

Abstract
In cross-coupling reactions, dihaloheteroarenes are usually most reactive at C─halide bonds adjacent to a heteroatom. This selectivity has been previously rationalized. However, no mechanistic explanation exists for anomalous reports in which specific ligands effect inverted selectivity with dihalopyridines and -pyridazines. Here we provide evidence that these ligands uniquely promote oxidative addition at 12e− Pd(0). Computations indicate that 12e− and 14e− Pd(0) can favor different mechanisms for oxidative addition due to differences in their HOMO symmetries. These mechanisms are shown to lead to different site preferences, where 12e− Pd(0) can favor oxidative addition at an atypical site distal to nitrogen.
Keywords: cross-coupling, site-selectivity, oxidative addition, N-heterocyclic carbenes, DFT
Graphical Abstract
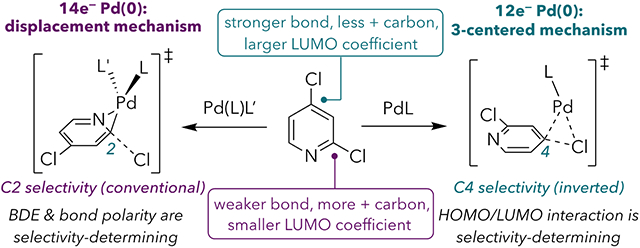
Heteroarenes are common motifs in high-value small molecules such as pharmaceutical drugs and agrochemicals.1 A popular method for their elaboration involves employing halogenated heteroarene substrates in cross-coupling reactions. Dihaloheteroarenes bearing two identical halides typically react with predictable selectivity at a C─X bond α to a heteroatom, when present (Scheme 1A).2 However, the ability to invert selectivity can open up underexplored synthetic space. Because of the importance of dihaloheteroarene cross-coupling reactions, the origin of their innate selectivity has been thoroughly studied. Houk and Merlic rationalized the typical selectivity with a distortion-interaction model.3 Relevant to the current work, oxidative addition of 6-membered nitrogen-containing dihaloheteroarenes takes place at the weaker C─X bond, which is the bond adjacent to nitrogen. Because this bond is weaker, it is easier to distort into the transition state geometry. Leitch et al. recently demonstrated that conventional selectivity is also correlated with more positive electrostatic potentials at the ipso carbon and with more negative potentials at an ortho atom.4
Scheme 1.

Ligands that Give Unconventional Selectivity
Although current models explain the conventional selectivity of 6-membered dihaloheteroarenes, there exist rare reports in which the use of specific ligands leads to inverted selectivity, resulting in cross-coupling at a site distal to nitrogen. The use of QPhos gives modest C4-selectivity with 2 and high C5-selectivity with 5 (Scheme 1B).5 Bulky NHC ligands (6 and IPr) can promote C4-selectivity with 2 and 3 and C5-selectivity with 5 (Scheme 1C-D).6,7,8,9 There is currently no mechanistic rationale for these ligand-controlled deviations from the conventional selectivity.10 The absence of mechanistic understanding precludes rational catalyst design, and thus cross-coupling reactions at distal C─X bonds remain unexplained curiosities. Here, we communicate the key discovery that two competing mechanisms for oxidative addition display different site preferences. Due to its HOMO π symetry, 14e− Pd(0) prefers to react with halogenated heteroarenes through a nucleophilic displacement mechanism at an α C─X bond. Conversely, 12e− Pd(0) prefers a concerted three-centered transition state due to its HOMO σ symmetry, and this mechanism is biased toward a distal C─X bond. As such, accessing 12e− Pd(0) is critical to achieving oxidative addition at the atypical site.
RESULTS AND DISCUSSION
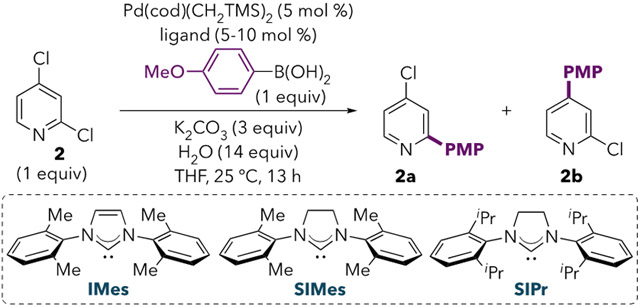
The reported C4- and C5-cross-couplings of 2, 3, and 5 employ very sterically hindered electron-rich monodentate ligands.5,6,7 To further evaluate ligand effects, we screened a number of monodentate phosphines and NHC ligands for the Suzuki coupling of 2 using Pd(cod)(CH2SiMe3)2 (Table 1).11 There is a clear correlation between ligand sterics and the C4:C2 ratio for triarylphosphines (entries 1-4), alkylphosphines (entries 5-13), and NHCs (entries 14-18). Bulky alkyl phosphines are more C4-selective than bulky triaryl phosphines, and unsaturated NHC ligands are more selective than their saturated analogues. Based on these trends, we hypothesized that sterically hindered strong σ-donor ligands favor unconventional selectivity by promoting oxidative addition at a low-coordinate species (i.e., 12e− PdL). Smaller ligands or ones that are better π-acceptors, such as PAr3 and saturated NHCs, would be more likely to favor coordination of a second ligand.14,15,16 Notably, among trialkylphosphines, selectivity switches near the %Vbur(min) threshold previously reported to determine whether PdL or PdL2 dominates as the active species for oxidative addition.17,18 Nevertheless, it was unclear why 12e− PdL might react at the stronger C4─X bond. To evaluate this question, we used density functional theory (DFT) calculations to closely examine the mechanisms of oxidative addition at 12e− and 14e− Pd(0) using a simple model system comprising PhCl and Pd(PMe3)n (n = 1 or 2). Three oxidative addition transition structures were located with Pd(PMe3) (Figure 1A). Two of these (TS7a and TS7b) represent classic 3-centered concerted mechanisms, wherein Pd simultaneously interacts with the ipso carbon and the chloride. The third structure is better described as a nucleophilic displacement mechanism (TS7c). In this mechanism, Pd interacts with both the ipso and the ortho carbons of the aromatic ring, as evidenced by the short Cortho---Pd distance (2.49 Å). Conversely, the long Pd---Cl distance indicates that there is no significant interaction between Pd and Cl. This type of mechanism has also been referred to as a nucleophilic substitution (SNAr-type) mechanism19 or a dissociative process.20
Table 1.
Evaluation of Monodentate Ligands for the Suzuki-Miyaura Coupling Using Pd(OAc)2a
| |||||
|---|---|---|---|---|---|
| entry | ligand (mol %) |
%Vbur (min)b |
2a (%) |
2b (%) |
2a : 2b |
| 1 | PPh3 (5) | (28.2) | 84.2 | 0.6 | >99 : 1 |
| 2 | PPh3 (10) | 81.6 | 0.4 | >99 : 1 | |
| 3 | P(o-tol)3 (5) | (34.4) | 42.0 | 21.5 | 2.0 : 1 |
| 4 | P(o-tol)3 (10) | 30.3 | 17.0 | 1.8 : 1 | |
| 5 | PMe3 (5) | (22.1) | 18.5 | 2.2 | 8.4 : 1 |
| 6 | PMe3 (10) | 9.9 | 0.9 | 11.0 : 1 | |
| 7 | P(n-Bu)3 (5) | (24.2) | 20.0 | 7.6 | 2.6 : 1 |
| 8 | P(n-Bu)3 (10) | 0.8 | 0.3 | 3.2 : 1 | |
| 9 | PCy3 (5) | (30.2) | 27.9 | 44.9 | 1 : 1.6 |
| 10 | PCy3 (10) | 27.2 | 39.9 | 1 : 1.5 | |
| 11 | PAd2(n-Bu) (5) | (32.8) | 24.1 | 51.8 | 1 : 2.2 |
| 12 | PtBu3 (5) | (36.3) | 27.0 | 47.3 | 1 : 1.8 |
| 13 | Q-Phos (5) | (47.6) | 24.1 | 50.1 | 1 : 2.1 |
| 14 | IMes (5) | 36.5 | 32.5 | 52.0 | 1 : 1.6 |
| 15 | SIMes (5) | 36.9 | 27.9 | 22.0 | 1.3 : 1 |
| 16 | IPr (5) | 44.5 | 9.5 | 66.8 | 1 : 7.0 |
| 17 | IPrc | 6.6 | 68.4 | 1 : 10.4 | |
| 18 | SIPr (5) | 47.0 | 9.5 | 36.4 | 1 : 3.8 |
GC yields calibrated against undecane as the internal standard. Average of two trials. 0–6.5% diarylation observed in all entries (see SI). PMP = p-methoxyphenyl.
Values in parentheses are minimum percent buried volumes obtained from the Kraken database.12 Percent buried volumes of NHCs reported for LAuCl complexes at a L–Au distance of 2.00 Å from reference 13.
With (η3-1-tBu-indenyl)Pd(IPr)(Cl) (3 mol %) as catalyst, 15.5 h.
Figure 1.

Calculated transition structures for oxidative addition of chlorobenzene at (A) Pd(PMe3) and (B) Pd(PMe3)2. Differences in Gibbs free energies of activation are listed in units of kcal mol−1 relative to the lowest-energy structure in each set (defined as ΔΔG‡ = 0.0).21 (C) Key frontier molecular orbitals of the relevant species, where Pd(PMe3)2 is distorted into a bent geometry as adopted in the arene—Pd(PMe3)2 pre-oxidative addition π complex.22
For monoligated Pd(PMe3), the lowest energy transition structure is the concerted mechanism TS7a. In contrast, a nucleophilic displacement mechanism (TS8b) is favored over a concerted mechanism (TS8a) for bisligated Pd(PMe3)2 (Figure 1B).23,24,25 This difference in the preferred mechanisms for PdL and PdL2 may be explained by frontier molecular orbital interactions. The HOMO of Pd(PMe3) is a σ-type orbital, so its symmetry is suited to the concerted mechanism in which Pd donates only into the ipso carbon (via overlap with chlorobenzene's lowest energy unoccupied orbital without a node through the ipso carbon, LUMO+1).26 In contrast, the HOMO of Pd(PMe3)2 has π-symmetry (Figure 1C), and thus can backbond into two carbons of PhCl during oxidative addition via a displacement mechanism. Notably, simple model ligands like PMe3 are not always adequate for describing the behavior of more complex ligands.27 Furthermore, Maseras and coworkers have shown that ligand identity and solvent can influence the predicted mechanism of oxidative addition at 14e− PdL2 with the substrate bromobenzene.19,28 However, in this case, our calculations using Pd(PMe3)n and PhCl prove to be consistent with those obtained using Pd(IPr) and 2,4-dichloropyridine (2).
The LUMO of 2,4-dichloropyridine has π-symmetry, with nodal planes on either side of nitrogen and of C4 (Figure 2, inset). The LUMO coefficient at C4 is substantially larger than at C2, with C4 accounting for 26% of the LUMO density and C2 contributing only 8%. Thus, it becomes apparent why PdL2 and PdL might exhibit different site-selectivity. Because the π-type HOMO of PdL2 would donate into two atoms of the pyridine ring during a nucleophilic displacement mechanism, strong orbital overlap can be achieved during reaction at C2 despite the small LUMO coefficient at that carbon, and bond dissociation energies become more important to selectivity. However, the σ-type HOMO of PdL can only donate into one atom of the pyridine ring. As such, the larger LUMO coefficient at C4 versus C2 is important during the reaction of PdL because it dictates the strength of the HOMO(Pd)/LUMO(substrate) overlap.
Figure 2.

Calculated free energy diagrams illustrating the oxidative addition of 2,4-dichloropyridine at Pd(NHC) or Pd(NHC)(2), where NHC = IPr (A) or IMes (B).21,22,34
Using monoligated Pd(IPr), a 3-centered mechanism for oxidative addition at C2 (TS10a-IPr, Figure 2A) is preferred over a displacement mechanism (see SI). Only a 3-centered mechanism could be located for oxidative addition at C4 (TS10b-IPr). DFT predicts that reaction at C4 via TS10b-IPr should be favored over C2 by 1.7 kcal/mol (C4:C2 = 18:1) using Pd(IPr). This prediction is in good agreement with the experimentally reported C4-selectivity with IPr (typically ~10:1),7 and stands in stark contrast to the prediction with bisligated Pd(IPr)(L). Unsurprisingly, considering the sterics of IPr, we were unable to find transition structures involving Pd(IPr)2. However, nucleophilic displacement transition structures were located with bisligated Pd(IPr)(L) where L is a second substrate molecule (2).29 With the bisligated complex, reaction at C2─Cl via TS13a-IPr is strongly favored over reaction at C4 (TS13b-IPr). Overall, the lowest energy path in Figure 2A is C4-oxidative addition at monoligated Pd(IPr) via TS10b-IPr, consistent with experimental selectivity with this ligand.
For comparison, analogous DFT calculations were performed with PtBu3, another bulky monodentate ligand that favors reaction at C4 but to a lesser extent than IPr (see Table 1, entry 12). Consistent with experiment, the DFT calculations predict worse C4-selectivity with Pd(PtBu3) (Figure 2B, predicted C4:C2 = 1:5, experimental C4:C2 = 1:2). The enhanced selectivity of IPr compared to PtBu3 is likely due to the stronger σ-donation by IPr,14d,30 which could enhance the PdL(HOMO)→2(LUMO) interaction (see SI for further discussion). Like IPr, PtBu3 is too bulky for a bisligated transition state to contribute significantly to the reaction mechanism.
DFT calculations with IMes reveal similar trends as seen with IPr: PdL favors reaction at C4 (Figure 2C, TS10b-IMes), while PdL2 favors reaction at C2 (TS13a-IMes). However, unlike IPr, the calculations with IMes predict that the bisligated path via TS13a-IMes, involving oxidative addition at C2, is energetically competitive with the monoligated path (C4:C2 = 1:13 based on TS10b-IMes and TS13a-IMes). This prediction is consistent with the experimental observation that IMes is much less C4-selective than IPr, and supports the hypothesis that smaller ligands lead to a higher proportion of bisligated active catalyst. The calculations do not quantitatively reproduce the experimentally observed 1.6:1 selectivity with IMes (Table 1 entry 14). However, the energy of the crowded transition structure TS13a-IMes was found to be extremely sensitive to DFT method (see SI), likely due in part to differences in how dispersion interactions are handled. This suggests the possibility of considerable error in quantitative analysis when comparing mono- to bisligated structures. Furthermore, these calculations cannot take into account the ability of 2 to coordinate to other acids that are present in higher concentrations than Pd under the catalytic conditions (e.g., boron, K+, H2O). Equilibria involving coordination of 2 to these species would detract from the overall concentration of Pd(IMes)(2).
The calculations suggest that IMes gives worse C4-selectivity than IPr due to a higher proportion of bisligated active catalyst. Experimental data support the hypothesis that an N-bound dichloropyridine group could serve as the second ligand on Pd in Suzuki couplings catalyzed by Pd/IMes. C4-selectivity is significantly enhanced for 15 compared to 2 (Scheme 2A), where 15 should be a worse ligand for Pd due to sterics.31 Furthermore, increased reaction is observed at C2 at higher substrate concentrations (Scheme 2B). The increased C2-selectivity at high [2] is consistent with a higher probability for reaction at bisligated Pd(IMes)(2).
Scheme 2.

(A) Influence of 6-Substituent on the Selectivity of Cross-Coupling Catalyzed by Pd/IMes; (B) Influence of Substrate Concentration on the Selectivity of Cross-Coupling Catalyzed by Pd/IMes; (C) With Pd/IPr, Selectivity Trends with Difference in LUMO Coefficients.
Based on the calculations, we anticipated that, under conditions that promote oxidative addition at 12e− PdL using IPr, selectivity should trend with the difference between LUMO coefficients at the two sites of a dihaloheteroarene. Indeed, this prediction bears out for the substrates in Scheme 2B. The best selectivity for the site remote to nitrogen is achieved with the substrate displaying the largest difference in LUMO coefficients (5), while preferential reaction at C2 is observed when the two sites have nearly identical LUMO coefficients (4).32,33 To our knowledge, this is the first report of C3-favored cross-coupling of 1 supported by spectroscopic characterization, although diarylation is competitive with monoarylation (see SI).
CONCLUSION
This work shows that 12- and 14-electron Pd(0) can favor different mechanisms for oxidative addition due to differences in their HOMO symmetries. The HOMO σ-symmetry of 12e− PdL means that LUMO coefficients at individual sites of dihaloheteroarenes are particularly important for determining selectivity. Conversely, the HOMO π-symmetry of 14e− PdL2 enables Pd to backbond into two atoms of the substrate during oxidative addition, rendering LUMO coefficients at individual atoms less important, and allowing selectivity to be dominated by other factors including C─X bond strengths. This work has implications for understanding oxidative addition reactivity trends and for rational design of site-selective catalysts. A systematic evaluation of the factors influencing the preferred mechanisms of oxidative addition will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
The experimental research reported in this publication was supported by the National Institute Of General Medical Sciences (NIGMS) of the NIH under Award Number R35GM137971. The computational work was supported by an NSF CAREER award (CHE-1848090). Calculations were performed on Comet and Expanse at SDSC through XSEDE (CHE-170089), which is supported by NSF (ACI-1548562). Support for MSU’s NMR Center was provided by the NSF (Grant No. NSF-MRI:CHE-2018388 and NSF-MRI:DBI-1532078), MSU, and the Murdock Charitable Trust Foundation (2015066:MNL). Funding for the mass spectrometry facility was provided in part by NIH NIGMS (P20GM103474 and S10OD28650), the Murdock Charitable Trust Foundation, and MSU. We are grateful to Umicore for a gift of (η3-1-tBu-indenyl)2(μ-Cl)2Pd2 and (η3-1-tBu-indenyl)Pd(IPr)(Cl)
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental and computational details, NMR spectra, and calculated energies (PDF)
Cartesian coordinates of minimum-energy calculated struc-tures (XYZ)
REFERENCES
- 1.Pennington LD; Moustakas DT The Necessary Nitrogen Atom: A Versatile High-Impact Design Element for Multiparameter Optimization. J. Med. Chem 2017, 60, 3552–3579. [DOI] [PubMed] [Google Scholar]
- 2.(a) For reviews, see: Schröter S; Stock C; Bach T Regioselective cross-coupling reactions of multiple halogenated nitrogen-, oxygen-, and sulfur-containing heterocycles. Tetrahedron 2005, 61, 2245–2267; [Google Scholar]; (b) Fairlamb IJS Regioselective (site-selective) functionalisation of unsaturated halogenated nitrogen, oxygen and sulfur heterocycles by Pd-catalysed cross-couplings and direct arylation processes. Chem. Soc. Rev 2007, 36, 1036–1045; [DOI] [PubMed] [Google Scholar]; (b) Manabe K; Yamaguchi M; Catalyst-Controlled Site-Selectivity Switching in Pd-Catalyzed Cross-Coupling of Dihaloarenes. Catalysts 2014, 4, 307–320; [Google Scholar]; (c) Almond-Thynne J; Blakemore DC; Prydeb DC; Spivey AC Site-selective Suzuki–Miyaura coupling of heteroaryl halides – understanding the trends for pharmaceutically important classes. Chem. Sci 2017, 8, 40–62; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Palani V; Perea MA; Sarpong R Site-Selective Cross-Coupling of Polyhalogenated Arenes and Heteroarenes with Identical Halogen Groups. Chem. Rev 2021, 122, 10126–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Legault CY; Garcia Y; Merlic CA; Houk KN Origin of Regioselectivity in Palladium-Catalyzed Cross-Coupling Reactions of Polyhalogenated Heterocycles. J. Am. Chem. Soc 2007, 129, 12664–12665. [DOI] [PubMed] [Google Scholar]; (b) Garcia Y; Schoenebeck F; Legault CY; Merlic CA; Houk KN Theoretical Bond Dissociation Energies of Halo-Heterocycles: Trends and Relationships to Regioselectivity in Palladium-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc 2009, 131, 6632–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J; Donnecke S; Paci I; Leitch DC A reactivity model for oxidative addition to palladium enables quantitative predictions for catalytic cross-coupling reactions. Chem. Sci 2022, 13, 3477–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai X; Chen Y; Garrell S; Liu H; Zhang L-K; Palani A; Hughes G; Nargund R Ligand-Dependent Site-Selective Suzuki Cross-Coupling of 3,5-Dichloropyridazines. J. Org. Chem 2013, 78, 7758–7763. [DOI] [PubMed] [Google Scholar]
- 6.Fowler JM; Britton E; Pask CM; Willans CE; Hardie MJ Cyclotriveratrylene-tethered trinuclear palladium(II)-NHC complexes; reversal of site selectivity in Suzuki-Miyaura reactions. Dalton Trans. 2019, 48, 14687–14695. [DOI] [PubMed] [Google Scholar]
- 7.Norman JP; Larson NG; Entz ED; Neufeldt SR Unconventional Site-Selectivity in Palladium-Catalyzed Cross-Couplings of Dichloroheteroarenes under Ligand-Controlled and Ligand-Free Systems. J. Org. Chem 2022, 87, 7414–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang M; Chen J; He C; Hu X; Ding Y; Kuang Y; Liu J; Huang Q Palladium-Catalyzed C-4 Selective Coupling of 2,4-Dichloropyridines and Synthesis of Pyridine-Based Dyes for Live-Cell Imaging. J. Org. Chem 2020, 85, 6498–6508. [DOI] [PubMed] [Google Scholar]
- 9.(a) The precatalysts used in ref 7 are of the type (η3-1-tBu-indenyl)Pd(NHC)(Cl), and were designed to release monoligated PdL: Melvin PR; Nova A; Balcells D; Dai W; Hazari N; Hruszkewycz DP; Shah HP; Tudge MT Design of a Versatile and Improved Precatalyst Scaffold for Palladium-Catalyzed Cross-Coupling: (η3-1-tBu-indenyl)2(μ-Cl)2Pd2. ACS Catal. 2015, 5, 3680–3688; [Google Scholar]; (b) Espinosa MR; Doppiu A; Hazari N Differences in the Performance of Allyl Based Palladium Precatalysts for Suzuki-Miyaura Reactions. Adv. Synth. Catal 2020, 362, 5062–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Multinuclear clusters are implicated in C4-selective cross-coupling of 2,4-dibromopyridine at low ligand loadings in a Pd/PPh3 system, although the reason for a C4-preference of multinuclear clusters is not yet known: Scott NWJ; Ford MJ; Jeddi N; Eyles A; Simon L; Whitwood AC; Tanner T; Willans CE; Fairlamb IJS A Dichotomy in Cross-Coupling Site Selectivity in a Dihalogenated Heteroarene: Influence of Mononuclear Pd, Pd Clusters, and Pd Nanoparticles—the Case for Exploiting Pd Catalyst Speciation. J. Am. Chem. Soc 2021, 143, 9682–9693. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Appleby KM; Dzotsi E; Scott NWJ; Dexin G; Jeddi N; Whitwood AC; Pridmore NE; Hart S; Duckett SB; Fairlamb IJS Bridging the Gap from Mononuclear PdII Precatalysts to Pd Nanoparticles: Identification of Intermediate Linear [Pd3(XPh3)4]2+ Clusters as Catalytic Species for Suzuki–Miyaura Couplings (X = P, As). Organometallics 2021, 40, 3560–3570. [Google Scholar]
- 11.(a) For examples of the use of Pd(COD)(CH2SiR3)2 + free ligand "L" as a precursor to putative PdLn see: Fors BP; Watson DA; Biscoe MR; Buchwald SL A Highly Active Catalyst for Pd-Catalyzed Amination Reactions: Cross-Coupling Reactions Using Aryl Mesylates and the Highly Selective Monoarylation of Primary Amines Using Aryl Chlorides. J. Am. Chem. Soc 2008, 130, 13552–13554; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Watson DA, Su M; Teverovskiy G; Zhang Y; Garcia-Fortanet J; Kinzel T; Buchwald SL Formation of ArF from LPdAr(F): Catalytic Conversion of Aryl Triflates to Aryl Fluorides. Science, 2009, 325, 1661–1664; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sergeev AG; Schultz T; Torborg C; Spannenberg A; Neumann H; Beller M Palladium-Catalyzed Hydroxylation of Aryl Halides under Ambient Conditions. Angew. Chem., Int. Ed, 2009, 48, 7595–7599; [DOI] [PubMed] [Google Scholar]; (d) Larini P; Kefalidis CE; Jazzar R; Renaudat A; Clot E; Baudoin O On the Mechanism of the Palladium-Catalyzed β-Arylation of Ester Enolates Chem.–Eur. J, 2012, 18, 1932–1944; [DOI] [PubMed] [Google Scholar]; (e) Andersen TL; Friis SD; Audrain H; Nordeman P; Antoni G; Skrydstrup T Efficient 11C-Carbonylation of Isolated Aryl Palladium Complexes for PET: Application to Challenging Radiopharmaceutical Synthesis. J. Am. Chem. Soc 2015, 137, 1548–1555; [DOI] [PubMed] [Google Scholar]; (f) Milner PJ; Yang Y; Buchwald SL In-Depth Assessment of the Palladium-Catalyzed Fluorination of Five-Membered Heteroaryl Bromides. Organometallics 2015, 34, 4775–4780; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Elias EK; Rehbein SM; Neufeldt SR Solvent coordination to palladium can invert the selectivity of oxidative addition. Chem. Sci 2022, 13, 1618–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gensch T; dos Passos Gomes G; Friederich P; Peters E; Gaudin T; Pollice R; Jorner K; Nigam A; D'Addario ML; Sigman MS; Aspuru-Guzik A A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis. J. Am. Chem. Soc 2022, 144, 1205–1217. [DOI] [PubMed] [Google Scholar]
- 13.Clavier H; Nolan SP Percent buried volume for phosphine and N-heterocyclic carbene ligands: steric properties in organometallic chemistry. Chem. Commun 2010, 46, 841–861. [DOI] [PubMed] [Google Scholar]
- 14.(a) Vummaleti SVC; Nelson DJ; Poater A; Gómez-Suárez A; Cordes DB; Slawin AMZ; Nolan SP; Cavallo L What can NMR spectroscopy of selenoureas and phosphinidenes teach us about the π-accepting abilities of N-heterocyclic carbenes? Chem. Sci 2015, 6, 1895–1904; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lummiss JAM; Higman CS; Fyson DL; McDonald R; Fogg DE The divergent effects of strong NHC donation in catalysis. Chem. Sci 2015, 6, 6739–6746; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Back O; Henry-Ellinger M; Martin CD; Martin D; Bertrand G 31P NMR Chemical Shifts of Carbene-Phosphinidene Adducts as an Indicator of the π-Accepting Properties of Carbenes. Angew. Chem., Int. Ed 2013, 52, 2939–2943; [DOI] [PubMed] [Google Scholar]; (d) Morán-González L; Pedregal JR-G; Besora M; Maseras F Understanding the Binding Properties of N-heterocyclic Carbenes through BDE Matrix App. Eur. J. Inorg. Chem 2022, e202100932. [Google Scholar]
- 15.(a) For examples of nitrogen-containing substrates/products acting as ligands for Pd during cross-couplings, see Alcazar-Roman LM; Hartwig JF; Rheingold AL; Liable-Sands LM; Guzei IA Mechanistic Studies of the Palladium-Catalyzed Amination of Aryl Halides and the Oxidative Addition of Aryl Bromides to Pd(BINAP)2 and Pd(DPPF)2: An Unusual Case of Zero-Order Kinetic Behavior and Product Inhibition. J. Am. Chem. Soc 2000, 122, 4618–4630; [Google Scholar]; (b) Singh UK; Strieter ER; Blackmond DG; Buchwald SL Mechanistic Insights into the Pd(BINAP)-Catalyzed Amination of Aryl Bromides: Kinetic Studies under Synthetically Relevant Conditions. J. Am. Chem. Soc 2002, 124, 14104–14114; [DOI] [PubMed] [Google Scholar]; (c) Jutand A.l Négri S; Principaud A Formation of ArPdXL(amine) Complexes by Substitution of One Phosphane Ligand by an Amine in trans-ArPdX(PPh3)2 Complexes. Eur. J. Inorg. Chem 2005, 631–635; [Google Scholar]; (d) Tougerti A; Negri S; Jutand A Mechanism of the Copper-Free Palladium-Catalyzed Sonagashira Reactions: Multiple Role of Amines. Chem. Eur. J 2007, 13, 666–676; [DOI] [PubMed] [Google Scholar]; (e) Perego LA; Grimaud L; Bellina F Mechanistic Studies on the Palladium-Catalyzed Direct C-5 Arylation of Imidazoles: The Fundamental Role of the Azole as a Ligand for Palladium. Adv. Synth. Catal 2016, 358, 597–609. [Google Scholar]
- 16.(a) Evidence has been reported for oxidative addition of aryl halides at both bisligated Pd(PPh3)2 and monoligated Pd(PPh3): Amatore C; Jutand A; Khalil F; M'Barki MA; Mottier L Rates and Mechanisms of Oxidative Addition to Zerovalent Palladium Complexes Generated in Situ from Mixtures of Pd0(dba)2 and Triphenylphosphine. Organometallics 1993, 12, 3168–3178; [Google Scholar]; (b) Joshi C; Macharia JM; Izzo JA; Wambua V; Kim S; Hirschi JS; Vetticatt MJ Isotope Effects Reveal the Catalytic Mechanism of the Archetypical Suzuki-Miyaura Reaction. ACS Catal. 2022, 12, 2959–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman-Stonebraker SH; Smith SR; Borowski JE; Peters E; Gensch T; Johnson HC; Sigman MS; Doyle AG Univariate classification of phosphine ligation state and reactivity in cross-coupling catalysis. Science 2021, 374, 301–308. [DOI] [PubMed] [Google Scholar]
- 18.(a) Evidence suggests that PCy3 can support oxidative addition at both Pd(PCy3) and Pd(PCy3)3: Mitchell EA; Jessop PG; Baird MC A Kinetics Study of the Oxidative Addition of Bromobenzene to Pd(PCy3)2 (Cy = cyclohexyl) in a Nonpolar Medium: The Influence on Rates of Added PCy3 and Bromide Ion. Organometallics 2009, 28, 6732–6738; [Google Scholar]; (b) Barrios-Landeros F; Carrow BP; Hartwig JF Effect of Ligand Steric Properties and Halide Identity on the Mechanism for Oxidative Addition of Haloarenes to Trialkylphosphine Pd(0) Complexes. J. Am. Chem. Soc 2009, 131, 8141–8154; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McMullin CL; Jover J; Harvey JN; Fey N Accurate modelling of Pd(0) + PhX oxidative addition kinetics. Dalton Trans. 2010, 39, 10833–10836. [DOI] [PubMed] [Google Scholar]
- 19.(a) Portnoy M; Milstein D Mechanism of Aryl Chloride Oxidative Addition to Chelated Palladium(0) Complexes. Organometallics 1993, 12, 1665–1673; [Google Scholar]; (b) Maes BUW; Verbeek S; Verhelst T; Ekomié A; von Wolff N; Lefèvre G; Mitchell EA; Jutand A Oxidative Addition of Haloheteroarenes to Palladium(0): Concerted versus SNAr-Type Mechanism. Chem. Eur. J 2015, 21, 7858–7865; [DOI] [PubMed] [Google Scholar]; (c) Besora M; Maseras F The diverse mechanisms for the oxidative addition of C─Br bonds to Pd(PR3) and Pd(PR3)2 complexes. Dalton Trans. 2019, 48, 16242–16248. [DOI] [PubMed] [Google Scholar]
- 20.Senn HM; Ziegler T Oxidative Addition of Aryl Halides to Palladium(0) Complexes: A Density-Functional Study Including Solvation. Organometallics 2004, 23, 2980–2988. [Google Scholar]
- 21.Relative free energies are reported after applying Cramer and Truhlar's quasi-harmonic approximation to vibrational entropy: Ribeiro RF, Marenich AV, Cramer CJ and Truhlar DG, Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. Phys. Chem. B, 2011, 115, 14556–14562. [DOI] [PubMed] [Google Scholar]
- 22.(a) 3D images of optimized structures were generated with CYLview: CYLview20; Legault CY, Université de Sherbrooke, 2020. (http://www.cylview.org) [Google Scholar]; (b) 3D images of molecular orbitals were generated with Avogadro: Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR “Avogadro: An advanced semantic chemical editor, visualization, and analysis platform” J. Cheminformatics 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For previous experimental studies supporting a displacement mechanism for PdL2 during reaction of PhCl, see ref 19a.
- 24.For previous computational studies showing that PdL and PdL2 can favor different mechanisms during reaction with PhBr, see ref 19c.
- 25.(a) For seminal examples of computational studies showing a 3-centered concerted mechanism for oxidative addition of ArCl at PdL (L = PtBu3), see Ahlquist M; Norrby P-O Oxidative Addition of Aryl Chlorides to Monoligated Palladium(0): A DFT-SCRF Study. Organometallics 2007, 26, 550–553; [Google Scholar]; (b) Schoenebeck F; Houk KN Ligand-Controlled Regioselectivity in Palladium-Catalyzed Cross Coupling Reactions. J. Am. Chem. Soc 2010, 132, 2496–2497. [DOI] [PubMed] [Google Scholar]
- 26.The LUMO and LUMO+1 of PhCl are nearly isoenergetic (~1 kcal/mol difference). In contrast, the LUMO and LUMO+1 of 2 differ by about 9 kcal/mol.
- 27.Entz ED; Russell JEA; Hooker LV; Neufeldt SR Small Phosphine Ligands Enable Selective Oxidative Addition of Ar─O over Ar─Cl Bonds at Nickel(0). J. Am. Chem. Soc 2020, 142, 15454–15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.We also find that the preferred mechanism of oxidative addition of the C4-Cl bond of 2,4-dichloropyridine at PdL2 depends on L. Ligand-dependent mechanistic differences might explain the variable magnitude of C2-selectivity with small triaryl vs. trialkyl phosphines (see SI).
- 29.For prior evidence supporting a displacement mechanism for PdL2 (L = PPh3) during reaction of 2-chloropyridine, see ref 19b.
- 30.(a) Huang J; Schanz H-J; Stevens ED; Nolan SP Stereoelectronic Effects Characterizing Nucleophilic Carbene Ligands Bound to the Cp*RuCl (Cp* = η5-C5Me5) Moiety: A Structural and Thermochemical Investigation. Organometallics 1999, 18, 2370–2375; [Google Scholar]; (b) Hopkinson MN; Richter C; Schedler M; Glorius F An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [DOI] [PubMed] [Google Scholar]
- 31.A recent publication demonstrates that a C6-substituent can also slow the rate of oxidative addition at C2 due to sterics, although the effect of C6-sterics on C4─X oxidative addition was not examined: see ref 4.
- 32.Notably, LUMO differences are not the only variable that trends with selectivity. The most selective substrate (pyridazine) also has the smallest difference in bond dissociation energies. Additionally, pyridazines are worse ligands than pyridines, which could further disfavor bisligated Pd.
- 33.With 2,4-dibromopyridine 3, overarylation is observed (see ref 7).
- 34.All thermodynamic quantities were computed with the GoodVibes code (298.15 K) applying corrections for initial concentrations ([Pd] = 0.0075 M and [2] = 0.25 M): Luchini G, Alegre-Requena JV, Funes-Ardoiz I and Paton RS, GoodVibes: Automated Thermochemistry for Heterogeneous Computational Chemistry Data, F1000Research, 2020, 9, 291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.