

Summary
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is a pleiotropic, severe autoinflammatory disease caused by somatic mutations in the ubiquitin-like modifier activating enzyme 1 (UBA1) gene. To elucidate VEXAS pathophysiology, we performed transcriptome sequencing of single bone marrow mononuclear cells and hematopoietic stem and progenitor cells (HSPCs) from VEXAS patients. HSPCs are biased toward myeloid (granulocytic) differentiation, and against lymphoid differentiation in VEXAS. Activation of multiple inflammatory pathways (interferons and tumor necrosis factor alpha) occurs ontogenically early in primitive hematopoietic cells and particularly in the myeloid lineage in VEXAS, and inflammation is prominent in UBA1-mutated cells. Dysregulation in protein degradation likely leads to higher stress response in VEXAS HSPCs, which positively correlates with inflammation. TCR usage is restricted and there are increased cytotoxicity and IFN-γ signaling in T cells. In VEXAS syndrome, both aberrant inflammation and myeloid predominance appear intrinsic to hematopoietic stem cells mutated in UBA1.
Keywords: VEXAS, single-cell RNA sequencing, inflammation, clonal hematopoiesis
Graphical abstract
Highlights
-
•
Inflammation and myeloid dominance originate in hematopoietic stem cells in VEXAS syndrome
-
•
mtUBA1 myeloid cells upregulate inflammatory pathways compared with wild-type cells
-
•
There are biased granulocytic differentiation and increased proliferation of mtUBA1 cells
-
•
Apoptosis of mtUBA1 lymphoid progenitors causes impaired lymphocytopoiesis in disease
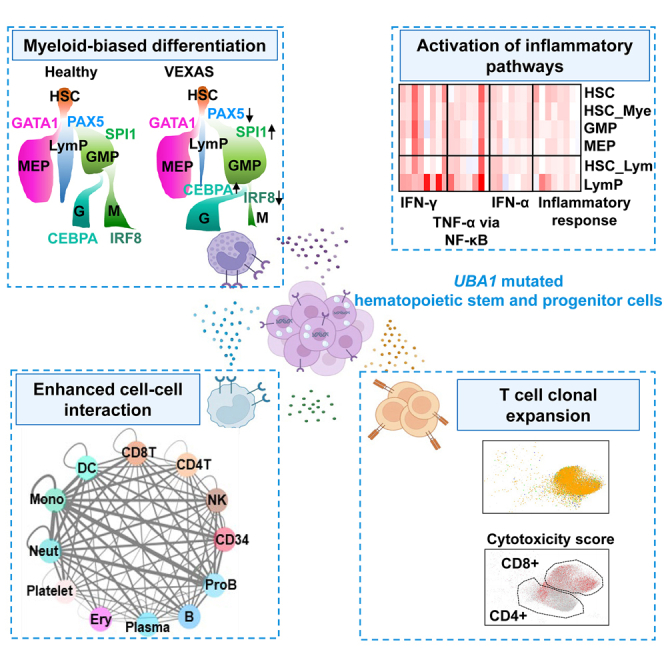
To address the link between UBA1 mutations and hyperinflammation in VEXAS syndrome, Wu et al. present single-cell transcriptome analyses on bone marrow of VEXAS patients. Myeloid lineage bias and inflammatory gene activation occur early in hematopoietic stem cells and appear intrinsic to UBA1 mutant cells highlighting mechanism of clonal dominance.
Introduction
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is a recently identified inflammatory disease caused by somatic mutations in UBA1, an X chromosome gene encoding the ubiquitin-like modifier-activating enzyme 1 (UBA1).1 VEXAS is an example of an emerging class of disorders with overlapping rheumatologic and hematologic manifestations caused by acquired mutations. VEXAS in particular may be relatively frequent, as the mutation has been identified in about 1 in 4,269 men older than 50 years.2 VEXAS features cytopenias, bone marrow (BM) dysplasia, and striking vacuolization of BM precursors, and patients have fever and a variety of organ-specific inflammatory manifestations.1,3,4,5,6,7 Following our initial report, UBA1 mutations have been discovered in such common clinical diagnoses as giant cell arteritis, relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, and myelodysplastic syndrome (MDS).3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21 However, the pathophysiology of VEXAS remains unclear, especially the relationship between a genetic defect in a hematopoietic stem cell and such diverse abnormalities of immunologic signaling, BM failure, myeloid neoplasm, and plasma cell dyscrasias.
Acquired mutations are age related and are originally implicated in cancer; they have now been recognized as key drivers of “benign” diseases across medical subspecialties.22 Benign (in the sense of not cancer) somatic mutation diseases are familiar to hematologists: paroxysmal nocturnal hemoglobinuria, a thalassemia-like syndrome in MDS, and histiocytoses are examples. Somatic mutations may also contribute to other complex multifactorial processes such as atherosclerosis and clonal hematopoiesis of indeterminate potential (CHIP).23
VEXAS is an example of a non-malignant disease secondary to acquired mutations. UBA1 “knockout” causes inflammation in zebrafish, but animal models of VEXAS are limited by germline, rather than somatic, loss of UBA1,1 and VEXAS cells are unusually fragile, intolerant of minimal in vitro manipulation, and poorly proliferative in culture. For these reasons, it has remained unclear how a specific genetic defect in hematopoietic cells results in refractory inflammation, marrow failure with dysplasia, and plasma cell dyscrasias. Alternatively, analogous to somatic mutations that are frequent in hematopoietic diseases, positive selection of clones containing somatic mutations by environmental factors might drive clonal dominance of mutated cells.21 Identification of differences between wild-type and mutant cell populations would be useful to understand pathologic mechanisms and to select and design treatments. Due to absent cellular and animal models, direct observation of patient cells is both advantageous and immediately relevant to elucidating the pathogenesis of VEXAS.
Single-cell genomic methods are appropriate to a disease like VEXAS: they require little sample manipulation, avoid culture artifacts, and allow for detection of rare cell types. Associated computational analyses are largely free of a priori bias in utilizing open-ended approaches to data collection and processing. Single-cell studies can directly detect genotypes and transcriptomes within cells and disclose altered pathways involved in disease.24,25,26,27,28,29,30,31,32,33,34
Here, we performed single-cell RNA sequencing (scRNA-seq) and single-cell T cell receptor/B cell receptor sequencing (scTCR/BCR-seq) of BM mononuclear cells (BMMNCs) and enriched hematopoietic stem and progenitor cells (HSPCs) from nine patients included in the original VEXAS report1 as an exploratory cohort. Myeloid dominance and inflammation (especially, tumor necrosis factor alpha [TNF-α] and interferon gamma [IFN-γ]) originated early in lineage-restricted progenitors and myeloid precursors in VEXAS. These findings were then validated in an independent cohort of patients using functional immunologic assays. UBA1-mutated (mtUBA1) myeloid cells exhibited upregulated inflammatory pathways and immune activation compared with wild-type UBA1 (wtUBA1). We implicate lineage bias toward myeloid, and granulocytic differentiation in particular, intrinsic increased cell cycling of mtUBA1 myeloid cells, and increased apoptosis of mtUBA1 lymphoid progenitors (LymPs) in clonal dominance of myeloid cells and loss of lymphocyte populations, respectively. Dysregulated protein degradation and therefore increased stress response were observed, and stress responses positively correlated with the VEXAS inflammatory signature. We also profiled cell-cell interactions of marrow myeloid cells with HSPCs and TCR and BCR repertoires in VEXAS. Our work presents detailed description of single hematopoietic cells in VEXAS, and our results should facilitate our understanding of hematopoiesis, clonal dominance, cell-cell interactions, and TCR/BCR repertoires in this newly defined syndrome. Furthermore, they suggest potential utility of TNF and IFN blockades in the treatment of VEXAS syndrome.
Results
Patient characteristics
An exploratory cohort composed of nine patients (all males and median age 65 years, and reported earlier) with confirmed UBA1 mutations underwent scRNA-seq, scTCR/BCR-seq, flow cytometry profiling, and colony formation. For validation, we enrolled another 11 patients (all males and medium age 67 years) for immunoassays and flow cytometry profiling (Figure 1A). Patient clinical characteristics are summarized in Table S1.
Figure 1.

Myeloid dominance and activation of the inflammatory pathways in VEXAS BMMNCs
(A) Experimental workflow. BMMNC samples from patients and healthy donors were subjected to multi-color flow cytometry to profile hematopoietic stem and progenitor cell (HSPC) subpopulations, and to ELISpot assay to quantify BMMNCs secreting TNF-α or IFN-γ. BMMNCs and FACS-sorted Lineage−CD34+ cells were subjected to colony forming assay and single-cell RNA sequencing (scRNA-seq) using the 10x Genomics platform. scRNA-seq libraries were sequenced on the Illumina NovaSeq system before data analysis, including single-cell transcriptome profiling (gene expression, gene mutation, and cell-cell interaction) and single-cell T cell receptor/B cell receptor (scTCR/BCR) profiling.
(B) A Uniform Manifold Approximation and Projection (UMAP) plot of single-cell gene expression in BMMNCs of all patients and healthy donors. Cells are colored by types (HSPC, erythroblast, neutrophil, monocyte, T cell, NK cell, B cell, plasma cell, eosinophil, and dendritic cell). A bar chart shows percentages of these cell populations in individual patients and healthy donors. The color legend is the same as that in the UMAP plot. A dot plot showing a myeloid (erythroblast, neutrophil, monocyte, and dendritic cell) vs. lymphoid (T cell, B cell, NK cell, and plasma cell) ratio in patients and healthy donors. Data are presented as mean values ± standard error of the mean (SEM). p values with the two-sided unpaired Mann-Whitney test are shown.
(C) Heatmap showing expression of representative differentially expressed genes grouped by their functional pathways in IFN-γ and IFN-α signaling, TNF-α via NF-κB signaling, inflammatory response, E2F targets, and apoptosis, between BMMNCs from VEXAS patients (n = 9) and healthy controls (n = 4). Values are presented as log2 fold-changes (log2FC).
(D) Gene set enrichment analysis (GSEA) of expressed genes in BMMNC subpopulations of VEXAS patients, including neutrophils, monocytes, erythroblasts, T cells, B cells, and NK cells. Normalized enrichment scores for the GSEA pathways are plotted, showing higher enrichment of the inflammatory pathways in neutrophils and monocytes than those in lymphoid cells.
(E) Representative ELISpot wells showing TNF-α secretion by BMMNCs from two VEXAS patients and two healthy donors in a second batch of the validation cohort, in triplicate. Bottom, quantification of TNF-α-, IFN-γ-, and TNF-α/IFN-γ-positive spots in BMMNCs plated (VEXAS patients n = 5 and healthy donors n = 2, in triplicate). Data are presented as mean values ± standard error of the mean (SEM). p values with the two-sided unpaired Mann-Whitney test are shown.
A distinct transcriptional profile of BM cells in VEXAS
We first sought to study gene expression of hematopoietic cells using scRNA-seq. After quality control, 84,401 single BMMNCs from nine patients and 36,425 from four healthy individuals were retained for further analyses.
Sequences from single BMMNCs were visualized in Uniform Manifold Approximation and Projection (UMAP). Cells formed clusters, imputed from similarity among transcriptomes with a graph-based approach,35 from which BMMNC subpopulations could be assigned computationally: CD34+ HSPCs, erythroblasts, neutrophils (a sum of neutrophil lineages at different differentiation stages), monocytes (a sum of monocytic lineages at different differentiation stages), CD4+ T cells, CD8+ T cells, B cells, plasma cells, natural killer (NK) cells, dendritic cells, and platelets (Figures 1B and S1A). By deconvoluting single cells of a heterogeneous BMMNC population, we determined that patients had increased myeloid cell (particularly granulocyte) proportions in their marrows (Figure 1B).
Gene expression of BMMNCs in patients was compared with that in healthy individuals: many genes were differentially expressed in VEXAS. By gene ontology (GO) analysis, aberrantly expressed genes were mainly involved in functions related to the immune response, leukocyte activation, cell communication, and protein metabolism (Figure S1B). IFN signaling, TNF-α signaling, and inflammatory response were upregulated in various cell types (Figures 1C and S1C) but predominately in myeloid cells (Figure 1D). These data were consistent with transcriptome changes described for peripheral blood cells.1 Cell cycling genes (e.g., E2F targets) were downregulated while apoptosis genes were upregulated in VEXAS. In an independent validation cohort (11 VEXAS patients and 8 healthy donors), we confirmed heterogeneous patterns of abundant TNF-α and IFN-γ secretion by VEXAS BMMNCs (Figures 1E and S1D).
Myeloid bias and activation of the inflammation pathways in VEXAS HSPCs
To characterize early hematopoiesis in VEXAS, flow cytometry using an established panel of cell surface markers36 was performed to profile BM stem cells and progenitors. The numbers of CD34+ HSPCs, CD34+CD38− stem cells, and multipotent progenitor cells were not different between patients and controls, but there was a marked reduction in LymP numbers and thus a significantly decreased lymphoid/myeloid cell progenitor ratio in VEXAS samples (Figures 2A and 2B), consistent with myeloid cell dominance in BM and peripheral blood in this disease.1,3 This result was confirmed in an independent cohort of patients (Figures S2A and S2B).
Figure 2.

Myeloid bias and activation of the inflammatory pathways in VEXAS HSPCs
(A) Phenotypes of HSPCs in healthy donors and VEXAS patients by flow cytometry. Cell populations were defined as reported36: HSC, Lineage−CD34+CD38−; CMP/MEP, Lineage−CD34+CD38+CD10−CD45RA−; GMP, Lineage−CD34+CD38+CD10−CD45RA+; LymP, Lineage−CD34+CD38+CD10+; HSC, hematopoietic stem cells and multipotent progenitors; CMP, multipotent common myeloid progenitor; MEP, megakaryocytic-erythrocytic progenitors; GMP, granulocytic-monocytic progenitors; LymP, lymphoid progenitors.
(B) Proportions of progenitor populations were compared between VEXAS patients (n = 9) and healthy donors (n = 4). Data are shown with mean values ± SEM. p values with the two-sided unpaired Mann-Whitney test are shown.
(C) A UMAP plot of single-cell gene expression in HSPCs of all patients and healthy donors. Cells are colored by cell types as HSC, MEP, GMP, LymP, HSC_Lym (HSC with lymphoid differentiation potential), and HSC_Mye (HSC with myeloid differentiation potential).
(D) Reconstruction of hematopoietic hierarchy pseudotime ordering with Palantir. The color legend is the same as in (C).
(E) Percentages of LymPs were compared between VEXAS patients (n = 9) and healthy donors (n = 4). Data are presented as mean values ± SEM. p values with the two-sided unpaired Mann-Whitney test are shown.
(F) Heatmap showing expression of representative differentially expressed genes grouped by their functional pathways in IFN-γ and IFN-α signaling, TNF-α via NF-κB signaling, inflammatory response, E2F targets, and apoptosis, between HSPCs from VEXAS patients (n = 9) and healthy controls (n = 4). Values are presented as log2FC.
(G) Relative inflammatory pathway scores (TNF-α signaling, IFN-γ signaling, and inflammatory response signaling scores) in VEXAS, CMML, CML, and MDS patients.37,38,39 p values with the two-sided unpaired t test are shown.
We next queried whether myeloid dominance and activation of inflammatory gene programs originated from HSPCs. We examined transcriptomes of enriched Lineage−CD34+ HSPCs by scRNA-seq; after quality control, 62,103 single HSPCs from patients and 52,272 from healthy individuals were retained for further analyses. From published cell type signatures,40 we deconvoluted single cells as stem cells and multipotent progenitors (HSCs), megakaryocyte-erythrocyte progenitors (MEPs), granulocyte-monocytic progenitors (GMPs), and LymPs (Figures 2C and S2C). When the hematopoietic hierarchy was reconstructed by pseudotemporal ordering, we observed the anticipated three major differentiation trajectories: from HSCs to MEPs, to myeloid cells, and to lymphoid cells (Figure 2D). Similar to flow cytometric phenotyping, there were markedly reduced LymPs in all VEXAS patients (Figure 2E).
Many differentially expressed genes were identified based on gene expression in stem cells and lineage-committed progenitors from patients with VEXAS syndrome. Gene set enrichment analysis was employed to characterize skewed gene sets. Genes involved in the immune response mediated by IFN-α, IFN-γ, and TNF-α, and the general inflammatory response, observed to be upregulated in VEXAS BMMNCs, were highly enriched in patients’ HSPCs (Figures S2D and S2F). Similar to BMMNCs, cell cycling genes (e.g., E2F targets) were downregulated and apoptosis genes were upregulated in HSPCs. From these striking changes, we inferred global effects of UBA1 mutations on early hematopoiesis, resulting in myeloid dominance and inflammatory gene pathway activation.
VEXAS syndrome shares overlapping clinical features with other hematologic diseases (such as MDS, chronic myelomonocytic leukemia [CMML], and chronic myeloid leukemia [CML]), including BM proliferation and dysplasia, myeloid cell dominance, and frequent co-occurrence of inflammatory or autoimmune disorders. CMML features Sweet syndrome and other inflammatory manifestations, and MDS has historically been associated with inflammation and frank autoimmunity in various organs.41,42,43,44,45,46,47 To investigate whether inflammation observed in early HSPCs was present in these other diseases, we integrated our scRNA-seq data from sorted Lineage−CD34+ cells with published data of HSPCs in MDS, CMML, and CML patients.37,38,39 We batch-corrected and integrated across samples, and then compared relative expression levels of the inflammatory pathways (activity scores of the IFN-γ response, TNF-α response, and inflammatory response pathways) in VEXAS patients with those in patients with the other three diseases; inflammation was indeed present in these myeloproliferative and MDSs but more extreme in VEXAS (Figure 2G).
mtUBA1 HSPCs exhibit active cell cycling and increased inflammatory gene expression
Within the scRNA-seq data, we were able to identify mtUBA1 single cells from their mRNA sequences. Due to “dropout” and other limitations of the platform, mtUBA1 transcripts were only captured in a fraction of total cells (∼9% of BMMNCs and 20% of CD34+ HSPCs) and preferentially in some patients (better in UPNs 14–17 [with 5′ capture] than in UPNs 1, 6, 10, 11, and 13 [with 3′ capture]). Captured UBA1 mutations were identical to concomitant Sanger sequences of bulk samples (Figure S3A). UBA1 transcripts were detected more readily in CD34+ cells than in BMMNCs, presumably due to higher UBA1 mRNA expression early in hematopoietic ontogeny (Figures S3B and S3C); there was higher frequency of mtUBA1 transcripts in CD34+ cells than in BMMNCs (Table S2), consistent with digital PCR data in our original report.1 As the UBA1 gene is located on the X chromosome and all patients were male, there is only a single UBA1 allele; we denoted single cells containing at least one mtUBA1 mRNA transcript as mtUBA1 cells, single cells with only wtUBA1 mRNA transcripts as wtUBA1 cells, and single cells with no UBA1 mRNA transcripts as “unknown.”48
To correlate UBA1 mutations with transcription, we first overlaid mutation data on a UMAP plot by highlighting mtUBA1 and wtUBA1 cells, assuming that UBA1 was expressed ubiquitously, and therefore that nonuniform distribution of mtUBA1 cells on UMAP would indicate distinct contributions of gene mutations to gene expression. mtUBA1 cells were most enriched in myeloid cells of BMMNCs and myeloid progenitors in HSPCs (Figures 3A and 3B), suggesting contributions of UBA1 mutations to myeloid dominance. We compared mtUBA1 and wtUBA1 myeloid cell gene expression to stratify potential effects of cell type distribution. Genes involved in the inflammatory pathways were upregulated in both mtUBA1 BMMNCs and mtUBA1 HSPCs; cell cycling genes also were upregulated in mtUBA1 cells (Figures 3C, S3D, and S3E; Table S2). A higher proportion of mtUBA1 cells were in S phase than were wtUBA1 (and unknown cells; Figure 3D). In summary, from a comparison between mtUBA1 cells with wtUBA1 cells, we inferred UBA1 mutations linked to immune activation, myeloid dominance, active cell cycling in stem cells and early progenitor cells, and myeloid lineage precursors in VEXAS BM.
Figure 3.

UBA1-mutated HSPCs exhibit increased inflammation and active cell cycling
(A) A UMAP plot of single-cell gene expression in BMMNCs of VEXAS patients, as in Figure 1B. Cells with expressed mutated UBA1 (mtUBA1) and wild-type UBA1 (wtUBA1) are colored red and blue, respectively, and all the other cells in gray. Lymphoid precursors are circled on the UMAP plot.
(B) A UMAP plot of HSPCs of VEXAS patients, the same as Figure 2C. Cells with expressed mtUBA1 and wtUBA1 are colored red and blue, respectively, and all the other cells in gray.
(C) A heatmap showing expression of representative differentially expressed genes grouped by their functional pathways in IFN-γ and IFN-α signaling, TNF-α via NF-κB signaling, inflammatory response, and cell cycling, between mtUBA1 and wtUBA1 BMMNCs (top) and HSPCs (bottom) in VEXAS patients (n = 9). Values are presented as log2FC.
(D) Bar plots showing percentages of BMMNCs (left) and HSPCs (right) in G1, G2/M, and S phases of cell cycle in mtUBA1, wtUBA1, and NULL cells in VEXAS patients.
(E) Immunoblotting results showing knockdown efficiency of UBA1 in cell lines (U937 and Raji).
(F) A dot plot showing top GO terms enriched in upregulated genes in UBA1 knockdown cell lines (U937, THP1, Raji, and Jurkat) compared with those in wild-type control cell lines.
To obtain direct evidence that loss of wtUBA1 resulted in major alterations in transcription, we “knocked down” in vitro UBA1 expression in two myeloid (U937 and THP1) and two lymphoid (Raji and Jurkat) cell lines (Figures 3E and S4A). Expression of inflammation genes, including genes involved in the TNF-α and IFN-γ pathways, increased in all four perturbed myeloid and lymphoid cell lines (Figure 3F; Table S3). These results supported a cell-autonomous mechanism of hyperinflammation due to deficiency of wtUBA1.
DNMT3A somatic mutations are frequent in myeloid neoplasms such as acute myeloid leukemia and MDS, and they are observed at unusually high frequency in VEXAS.18,19,20,21 UPN1 and UPN5 had DNMT3A mutations at variant allele frequency 40%–50%. To understand DNMT3A-mutated (mtDNMT3A) clones in the context of VEXAS syndrome, we identified single cells expressing DNMT3A mutations from mRNA sequencing (Figure S4B). As there are two alleles of the DNMT3A gene, we denoted single cells with at least one mtDNMT3A mRNA transcript as mtDNMT3A cells, single cells with only wild-type DNMT3A (wtDNMT3A) mRNA transcripts as wt-likely DNMT3A cells, and single cells with no DNMT3A mRNA transcripts as unknown.48 As for UBA1 mutations, expressed DNMT3A mutations were mainly in myeloid cells (Figures S4C–S4F). In both BMMNCs and HSPCs, the immune response- and inflammation-related pathways were upregulated, but the cell-cycle-related pathways were downregulated in mtDNMT3A cells compared with wt-likely DNMT3A cells (Table S4).
Perturbed protein degradation and unfolded protein response in VEXAS
An unfolded protein response gene set was upregulated only in the CD34+ HSPC compartment in VEXAS BMMNCs (Figure 4A). To assess functional changes in protein degradation (the protein ubiquitination/proteasome pathway and the autophagy pathway), we compared the expression of these genes in VEXAS HSPCs with that in healthy HSPCs; there was decreased expression of the protein ubiquitination/proteasome pathway, no significant changes of the autophagy pathway, but a marked increase of ER stress response genes (Figure 4B). Upregulation of unfolded protein response genes (including CALR, HSP90B1, XBP1, BANF1, and HSPA5) was observed in HSPC subtypes (Figure 4C). With observation of consistent upregulation of the IFN-γ response, TNF-α response, and inflammatory response pathways in BMMNCs, HSPCs, and mtUBA1 cells in VEXAS, we provisionally defined expression of genes involved in these four pathways as a “VEXAS inflammatory signature” and sought correlation of a VEXAS inflammatory score with ER stress in HSPCs. There was a strong positive correlation of the VEXAS inflammatory score with ER stress at a single-cell level (Figure 4D, left), and the same trend for individual patients (Figure 4D, right). More broadly, there was a positive correlation of a more general inflammatory score49,50 with ER stress in VEXAS HSPCs (Figure 4E). These results suggest a dysregulated protein ubiquitination/proteasome pathway due to UBA1 mutations, and a lack of the compensatory autophagy pathway for protein degradation leads to an increased unfolded protein stress, which likely contributes to enhanced inflammation in VEXAS HSPCs.
Figure 4.

Dysregulated protein degradation and stress response in VEXAS HSPCs
(A) A GSEA enrichment plot for a hallmark_unfolded protein response gene set for differentially expressed genes of mtUBA1 HSPCs compared with wtUBA1 HSPCs in VEXAS patients. GSEA was based on the Kolmogorov-Smirnov test.
(B) Expression levels of pathways (protein ubiquitination, proteasome, autophagy, and response to ER stress) in HSPCs of VEXAS patients (n = 9) and healthy controls (n = 4). p values with the two-sided unpaired t test are shown.
(C) Bubble plot showing expression of genes in the unfolded protein response pathway in HSPC subsets in VEXAS patients. Dot sizes correspond to percentages of cells expressing genes, and dot colors correspond to expression levels of genes.
(D) Correlation of a VEXAS inflammatory score (calculated based on a gene list of IFN-γ and IFN-α signaling, TNF-α via NF-κB signaling, and the inflammatory response pathways) and ER stress on single-cell levels (left, each dot indicates one cell) and in individual patients (right, each dot indicates one patient). p values and R value estimated using a Pearson correlation test are shown.
(E) Correlation of an inflammatory score49,50 and ER stress on single-cell levels. Each dot indicates one cell. p values and R value estimated using a Pearson correlation test are shown.
Biased lineage specification, increased apoptosis in mtUBA1 LymPs, and progressive loss of lymphocytic cells
To determine whether loss of lymphoid cells occurred early or late in differentiation, we plotted the ratios of patients’ versus healthy donors’ cells along pseudotime differentiation trajectories of different lineages. There was progressive loss of lymphoid cells with differentiation in comparison with normal hematopoiesis, while the numbers of GMP and MEP were stable (Figure 5A). Indeed, a potential of HSC differentiation to lymphocytes progressively decreased; a differentiation potential to myeloid cells remained stable and equivalent to normal hematopoiesis, while a potential to erythroid/megakaryocytes varied but was lower than in normal hematopoiesis (Figure 5B); results were consistent with frequent anemia and lymphocytopenia in VEXAS. We next assessed expression of master transcription factors in hematopoietic lineage specification in VEXAS. At the first lineage specification from stem cells and multipotent progenitors, GATA1 (to MEP) appeared equivalent to healthy donors, while SPI1 (encoding PU.1, to GMP) remained higher than in healthy donors, and PAX5 (to LymP) progressively decreased and was much lower than in normal hematopoiesis (Figure 5C). In the second lineage specification from GMP, CEBPA (to G) progressively increased to a level higher than normal, while IRF8 (to M) appeared normal. Indeed, expression of PAX5 and GATA1 were decreased while SPI1 increased in VEXAS HSCs, and expression of IRF8 was lower and CEBPA higher in VEXAS GMP compared with healthy donors (Figures S4G and S4H). These results indicate biased lineage specification in VEXAS HSPCs toward myeloid and against erythroid and lymphoid differentiation, and relative dominance of neutrophil over monocyte differentiation (Figure 5D). Single-cell results were consistent with clinical and laboratory evidence of myeloid dominance and normal neutrophil number, but monocytopenia in VEXAS.
Figure 5.

Lineage bias, increased cell apoptosis in mtUBA1 LymPs, and progressive loss of lymphocytic cells with differentiation
(A) Dynamic changes of LymP, GMP, and MEP ratios in VEXAS patients and healthy donors along pseudotime differentiation. x axis: pseudotime ordering from HSCs to LymP, GMP, and MEP, respectively, estimated by Palantir. y axis: Log2(percentages of corresponding cells in VEXAS patients) − log2(percentages of corresponding cells in healthy donors).
(B) Dynamic changes of lineage priming of HSCs to LymP, GMP, and MEP, along pseudotime differentiation. x axis: pseudotime ordering from HSCs to lineage-restricted progenitors estimated by Palantir. y axis: Log(lineage signature gene expression in patients/lineage signature gene expression in healthy donors). Lineage signature gene expression represented area under the receiver operating characteristic curve (AUC) values calculated with AUCell.
(C) Dynamic changes of expression levels of transcription factors (GATA1, SPI1, PAX5, CEBPA, and IRF8) along pseudotime differentiation. x axis: pseudotime ordering from HSCs to MEP, GMP, and LymP, respectively, estimated by Palantir. y axis: expression of transcription factors in patients normalized by that in healthy donors.
(D) A schematic diagram showing hematopoietic lineage specification and relative quantity of cell types in VEXAS patients and healthy donors.
(E) Dynamic changes of wtUBA1 LymP and mtUBA1 LymP ratios in HSPCs in VEXAS patients along differentiation. x axis: pseudotime ordering from HSCs to lineage-restricted progenitors estimated by Palantir. y axis: ratios of wtUBA1 LymP or mtUBA1 LymP in HSPCs.
(F) Dynamic changes of mtUBA1 cell ratios in LymP, GMP, and MEP in VEXAS patients along differentiation. x axis: pseudotime ordering from HSCs to lineage-restricted progenitors estimated by Palantir. y axis: Log(cell numbers of mutant/all HSPCs).
(G) Apoptosis gene expression scores (calculated by the AddModuleScore function in Seurat) of mtUBA1 cells normalized by wtUBA1 cells in myeloid and lymphoid BMMNCs were compared. y axis: normalized expression levels of apoptosis genes. A heatmap of apoptosis genes upregulated in mtUBA1 cells is shown on the right.
mtUBA1 frequency also decreased in lymphoid cells with differentiation (Figure 5E), but was stable or increased in myeloid and erythroid/megakaryocytic lineages (Figure 5F). Upregulation of apoptosis genes occurred in almost all lineages in patients’ HSPCs and BMMNCs (Figures 1C and 2F). To correlate mtUBA1 and wtUBA1 cells in myeloid and lymphoid lineages with loss of lymphoid cells and myeloid dominance in VEXAS, we compared ratios of apoptosis gene expression in mutated compared with wild-type cells. mtUBA1 lymphoid cells had a higher ratio than did mtUBA1 myeloid cells (Figure 5G), suggesting that mutated lymphoid cells were more susceptible to apoptosis than were mutated myeloid cells. Expression of cell apoptosis genes, including BCL2, JUN, CASP10, PARP1, and ATM, was higher in mtUBA1 cells.
Enhanced cell-cell interactions between activated myeloid cells and HSPCs
We first examined interactions among cell types in BMMNCs. Among 149 ligand-receptor pairs expressed in VEXAS and healthy donors, there were in total 2,488 cell type ligand-receptor pairs among BMMNC cell populations in VEXAS and 2,014 in healthy donors. In general, cell-cell interactions among populations of BM were higher in VEXAS (Figure 6A). The most differentially present ligand-receptor pairs included TNFSF13-TNFRSF1A, TNFSF13-TNFRSF14, CD47-SIRPG, APP-FPR2, TNFSF13-FAS, HLA-F-KIR3DL1, and HLA-A-KIR3DL1. Among them, most were uniquely present in VEXAS and absent in healthy donors (Figure 6B). The most frequent ligand receptors were among immune cells including neutrophils, monocytes, dendritic cells, NK cells, and T cells. HSPCs appeared to interact frequently with diverse immune cells, and interactions of HSPCs with most cell types were higher in VEXAS than in healthy donors (Figure 6C).
Figure 6.

Enhanced cell-cell interactions of activated myeloid cells with HSPCs in VEXAS patients
(A) Ligand-receptor pairs among cell types in BMMNCs were estimated by CellPhoneDB.51 Color legends for cell types are the same as in Figure 1B. Thickness of lines connecting cell types indicates total number of ligand-receptor pairs between two cell types estimated by CellPhoneDB.52 In general, there were more ligand-receptor interactions between cell types in BMMNCs of VEXAS (bottom) than in those of healthy donors (top). CD8T, CD8+ T cell; CD4T, CD4+ T cell; NK, natural killer cell; CD34, CD34+ cell; ProB, pro-B cell; B_Plasma, B cell_Plasma cell; Ery, erythroblast; Neut, neutrophil; Mono, monocyte; DC, dendritic cell.
(B) Among the 149 ligand-receptor pairs expressed in VEXAS patients and healthy donors, the number of ligand-receptor pairs presenting in BMMNCs in VEXAS patients and healthy donors were counted. A pink box indicates ligand-receptor pairs most represented in VEXAS; most of them were unique in VEXAS BMMNCs. A blue box indicates ligand-receptor pairs that are less represented in VEXAS than in healthy donors.
(C) Summary of ligand-receptor pairs between HSPCs with many immune cell types in VEXAS patients and in healthy donors. Highlights in pink for patients and blue for healthy donors indicate that the number of ligand-receptor pairs is higher in patients than in healthy donors.
(D) Potential target genes were identified as differentially expressed in VEXAS patients compared with healthy donors with an adjusted p value < 0.05 and a log fold change > 0.1 or < −0.1. Summary of ligand-receptor differential interactions identified in VEXAS patients by NicheNetr based on differentially expressed genes. Blue segments, molecules expressed by monocytes (left) and neutrophils (right); red segments, molecules expressed by CD34+HSPCs; blue arcs, interactions of ligands from monocytes/neutrophils with receptors on CD34+HSPCs; red arcs, interactions of ligands from CD34+HSPCs with receptors on monocytes/neutrophils; gray arcs, interactions of ligands and receptors in the same cell types. Left and right panels show interactions between monocytes and CD34+HSPCs, and between neutrophils and CD34+HSPCs, respectively.
(E) Cell-cell interactions were defined by NicheNetr.53 Ligands expressed by BMMNCs were ranked by likelihood that ligands would affect gene expression changes in CD34+HSPCs. Receptors expressed on CD34+HSPCs were selected based on their known potentials to interact with prioritized ligands. Finally, target genes were selected based on their differential expression in CD34+HSPCs and their potentials to be regulated by ligand-receptor interactions identified between BMMNCs and CD34+HSPCs.
(F) Interaction scores of patients’ CD34+HSPCs with myeloid cells were compared with those in healthy donors. p values with the two-sided unpaired t test are shown.
(G) Correlation of interaction scores (CD34+HSPCs with myeloid cells in patients) with inflammatory scores (left) and cytokine scores (right) were analyzed. p values and R values with the Pearson correlation test are shown.
NicheNet is a novel algorithm that employs gene expression data to impute ligand-receptor interactions that mediate downstream transcriptional changes by integrating pre-existing knowledge of signaling and regulatory networks.53 NicheNet was applied to model interactions between monocytes/neutrophils and CD34+ HSPCs, which could potentially induce differential expression of target genes in VEXAS (Figure 6D). For monocyte/neutrophil (ligand)-HSPC (receptor) interactions, top predicted ligands expressed by monocytes/neutrophils were ADAM17, SEMA4D, HLA-DRA, and TNFSF13B, and top receptors expressed by HSPCs were P2RY13, ITGA4, and PLXNB2; IL-6R and IL-1B also were highly expressed in monocytes/neutrophils in interactions with HSPCs. From a map of predicted target genes that were differentially expressed in HSPCs in VEXAS patients to a ligand-receptor activity heatmap (Figure 6E), ligand-receptor interactions between immune cells and HSPCs were consistent with upregulation of genes including STAT1, TNFAIP3, ITGAL, and GZMA involved in inflammation in HSPCs. In summary, cell-cell interaction analysis revealed enhanced interactions of myeloid cells with HSPCs and across most cell types in the BM, and involving frequent IFN and TNF interactions with their receptors. Interaction scores of HSPCs with myeloid cells were elevated in VEXAS (Figure 6F). Inflammatory scores and cytokine scores49,50 for each cell were calculated to evaluate inflammation in single progenitor cells: HSPCs with higher interaction scores with myeloid cells had higher inflammatory and cytokine scores (Figure 6G).
Lymphocyte clonal expansion in VEXAS
We unexpectedly observed clonal TCR rearrangements in UPNs 14–17, despite patients in our cohort not manifesting clinical evidence of T cell clonal expansion. Skyscraper plots showed TCR Vβ/Vα and matching Jβ/Jα in UPNs 14–17 (Figure 7A). The Gini index measures equality of distribution,54,55 and, for TCR/BCR diversity, the Gini index correlates positively with T/B cell clonality. We calculated Gini indexes of distribution of Vβ sequence clone sizes to quantify TCR clonality in UPNs 14–17, and compared them with published data.56 Gini indexes of TCRs in UPNs 14–17 were higher than in healthy donors (Figure 7B), indicating restricted TCR usage in UPNs 14–17, but there was little sharing of TCR clones among these four VEXAS patients, nor with healthy donors or T-large granular lymphocytic leukemia patients (Figure S5).56 T cell clonal expansion was mainly among CD8+ T cells compared with CD4+ T cells (Figures 7B and 7C). Cytotoxicity and IFN-γ response in VEXAS T cells were increased (Figure 7D) and predominantly in CD8+ T cells (Figure 7E). Using GLIPH2, we sought similar TCRs among these four patients, and the top TCR clusters were composed of TCR sequences from the patients, suggestive of common antigens in VEXAS, but this analysis was limited by a the small sample size.
Figure 7.

TCR and BCR usage in VEXAS
(A) Skyscraper plots showing Vβ/Vα and matching Jβ/Jα in VEXAS patients (UPNs 14–17).
(B) Gini indexes of TCR clonality in CD4+ T cells, CD8+ T cells, and total T cells of BM of VEXAS patients (n = 4), peripheral blood T cells of T-large granular lymphocytic leukemia (T-LGLL) patients pre-aleumtuzmab treatment (n = 13), T-LGLL patients post-aleumtuzumab treatment (n = 12), and healthy donors (n = 7).56
(C) Clone size information was projected to the UMAP of CD4+ T and CD8+ T cells in VEXAS patients. Clones with size 2 (two cells with identical TCR sequences) are in blue color, clones with sizes 3–9 (three to nine cells with identical TCR sequences) in green, and highly expanded clones with sizes ≥ 10 (at least 10 cells with identical TCR sequences) in red, and all other cells in gray.
(D) Expression of T cell activation score, cytotoxicity score, IFN-γ signaling score, and exhaustion score were plotted by individual for patients (n = 9) and healthy donors (n = 4). p values with the two-sided unpaired Mann-Whitney test were shown.
(E) CD4+ and CD8+ T cells from VEXAS patients were plotted in UMAP. Top 10% cells expressing the highest cytotoxicity score and IFN-γ signaling score are highlighted in red and all the rest in gray.
(F) TCRs identified in VEXAS patients were clustered by GLIPH2, and clusters with at least four TCRs are shown. Colors indicate TCR sequences originated from individual patients. CDR3 sequences for the top two largest TCR clusters are listed, encompassing TCRs from all four patients.
(G) Skyscraper plots showing VH/VK/VL and matching JH/JK/JL in VEXAS patients (UPNs 14–17).
(H) Clone size information was projected to the UMAP of B cells in VEXAS patients. Clones with size 2 (two cells with identical BCR sequences) are in blue color, clones with sizes 3–9 (three to nine cells with identical BCR sequences) in green, highly expanded clones with sizes ≥ 10 (at least 10 cells with identical BCR sequences) in red, and all other cells in gray.
(I) Gini indexes of BCR clonality in UPNs 14–17, and healthy donors in a reference study (n = 71).57 Data are presented as mean values ± SEM. p values with two-sided unpaired Mann-Whitney test are shown.
(J) B cells from the two largest clones are plotted on UMAP. Clone CAKVYSGEMATMFGFDHSHYYGMDVW (size 449) and clone CARNLLMWFGEFYPW (size 186). B cells with captured UBA1 mutations are highlighted in red; B cells with captured wild-type UBA1 transcripts and all the rest are highlighted in gray.
We examined BCR rearrangement in UPNs 14–17 (Figure 7G): UPNs15 and 16 had plasma cell myeloma and a small CD5+ B cell clone, respectively. B cell clonal expansion was observed in VEXAS (Figure 7H) but to a similar level as in healthy donors. Gini indexes of BCRs in UPNs 14 and 17 were not different from those in healthy donors57 and Gini indexes of BCRs in UPNs 15 and 16 were not higher than those in UPNs 14 and 17, despite clinical evidence of plasma cell or B cell dyscrasia (Figure 7I). We examined BCR usage in UPNs 14–17: there was no overlap among UPNs 14–17 nor with BCRs of healthy individuals57 (Figure S6). When we linked UBA1 mutation information with BCR sequences in the same cell, BCR expression was detected in hundreds of cells in UPNs 14 and 17, but expressed wtUBA1 and mtUBA1 transcripts were present in only a few cells, due to the low mutation frequency in lymphoid cells and technical dropout. mtUBA1 and wtUBA1 cells were present in the same BCR clones (Figures 7J and S7).
Discussion
VEXAS has been described only recently, but its unusual etiology was clear in the original report1: an acquired mutation in hematopoietic stem cells, found only in men due to the X chromosome location of the UBA1 gene, resulting in severe, multiorgan autoinflammation that manifested in a range of familiar rheumatologic diagnoses. Zebrafish knockouts of the homologous gene suggested that inflammation was a direct consequence of the mutations,1 but other attempts to model VEXAS in animals and in vitro have proven difficult in practice, perhaps due to the fundamental cellular role of the ubiquitylation pathway disrupted by an altered UBA1 gene product. Nevertheless, pathophysiologic mechanisms have been unclear: how gene mutations lead to inflammation, which cell types are important in effecting tissue damages, and the routes to both lymphocyte depletion and malignant plasma cell proliferation. Other basic questions are the discordance between mtUBA1 cell clonal dominance in patients and deficient hematopoietic cell proliferation in vitro, and how altered protein degradation relates to the initiation of profound upregulation of multiple innate immune pathways and globally elevated cytokine production.
Here, we utilize single-cell methodologies to characterize transcriptomes and expressed mutations of BM precursors and HSPCs from VEXAS patients, in a comprehensive and unbiased approach to characterize this poorly understood disease. At the high resolution of scRNA-seq, activation of multiple different inflammatory pathways were striking in primitive stem and progenitor cells. The protein ubiquitination and proteasome pathway were dysregulated, with no apparent compensation by autophagy to allow protein degradation, likely leading to elevated stress response in VEXAS, as suggested by our data and previous studies.58 Mutations in UBA1 not only resulted in marked activation of the innate immune pathways (implying a cell-autonomous mechanism resulting from UBA1 disruption), but they altered cell cycling, potentially providing a mechanism for clonal dominance. Lineage specification of HSCs, governed by several master transcription factors, was skewed toward myeloid, and especially granulocytic differentiation and against lymphoid differentiation in HSCs in VEXAS, and there was progressive loss of lymphocytes with differentiation, accompanied by increased apoptosis restricted to the lymphoid trajectory. Myeloid lineage cells had the most inflammatory activation, but lymphoid cells, predominantly wtUBA1, also had elevated expression to a lesser extent of the same genes. Increased cell-cell interactions between myeloid cells and HSPCs and among major cell types in BM in VEXAS revealed enhanced “crosstalk” in this inflammatory environment, particularly for the IFN and TNF-α pathways. Taken together, our results begin to define the characteristics of inflammation, lineage disequilibrium, and genotype association at single-cell molecular resolution for VEXAS syndrome.
One general hypothesis to explain clonal hematopoiesis is that cells with pre-existing somatic mutations are selected due to fitness in their microenvironments, as for example, the familiar loss of HLA gene expression as an escape from immune destruction.59,60,61 An alternative, not mutually exclusive mechanism, is that an acquired mutation contributes to or even drives chronic inflammation, as has been proposed for proinflammatory TET2 mutations in CHIP, with consequent proinflammatory tissue-resident macrophages and accelerated atherosclerotic events.62,63,64 Conversely, chronic infection and inflammation appear to secondarily favor expansion of mtDNMT3A somatic clones, perhaps due to their bias toward self-renewal over terminal differentiation in a stressed or regenerating environment, as has been inferred from murine models.65,66 DNMT3A mutations also alter immune phenotypes67; in a recently published single-cell multi-omics study, upregulation of a few genes involved in proinflammatory signaling is observed in mtDNMT3A HSPCs.33 Our data strongly favor UBA1 mutations in HSPCs as immediate drivers of both myeloid lineage dominance and the origin of inflammation in VEXAS syndrome. Nevertheless, with apoptosis genes upregulated and cell cycling genes downregulated in BMMNCs and HSPCs in VEXAS, and functionally, both BMMNCs and CD34+ HSPCs from patients formed fewer colonies than did cells from healthy individuals (Figure S7B), myeloid progenitors and HSPCs in VEXAS likely exhibited defective proliferation and differentiation, reflecting the ineffective hematopoiesis typical of MDS.
In addition, the hyperinflammatory microenvironment in VEXAS syndrome likely does positively select other somatically mutated clones: DNMT3A and TET2 clones are frequent in VEXAS18,19,20,21 as they are in other inflammatory clinical conditions.68 We also characterized the transcriptome of mtDNMT3A cells in the setting of VEXAS. Upregulation of the inflammation and immune response pathways in mtDNMT3A cells in our VEXAS cases indicated a proinflammatory phenotype associated with this epigenetic genotype, in accord with previous reports33,67; cell cycling of mtDNMT3A cells appeared to be decreased compared with wtDNMT3A cells. Whether mtUBA1 cells might also have a selective advantage in an inflammatory environment—possibly creating a deleterious autocrine/paracrine loop—has been unclear, but we noted marked activation of multiple inflammatory pathways early in hematopoietic ontogeny, in contrast to CHIP-mutated inflammatory terminal cells. In addition, the allele frequency of mtUBA1 clones did not correlate with clinical severity of inflammation in patients. Our analysis demonstrated a positive correlation between interaction of mtUBA1 HSPCs with myeloid cells and inflammation: with enhanced cell-cell interactions in VEXAS, malicious feedback may exaggerate inflammation in the disease. The marked involvement of the IFN-γ and TNF-α pathways, and cell-cell interactions mediated by IFNs, TNF-α, IL-1β, and IL-6 suggest that IFN and TNF blockers or disruption of cell-cell interactions are potential targets of therapies in VEXAS. In addition, diagnostic distinctions may be aided by application of single-cell genomic results: both marked activation of several critical inflammatory pathways and activation in primitive HSPCs may be useful in distinguishing among VEXAS syndrome and other hematologic diseases with overlapping clinical features.
Plasma cell dyscrasias are frequent in VEXAS,1,3,4 despite absence of UBA1 mutations in lymphocytes. Diversity of BCR repertoires in two of our patients with clinical evidence of B/plasma cell clonal expansion was similar to that in other VEXAS patients and both were similar to BCR clonality reported in healthy individuals,57 indicating that BCR usage was preserved in disease, but with reduced B cell numbers and distinct BCR repertoires across individuals. Linking UBA1 mutations with BCR sequences in single cells showed coexistence of mtUBA1and wtUBA1 cells within the same BCR clone, implying antigenic drivers rather than the UBA1 mutation itself were driving B cell expansion. Unexpected was reduced TCR diversity detected by scRNA-seq in four patients, as to date there are no reports of TCR clonal expansion in VEXAS. Furthermore, T cells, especially clonally expanded CD8+ T cells in VEXAS marrow had increased cytotoxicity and IFN-γ signaling, and one inference is T cells targeting unknown antigens could be actively involved in disease pathogenesis. A higher percentage of activated CD8+ T cells was observed in relapsing polychondritis patients with VEXAS compared with polychondritis patients without VEXAS and healthy controls.5 Oligoclonal B and T cells (reduced in number and mainly unmutated) may be secondary to immune cell recognition of novel, aberrant, or overexpressed antigens, and specific presentation of immunogenic epitopes due to abnormal protein degradation.
In conclusion, our study of single-BM hematopoietic cells facilitates understanding of the pathophysiology of the newly defined disease VEXAS. Specifically, simultaneous genotyping and phenotyping, and direct comparison of mutated and wild-type human HSPCs provide fundamental insights in the direct and indirect roles of UBA1 mutations in VEXAS pathophysiology. Our results expand our knowledge of distinct transcriptome signatures and crosstalk among BM hematopoietic cells, UBA1 and DNMT3A mutations, and TCR/BCR repertoires in VEXAS.
Limitations of the study
Our study has limitations. First, the sample size of the explorative cohort was relatively small (due to the high cost of experiments) and potentially biased by patients with rheumatologic manifestations (due to referral patterns at our center). Nevertheless, observations from scRNA-seq were reproducible in an independent validation cohort with traditional immunological methods. However, attempts at confirmation by flow cytometry and ELISpot with HSPCs were not successful due to low cell numbers. Second, detection of UBA1 and DNMT3A mutations with scRNA-seq data was limited by low transcript abundance, allelic and technical dropout, and incomplete transcript coverage inherent to the platform.48 Indeed, very few experimental protocols have been published that allow reliable simultaneous RNA and DNA sequencing of single cells.31,32,33 Due to the prospective nature of the study and the limitation of 3′- or 5′-biased sequencing reagents, completely reliable simultaneous detection of UBA1 and DNMT3A mutations in single cells was not achieved. Technologies under development that enable simultaneous sequencing of both RNA and DNA of single cells at high throughput31,32,33 may help to define the clonal architecture of UBA1 and other somatic mutations frequent in VEXAS, and allow correlation of transcriptome signatures in multiply mutated single cells. Third, although we performed knockdown in vitro experiments with human cell lines, validation in primary human cells and animal models is desirable, if not yet achievable.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human lineage cocktail (Clones UCHT1, HCD14, 3G8, HIB19, 2H7, and HCD56) Pacific blue | BioLegend | Cat# 348805; RRID: AB_2889063 |
| Mouse anti-human CD34 (Clone 581) PE | BD Biosciences | Cat# 555822; RRID: AB_396151 |
| Mouse anti-human CD38 (Clone HIT2) APC | BD Biosciences | Cat# 555462, RRID: AB_398599 |
| Anti-human CD90 (Clone 5E10) FITC | BioLegend | Cat# 328108, RRID: AB_893429 |
| Anti-human CD10 (Clone HI10A) BV605 | BD Biosciences | Cat# 562978, RRID: AB_2737929 |
| Anti-human CD135 (Clone BV10A4H2) PE/Cyanine7 | BioLegend | Cat# 313314, RRID: AB_2565478 |
| Anti-human CD45RA (Clone HI100) BV510 | BioLegend | Cat# 304142, RRID: AB_2561947 |
| Human IFN-γ/TNF-α Double-Color Enzymatic ELISPOT Assay kit | ImmunoSpot | Cat# SKU:hIFNgTNFa-2M |
| Biological samples | ||
| 10% fetal bovine serum | Sigma-Aldrich | Cat# 12306C |
| Healthy bone marrow sample | National Institutes of Health | Healthy bone marrow sample |
| VEXAS patients’ bone marrow sample | National Institutes of Health | VEXAS patients’ bone marrow sample |
| Chemicals, peptides, and recombinant proteins | ||
| LSM Lymphocyte Separation Medium | MP Biomedicals | Cat# 50494X |
| Phosphate buffered saline | Lonza | Cat# 17-516Q |
| ACK lysing buffer | Quality Biological | Cat# 118-156-101 |
| IMDM | Thermo Fisher Scientific | Cat# 12440053 |
| MethoCult™ H4434 Classic semisolid methylcellulose medium | STEMCELL Technologies | Cat# 04444 |
| MethoCult™ H4435 Enriched semisolid methylcellulose medium | STEMCELL Technologies | Cat# 04445 |
| RPMI 1640 | Thermo Fisher Scientific | Cat# 11875093 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003 |
| Critical commercial assays | ||
| 10x Genomics System using the Chromium Single Cell 3′ Reagent Kit v2 | 10x Genomics | Cat# 120237 |
| 10x Genomics Single Cell Immune Profiling Solution v 1.1 | 10x Genomics | Cat# 1000165 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE196052 |
| Published scRNA-seq data of HSPCs in MDS patients | Compare inflammatory signaling scores with published data Ganan-Gomez et al. 202237 | GEO: GSE137429 |
| Published scRNA-seq data of HSPCs in CMML patients | Compare inflammatory signaling scores with published data Wiseman et al. 202038 | Array Express: E-MTAB-8884 |
| Published scRNA-seq data of HSPCs in CML patients | Compare inflammatory signaling scores with published data Giustacchini et al. 201739 | GEO: GSE76312 |
| A public database of B cell receptor sequences | Compare BCR usage with published data DeWitt et al. 201657 |
http://adaptivebiotech.com/pub/robins-bcell-2016 |
| Published T cell receptor sequences in T-LGLL patients and healthy donors | Compare TCR usage with published data Gao et al. 202256 |
GEO: GSE168859 |
| Experimental models: Cell lines | ||
| 293T cells | ATCC | CRL-3216 |
| U937 cells | ATCC | CRL-3253 |
| THP-1 cells | ATCC | TIB-202 |
| Raji cells | ATCC | CCL-86 |
| Jurkat cells | ATCC | TIB-152 |
| Oligonucleotides | ||
| pLKO1-Puro Mission shRNA constructs | Sigma-Aldrich | KD1 TRCN0000004003 |
| pLKO1-Puro Mission shRNA constructs | Sigma-Aldrich | KD2 TRCN0000277770, |
| pLKO1-Puro Mission shRNA constructs | Sigma-Aldrich | KD3 TRCN0000277769 |
| pLKO1-Puro Mission shRNA constructs | Sigma-Aldrich | KD4 TRCN0000004004 |
| Recombinant DNA | ||
| Lentiviral construct pLKO-1 puro plasmid | Addgene | Cat# 8453 |
| Packaging plasmid pCMV-VSV-G | Addgene | Cat# 8454 |
| Packaging plasmid pRSV-Rev | Addgene | Cat# 12253 |
| Packaging plasmid pHM-Tat1b | Addgene | Cat# 164442 |
| Software and algorithms | ||
| cellranger count version 3.0.1 | 10x Genomics | https://support.10xgenomics.com/singlecell-gene expression/software/downloads/3.0/ |
| cellranger vdj version 3.0.1 | 10x Genomics | https://support.10xgenomics.com/singlecell-gene-expression/software/downloads/3.1/ |
| Seurat version 2.3.4 | Stuart et al., 201970 | https://cran.r-project.org/web/packages/Seurat/index.html |
| R version 3.5.0 | R Core Team, 202170 | https://www.r-project.org/ |
| cb_sniffer version 1.0 | Pettti et al., 201948 | https://github.com/sridnona/cb_sniffer |
| CellPhoneDB version 3.1.0 | Garcia-Alonso L et al. 202271 | https://www.cellphonedb.org/ |
| NicheNet version 1.0.0 | Browaeys et al., 202053 | https://github.com/saeyslab/nichenetr |
| fGSEA version 1.16.0 | Korotkevich et al., 201972 |
http://bioconductor.org/packages/release/bioc/ html/fgsea.html |
| Palantir version 1.0.0 | Setty et al.201973 | https://github.com/dpeerlab/Palantir |
| topGO version 2.34.0 | Alex, A., and Rahnenfuthrer, J. 201974 | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| AUCell version 1.4.1 | Aibar et al., 201775 | https://bioconductor.org/packages/release/bioc/html/AUCell.html |
| tCR version 2.3.2 | Github repository | https://imminfo.github.io/tcr/ |
| Genomatix Generanker | Werner et al., 201376 | http://www.genomatix.de |
| STRING version 9 | Szklarczyk et al., 201577 | https://string-db.org/ |
| GraphPad Prism 9.02 | GraphPad software | https://www.graphpad.com/scientificsoftware/prism/ |
| FlowJo v7.6.4 | Tree Star | https://www.flowjo.com |
| Analysis scripts | This paper | https://github.com/shouguog/UBA1 |
| Other | ||
| Analysis and visualization of the scRNA-seq datasets in this study can be performed at the interactive website | This paper | https://shouguog.shinyapps.io/vexas_cd34_bm/ |
Resource availability
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact Zhijie Wu (zhijie.wu@nih.gov).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Human samples
Bone marrow (BM) samples were obtained from VEXAS patients after written informed consent under protocol (www.clinicaltrials.gov NCT00001373) approved by the Institutional Review Board of the National Human Genome Research Institute, in accordance with the Declaration of Helsinki. Healthy donors were enrolled as controls under protocol NCT00001620 in National Heart, Lung, and Blood Institute. Four healthy donors (male, 57/61/62/68 years old) were age- and gender-matched with patients in explorative cohort for scRNA-seq analyses.
Method details
Bone marrow processing and cell sorting
BM mononuclear cells (BMMNCs) were isolated, followed by flow cytometric sorting to enrich lineage CD34+ hematopoietic stem and progenitor cells (HSPCs); both BMMNCs and HSPCs were used for single-cell RNA sequencing (scRNA-seq). Fresh BM samples were processed within 16 h, followed by either direct analyses (flow cytometry and colony forming assay for all individuals; scRNA-seq for UPNs 1, 6, 10, and 11) or cryopreserved until use (UPNs 14–17) to enrich lineage-CD34+ HSPCs and for scRNA-seq. Another 11 patients and 8 healthy donors were enrolled in a validation cohort, with cryopreserved BMMNC samples primarily for immunophenotyping and ELISpot assays.
BM specimens were obtained from patients and healthy donors and kept in heparin tubes, and processed within 16 h after collection. BMMNCs from each person were isolated by density centrifugation using LSM Lymphocyte Separation Medium (Cat# 50494X, MP Biomedicals). Briefly, BM was diluted 2-fold using phosphate buffered saline (PBS) (Cat# 17-516Q, Lonza), layered on top of 1 volume LSM Lymphocyte Separation Medium in a 50-mL Falcon tube, and spun down at 1,140g for 25 min at room temperature with brake off. A BMMNC layer was isolated and washed with PBS after red blood cell lysing with ACK lysing buffer (Cat# 118-156-101, Quality Biological). BMMNCs were resuspended in the IMDM (Cat# 12440053, Thermo Fisher Scientific) + 2% fetal bovine serum (Cat# 12306C, Sigma-Aldrich) before fluorescence-activated cell sorting (FACS) to enrich lineage−CD34+ hematopoietic stem and progenitor cells (HSPCs). BMMNCs were stained with monoclonal antibodies (Abs) for 30 min on ice: anti-human lineage cocktail (CD3, CD14, CD16, CD19, CD20, and CD56; clones UCHT1, HCD14, 3G8, HIB19, 2H7, and HCD56, respectively, Cat# 348805, BioLegend) in Pacific Blue; anti-CD34 Ab (clone 581, Cat# 555822, BD Biosciences) in PE, and anti-CD38Ab (clone HIT2, Cat# 555462, BD Biosciences) in APC. Cells were sorted using the FACSAria Fusion Flow Cytometer (BD Biosciences). Aliquots of BMMNCs were subjected to multi-color flow cytometry to profile HSP subpopulations. BMMNCs and purified lineage−CD34+ cells were subjected to colony forming assay and scRNA-seq analysis.
Human primary cell culture
BMMNC isolated from fresh BM were used for flow cytometry, cell sorting, and scRNA-seq were proceeded without cell culture. Cell culture conditions for primary BM cells used for colony forming assay and ELISpot assay were described in “Colony forming assay” and “ELISpot assay to check IFN-γ and TNF-α secreted by human BMMNCs” sections below, respectively, with different cell culturing conditions per experiments. In brief, for colony forming assay, isolated BMMNCs and sorted CD34+ cells were cultured in semisolid methylcellulose medium at 37°C with 5% CO2 for 14 days. For ELISpot assay, BMMNCs were cultured in CTL-Test Medium in 96-well plates and incubated in a 37°C humidified incubator, 5–9% CO2 for 20 h.
Colony forming assay
BMMNCs from individuals were mixed in semisolid methylcellulose medium (MethoCult H4434 Classic, Cat# 04444, STEMCELL Technologies) containing interleukin (IL)-3, stem cell factor (SCF), erythropoietin (EPO), and granulocyte-macrophage colony-stimulating factor (GM-CSF) at 2 x 104 cells/plate. Sorted CD34+ cells from individuals were mixed in semisolid methylcellulose medium (MethoCult H4435 Enriched, Cat# 04445, STEMCELL Technologies) containing IL-3, IL-6, SCF, EPO, granulocyte colony-stimulating factor (G-CSF), and GM-CSF at 500 cells/plate. Cells were cultured at 37°C with 5% CO2. Colonies were counted at day 14.
Flow cytometry profiling of HSPCs
Flow cytometric sorting of lineage−CD34+ HSPCs following isolation of BMMNCs. BMMNCs were stained with antibody mixtures on ice for 30 min in RPMI 1640 (Cat# 11875093, Thermo Fisher Scientific). Samples were subsequently acquired using the BD LSR Fortessa cytometer (BD Biosciences), and post-acquisition analysis was performed using Flowjo software (v.7.6.4; Flowjo LLC, BD Biosciences). Antibodies used for flow cytometry analyses were: anti-human lineage cocktail (CD3, CD14, CD16, CD19, CD20, and CD56; clones UCHT1, HCD14, 3G8, HIB19, 2H7, and HCD56, respectively, Cat# 348805, BioLegend) in Pacific Blue; anti-human CD34 in PE (clone 581, Cat# 550761, BD Biosciences), anti-human CD38 in APC (clone HIT2, Cat# 555462, BD Biosciences), anti-CD90 in FITC (clone 5E10, Cat# 328108, BioLegend), anti-human CD10 in BV605 (clone HI10A, Cat# 562978, BD Biosciences), anti-human CD135 in PE/Cy7 (clone BV10A4H2, Cat# 313314, BioLegend), and anti-human CD45RA in BV510 (clone HI100, Cat# 304142, BioLegend).
ELISpot assay to check IFN-γ and TNF-α secreted by human BMMNCs
IFN-γ and TNF-α secretion from BMMNCs of VEXAS patients and healthy donors were measured using the Human IFN-γ/TNF-α Double-Color Enzymatic ELISPOT Assay kit (Cat# SKU:hIFNgTNFa-2M, ImmunoSpot) in two separate experiments in triplicate (4 patients versus 3 healthy donors for a 1st batch, and 5 patients versus 2 healthy donors for a 2nd batch), according to the manufacturer’s protocol. In brief, pre-coated 96-well plates were activated with Human IFN-γ/TNF-α Capture Solution and 70% ethanol on Day 0, and incubated at 4°C overnight. On Day 1, BMMNCs were suspended in CTL-Test Medium and seeded in 96-well plates at a density of 90,000 cells/well (1st batch) or 400,000 cells/well (2nd batch), and incubated in a 37°C humidified incubator, 5–9% CO2 for 20 h. On Day 2, 96-well plates were washed and incubated sequentially with Anti-human IFN-γ/TNF-α Detection Solution, Tertiary Solution, and Blue and Red Developer Solutions. 96-well plates were then air-dried and face down on paper towels on a bench top for more than 24 h before scanning and counting with the CTL ImmunoSpot Analyzers and ImmunoSpot Software.
Human leukemic cell lines culture
Human leukemic cell lines U937, THP-1, Raji, and Jurkat were purchased from the American Type Culture Collection (ATCC). U937, THP-1, Raji and Jurkat cell lines were maintained in RPMI-1640 (Cat# 11875093, Thermo Fisher Scientific) in 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 1% L-glutamine, 100 units/ml penicillin and 100 μg/mL streptomycin (Thermo Fisher) at 37°C with 5–9% CO2.
Knockdown of UBA1 in human leukemic cell lines
pLKO1-Puro Mission shRNA constructs (Sigma-Aldrich) targeting UBA1 included KD1 TRCN0000004003, KD2 TRCN0000277770, KD3 TRCN0000277769, and KD4 TRCN0000004004 along with control scramble shRNA were used for producing shRNA knockdown. Lentiviruses were produced in 293T cells by co-transfection of the lentiviral construct pLKO-1 puro plasmid (Cat# 8453, Addgene) with packaging plasmids (pCMV-VSV-G, Cat# 8454; pRSV-Rev, Cat# 12253; pHM-Tat1b, Cat# 164442, Addgene) for 48 to 72 h. Infection was carried out with 2 x 106 of U937, THP-1, Raji, or Jurkat cells (ATCC) in a 6-well plate with lentiviruses in the presence of Polybrene (6 μg/mL; Cat# TR-1003, Sigma-Aldrich). For infection of lentiviruses carrying ectopic expression vectors, cells were centrifuged at 1,000 g at 30°C for 90 min. After 4 to 6 days of selection with 0.5–1 μg/mL of puromycin for pLKO1-puro-shRNA constructs, cells were analyzed by immunoblotting with UBA1 (Cat# 4891, Cell Signaling Technology) and actin (Cat# 3700, Cell Signaling Technology) antibodies using the protocol previously described (Beck et al., 2020). RNA was extracted from pooled antibiotic resistant clones for each respective shRNA, after confirmation of knockdown by immunoblotting.
Cell preparation, whole transcriptome amplification (WTA), cDNA library preparation, and sequencing
scRNA-seq analysis for patients (UPNs 6, 11, 1, 10, and 13) and healthy donors was performed with the 10x Genomics System using the Chromium Single Cell 3′ Reagent Kit v2 (Cat# 120237), according to the manufacturer’s protocol (www.10xgenomics.com).78 scRNA-seq coupled with single-cell T cell receptor/B cell receptor sequencing (scTCR/BCR-seq) analysis for UPNs 14–17 was performed with the 10x Genomics System using the 10x Genomics Single Cell Immune Profiling Solution v 1.1 (Chromium Single Cell 5′ Reagent Kit v1.1, Cat# 1000165, 10x Genomics), following the manufacturer’s protocol (www.10xgenomics.com).78 Briefly, BMMNCs and FACS-sorted BM lineage−CD34+ cells were washed with 1X PBS with 0.04% (w/v) bovine serum albumin. Cell concentration and viability were determined using the Countess II Automatic Cell Counter and the trypan blue staining method. Cell loading and capturing were done on the Chromium Controller (10x Genomics). Following reverse transcription and cell barcoding in droplets, emulsions were broken, and cDNA was purified using Dynabeads MyOne SILANE, followed by PCR amplification. Amplified cDNA was then used for both 3′ and 5′ gene expression library construction and TCR/BCR enrichment. For gene expression library construction, the amplified cDNA was fragmented, end-repaired, and double-sided size-selected with SPRIselect beads. For TCR/BCR library construction, TCR/BCR transcripts were enriched from amplified cDNA by PCR. Subsequently, the enriched PCR product was fragmented, end-repaired, and size-selected with SPRIselect beads. The scRNA libraries were pooled together and sequenced on the Illumina NovaSeq system using read lengths of 26-bp read 1, 8 bp i7 index, 98-bp read 2. The single-cell TCR/BCR libraries were sequenced on the Illumina NovaSeq system using read lengths of 150-bp read 1, 8 bp i7 index, 150-bp read 2. Sequencing metrics were summarized in Table S5.
scRNA-seq data analysis
Preprocessing of scRNA-seq and scTCR/BCR-seq data
Alignment, barcode assignment, and Unique Molecular Identifier (UMI) counting were performed using the cellranger pipeline (http://software.10xgenomics.com/single-cell/overview/welcome).78.
After single-cell libraries were sequenced using the Illumina system, cellranger pipeline (support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger" title="https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger">https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger) was used to process scRNA-seq raw data in order to align reads to the genome, and to generate gene–cell expression matrices. Specifically, sequencing reads were aligned to the hg19 reference genome by STAR with annotation of ENSEMBL. Uniquely aligned reads were used to quantify gene expression levels for all ENSEMBL genes with UMIs. We filtered and removed low-quality cells from further analysis if the number of genes detected was fewer than 500 (low quality, potential fragments) or more than 3,000 (potential doublets). We also excluded those cells with a high percentage of mitochondrial gene reads (>10%),79 and remaining single cells were subjected to subsequent data analyses. Sequencing metrics and detailed information are provided in Table S1.
TCR reads were aligned to the GRCh38 reference genome and consensus TCR annotation was performed using the cellranger vdj program (10x Genomics, version 3.0.1). TCR libraries were sequenced at depth of over 2,000 reads/cell, with a final 33418 mean read pairs/cell. On average, 27,053 reads mapped to either the TRA or TRB loci in each cell. TCR annotation was performed using the 10x cellranger vdj pipeline as described at support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/using/vdj" title="https://support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/using/vdj">https://support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/using/vdj. Barcodes with a higher number of UMI counts than those of simulated background were considered as cell barcodes. V(D)J read filtering and assembly were implemented as a previous study.80 cellranger firstly trimmed known adaptor and primer sequences from the 5′ and 3′ ends of reads, and then filtered away reads lacking at least one 15-bp exact match against at least one reference segment (TCR, TRA, and TRB gene annotations in Ensembl version 87). Next, cellranger performed de novo assembly for each barcode by building a De Bruijn graph of reads independently. The assembler output contig sequences which were assigned at least one UMI. Finally, each assembled contig was aligned against all of the germline segment reference sequences of the V, D, J, C, and 5′ UTR regions. cellranger searched a CDR3 motif (Cys-to-FGXG/WGXG) in a frame defined by a start codon in the L + V region or all 6 frames when the L + V region was absent. A contig was kept and considered as productive if: 1) it fully spanned the V and J segments; 2) there was a start codon in the V region; 3) it contained a CDR3 region in-frame with a V start codon; 4) there were no stop codons in the V-J spanning region. Most cell barcodes contained two matching productive contigs, comprising either a TCRA or a TCRB though it was of biological possibility that fewer productive contigs (low sensitivity) or >2 productive contigs (some cells do contain more than one TCRB or TCRA chain) were associated with one cell barcode.81 Similarly, BCR reads were also processed using the cellranger vdj program, with the IMGT database of GRCh38 genome as reference. Only productive contigs of BCR were kept for analysis.
Downstream analyses were performed using the R software82 package in Seurat (Stuart et al., 2019; http://satijalab.org/seurat/, v2.3.4)70 on BMMNCs and lineage−CD34+ cells separately (Satija et al., 2015).83 Raw reads in each cell were first scaled by a library size to 10,000, and then log-transformed. To improve downstream dimensionality reduction and clustering, regressionOut in the Seurat package84 was used to remove unwanted sources of variation based on the number of UMIs and percentages of mitochondrial reads. Highly variable genes (∼1,600 for BM cells and ∼1,900 for CD34+ cells, identified with y.cutoff = 0.5) were used for Principal Component Analysis (PCA) of high-dimensional data. Top 30 principal components were selected for unsupervised clustering of cells with a graph-based clustering approach.
Downstream analysis
Dimensionality reduction and clustering were performed by PCA and visualized with Uniform Manifold Approximation and Projection (UMAP). Cell type identity was assigned to each cluster based on significance in overlap between signature genes of BMMNCs35 and HSPCs40 and cluster-specific genes (Fisher’s exact test). Palantir73 was used to reconstruct a differentiation continuum of cells and to order individual cells’ differentiation for pseudotime analysis. Gene Set Enrichment Analysis (GSEA; http://software.broadinstitute.org/gsea) and Gene Ontology (GO)85,86 were used to interpret gene set enrichment and pathways of defined differentially expressed genes. Single-nucleotide variations in UBA1 and DNMT3A were identified in single cells using a Pysam-based tool, cb_sniffer with default parameters.48 A scoring algorithm to calculate interaction scores,87 CellPhoneDB52,71 and NicheNet51 were used to examine ligand-receptor interactions.
Quantification and statistical analysis
Unsupervised dimensionality reduction and UMAP visualization
PCA was used to reduce feature dimensions on the pooled cells of all patients and healthy donors, and top 30 principal components were input into t-SNE for further dimensional reduction. We found that cells of individuals clustered together, due to subject specificity and batch effects. The canonical correlation analysis (CCA) algorithm83 implemented in Seurat is a multivariate statistical technique for detecting the statistically common factors among digital gene expression (DGE) matrices, which varies from each other due to batch effects. After alignment with CCA, cells from different subjects were mixed well and separated by cell type categories. Resolution in the FindClusters function in Seurat88 was set to 2 for BMMNCs and 1 for HSPCs, and clustering results were shown in PCA and UMAP plots. Accordingly, marker genes in each cluster were identified using the Wilcoxon Rank-Sum test implemented in the Seurat v.2.3.4 package.
Cell type assignment
For lineage−CD34+ cells, an HSPC type was assigned to each cluster based on significance in overlapping between HSPCs and cluster-specific genes (the Fisher’s exact test).28,40 More specifically, top 250 overexpressed genes in each HSPC population were downloaded from http://www.jdstemcellresearch.ca/node/32, and were denominated as cell-type specific signature genes. Subsequently, the one-tailed Fisher’s exact test was utilized to assert enrichment of HSPC signature genes in the cluster marker gene list for each cluster,69 and a top associated cell type was assigned to each cluster. Cell types of BM cells were assigned with the same strategies using the Human Cell Atlas as ref. 35
Single-cell mutation identification and analysis
Aligned sequence data were generated by cellranger, and single-nucleotide variations in UBA1 and DNMT3A were identified in single cells using the Pysam-based tool, cb_sniffer (https://github.com/sridnona/cb_sniffer), with default parameters.48 Reads that had no Chromium Cellular Barcode (CB) tag or no Chromium Molecular Barcode (UB) tag were filtered out. Then, cell-associated tags for downstream analyses of UMIs were obtained. Usually, duplicate reads existed for a given UB and a base at a mutant position were identical across all reads. In rare cases when there were inconsistent reads, the most common base was chosen if a mutation was present in at least 75% of the reads. All reads corresponding to the UB were discarded when there was no common base at a mutation position (>75% reads).
Reconstruction of hematopoiesis trajectories using scRNA-seq data and dynamic gene expression
We used Palantir,73 a recently published trajectory-detection algorithm for pseudotime ordering. Palantir is based on results of diffusion maps, which is suitable to explore a differentiation trajectory. It firstly uses diffusion maps to focus on developmental trends and avoid spurious edges resulting from the sparsity and noise in scRNA-seq. Projecting the data onto top diffusion components effectively focuses edges in directions with high cell densities and reweighs similarity along these directions. Then, Palantir estimates probability of a cell in an intermediate state to reach any of terminal states of differentiation. It thus provides a quantitative measure of differentiation potential, in which multipotent cells have the highest differentiation potential and mature terminal states have the lowest potential. A high resolution achieved by Palantir allows detailed mapping of gene expression trends and dynamics that correlate with changes in lineage potential. After calculating diffusion components by using the Harmony augmented affinity matrix, Palantir orders cells along pseudotime that recapitulates known marker trends in development. Tracking gene expression changes along pseudotime enable determination of the differentiation change for each of the terminal fates. We used Palantir to estimate the differentiation status in order to characterize cells in patients with UBA1 mutations.
Projection of patients’ cells to the map of normal hematopoiesis
To characterize early hematopoiesis in VEXAS patents, individual cells were projected onto the map of normal hematopoietic differentiation based on cell-by-cell comparison of the patterns of global gene expression and localization to the most similar healthy donor cells (by Pearson correlation). This strategy was used for mapping patients’ cells on t-SNE and diffusion map plots, locating pseudotime estimation for Palantir.
Comparison of lineage gene Area Under the Receiver Operating Characteristic Curve (AUC) scores
We calculated AUC scores of lineage-specific gene expression (HSC, MEP, GMP, and LymP) of single cells in individual patients, and average AUC scores75 of specific lineages of all cells in each patient were compared. Comparison between two groups was performed using Prism (v.7.02; the GraphPad Software), and results were shown as mean ± standard derivation. Statistical analysis was performed using the two-sided unpaired Mann-Whitney test for two groups. p < 0.05 was considered statistically significant.
Differential expression of genes and generation of heatmaps
Differentially expressed genes were defined with the FindMarkers function in Seurat, by comparing gene expression in one cell subset with expression in all others. Genes with p value <0.05 and Log2(average fold change) > 0.1 were regarded as differentially expressed genes. Heatmaps and network visualization were generated with ggplot2 and heatmap2 in the R package.
GO and pathway analysis
GO was assessed with the R package topGO v2.26 using the algorithm elim,74 a minimum node size of 10, and genes that were expressed over 100 cells as the background gene list. p values derived from the GO analysis were not corrected for multiple testing. We examined the biological processes GO terms (The Gene Ontology Consortium, 2019) and the KEGG pathways.89,90
GSEA is the widely used pathway analysis tool that determines whether pre-defined gene sets show statistically significant, concordant differences between two biological states. GSEA is based on fold changes of all detected genes. To create gene sets for a genome with custom annotations, we associated our genes with known KEGG pathways and manually created gene sets. Fgsea51 was used for GSEA and to plot the running normalized enrichment scores along the ranked gene list.
Inflammatory gene pathway activity score analysis
To compare inflammation in HSPCs of VEXAS with several other hematopoietic diseases with overlapping clinical features, we calculated activity scores (expression levels) of several inflammatory response pathways in HSPCs of VEXAS patients in the current study, with data from published datasets (E-MTAB_8884 for chronic myelomonocytic leukemia (CMML) patients, GSE137429 for myelodysplastic syndrome (MDS) patients, and GSE76312 for chronic myelogenous leukemia (CML) patients). Briefly, we downloaded the fastq files from ArrayExpress with access number E-MTAB_8884, and used Cellranger 2.0 to analyze gene expression for this dataset. We also downloaded the processed scRNA-seq data for MDS (GSE137429) and CML (GSE76312) patients. We then downloaded the gene lists of HALLMARK_INFLAMMATORY_RESPONSE, HALLMARK_TNFA_SIGNALING VIA NFKB, and HALLMARK_INTERFERON_GAMMA_RESPONSE from MSigDB of GSEA, and their activity scores (expression levels) in our and these three datasets were calculated with the addModuleScore function built in the Seurat (http://satijalab.org/seurat/). The activity scores were normalized with healthy donors included in individual studies, and the double-sided t-test was used to assess the difference between VEXAS and three other diseases (MDS, CML, and CMML).
Inflammatory and cytokine score calculation
Inflammatory and cytokine scores were defined based on the published reference gene list,43,49 and were evaluated with the AddModuleScore function built in Seurat.70
Cell cycle stages calculation
Cell cycle stages were assigned with the CellCycleScoring function in Seurat.70,91 In specific, it calculated the mean expression levels of 43 “S phase" marker genes and 54 “G2/M phase" marker genes to obtain standardized scores for S and G2/M phases for each cell, and then assigns each cell to a specific phase with the highest score.
Ligand receptor analysis
Cell-cell interactions based on the expression of known ligand-receptor pairs in different cell types were calculated using the CellPhone DB version 3.1.0.52,71 Sorted CD34+ HSPCs were merged with defined HSPCs in BMMNCs as one population, and the algorithm was run on log-normalized expression values for cell populations of BMMNCs with default parameters and no subsampling to identify the enriched ligand-receptor pairs in VEXAS patients and healthy controls.
To examine and quantify a ligand–receptor interaction between different cell types, we implemented the established scoring algorithm proposed by Kumar et al. (2018)87 to calculate an interaction score based on ligand and receptor expression abundance. First, we collected 1,141 curated ligand-receptor pairs from the KEGG database for analysis. Then, for each cell, the gene expression of ligand ( and receptor ( were normalized to ( and by subtracting an average housekeeping expression value. A score of a given ligand-receptor interaction between cell types A and B was calculated as a product of average ligand expression across all cells of type A and average receptor expression across all cells of type B (. The one-sided Wilcoxon rank-sum test was applied on the hypothesis that the interaction score was greater than 0. For each HSPC, the interaction score was defined as the sum of its L-R scores with all monocytes/granulocytes in BMMNCs ).
NicheNet analysis
We used the R package NicheNet53 to predict ligand-receptor interactions that might drive gene expression changes in our cell types of interest. We combined all HSPCs, monocytes, and neutrophils for this analysis. All default parameters were used with an exception of setting a lower cutoff threshold of 0.3 and 0.6 for “prepare_ligand_target_visualization”.
Diversity index calculation
There are many ways of defining the diversity of a population, clonal types in this study, with each method providing a different representation of the number of clones (identical TCR/BCR chains) present (richness) and of their relative frequency (evenness). The Shannon entropy weighs both of these aspects of diversity equally, and it is an intuitive measure whereby the maximum value is determined by a total size of the repertoire. Entropy values decreases with increasing inequality of frequency as a result of clonal expansion. The Shannon entropy in a population of N clones with nucleotide frequency pi is defined by the following equation:
The Gini coefficient is a number aimed at measuring the inequality in a distribution. It is most often used in economics to measure a country’s wealth distribution and has been widely used in diversity assessment of TCRs/BCRs.55 The Gini coefficient is usually defined mathematically based on the Lorenz curve or Relative mean absolute difference.92 The Gini index and Shannon entropy for diversity and clonality analysis were calculated with the R package of tCR (https://imminfo.github.io/tcr/).
Identification of TCR motifs with shard antigen specificity using GLIPH2
GLIPH293 was applied to T cells of VEXAS patients to identify clusters of TCRs that recognized the same epitope based on CDR3β amino acid sequence similarities, with default parameters. CDR3β amino acid sequences of the top 1,000 most abundant CDRs were used to identify significant motif lists and associated TCR convergence groups. The motif-shared TCRs network was visualized using Cytoscape version 3.9.1.94
Statistical analysis
Pearson correlations between interaction scores and inflammatory and cytokine scores were calculated with the R package. Comparison between groups was performed using the GraphPad Prism (v.9.5.1; GraphPad software, La Jolla, CA), and results were shown as mean ± standard derivation.
Additional resources
Analysis and visualization of the scRNA-seq datasets in this study can be performed at the interactive website https://shouguog.shinyapps.io/vexas_cd34_bm/. DOIs are listed in the key resources table.
Acknowledgments
The authors thank Dr. Bettina Nadorp (Departments of Medicine and Pathology, New York University) for critically reading our manuscript. We thank Bretagne Cowling (National Heart, Lung, and Blood Institute/National Institutes of Health, NHLBI/NIH), Olga Rios (NHLBI/NIH), Katherine Roskom (NHLBI/NIH), and Janet Valdez (NHLBI/NIH) for assistance in obtaining samples, and patients and healthy volunteers who donated BM. Sequencing and technical supports were provided by the DNA Sequencing and Genomics Core of NHLBI. FACS was provided by the NHLBI flow cytometry core. This research was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute. D.B.B. is supported by the Jeffrey Modell Foundation, the Relapsing Polychondritis Foundation, the Department of Defense (BMFRP- IDA HT9425-23-1-0507), and funding from the NIH (R00AR078205).
Author contributions
Z.W. designed and performed the experiments, analyzed data, and wrote the manuscript. S.G. carried out bioinformatics analysis, set up the interactive website, and wrote the manuscript. H.L. did bioinformatics analysis and set up the interactive website. Q.G., X.F., A.L.M., D.O.C., L.A., and D.Q.R. performed the experiments. B.A.P., E.M.G., A.K.O., M.A.F., P.C.G., and K.R.C. collected and analyzed clinical data. S.K. and D.L.K. supervised data analysis and edited the manuscript. D.B.B. designed and performed experiments, analyzed data, and edited the manuscript. N.S.Y. conceived, designed, and supervised the experiments, analyzed results, and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 15, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101160.
Contributor Information
Zhijie Wu, Email: zhijie.wu@nih.gov.
David B. Beck, Email: david.beck@nyulangone.org.
Supplemental information
Data and code availability
The raw and analyzed sequencing data in this study have been deposited in the NCBI’s Gene Expression Omnibus (Database: GSE196052) and Sequence Read Archive (Database: SRP358093), and are publicly available. Accession numbers are listed in the key resources table. Code supporting this study is available at a dedicated Github repository [https://github.com/shouguog/UBA1]. Analysis and visualization of the scRNA-seq datasets in this study can be performed at the interactive website https://shouguog.shinyapps.io/vexas_cd34_bm/. DOIs are listed in the key resources table. All other relevant data supporting the key findings of this study are available within the article, and any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Beck D.B., Ferrada M.A., Sikora K.A., Ombrello A.K., Collins J.C., Pei W., Balanda N., Ross D.L., Ospina Cardona D., Wu Z., et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N. Engl. J. Med. 2020;383:2628–2638. doi: 10.1056/NEJMoa2026834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck D.B., Bodian D.L., Shah V., Mirshahi U.L., Kim J., Ding Y., Magaziner S.J., Strande N.T., Cantor A., Haley J.S., et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA. 2023;329:318–324. doi: 10.1001/jama.2022.24836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Obiorah I.E., Patel B.A., Groarke E.M., Wang W., Trick M., Ombrello A.K., Ferrada M.A., Wu Z., Gutierrez-Rodrigues F., Lotter J., et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. 2021;5:3203–3215. doi: 10.1182/bloodadvances.2021004976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Georgin-Lavialle S., Terrier B., Guedon A.F., Heiblig M., Comont T., Lazaro E., Lacombe V., Terriou L., Ardois S., Bouaziz J.-D., et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case-series of 116 French patients. Br. J. Dermatol. 2022;186:564–574. doi: 10.1111/bjd.20805. [DOI] [PubMed] [Google Scholar]
- 5.Ferrada M.A., Sikora K.A., Luo Y., Wells K.V., Patel B., Groarke E.M., Ospina Cardona D., Rominger E., Hoffmann P., Le M.T., et al. Somatic mutations in UBA1 defined a distinct subset of relapsing polychondritis patients with VEXAS. Arthritis Rheumatol. 2021;73:1886–1895. doi: 10.1002/art.41743. [DOI] [PubMed] [Google Scholar]
- 6.Grayson P.C., Patel B.A., Young N.S. VEXAS syndrome. Blood. 2021;137:3591–3594. doi: 10.1182/blood.2021011455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel B.A., Ferrada M.A., Grayson P.C., Beck D.B. VEXAS syndrome: an inflammatory and hematologic disease. Semin. Hematol. 2021;58:201–203. doi: 10.1053/j.seminhematol.2021.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourbon E., Heiblig M., Gerfaud Valentin M., Barba T., Durel C.-A., Lega J.C., Barraco F., Sève P., Jamilloux Y., Sujobert P., et al. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. 2021;137:3682–3684. doi: 10.1182/blood.2020010177. [DOI] [PubMed] [Google Scholar]
- 9.Zhao L.-P., Schell B., Sébert M., Kim R., Lemaire P., Boy M., Mathis S., Larcher L., Chauvel C., Dhouaieb M.B., et al. Prevalence of UBA1 mutations in MDS/CMML patients with systemic inflammatory and auto-immune disease. Leukemia. 2021;35:2731–2733. doi: 10.1038/s41375-021-01353-8. [DOI] [PubMed] [Google Scholar]
- 10.Poulter J.A., Collins J.C., Cargo C., De Tute R.M., Evans P., Ospina Cardona D., Bowen D.T., Cunnington J.R., Baguley E., Quinn M., et al. to somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood. 2021;137:3676–3681. doi: 10.1182/blood.2020010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Templé M., Templé M., Croizier C., Rossignol J., Huet T., Friedrich C., Zalmai L., Priollet P., Hayem G., Tournillhac O., et al. Atypical splice-site mutations causing VEXAS syndrome. Rheumatology. 2021;60:e435–e437. doi: 10.1093/rheumatology/keab524. [DOI] [PubMed] [Google Scholar]
- 12.Barba T., Jamilloux Y., Durel C.-A., Bourbon E., Mestrallet F., Sujobert P., Hot A., Barba T. VEXAS syndrome in a woman. Rheumatology. 2021;60:e402–e403. doi: 10.1093/rheumatology/keab392. [DOI] [PubMed] [Google Scholar]
- 13.Tsuchida N., Kunishita Y., Uchiyama Y., Kirino Y., Enaka M., Yamaguchi Y., Taguri M., Yamanaka S., Takase-Minegishi K., Yoshimi R., et al. Pathogenic UBA1 variants associated with VEXAS syndrome in Japanese patients with relapsing polychondritis. Ann. Rheum. Dis. 2021;80:1057–1061. doi: 10.1136/annrheumdis-2021-220089. [DOI] [PubMed] [Google Scholar]
- 14.Kao R.L., Jacobsen A.A., Billington C.J., Jr., Yohe S.L., Beckman A.K., Vercellotti G.M., Pearson D.R. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol. Dis. 2022;93 doi: 10.1016/j.bcmd.2021.102636. [DOI] [PubMed] [Google Scholar]
- 15.Koster M.J., Warrington K.J. VEXAS within the spectrum of rheumatologic disease. Semin. Hematol. 2021;58:218–225. doi: 10.1053/j.seminhematol.2021.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Sharma A., Naidu G., Deo P., Beck D.B. VEXAS syndrome with systemic lupus erythematosus: expanding the spectrum of associated conditions. Arthritis Rheumatol. 2022;74:369–371. doi: 10.1002/art.41957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaukat F., Hart M., Burns T., Bansal P. UBA1 and DNMT3A mutations in VEXAS syndrome. A case report and literature review. Mod. Rheumatol. Case Rep. 2022;6:134–139. doi: 10.1093/mrcr/rxab021. [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez-Rodrigues F., Kusne Y., Fernandez J., Lasho T., Shalhoub R., Ma X., Alessi H., Finke C., Koster M.J., Mangaonkar A., et al. Spectrum of clonal hematopoiesis in VEXAS syndrome. Blood. 2023;142:244–259. doi: 10.1182/blood.2022018774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Staels F., Betrains A., Woei-A-Jin F.J.S.H., Boeckx N., Beckers M., Bervoets A., Willemsen M., Neerinckx B., Humblet-Baron S., Blockmans D.E., et al. Case report: VEXAS syndrome: from mild symptoms to life-threatening macrophage activation syndrome. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.678927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pagliuca S., Gurnari C., Durkin L., Terkawi L., Awada H., Kongkiatkamon S., Zawit M., Hsi E.D., Carraway H.E., Rogers H.J., et al. Vacuolization of hematopoietic precursors: an enigma with multiple etiologies. Blood. 2021;137:3685–3689. doi: 10.1182/blood.2021010811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oganesyan A., Jachiet V., Chasset F., Hirsch P., Hage-Sleiman M., Fabiani B., Duriez P., Georgin-Lavialle S., Delhommeau F., Hakobyan Y., et al. VEXAS syndrome: still expanding the clinical phenotype. Rheumatology. 2021;60:e321–e323. doi: 10.1093/rheumatology/keab225. [DOI] [PubMed] [Google Scholar]
- 22.Mustjoki S., Young N.S. Somatic mutations in “benign” disease. N. Engl. J. Med. 2021;384:2039–2052. doi: 10.1056/NEJMra2101920. [DOI] [PubMed] [Google Scholar]
- 23.Jaiswal S., Natarajan P., Ebert B.L. Clonal hematopoiesis and atherosclerosis. N. Engl. J. Med. 2017;377:1401–1402. doi: 10.1056/NEJMc1710381. [DOI] [PubMed] [Google Scholar]
- 24.Andor N., Simonds E.F., Czerwinski D.K., Chen J., Grimes S.M., Wood-Bouwens C., Zheng G.X.Y., Kubit M.A., Greer S., Weiss W.A., et al. Single-cell RNA-Seq of follicular lymphoma reveals malignant B-cell types and coexpression of T-cell immune checkpoints. Blood. 2019;133:1119–1129. doi: 10.1182/blood-2018-08-862292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stetson L.C., Balasubramanian D., Ribeiro S.P., Stefan T., Gupta K., Xu X., Fourati S., Roe A., Jackson Z., Schauner R., et al. Single cell RNA sequencing of AML initiating cells reveals RNA-based evolution during disease progression. Leukemia. 2021;35:2799–2812. doi: 10.1038/s41375-021-01338-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Z., Gao S., Diamond C., Kajigaya S., Chen J., Shi R., Palmer C., Hsu A.P., Calvo K.R., Hickstein D.D., et al. Sequencing of RNA in single cells reveals a distinct transcriptome signature of hematopoiesis in GATA2 deficiency. Blood Adv. 2020;4:2656–2670. doi: 10.1182/bloodadvances.2019001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z., Gao S., Watanabe N., Batchu S., Kajigaya S., Diamond C., Alemu L., Raffo D.Q., Feng X., Hoffmann P., et al. Single-cell profiling of T lymphocytes in deficiency of adenosine deaminase 2. J. Leukoc. Biol. 2022;111:301–312. doi: 10.1002/JLB.5A0621-314R. [DOI] [PubMed] [Google Scholar]
- 28.Zhao X., Gao S., Wu Z., Kajigaya S., Feng X., Liu Q., Townsley D.M., Cooper J., Chen J., Keyvanfar K., et al. Single-cell RNA-seq reveals a distinct transcriptome signature of aneuploid hematopoietic cells. Blood. 2017;130:2762–2773. doi: 10.1182/blood-2017-08-803353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pellin D., Loperfido M., Baricordi C., Wolock S.L., Montepeloso A., Weinberg O.K., Biffi A., Klein A.M., Biasco L. A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 2019;10:2395. doi: 10.1038/s41467-019-10291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ranzoni A.M., Tangherloni A., Berest I., Riva S.G., Myers B., Strzelecka P.M., Xu J., Panada E., Mohorianu I., Zaugg J.B., Cvejic A. Integrative single-cell RNA-seq and ATAC-seq analysis of human developmental hematopoiesis. Cell Stem Cell. 2021;28:472–487.e7. doi: 10.1016/j.stem.2020.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macaulay I.C., Haerty W., Kumar P., Li Y.I., Hu T.X., Teng M.J., Goolam M., Saurat N., Coupland P., Shirley L.M., et al. G&T: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods. 2015;12:519–522. doi: 10.1038/nmeth.3370. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Meira A., Buck G., Clark S.-A., Povinelli B.J., Alcolea V., Louka E., McGowan S., Hamblin A., Sousos N., Barkas N., et al. Unravelling intratumoral heterogeneity through high-sensitivity single cell mutational analysis and parallel RNA sequencing. Mol. Cell. 2019;73:1292–1305.e8. doi: 10.1016/j.molcel.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nam A.S., Dusaj N., Izzo F., Murali R., Myers R.M., Mouhieddine T.H., Sotelo J., Benbarche S., Waarts M., Gaiti F., et al. Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation. Nat. Genet. 2022;54:1514–1526. doi: 10.1038/s41588-022-01179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Galen P., Hovestadt V., Wadsworth Ii M.H., Hughes T.K., Griffin G.K., Battaglia S., Verga J.A., Stephansky J., Pastika T.J., Lombardi Story J., et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176:1265–1281.e24. doi: 10.1016/j.cell.2019.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hay S.B., Ferchen K., Chetal K., Grimes H.L., Salomonis N. The human cell atlas bone marrow single-cell interactive web portal. Exp. Hematol. 2018;68:51–61. doi: 10.1016/j.exphem.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Galen P., Kreso A., Mbong N., Kent D.G., Fitzmaurice T., Chambers J.E., Xie S., Laurenti E., Hermans K., Eppert K., et al. The unfolded protein response governs integrity of the haematopoietic stem cell pool during stress. Nature. 2014;510:268–272. doi: 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- 37.Ganan-Gomez I., Yang H., Ma F., Montalban-Bravo G., Thongon N., Marchica V., Richard-Carpentier G., Chien K., Manyam G., Wang F., et al. Stem cell architecture drives myelodysplastic syndrome progression and predicts response to venetoclax-based therapy. Nat. Med. 2022;28:557–567. doi: 10.1038/s41591-022-01696-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiseman D.H., Baker S.M., Dongre A.V., Gurashi K., Storer J.A., Somervaille T.C., Batta K. Chronic myelomonocytic leukaemia stem cell transcriptomes anticipate disease morphology and outcome. EBioMedicine. 2020;58 doi: 10.1016/j.ebiom.2020.102904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giustacchini A., Thongjuea S., Barkas N., Woll P.S., Povinelli B.J., Booth C.A.G., Sopp P., Norfo R., Rodriguez-Meira A., Ashley N., et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017;23:692–702. doi: 10.1038/nm.4336. [DOI] [PubMed] [Google Scholar]
- 40.Laurenti E., Doulatov S., Zandi S., Plumb I., Chen J., April C., Fan J.B., Dick J.E. The transcriptional architecture of early human hematopoiesis identifies multilevel control of lymphoid commitment. Nat. Immunol. 2013;14:756–763. doi: 10.1038/ni.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ambinder A.J., Miller J., DeZern A.E. Autoimmune disease in CMML-the chicken or the egg? Best Pract. Res. Clin. Haematol. 2020;33 doi: 10.1016/j.beha.2019.101136. [DOI] [PubMed] [Google Scholar]
- 42.Bai Z., Chen Y., Chen X., Dong L. A medical mirroring: chronic myelomonocytic leukemia mimicking immunoglobulin G4-related disease. Am. J. Transl. Res. 2019;11:4561–4567. [PMC free article] [PubMed] [Google Scholar]
- 43.Saif M.W., Hopkins J.L., Gore S.D. Autoimmune phenomena in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk. Lymphoma. 2002;43:2083–2092. doi: 10.1080/1042819021000016186. [DOI] [PubMed] [Google Scholar]
- 44.Mekinian A., Grignano E., Braun T., Decaux O., Liozon E., Costedoat-Chalumeau N., Kahn J.E., Hamidou M., Park S., Puéchal X., et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: a French multicentre retrospective study. Rheumatology. 2016;55:291–300. doi: 10.1093/rheumatology/kev294. [DOI] [PubMed] [Google Scholar]
- 45.Hamamyh T., Yassin M.A. Autoimmune Hemolytic Anemia in Chronic Myeloid Leukemia. Pharmacology. 2020;105:630–638. doi: 10.1159/000507295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steegmann J.L., Requena M.J., Martín-Regueira P., De La Cámara R., Casado F., Salvanés F.R., Fernández Rañada J.M. High incidence of autoimmune alterations in chronic myeloid leukemia patients treated with interferon-alpha. Am. J. Hematol. 2003;72:170–176. doi: 10.1002/ajh.10282. [DOI] [PubMed] [Google Scholar]
- 47.Montoro J., Gallur L., Merchán B., Molero A., Roldán E., Martínez-Valle F., Villacampa G., Navarrete M., Ortega M., Castellví J., et al. Autoimmune disorders are common in myelodysplastic syndrome patients and confer an adverse impact on outcomes. Ann. Hematol. 2018;97:1349–1356. doi: 10.1007/s00277-018-3302-0. [DOI] [PubMed] [Google Scholar]
- 48.Petti A.A., Williams S.R., Miller C.A., Fiddes I.T., Srivatsan S.N., Chen D.Y., Fronick C.C., Fulton R.S., Church D.M., Ley T.J. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat. Commun. 2019;10:3660. doi: 10.1038/s41467-019-11591-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184 doi: 10.1016/j.cell.2021.10.023. 5838-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sergushichev A.A. An Algorithm for Fast Preranked Gene Set Enrichment Analysis Using Cumulative Statistic Calculation. bioRxiv. 2016 doi: 10.1101/060012. [DOI] [Google Scholar]
- 52.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 53.Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 54.Desponds J., Mora T., Walczak A.M. Fluctuating fitness shapes the clone-size distribution of immune repertoires. Proc. Natl. Acad. Sci. USA. 2016;113:274–279. doi: 10.1073/pnas.1512977112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosati E., Dowds C.M., Liaskou E., Henriksen E.K.K., Karlsen T.H., Franke A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol. 2017;17:61. doi: 10.1186/s12896-017-0379-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao S., Wu Z., Arnold B., Diamond C., Batchu S., Giudice V., Alemu L., Raffo D.Q., Feng X., Kajigaya S., et al. Single-cell RNA sequencing coupled to TCR profiling of large granular lymphocyte leukemia T cells. Nat. Commun. 2022;13:1982. doi: 10.1038/s41467-022-29175-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeWitt W.S., Lindau P., Snyder T.M., Sherwood A.M., Vignali M., Carlson C.S., Greenberg P.D., Duerkopp N., Emerson R.O., Robins H.S. A public database of memory and naïve B-cell receptor sequences. PLoS One. 2016;11 doi: 10.1371/journal.pone.0160853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grootjans J., Kaser A., Kaufman R.J., Blumberg R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016;16:469–484. doi: 10.1038/nri.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young N.S. Aplastic anemia. N. Engl. J. Med. 2018;379:1643–1656. doi: 10.1056/NEJMra1413485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zaimoku Y., Patel B.A., Adams S.D., Shalhoub R., Groarke E.M., Lee A.A.C., Kajigaya S., Feng X., Rios O.J., Eager H., et al. HLA associations, somatic loss of HLA expression, and clinical outcomes in immune aplastic anemia. Blood. 2021;138:2799–2809. doi: 10.1182/blood.2021012895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katagiri T., Sato-Otsubo A., Kashiwase K., Morishima S., Sato Y., Mori Y., Kato M., Sanada M., Morishima Y., Hosokawa K., et al. Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood. 2011;118:6601–6609. doi: 10.1182/blood-2011-07-365189. [DOI] [PubMed] [Google Scholar]
- 62.Fuster J.J., MacLauchlan S., Zuriaga M.A., Polackal M.N., Ostriker A.C., Chakraborty R., Wu C.-L., Sano S., Muralidharan S., Rius C., et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sano S., Oshima K., Wang Y., MacLauchlan S., Katanasaka Y., Sano M., Zuriaga M.A., Yoshiyama M., Goukassian D., Cooper M.A., et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP inflammasome. J. Am. Coll. Cardiol. 2018;71:875–886. doi: 10.1016/j.jacc.2017.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y., Sano S., Yura Y., Ke Z., Sano M., Oshima K., Ogawa H., Horitani K., Min K.-D., Miura-Yura E., et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight. 2020;5 doi: 10.1172/jci.insight.135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hormaechea-Agulla D., Matatall K.A., Le D.T., Kain B., Long X., Kus P., Jaksik R., Challen G.A., Kimmel M., King K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFN signaling. Cell Stem Cell. 2021;28:1428–1442.e6. doi: 10.1016/j.stem.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao M., Chen R., Yang Y., He H., Xu L., Jiang Y., Guo Z., He W., Jiang H., Wang J., et al. Aging-elevated inflammation promotes DNMT3A R878H-driven clonal hematopoiesis. Acta Pharm. Sin. B. 2022;12:678–691. doi: 10.1016/j.apsb.2021.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abplanalp W.T., Cremer S., John D., Hoffmann J., Schuhmacher B., Merten M., Rieger M.A., Vasa-Nicotera M., Zeiher A.M., Dimmeler S. Clonal hematopoiesis-driver DNMT3A mutations alter immune cells in heart failure. Circ. Res. 2021;128:216–228. doi: 10.1161/CIRCRESAHA.120.317104. [DOI] [PubMed] [Google Scholar]
- 68.Dharan N.J., Yeh P., Bloch M., Yeung M.M., Baker D., Guinto J., Roth N., Ftouni S., Ognenovska K., Smith D., et al. HIV is associated with an increased risk of age-related clonal hematopoiesis among older adults. Nat. Med. 2021;27:1006–1011. doi: 10.1038/s41591-021-01357-y. [DOI] [PubMed] [Google Scholar]
- 69.Guo M., Wang H., Potter S.S., Whitsett J.A., Xu Y. SINCERA: A pipeline for single-cell RNA-seq profiling analysis. PLoS Comput. Biol. 2015;11 doi: 10.1371/journal.pcbi.1004575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Alonso L., Lorenzi V., Mazzeo C.I., Alves-Lopes J.P., Roberts K., Sancho-Serra C., Engelbert J., Marečková M., Gruhn W.H., Botting R.A., et al. Single-cell roadmap of human gonadal development. Nature. 2022;607:540–547. doi: 10.1038/s41586-022-04918-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fast Gene Set Enrichment Analysis Gennady Korotkevich, Vladimir Sukhov, Alexey Sergushichev. bioRxiv 060012.
- 73.Setty M., Kiseliovas V., Levine J., Gayoso A., Mazutis L., Pe'er D. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol. 2019;37:451–460. doi: 10.1038/s41587-019-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alex A., Rahnenfuthrer J. 2019. topGO: Enrichment Analysis for Gene Ontology. R package version 2.36.0. [Google Scholar]
- 75.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Werner T., Dombrowski S.M., Zgheib C., Zouein F.A., Keen H.L., Kurdi M., Booz G.W. Elucidating functional context within microarray data by integrated transcription factor-focused gene-interaction and regulatory network analysis. Eur. Cytokine Netw. 2013;24:75–90. doi: 10.1684/ecn.2013.0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P., et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ilicic T., Kim J.K., Kolodziejczyk A.A., Bagger F.O., McCarthy D.J., Marioni J.C., Teichmann S.A. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 2016;17:29. doi: 10.1186/s13059-016-0888-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Azizi E., Carr A.J., Plitas G., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M., et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293–1308.e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Redmond D., Poran A., Elemento O. Single-cell TCRseq: paired recovery of entire T-cell alpha and beta chain transcripts in T-cell receptors from single-cell RNAseq. Genome Med. 2016;8:80. doi: 10.1186/s13073-016-0335-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.R Core Team . R Foundation for Statistical Computing; 2021. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 83.Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.The Gene Ontology Consortium The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47:D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kumar M.P., Du J., Lagoudas G., Jiao Y., Sawyer A., Drummond D.C., Lauffenburger D.A., Raue A. Analysis of single-cell RNA-seq identifies cell-cell communication associated with tumor characteristics. Cell Rep. 2018;25:1458–1468.e4. doi: 10.1016/j.celrep.2018.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kanehisa M., Goto S. KEGG Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kanehisa M., Sato Y., Furumichi M., Morishima K., Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47:D590–D595. doi: 10.1093/nar/gky962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oakes T., Heather J.M., Best K., Byng-Maddick R., Husovsky C., Ismail M., Joshi K., Maxwell G., Noursadeghi M., Riddell N., et al. Quantitative characterization of the T cell receptor repertoire of naïve and memory subsets using an integrated experimental and computational pipeline which is robust, economical, and versatile. Front. Immunol. 2017;8:1267. doi: 10.3389/fimmu.2017.01267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang H., Wang C., Rubelt F., Scriba T.J., Davis M.M. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat. Biotechnol. 2020;38:1194–1202. doi: 10.1038/s41587-020-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw and analyzed sequencing data in this study have been deposited in the NCBI’s Gene Expression Omnibus (Database: GSE196052) and Sequence Read Archive (Database: SRP358093), and are publicly available. Accession numbers are listed in the key resources table. Code supporting this study is available at a dedicated Github repository [https://github.com/shouguog/UBA1]. Analysis and visualization of the scRNA-seq datasets in this study can be performed at the interactive website https://shouguog.shinyapps.io/vexas_cd34_bm/. DOIs are listed in the key resources table. All other relevant data supporting the key findings of this study are available within the article, and any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.