

Summary
Immune checkpoint immunotherapy (ICI) can re-activate immune reactions against neoantigens, leading to remarkable remission in cancer patients. Nevertheless, only a minority of patients are responsive to ICI, and approaches for prediction of responsiveness are needed to improve the success of cancer treatments. While the tumor mutational burden (TMB) correlates positively with responsiveness and survival of patients undergoing ICI, the influence of the subcellular localizations of the neoantigens remains unclear. Here, we demonstrate in both a mouse melanoma model and human clinical datasets of 1,722 ICI-treated patients that a high proportion of membrane-localized neoantigens, particularly at the plasma membrane, correlate with responsiveness to ICI therapy and improved overall survival across multiple cancer types. We further show that combining membrane localization and TMB analyses can enhance the predictability of cancer patient response to ICI. Our results may have important implications for establishing future clinical guidelines to direct the choice of treatment toward ICI.
Keywords: tumor mutational burden, cell membrane, neoantigens, immunotherapy, cancer, biomarkers
Graphical abstract
Highlights
-
•
Subcellular localization of antigens modulates anti-tumor immune responses
-
•
Membrane-localized antigens (mAgs) improve response to immune checkpoint immunotherapy (ICI)
-
•
High proportion of mAgs extend survival of ICI-treated mice and cancer patients
-
•
mAgs and TMB analyses can be combined to improve prediction of response to ICI
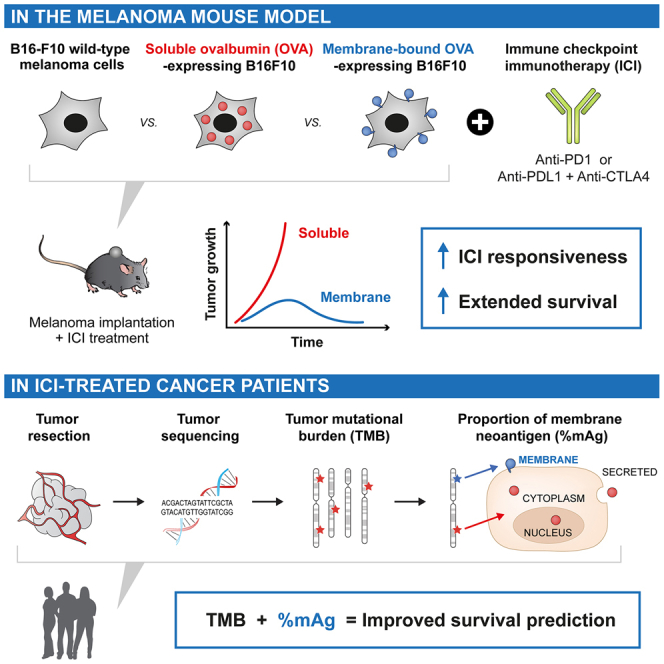
Goldberger et al. highlight that the subcellular localizations of tumor antigens modulate anti-cancer immune responses. Membrane-localized antigens (mAgs) correlate with improved response and extended survival upon immune checkpoint immunotherapy (ICI) in mice and cancer patients. Consideration of mAgs enhances the predictability of response to ICI when combined with TMB analysis, currently performed in the clinic.
Introduction
Immunotherapies have revolutionized the landscape of clinical oncology, being established as first-line treatments for multiple advanced cancer types, including melanoma, non-small cell lung cancer (NSCLC), and renal cell carcinoma.1,2,3 Despite the strong efficacy of immune checkpoint immunotherapy (ICI), less than 20% of patients show a complete or durable response.4,5 While infiltration of immune cells in the tumors6 and high tumor mutational burden (TMB) are key correlates of response to ICI,7,8,9,10,11,12 more accurate prediction of patient responsiveness to ICI remains an important challenge.13 Greater predictivity would enhance patient survival and quality of life and reduce the burden of treatments, including their number, duration, side effects, and associated costs.
Here, we hypothesized that the potency of immune response against tumor mutated proteins not only depends on the total TMB but also on the subcellular localization of these proteins in the tumor cell. Indeed, the efficiency of presentation of neoantigens on major histocompatibility complex (MHC) class I by the tumor cell, required for recognition and killing by CD8+ T cells,14 might vary for cytoplasmic, nuclear, membrane-localized, or secreted proteins because of their specific intracellular processing and trafficking routes.15,16,17 In addition, the efficiency of collection and presentation of neoantigens by antigen-presenting cells (APCs) on MHC class I and II to generate CD8+ and CD4+ T cell responses, respectively, could similarly be impacted by these different forms of proteins upon release in debris, vesicles, or the extracellular milieu. Apart from antigen presentation, membrane-localized antigens (mAgs) can be recognized by antibodies, induced via B cell immunity, which could allow antibody-dependent cytotoxic mechanisms that kill tumor cells by activating natural killer (NK) cells, macrophages, or the immune complement cascade.18,19 Of note, tumor mutated proteins that successfully activate an adaptive immune response are commonly defined as tumor neoantigens.
To date, very few reports have examined how the subcellular localization of tumor neoantigens modulates anti-cancer immunity. In this study, we show that high proportion of mAgs increase tumor immunogenicity and improve responsiveness to ICI therapies. We first demonstrated in a melanoma mouse model that membrane-localization of ovalbumin (mOVA; used as a model tumor antigen) in B16-F10 cells increased local and systemic immunity compared with soluble OVA and rendered these tumors highly susceptible to ICI in a manner that did not depend on immunoglobulin G (IgG) antibody-mediated cytotoxicity but rather on T cell immune responses. We then questioned whether a high proportion of mAgs improves responsiveness to ICI in cancer patients. We developed a simple algorithm that extracts the subcellular localizations associated with tumor mutated genes from the UniProtKB/Swiss-Prot database20 and analyzed the publicly available sequencing data of 4,864 patients, treated or not with ICI, from studies by Samstein et al.,7 Hellman et al.,8 and Hugo et al.9 We demonstrated that a high mAg proportion correlates with increased patient survival and responsiveness to ICI across multiple cancer types. Moreover, we highlighted that mutated genes encoding for some particular membrane-localized proteins may serve as potent biomarkers to predict extended survival of patients upon ICI, such as NOTCH1 or NTRK3 in NSCLC. Together, our results highlight the importance of considering the subcellular localization of tumor neoantigens, in particular mAg, in addition to the total TMB to improve the predictivity of patient responsiveness to ICI therapy and, potentially, clinical guidelines for selection of the most appropriate cancer treatment.
Results
mAgs increase tumor immunogenicity
We began by studying the effect of cell mAgs in the B16-F10 murine melanoma model. We first modified B16-F10 cells for expression of membrane-bound OVA (B16mOVA) by fusing the full-length OVA sequence to the transmembrane domain of H-2DB (Figure S1A).21 As a control, we used B16-F10 cells that expresses full-length OVA in a soluble form (i.e., not membrane bound; B16-OVA). For both designs, we generated cell lines with matching high (HI) and low (LO) levels of OVA expression, as quantified by qPCR in vitro and in vivo (Figures S1B and S1C). The presence of OVA at the surface of the B16mOVA cells was confirmed by flow cytometry and fluorescence (Figures S1D and S1E).
Upon intradermal injection in C57BL6 wild-type (WT) mice, all cell lines were tumorigenic. B16mOVAHI tumors grew significantly slower than B16-OVAHI and the parental B16 WT, which resulted in extended survival of mice bearing B16mOVAHI tumors (Figures 1A and 1B). This effect was antigen dose dependent, as seen by an intermediate growth rate of the B16mOVALO tumors. To confirm that this difference was due to immune-mediated rejection of the tumor rather than a difference in cell growth/division rate, we evaluated B16mOVAHI tumor growth in transgenic Act-mOVA mice, which are immune tolerant to mOVA. In these mice, B16mOVAHI tumors grew faster than B16 WT tumors, demonstrating an intact proliferation capacity of the B16mOVAHI cells (Figure 1C).
Figure 1.

Effects of mAgs on melanoma anti-tumor immune responses
B16-F10 melanoma cells modified to express membrane-bound or soluble full-length OVA (B16mOVA and B16-OVA, respectively), at high (HI) or low (LO) levels, were injected intradermally into C57BL6 mice. The parental B16-F10 wild-type (WT) cells were used as a control.
(A and B) Tumor growth (A) and survival (B) of the different OVA-expressing B16 cell lines upon injection in vivo (n ≥ 8, mean ± SEM, Kruskal-Wallis with Dunn’s post test on day 12 in A, log rank tests with Holm-Bonferroni p value adjustment in B).
(C) Tumor growth of B16mOVAHI in Act-mOVA mice compared with growth in WT mice (n ≥ 3, mean ± SEM, Kruskal-Wallis with Dunn’s post-test on day 10).
(D) Immune cell populations infiltrated in the different tumors on day 10 post injection, analyzed by flow cytometry, in percentage and absolute number per weight (n ≥ 4, mean ± SD, ANOVA with Tukey’s post-test and Brown-Forsythe correction when appropriate).
(E) CD8+ T cells, CD4+ T cells, and NK1.1+ cell populations infiltrated into the different tumors on day 10 post injection, analyzed by flow cytometry (n ≥ 4, mean ± SD, ANOVA with Tukey’s post-test and Brown-Forsythe correction when needed).
(F) Intratumoral cytokine quantification via Legendplex in the different tumors in vivo on day 10 (n ≥ 6, mean ± SEM, ANOVA with Tukey’s post-test).
(G) Increase in fluorescence intensity of SIINFEKL-loaded MHC class I for B16mOVA and B16-OVA cell lines in the presence of IFNγ (mean ± SEM, ANOVA with Tukey’s post-test at IFNγ doses ≥ 1 ng/mL).
(H) Proliferation of OVA-specific CD8+ T cells (OT-I) in the presence of in vitro-induced CD103+ DCs stimulated by EVs or non-EVs derived from B16mOVA and B16-OVA cell lines (mean ± SD, ANOVA with Brown-Forsythe correction with Dunnett’s post-test).
(I) Ex vivo restimulation of OVA-specific CD8+ and CD4+ T cells in the spleen of mice bearing the different OVA-expressing tumors on day 10 post injection (n ≥ 4, mean ± SD, Kruskal-Wallis with Dunn’s post-test).
(J) Anti-OVA antibody quantification per IgG subtype in the plasma of tumor-bearing mice on day 10 post-injection (AUC, area under the curve; n ≥ 4, mean ± SD, Kruskal-Wallis with Dunn’s post-test).
Therefore, we analyzed immune cell infiltrates in the different OVA-expressing B16-F10 tumors (Figures S2 and S3). We found a significant increase in CD45+ immune cells in tumors expressing mOVA compared with dose-matched soluble OVA, about 2-fold in the case of B16mOVAHI vs. B16-OVAHI tumors, in terms of percentage of tumor-infiltrating cells and total number of cells per tumor weight (Figure 1D). Particularly, CD8+ T cells and NK cells were more numerous in this tumor but not CD4+ T cells (Figures 1E, S4A, and S4B). No difference in PD-1 expression was observed in T cells in tumors expressing mOVA versus soluble OVA (Figure S4C). Among the other immune cell types screened, NK T cells were slightly increased, and dendritic cells (DCs) and B cells were slightly decreased in B16mOVA tumors when quantified relative to the total CD45+ immune cell population (Figure S4D). No differences were observed in macrophages or myeloid-derived suppressor cells (MDSCs).
In addition, we characterized the inflammatory environment of the tumors via intratumoral cytokine quantification. We detected a 6.4-fold increase in interferon-γ (IFNγ) and a 2.6-fold increase in tumor necrosis factor alpha (TNF-α) between B16mOVAHI and B16-OVAHI tumors. We also observed a significant increase in the chemokines CCL-3, CCL-4, CXCL-9, and CXCL-10 (Figure 1F) but not in IFNα, CCL-2, interleukin-4 (IL-4), IL-10, granulocyte-macrophage colony-stimulating factor (GM-CSF), or vascular endothelial growth factor (VEGF) (Figure S4E).
Together, there are consistent cellular and molecular pro-inflammatory microenvironments in B16mOVA. CXCL-9 and CXCL-10 are critically involved in T cell recruitment in the tumor microenvironment and are widely expressed in so-called “hot” inflamed tumors.22 Interestingly, CXCL-9 and CXCL-10 have also been shown to colocalize with CCL-4, and CCL-3 and CCL-4 recruit CD8+ T cells.23 In addition to affecting T cells, CXCL-10 also increases NK cell recruitment, and CCL-3 and CCL-4 enhance their cytotoxic activity.24 Upon activation, CD8+ T cells and NK cells secrete high levels of IFNγ and TNF-α in the tumor microenvironment (Figures 1E and 1F).
Because antigen presentation on the MHC of tumor cells is needed for specific recognition and killing by T cells, we next questioned whether presentation of OVA-derived antigen is particularly enhanced in the more immunogenic B16mOVA cell lines. As suggested by previous studies,25,26 we showed in vitro that all modified B16-F10 cell lines highly increase their expression of MHC class I and MHC class II in the presence of IFNγ (Figures S5A and S5B). Consistent with this, presentation of the immunodominant OVA peptide SIINFEKL on MHC class I was enhanced in all cell lines in an IFNγ dose-dependent fashion. Moreover, the level of SIINFEKL-loaded MHC class I was strikingly elevated in the B16mOVAHI cell line compared with the B16mOVALO and B16-OVA lines (Figure 1G). The increased level of IFNγ associated with increased presentation of OVA on MHC class I can further explain the higher immunogenicity of B16mOVAHI/LO tumors.
Next, we assessed whether immunity against OVA in B16mOVA-bearing mice was strong enough to induce systemic immunity. We highlighted that membrane-bound OVA was secreted on extracellular vesicles (EVs) produced by B16mOVA (Figure S5C), whereas OVA was not detected in EVs derived from B16-OVA but, rather, was enriched in the non-EV fraction as soluble proteins. This difference is likely to impact antigen transport and availability to APCs in vivo because it has been shown that EVs carrying cancer-associated antigens are more effectively taken up by DCs than soluble antigens, which enhances cross-presentation.27,28 Indeed, we then confirmed that in vitro-induced CD103+ DCs exposed to EVs derived from B16mOVAHI enhanced OVA-specific OT-I CD8+ T cell proliferation compared with EVs or non-EV proteins derived from B16-OVAHI cells (Figure 1H). These data suggest that EVs shed by B16mOVA tumors could enhance the antigen-specific immune response of mAgs.
Finally, we used ex vivo restimulation of splenocytes by OVA-derived MHC class I and MHC class II immunodominant peptides to quantify the systemic cellular immunity raised in mice bearing B16mOVA and B16-OVA tumors. CD8+ and CD4+ T cell responses were increased in mice with B16mOVA versus B16-OVA, as revealed by higher secretion of IFN-γ (Figure 1I). In addition to cellular immunity, OVA-specific antibody responses were detected in the plasma of tumor-bearing mice for B16mOVA and B16-OVA, but different subtypes of IgG were generated, depending on antigen localization. Particularly, OVA-specific IgG2b and IgG2c were detected in mice bearing B16mOVA tumors but were largely absent in those bearing B16-OVA tumors (Figure 1J). Thus, antigen-specific systemic immune responses were enhanced in mice bearing B16mOVA tumors.
Together, these results showed that tumor mAgs, here modeled by mOVA, strongly enhanced tumor immunogenicity locally and systemically, resulting in slowed tumor growth and extended mouse survival.
mAgs restore responsiveness to ICI
While B16-F10 WT melanoma does not respond ICI, we examined whether the increased immunogenicity of B16mOVA, particularly the enhanced presence of intratumoral T cells, would render them more susceptible to ICI. Remarkably, all mice (5 out of 5) bearing B16mOVAHI tumors and treated with anti-PD1 therapy showed complete responses to ICI, whereas B16-OVAHI- and B16 WT-bearing mice were unresponsive (Figure 2A). Lowering the antigen dose in the B16mOVALO group reduced the efficacy of ICI but resulted in 2 out of 5 tumor eradications and otherwise slowed tumor growth. Such effects were also confirmed using the combination therapy anti-PD-L1 and anti-CTLA-4 (Figure 2B). In both therapies, responsiveness to ICI significantly extended survival.
Figure 2.

Effects of mAgs on the responsiveness and survival of melanoma-bearing mice to ICI
The different OVA-expressing or WT B16-F10 melanoma cells were injected intradermally into C57BL6 mice. Mice were treated with anti-PD1 or a combination of anti-PD-L1 + anti-CTLA-4 intraperitoneally when the tumor volume reached 20–50 mm3 (gray thresholds).
(A) Tumor growth and associated survival of OVA-expressing tumor-bearing mice treated with anti-PD-1 (n ≥ 5, mean ± SEM, log rank tests with Holm-Bonferroni p value adjustment).
(B) Tumor growth and associated survival of OVA-expressing tumor-bearing mice treated with anti-PD-L1 + anti-CTLA-4 (n ≥ 3, mean ± SEM, log rank tests with Holm-Bonferroni p value adjustment).
(C) B16mOVAHI tumor growth upon depletion of CD8+ or/and CD4+ T cells with anti-PD-1 treatment (n ≥ 5, mean ± SEM, Kruskal-Wallis with Dunn’s post-test on day 14).
(D) B16mOVAHI tumor growth in BatF3−/− mice upon anti-PD1 treatment (n ≥ 5, mean ± SEM, Mann-Whitney test on day 11).
(E) B16mOVAHI tumor growth upon depletion of NK1.1+ or/and CD8+ T cells with anti-PD1 treatment (n ≥ 5, mean ± SEM, Kruskal-Wallis with Dunn’s post-test on day 20).
(F) B16mOVAHI tumor growth in MuMt− mice with treatment with anti-PD-1 (n ≥ 4, mean ± SEM, Kruskal-Wallis with Dunn’s post-test on day 17).
(G) Tumor growth in mice that survived B16mOVAHI tumors treated with anti-PD-1 upon rechallenge with B16-F10 WT cells (n ≥ 8, mean ± SEM, Mann-Whitney test on day 14) and associated survival (n ≥ 8, mean ± SEM, log rank test).
We then characterized which cell types were predominantly involved in B16mOVAHI tumor rejection by depleting specific immune cell populations upon ICI treatment. In the absence of the CD4+ T cells, CD8+ T cells were still capable of controlling tumor growth and led to rejection in 3 out of 5 mice, thus showing slightly lower efficacy than with proper help from the CD4+ T cells, as highlighted by the isotype control group, in which all tumors were rejected (Figure 2C). In contrast, CD4+ T cells alone were insufficient to eradicate tumors, although they slightly slowed tumor growth compared with tumors depleted of CD8+ and CD4+ T cells. Because education of antigen-specific CD8+ T cells strongly relies on the activity of cross-presenting DCs, we used the BatF3−/− knockout mouse model to evaluate their role in B16mOVAHI rejection upon anti-PD1 therapy. Surprisingly, we observed that BatF3−/− mice were capable of fully rejecting B16mOVAHI tumors when treated with anti-PD-1 (Figure 2D), highlighting that conventional cross-presenting DCs were not necessary for tumor rejection. While the BatF3−/− knockout mouse is the gold standard model for defective antigen cross-presentation, this mouse still contains DC subsets and other cells (e.g., macrophages) capable of cross-presentation,29,30,31 which might have been sufficient to educate enough CD8+ T cells, further proliferating upon help from CD4+ T cells. In addition, NK cells can cross-talk with DCs, acting as helpers to promote maturation and cross-presentation.32,33 Therefore, in our model, the significant increase in NK cells in B16mOVA tumors may have successfully provided a compensatory mechanisms for effective development of CD8+ T cells in BatF3−/− mice. Similarly, we found that NK1.1+ cells were not required for responsiveness to ICI in WT mice (Figure 2E). Last, we found that muMT− transgenic mice, which lack mature B cells and cannot produce IgG, were also able to completely reject B16mOVAHI tumors upon ICI, importantly highlighting that IgG-based antibody-dependent cytotoxicity mechanisms were not necessary for tumor eradication (Figure 2F). Together, these data suggest that tumor rejection is mainly mediated by CD8+ T cell cytotoxic activity, with help from the CD4+ T cells needed for optimal efficacy. While other mechanisms of BatF3−/− cross-presenting DCs, NK cells, and antibodies have not been found to be necessary, we do not exclude that they might take place in WT mice.
Finally, we investigated whether immune rejection of B16mOVA tumors upon ICI was solely directed against mOVA or whether immune reactions against other tumor-associated antigens were at play. Upon re-challenge, mice that had rejected B16mOVA tumors showed delayed growth of B16 WT tumors, suggesting the presence of pre-existing immune reactions against B16 WT neoantigens induced during initial rejection of B16mOVA (Figure 2G). Secondary B16 WT tumors remained non-responsive to ICI. Therefore, while mOVA was necessary to eradicate the primary tumor upon ICI, its loss in secondary tumors still resulted in delayed tumor growth.
mAgs increase patient survival upon ICI
The remarkable ability of an mAg (i.e., mOVA) to restore responsiveness to ICI in the murine melanoma model encouraged us to validate this hypothesis in cancer patients. Therefore, we analyzed publicly available tumor mutation sequencing data of patients treated or not with ICI from 3 independent studies by Samstein et al.,7 Hellman et al.,8 and Hugo et al.9 in which the tumors were sequenced pre ICI. For each tumor mutated gene detected in patients, we extracted the subcellular localization of its encoded protein from the UniProtKB/Swiss-Prot database20 (Data S1). We then quantified, per patient, the number of mutated genes that encode for membrane, cytoplasmic, nuclear, or secreted proteins. Genes that encode proteins expressed at several localizations were classified in all locations in a non-exclusive manner. We last normalized the number of protein-encoding mutated genes at a specific subcellular location to the total number of mutated genes, obtaining proportions of protein-encoding mutated genes per subcellular location (Figure 3A), here referred as the proportion of mAgs, cytoplasmic neoantigens (cAgs), nuclear neoantigens (nAgs), or secreted neoantigens (sAgs).
Figure 3.

Proportion of mAgs correlates with increased survival in cancer patients treated with ICI in a pan-cancer analysis
Data are available from Samstein et al.7 Patients suffering from 9 different cancer types were treated with immune checkpoint inhibitor (ICI) immunotherapy, and their survival was evaluated from the first day of treatment (n = 1,609 patients). A control cohort of patients not treated with ICI was used for comparison (n = 3,142 patients). All Kaplan-Meier survival curves and Cox hazard ratios (HRs) for survival were statistically compared using log rank tests.
(A) Workflow for analysis of subcellular localizations associated with the tumor mutations.
(B) Survival of patients with a high (top 25% group) or low (bottom 50%, 25%, or 10% groups) proportion of mAgs.
(C) Survival of patients having a high (top 25% group) or low (bottom 25% group) proportion of cAgs, nAgs, or sAgs.
(D) Survival of patients as a function of their predominant subcellular location of mutated genes (top 25% groups of membrane, nucleus, or cytoplasm mutations; p values adjusted using Holm-Bonferroni correction).
(E) Survival of non-ICI-treated patients that have a high (top 25%) or low (bottom 25%) proportion of mAgs.
(F) HR for survival of patients having a high (top 25%) versus low (bottom 25%) proportion of mAgs upon ICI treatment, not treated with immunotherapy (non-ICI), or depending on the type of ICI received (i.e., PD-1/PDL-1, CTLA-4, or combination) (HR ± 95% confidence interval [CI]).
We first analyzed the dataset by Samstein et al.,7 comprising 1,609 patients with 9 different types of advanced cancers treated with ICI whose tumor mutations were determined using targeted next-generation sequencing (MSK-IMPACT) (Data S2). In total, 424 genes out of the 469 sequenced were classified in the 4 subcellular locations of interest (Figure S6A). We compared groups of patients with high and low proportions of neoantigens for each specific location using the cutoff values of the top and bottom group quartiles (top 25% vs. bottom 25%; Figure S6B). A high proportion of mAgs was found to correlate with significantly increased patient survival (Figure 3B). This effect was also conserved at percentiles other than 25% (Figure S6C). Interestingly, an insufficient proportion of mAgs was strongly associated with worsened survival, as highlighted by the gradual decrease between the groups bottom 50%, 25%, and 10%, with the bottom 10% group being patients with no membrane-localized mutation (Figure 3B). None of the other subcellular locations consistently correlated with a significant improvement in survival (Figures 3C and S6D–S6F). Instead, trends toward reduced survival were observed for high cAgs and nAgs, and no difference was seen for sAgs. Further division into exclusive patient groups with high proportions of neoantigens at a single location highlighted that membrane localization provides higher survival benefits than the cytoplasmic and nuclear localizations (Figures 3D, S6G, and S6H).
We then questioned whether the survival advantage that correlated with the high proportion of mAgs was present in non-ICI-treated patients. We analyzed 3,142 patients from the non-ICI-treated control cohort of Samstein et al.7,34 (Data S3) and found that no survival benefit was associated with membrane localization in the absence of ICI (Figures 3E and 3F). However, all types of ICI therapies (PD-1/PD-L1, CTLA-4, or the combination PD-1/PD-L1 + CTLA-4) correlated with extended survival in patients harboring a high proportion of mAgs, as indicated by a hazard ratio (HR) for survival inferior to 1. This effect did not reach statistical significance for CTLA-4, likely because of the limited number of patients in this group (Figure 3F).
Together, these findings suggest that a high proportion of mAgs improves cancer patient survival with different types of ICI treatments.
mAgs improve TMB-based survival prediction upon ICI
In the clinic, tumors of patients are sequenced to determine the TMB, which is currently used as a predictive marker for responsiveness to ICI and as a criterion for patient eligibility for ICI.35 Because determination of the proportion of mAgs from these sequencing data would require only a simple algorithm but no additional clinical or laboratory procedures as used for determination of the TMB, we examined the benefit of combining mAg analysis with the standard total TMB analysis to predict survival upon ICI. To do so, we separated the ICI-treated patients from Samstein et al.7 into 4 exclusive groups of low vs. high TMB and low vs. high proportion of mAgs, using the respective medians as cutoffs. We showed that, in each category of TMB, patients with a high mAg proportion have significantly extended survival compared with patients with a low mAg proportion (Figure 4A). No correlation was found between the proportion of tumor mAgs and the TMB (Figure 4B). In addition, a multivariate Cox regression analysis indicated that mAg proportion significantly improves prediction of ICI-treated patient survival after adjustment for TMB (Figure 4B).
Figure 4.

Proportion of mAgs improves TMB-based patient survival prediction
All comparisons for survival were tested using log rank tests.
(A) Comparison of survival of patients with a high (>median) or low (<median) proportion of mAgs and TMB.
(B) Correlation between proportion of mAgs and total TMB (Spearman correlation) and multivariate Cox regression analysis.
(C) Comparison of survival of patients with a high (>median) or low (<median) proportion of mAgs for different levels of TMB (with TMB ≥ 10 mut/Mbp being the FDA-validated criterion for ICI treatment for solid tumors35) in a pan-cancer analysis.
(D) Survival of patients with low TMB (<10 mut/Mbp) and high proportion of mAgs or with high TMB (between 10 and 20 mut/Mbp) and low proportion of mAgs in a pan-cancer analysis.
Currently, the TMB threshold defined by the US Food and Drug Administration (FDA) for patient eligibility to ICI is TMB ≥10 mutations (mut)/Mbp, for adults and children with unresectable or metastatic solid tumors that failed to respond to prior therapies.28 We therefore repeated the analysis, taking into account this FDA criterion. We found that a high mAg proportion enhanced prediction of survival in patients with low TMB (<10 mut/Mbp) and showed positive trends in patients with high TMB (≥10 mut/Mbp) (Figure 4C). In this cohort, we noticed that patients with TMB ≥10 mut/Mbp did not have homogeneous survival. Indeed, patients with a TMB between 10 and 20 mut/Mbp had significantly lower survival than patients with >20 mut/Mbp upon treatment with anti-PD1 (Figure S7A), although they are eligible for immunotherapy according to the FDA criterion. Each of these two groups represents 15% of the cohort. Interestingly, we observed that patients with a TMB of 10–20 mut/Mbp and low proportion of mAgs, for whom ICI is indicated, had similar survival as patients with a low TMB but high proportion of mAgs (Figure 4D), who may not currently qualify for ICI. The latter represent 30.5% of the patients in the Samstein et al. dataset, which could thus be considered for ICI when taking the proportion of mAgs into account but not otherwise. This suggests that the proportion of mAgs could be a valuable parameter to include, in addition to current TMB analysis, to extend the inclusion criteria for ICI in the clinic and improve prediction of patient survival upon treatment.
Impact of mAgs in different cancer types
The ICI-treated cohort analyzed above included patients with 9 different types of cancers, distributed non-equally (Figure S7B). When comparing the distribution of cancer types within the top 25% and bottom 25% of the mAg groups, we noticed that the population with a high proportion of mAgs was enriched in melanoma, renal cell carcinoma, and colorectal cancer patients and depleted from bladder cancer, glioma, and head-and-neck cancer patients (Figure S7C). This implied that not all cancer types had the same distribution of mAg proportion. In fact, glioma, bladder, and head-and-neck cancers had a significantly lower mAg proportion than the pan-cancer group, whereas colorectal cancer (with high-microsatellite instability) and melanoma cancers had significantly more (Figure 5A).
Figure 5.

High mAg proportion correlates with increased survival in multiple cancer types
(A) Distribution of mAg proportion by cancer type (blue line, cutoff value [CV] for the pan-cancer upper quartile; red line, CV for the pan-cancer lower quartile; Kruskal-Wallis test with Dunn’s post-tests for comparisons with the pan-cancer group).
(B) Heatmap of the HR for survival of patients harboring a high versus low proportion of mAgs, cAgs, nAgs, or sAgs per cancer type (#p < 0.1, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). High and low groups are determined using either the CVs from the pan-cancer group or the upper and lower quartiles (25%) specific to each cancer type (log rank tests).
Therefore, we detailed the effects of a high proportion of mAgs and other subcellular localizations per cancer type. We computed the HR for survival to compare patients with high versus low proportions of neoantigens at a specific location (Figure 5B; Data S4), using 2 different strategies: (1) keeping the same cutoff values that we used for the pan-cancer group analysis in Figure 3, reasoning that a “universal” threshold might be determined across cancers, or (2) using the upper and lower quartile values specific to each cancer type. Overall, a high proportion of mAgs correlates with statistically significant better survival in 2 out of 9 individual cancers, namely in renal cell carcinoma and head-and-neck cancer. Trends toward improved survival were observed in 4 additional cancer types, with a close-to-significant p value of 0.072 in esophagogastric cancer (Figure 5B). The lack of statistical significance in these cancer types might be due to smaller effects or limited numbers of patients in each subcohort. On the other hand, a high proportion of nAgs and cAgs was associated with significantly worsened survival in 1 and 2 cancer types, respectively, and negative trends were observed in 4 or 5 additional cancer types. Besides, a high proportion of sAgs did not strongly impact patient survival. Interestingly, both thresholding methods for selection of high vs. low groups showed very similar results, except for glioma and bladder cancers at the membrane locations. Further analysis with a higher number of patients would clarify whether an absolute threshold for mAg proportion can be determined to predict increased survival upon ICI across cancers.
mAgs predict patient response to ICI
While the metric of survival is a relevant measure to evaluate effectiveness of ICI, response rate and survival do not always correlate well. Hence, we searched for published datasets in which the patient response to ICI was reported. We found 2 such studies, from Hellman et al.8 and Hugo et al.,9 which, respectively, focused on patients with NSCLC treated with anti-PD1 and anti-CTLA-4 (75 patients) and metastatic melanoma treated with anti-PD1 (38 patients). Both studies used whole-exome sequencing (WES) to determine tumor mutations in patients prior to treatment. We thus repeated the subcellular localization analysis using the same algorithm to categorize tumor mutations according to their possible expression in the membrane, cytoplasm, or nucleus or as secreted proteins (Figure S8A; Data S5 and S6). Because more genes were sequenced by WES than by MSK-IMPACT, the detected variation range of the mAg proportion in the WES-sequenced patients was smaller, but the overall median remained similar (Figure S8B). The lowered mAg proportion found in the cohort from Hellman et al.8 might be due to the increased number of genes for which the subcellular locations could not be determined.
In this NSCLC cohort,8 patients who responded to ICI had a significantly higher proportion of neoantigens at the membrane but not at the other studied locations (Figures 6A and S8C). Impressively, the response rate was 61% in the group with a high mAg proportion (25% top) vs. 23.5% in patients with a low mAg proportion (25% bottom) (Figure 6B). In addition, patients with a high mAg proportion tended to survive longer, although statistical significance was not obtained, highlighting the potential discrepancy between ICI responsiveness and overall survival readouts (Figure S8D). Importantly, Hellmann et al.8 highlighted in their cohort that the responsiveness to anti-PD-1 plus anti-CTLA4 associated with TMB was independent of tumor PD-L1 expression; here, we similarly found that the proportion of mAgs did not correlate with expression of PD-L1 in the tumor (Figure S8E).
Figure 6.

Proportion of mAg correlates with better responsiveness to cancer ICI
Data are from Hellman et al.8 Patients (n = 75) with NSCLC were treated with anti-PD-1 + anti-CTLA-4, and their responsiveness to treatment was evaluated (responders, complete response [CR] or partial response [PR]; non-responders, stable disease [SD] or progressive disease [PD]).
(A) mAg proportion in patients who responded or not to the immunotherapy (Mann-Whitney test).
(B) Proportion of responders and non-responders among patients with a high (top 25%) or low (bottom 25%) proportion of mAgs (Fisher’s exact test).
(C) Proportion of mAgs at the cell plasma membrane or in other specific membrane-containing cell organelles in responders and non-responders to immunotherapy.
(D) Heatmap of the HR for survival, comparing the top vs. bottom 50%, 25%, or 10% groups having mutations at the plasma membrane or in other membrane-containing organelles, from the cohorts from Samstein et al.7 (pan-cancer group), Hellman et al.,8 and Hugo et al.9 (∗p < 0.05).
Last, we found in the melanoma cohort from Hugo et al.9 that a high proportion of mAgs enhanced overall survival (top 25% vs. bottom 25%) (Figure S8F) despite the low number of patients available. In addition, positive trends toward enhanced responsiveness were observed in patients with a high proportion of mAgs (8 out of 10 responders) compared with patients with a low proportion of mAgs (3 out of 10 responders) (p = 0.07; Figures S8G and S8H). Interestingly, Hugo et al.9 also showed in their study that high TMB correlated with enhanced survival but not significantly with tumor responsiveness to ICI.9
These additional studies further support the hypothesis that a high proportion of mAgs correlates with ICI responsiveness, consistent with the survival results obtained in the larger, multicancer cohort from Samstein et al.7 Importantly, they point out that this effect is conserved independent of the sequencing methods.
Predicted immunogenicity of mAgs
To get further insights into why mAgs would improve responsiveness to ICI, we compared the predicted number of epitopes in the proteins that were exclusively expressed at the membrane with the ones that were exclusively expressed in the cytoplasm, in the nucleus, or secreted, using the restricted gene list from the MSK-IMPACT sequencing by Samstein et al.7 We used the tools available from The Immune Epitope Database (IEDB)36 to predict the number of epitopes on MHC class I from each protein. We considered a possible epitope any peptide with <50 nM affinity for the MHC using the artificial neural network (ANN) prediction-based method.8,9,37 We found that membrane-localized proteins can generate more epitopes on MHC class I; indeed, 2.51% of peptides derived from membrane proteins were predicted to be immune epitopes compared with 1.60% for cytoplasmic proteins, 1.90% for nuclear proteins, and 1.45% for secreted proteins (Figure S9A). Moreover, the MHC class I immunogenicity prediction tool developed by IEDB highlighted that peptides derived from membrane proteins were significantly more immunogenic than peptides derived from the other localizations (Figure S9A). This suggested that mAgs might be better mounted on MHC class I on tumor cells and, thus, can potentially be better recognized by CD8+ T cells for subsequent killing.38
In addition, we used the datasets from Hellman et al.8 and Hugo et al.9 to analyze correlations between the predicted load of neoantigens and the proportion of mAgs using the patients’ predicted neoantigen burden reported by the authors in their original studies. We observed a statistically significant positive and strong correlation in the cohort from Hellman et al.8 but not in the cohort from Hugo et al.9 (Figures S9B and S9C). This difference between the datasets could be due to the difference in cancer type or the limited number of patients. Future studies would be necessary to comprehend the relations between the neoantigen burden and the mAg proportion.
mAgs at the plasma membrane
Next, we questioned the effects of mAgs depending on their presence in particular membranes in the cell. We refined our algorithm to segregate for cell membrane (i.e., plasma membrane), endoplasmic reticulum, Golgi apparatus, or endosome localizations. Proteins for which the subcellular localization was only labeled as “membrane” in the UniProtKB/Swiss-Prot database, with no further specification of the particular membrane type, were not included in this analysis (about 21% of all membrane proteins). Using the data on ICI responders from the NSCLC cohort,8 we found that only the proportion of mutated genes expressing proteins at the cell plasma membrane was significantly increased in ICI responders, while localization at the membranes of organelles did not correlate with ICI response (Figure 6C). Similar trends were observed for the melanoma cohort (Figure S10). In addition, consistent trends toward improvement of survival for patients with increased cell plasma-localized neoantigens was observed across the pan-cancer, NSCLC, and melanoma cohorts (Figure 6D).
mAgs as clinical biomarkers for ICI
In a more exploratory way, we analyzed which mAgs most impact survival upon ICI and could serve as biomarkers in the clinic. Using the dataset from Samstein et al.,7 we computed the HR of survival between patients bearing mutated and WT membrane protein-encoding genes within each cancer type (Figure 7A, Data S7). We observed that most mAgs seemed to correlate with improved survival. We particularly highlighted a subset of 1–13 genes per cancer type for which mutations could serve as biomarkers to predict extended survival upon ICI, as indicated by low HRs (Figure 7A, blue; Data S7).
Figure 7.

Specific membrane protein-encoding mutated genes as potent biomarkers for ICI in the clinic
The ICI- and non-ICI-treated cohorts from Samstein et al.7 were analyzed to determine which membrane protein-encoding mutations were the most potent to predict survival upon ICI.
(A) HR of survival associated with specific membrane protein-encoding genes per cancer type. An HR less than 1 indicates that the mutated version of the gene correlates with increased patient survival compared with the WT gene. Genes for favorable prognosis are labeled in blue, whereas the ones for poor prognosis are in red, for the ICI-treated cohort. Corresponding gene-specific HRs from the non-ICI-treated cohort are in gray (log rank test, ∗p < 0.05, non-adjusted p values). Patient coverage indicates the proportion of patients that contains at least one of the mutated genes in blue.
(B and C) Survival of ICI and non-ICI-treated NSCLC patients bearing NTRK3 (B) or NOTCH1 (C) mutations and associated TMB burden.
Further seeking ICI-specific membrane-localized biomarkers, we compared the HRs obtained upon ICI with the ones from the non-ICI-treated cohort for each gene for which enough patients were available (Figure 7A; Data S7). In most cases, gene mutations did not seem to improve survival in the non-ICI-treated cohort to the same extent than in the ICI-treated cohort, suggesting that these biomarkers could be specific for prediction of ICI efficacy. One exception was VHL in renal cell carcinoma, for which mutations appeared to be beneficial in both cohorts. We also found that some mutated genes correlated with very high survival in the ICI-treated cohort but with worsened survival in the non-ICI-treated one, such as NTRK3 and NOTCH1 in NSCLC or NOTCH3 or RNF43 in colorectal cancer (the latter was recently elucidated by Zhang et al.39) (Figures 7B, 7C, S11A, and S11B). Because patients with a high TMB have a higher chance of bearing mutations in these specific genes, we checked the TMB level of patients with mutation or not in the ICI-treated or non-treated cohorts. Interestingly, we observed that the TMB of patients with mutated NTRK3 or NOTCH1 in the ICI-treated cohort was similar to the ones of the non-ICI-treated cohort (Figures 7B and 7C). In addition, in the example of NOTCH1, we found that patients with the mutated gene did not have a significantly higher TMB compared with patients with the WT gene (Figure 7C), suggesting that membrane-specific markers could be used to improve survival prediction in a way complementary to use of TMB. Therefore, upon confirmation by future studies, such genes could constitute relevant biomarkers to guide medical choice toward ICI rather than other treatments for specific cancer types.
Discussion
This study focused on the influence of subcellular localization of tumor mutations for responsiveness to cancer immunotherapy. We demonstrated, in the B16-F10 melanoma mouse model and on a large clinical dataset of 4864 ICI- and non-ICI-treated cancer patients, that responsiveness to ICI and extended survival correlated with a high proportion of tumor mAgs, especially of neoantigens localized at the plasma membrane. Interestingly, this effect was not seen for an increased load of cAgs, nAgs, or sAgs, nor was it seen in patients who were not treated with ICI. This conclusion was supported in a pan-cancer analysis gathering 9 different types of cancer. Although a pan-cancer analysis merges heterogeneous cancer types, it has the strong advantage of including a large number of patients, therefore increasing statistical power, and mirrors the design of basket clinical trials.40,41 Further analyses per individual cancer type similarly correlated a high mAg proportion with extended survival in renal cell carcinoma and head and neck cancer in the cohorts from Samstein et al.,7 in a melanoma cohort from Hugo et al.,9 and in an NSCLC cohort from Hellmann et al.8 In contrast, we observed that a high proportion of cytoplasmic or nuclear mutations may lead to poorer patient survival in some cancer types. Because mutations at these locations are less immunogenic than mutations at the membrane, and because a high proportion of cytoplasmic or nuclear mutations implies a lower proportion of membrane mutations, these tumors may better escape the immune system. In-depth analysis per cancer type would be needed on a larger number of patients to further elaborate these conclusions.
In our analysis, we found consistent results from clinical datasets published by three independent research groups using two different methods of tumor mutation sequencing, namely MSK-IMPACT and WES, both recently approved by the FDA and rapidly emerging in the clinic.42,43,44,45 While these sequencing methods aim to quantify the TMB, a high load of which is approved as an inclusion criterion for treatment with ICI, our work provides a complementary, simple, algorithm-based method that can further filter the sequencing data to improve the prediction accuracy of ICI responsiveness and survival extension. Particularly, we found that our mAg-based criterion may indicate 30.5% more patients for inclusion into ICI than the current FDA standard of TMB, based on the Samstein et al.7 dataset. This threshold would be important to validate further in larger cohorts as well as in specific cancer types or in relation to other patient conditions (e.g., pre-treatment). For example, we have noticed in another cohort of melanoma patients46 that the proportion of mAgs was not predictive of ICI response in patients receiving prior steroid treatment or mitogen-activated protein kinase (MAPK) inhibitors. In this cohort, neither the TMB nor the proportion of mAgs significantly improved prediction of responsiveness to ICI in the different subtypes of melanoma. Last, our study highlighted particular membrane protein-encoding mutated genes that may be used as predictive biomarkers to guide the choice toward treatment by ICI in certain cancer types, such as NTRK3 or NOTCH1 in NSCLC.
Moreover, we importantly highlighted that the membrane proteins listed in the MSK-IMPACT method were predicted to generate more epitopes on MHC class I and be more immunogenic than the proteins localized in the cytoplasm or the nucleus or secreted. This could give some insight into the mechanisms by which a high proportion of mAgs can improve responsiveness to ICI and extend patient survival. Indeed, this might reveal a bias for membrane proteins to be better presented on MHC class I by tumor cells, allowing better recognition and killing by CD8+ T cytotoxic cells, whose exhaustion is reversed by ICI.
Further mechanistic insights were provided by our data in the B16-F10 mouse model, which consistently support that rejection of tumors with a high load of mAg strongly relies on T cells rather than on NK cells or on IgG-dependent cytotoxic mechanisms. While CD8+ T cells were necessary and sometimes sufficient to eradicate the tumors, our data suggest that CD4+ T cells provided important help to the CD8+ T cells, consistent with other reports that stressed the key role of MHC class II-restricted neoantigens in responsiveness to ICI.47 In addition, some previous research has highlighted that sAgs,48 mAgs,49 or extravesicular-bound50 antigens enhance CD4+ T cell responses and strengthen antigen-specific immunity. Moreover, the increase in CXCL9 and CXCL10 observed in tumors with a high load of mAgs are common hallmarks of “hot” tumors, which have been associated with enhanced responsiveness to ICI.22 Interestingly, non-ICI-treated tumors with a high proportion of mAgs in the mouse resulted in delayed tumor growth and extended survival (Figure 1), whereas in humans, no extension of survival was observed in the non-ICI-treated cohort (Figure 3E). This is likely due to the fact that the onset of a tumor in humans is not defined, in contrast to experimentation in the mouse. Moreover, in the mouse, while a growth delay was observed between B16mOVAHI and B16-OVAHI, the growth speed (i.e., slope) was comparable when the tumor started to eventually grow.
Apart from being immune targets, mutations of membrane proteins on tumor cells can also impact the primary biological functions of the membrane proteins (e.g., of growth factor receptors) and their downstream signaling, which could have direct effects on tumor biology, growth, and aggressiveness. This is likely not the case in the OVA-based mouse model but could take place in cancer patients.
Recently, reports have also highlighted that the subcellular localization of a protein plays a key role in the way it is mounted on MHC and on modulating the immune response.51,52,53 Particularly, Castro et al.52 have demonstrated that neoepitopes derived from proteins from accessible locations were associated with a better neoantigen vaccination response and enhanced success of ICI.
Besides the basic immunology perspective, our work provides a rationale for therapeutic immunomodulation by neoantigen selection at different subcellular locations. In particular, personalized cancer vaccines currently target neoantigens based on prediction of MHC binding neoepitopes for optimized T cell activation, with little consideration of the subcellular localization of the neoantigen.13 Adding vaccinal antigen selection criteria for preferential targeting of plasma mAgs might improve the therapeutic efficacy of such vaccines. Taken together, we believe that considering the neoantigens’ subcellular localizations, particularly the mAg proportion, for increased predictability to ICI response, to guide medical decisions of cancer treatments, as well as in the design of future immunotherapies will be valuable in the fight against cancer.
Limitations of the study
While the mAg proportion could be useful to extend the inclusion criteria of patients to ICI, particularly for patients that bear <10 mut/Mbp, it is important to note that ICI is already approved as a first-line treatment in some cancer types, independent of TMB stratification. For example, anti-PD1 is approved in melanoma even for patients with <10 mut/Mbp. Therefore, including melanoma patients in our analyses in Figures 4C and 4D may constitute a limitation. However, we noticed that removing them from the analysis does not alter our conclusions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-PD-1 clone 29F.1A12 | Bio X Cell | Cat # BE0273; RRID:AB_2687796 |
| Mouse anti-PD-L1 (clone 10F.9G2) | Bio X Cell | Cat # BE0101; RRID:AB_10949073 |
| Mouse anti-CTLA-4 (clone 9H10) | Bio X Cell | Cat # BE0131; RRID:AB_10950184 |
| Mouse anti-CD8α (clone 2.43) | Bio X Cell | Cat # BE0061; RRID:AB_1125541 |
| Mouse anti-CD4 (clone GK1.5) | Bio X Cell | Cat # BE0003-1; RRID:AB_1107636 |
| Mouse anti-NK1.1 (clone PK136) | Bio X Cell | Cat # BE0036; RRID:AB_1107737 |
| Mouse Isotype IgG2a (clone C1.18.4) | Bio X Cell | Cat # BE0085; RRID:AB_1107771 |
| Mouse Isotype IgG2b (clone LTF-2) | Bio X Cell | Cat # BE0090; RRID:AB_1107780 |
| Mouse anti-CD16/32 | BioLegend | Cat #101302; RRID:AB_312801 |
| Mouse anti-CD45 (30-F11) | BioLegend | Cat # 103101; RRID:AB_312966 |
| Mouse anti-CD8a (53-6.7) | BioLegend | Cat # 100711; RRID:AB_312750 |
| Mouse anti-PD-1 (29F.1A12) | BioLegend | Cat # 135202; RRID:AB_1877121 |
| Mouse anti-NK1.1 (PK136) | BioLegend | Cat # 108707; RRID:AB_313394 |
| Mouse anti-Ly6G (1A8) | BioLegend | Cat # 127601; RRID:AB_1089179 |
| Mouse anti-Ly6C (HK1.4) | BioLegend | Cat # 128015; RRID:AB_1732087 |
| Mouse anti-CD11b (M1/70) | BioLegend | Cat # 101201; RRID:AB_312784 |
| Mouse anti-F4/80 (BM8) | BioLegend | Cat # 123115; RRID:AB_893493 |
| Mouse anti-I-A/I-E (M5/114.15.2) | BioLegend | Cat # 107601; RRID:AB_313316 |
| Mouse anti-CD3ε (145-2C11) | BioLegend | Cat # 100307; RRID:AB_312672 |
| Mouse anti-CD4 (GK1.5) | BioLegend | Cat # 100401; RRID:AB_312686 |
| Mouse anti-CD62L (MEL-14) | BioLegend | Cat # 104407; RRID:AB_313094 |
| Mouse anti-CTLA-4 (UC10-4F10-11) | BD Biosciences | Cat # 561718; RRID:AB_10895585 |
| Mouse anti-CD25 (PC61) | BD Biosciences | Cat # 561780; RRID:AB_10893596 |
| Mouse anti-CD80 (16-10A1) | BD Biosciences | Cat # 560523; RRID:AB_1727515 |
| Mouse anti-B220 (RA3-6B2) | BD Biosciences | Cat # 562290; RRID:AB_11151901 |
| Mouse anti-CD19 (1D3) | BD Horizon | Cat # 563333; RRID:AB_2738141 |
| Mouse anti-CD11c (HL3) | BD Pharmingen | Cat # 561022; RRID:AB_2033997 |
| Mouse anti-CD44 (IM7) | eBioscience | Cat # 45-0441-82; RRID:AB_925746 |
| Mouse anti-CD103 (2E7) | BD Horizon | Cat # 566297; RRID:AB_2739670 |
| Mouse anti-FoxP3 | BD Biosciences | Cat # 566881; RRID:AB_2869932 |
| Goat anti-mouse IgG1 | Southern Biotech | Cat # 1070-05 |
| Goat anti-mouse IgG2a | Southern Biotech | Cat # 1080-05 |
| Goat anti-mouse IgG2b | Southern Biotech | Cat # 1090-05 |
| Goat anti-mouse IgG3 | Southern Biotech | Cat # 1100-05 |
| Hamster anti-mouse-CD3 (BUV396) | BD Horizon | Cat # 563565; RRID:AB_2738278 |
| Rat anti-mouse-CD4 (PE-cy7) | Biolegend | Cat # 100528; RRID:AB_312729 |
| Rat anti-mouse-CD8 (APC-cy7) | Biolegend | Cat # 100714; RRID:AB_312753 |
| Bacterial and virus strains | ||
| Envelop-expressing plasmid | Addgene | Cat # 12259; RRID:Addgene_12259 |
| pMDLg/pRRE | Addgene | Cat # 12251; RRID:Addgene_12251 |
| pRSV-Rev | Addgene | Cat # 12253; RRID:Addgene_12253 |
| Chemicals, peptides, and recombinant proteins | ||
| Live dead- violet | Invitrogen | Cat #L34958 |
| Murine recombinant GM-CSF | Peprotech | Cat # 315-03 |
| Murine recombinant IFNγ | Peprotech | Cat # 315-05 |
| Critical commercial assays | ||
| LEGENDplex MU Cytokine Release Syndrome Panel (13-plex) | BioLegend | Cat # 741026 |
| IFNγ quantikine ELISA kit | R&D systems | Cat # MIF00 |
| EasySep Mouse CD8+ T cell isolation kit | StemCell Technologies | Cat # 19853 |
| Deposited data | ||
| Human dataset | Samstein et al.7 | http://www.cbioportal.org/study/clinicalData?id=tmb_mskcc_2018 |
| Human dataset | Hellman et al.8 | PRJEB24995 https://www.ebi.ac.uk/eva/?eva-study=PRJEB24995 |
| Human dataset | Hugo et al.9 | GEO: GSE78220https://www.ncbi.nlm.nih.gov/sra |
| Experimental models: Cell lines | ||
| Human: melanoma B16F10 cells | American Type Culture Collection | ATCC # CRL-6475 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | JAX: 000664 |
| Mouse: B6.129S2-Ighmtm1Cgn/J | Jackson Laboratory | JAX: 002288 |
| Mouse: C57BL/6-Tg(CAG-OVAL)916Jen/J | Jackson Laboratory | JAX: 005145 |
| Mouse: B6.129S(C)-Batf3tm1Kmm/J | Jackson Laboratory | JAX: 013755 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | JAX: 003831 |
| Software and algorithms | ||
| FlowJo | FlowJo LLC | N/A |
| MHC-I binding prediction | Immune Epitope DataBase (IEDB) | N/A |
| Class I Immunogenicity | Immune Epitope DataBase (IEDB) | N/A |
| UniProtKB/Swiss-Prot database | UniProtKB/Swiss-Prot database | N/A |
| Other | ||
| BD Cytofix/Cytoperm Plus kit | BD Bioscience | BD 555028 |
| Illustrator CS5 | N/A | |
| Prism 9 | N/A | |
Resource availability
Lead contact
Further information and requests for resources, datasets, or protocols should be directed to the lead contact, Priscilla S. Briquez (priscilla.briquez@uniklinik-freiburg.de).
Materials availability
Materials generated in this study that are not commercially available will be made available on request addressed to the lead contacts. They may require completed materials transfer agreement.
Experimental models
Mice
All animal experimentation was approved by the University of Chicago Institutional Animal Care and Use Committee in compliance with local ethical and procedural regulations. Mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Female C57BL/6J (No 000664), female MuMt− mice (B6.129S2-Ighmtm1Cgn/J, No 002288) or female Batf3−/− mice (B6.129S(C)-Batf3tm1Kmm/, No 013755) were between 8 and 12 weeks old at the start of the experiments, with mice being aged-matched within an experiment. Act-mOVA mice (C57BL/6-Tg(CAG-OVAL)916Jen/J, No 005145) were bred in-house and female mice of 25–35 week old were used for experimentation. Mice were housed at the Animal Resources Center Facility at the University of Chicago, had water and food ad libitum, and were daily monitored for health care.
Cell lines
B16F10 (B16) melanoma cells (American Type Culture Collection, Manassas, VA, USA) were genetically modified by transduction with OVA-encoding lentivirus, with the approval and in accordance with the biological and safety institutional guidelines of the University of Chicago. Briefly, OVA-encoding DNA sequences were purchased from GenScript (Piscataway, NJ, USA). In one design, full-length OVA (UniprotKB P01012) was fused at the N-terminus to the signal peptide of mouse H-2KB (aa1-aa21, UniprotKB P01901) and at the C-terminus to the transmembrane domain of mouse H-2DB (aa299-aa331, UniProtKB P01899). Sequences were subcloned in the pLV-mCherry backbone (Addgene #36804) in place of mCherry. Lentiviruses were made by polyethylenimine (PEI)-mediated transfection of human embryonic kidney (HEK) 293-T cells using OVA-encoding plasmid with the packaging plasmids pMD2.G (Addgene #12259), pMDLg/pRRE (Addgene #12251) and pRSV-Rev (Addgene #12253). Twelve hours after transfection, the cell culture medium was refreshed and 36 h later, the medium was collected and filtered at 0.22 μm. Lentiviruses were concentrated by ultracentrifugation at 100,000 xg for 2 h at 4°C and resuspended in phosphate-buffered saline (PBS). B16 cells cultured in 48-well plates were transduced by adding OVA-encoding lentiviruses in the culture medium and centrifuging at 1150 xg for 30 min at room temperature, and then were cultured for 24 h, after which the medium was refreshed. For B16mOVAHI/LO and B16-OVAHI, monoclonal selection was performed by limiting dilution, and OVA-expression was quantified by quantitative polymerase chain-reaction (qPCR). The B16-OVALO cell line was a gift from B. Huard (University of Geneva, Switzerland). All cell lines were tested as negative for mycoplasma contamination by PCR. All cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco) with 10% heat-inactivated fetal bovine serum (Gibco) with no antibiotic.
Method details
Quantitative PCR for OVA expression
Expression of OVA in B16 cell lines or tumors was quantified by qPCR. Prior to RNA extraction, 30–50 mg of tumor tissues were homogenized (FastPrep-24 5G, MP Biomedicals, Santa Ana, CA, USA) in RLT lysis buffer (Qiagen, Hilden, Germany), spun down at 10,000 xg for 10 min and the supernatant was collected. For cells in culture, 1–2 million cells were pelleted, washed with PBS and lysed in RLT buffer. RNA was extracted using the RNeasy Plus Mini kit (Qiagen). The extracted RNA (1 μg) was then converted to cDNA using SuperScript IV VILO Master Mix (ThermoFisher Scientific, Waltham, MA, USA). All kits were used according to manufacturers instructions. TaqMan qPCR were finally performed using TaqMan Universal PCR Master Mix, OVAL primer (Gg03366807_m1) and ActB primer (Mm02619580_g1) (ThermoFisher Scientific), in a LightCycler 96 real-time PCR system (Roche Life Science, Basel, Switzerland).
Detection of membrane-bound OVA
Surface-expression of OVA was verified by flow cytometry and microscopy. Single cell suspensions of the different OVA-expressing B16 were incubated for 30 min on ice with anti-OVA (ab181688, Abcam, Cambridge, UK) in PBS +2% fetal bovine serum (FBS). Cells were washed twice and stained using an anti-rabbit secondary antibody (A315723, Invitrogen, Carlsbad, CA, USA) for 20 min on ice in the dark. Cells were washed and analyzed by flow cytometry (BD LSRFortessa, BD Biosciences, Franklin Lakes, NJ, USA) or imaged by fluorescence microscopy (Leica DMi8, Wetzlar, Germany). Flow cytometry data were analyzed using FlowJo (FlowJo LLC) and microscopy images were processed using Fiji (ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA).
Extracellular vesicles (EV) isolation
EV from the B16mOVAHI and B16-OVAHI cell lines were harvested using the CLAD1000 system (2440655, Cole-Palmer, Vernon Hills, IL, USA) as described by Mitchell et al.54 Briefly, 16 million cells were suspended in 15 mL complete EV-depleted DMEM (DMEM +1% penicillin/streptomycin (P/S) + 10% exosome-depleted FBS (A2720801, Thermo Fisher Scientific)) and loaded into the lower chamber of the CLAD flask. The upper chamber was then loaded with DMEM +1% P/S, and cells were allowed to recover for 4 days. On the 4th day, the upper reservoir was emptied and the media in the lower chamber was collected. The lower chamber was washed twice with DMEM, collecting only the first wash. The lower chamber was then refilled with 15 mL of complete EV-depleted DMEM. This harvesting process was repeated every 4 days. Collected media was first spun at 300 xg for 10 min to remove cells, then centrifuged at 3000 xg for 10 min to remove large cell debris and finally at 10,000 xg for 30 min to further remove debris. The final supernatant was concentrated using 100,000 MWCO concentrator tubes (UFC910024, EMD Millipore, Burlington, MA, USA) before processing via size exclusion. Size exclusion was performed using the Izon qEV10 system (IZON SP3) according to the manufacturer’s instructions to collect separately the EV fractions, containing particulates of 70–1000 nm in size, and the non-particulates non-EV fractions. Once purified, EV harvests were pooled and re-concentrated. Total protein content of the purified EV was quantified using a Micro BCA kit (Thermo Fisher) before storage at −20°C. Equal amount of proteins (34 μg) were loaded on SDS-PAGE gels for further analysis by Western blot.
Western blot analysis
Samples were run on SDS-PAGE gels for 45 min at 140 V (Mini-PROTEAN gel system, Bio-Rad Laboratories, Hercules, CA, USA) in Laemmli loading buffer before being transferred onto Western blot membranes (Immobilon-P PVDF membrane, EMD Millipore; Mini Trans-Blot cell, Bio-Rad) for 1 h at 90 V. Membranes were blocked using 5% milk in PBS +0.05% Tween 20 (PBST) overnight at 4°C under agitation and probed with anti-OVA (ab181688) for 4 h at room temperature. Membranes were washed in PBST thrice and incubated with a horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody for 1 h at room temperature. Membranes were washed at least 3 times for 5 min in PBST, revealed using the Clarity Western ECL substrate (Bio-Rad) and imaged using a gel imaging system (Universal Hood III, Bio-Rad).
In vitro OT-I CD8+ T cell proliferation
Induced iCD103+ DCs were generated as published in Mayer et al.55 Briefly, bone marrow cells were isolated from the long bones of C57BL/6 mice and plated at 15.106 cells in 10 mL of complete Lutz media (RPMI supplemented with 10% FBS, 1% Pen/Strep, 25 mM HEPES, and 50 μM β-mercaptoethanol) supplemented with 200 ng/mL Flt3L (made in-house) and 5 ng/mL GM-CSF (Peprotech). On day 5 after plating, 5 mL of complete Lutz media (without added cytokines) was added to each dish. Cells were then harvested on day 9 and replated at 3.106 cells in 10mL of complete Lutz media with added 200 ng/mL Flt3L and 5 ng/mL GM-CSF. Finally, iCD103+ DCs were harvested for use on day 16. For the presentation assay, iCD103+ DCs were plated (2.104 cells per well in a 96 well plate) with antigen at the given concentration with 5 ng/mL LPS and incubated at 37°C for 9 h. During that time, OT-I cells were isolated using CD8 T cell negative selection kits (STEMCELL Technologies) and labeled using 5 μM CFSE. After the incubation, the antigen loaded DCs were then washed in complete Lutz media before 1.105 CFSE labeled T cells were added to each well. Cells were then incubated for 3 days before staining and analysis by flow cytometry on a BD Fortessa.
In vivo antibodies
All antibodies used in vivo were the InVivoMAb grade antibodies purchased from Bio X Cell (Lebanon, NH, USA). Antibodies used as immune checkpoint therapies were anti-PD-1 (clone 29F.1A12), anti-PD-L1 (clone 10F.9G2) and anti-CTLA-4 (clone 9H10). Antibodies used for immune cell depletion were anti-CD8α (clone 2.43), anti-CD4 (clone GK1.5), anti-NK1.1 (clone PK136), Isotype IgG2a (clone C1.18.4), Isotype IgG2b (clone LTF-2).
Tumor injections
Mice were anesthetized by isoflurane inhalation and were injected intradermally with 1.5 million of the different OVA-expressing or WT B16 cell lines. The tumor was measured using a digital caliper every 2 days, and tumor volume was calculated as follows: volume = length∗width∗height∗(π/6). Mice were euthanized if sick or when the tumor volume reached 1 cm3. When indicated, mice were treated with immunotherapy, i.e., anti-PD-1 (200 μg) or the combination anti-PDL-1 + anti-CTLA-4 (100 μg each), once by intraperitoneal injection when the tumor volume was between 20 and 50 mm3 (day 5–8 post-tumor injection). When needed, 500 μg of depletion antibodies (anti-CD8α, anti-CD4, anti-NK1.1 or isotype control) were injected intraperitoneally 24 h after the checkpoint inhibitor therapy and repeated 7 days later. In the re-challenge experiments, 250k WT B16 cells were injected intradermally on the contralateral side on the mice 1 month after they cleared the primary tumor. In the BatF3−/− knock-out mice experiment, one mouse was a statistical outlier and was removed from the experiment.
Flow cytometry analysis of tumor
Ten days after tumor injection, tumor were harvested on euthanized mice. Tumors were weighed, and about 300 mg were processed. Tumors were cut into small pieces, digested for 45 min in collagenase IV (1 mg/mL), DNAse I (40 μg/mL) in DMEM +2% FBS +1.2 mM CaCl2 at 37°C under magnetic stirring. The samples were pipetted 100 times to dissociate tumor pieces, and single cell suspensions were obtained by using 70 μm cell strainer. Cells were kept on ice. Undigested pieces were further mixed with collagenase D (3.3 mg/mL), DNAse I (40 μg/mL) in DMEM +2% FBS +1.2 mM CaCl2 for 30 min at 37°C and collected as above. EDTA (5 mM) was added to the single cell suspension. The equivalent of 20 mg of tumor was used for staining for flow cytometry analysis. Tumor samples were washed in PBS and stained for cell viability for 15 min using Fixable Viability Dye eFluor 455UV (eBioscience, San Diego, CA, USA). The cells were washed and Fc receptors were blocked using anti-CD16/32 (#101302, BioLegend) for 20 min. Cells were then stained for 20 min on ice using the following antibodies: anti-CD45 (30-F11), anti-CD8α (53-6.7), anti-PD-1 (29F.1A12), anti-NK1.1 (PK136), anti-Ly6G (1A8), anti-Ly6C (HK1.4), anti-CD11b (M1/70), anti-F4/80 (BM8), anti-I-A/I-E (M5/114.15.2), from BioLegend (San Diego, CA, USA); anti-CD3ε (145-2C11), anti-CD4 (GK1.5), anti-CD62L (MEL-14), anti-CTLA-4 (UC10-4F10-11), anti-CD25 (PC61), anti-CD80 (16-10A1), anti-B220 (RA3-6B2), anti-CD19 (1D3), anti-CD11c (HL3), from BD Biosciences; anti-CD44 (IM7), anti-CD103 (2E7), from eBioscience. Cells were washed before analysis. When needed, intracellular staining with anti-FoxP3 (MF23, BD Biosciences) was performed using the BD Cytofix/Cytoperm Plus kit (BD Biosciences) according to the manufacturer’s instruction. All staining procedures were done on ice with samples protected from light, in PBS +2% FBS +1 mM EDTA when not stated otherwise. Cells were analyzed using a LSRFortessa flow cytometer (BD Biosciences). Data were processed using FlowJo (FlowJo LLC). Gating strategies for the flow analysis and biomarkers used to define cell populations are detailed in Data S1.
Ex vivo antigen-specific T cell restimulation
Ten days after tumor injection, spleens were harvested on euthanized mice. Single cell suspensions of splenocytes were obtained using a 70 μm cell strainer. Cells were washed in PBS before the red blood cells were lysed in ACK buffer (Lonza, Basel, Switzerland) for 4 min and blocked with complete media (IMDM +10% FBS +1% P/S). Cells were centrifuged, resuspended in complete media, and 0.5 million were plated in in 96 U-bottom plate. OVA257-264 (SIINFEKL; GenScript) and OVA323-339 (ISQAVHAAHAEINEAGR; GenScript) were added to the splenocytes at a final concentration of 1 μg/mL to restimulate CD8+ and CD4+ T cells, respectively. Unstimulated controls were tested using complete media without peptide, and positive controls were tested using ionomycin (1 μg/mL) + PMA (50 ng/mL). After 4 days in culture, the cell supernatant was collected and the amount of ΙFNγ secreted was quantified using mouse IFNγ quantikine ELISA kit (R&D systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Data represent the concentration of IFNγ secreted in restimulated culture supernatants subtracted with the amount detected in unstimulated supernatants.
IgG titration in plasma
Ten days after tumor injection, mice were bled by intracardiac puncture upon euthanasia. The blood was collected in EDTA-containing tubes, spun down at 1000 xg for 5 min and the plasma was collected and stored at −80°C until analysis. ELISA plates (Maxisorp, Nunc, Roskilde, Denmark) were coated with 10 μg/mL OVA (Sigma-Aldrich, St. Louis, MO, USA) in PBS overnight at 4°C, and blocked with casein (Sigma-Aldrich, St. Louis, MO, USA) for 2 h at room temperature. The plates were washed with PBST, and plasma diluted in casein was added to the wells, starting at a concentration of 1:100 and serially diluted by 10, for 2 h at room temperature. The plates were washed again, and the following HRP-conjugated antibodies were used for detection: anti-mouse IgG1 (#1070-05), anti-mouse IgG2a (#1080-05), anti-mouse IgG2b (#1090-05) and anti-mouse IgG3 (#1100-05) from Southern Biotech (Birmingham, AL, USA). The plates were revealed with TMB substrate (EMD Millipore) and stopped with 2N H2SO4. Absorbance at 450 nm was read using an Epoch ELISA reader (BioTek, Winooski, VT, USA), and corrected by the absorbance at 570 nm. Antibody titers were determined as the highest plasma dilution for which the corrected absorbance was twice the background level. The area under curve (AUC) was calculated as area under the titration curve of the log10(corrected absorbance over background).
Human data analysis
Processed sequencing data of tumor mutations (list of tumor mutated genes and tumor mutational burden score) and corresponding patient clinical data were obtained from the studies by Samstein et al.,7 Hellman et al.,8 and Hugo et al.9 Subcellular locations associated with Homo Sapiens genes (taxon ID = 9606) were uploaded from UniProtKB/Swiss-Prot database on August 2, 2020 and are provided in Data S2 (Gene subcellular locations inventory). Algorithms for data processing and analysis (Methods S1) were coded in R (Rstudio, Boston, MA, USA).
For each distinct tumor mutated gene of a patient, we searched the gene name that matches in the gene subcellular locations inventory file. Genes that were not found were categorized as "Unfound genes". Genes that were found but for which the subcellular location was unfound were further checked on the online UniprotKB/Swiss-Prot database20 using the GetSubcellular_location() function from the R package 'UniprotR'. If the gene subcellular location remained unfound, it was then categorized as "Unknown location". When multiple locations were found for a specific gene name, they were concatenated to obtain a single subcellular location entry per gene name. The gene subcellular locations were then categorized as membrane, cytoplasmic, nuclear or secreted by checking the presence of the character sequence: membrane = "Membrane" or "Cell membrane", cytoplasmic = "Cytoplasm", nuclear = "Nucleus", secreted = "Secreted" in the subcellular location entry associated with the gene. When indicated, the categories were extended to the cell membrane = "Cell membrane", the endoplasmic reticulum = "Reticulum" or "reticulum", the Golgi apparatus = "Golgi" or "golgi", or endosomal location = "Endosome" or "endosome" or "Endosomal" or "endosomal". Oftentimes, a single gene name was associated with several subcellular locations, in which case the gene was included in several category in a non-exclusive way. For each patient, we counted the number of mutated genes at each specific subcellular locations, and the proportion of mutated genes at a specific location was computed as the "number of tumor mutated genes at a location divided by the total number of tumor mutated genes in the patient". Patients with no tumor mutated genes were removed from the analysis. In the presented data, groups of patients were determined using inclusive percentiles, except in groups separated at the median, for which the group below median was inclusive and the group above median was exclusive.
In R, survival analysis were performed using the libraries 'survival', 'survminer' and 'survcomp' and the functions survfit() and surv_pvalue(). Hazard ratios were computed using the function hazard.ratio(). All these functions were used using log rank tests when asked. In case of multiple comparisons, p values were adjusted using the function p.adjust().
Analysis of the MHC-I binding prediction
The Immune Epitope DataBase (IEDB) was used to calculate predictions for MHC-I binding across candidate antigens. Specifically, predict_binding.py was used with the consensus method and the list of alleles provided by IEDB as the most frequently occurring in human populations. Predictions were generated for antigens ranging from 8 to 11 amino acids.
Analysis of the immunogenicity score
The Immune Epitope Database (IEDB) was used to calculate immunogenicity scores. Specifically, predict_immunogenicity.py was used across all available alleles for results from the MHC binding prediction. The binding results were first filtered for IC50 < 50 nM, as determined by the ANN (artificial neural network) method. The distributions of immunogenicity scores were compared using a one-way ANOVA, and Scheffe’s post hoc test for pairwise evaluation of groups.
Analysis of the membrane-localized biomarkers
HR of survival was computed for each membrane protein-encoding gene, between patients that bear mutated version of the gene vs. patients that bear the wild-type version of the gene. The analysis was done independently for each cancer type. Results were considered relevant to report when at least 7 patients with a mutated version of a gene were available, and 1) when the HR was ≤0.5 or statistically significance by the log rank test was reached, or 2) when the HR ratio was ≥1.3 and close to statistically significance (p value <0.2). The non ICI-treated cohort results were reported when at least 7 patients had the mutated version of the gene of interest.
Quantification and statistical analysis
Graphs were plotted using Prism 9 (GraphPad, San Diego, CA, USA). Statistical analyses were run on Prism 9 or on R (RStudio). Used statistical tests are described in the respective figure legends. Overall threshold for statistical significance was considered as p value <0.05. Figures were made us on Illustrator CS5 (Adobe, San Jose, CA, USA).
Acknowledgments
The authors would like to thank Prof. Luc G.T. Morris and Prof. David B. Solit for their help in the use of their clinical dataset, Dr. Alexandre de Titta and Dr. Sandra Gribi for advice on data analysis, and Dr. Jialu Liu for technical assistance. This article is dedicated to the memory of Anthony Gomes. This work was funded by the Chicago Immunoengineering Innovation Center of the University of Chicago, NIH R01 CA219304 (to M.A.S.), and the AbbVie-UChicago collaborative grant (to J.A.H. and P.S.B.). Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia LLC, a wholly owned subsidiary of Honeywell Inc. for the US Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525.
Author contributions
P.S.B., S.H., M.A.S., and J.A.H. conceived the project and designed the experiments. P.S.B., S.H., Z.G., K.C., T.K., A.T.A., G.R., Y.W., and S.G. performed the experiments. P.S.B., S.H., Z.G., K.C., T.K., A.T.A., R.K., P.S., and J.A.H. analyzed and interpreted the data. P.S.B., J.A.H., and Z.G. wrote the manuscript, and S.H., P.S., R.K., and M.A.S. corrected it.
Declaration of interests
The University of Chicago has filed for patent protection for biomarkers described in this work, of which P.S.B., Z.G., S.H., and J.A.H. are inventors.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101145.
Contributor Information
Jeffrey A. Hubbell, Email: jhubbell@uchicago.edu.
Priscilla S. Briquez, Email: priscilla.briquez@uniklinik-freiburg.de.
Supplemental information
Data and code availability
-
•
Tumor mutation sequencing data for the human cohorts used in this study are publicly available from Samstein et al.,7 Hellman et al.8 and Hugo et al.9 Subcellular locations associated to Homo Sapiens genes are provided in Data S1 and updated versions can be downloaded from the UniProtKB/Swiss-Prot database. Proportion of mutation subcellular locations per patient and corresponding selected clinical data are provided in Data S2. Proportion of mutations at specific locations for the ICI-treated cohort from Samstein et al., related to Figures 3, 4, and 5, Data S6. Proportion of mutations at specific locations for the melanoma patient cohort from Hugo et al., related to Figure 6.
-
•
All original code is available in this paper’s supplemental information (Methods S1).
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Peters S., Reck M., Smit E.F., Mok T., Hellmann M.D. How to make the best use of immunotherapy as first-line treatment of advanced/metastatic non-small-cell lung cancer. Ann. Oncol. 2019;30:884–896. doi: 10.1093/annonc/mdz109. [DOI] [PubMed] [Google Scholar]
- 2.Murciano-Goroff Y.R., Warner A.B., Wolchok J.D. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30:507–519. doi: 10.1038/s41422-020-0337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaddepally R.K., Kharel P., Pandey R., Garje R., Chandra A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers. 2020;12:738. doi: 10.3390/cancers12030738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C., Ribas A., Hamid O., Daud A., Wolchok J.D., Joshua A.M., Hwu W.-J., Weber J.S., Gangadhar T.C., Joseph R.W., et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients With Metastatic Melanoma. J. Clin. Oncol. 2017;36:1668–1674. doi: 10.1200/jco.2017.75.6270. [DOI] [PubMed] [Google Scholar]
- 5.Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020;11:3801. doi: 10.1038/s41467-020-17670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonaventura P., Shekarian T., Alcazer V., Valladeau-Guilemond J., Valsesia-Wittmann S., Amigorena S., Caux C., Depil S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019;10:168. doi: 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samstein R.M., Lee C.-H., Shoushtari A.N., Hellmann M.D., Shen R., Janjigian Y.Y., Barron D.A., Zehir A., Jordan E.J., Omuro A., et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hellmann M.D., Nathanson T., Rizvi H., Creelan B.C., Sanchez-Vega F., Ahuja A., Ni A., Novik J.B., Mangarin L.M.B., Abu-Akeel M., et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell. 2018;33:843–852.e4. doi: 10.1016/j.ccell.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keenan T.E., Burke K.P., Van Allen E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019;25:389–402. doi: 10.1038/s41591-019-0382-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tran E., Robbins P.F., Rosenberg S.A. Final common pathway” of human cancer immunotherapy: targeting random somatic mutations. Nat. Immunol. 2017;18:255–262. doi: 10.1038/ni.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galluzzi L., Chan T.A., Kroemer G., Wolchok J.D., López-Soto A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aat7807. [DOI] [PubMed] [Google Scholar]
- 13.Wells D.K., van Buuren M.M., Dang K.K., Hubbard-Lucey V.M., Sheehan K.C.F., Campbell K.M., Lamb A., Ward J.P., Sidney J., Blazquez A.B., et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell. 2020;183:818–834.e13. doi: 10.1016/j.cell.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yarchoan M., Johnson B.A., Lutz E.R., Laheru D.A., Jaffee E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer. 2017;17:209–222. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blum J.S., Wearsch P.A., Cresswell P. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neefjes J., Jongsma M.L.M., Paul P., Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011;11:823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 17.Vyas J.M., Van der Veen A.G., Ploegh H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Briquez P.S., Hauert S., de Titta A., Gray L.T., Alpar A.T., Swartz M.A., Hubbell J.A. Engineering Targeting Materials for Therapeutic Cancer Vaccines. Front. Bioeng. Biotechnol. 2020;8:19. doi: 10.3389/fbioe.2020.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almagro J.C., Daniels-Wells T.R., Perez-Tapia S.M., Penichet M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2017;8:1751. doi: 10.3389/fimmu.2017.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bateman A., Martin M.J., Orchard S., Magrane M., Agivetova R., Ahmad S., Alpi E., Bowler-Barnett E.H., Britto R., Bursteinas B., et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2020;49:D480–D489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiLillo D.J., Yanaba K., Tedder T.F. B Cells Are Required for Optimal CD4+ and CD8+ T Cell Tumor Immunity: Therapeutic B Cell Depletion Enhances B16 Melanoma Growth in Mice. J. Immunol. 2010;184:4006–4016. doi: 10.4049/jimmunol.0903009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reschke R., Gajewski T.F. CXCL9 and CXCL10 bring the heat to tumors. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abq6509. [DOI] [PubMed] [Google Scholar]
- 23.Castellino F., Huang A.Y., Altan-Bonnet G., Stoll S., Scheinecker C., Germain R.N. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell–dendritic cell interaction. Nature. 2006;440:890–895. doi: 10.1038/nature04651. [DOI] [PubMed] [Google Scholar]
- 24.Robertson M.J. Role of chemokines in the biology of natural killer cells. J. Leukoc. Biol. 2002;71:173–183. doi: 10.1189/jlb.71.2.173. [DOI] [PubMed] [Google Scholar]
- 25.Das K., Eisel D., Lenkl C., Goyal A., Diederichs S., Dickes E., Osen W., Eichmüller S.B. Generation of murine tumor cell lines deficient in MHC molecule surface expression using the CRISPR/Cas9 system. PLoS One. 2017;12 doi: 10.1371/journal.pone.0174077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merritt R.E., Yamada R.E., Crystal R.G., Korst R.J. Augmenting major histocompatibility complex class I expression by murine tumors in vivo enhances antitumor immunity induced by an active immunotherapy strategy. J. Thorac. Cardiovasc. Surg. 2004;127:355–364. doi: 10.1016/j.jtcvs.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 27.Marar C., Starich B., Wirtz D. Extracellular vesicles in immunomodulation and tumor progression. Nat. Immunol. 2021;22:560–570. doi: 10.1038/s41590-021-00899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dionisi M., De Archangelis C., Battisti F., Rahimi Koshkaki H., Belleudi F., Zizzari I.G., Ruscito I., Albano C., Di Filippo A., Torrisi M.R., et al. Tumor-Derived Microvesicles Enhance Cross-Processing Ability of Clinical Grade Dendritic Cells. Front. Immunol. 2018;9:2481. doi: 10.3389/fimmu.2018.02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muntjewerff E.M., Meesters L.D., van den Bogaart G. Antigen Cross-Presentation by Macrophages. Front. Immunol. 2020;11:1276. doi: 10.3389/fimmu.2020.01276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joffre O.P., Segura E., Savina A., Amigorena S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 31.Gutiérrez-Martínez E., Planès R., Anselmi G., Reynolds M., Menezes S., Adiko A.C., Saveanu L., Guermonprez P. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Front. Immunol. 2015;6:363. doi: 10.3389/fimmu.2015.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferlazzo G., Morandi B. Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front. Immunol. 2014;5:159. doi: 10.3389/fimmu.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deauvieau F., Ollion V., Doffin A.C., Achard C., Fonteneau J.F., Verronese E., Durand I., Ghittoni R., Marvel J., Dezutter-Dambuyant C., et al. Human natural killer cells promote cross-presentation of tumor cell-derived antigens by dendritic cells. Int. J. Cancer. 2015;136:1085–1094. doi: 10.1002/ijc.29087. [DOI] [PubMed] [Google Scholar]
- 34.Zehir A., Benayed R., Shah R.H., Syed A., Middha S., Kim H.R., Srinivasan P., Gao J., Chakravarty D., Devlin S.M., et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.U. S. Food and Drug Administration . 2021. FDA Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors _ FDA.Pdf. [Google Scholar]
- 36.Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao W., Sher X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 2018;14 doi: 10.1371/journal.pcbi.1006457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calis J.J.A., Maybeno M., Greenbaum J.A., Weiskopf D., De Silva A.D., Sette A., Keşmir C., Peters B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013;9 doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang N., Shi X., Ju W., Lou Y., Luo X. 2021. Rnf43 Mutation as A Biomarker for Immune Checkpoint Inhibitor Efficacy in Colorectal Cancer. [DOI] [Google Scholar]
- 40.Subbiah V., Puzanov I., Blay J.-Y., Chau I., Lockhart A.C., Raje N.S., Wolf J., Baselga J., Meric-Bernstam F., Roszik J., et al. Pan-Cancer Efficacy of Vemurafenib in BRAFV600-Mutant Non-Melanoma Cancers. Cancer Discov. 2020;10:657–663. doi: 10.1158/2159-8290.cd-19-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lengliné E., Peron J., Vanier A., Gueyffier F., Kouzan S., Dufour P., Guillot B., Blondon H., Clanet M., Cochat P., et al. Basket clinical trial design for targeted therapies for cancer: a French National Authority for Health statement for health technology assessment. Lancet Oncol. 2021;22:e430–e434. doi: 10.1016/s1470-2045(21)00337-5. [DOI] [PubMed] [Google Scholar]
- 42.Allegretti M., Fabi A., Buglioni S., Martayan A., Conti L., Pescarmona E., Ciliberto G., Giacomini P. Tearing down the walls: FDA approves next generation sequencing (NGS) assays for actionable cancer genomic aberrations. J. Exp. Clin. Cancer Res. 2018;37:47. doi: 10.1186/s13046-018-0702-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rusch M., Nakitandwe J., Shurtleff S., Newman S., Zhang Z., Edmonson M.N., Parker M., Jiao Y., Ma X., Liu Y., et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat. Commun. 2018;9:3962. doi: 10.1038/s41467-018-06485-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.U. S. Food and Drug Administration FDA 510(k) Clearance K190661: Omics Core NantHealth. https://www.accessdata.fda.gov/cdrh_docs/reviews/K190661.pdf.
- 45.U. S. Food and Drug Administration FDA 510(k) Clearance K192073: Helix OpCo, LLC. https://www.accessdata.fda.gov/cdrh_docs/pdf19/K192073.pdf.
- 46.Liu D., Schilling B., Liu D., Sucker A., Livingstone E., Jerby-Arnon L., Zimmer L., Gutzmer R., Satzger I., Loquai C., et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019;25:1916–1927. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alspach E., Lussier D.M., Miceli A.P., Kizhvatov I., DuPage M., Luoma A.M., Meng W., Lichti C.F., Esaulova E., Vomund A.N., et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696–701. doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corthay A., Lundin K.U., Lorvik K.B., Hofgaard P.O., Bogen B. Secretion of Tumor-Specific Antigen by Myeloma Cells Is Required for Cancer Immunosurveillance by CD4+ T Cells. Cancer Res. 2009;69:5901–5907. doi: 10.1158/0008-5472.can-08-4816. [DOI] [PubMed] [Google Scholar]
- 49.Henning P., Gustafsson T., Flach C.F., Hua Y.J., Strömbeck A., Holmgren J., Lindholm L., Yrlid U. The subcellular location of antigen expressed by adenoviral vectors modifies adaptive immunity but not dependency on cross-presenting dendritic cells. Eur. J. Immunol. 2011;41:2185–2196. doi: 10.1002/eji.201041009. [DOI] [PubMed] [Google Scholar]
- 50.Sedlik C., Vigneron J., Torrieri-Dramard L., Pitoiset F., Denizeau J., Chesneau C., de la Rochere P., Lantz O., Thery C., Bellier B. Different immunogenicity but similar antitumor efficacy of two DNA vaccines coding for an antigen secreted in different membrane vesicle-associated forms. J. Extracell. Vesicles. 2014;3 doi: 10.3402/jev.v3.24646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fessenden T.B., Stopfer L.E., Chatterjee F., Zulueta J., Mesfin J., Cordero Dumit T., Reijers I., Hoefsmit E.P., Blank C., White F., Spranger S. Dendritic cell-mediated cross presentation of tumor-derived peptides is biased against plasma membrane proteins. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-004159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castro A., Kaabinejadian S., Yari H., Hildebrand W., Zanetti M., Carter H. Subcellular location of source proteins improves prediction of neoantigens for immunotherapy. EMBO J. 2022;41 doi: 10.15252/embj.2022111071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bianchi F., Textor J., van den Bogaart G. Transmembrane Helices Are an Overlooked Source of Major Histocompatibility Complex Class I Epitopes. Front. Immunol. 2017;8:1118. doi: 10.3389/fimmu.2017.01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mitchell J.P., Court J., Mason M.D., Tabi Z., Clayton A. Increased exosome production from tumour cell cultures using the Integra CELLine Culture System. J. Immunol. Methods. 2008;335:98–105. doi: 10.1016/j.jim.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 55.Mayer C.T., Ghorbani P., Nandan A., Dudek M., Arnold-Schrauf C., Hesse C., Berod L., Stüve P., Puttur F., Merad M., Sparwasser T. Selective and efficient generation of functional Batf3-dependent CD103+ dendritic cells from mouse bone marrow. Blood. 2014;124:3081–3091. doi: 10.1182/blood-2013-12-545772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Tumor mutation sequencing data for the human cohorts used in this study are publicly available from Samstein et al.,7 Hellman et al.8 and Hugo et al.9 Subcellular locations associated to Homo Sapiens genes are provided in Data S1 and updated versions can be downloaded from the UniProtKB/Swiss-Prot database. Proportion of mutation subcellular locations per patient and corresponding selected clinical data are provided in Data S2. Proportion of mutations at specific locations for the ICI-treated cohort from Samstein et al., related to Figures 3, 4, and 5, Data S6. Proportion of mutations at specific locations for the melanoma patient cohort from Hugo et al., related to Figure 6.
-
•
All original code is available in this paper’s supplemental information (Methods S1).
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.