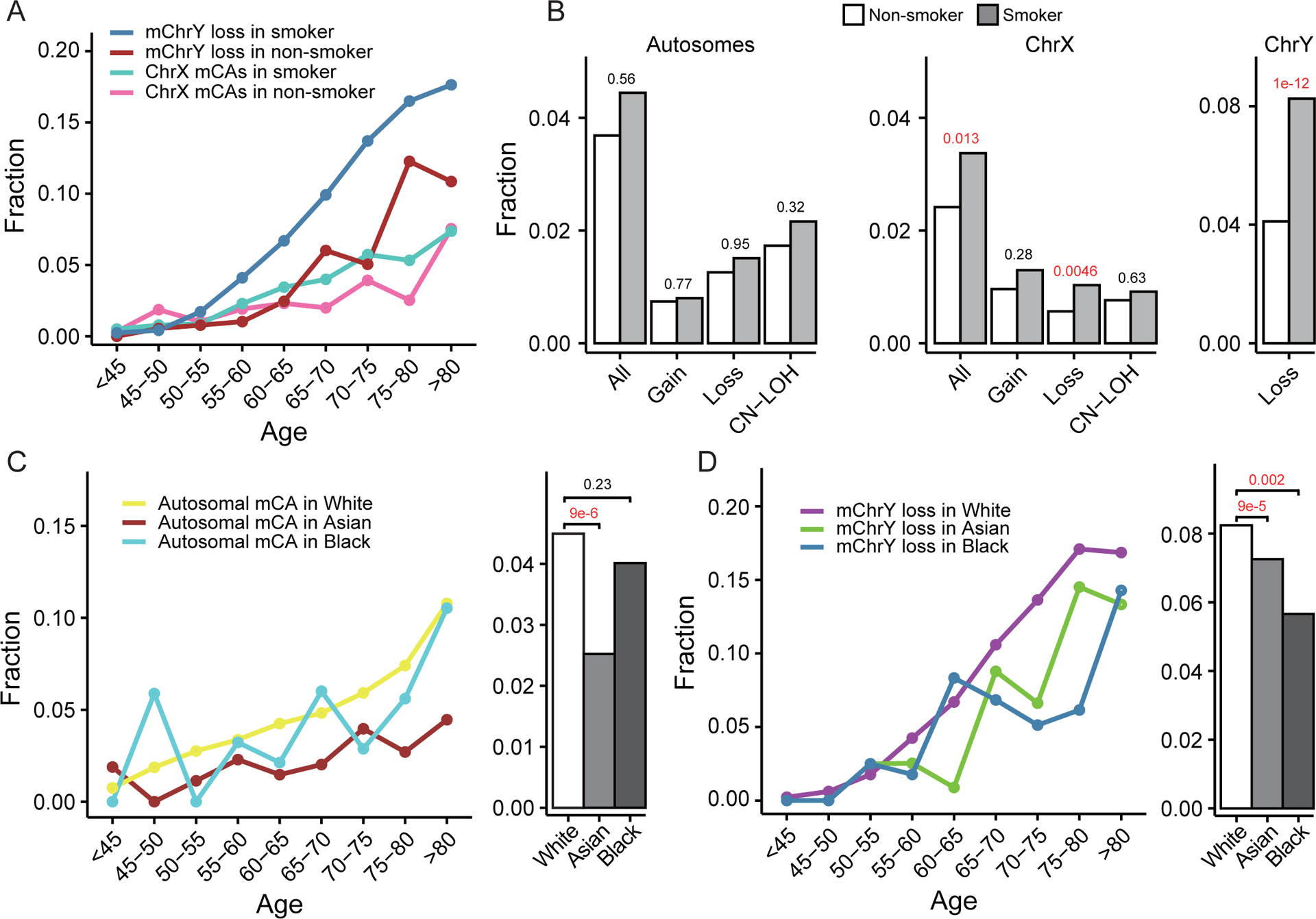
(A) Distribution of mChrX and mChrY losses across age in smokers and non-smokers. (B) Smokers show a significantly higher rate of overall ChrX mCAs (mainly losses) in females and mChrY losses in males. (C-D) Racial difference in the rate of autosomal mCAs and mChrY losses. Compared with Whites, Asians tend to have less autosomal mCAs and mChrY, and Blacks tend to have less mChrY losses.
Racial disparities in the rate of mCAs
We observed racial differences in the rate of mCA after adjusting for age, sex and smoking staus using logistic regression (Model I) (Supplementary Table S1). Specifically, Asians tended to have a lower rate of autosomal mCAs (OR=0.46, p=9.3e-6, Fig. 4C), ChrX mCAs (OR=0.48, p=0.027) and mChrY loss (OR=0.57, p=9e-5, Fig. 4D) compared to Whites. A similar tread was observed when the analysis was restricted to non-cancer controls (Supplementary Table S4). In addition, Blacks have a significantly lower rate of mChrY loss than Whites (OR=0.55, p=0.002, Fig. 4D) but no significant difference in the rate of autosomal (Fig. 4C) or ChrX mCAs (Supplementary Table S1). Of note, the significantly lower rate of mChrY loss in Asians and Blacks compared to Whites is consistent with a previous study based on the UKBB data43.
Genetic variants associated with mCA phenotypes
We performed genome-wide association analysis to identify genetic variants associated with the presence of different types of mCA events. At the significance level of p<5e-8, we did not identify any genetic loci that are associated with the presence of autosome mCA events (Fig. 5A and Fig. S2). However, we did find that a locus on Chr1q23.3 is significantly associated with the presence of ChrX mCAs (Fig. 5B) while a locus on Chr14q32.13 is significantly associated with mChrY loss (Fig. 5C). These results suggest that the occurrence of autosome mCAs might be a complex phenotype with different genetic loci contributing to mCAs of different types or different chromosomes. In contrast, the mCAs on sex chromosomes are relatively simple phenotypes, but ChrX mCAs and mChrY loss seem to be controlled by different genetic loci, as also revealed in previous studies12,22,25. In particular, the Chr1q23.3 locus is located at ~300kb upstream of the PBX1 gene (Fig. 5D), a cancer hallmark gene which is associated with leukemia44, non-small cell lung cancer45 and breast cancer46. In addition, the link between Chr14q32.13 locus and mChrY loss has also been identified from independent datasets, with the most significant variant rs2887399 mapping to the 5’ end of the TCL1A gene (Fig. 5E)22,25. In addition, we divided autosomal and ChrX mCAs into Gains, Losses, and CN-LOHs, and determined genetic variants associated with these more specific mCA phenotypes. We identified several loci associated with mosaic autosomal Gains (Chr3p23), ChrX Gains (Chr3q29), and ChrX CN-LOHs (Chr11p15.5) (Fig. S3A and Supplementary Table S6). All the significant variants of locus Chr3p23 are located in the intronic region of OSBPL10 (Fig. S3B). Circular RNAs derived from OSBPL10 were found correlated with cell proliferation in cervical and gastric cancers47,48. The nearest gene of significant variants at locus Chr3q29 is XXYLT1 (Fig. S3C), which has been found associated with lung cancer by GWAS49. Interestingly, the most significant variant rs76313919 at Chr11p15.5 maps to the 5’ end of MOB2 (Fig. S3D), a gene involved in DNA damage response and cell cycle regulation50.
Figure 5. Genetic variants associated with mCA phenotypes.
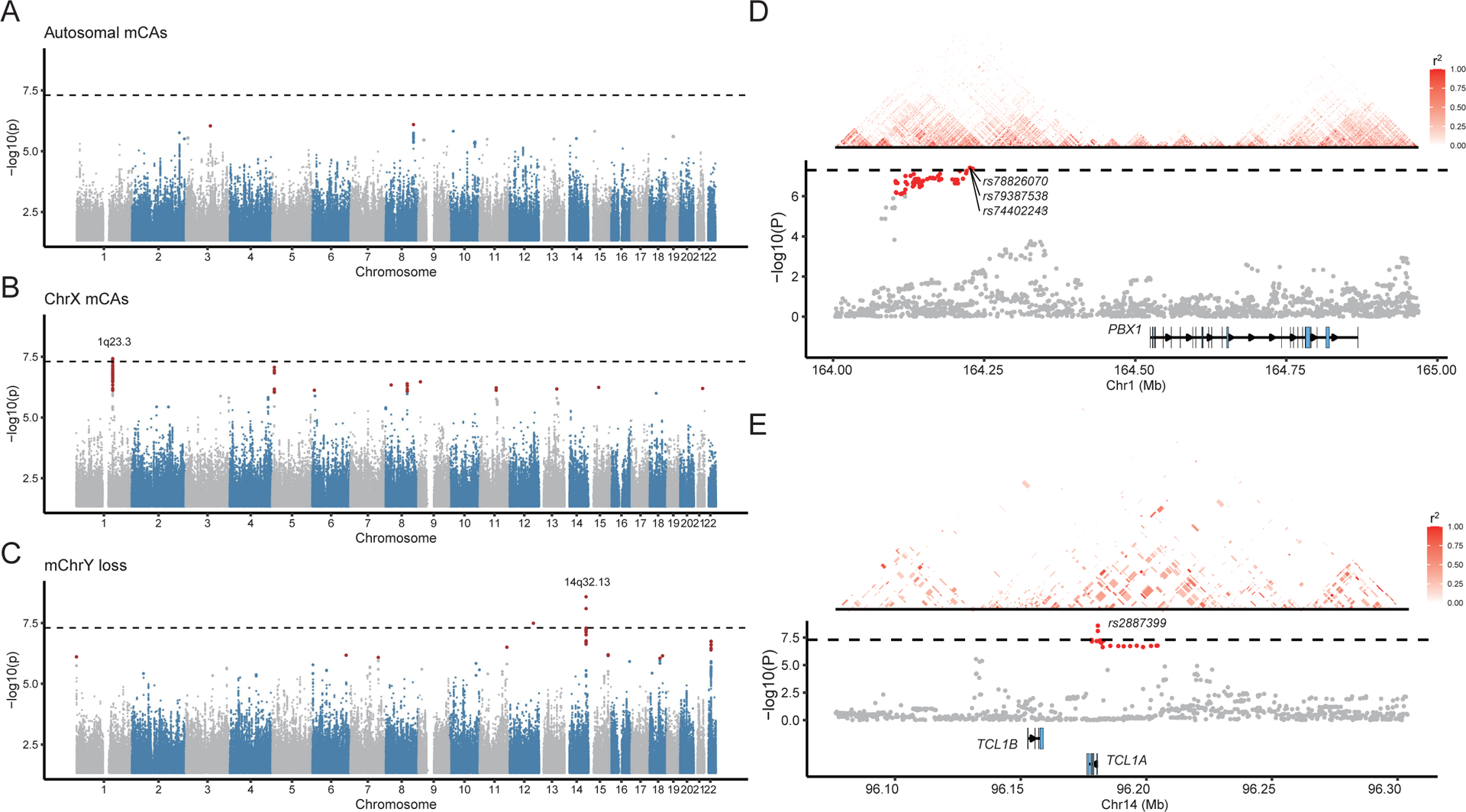
(A-C) Genetic variants associated with autosomal mCAs, ChrX mCAs, and mChrY losses. The dashed line indicates p-value cutoff 5e-8. Genetic variants with p<1e-6 were marked in red. (D-E) The nearest protein coding genes for loci Chr1q23.3 and Chr14q32.13, respectively. Variants with the lowest p-values in each locus were labeled. Heatmaps indicate the pairwise LD r2 score between variants.
Discussion
In this study, we investigated the association between mCAs and lung cancer risk using the OncoArray dataset generated by the INTEGRAL-ILCCO cohort. As the largest lung cancer genetics cohort, this dataset contains 18,221 lung cancer cases and 14,825 non-cancer controls. We identified a comprehensive list of mCAs, including mosaic autosomal/ChrX gain, loss, and CN-LOH as well as mChrY loss. Our analysis indicated that the presence of mCAs was associated with increased lung cancer risk, which was driven by the autosomal CN-LOH events. Stratified analysis confirmed that this association was significant in both lung adenocarcinoma and squamous lung cancer subjects.
Using the same pipeline, we identified more mCAs in ChrX (with a rate of 3.6% in females) than in each individual autosomal chromosomes (with an average rate of 0.25% in all subjects). A similar observation has been reported in previous studies12,51. Moreover, ChrX mCAs are more likely to be a whole-chromosome event compared to autosomal mCAs (67.5% vs. 8.2%), suggesting a potential mechanistic difference between the two types of mCAs. While ChrX is a large chromosome and hosts many housekeeping genes, only one copy is active and transcribed in females. Most genes on the inactivated copy of ChrX are packed into heterochromatin, which is not active for transcription. As such, alterations on the ChrX might be less harmful and more likely to accumulate in blood cells than those on autosomal chromosomes. As a matter of fact, it has been experimentally shown that genomic alterations on the inactive ChrX were more likely to be accumulated in the blood51. In addition, some genomic alterations on ChrX may contribute to the clonal fitness of the host blood cells, which increases their chance to be detected as mCAs12,52,53. In addition to the simple copy number variation events, complex chromosomal rearrangement events such as chromothripsis can be also found in blood cancer such as leukemia54. Due to technical difficulties, we were not able to distinguish chromothripsis from other mCAs as mosaic altercations. Nevertheless, we did observe several samples with multiple mCAs on the same chromosome, which might result from chromothripsis or other type of complex genomic rearrangement.
This study confirmed previous reports on the association between mChrY loss and smoking staus16,23,25. Interestingly, our analysis also revealed a significant association between ChrX mCAs and smoking status. Specifically, smokers had a significantly higher rate of mChrX loss, but such a correlation was not detected for autosomal mCAs. Associations between mChrY loss and lung cancer risk have been investigated in previous studies but reported contradictory results. Qin et al. reported that mChrY loss was associated with reduced lung cancer risk in non-smoking Chinese15. In contrast, using the UKBB dataset Loftfield et al. found that individuals with mChrY loss in a high fraction of blood cells were more likely to have lung cancer32. As shown in Table 2, no significant association between mChrY loss and lung cancer was observed in the OncoArray data in the present study. We also stratified samples based on the blood cell fraction of mCAs using the same threshold setting as Loftfild et al32, but did not identify the association in either group (Supplementary Table S2). Stratified analysis based on smoking status indicated a protective effect of mCAs in current smokers but not in ever- or non-smokers (Supplementary Table S2).
GWAS analyses failed to identify genetic loci associated with overall autosome mCA phenotype but identified different genetic loci linked with ChrX mCAs and mChrY loss. Specifically, we verified in our cohort the previously reported association between Chr14q32.13 and mChrY loss22,25. In another study, Loh et al. performed GWAS to investigate different mCA phenotypes using the UKBB dataset12. Similar to our results, no genetic variants were found to be associated with the overall autosome mCA phenotype, but they identified two genetic loci (SP140L locus on Chr2q37.1 and HLA locus on Chr6p21.33) linked with mChrX losses. While these two loci were not identified in our analysis, we uncovered several genetic loci associated with ChrX mCAs (Chr1q23.3), ChrX Gains (Chr3q29) and ChrX CN-LOHs (Chr11p15.5), respectively. Altogether, our and previous studies may suggest the following insights on genetic regulation of mCAs: i) the autosome and sex chromosome mCAs might be affected by different genetic factors, ii) the overall autosome mCA may be a more complex phenotype compared with ChrX mCA and mChrY loss phenotypes, and iii) the ChrX mCA and mChrY loss phenotypes are linked with different genetic loci.
In summary, we performed a systematic analysis to identify different types of mCAs in white blood cells and investigated their association with lung cancer risk while adjusting for clinical factors. By using the large cohort data from INTEGRAL-ILCCO, our analysis confirmed previously reported associations between mCAs and clinical factors (e.g., age and smoking status). Moreover, we revealed a significant association between mCAs and increased lung cancer risk in both lung adenocarcinoma and squamous lung cancers.
Supplementary Material
Figure S1. Examples of mosaic gain, loss and CN-LOH events. A-C) Distributions of cell fraction in mosaic A) gain, B) loss and C) CN-LOH. All the events have low cell fraction. D-F) Examples of whole-chromosomal mosaic D) gain, E) loss and F) CN-LOH. G-I) Examples of partial-chromosomal mosaic D) gain, E) loss and F) CN-LOH. Dashed lines indicate imputed boundary of events. LRR: log-R ratio; BAF: B-allele frequency; pBAF: phased BAF.
Figure S2. Quantile-quantile plots and inflation factors (λ) of genome-wide association analyses on nine mCA phenotypes.
Figure S3. Manhattan plot of genetic variants associated with mosaic gain, loss and CN-LOH in autosomes and ChrX. A) Dashed line indicated p-value cutoff 5e-8. Genetic variants with p-value<1e-6 were marked in red. B-D) show the nearest protein coding genes of loci Chr3p23, Chr3q29 and Chr11p15.5, respectively. Variants with the lowest p-values in each locus were labeled. Heatmaps indicate the pairwise LD r2 score between variants.
Table S2. The association of mChrY loss with lung cancer when stratified by cell fraction and smoking status. Results are based on the logistic regression model II, adjusting for age, sex, race and smoking status (removed when stratified by smoking status). Significant associations were highlighted in bold. No. of samples: number of lung cancer cases/controls; Freq.: percentage of samples with mChrY loss in lung cancer cases/controls.
Table S1. The associations between different types of mCA and clinical phenotypes in all samples (cases and controls). Results are based on the logistic regression model I. Significant associations were highlighted in bold.
Table S3. Genes that were more frequently covered by mosaic Gain, Loss or CN-LOH events in lung cancer cases than in controls. Genes with p-value<0.05 were included.
Table S4. The associations between different types of mCA and clinical phenotypes in non-cancer controls. Results are based on the logistic regression model I. Significant associations were highlighted in bold.
Table S5. The associations between different types of mCA and smoking status while adjusting for age, sex, race, and lung cancer. Results are based on the logistic regression model I. Significant associations were highlighted in bold. mCA freq: percentage of samples with mCA events in corresponding smoking status.
Table S6. Genetic variants associated with autosomal mCA, ChrX mCA, and mChrY loss phenotypes. Genetic variants with p-value<1e-6 were included.
Acknowledgments
This study is supported by the Cancer Prevention Research Institute of Texas (CPRIT) (RR180061 to Chao Cheng and RR170048 to Christopher Amos), the National Cancer Institute of the National Institute of Health (1R01CA269764 to Chao Cheng), the National Natural Science Foundation of China (81820108028 to Hongbin Shen). CC and CA are CPRIT Scholars in Cancer Research. Vanderbilt University Medical Center’s BioVU is supported by institutional funding and by the Vanderbilt CTSA grant UL1 TR000445 from National Center for Advancing Translational Sciences of the National Institute of Health.
Footnotes
Conflict of Interest
Dr. Aldrich discloses having consultant roles with Guardant Health; having leadership roles in American College of Epidemiology, American Society of Human Genetics, and International Lung Cancer Consortium. Dr. Schabath discloses having consultant roles with Bristol Myers Squibb. The remaining authors declare no conflict of interest.
Disclosures
Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer /World Health Organization.
References
- 1.Jaiswal S & Ebert BL Clonal hematopoiesis in human aging and disease. Science 366, eaan4673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu X, Kamatani Y & Terao C Genetics of autosomal mosaic chromosomal alteration (mCA). J. Hum. Genet 66, 879–885 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Guo X et al. Mosaic loss of human Y chromosome: what, how and why. Hum. Genet 139, 421–446 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Loh P-R, Genovese G & McCarroll SA Monogenic and polygenic inheritance become instruments for clonal selection. Nature 584, 136–141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terao C et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature 584, 130–135 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sano S et al. Hematopoietic loss of Y chromosome leads to cardiac fibrosis and heart failure mortality. Science 377, 292–297 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sherman MA et al. Large mosaic copy number variations confer autism risk. Nat. Neurosci 24, 197–203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zekavat SM et al. Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection. Nat. Med 27, 1012–1024 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laurie CC et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet 44, 642–650 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobs KB et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet 44, 651–658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niroula A et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med 27, 1921–1927 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loh P-R et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559, 350–355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kar SP et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet 54, 1155–1166 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coombs CC et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers Is common and associated with adverse clinical outcomes. Cell Stem Cell 21, 374–382.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin N et al. Association of mosaic loss of chromosome Y with lung cancer risk and prognosis in a Chinese population. J. Thorac. Oncol 14, 37–44 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Thompson DJ et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 575, 652–657 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byun J et al. Trans-ethnic genome-wide meta-analysis of 35,732 cases and 34,424 controls identifies novel genomic cross-ancestry loci contributing to lung cancer susceptibility. medRxiv 2020.10.06.20207753 (2020) doi: 10.1101/2020.10.06.20207753. [DOI] [Google Scholar]
- 18.McKay JD et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet 49, 1126–1132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amos CI et al. The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomarkers Prev 26, 126–135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delaneau O, Zagury J-F, Robinson MR, Marchini JL & Dermitzakis ET Accurate, scalable and integrative haplotype estimation. Nat. Commun 10, 5436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alves JM et al. Reassessing the evolutionary history of the 17q21 inversion polymorphism. Genome Biol. Evol 7, 3239–3248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright DJ et al. Genetic variants associated with mosaic Y chromosome loss highlight cell cycle genes and overlap with cancer susceptibility. Nat. Genet 49, 674–679 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dumanski JP et al. Smoking is associated with mosaic loss of chromosome Y. Science 347, 81–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loftfield E et al. Predictors of mosaic chromosome Y loss and associations with mortality in the UK Biobank. Sci. Rep 8, 12316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou W et al. Mosaic loss of chromosome Y is associated with common variation near TCL1A. Nat. Genet 48, 563–568 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byun J et al. Cross-ancestry genome-wide meta-analysis of 61,047 cases and 947,237 controls identifies new susceptibility loci contributing to lung cancer. Nat. Genet 54, 1167–1177 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forsberg LA et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat. Genet 46, 624–628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirata T et al. Investigation of chromosome Y loss in men with schizophrenia. Neuropsychiatr. Dis. Treat 14, 2115–2122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graham EJ et al. Somatic mosaicism of sex chromosomes in the blood and brain. Brain Res. 1721, 146345 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loftfield E et al. Mosaic Y loss is moderately associated with solid tumor risk. Cancer Res. 79, 461–466 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenman C et al. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loh ML et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114, 1859–1863 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niemeyer CM et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet 42, 794–800 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Westphal CH et al. Genetic interactions between atm and p53 influence cellular proliferation and irradiation-induced cell cycle checkpoints. Cancer Res. 57, 1664–1667 (1997). [PubMed] [Google Scholar]
- 37.Kou F, Wu L, Ren X & Yang L Chromosome abnormalities: new insights into their clinical significance in cancer. Mol. Ther. - Oncolytics 17, 562–570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tate JG et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ong DCT et al. LARG at chromosome 11q23 has functional characteristics of a tumor suppressor in human breast and colorectal cancer. Oncogene 28, 4189–4200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gai M, Bo Q & Qi L Epigenetic down-regulated DDX10 promotes cell proliferation through Akt/NF-κB pathway in ovarian cancer. Biochem. Biophys. Res. Commun 469, 1000–1005 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Phan RT & Dalla-Favera R The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432, 635–639 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Ngan E et al. LPP is a Src substrate required for invadopodia formation and efficient breast cancer lung metastasis. Nat. Commun 8, 15059 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin SH et al. Mosaic chromosome Y loss is associated with alterations in blood cell counts in UK Biobank men. Sci. Rep 10, 2–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimabe M et al. Pbx1 is a downstream target of Evi-1 in hematopoietic stem/progenitors and leukemic cells. Oncogene 28, 4364–4374 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Mo M-L et al. Detection of E2A-PBX1 fusion transcripts in human non-small-cell lung cancer. J. Exp. Clin. Cancer Res 32, 29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ao X et al. PBX1 is a valuable prognostic biomarker for patients with breast cancer. Exp. Ther. Med 20, 385–394 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang S et al. FOXA1-induced circOSBPL10 potentiates cervical cancer cell proliferation and migration through miR-1179/UBE2Q1 axis. Cancer Cell Int. 20, 389 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S et al. Circular RNA profile identifies circOSBPL10 as an oncogenic factor and prognostic marker in gastric cancer. Oncogene 38, 6985–7001 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Yoon K-A et al. A genome-wide association study reveals susceptibility variants for non-small cell lung cancer in the Korean population. Hum. Mol. Genet 19, 4948–4954 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Gomez V et al. Regulation of DNA damage responses and cell cycle progression by hMOB2. Cell. Signal 27, 326–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Machiela MJ et al. Female chromosome X mosaicism is age-related and preferentially affects the inactivated X chromosome. Nat. Commun 7, 11843 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skowyra A, Allan LA, Saurin AT & Clarke PR USP9X limits mitotic checkpoint complex turnover to strengthen the spindle assembly checkpoint and guard against chromosomal instability. Cell Rep. 23, 852–865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunford A et al. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat. Genet 49, 10–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fontana MC et al. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia 32, 1609–1620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Examples of mosaic gain, loss and CN-LOH events. A-C) Distributions of cell fraction in mosaic A) gain, B) loss and C) CN-LOH. All the events have low cell fraction. D-F) Examples of whole-chromosomal mosaic D) gain, E) loss and F) CN-LOH. G-I) Examples of partial-chromosomal mosaic D) gain, E) loss and F) CN-LOH. Dashed lines indicate imputed boundary of events. LRR: log-R ratio; BAF: B-allele frequency; pBAF: phased BAF.
Figure S2. Quantile-quantile plots and inflation factors (λ) of genome-wide association analyses on nine mCA phenotypes.
Figure S3. Manhattan plot of genetic variants associated with mosaic gain, loss and CN-LOH in autosomes and ChrX. A) Dashed line indicated p-value cutoff 5e-8. Genetic variants with p-value<1e-6 were marked in red. B-D) show the nearest protein coding genes of loci Chr3p23, Chr3q29 and Chr11p15.5, respectively. Variants with the lowest p-values in each locus were labeled. Heatmaps indicate the pairwise LD r2 score between variants.
Table S2. The association of mChrY loss with lung cancer when stratified by cell fraction and smoking status. Results are based on the logistic regression model II, adjusting for age, sex, race and smoking status (removed when stratified by smoking status). Significant associations were highlighted in bold. No. of samples: number of lung cancer cases/controls; Freq.: percentage of samples with mChrY loss in lung cancer cases/controls.
Table S1. The associations between different types of mCA and clinical phenotypes in all samples (cases and controls). Results are based on the logistic regression model I. Significant associations were highlighted in bold.
Table S3. Genes that were more frequently covered by mosaic Gain, Loss or CN-LOH events in lung cancer cases than in controls. Genes with p-value<0.05 were included.
Table S4. The associations between different types of mCA and clinical phenotypes in non-cancer controls. Results are based on the logistic regression model I. Significant associations were highlighted in bold.
Table S5. The associations between different types of mCA and smoking status while adjusting for age, sex, race, and lung cancer. Results are based on the logistic regression model I. Significant associations were highlighted in bold. mCA freq: percentage of samples with mCA events in corresponding smoking status.
Table S6. Genetic variants associated with autosomal mCA, ChrX mCA, and mChrY loss phenotypes. Genetic variants with p-value<1e-6 were included.