

Summary
The annotation of microRNAs depends on the availability of transcriptomics data and expert knowledge. This has led to a gap between the availability of novel genomes and high-quality microRNA complements. Using >16,000 microRNAs from the manually curated microRNA gene database MirGeneDB, we generated trained covariance models for all conserved microRNA families. These models are available in our tool MirMachine, which annotates conserved microRNAs within genomes. We successfully applied MirMachine to a range of animal species, including those with large genomes and genome duplications and extinct species, where small RNA sequencing is hard to achieve. We further describe a microRNA score of expected microRNAs that can be used to assess the completeness of genome assemblies. MirMachine closes a long-persisting gap in the microRNA field by facilitating automated genome annotation pipelines and deeper studies into the evolution of genome regulation, even in extinct organisms.
Keywords: microRNAs, genome annotation, machine learning, evolution, genomics
Graphical abstract
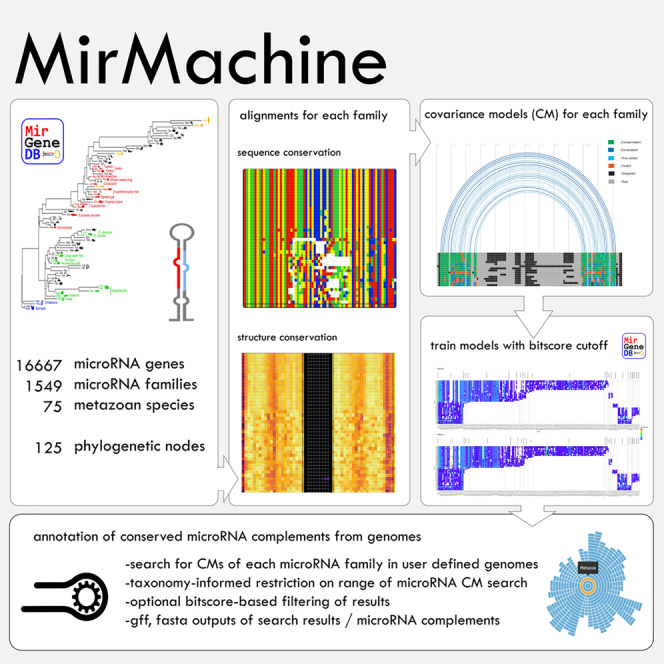
Highlights
-
•
An annotation pipeline using trained covariance models of microRNA families
-
•
Enables massive parallel annotation of microRNA complements of genomes
-
•
MirMachine creates meaningful annotations for very large and extinct genomes
-
•
microRNA score to assess genome assembly completeness
By building and training covariance models from ∼16,000 manually curated microRNA genes, Umu et al. developed the microRNA annotation tool MirMachine. MirMachine can accurately annotate conserved microRNA complements directly from hundreds of genomes. This timely development opens the field of comparative regulatory genomics tapping into the explosion of genome sequencing efforts.
Introduction
MicroRNAs are among the most conserved regulatory elements in animal genomes and have crucial roles in development and disease.1,2 They have been proposed as disease biomarkers,3,4,5 phylogenetic markers for studying animal systematics,6,7 and for understanding the evolution of complexity in metazoans.8,9 Currently, however, the annotation and naming of bona fide microRNA complements requires assembled genome references, small RNA sequencing (small RNA-seq) data from different tissues and developmental stages and substantial hands-on curation of the outputs from microRNA prediction tools.10,11,12 These tools were not designed to handle the amount of sequencing data or genome assembly sizes available today and often have high false-positive rates. Thus, annotating microRNAs is a tedious process that requires years of training as well as extensive computational resources and experience and substantial amounts of time.13 In the case of larger projects that are not focused on microRNAs, those without the appropriate background might annotate them along with other coding and non-coding genes without the required level of attention to detail. Such efforts suffer from biologically meaningless microRNA results,13,14,15,16,17 for instance when using a non-bona fide microRNA from mouse as a template for the search for partially homologous sequences with supposed biomarker potential in human,16,18 when conducting sequence motif predictions on a subset of supposed microRNAs that include other RNA fragments,19 or when interpreting the functions of a small nucleolar RNA (snoRNA) in the light of microRNA biology,20 as well as thousands of spurious microRNA annotations.21,22,23,24 These shortcomings coupled with the availability of high-quality and publicly available microRNA annotations suited for comparative genomics studies have led to the construction of the curated microRNA gene database MirGeneDB.1,25,26
MirGeneDB v.2.1 (2022) now contains microRNA complements for 75 metazoan species spanning all major metazoan phyla representing over ∼850 million years of animal evolution.26 Since each gene and family was manually curated in all species in MirGeneDB, highly accurate alignments that capture a high proportion of the sequence variability for each family are available across animal evolution. Importantly, each microRNA gene and family is associated with a detailed phylogenetic reconstruction of the evolutionary node of origin and estimated age. This dataset, hence, represents a starting point to better understand features of microRNAs27 and to generate better tools for the prediction of microRNAs. Despite MirGeneDB curating a relatively large number of phyla, the number of species currently covered (75 species) is a far cry from the thousands of high-quality animal genomes currently available28 (Figure 1).
Figure 1.

The number of available animal genome assemblies grows exponentially and, with more than 5,250 currently (as of December 31, 2022) available datasets, has dramatically grown
Very few of these species have been annotated for microRNAs or have had small RNA-seq data published; thus, comparatively little progress has been made on the suggested microRNA applications (but see Wheeler et al.,12 Fromm et al.,29 Peterson et al.,30 and Zolotarov et al.31 for examples generated with manual curation). This discrepancy persists because no reliable in silico method currently exists to annotate conserved or species-specific microRNA complements solely from genomic references. Previously, “lift-over” approaches based on whole-genome alignments in model organisms have been used to identify microRNA loci across species,32,33 but it is unclear how accurate these predictions are on the level of the full microRNA complement or how they computationally scale with size or number of aligned genomes in, for instance, mammals. Despite the availability of computational methods for the search of short RNAs such as microRNAs34 and sophisticated machine-learning-based tools for non-coding RNA applications,35 there is currently no approach satisfying the demands of high precision, low false discovery rates, and minimized computational demand in a fully automated and user-friendly pipeline.36 It is a widely acknowledged problem for machine-learning applications in genomics, and in general, that existing tools are based on incomplete models.37,38 This is the case for microRNA families from miRBase.39 Such models, including covariance models (CMs) of individual RNA classes, families, or genes, as used to group all RNA families in the Rfam database,39 are technically quite accurate in detection of many non-coding RNA families.40 However, these probabilistic models that flexibly describe the secondary structure and primary sequence consensus of an RNA sequence family require high-quality alignments from curated RNAs, ideally coupled with detailed evolutionary information to distinguish families and genes over evolutionary time. This information, until recently, did not exist for microRNAs.
Taking advantage of the manually curated and evolutionarily informed microRNA complements of 75 metazoan organisms in MirGeneDB 2.1,26 we built and trained high-quality CMs for 508 conserved microRNA families and integrated them into a fully automated pipeline for microRNA annotation: MirMachine. MirMachine produces highly accurate microRNA annotations in a time-efficient manner from animal genomes of all classes, including those with very large and recently duplicated genomes, as well as from genomes of extinct species. Using 88 eutherian genomes as an example, we show that MirMachine predictions can be summarized in a microRNA score that can be used to assess low contiguity or completeness of genome assemblies. MirMachine is freely available (10.5281/zenodo.7897616, https://github.com/sinanugur/MirMachine) and has also been implemented as a user-friendly web application (www.mirmachine.org).
Results
Accurate CMs of 508 conserved microRNA families
16,670 microRNA precursor sequences from 75 species were downloaded from MirGeneDB, and all variants from the same genes, antisense loci, and species-specific microRNAs (i.e., not conserved in any other species) were removed, resulting in a total of 14,953 genes representing 508 families (Figure 2A).
Figure 2.

Developing MirMachine covariance models (CMs)
(A) The MirMachine workflow uses microRNA family-based precursor sequence alignments and structural information to build CMs that (B) show very good overall prediction performances when models are run on (C) 75 MirGeneDB species using distinct models for protostomes (yellow) and deuterostomes (green) or combined models (not shown). Silhouette in (B) depicts the turtle Chrysemys picta bellii, in (C) human (top) representing deuterostomes and Drosophila representing protostomes.
All microRNA genes for each family were aligned, and CMs were built using all species, which are referred to as combined models. However, given the evolutionary microRNA family definition used by MirGeneDB, microRNA families can include nucleotide differences in mature and seed that are captured and summarized in the models. Thus, to get a finer resolution of our models, we then split deuterostome (N = 42) and protostome (N = 29) representatives and repeated the process to arrive at 388 microRNA family models for deuterostomes and 143 microRNA family models for protostomes. Depending on the age of a given microRNA family, the number of species that shared the family, the number of existing paralogs, and the degree of conservation between orthologs and paralogs, these models contain between very few and many hundreds of individual sequences (see Figure S1 for representative examples).
Using our workflow (see STAR Methods), CMs were subsequently trained on the full MirGeneDB dataset to derive optimal cutoffs for their prediction. We used the models on all MirGeneDB species comparing the predictions with the actual complements. We obtained an overall very high mean prediction accuracy of 0.975 (Matthews correlation coefficient [MCC]) for combined models, 0.975 for deuterostomes, and 0.966 for protostome models (Figures 2B, left, and 2C). Two microRNA families, MIR-430 and MIR-1677 from the deuterostome models, showed substantially lower MCC scores due to a well-known variability within the MIR-430 family41,42,43 and a combination of a low level of complexity and high variation between orthologs in the Diapsida-specific MIR-1677 (Figure S2).
Conversely, we observe high mean species accuracies of 0.91 for combined models, 0.92 for deuterostomes and 0.92 for the protostome models (Figure 2B, right). We found that the turtle (Chrysemys picta bellii) has a low MCC due to the identification of nearly 2,000 likely artifactual hits for MIR-1677.
MirMachine CMs are largely independent of any single species
To identify potential effects from circular logic of predicting microRNAs of a species that were included to build the query models, we retrained all models for deuterostomes without including human and all protostome models without including the polychaete Capitella teleta. Those were chosen because of their relatively complete microRNA complements relative to their respective phylogenetic nodes and given the fact that neither has a sister species in our database (unlike, e.g., Drosophila or Caenorhabditis), which would have heavily biased microRNA recovery. We then used the new deuterostome and protostome CMs to predict microRNA complements in human and C. teleta, respectively. We found that the MCC for Homo sapiens only very slightly decreased in accuracy from 0.97 to 0.96, highlighting the robustness of MirMachine CMs in deuterostomes. In protostomes, the effect on MCC was stronger, as leaving out C. teleta resulted in a decrease from 0.92 to 0.76. Specifically, some families were not found, including the bilaterian families MIR-193, MIR-210, MIR-242, MIR-278, MIR-281, and MIR-375, the protostome families MIR-12 and MIR-1993, and the lophotrochozoan family MIR-1994, which were still predicted but fell below a newly defined threshold. This highlights the higher sequence divergence within protostomes, which is likely due to the age of the group, a lower number of representative clades, a lower number of paralogs and orthologs per family, and a lower number of sequenced species in general. The annelid families MIR-1987, MIR-1995, MIR-2000, MIR-2685, MIR-2687, MIR-2689, and MIR-2705 were not searched because no models were built given the absence of a second annelid species, highlighting the importance of including at least two representative species for each clade in MirGeneDB.26
Performance of MirMachine prediction versus MirGeneDB complement
To get a comprehensive understanding of the performance of MirMachine on the microRNA complements of MirGeneDB species, we looked in more detail at the performance of CMs and their respective cutoffs. This resulted in us examining a selection of major microRNA families (N = 305), including all gene copies (N = 12,430) (Figure 3). When comparing the MirGeneDB complements (Figure 3A) with the predictions from MirMachine (Figure 3B), we observed striking similarities, with overall differences limited to few families (Figure 3C). This result indicates a return of either false positives (231) or false negatives (421), respectively (Table S1). These are of further interest as they either represent missed microRNAs in MirGeneDB or significant deviations from the general CMs and, hence, possibly incorrectly assigned microRNA paralogs in MirGeneDB.
Figure 3.

Detailed comparison of MirMachine predictions on 75 MirGeneDB species and 305 representative microRNA families in the form of banner-plots
Columns are microRNA families sorted by phylogenetic origin and rows are species. Heatmap indicates number of paralogs/orthologs per family.
(A) The currently annotated microRNA complements in MirGeneDB 2.1.26
(B) MirMachine predictions for the same species and families show very high similarity to (A).
(C) Differences between (A) and (B) highlighted as potential false positives (pink) or false negatives (gray).
(D) MirMachine predictions below cutoff based on training of CMs on MirGeneDB show a range of potential random predictions and pseudogenes, highlighting the effect of curation and machine learning on models.
Finally, we found a substantial number of low-scoring MirMachine predictions of microRNA families that did not reach the cutoff based on trained CMs (Figure 3D) and therefore are not considered bona fide microRNAs. However, we found that these also contain pseudogenized microRNA orthologs (or paralogs) exemplified by a hitherto unknown human LET-7 sequence with similarity to functional microRNAs that is not expressed in any MirGeneDB sample (Figure 4). To our knowledge, this is the first report of, and MirMachine the respective tool for, a pseudogene-like process predicted for microRNAs. Studying sequences such as those observed here across organisms should inform upon the evolution of microRNAs, their tolerance for mutations, and the cause and consequences of duplications on microRNAs.30
Figure 4.

The human Chr.17 LET-7 pseudogene-like sequence
(A) Sequence alignment of the currently annotated 12 bona fide LET-7 family members in human and the pseudogene candidate discovered by MirMachine. Non-random sequence similarities, including LIN28 binding sites (pink), are apparent with few noteworthy differences (asterisks) such as in position 2 on the 5′ end (red box indicates mature annotation, position 2 equals seed sequence) or a triplet insertion at the 3′ end (blue box indicates star sequence annotation) are indications for non-functionality.
(B) Structural comparison of a representative bona fide LET-7-member (Hsa-Let-7-P2c1, green triangle) with the pseudogene (yellow triangle) highlights similarities of pseudogene candidate to bona fide microRNA but points out the disruptive nature of nucleotide changes for the structure (asterisks), very likely affecting a potential Drosha processing pathway.44
(C) Sequence conservation of bona fide Hsa-Let-7-P2c1 (top) and the pseudogene (bottom) in 24 primate genome (ENSEMBL v.100) highlights the sequence conservation of bona fide microRNAs from the loop showing some changes, the star (blue) showing few changes, and the mature (red) showing none, while the pseudogene shows many more changes and seems to be enriched in disruptive changes in the mature/seed region.
The microRNA complements of eutherians reveal the microRNA score as a simple feature for genome contiguity
Applying MirMachine to a test case, we downloaded 89 eutherian genomes currently available in Ensembl that are not curated in MirGeneDB and annotated their conserved microRNA complements. Altogether, 38,550 genes in 260 families, in about 4,400 CPU hours, were found that overall show very high concordance between species (Figure 5A). As expected, Catharrini- (pink) and Muridae-specific (light green) microRNAs were only found in the respective representatives, but surprisingly, six species (Figure 5, yellow arrows) showed substantial absences of microRNA families. We therefore wondered whether these absences indicate microRNA losses due to biological simplifications (see Fromm et al.29) or proposed random events45,46 or whether they might be due to technical reasons.7 Given that the outlier species (alpaca, shrew, hedgehog, tree shrew, pika, and sloth) have no particularly reduced morphology, we reasoned that the source might be technical and recovered N50 contiguity values for all genomes. We found that these six genomes had substantially lower N50 values than all other genomes, indicating that microRNAs might be able to predict completeness of genome assemblies (Figure 5B). Therefore, we developed a simple microRNA scoring system defined as the percentage of expected conserved microRNA families found in a genome (in this case including 175 microRNA families found in most eutherians according to MirGeneDB)26 and showed that microRNA scores below 80% correlate with very poor N50 values <10 kb and that N50 values of 100 kb indicate microRNA scores of 90% and higher (Figure 5C, red and blue lines). A noteworthy exception is the microbat Myotis lucifugus with an N50 of 64 kb and a microRNA score of 74%, which might be explainable by previously suggested genome evolution mode through loss.47,48
Figure 5.

MirMachine predicts conserved microRNA complements of 89 eutherian mammals available on Ensembl and not currently represented in MirGeneDB
(A) Banner plot of results for MirMachine predictions on 88 eutherian mammalian species for selected range of major microRNA families and genes showed very strong homogeneity of microRNA complements in general and identified several clear outliers (yellow arrows, including alpaca, shrew, hedgehog, tree shrew, pika, and sloth).
(B) Stacked histogram sorted by N50 values. Outlier species (yellow arrows: same as in A) all have very low N50 values, indicating an artificial absence of these phylogenetically expected microRNA families.
(C) The microRNA score predicts the assembly contingency and is the proportion of phylogenetically expected microRNA families that are found in respective genomes (here, eutherians). MicroRNA scores below 80% (red horizontal line) tend to have low N50 values (red vertical line indicates N50 below 10,000 nucleotides), while scores above 90% indicate an N50 value higher than 10,000 nucleotides. A noteworthy exception is the bat Myotis lucifugus (represented by an outline), which might be explained by a mode of genomic evolution involving gene loss.47,48
MirMachine predicts microRNAs from extinct organisms and very large genomes
High-quality in silico annotation of genomes is particularly important for organisms where no high-quality RNA is likely to ever become available. This is the case for species such as mammoths, which went extinct within the past 40,000 years ago (but see Fromm et al.49). Using available data from extinct and extant elephantids,50,51 we ran MirMachine on 16 afrotherian genomes, including a close extant relative, the hyrax (Procavia capensis), from Ensembl and the more distantly related tenrec (Echinops telfairi) from MirGeneDB, and 14 elephantids including extant savanna elephants (Loxodonta africana), forest elephants (Loxodonta cyclotis), and Asian elephants (Elephas maximus), respectively (Figure 6A, green elephantid silhouettes), but also the extinct American mastodon (Mammuthus americanum), straight-tusked elephants (Palaeoloxodon antiquus), the Columbian mammoth (Mammuthus columbi), and the woolly mammoths (Mammuthus primigenius) (Figure 6A, red elephantid silhouettes). We find a very high degree of similarities between afrotherians and striking congruence between extinct and extant species, which indicates the high accuracy of the MirMachine workflow. More so, we find patterns of microRNA losses that could be phylogenetically informative (Figure 6A, arrows). For instance, we do not find MIR-210 in any of the elephant species, which might be an elephantid-specific loss (Figure 6A, pink arrow); we further find that P. antiquus and L. cyclotis have both lost MIR-1251 (Figure 6A, light blue arrow) and have a shared loss of MIR-675 and MIR-1343 (Figure 6A, purple arrows), both results supporting previously identified sister group relationships.50
Figure 6.

MirMachine enables microRNA complement annotations from extinct and very large genomes
(A) MirMachine predictions from afrotherians show no clear differences between extinct and extant genomes but likely phylogenetically informative losses of microRNA families (colored arrows).
(B) MirMachine predictions in organisms with extensive genome expansions (pink arrows) show no expansion of microRNAs but organisms with known genome duplications (green arrows) do. Several shared microRNA copies in sterlet (A. ruthenus) and paddlefish (P. spatula) (outlined on the figure) support a common genome duplication event in the last common ancestor of Acipenseriformes (yellow arrows).
A challenge for microRNA prediction and annotation of extant species is the occurrence of additional whole-genome duplication events and, although not necessarily connected events, extreme genome expansions. This often leads to computational challenges because identical copies are hard to distinguish from read mappings or because genomes are so large that existing pipelines need extensive computational resources and may face programmatic limits. Therefore, we investigated the performance of MirMachine in vertebrate species with very large genomes and of known multiple rounds of genome duplications.
We included the axolotl (Ambystoma mexicanum) with a genome of 28 Gbp and the African lungfish (Protopterus annectens) with a genome larger than 40 Gbp into our analysis. For the second group, we included the African clawed frosh (Xenopus laevis) with an allotetraploid genome52 and the zebrafish (Danio rerio) from MirGeneDB, the sterlet (Acipenser ruthenus) with a sturgeon specific genome duplication and segmental rediploidization,53 as well as the American paddlefish (Polyodon spathula) with a recently shown genome duplication independent of the sturgeon.54 We combined these species with the gray bichir (Polypterus senegalus), which has a moderately sized (e.g., human-sized) genome and no unique known genome duplication events, along with 13 other MirGeneDB species representing a range of Olfactores, vertebrates, gnathostomes, Osteichthyes, Sarcopterygii, and Tetrapoda representatives (Figure 6B).
We found that MirMachine ran well on all genomes using 32 cores and completed most runs in under 2 h per species, with the lungfish run taking the longest but finishing in 3 h 45 min. As expected, we find that the size of the genomes does not affect the microRNA complements (Figure 6B, pink arrows) but that organisms with additional whole-genome duplications (Figure 6B, green arrows) show clear traces of duplications in that they show multiple additional paralogs in the heatmap (also see Peterson et al.30). A curious observation was that sterlet and paddlefish showed very consistent microRNA copy-number patterns, in particular in the retention of additional MIR-138, MIR-146, MIR-148, MIR-192, and MIR-208 copies (Figure 6B, orange arrows), indicating a likely common origin of genome duplication at the last common ancestor (Acipenseriformes) or else similar retention pressure in the more unlikely case of independent duplication events. Altogether, these results indicate that MirMachine is a suitable tool for the annotation of microRNA complements from both extinct and very large genomes.
MirMachine models outperform existing Rfam models
In the most recent Rfam update (v.14), an expanded assembly of microRNA models based on miRBase was released.39 As mentioned here before and stated elsewhere, a major concern in microRNA research has been the quality of this online repository of published microRNA candidates,1,55,56,57,58,59,60,61,62,63,64,65,66 with estimates that two out of three entries are false positives. These contain numerous tRNA, rRNA, or other fragments but also incorrectly annotated bona fide microRNAs that can influence interpretations of data. In addition to the false positives, numerous miRBase annotations are imprecise and have varying precursor annotation forms (with or without flanking regions of varying lengths). We observe cases where only one arm is annotated, incorrect 3′ ends, and, in a few cases, even 5′ regions are not correctly annotated. These errors can substantially affect target predictions (for details, see Fromm et al.1). Further, miRBase uses an outdated nomenclature that is inconsistent in naming members of the same microRNA family, making the identification of family members cumbersome. This problem has, to a large extent, been transferred to Rfam and their microRNA family models in particular (e.g., MIR-95 family member Hsa-Mir-95-P4 [https://mirgenedb.org/show/hsa/Mir-95-P4] with our own model [https://rnacentral.org/rna/URS0002313758/9606] or MIR-15 member Hsa-Mir-15-P1d [https://mirgenedb.org/show/hsa/Mir-15-P1d] with our own model [https://rnacentral.org/rna/URS000062BB4A/9606]). This has been addressed in the manually curated microRNA gene database MirGeneDB.org1,26 and MirMachine, respectively.
We tested the performance of curated animal origin 523 Rfam microRNA models on the 75 MirGeneDB species and found that 36,931 microRNAs were predicted (compared with 16,913 MirMachine and the 15,846 microRNA annotations in MGDB 2.1). Given that the number of conserved microRNA families is a focus of MirGeneDB and very unlikely to be expanded in the future,13 this much higher number of predictions suggests that Rfam predictions contain thousands of false positives (FPs). We further looked for performance of highly conserved families (see STAR Methods). Rfam models had MCCs of 0.96, 0.94, 0.96, and 0.89 for microRNA families LET-7, MIR-1, MIR-196, and MIR-71, respectively. The same family performances for MirMachine were 0.97, 0.98, 0.97, and 0.97. Thus, as expected, the Rfam model had comparable performance for these correctly assigned and deeply conserved families but performed poorly for incorrectly assigned microRNAs.
MirMachine outperforms whole-genome alignment approaches
We compared the performance of MirMachine with a whole-genome alignment approach as used previously in “lift-over” approaches in, e.g., Drosophila genus.32,33 Using the 470-way mammalian species MULTIZ genome alignment based on the human genome, we tested how accurate these predictions are on the level of the full microRNA complement and how they computationally scale with size or number of aligned genomes. Testing mammals, we found that most human microRNA loci produced alignments in most species but with (1) a substantial number of missing families and genes and (2) a high number of FP calls in these microRNA alignments (Figure S3 and see Umu and Fromm67).
Specifically, on average, for the 90 eutherian genomes we previously analyzed with MirMachine, more than 90 FPs per species were reported from whole-genome analysis (WGA) on average (Figure S4).
To investigate the nature of these likely false calls, we selected 10 microRNA families (see Figure S3, small pink arrows at the bottom) with origins in eutherians and Catharrini that were reported in non-eutherians and outside Catharrini, respectively, and carefully checked all alignments to investigate sequence conservation (Figure S5). We found that alignments reported from outside the expected groups are too distinct from the reference and are likely not microRNAs. In an attempt to verify the effect of nucleotide difference between bona fide genes and the aligned regions bearing substantial changes, we took the example of Catharrini-specific MIR-4677 (Figures S5B and S5C) and, for a subset of representative mammals, made structure predictions. From these predictions, we were able to show that slightly different loci in other primate species created structures less likely to be processed as microRNAs (middle structure), with the non-primate mammals showing almost random structures (yellow bar). These results indicate that WGA-based approaches have pitfalls that the MirMachine pipeline avoids.
MirMachine functions and options
All models (combined, protostome, and deuterostome) were implemented into the standalone MirMachine workflow, which is available under https://github.com/sinanugur/MirMachine, and the web app www.mirmachine.org. MirMachine also contains the curated “node of origin” information from MirGeneDB that can be used to limit the microRNA gene search to phylogenetically expected microRNA families, substantially reducing the search space and shortening the necessary run time. Several other options, such as the search for single families (e.g., LET-7) or families of a particular node (e.g., Bilateria) are also available. In the web app, genome accession numbers can be provided.
Discussion
The existence of thousands of animal genome assemblies is massively mismatched by the availability of annotations of important gene-regulatory elements such as microRNAs. Here, we have presented MirMachine as an important first step to overcome this discrepancy and highlight the need for small RNA-seq data or extensive expert manual curation. This is particularly valuable for organisms, tissues, or developmental time points, where expression datasets will be difficult to acquire and, hence, difficult to obtain microRNA detection based on small RNA-seq.
MirMachine’s ability to accurately predict full conserved microRNA complements from genome assemblies, as exemplified by our analysis of nearly 90 eutherian genomes from Ensembl, not only enables large comparative microRNA studies and automated genome annotation for microRNAs but also shows the potential of microRNAs for the assessment of genome assembly completeness (Figure 5).
Because of their near-hierarchical evolution and rare loss events, microRNAs are already used as taxonomic markers, e.g. miRTrace68 or sRNAbench,69 and we have shown that the microRNA score has a strong potential to outperform existing approaches to assess genome completeness based on protein-coding genes such as BUSCO70 or OMArk.71 This might have wide-reaching consequences for future applications, as a microRNA score could become a standard measure for genome annotation pipelines.
MirMachine currently provides predictions as the community standard file formats GFF or FASTA that are named by family and coordinates. MirMachine predictions are a solid foundation for future small RNA-seq-driven annotation efforts of novel microRNAs and synteny-supported annotation of paralogs and orthologs.
MirMachine is freely available as a standalone tool or web application. It enables even non-microRNA experts to annotate conserved microRNA complements regardless of the availability of small RNA-seq data. Thus, it has a strong potential to close the ever-increasing gap between existing high-quality genomes72,73 and their microRNA annotations. A possible addition of MirMachine into the standard genome annotation pipelines of Refseq and Ensembl is currently being discussed. The availability of thousands of metazoan genomes and their microRNA annotations will pave the way toward the promise of microRNAs and a true postgenomic era.
Limitations of the study
Despite the major leap toward fully automatized microRNA complement annotation, several major challenges remain for the future. (1) Per design, MirMachine can only predict conserved microRNAs based on MirGeneDB-derived CMs. While there are a number of tools for the prediction of novel microRNA candidates from genomes available today, these are not based on a curated reference and, hence, are of limited value (see Stegmayer et al.74 and Saçar Demirci et al.75). (2) Conserved microRNA annotations accurately identify and name all family members of a genome but do not differentiate according to their possible paralog or ortholog identity. Recent approaches that can automatically analyze syntenic information for ortholog and paralog assignment, such as TOGA,76 could be implemented in the future. (3) Limited sampling of several animal phyla and within large groups of invertebrates in MirGeneDB might affect MirMachine performance for these groups as branch- or clade-specific microRNA families, or deviation of a consensus sequence and structure cannot be captured by CMs. (4) We stress that for pre-bilaterian groups of Cnidaria and Porifera, MirMachine currently only provides a small set of microRNA models, as these groups show comparably little conservation of their microRNA complements and aberrant microRNA structures.77,78,79,80 (5) Another important area of possible expansion clearly are plant microRNAs, which currently suffer from multiple non-overlapping available databases and potentially stronger curation problems than observed in animals (see Fromm et al.66 and Taylor et al.81).
We strive to address those issues in the future but would like to stress, in the meantime and in general, that manual curation is a crucial step that should never be disregarded, even though MirMachine heavily reduces the need for extensive and time-consuming efforts.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Bastian Fromm (bastian.fromm@uit.no).
Materials availability
This study did not generate new unique reagents.
Method details
Creation of high-quality CMs
MicroRNA precursor sequences were downloaded from MirGeneDB as FASTA files. We separated them into separate files based on microRNA family and we then aligned each microRNA family using the mafft v7.475 aligner (mafft-xinsi)83 and created multiple sequence alignments (MSAa) of microRNA families. We chose mafft since it considers secondary structure. We filtered out identical or highly similar sequences using the esl-weight v0.48 tool (-f --idf 0.90 --rna) from HMMER package84 to reduce bias due to overrepresentation of highly similar sequences. RNAalifold also expects non-identical sequences. The secondary structures of the MSAs were predicted by RNAalifold v2.4.17 (-r --noPS).85 Lastly, CMs for each microRNA family were generated (cmbuild) and calibrated (cmcalibrate) using Infernal86 and the default setting. Cmcalibrate is a necessary step to calibrate E-value parameters of CMs. We used the same workflow to create deuterostome and protostome specific CMs. In short, the MirGeneDB FASTA sequences were subsetted for deuterostome and protostome species.
Determining accuracy of MirMachine predictions
First, we used the cmsearch function of Infernal to predict microRNA regions. In this study, true positives (TPs) are correctly predicted microRNA families and false positives (FPs) are false predictions. False negatives (FNs) refer to microRNA annotations available in MirGeneDB but not predicted by MirMachine. Using MirGeneDB and MirMachine, we extracted all true positives, false positives, and false negative predictions. We can calculate an approximation to the Matthews correlation coefficient (MCC) by using the geometric mean of sensitivity and precision. This metric is sensitive to both false negatives and false positives.
A standard cmsearch run reports bit score value of each prediction, which is a statistical indicator measuring the quality of an alignment score. We determined an optimal bit score value for each microRNA family to maximize MCC scores. We then filtered any MirMachine hits lower than the optimal cut-off points. We reported MCC values (and other metrics) before and after filtering. See Figure S6 for an overview of MirMachine training workflow.
Benchmarking MirMachine models
We retrained MirMachine CM models by excluding two species: H. sapiens and C. teleta and compared MirMachine performance on these species. Another benchmarking was done using Rfam models. We downloaded all microRNA models (523 in total) from the Rfam database (v 14).39 We predicted microRNA families using Rfam models and compared their model performance with MirMachine on selected families (e.g. LET-7, MIR-1, MIR-71, MIR-196). These families were selected because they are highly conserved and contain low false-positives or false negatives in Rfam. We also reported the total number of microRNA predictions done by both methods.
MirMachine command line tool
The main MirMachine engine was written in Snakemake87 and the command line tool (CLI) wrapper in Python and R. The documentation of the MirMachine CLI tool is available at our GitHub repository. It is also available as a BioConda package91 for easy installation.
MirMachine WebApplication implementation
We implemented the web application using a software stack primarily composed of Django, React and Nginx. The application wraps the MirMachine CLI tool to provide a simpler, interactive interface for users. It is hosted at the Norwegian Research and Education Cloud (NREC), utilizing their sHPC (shared High Performance Computing) resources.88 It is available at https://mirmachine.org.
Available genome assemblies
Lists of reference genomes of invertebrates, vertebrate mammalians and other vertebrates were downloaded from NCBI GenBank on 1/24/2022.92 Analysis of yearly submitted reference genomes was conducted using Python and customized scripts.
Covariance-model-based structure plots
The covariance-model-based plots were generated using the R4RNA-package in R-chie90 run on R Studio version 4.2.0. The arc diagrams along with the grid-based alignment, were created with a multiple sequence alignment of all respective microRNA family members and its corresponding secondary structure as input. Within the R4RNA package, covariation was plotted, and the arc was colored based on the conservation status relative to the multiple sequence alignment provided.
Whole-genome alignment comparisons
Multiple genome alignment of 470 mammals generated with multiz as described in Hecker et al.,93 which was kindly provided by Michael Hiller (available at http://hgdownload.soe.ucsc.edu/goldenPath/hg38/multiz470way/), was intersected with human microRNA annotations from MirGeneDB.
Acknowledgments
We are grateful to Michael Hiller for help with intersecting MirGeneDB with the 470 MULTIZ alignment and to Oleg Simakov for discussions on whole-genome alignments. We thank Wenjing Kang for help with establishing the banner plots and Eirik Høye for the structure heatmap. We are grateful to Love Dalén and David Diez for help with mammoth genomes. We would like to thank Fergal Martin and Leanne Haggerty (Ensembl), Terence Murphy (Refseq), Mark Blaxter (Darwin Tree of Life), and Blake Sweeney (RNAcentral, Rfam) for discussion on the integration of MirMachine into their services and useful comments. We would like to acknowledge Torbjørn Rognes and Eivind Hovig for administrative help, and we are grateful to Norwegian Research and Education Cloud (NREC) for hosting MirMachine.org. B.F. is supported by the Tromsø Research Foundation (Tromsø forskningsstiftelse [TFS]) (20_SG_BF “MIRevolution”) and the UiT Aurora Outstanding program 2020-2022. S.U.U. and T.B.R. were supported by the Research Council of Norway under the Program Human Biobanks and Health Data (grant numbers 229621/H10 and 248791/H10).
Author contributions
S.U.U., T.B.R., K.J.P., and B.F. designed the study. S.U.U. performed CM generation and training. S.U.U. and B.F. wrote and designed the MirMachine workflow, and H.T. implemented the MirMachine webpage with feedback by T.B.R. and K.J.P. T.B. generated queries on available genomes and figures. V.M.P generated CM visualizations. S.U.U. and B.F. performed all analyses, created the main display items, and drafted the manuscript with input from all other authors.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: June 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100348.
Supplemental information
Data and code availability
-
•
All original code has been deposited at GitHub, archived at Zenodo and is publicly available as of the date of publication. GitHub URLs and DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Fromm B., Billipp T., Peck L.E., Johansen M., Tarver J.E., King B.L., Newcomb J.M., Sempere L.F., Flatmark K., Hovig E., Peterson K.J. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet. 2015;49:213–242. doi: 10.1146/annurev-genet-120213-092023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel D.P. Metazoan MicroRNAs. Cell. 2018;173:20–51. doi: 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendell J.T., Olson E.N. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J., Chen J., Sen S. MicroRNA as biomarkers and diagnostics. J. Cell. Physiol. 2016;231:25–30. doi: 10.1002/jcp.25056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Umu S.U., Langseth H., Zuber V., Helland Å., Lyle R., Rounge T.B. Serum RNAs can predict lung cancer up to 10 years prior to diagnosis. Elife. 2022;11:e71035. doi: 10.7554/eLife.71035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarver J.E., Sperling E.A., Nailor A., Heimberg A.M., Robinson J.M., King B.L., Pisani D., Donoghue P.C.J., Peterson K.J. miRNAs: small genes with big potential in metazoan phylogenetics. Mol. Biol. Evol. 2013;30:2369–2382. doi: 10.1093/molbev/mst133. [DOI] [PubMed] [Google Scholar]
- 7.Tarver J.E., Taylor R.S., Puttick M.N., Lloyd G.T., Pett W., Fromm B., Schirrmeister B.E., Pisani D., Peterson K.J., Donoghue P.C.J. Well-annotated microRNAomes do not evidence pervasive miRNA loss. Genome Biol. Evol. 2018;10:1457–1470. doi: 10.1093/gbe/evy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heimberg A.M., Sempere L.F., Moy V.N., Donoghue P.C.J., Peterson K.J. MicroRNAs and the advent of vertebrate morphological complexity. Proc. Natl. Acad. Sci. USA. 2008;105:2946–2950. doi: 10.1073/pnas.0712259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson K.J., Dietrich M.R., McPeek M.A. MicroRNAs and metazoan macroevolution: insights into canalization, complexity, and the Cambrian explosion. Bioessays. 2009;31:736–747. doi: 10.1002/bies.200900033. [DOI] [PubMed] [Google Scholar]
- 10.Friedländer M.R., Chen W., Adamidi C., Maaskola J., Einspanier R., Knespel S., Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 11.Hackenberg M., Sturm M., Langenberger D., Falcón-Pérez J.M., Aransay A.M. miRanalyzer: a microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res. 2009;37:W68–W76. doi: 10.1093/nar/gkp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheeler B.M., Heimberg A.M., Moy V.N., Sperling E.A., Holstein T.W., Heber S., Peterson K.J. The deep evolution of metazoan microRNAs. Evol. Dev. 2009;11:50–68. doi: 10.1111/j.1525-142X.2008.00302.x. [DOI] [PubMed] [Google Scholar]
- 13.Fromm B., Zhong X., Tarbier M., Friedländer M.R., Hackenberg M. The limits of human microRNA annotation have been met. RNA. 2022;28:781–785. doi: 10.1261/rna.079098.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witwer K.W., Halushka M.K. Toward the promise of microRNAs - enhancing reproducibility and rigor in microRNA research. RNA Biol. 2016;13:1103–1116. doi: 10.1080/15476286.2016.1236172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fromm B., Tosar J.P., Yu L., Halushka M.K., Witwer K.W. Human and Cow Have Identical miR-21-5p and miR-30a-5p Sequences, Which Are Likely Unsuited to Study Dietary Uptake from Cow Milk. The Journal of Nutrition. 2018;148:1506–1507. doi: 10.1093/jn/nxy144. [DOI] [PubMed] [Google Scholar]
- 16.Blanco-Domínguez R., Sánchez-Díaz R., Martín P. A novel circulating MicroRNA for the detection of acute myocarditis. N. Engl. J. Med. 2022;387:1240–1241. doi: 10.1056/NEJMc2115639. [DOI] [PubMed] [Google Scholar]
- 17.Fromm B., Kang W., Rovira C., Cayota A., Witwer K., Friedländer M.R., Tosar J.P. Plant microRNAs in human sera are likely contaminants. J. Nutr. Biochem. 2019;65:139–140. doi: 10.1016/j.jnutbio.2018.07.019. [DOI] [PubMed] [Google Scholar]
- 18.Blanco-Domínguez R., Sánchez-Díaz R., de la Fuente H., Jiménez-Borreguero L.J., Matesanz-Marín A., Relaño M., Jiménez-Alejandre R., Linillos-Pradillo B., Tsilingiri K., Martín-Mariscal M.L., et al. A novel circulating noncoding small RNA for the detection of acute myocarditis. N. Engl. J. Med. 2021;384:2014–2027. doi: 10.1056/NEJMoa2003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Martin R., Wang G., Brandão B.B., Zanotto T.M., Shah S., Kumar Patel S., Schilling B., Kahn C.R. MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature. 2022;601:446–451. doi: 10.1038/s41586-021-04234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chinnappa K., Cárdenas A., Prieto-Colomina A., Villalba A., Márquez-Galera Á., Soler R., Nomura Y., Llorens E., Tomasello U., López-Atalaya J.P., Borrell V. Secondary loss of miR-3607 reduced cortical progenitor amplification during rodent evolution. Sci. Adv. 2022;8:eabj4010. doi: 10.1126/sciadv.abj4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jha A., Panzade G., Pandey R., Shankar R. A legion of potential regulatory sRNAs exists beyond the typical microRNAs microcosm. Nucleic Acids Res. 2015;43:8713–8724. doi: 10.1093/nar/gkv871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Londin E., Loher P., Telonis A.G., Quann K., Clark P., Jing Y., Hatzimichael E., Kirino Y., Honda S., Lally M., et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc. Natl. Acad. Sci. USA. 2015;112:E1106–E1115. doi: 10.1073/pnas.1420955112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alles J., Fehlmann T., Fischer U., Backes C., Galata V., Minet M., Hart M., Abu-Halima M., Grässer F.A., Lenhof H.-P., et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019;47:3353–3364. doi: 10.1093/nar/gkz097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenzi L., Chiu H.-S., Cobos F.A., Gross S., Volders P.-J., Cannoodt R., Nuytens J., Vanderheyden K., Anckaert J., Lefever S., et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat. Biotechnol. 2021:1–13. doi: 10.1038/s41587-021-00936-1. [DOI] [PubMed] [Google Scholar]
- 25.Fromm B., Domanska D., Høye E., Ovchinnikov V., Kang W., Aparicio-Puerta E., Johansen M., Flatmark K., Mathelier A., Hovig E., et al. MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res. 2020;48:D132–D141. doi: 10.1093/nar/gkz885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fromm B., Høye E., Domanska D., Zhong X., Aparicio-Puerta E., Ovchinnikov V., Umu S.U., Chabot P.J., Kang W., Aslanzadeh M., et al. MirGeneDB 2.1: toward a complete sampling of all major animal phyla. Nucleic Acids Res. 2022;50:D204–D210. doi: 10.1093/nar/gkab1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang W., Fromm B., Houben A.J., Høye E., Bezdan D., Arnan C., Thrane K., Asp M., Johnson R., Biryukova I., Friedländer M.R. MapToCleave: high-throughput profiling of microRNA biogenesis in living cells. Cell Rep. 2021;37:110015. doi: 10.1016/j.celrep.2021.110015. [DOI] [PubMed] [Google Scholar]
- 28.Hotaling S., Kelley J.L., Frandsen P.B. Toward a genome sequence for every animal: where are we now? Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2109019118. e2109019118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fromm B., Worren M.M., Hahn C., Hovig E., Bachmann L. Substantial loss of conserved and gain of novel MicroRNA families in flatworms. Mol. Biol. Evol. 2013;30:2619–2628. doi: 10.1093/molbev/mst155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peterson K.J., Beavan A., Chabot P.J., McPeek M.A., Pisani D., Fromm B., Simakov O. microRNAs as indicators into the causes and consequences of whole genome duplication events. Mol. Biol. Evol. 2022;39:msab344. doi: 10.1093/molbev/msab344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zolotarov G., Fromm B., Legnini I., Ayoub S., Polese G., Maselli V., Chabot P.J., Vinther J., Styfhals R., Seuntjens E., et al. MicroRNAs are deeply linked to the emergence of the complex octopus brain. Sci. Adv. 2022;8 doi: 10.1126/sciadv.add9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohammed J., Flynt A.S., Siepel A., Lai E.C. The impact of age, biogenesis, and genomic clustering on Drosophila microRNA evolution. RNA. 2013;19:1295–1308. doi: 10.1261/rna.039248.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohammed J., Flynt A.S., Panzarino A.M., Mondal M.M.H., DeCruz M., Siepel A., Lai E.C. Deep experimental profiling of microRNA diversity, deployment, and evolution across the Drosophila genus. Genome Res. 2018;28:52–65. doi: 10.1101/gr.226068.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Velandia-Huerto C.A., Fallmann J., Stadler P.F. miRNAture—computational detection of microRNA candidates. Genes. 2021;12:348. doi: 10.3390/genes12030348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amin N., McGrath A., Chen Y.-P.P. Evaluation of deep learning in non-coding RNA classification. Nat. Mach. Intell. 2019;1:246–256. [Google Scholar]
- 36.Yazbeck A.M., Tout K.R., Stadler P.F., Hertel J. Towards a consistent, quantitative evaluation of MicroRNA evolution. J. Integr. Bioinform. 2017;14:20160013. doi: 10.1515/jib-2016-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whalen S., Schreiber J., Noble W.S., Pollard K.S. Navigating the pitfalls of applying machine learning in genomics. Nat. Rev. Genet. 2022;23:169–181. doi: 10.1038/s41576-021-00434-9. [DOI] [PubMed] [Google Scholar]
- 38.Saçar M.D., Hamzeiy H., Allmer J. Can MiRBase provide positive data for machine learning for the detection of MiRNA hairpins? J. Integr. Bioinform. 2013;10:215. doi: 10.2390/biecoll-jib-2013-215. [DOI] [PubMed] [Google Scholar]
- 39.Kalvari I., Nawrocki E.P., Ontiveros-Palacios N., Argasinska J., Lamkiewicz K., Marz M., Griffiths-Jones S., Toffano-Nioche C., Gautheret D., Weinberg Z., et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021;49:D192–D200. doi: 10.1093/nar/gkaa1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eddy S.R., Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giraldez A.J., Mishima Y., Rihel J., Grocock R.J., Van Dongen S., Inoue K., Enright A.J., Schier A.F. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–79. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- 42.Choi W.-Y., Giraldez A.J., Schier A.F. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- 43.Bazzini A.A., Lee M.T., Giraldez A.J. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336:233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bofill-De Ros X., Kasprzak W.K., Bhandari Y., Fan L., Cavanaugh Q., Jiang M., Dai L., Yang A., Shapiro B.A., Wang Y.-X., Gu S. Structural Differences between Pri-miRNA Paralogs Promote Alternative Drosha Cleavage and Expand Target Repertoires. Cell Rep. 2019;26:447–459.e4. doi: 10.1016/j.celrep.2018.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomson R.C., Plachetzki D.C., Mahler D.L., Moore B.R. A critical appraisal of the use of microRNA data in phylogenetics. Proc. Natl. Acad. Sci. USA. 2014;111:E3659–E3668. doi: 10.1073/pnas.1407207111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunn C.W. Reconsidering the phylogenetic utility of miRNA in animals. Proc. Natl. Acad. Sci. USA. 2014;111:12576–12577. doi: 10.1073/pnas.1413545111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang Z., Jebb D., Teeling E.C. Blood miRNomes and transcriptomes reveal novel longevity mechanisms in the long-lived bat, Myotis myotis. BMC Genom. 2016;17:906. doi: 10.1186/s12864-016-3227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jebb D., Huang Z., Pippel M., Hughes G.M., Lavrichenko K., Devanna P., Winkler S., Jermiin L.S., Skirmuntt E.C., Katzourakis A., et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature. 2020;583:578–584. doi: 10.1038/s41586-020-2486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fromm B., Tarbier M., Smith O., Dalén L., Gilbert M.T.P., Friedländer M.R. Ancient microRNA profiles of a 14,300-year-old canid are taxonomically informative and give glimpses into gene regulation from the Pleistocene. RNA. 2019;27:324–334. doi: 10.1261/rna.078410.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palkopoulou E., Lipson M., Mallick S., Nielsen S., Rohland N., Baleka S., Karpinski E., Ivancevic A.M., To T.-H., Kortschak R.D., et al. A comprehensive genomic history of extinct and living elephants. Proc. Natl. Acad. Sci. USA. 2018;115:E2566–E2574. doi: 10.1073/pnas.1720554115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palkopoulou E., Mallick S., Skoglund P., Enk J., Rohland N., Li H., Omrak A., Vartanyan S., Poinar H., Götherström A., et al. Complete genomes reveal signatures of demographic and genetic declines in the woolly mammoth. Curr. Biol. 2015;25:1395–1400. doi: 10.1016/j.cub.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Session A.M., Uno Y., Kwon T., Chapman J.A., Toyoda A., Takahashi S., Fukui A., Hikosaka A., Suzuki A., Kondo M., et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538:336–343. doi: 10.1038/nature19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Du K., Stöck M., Kneitz S., Klopp C., Woltering J.M., Adolfi M.C., Feron R., Prokopov D., Makunin A., Kichigin I., et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 2020;4:841–852. doi: 10.1038/s41559-020-1166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng P., Huang Y., Lv Y., Du H., Ruan Z., Li C., Ye H., Zhang H., Wu J., Wang C., et al. The American paddlefish genome provides novel insights into chromosomal evolution and bone mineralization in early vertebrates. Mol. Biol. Evol. 2021;38:1595–1607. doi: 10.1093/molbev/msaa326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Castellano L., Stebbing J. Deep sequencing of small RNAs identifies canonical and non-canonical miRNA and endogenous siRNAs in mammalian somatic tissues. Nucleic Acids Res. 2013;41:3339–3351. doi: 10.1093/nar/gks1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiang H.R., Schoenfeld L.W., Ruby J.G., Auyeung V.C., Spies N., Baek D., Johnston W.K., Russ C., Luo S., Babiarz J.E., et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones-Rhoades M.W. Conservation and divergence in plant microRNAs. Plant Mol. Biol. 2012;80:3–16. doi: 10.1007/s11103-011-9829-2. [DOI] [PubMed] [Google Scholar]
- 58.Ludwig N., Becker M., Schumann T., Speer T., Fehlmann T., Keller A., Meese E. Bias in recent miRBase annotations potentially associated with RNA quality issues. Sci. Rep. 2017;7:5162. doi: 10.1038/s41598-017-05070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langenberger D., Bartschat S., Hertel J., Hoffmann S., Tafer H., Stadler P.F. Advances in Bioinformatics and Computational Biology. Springer Berlin Heidelberg); 2011. MicroRNA or not MicroRNA? pp. 1–9. [Google Scholar]
- 60.Meng Y., Shao C., Wang H., Chen M. Are all the miRBase-registered microRNAs true? A structure- and expression-based re-examination in plants. RNA Biol. 2012;9:249–253. doi: 10.4161/rna.19230. [DOI] [PubMed] [Google Scholar]
- 61.Tarver J.E., Donoghue P.C.J., Peterson K.J. Do miRNAs have a deep evolutionary history? Bioessays. 2012;34:857–866. doi: 10.1002/bies.201200055. [DOI] [PubMed] [Google Scholar]
- 62.Taylor R.S., Tarver J.E., Hiscock S.J., Donoghue P.C.J. Evolutionary history of plant microRNAs. Trends Plant Sci. 2014;19:175–182. doi: 10.1016/j.tplants.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 63.Wang X., Liu X.S. Systematic curation of miRBase annotation using integrated small RNA high-throughput sequencing data for C. elegans and Drosophila. Front. Genet. 2011;2:25. doi: 10.3389/fgene.2011.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Axtell M.J., Meyers B.C. Revisiting criteria for plant MicroRNA annotation in the era of big data. Plant Cell. 2018;30:272–284. doi: 10.1105/tpc.17.00851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo Z., Kuang Z., Wang Y., Zhao Y., Tao Y., Cheng C., Yang J., Lu X., Hao C., Wang T., et al. PmiREN: a comprehensive encyclopedia of plant miRNAs. Nucleic Acids Res. 2020;48:D1114–D1121. doi: 10.1093/nar/gkz894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fromm B., Keller A., Yang X., Friedlander M.R., Peterson K.J., Griffiths-Jones S. Quo vadis microRNAs? Trends Genet. 2020;36:461–463. doi: 10.1016/j.tig.2020.03.007. [DOI] [PubMed] [Google Scholar]
- 67.Umu S.U., Fromm B. 2023. MirMachine, a command line tool to detect microRNA homologs in genome sequences. [Google Scholar]
- 68.Kang W., Eldfjell Y., Fromm B., Estivill X., Biryukova I., Friedländer M.R. miRTrace reveals the organismal origins of microRNA sequencing data. Genome Biol. 2018;19:213. doi: 10.1186/s13059-018-1588-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aparicio-Puerta E., Gómez-Martín C., Giannoukakos S., Medina J.M., Scheepbouwer C., García-Moreno A., Carmona-Saez P., Fromm B., Pegtel M., Keller A., et al. sRNAbench and sRNAtoolbox 2022 update: accurate miRNA and sncRNA profiling for model and non-model organisms. Nucleic Acids Res. 2022;50:W710–W717. doi: 10.1093/nar/gkac363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seppey M., Manni M., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019;1962:227–245. doi: 10.1007/978-1-4939-9173-0_14. [DOI] [PubMed] [Google Scholar]
- 71.Nevers Y., Rossier V., Train C.M., Altenhoff A., Dessimoz C., Glover N. Multifaceted quality assessment of gene repertoire annotation with OMArk. bioRxiv. 2022 doi: 10.1101/2022.11.25.517970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lewin H.A., Robinson G.E., Kress W.J., Baker W.J., Coddington J., Crandall K.A., Durbin R., Edwards S.V., Forest F., Gilbert M.T.P., et al. Earth BioGenome project: sequencing life for the future of life. Proc. Natl. Acad. Sci. USA. 2018;115:4325–4333. doi: 10.1073/pnas.1720115115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Formenti G., Theissinger K., Fernandes C., Bista I., Bombarely A., Bleidorn C., Ciofi C., Crottini A., Godoy J.A., Höglund J., et al. The era of reference genomes in conservation genomics. Trends Ecol. Evol. 2022;37:197–202. doi: 10.1016/j.tree.2021.11.008. [DOI] [PubMed] [Google Scholar]
- 74.Stegmayer G., Di Persia L.E., Rubiolo M., Gerard M., Pividori M., Yones C., Bugnon L.A., Rodriguez T., Raad J., Milone D.H. Predicting novel microRNA: a comprehensive comparison of machine learning approaches. Brief. Bioinform. 2019;20:1607–1620. doi: 10.1093/bib/bby037. [DOI] [PubMed] [Google Scholar]
- 75.Saçar Demirci M.D., Baumbach J., Allmer J. On the performance of pre-microRNA detection algorithms. Nat. Commun. 2017;8:330. doi: 10.1038/s41467-017-00403-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kirilenko B.M., Munegowda C., Osipova E., Jebb D., Sharma V., Blumer M., Morales A.E., Ahmed A.-W., Kontopoulos D.-G., Hilgers L., et al. Integrating gene annotation with orthology inference at scale. Science. 2023;380:eabn3107. doi: 10.1126/science.abn3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Praher D., Zimmermann B., Dnyansagar R., Miller D.J., Moya A., Modepalli V., Fridrich A., Sher D., Friis-Møller L., Sundberg P., et al. Conservation and turnover of miRNAs and their highly complementary targets in early branching animals. Proc. Biol. Sci. 2021;288:20203169. doi: 10.1098/rspb.2020.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nong W., Cao J., Li Y., Qu Z., Sun J., Swale T., Yip H.Y., Qian P.Y., Qiu J.-W., Kwan H.S., et al. Jellyfish genomes reveal distinct homeobox gene clusters and conservation of small RNA processing. Nat. Commun. 2020;11:3051–3111. doi: 10.1038/s41467-020-16801-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grimson A., Srivastava M., Fahey B., Woodcroft B.J., Chiang H.R., King N., Degnan B.M., Rokhsar D.S., Bartel D.P. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature. 2008;455:1193–1197. doi: 10.1038/nature07415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liew Y.J., Ryu T., Aranda M., Ravasi T. miRNA Repertoires of demosponges stylissa carteri and xestospongia testudinaria. PLoS One. 2016;11:e0149080. doi: 10.1371/journal.pone.0149080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taylor R.S., Tarver J.E., Foroozani A., Donoghue P.C.J. MicroRNA annotation of plant genomes- Do it right or not at all. Bioessays. 2017;39:1600113. doi: 10.1002/bies.201600113. [DOI] [PubMed] [Google Scholar]
- 82.Fromm B., Hoye E., Domanska D., Zhong X., Aparicio-Puerta E., Ovchinnikov V., Umu S.U., Chabot P.J., Kang W., Aslanzadeh M., et al. MirGeneDB 2.1: toward a complete sampling of all major animal phyla. Nucleic Acids Res. 2022;50:D204–D210. doi: 10.1093/nar/gkab1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Katoh K., Rozewicki J., Yamada K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019;20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wheeler T.J., Eddy S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013;29:2487–2489. doi: 10.1093/bioinformatics/btt403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nawrocki E.P., Eddy S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mölder F., Jablonski K.P., Letcher B., Hall M.B., Tomkins-Tinch C.H., Sochat V., Forster J., Lee S., Twardziok S.O., Kanitz A., et al. Sustainable data analysis with Snakemake. F1000Res. 2021;10:33. doi: 10.12688/f1000research.29032.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Trondsen H.T. 2022. A Web Application for MirMachine, a MicroRNA Annotation Tool. [Google Scholar]
- 89.R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2022. https://www.R-project.org/ [Google Scholar]
- 90.Lai D., Proctor J.R., Zhu J.Y.A., Meyer I.M. R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 2012;40:e95. doi: 10.1093/nar/gks241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grüning B., Dale R., Sjödin A., Chapman B.A., Rowe J., Tomkins-Tinch C.H., Valieris R., Köster J., Bioconda Team Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods. 2018;15:475–476. doi: 10.1038/s41592-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. Nucleic Acids Res. 2016;44:D67–D72. doi: 10.1093/nar/gkv1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hecker N., Hiller M. A genome alignment of 120 mammals highlights ultraconserved element variability and placenta-associated enhancers. GigaScience. 2020;9:giz159. doi: 10.1093/gigascience/giz159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All original code has been deposited at GitHub, archived at Zenodo and is publicly available as of the date of publication. GitHub URLs and DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.