

Abstract
Purpose
This study aimed to characterize the gut microbiota in obese adolescents from Shenzhen (China), and evaluate influence of gender on BMI-related differences in the gut microbiome.
Methods
Evaluation of physical examination, blood pressure measurement, serological assay and body composition were conducted in 205 adolescent subjects at Shenzhen. Fecal microbiome composition was profiled via high-throughput sequencing of the V3–V4 regions of the 16S rRNA gene. A Random Forest (RF) classifier model was built to distinguish the BMI categories based on the gut bacterial composition.
Results
Fifty-six taxa consisting mainly of Firmicutes were identified that having significant associations with BMI; 2 OTUs belonging to Ruminococcaceae and 1 belonging to Lachnospiraceae had relatively strong positive correlations with body fate rate, waistline and most of serum biochemical properties. Based on the 56 BMI-associated OTUs, the RF model showed a robust classification accuracy (AUC 0.96) for predicting the obese phenotype. Gender-specific differences in the gut microbiome composition was obtained, and a lower relative abundance of Odoribacter genus was particularly found in obese boys. Functional analysis revealed a deficiency in bacterial gene contents related to peroxisome and PPAR signaling pathway in the obese subjects for both genders.
Conclusions
This study reveals unique features of gut microbiome in terms of microbial composition and metabolic functions in obese adolescents, and provides a baseline for reference and comparison studies.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13755-023-00236-9.
Keywords: Adolescent obesity, Gut microbiota, 16S rRNA, Peroxisome signaling
Introduction
Obesity during childhood and adolescence is associated with cardiovascular disease and metabolic syndrome later in life, and has become a significant public health concern worldwide [1]. Human gut microbiome, highly involved in the host calorie harvest and energy homeostasis [2], has increasingly been recognized as an important factor in the development of obesity [3, 4]. For example, alterations in the gut microbiota, such as antibiotic exposure, are linked with a variety of metabolic diseases including obesity, type 1 and type 2 diabetes [5]. Both animal and human studies showed divergences in the gut microbiome composition resulted in different post-dieting weight gain [6, 7]. Gut microbes also showed to stimulate chronic low-grade inflammation by producing lipopolysaccharides and contributed to obesity and insulin resistance [8]. On the other hand, modulation of gut microbiota by fiber supplementation or fecal microbiota transplantation suppressed inflammation and improved insulin sensitivity, demonstrating gut microbiota played key role in etiology of the metabolic syndrome [9].
Investigations of gut microbiota markers of obesity were performed in different previous studies, but no consistent pattern has not been obtained. For example, both Bäckhed et al. [10] and Turnbaugh et al. [11] identified lower ratio of Bacteroidetes to Firmicutes in people with obesity than in those with normal body weight, but this characteristic was not supported by other studies [12, 13]. Gut microbiome composition appears to be affected by host genetics, age, gender, geographical locations, and other environmental factors [14]. By characterizing the gut microbiota of 7009 individuals from 14 districts within the Guangdong province of China, host location is shown to be the strongest explanatory factor of the microbiota variations [15]. Upon analyzing gut microbiota sequencing data collected from 516 Chinese adults, our previous study showed that BMI differences in the gut microbiome composition are gender specific [16]. Since age has also been reported as a confounding factor influencing the gut microbiome, it is uncertain whether the gender-dependent differences identified in adult also present in adolescents. Therefore, factors, including region, age and gender, seem to be import considerations for investigating gut microbiome, and identification of gut microbial signatures responsible for obesity has great potential in prevention and treatment of obesity related diseases.
Taking geographical location and age into account as confounding factors, this study characterizes the composition and functions of the gut microbiota in obese adolescents in Shenzhen city, and evaluated the influence of gender on the BMI-related differences in the gut microbiome.
Materials and methods
Study cohort
The study started after we received approval from the Institutional Review Board of the Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences (SIAT-IRB-131115-H0032), and was registered at ClinicalTrials.gov (number NCT02539836). All individuals participating in the study received informed consent from their guardians. Two hundred and five Chinese adolescents were recruited at Shenzhen Children’s Hospital, between September and December 2015 (Fig. 1). Written informed consent was obtained from the guardians of the subjects before participation. The subjects with any of the criteria below were excluded from the present study:
Type 1 or type 2 diabetes.
Antibiotic use in the 2 months prior to sampling.
Long-term use of medication (e.g. antihypertensive drugs).
Diarrhea: had three or more loose or liquid stools a day in the past week.
Chronic constipation: have infrequent (no bowel movement in 3 days) or difficult evacuation of the bowels.
According to the WHO BMI-for-age percentile growth charts (growth reference 5–19 years; https://www.who.int/growthref/who2007_bmi_for_age/en/), we classified each participant into normal-weight (N), overweight (OW) or obese (OB). General characteristics of the subjects, body composition, and serological test results for each BMI group are shown in Table 1.
Fig. 1.
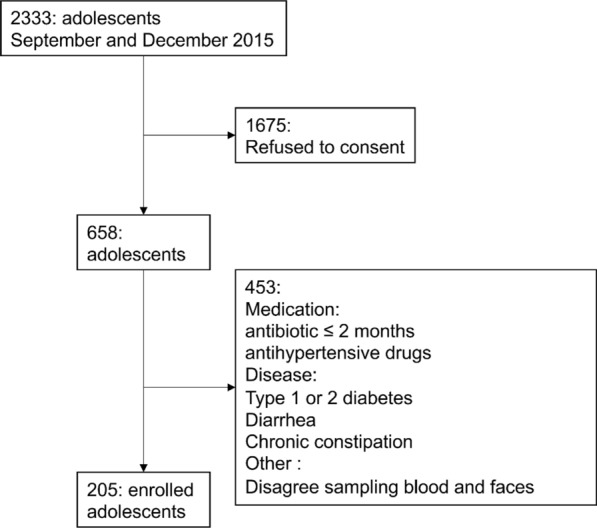
The diagram of subjects’ recruitment
Table 1.
Characteristics of the studied adolescent population
| Characteristics | Normal weight | Overweight | Obesity | p-Value | CRV | |
|---|---|---|---|---|---|---|
| Number (F/M) | 65 (30/35) | 53(19/34) | 87 (19/68) | – | ||
| Age (year) | 13.91 ± 1.61 | 13.26 ± 1.50 | 12.90 ± 1.41 | 2.75E−04 | – | |
| Age Range (year) | 11–16 | 11–16 | 11–16 | – | ||
| Anthropometric data | ||||||
| Weight (kg) | 50.60 ± 9.67 | 62.88 ± 8.85 | 74.62 ± 11.50 | 2.99E−31 | – | |
| Height (cm) | 163.03 ± 9.31 | 162.81 ± 8.94 | 163.92 ± 8.79 | 7.34E−01 | – | |
| BMI | 18.87 ± 1.96 | 23.60 ± 1.42 | 27.65 ± 2.53 | 1.78E−63 | – | |
| Waistline (cm) | 66.91 ± 6.84 | 78.19 ± 6.81 | 88.36 ± 7.16 | 9.40E−45 | – | |
| Blood pressure | ||||||
| Systolic blood pressure (mm Hg) | 114.28 ± 14.48 | 121.87 ± 11.93 | 125.76 ± 12.48 | 1.20E−06 | 90–120 | |
| Diastolic blood pressure (mm Hg) | 70.78 ± 8.17 | 72.79 ± 10.24 | 76.59 ± 8.74 | 3.84E−04 | 60–80 | |
| Body composition | ||||||
| Body fat (kg) | 7.67 ± 3.63 | 14.91 ± 3.70 | 21.81 ± 5.37 | 8.69E−40 | ||
| Body fat rate | 0.15 ± 0.06 | 0.23 ± 0.05 | 0.29 ± 0.41 | 6.94E−34 | ||
| Body muscle (kg) | 40.67 ± 7.50 | 45.08 ± 6.77 | 49.55 ± 7.83 | 2.95E−09 | ||
| Body muscle rate | 0.8 ± 0.06 | 0.71 ± 0.04 | 0.66 ± 0.04 | 8.19E−34 | ||
| Body mineral salt (kg) | 3.04 ± 0.60 | 3.66 ± 0.50 | 4.32 ± 0.71 | 8.49E−22 | ||
| Body mineral salt rate | 0.06 ± 0.01 | 0.06 ± 0.00 | 0.06 ± 0.00 | 1.62E−03 | ||
| Body water (L) | 31.86 ± 5.92 | 35.10 ± 5.21 | 38.78 ± 6.16 | 4.09E−09 | ||
| Body water rate | 0.62 ± 0.06 | 0.55 ± 0.03 | 0.51 ± 0.03 | 1.69E−27 | ||
| Body protein (kg) | 9.28 ± 1.73 | 9.98 ± 1.56 | 10.76 ± 1.74 | 1.62E−05 | ||
| Body protein rate | 0.18 ± 0.02 | 0.16 ± 0.01 | 0.14 ± 0.01 | 2.01E−31 | ||
| Serological investigation | ||||||
| ALB (g/L) | 47.62 ± 4.33 | 46.86 ± 3.99 | 47.69 ± 4.46 | 5.06E−01 | 35–55 | |
| ALT (U/L) | 13.55 ± 8.60 | 17.06 ± 12.79 | 27.66 ± 22.58 | 1.26E−06 | 0–40 | |
| ApoA1 (g/L) | 1.38 ± 0.17 | 1.31 ± 0.17 | 1.33 ± 0.17 | 9.49E−02 | 1.05–1.75 | |
| ApoB (g/L) | 0.60 ± 0.15 | 0.64 ± 0.16 | 0.70 ± 0.16 | 4.18E−04 | 0.60–1.40 | |
| BUN (mmol/L) | 3.85 ± 1.07 | 3.95 ± 0.86 | 4.08 ± 0.83 | 3.03E−01 | 2.5–6.0 | |
| CER/CP (mg/L) | 24.35 ± 4.64 | 28.07 ± 5.60 | 29.06 ± 6.39 | 3.39E−06 | 21–65 | |
| CK (U/L) | 123.83 ± 59.79 | 133.43 ± 68.69 | 179.41 ± 184.45 | 2.00E−02 | 24–229 | |
| CK-MB (U/L) | 1.52 ± 0.67 | 1.60 ± 0.80 | 1.90 ± 1.12 | 3.35E−02 | 0–6.8 | |
| C-peptide (ng/mL) | 1.88 ± 1.13 | 2.48 ± 1.80 | 2.82 ± 1.93 | 3.47E−03 | 0.78–5.19 | |
| Cr (μmol/L) | 55.93 ± 13.71 | 54.46 ± 11.00 | 54.08 ± 11.47 | 6.32E−01 | 21–65 | |
| CRP (mg/L) | 0.46 ± 0.55 | 1.39 ± 1.91 | 2.23 ± 3.16 | 2.94E−05 | 0–10 | |
| Hb (g/L) | 134.95 ± 18.06 | 137.30 ± 11.03 | 136.94 ± 14.66 | 6.35E−01 | 110–160 | |
| HDL (mmol/L) | 1.22 ± 0.23 | 1.12 ± 0.23 | 1.09 ± 0.21 | 1.18E−03 | > 1.04 | |
| INS (pmol/mL) | 10.35 ± 8.00 | 16.28 ± 15.91 | 22.04 ± 17.61 | 1.56E−05 | 1.9–23 | |
| LDL (mmol/L) | 2.34 ± 0.56 | 2.55 ± 0.68 | 2.69 ± 0.55 | 2.15E−03 | < 3.37 | |
| PLC (109/L) | 291.12 ± 64.73 | 312.93 ± 57.37 | 329.56 ± 63.03 | 1.04E−03 | 100–300 | |
| PUFA (μg/mL) | 0.45 ± 0.20 | 0.50 ± 0.21 | 0.57 ± 0.27 | 6.06E−03 | 0.1–0.9 | |
| RBC (1012/L) | 4.91 ± 0.53 | 4.90 ± 0.36 | 5.02 ± 0.42 | 1.94E−01 | 3.5–5.5 | |
| TBIL (μmol/L) | 9.85 ± 4.52 | 8.72 ± 4.92 | 7.23 ± 3.11 | 5.64E−04 | 0.9–17.1 | |
| TC (mmol/L) | 3.93 ± 0.83 | 4.07 ± 0.85 | 4.37 ± 0.77 | 3.47E−03 | < 5.18 | |
| TG (mmol/L) | 1.19 ± 0.60 | 1.59 ± 1.17 | 1.76 ± 0.94 | 1.02E−03 | < 1.7 | |
| TP (g/L) | 76.90 ± 7.98 | 75.70 ± 6.00 | 76.41 ± 5.50 | 6.11E−01 | 46–80 | |
| UA (μmol/L) | 337.71 ± 87.60 | 398.79 ± 120.42 | 425.78 ± 102.84 | 2.55E−06 | 90–420 | |
| WBC (109/L) | 7.61 ± 1.76 | 8.60 ± 2.05 | 8.70 ± 1.67 | 6.79E−04 | 5–12 | |
Values represent mean ± SD
CRV clinical reference value, ALB albumin, ALT alanine transaminase, Apo-A1 apolipoprotein A1, Apo-B apolipoprotein B, BUN blood urea nitrogen, CER/CP ceruloplasmin, CK creatine kinase, CK-MB creatine kinase isoenzyme-MB, Cr creatinine, CRP C-reactive protein, Hb hemoglobin, HDL high-density lipoprotein, INS insulin, LDL low-density lipoprotein, PLC platelet, PUFA polyunsaturated fatty acid, RBC red blood cell, TBIL total bilirubin, TC total cholesterol, TG triglyceride, TP total protein, UA blood uric acid, WBC white blood cell
Sample collection, DNA extraction, and 16S rRNA gene sequencing
Body compositions of the participants were measured by using GAIA KIKO body composition analyzer (Jawon Medical, Korea). Venous blood samples were collected at Shenzhen Children’s Hospital during annual physical examination organized by the Bureau of Education of Shenzhen Municipality. Serological assays were performed to determine white blood cell (WBC), red blood cell (RBC), Hemoglobin (Hb), proinsulin-like component (PLC), alanine aminotransferase (ALT), total bilirubin (TBIL), total protein (TP), albumin (ALB), Cerutoplasmin (CER/CP), ceruloplasmin (CRP), blood urea nitrogen (BUN), creatinine (Cr), urine acid (UA), creatine kinase (CK), creatine kinase- myoglobin (CK-MB), insulin (INS), C-peptide, triglycerides (TG), total cholesterol (TC), high-density lipoprotein (HDL), low-density lipoprotein (LDL), apolipoprotein A1 (Apo-A1), apolipoprotein B (Apo-B), and polyunsaturated fatty acid (PUFA).
Each participant donated about 10 g of fresh stool and placed inside a sterile plastic bag with ice pack. These samples were homogenized to a uniform consistency, and DNA was routinely extracted from 0.3 g fecal material using TIANamp Stool DNA Kit (TIANGEN BIOTECH, cat. #DP328-02, Beijing, China), following the manufacturer’s instructions. DNA was quantified using a dsDNA HS assay on a Qubit 3.0 (Thermo Fisher Scientific, USA). Universal primers (forward: 5′-AYTGGGYDTAAAGNG-3′, reverse: 5′-TACNVGGGTATCTAATCC-3′) were used to PCR amplify the isolated genomic DNA for the V3–V4 16S rDNA hypervariable regions. The PCR products were sequenced by an Illumina MiSeq (Illumina, Inc, San Diego, CA) using the 2 × 300 bp paired-end protocol.
16S rRNA gene sequencing analysis
The raw sequencing data was quality-filtered and demultiplexed using QIIME 2 [16]. Chimeric sequences were identified and removed using UCHIME [17]. A total of 205 samples that passed the quality control were incorporated into the analysis. Operational taxonomic units (OTUs) were clustered using a closed-reference picking protocol with the UCLUST algorithm based on 97% nucleotide similarity. Microbial OTUs were annotated with the GreenGenes reference database (version 13.8). OTU counts were processed with total sum normalization (TSS) followed by cumulative-sum scaling (CSS) by using Calypso [18]. The relative abundances were log2 transformed to account for the non-normality of taxonomic counts data.
The biodiversity was measured by the number of OTUs, Chao1 index, Shannon index, and Inverse Simpson index. Richness is the number of different species present in the gut microbial communities, which are measured with the number of observed OTUs and Chao1 index. We performed Principal Coordinate Analysis (PCoA) to determine whether the samples could be separated on the basis of BMI and gender. The significance of composition difference among groups was determined using Kruskal–Wallis nonparametric test followed by post-hoc Tukey's HSD (honestly significant difference) test. We used Spearman correlations to identify the BMI-associated taxa, which appeared in more than 25% of the samples that had a significant correlation (P < 0.01) with BMI. PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) [19] was applied to obtain the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways by predicting metagenome content from 16S rRNA gene surveys.
A random forest (RF) model was constructed by using R-package randomForest [20] to perform supervised classification of the three BMI categories. Half of the dataset was used for building and training the RF model, and the other half for testing. The optimal number of variables (mtry) randomly sampled as candidates at each split was accessed by tuneRF function. We calculated the interpolated area under the receiver operating characteristic (ROC) curve (area under the receiver operating characteristic curve, AUC) for each classifier based on the cross-validation testing results.
Results
BMI is associated with compositional changes in the gut microbiome of adolescent
After quality filtering, a total of 17,323,193 sequencing reads were obtained from the 205 fecal samples. Taxa with less than 0.01% relative abundance across all the samples were excluded. A total of 518 OTUs were identified and grouped in 9 phyla and 69 genera. The gut microbiota richness and diversity were estimated at the OTU level with samples rarefied to the depth of 37,027 reads (the lowest number of sequences). Richness was higher in the overweight subjects than those with normal weight (Supplementary Fig. S1A, S1B). No significance in alpha diversity was detected among the three BMI groups (Supplementary Fig. S1C, S1D). With respect to beta diversity, the result of PCoA of the Bray–Curtis dissimilarity index revealed that the overall gut microbiome composition was not able to be stratified by BMI (Supplementary Fig. S2).
The gut microbiota composition in the adolescent population was dominated by Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria phyla, accounting for > 97% of the community (Supplementary Fig. S3). The relative abundance of Verrucomicrobia phylum was found to be reduced in the obese subjects (Fig. 2A). At the family level (Fig. 2B), the obese subjects had greater abundance of Actinomycetaceae (including Actinomyces genus; Fig. 2C), Clostridiaceae, Cytophagaceae (including Rhodococcus genus; Fig. 2C), Lactobacillaceae, Streptococcaceae (including Streptococcus genus; Fig. 2C), and Veillonellaceae (including Megasphaera genus; Fig. 2C), while had decreased abundance of Odoribacteraceae (including Odoribacter genus; Fig. 2C) and Rikenellaceae (including an unclassified genus of Rikenellaceae; Fig. 2C). The relative abundance of Nocardiaceae family was particularly higher in the overweight subjects.
Fig. 2.

Differences in fecal microbiota constituents of normal-weight, overweight, and obese Shenzhen adolescents. Bacterial taxa at the A phylum, B family, and C genus levels demonstrated significantly different abundances across the three BMI groups. Each bar plot indicates the mean proportion of sequences assigned to a feature in each group. Whiskers represent 1.5* inter-quartile range. Relative abundances were analyzed by Kruskal–Wallis test followed by post-hoc Tukey HSD test (*P < 0.05, **P < 0.01, and ***P < 0.001). N, normal-weight; OW, overweight; OB, obese
Fifty-six taxa correlated significantly with the BMI, and most of the taxa members belonged to Firmicutes phylum (48 OTUs), with 17 to Ruminococcaceae family and 12 to Lachnospiraceae family (Supplementary Tables S1, S2, S3). As to the relationship between the 56 BMI-associated taxa and serological or body composition properties, the result showed that 3 Firmicutes taxa (2 OTUs belonging to Ruminococcaceae, 1 OTU to Lachnospiraceae) strongly positively correlated with BMI, body fat rate, waistline, but negatively correlated with body water, protein, muscle, and mineral salts rates (Fig. 3A). Notably, these 3 taxa also positively correlated with most of the serum biochemical properties (except TBIL). Yet, a few of Ruminococcaceae bacteria, including Faecalibacterium prausnitzii, were inversely associated with BMI.
Fig. 3.

Identifying BMI-associated bacterial taxonomic biomarkers Shenzhen adolescents. A Heatmap of Spearman correlations between 56 BMI-associated bacterial taxa and serum biochemical parameters and body composition. Significance is given as **P < 0.01 and ***P < 0.001. B Thirty BMI-discriminatory bacterial taxa were identified by the RF model which were listed in rank order of their contribution to the classification accuracy (mean decrease accuracy). C ROC curve of the RF model
In order to identify the BMI-associated bacterial taxonomic markers in the gut microbiota in adolescents, we built a RF model to classify the phenotypes based on the 56 OTUs identified in the Spearman correlation analyses. The RF model tuning resulted in mtry = 14 for the number of variables when setting the number of trees (ntree) to 500. The 30 most BMI-discriminatory taxa, following rank order of their contribution to the predictive accuracy, were shown in Fig. 3B, among which 5 Ruminococcaceae and 8 Lachnospiraceae were included. We calculated the interpolated area under the ROC curves (AUC) for the classifier based on the cross-validation testing results, and successfully classified the obese adolescents with a high classifiability (AUC = 0.976; Fig. 3C). The normal-weight and overweight adolescents were also well classified with high AUC values of 0.886 and 0.740, respectively.
Gut microbiota gene content associated with PPAR signaling is reduced in obese adolescents
We analyzed BMI-associated functional profile of microbial community 16S rRNA sequence data using Spearman correlations via employing PICRUSt [21]. We selected seven KEGG pathways significantly correlated (P < 0.01) with BMI (Supplementary Tables S4–6), and their links with the serological properties and body composition were further examined using Spearman correlations (Fig. 4A). The predicted gene content related to pathways of peroxisome, PPAR signaling and adipocytokine signaling showed negative associations with BMI, body fat rate and waistline, but positively correlated with rates of body water, protein, muscle, and mineral salts. These three pathways also inversely correlated with CER/CP, blood sugar level, blood pressure (both systolic and diastolic), CRP, WBC, PUFA and ALT. In particular, the two pathways of peroxisome and PPAR signaling were significantly downregulated in the obese adolescents compared to the normal-weight and overweight counterparts (Fig. 4B). The predicted gene content related to other pathways, including synthesis and degradation of ketone bodies, beta Alanine metabolism, glycosyltransferases and other ion coupled transporters, positively associated with BMI, body fat rate and waistline but negatively associated with rates of body water, protein, muscle, and mineral salts.
Fig. 4.

Functional divergence of gut microbiota across different BMI groups. A Predicted KEGG pathways that significantly correlated with BMI, and their associations with the boy composition and serum biochemical parameters. B KEGG pathways that are differentially expressed by the gut microbiome of the BMI categories. Each bar plot indicates the mean proportion of sequences assigned to a feature in each group. Whiskers represent 1.5* inter-quartile range. Relative abundances of were analyzed by Kruskal–Wallis test followed by post-hoc Tukey HSD test (*P < 0.05; **P < 0.01, and ***P < 0.001). N, normal-weight; OW, overweight; OB, obese
BMI differences in the gut microbiota of adolescents are influenced by gender
Neither alpha nor beta diversities were found significantly different between the boys and girls (Supplementary Figs. S1, S2). At the family level, the relative abundances of Alcaligenes, Bacteroidaceae, Brucellaceae, Clostridiaceae, Lachnospiraceae, Planococcaceae, and Streptococcaceae were greater in boys than girls (Supplementary Fig. S4A). We then compared the relative abundances of the gut bacteria among the three BMI categories with gender stratification (Supplementary Fig. S4B, S4C). At the family level, the highest abundance of Cytophagaceae was observed in obese subjects of both genders. Overweight boys had a lower abundance of Actinomycetaceae, and a higher abundance of Nocardiaceae than those of normal-weight and obesity. At the genus level, both genders had higher relative abundances of Hymenobacter and Megasphaera. A lower relative abundance of Odoribacter was particularly detected in obese boys. In addition, overweight boys had significantly lower Actinomyces and higher Rhodococcus (Supplementary Fig. S4C).
PICRUSt analysis revealed a higher level of translation factor in girls and a higher level of bisphenol degradation in boys (Supplementary Fig. S5A, Table S7). A lower level of PPAR signaling pathway was detected in the obese subjects of both genders (Supplementary Fig. S5B, Tables S8, S9). A reduced biosynthesis level of tropane piperidine and pyridine alkaloid was only observed in obese boys (Supplementary Fig. S5C, Table S9).
Discussion
Identification of gut microbial signatures responsible for obesity has great potential in prevention and treatment of overweight or obesity related diseases. In this study, we recognized several BMI-associated patterns in the adolescent gut microbiome composition and functions. Previously, we found Bacteroidetes was enriched in obese adults compared to the lean [22], and the imbalance of Bacteroidetes to Firmicutes ratio was also reported in other studies [13, 23, 24]. However, the pattern was not supported in this study for the adolescent subjects, that no difference was detected in the relative abundances of Bacteroidetes to Firmicutes upon comparing between obese and normal-weight subjects (data no shown). The present data agreed that the ratio of Bacteroidetes to Firmicutes was not a microbial marker associated with obesity [13, 20]. On the other hand, we identified 56 microbial taxa at the OTU level significantly (P < 0.01) correlated with BMI, and a large proportion of which belonged to Firmicutes phylum, especially families Ruminococcaceae and Lachnospiraceae. Further supervised learning algorithm constructed based on the 56 BMI-associated OTUs resulted in the successful classification of the obese adolescents with a high accuracy exceeding 90%.
The enrichment of Ruminococcaceae has been observed in animals fed by high-fat diet [24], but some Ruminococcaceae taxa were linked to a lower risk of weight gain [25]. In human studies, higher abundances of Ruminococcus species (such as Ruminococcus bromii and Ruminococcus obeum) were also observed in obese subjects [26], but some Ruminococcaceae bacteria (e.g. Dialister, Methanobrevibacter and Oscillospira) were associated with lower BMI [23, 27]. In this study, we identified 17 OTUs assigned to Ruminococcaceae were significantly correlated with BMI, and many of which were inversely correlated with BMI including the well-studied butyrate producer F. prausnitzii. The result is consistent with several previous studies [28, 29]. Butyrate has been known to exert a profound effect in regulating metabolic inflammation, and reduced level of butyrate may contribute to low-grade chronic inflammation that participating the development of obesity.
Previous evidence suggests that Lachnospiraceae may play a pivotal role in the development of obesity and type 2 diabetes [29]. Animal studies demonstrated that the abundance of family Lachnospiraceae increased along with body weight of mice fed with high-fat diet [30]; colonization of Lachnospiraceae in germ-free mice induced significant increases in fasting blood glucose concentrations as well liver and mesenteric adipose weights, and reductions in plasma insulin levels. A human study with 190 Mexican children showed that the Lachnospiraceae family was significantly increased in overweight and obese children [31]. In this study, 12 taxa of Lachnospiraceae were identified to be significantly associated with BMI. Similar to the Ruminococcaceae family, both positive (8 OTUs) and negative (4 OTUs) correlations were detected between Lachnospiraceae taxa and BMI. Overall, investigations at higher phylogenetic level and animal experiments are needed to identify the specific bacteria species and their functions in influencing host body fat and blood glucose level.
Although the exact mechanisms by which the gut microbiota contribute to obesity are unclear, it is well established that modification of the gut microbiota can increase energy production, trigger low-grade inflammation, induce insulin resistance and affect fatty acid tissue composition [30]. Based on the predicted functional profiles of the fecal microbiota, we found that genes associated peroxisome and PPAR signaling pathways were inversely correlated with CER/CP, blood sugar level, blood pressure (both systolic and diastolic), CRP, WBC, PUFA, and ALT, and their levels were significantly upregulated in obese adolescents. Peroxisomes are important regulators of energy and their disruption influences the risk for obesity and associated metabolic disorders [31]. It was demonstrated that decreases in a set of peroxisomal genes in white adipose tissue of both humans and mice with obesity [32]. PPARs are a member of the nuclear receptor superfamily of ligand-dependent transcription factors, which regulate peroxisomal proteins by binding to promoters of peroxisomal genes. Lower levels of PPARs have been observed in obese patients, and activation of PPARs could decrease fibro-inflammation and ectopic fat accumulation in the adipose tissue [33]. A decline in bacterial genes capable of altering PPAR signaling may reflect a reduction of PPARs expression in the host. In fact, some gut bacteria have capacities in regulating PPARs. For example, mice artificially infected with Trichinella spiralis could induce a decrease in the levels of PPARγ in the colon, which is accompanied by a decline in beneficial species (such as Akkermansia) and an increase in pathogenic bacteria (such as Escherichia/Shigella) [34]. Another pathway of importance significantly associated with BMI was adipocytokine signaling. Adipocytokines derived from adipose tissue, including various hormones (such as leptin, adiponectin, resistin, and visfatin) and cytokines (such as interleukin-6 and tumor necrosis factor α), are important regulators of energy homeostasis and mediators of inflammation and immunity [35]. There is overwhelming evidence that deficiencies in adipocytokines contribute to the development of obesity and associated comorbidities [36]. Importantly, some adipocytokines are able to modulate gut microbial composition independently of dietary [37]. In addition, hydroxy fatty acids produced from gut microbiota affect host lipid metabolism by modulating peroxisomal β-oxidation activity [38]. Although the relationship between gut microbiota and adipose tissue remains unclear, our result implies that gut bacteria may contribute to the development of obesity via impairing the host peroxisomal fitness. Further investigations are needed to identify the exact bacterial products that affect these two pathways in order to determine their roles in adiposity.
In our previous study with the fecal microbiota profiles Chinese adults (527 adults aged 37.3 ± 16.3), enrichment of Fusobacteria and Actinobacteria were observed in the male and female obese subjects, respectively [22]. Bacterial genes associated with butyrate-acetoacetate CoA-transferase were also found to be enriched in the gut microbiome of obese Chinese adults [22]. However, these patterns were not obtained from the adolescent data (205 adolescents aged 13.31 ± 1.55 years old ranging from 11 to 15) in this study. The inconsistency may relate to differences of age.
Moreover, the influence of gender on gut microbiota have been reported previously. For example, members of Bacteroides were found at a lower level in adult females than males in surveys of European [39] and US populations [40]. The pattern was supported by the present result of Shenzhen (China) adolescents, but was not in our previous study on Chinese adults [22]. We believe geographical location exerted a strong effect on human gut microbiota variations, as previously reported [15]. Shenzhen is one of the most developed cities in China, and lifestyles of the residents (especially diet) are quite similar to those in western countries. Hence, some common patterns in the gut microbiota composition may share between Shenzhen citizens and westerners. Overall, the influence of geographical location, age, and gender partly explained the inconsistent patterns across the studies.
In conclusion, the BMI-associated differences in gut microbiota profiles were able to be used as biomarkers for characterizing obese adolescents. The gene contents associated with the peroxisome signaling pathway are significantly reduced in the gut microbiome of obese adolescents, and determining the exact metabolites produced by specific species may provide invaluable microbial targets for the prevention, assessment, and treatment of obesity for adolescents. Our result reinforced a need to consider the influence of age, gender and geographical location when choosing controls for investigating gut microbiome and the association with human diseases.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We acknowledge the contributions of the nursing staff of Shenzhen Children’s Hospital for their help with blood, stool, and metadata collection. We thank all volunteers for their participation in this study.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (42077395 and 81900474), the Shenzhen key technology R&D program (JSGG20170413152936281, 20170502165510880), University of Electronic Science and Technology of China (Y03019023601008022), and Shenzhen Science and Technology Project (JCYJ20220530165012027), Shenzhen Peacock Talent Program (802-012677 to X.G.), Shenzhen-Hongkong Join Research Funding (SGDX20201103095603009), National Key R&D Program of China (2021YFA0717001), and Shenzhen Key Laboratory of Gastrointestinal Microecology and Diseases (ZDSYS20220606100800002).
Data availability
Raw sequencing data have been deposited on the European Nucleotide Archive server, with accession number PRJEB33385.
Declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Ethical approval
The study started after we received approval from the Institutional Review Board of the Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences (SIAT-IRB-131115-H0032). All individuals participating in the study received informed consent from their guardians. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xue-Feng Gao and Bin-Bin Wu have contributed equally to this work.
Contributor Information
Xue-Feng Gao, Email: xfgao@smu.edu.cn.
Yun-Peng Cai, Email: yp.cai@siat.ac.cn.
Yan Liang, Email: yliang@uestc.edu.cn.
References
- 1.Collaboration NCDRF. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet. 2017;390(10113):2627–2642. doi: 10.1016/S0140-6736(17)32129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rios-Covian D, Ruas-Madiedo P, Margolles A, Gueimonde M, de Los Reyes-Gavilan CG, Salazar N. Intestinal short chain fatty acids and their link with diet and human health. Front Microbiol. 2016;7:185. doi: 10.3389/fmicb.2016.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 4.Baothman OA, Zamzami MA, Taher I, Abubaker J, Abu-Farha M. The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis. 2016;15:108. doi: 10.1186/s12944-016-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox LM, Blaser MJ. Antibiotics in early life and obesity. Nat Rev Endocrinol. 2015;11(3):182–190. doi: 10.1038/nrendo.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 7.Thaiss CA, Itav S, Rothschild D, Meijer MT, Levy M, Moresi C, Dohnalova L, Braverman S, Rozin S, Malitsky S, et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature. 2016;540(7634):544–551. doi: 10.1038/nature20796. [DOI] [PubMed] [Google Scholar]
- 8.Boulange CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016;8(1):42. doi: 10.1186/s13073-016-0303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 10.Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 12.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, Flint HJ. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 2008;32(11):1720–1724. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- 14.Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, Tremaroli V, Bakker GJ, Attaye I, Pinto-Sietsma SJ, et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat Med. 2018;24(10):1526–1531. doi: 10.1038/s41591-018-0160-1. [DOI] [PubMed] [Google Scholar]
- 15.He Y, Wu W, Zheng HM, Li P, McDonald D, Sheng HF, Chen MX, Chen ZH, Ji GY, Zheng ZD, et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat Med. 2018;24(10):1532–1535. doi: 10.1038/s41591-018-0164-x. [DOI] [PubMed] [Google Scholar]
- 16.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zakrzewski M, Proietti C, Ellis JJ, Hasan S, Brion MJ, Berger B, Krause L. Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics. 2017;33(5):782–783. doi: 10.1093/bioinformatics/btw725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MGI. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38(6):685–688. doi: 10.1038/s41587-020-0548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2(3):18–22. [Google Scholar]
- 21.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao X, Zhang M, Xue J, Huang J, Zhuang R, Zhou X, Zhang H, Fu Q, Hao Y. Body mass index differences in the gut microbiota are gender specific. Front Microbiol. 2018;9:1250. doi: 10.3389/fmicb.2018.01250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, Krakoff J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr. 2011;94(1):58–65. doi: 10.3945/ajcn.110.010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, Hardt PD. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 2010;18(1):190–195. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- 25.Menni C, Jackson MA, Pallister T, Steves CJ, Spector TD, Valdes AM. Gut microbiome diversity and high-fibre intake are related to lower long-term weight gain. Int J Obes (Lond) 2017;41(7):1099–1105. doi: 10.1038/ijo.2017.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasai C, Sugimoto K, Moritani I, Tanaka J, Oya Y, Inoue H, Tameda M, Shiraki K, Ito M, Takei Y, et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015;15:100. doi: 10.1186/s12876-015-0330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borgo F, Garbossa S, Riva A, Severgnini M, Luigiano C, Benetti A, Pontiroli AE, Morace G, Borghi E. Body mass index and sex affect diverse microbial niches within the gut. Front Microbiol. 2018;9:213. doi: 10.3389/fmicb.2018.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut. 2014;63(9):1513–1521. doi: 10.1136/gutjnl-2014-306928. [DOI] [PubMed] [Google Scholar]
- 30.Musso G, Gambino R, Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded? Diabetes Care. 2010;33(10):2277–2284. doi: 10.2337/dc10-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleiboeker B, Lodhi IJ. Peroxisomal regulation of energy homeostasis: Effect on obesity and related metabolic disorders. Mol Metab. 2022;65:101577. doi: 10.1016/j.molmet.2022.101577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piao L, Dorotea D, Jiang S, Koh EH, Oh GT, Ha H. Impaired peroxisomal fitness in obese mice, a vicious cycle exacerbating adipocyte dysfunction via oxidative stress. Antioxid Redox Signal. 2019;31(18):1339–1351. doi: 10.1089/ars.2018.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corrales P, Vidal-Puig A, Medina-Gomez G. PPARs and metabolic disorders associated with challenged adipose tissue plasticity. Int J Mol Sci. 2018;19(7):2124. doi: 10.3390/ijms19072124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, Ren Y, Lu J, Bartlett M, Chen L, Zhang Y, Guo X, Liu C. A novel prebiotic blend product prevents irritable bowel syndrome in mice by improving gut microbiota and modulating immune response. Nutrients. 2017;9(12):1341. doi: 10.3390/nu9121341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6(10):772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 36.Cao H. Adipocytokines in obesity and metabolic disease. J Endocrinol. 2014;220(2):T47–59. doi: 10.1530/JOE-13-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajala MW, Patterson CM, Opp JS, Foltin SK, Young VB, Myers MG., Jr Leptin acts independently of food intake to modulate gut microbial composition in male mice. Endocrinology. 2014;155(3):748–757. doi: 10.1210/en.2013-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morito K, Shimizu R, Kitamura N, Park SB, Kishino S, Ogawa J, Fukuta T, Kogure K, Tanaka T. Gut microbial metabolites of linoleic acid are metabolized by accelerated peroxisomal beta-oxidation in mammalian cells. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(11):1619–1628. doi: 10.1016/j.bbalip.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T, Cresci A, Silvi S, Orpianesi C, Verdenelli MC, et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol. 2006;72(2):1027–1033. doi: 10.1128/AEM.72.2.1027-1033.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dominianni C, Sinha R, Goedert JJ, Pei Z, Yang L, Hayes RB, Ahn J. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS ONE. 2015;10(4):e0124599. doi: 10.1371/journal.pone.0124599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data have been deposited on the European Nucleotide Archive server, with accession number PRJEB33385.