

Abstract
D2C7-immunotoxin (IT), a dual-specific IT targeting wild-type epidermal growth factor receptor (EGFR) and mutant EGFR variant III (EGFRvIII) proteins, demonstrates encouraging survival outcomes in a subset of patients with glioblastoma -. We hypothesized that immunosuppression in glioblastoma limits D2C7-IT efficacy. To improve the response rate and reverse immunosuppression, we combined D2C7-IT tumor cell killing with αCD40 costimulation of antigen-presenting cells. In murine glioma models, a single intratumoral injection of D2C7-IT+αCD40 treatment, activated a proinflammatory phenotype in microglia and macrophages, promoted long-term tumor-specific CD8+ T cell immunity, and generated cures. D2C7-IT+αCD40 treatment increased intratumoral Slamf6+CD8+ T cells with a progenitor phenotype and decreased terminally exhausted CD8+ T cells. D2C7-IT+αCD40 treatment stimulated intratumoral CD8+ T cell proliferation and generated cures in glioma-bearing mice despite FTY720-induced peripheral T cell sequestration. Tumor transcriptome profiling established CD40 upregulation, pattern recognition receptor, cell senescence, and immune response pathway activation as the drivers of D2C7-IT+αCD40 antitumor responses. In order to determine potential translation, immunohistochemistry staining confirmed CD40 expression in human GBM tissue sections. These promising preclinical data allowed us to initiate a phase I study with D2C7-IT+αhCD40 in patients with malignant glioma (NCT04547777) to further evaluate this treatment in humans.
One Sentence Summary:
Immunotoxin-mediated tumor cell killing and αCD40-mediated immune cell activation generate cures in brain tumor models.
INTRODUCTION
Glioblastoma (GBM) is the most frequent and malignant primary brain tumor in adults (1). The current standard of care comprises maximum safe surgical resection and radiotherapy with concomitant and adjuvant temozolomide (2). Despite these interventions, GBMs are extremely invasive and highly resistant to therapy resulting in poor prognosis, with a median survival (MS) of 15–18 months (3). Therapeutic strategies that harness the immune system demonstrate clinical benefits for a minority of patients, but the overall patient survival remains low (4, 5). Clinical success of immunotherapy has been hindered by GBM-intrinsic (inter and intratumoral heterogeneity, lack of neoantigens, poor antigen presentation and priming, and secretion of immunosuppressive cytokines) (6–8) and -extrinsic immunosuppression (decreased frequency of T cells, local and systemic T cell exhaustion and suppression, and increased frequency of immunosuppressive tumor-associated macrophages [TAMs]) (9–12). Innovative therapeutic approaches are urgently needed to combat GBM immunosuppression, generate productive antitumor immunity, and prolong the survival rates of patients beyond that given by the current standard of care. To that end, our group has examined immunotoxin (IT)-based targeted therapies administered using convection-enhanced delivery (CED) for GBM. During CED, a catheter inserted into the tumor mass facilitates drug distribution, leveraging the positive pressure generated by an infusion pump. Thus, intratumoral (i.t.) infusion using CED enables the direct delivery of macromolecular therapeutics that are too large to cross the blood-brain barrier or too toxic when administered systemically (13).
D2C7-immunotoxin (IT) (D2C7) is a recombinant antibody (Ab) fragment-derived Pseudomonas exotoxin-based IT that targets the wild-type epidermal growth factor receptor (EGFRwt) and its mutant EGFR variant III (EGFRvIII), two dominant driver oncogenes in GBM (14, 15). In preclinical mouse glioma models, delivery of D2C7 using CED induces tumor cell killing and elicits secondary immune responses by activating CD4+ and CD8+ T cells (16). A phase I clinical trial of D2C7 in patients with recurrent WHO grade 3 and 4 malignant glioma (NCT02303678) showed encouraging survival outcomes and radiological responses in a subset of patients (17). The limited clinical efficacy suggests an inhibitory role for the immunosuppressive tumor microenvironment (TME) in D2C7 antitumor immunity. Hence, we hypothesized that modulating the immunosuppressive TME would improve GBM patient response to D2C7.
CD40 is a costimulatory molecule belonging to the tumor necrosis factor receptor superfamily (18). CD40 is expressed on a wide range of cells including microglia, monocytes, macrophages , dendritic cells (DCs), B cells, platelets, fibroblasts, endothelial cells, smooth muscle cells, and tumor cells (19). CD40 ligation activates antigen-presenting cells (APCs), enhances antigen processing and presentation, and consequently T cell stimulation (20–22). Thus, CD40 is a key molecule involved in bridging innate and adaptive immune responses. Studies in mouse models of cancer have demonstrated the importance of CD40 in driving antitumor immunity, wherein administration of an agonistic CD40 monoclonal Ab (mAb) activated DCs and engendered protective T cell responses (23–25). T cell-independent and macrophage-dependent antitumor effects of αCD40 have also been described and involve TAM re-education from a protumor to an antitumor phenotype leading to tumor and stromal destruction (26–28). Furthermore, CD40 is also prevalent in grade 3 and 4 gliomas (29, 30). Since CD40 is highly expressed in GBM, we hypothesized that CD40 costimulation would activate APCs and modulate TAMs in the TME. Therefore, we investigated whether CD40 agonism in combination with D2C7 cytotoxicity could invoke innate and tumor-specific T cell immunity and improve the survival outcomes in GBM models.
RESULTS
D2C7 and agonistic αCD40 combination therapy generates cures and induces immunological memory in syngeneic glioma models
When administered systemically, agonistic αCD40 therapy induces transaminitis (elevated liver enzymes called transaminases) in murine models and patients (31, 32). To overcome dose-limiting toxicities associated with the systemic delivery of αCD40 in the clinic (31) and to determine a biologically effective dose for combination with D2C7, we performed a dose-finding study with intratumoral CED of αCD40 (3–100 μg) in the CT-2A-dmEGFRvIII-Firefly Luciferase (CT-VIII) glioma model (fig. S1A–S1C). Doses up to 100 μg of αCD40 were safe and effective. The greatest increase in survival, compared to control (MS=11-d), was noted with the 30 μg (MS=22-d; P=0.12) and 100 μg doses (MS=21.5-d, P=0.05) (fig. S1A). We next tested the 300 μg dose of αCD40 in combination with D2C7 (D2C7+αCD40 [D+C]) in the CT-VIII glioma model and identified treatment-associated hepatic toxicity, such as enlarged spleen and mottled liver discoloration, and physical signs of illness, such as hunched posture and lethargy, upon completion of CED on day 9 post tumor implantation (fig. S1B–S1C), suggesting that the higher dose was attaining saturation in the brain and excess Ab was reaching the circulation.
We next examined if the 100 μg αCD40 dose was safe when combined with D2C7. Control and D2C7-treated mice exhibited no symptoms of hepatic toxicity at any observed timepoint (fig. S1D–S1I). Mice treated with αCD40 (100 μg) and D+C (0.2 μg + 100 μg) therapies did not show physical signs of illness, but exhibited liver discoloration to a lesser degree than the D+C (0.2 μg + 300 μg) group upon completion of CED. The liver discoloration in the 100 μg groups (αCD40 and D+C), gradually decreased and returned to normal at 24 and 72 hr post-CED, respectively (fig. S1J–S1O). Consistent with these data, the control and D2C7 treated mice presented normal aspartate transaminase (AST) and alanine transaminase (ALT) amounts at each time point (fig. S1P and S1Q) (33). Higher amounts of AST and ALT were observed in the αCD40 and D+C groups upon completion of CED, but gradually returned to the normal range by 24 and 72 hr post-CED (fig. S1P and S1Q). Platelet concentrations were consistently elevated in the control and D2C7 mice at all time points (fig. S1R) (33). Contrarily, platelet concentrations were lower in the αCD40 and D+C groups upon completion of CED, but gradually reached concentrations similar to the control and D2C7 groups 72 hr post-CED (fig. S1R). These data indicate that the 100 μg αCD40 dose in combination with D2C7 results in reduced and transient toxicity while demonstrating efficacy (fig. S1A). Thus, we chose 100 μg αCD40 as the working dose for subsequent experiments.
Next, we evaluated the in vivo efficacy of D2C7 and αCD40 mono and combination therapies in the CT-VIII model. Compared to control (MS=16 days), we observed significant increases in survival with D2C7 (MS=35 days, P<0.05) and D+C (MS>75 days, P<0.0001) therapies (Fig. 1A). At the end of the study (>75 days), there were tumor-free mice in the D2C7 (2/10), αCD40 (1/10), and D+C (8/10) groups (Fig. 1A). The surviving and naïve control mice were rechallenged on day 76 (time point for rechallenge; MS>75 days) in the contralateral hemisphere with parental CT-2A cells that do not express dmEGFRvIII. All control mice developed tumors whereas all therapy mice survived and remained tumor-free until study termination (87 days post rechallenge) (Fig. 1B), indicating the development of immunologic memory that prevents tumor recurrence.
Fig. 1. Intratumoral D2C7 and αCD40 combination elicit robust antitumor immunity in orthotopic glioma models.
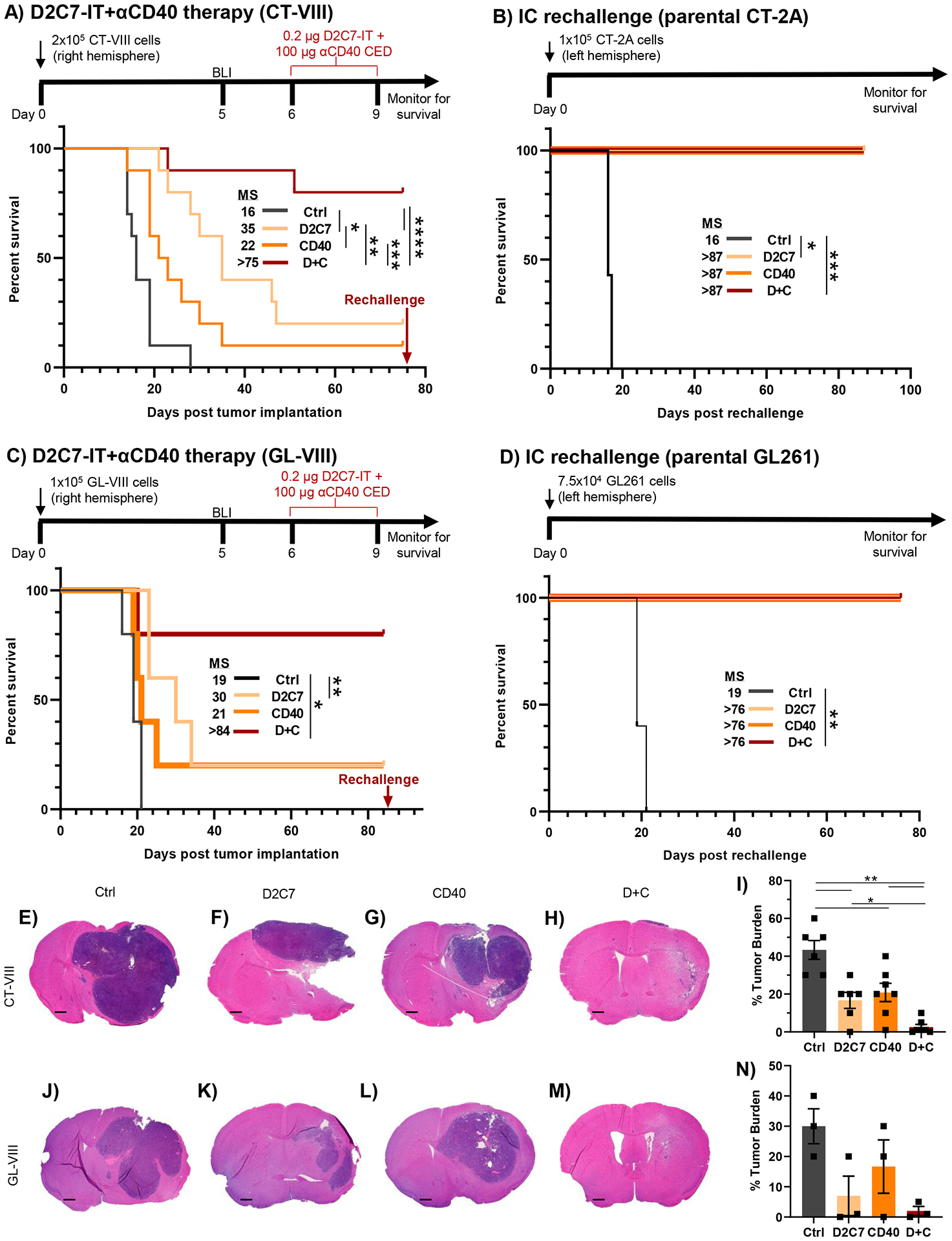
(A) Survival of C57BL/6J mice implanted with CT-VIII (N=10/group) cells and treated with Ctrl, D2C7, αCD40, or D+C as indicated. (B) Treated mice from A that experienced complete tumor regression for >75-d, were rechallenged with the CT-2A tumor cells. Tumor naïve mice (n=7) served as controls. (C) Survival of mice implanted with GL-VIII (n=5/group) cells and treated with Ctrl, D2C7, αCD40, or D+C as indicated. (D) Treated mice from C that experienced complete tumor regression for >83-d, were rechallenged with the GL261 tumor cells. Tumor naïve mice (n=5) served as controls. (E-H and J-M) Representative images of H&E stained tumor sections from CT-VIII (E-H) brains (n=6–7/group, harvested 6–7-d post-therapy) and GL-VIII (J-M) brains (n=3/group, harvested 6-d post-therapy), treated with control (E, J), D2C7 (F, K), αCD40 (G, L), or D+C (H, M). Scale bar: 600 μm. (I and N) Percentage compositions of tumor cells of entire CT-VIII (I) and GL-VIII (N) tumor section cohorts from E-H and J-M are presented. Data are mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. BLI, Bioluminescence imaging.
The efficacy determination of D+C in a second syngeneic glioma model, GL261-dmEGFRvIII-Firefly Luciferase (GL-VIII), showed significant increases in survival with D2C7 (MS=30 days, P<0.01) therapy with one surviving mouse remaining, whereas the control group had an MS of 19 days, with no survivors (Fig. 1C). The D+C group had 4/5 survivors and an MS of >84 days (P<0.05). All survivors and naïve controls were rechallenged contralaterally on day 85 (time point for rechallenge; MS>75 days) with parental GL261 cells. The control mice died from tumors. The initial therapy mice remained tumor-free when the study ended (76 days post rechallenge) (Fig. 1D), demonstrating that the immunologic memory persists for up to five months post-therapy.
Brains from CT-VIII or GL-VIII tumor-bearing mice, treated as in Fig. 1A or 1C and harvested 6–7-d or 6-d post-therapy, respectively, were histologically examined using coronal sections from the tumor injection sites. In both CT-VIII (Fig. 1E–1I) and GL-VIII (Fig. 1J–1N) models, the control mice (Fig. 1E and 1J) had the largest tumor burden (20–60%) (Fig. 1I and 1N), D2C7 (Fig. 1F and 1K) and αCD40 (Fig. 1G and 1L) treated mice exhibited smaller tumor burdens than control (20–40%) (Fig. 1I and 1N), and the D+C treated mice (Fig. 1H and 1M) showed minimal tumor burden (1–10%) (Fig. 1I and 1N). Thus, both D2C7 and αCD40 monotherapies slow tumor growth and improve survival, D+C therapy effectively destroys malignant gliomas, generates a systemic antitumor immune response, and results in robust survival outcomes, including cures.
Systemic αCD40 combinations have been evaluated for antitumor efficacy in glioma models (34, 35); therefore, we compared the efficacy of our intratumoral approach to systemic αCD40 dosing in the CT-VIII model. Compared to the control, all four treatment groups showed increased survival. The CED co-delivery group (D+C CED: >76 days, 7/10 survivors and D+C systemic: >62.5 days, 5/10 survivors) had the largest MS and survivors (fig. S2A). These results indicate that D+C co-infusion by CED generates an improved antitumor response and a safe therapeutic option for clinical translation.
D2C7 and αCD40 therapy is efficacious against heterogeneous and metastatic gliomas, and the antitumor response requires D2C7-mediated tumor killing
GBMs exhibit a high degree of intra and intertumoral heterogeneity (7), which often leads to therapy resistance and disease relapse. Hence, we examined the effectiveness of D+C against phenotypically heterogeneous gliomas with varying degrees of target antigen expression. Intracranial gliomas containing a mix of CT-VIII (antigen-positive) and parental CT-2A (antigen-negative) cells in a 2:1 ratio, based on the number of cells used during homogenous implantation, were established and treated as outlined (Fig. 2A). Compared to control (MS=19 days), we observed an increase in survival with D2C7 (MS=26 days), αCD40 (MS=24.5 days), and D+C (MS>99 days) therapies (Fig. 2A). At the end of the study, there were 2/10 (D2C7), 3/10 (αCD40), and 7/10 (D+C) survivors, demonstrating that D+C treatment is also efficacious against heterogeneous gliomas.
Fig. 2. D2C7+αCD40 therapy is antigen-dependent and demonstrates local and systemic antitumor immunity.
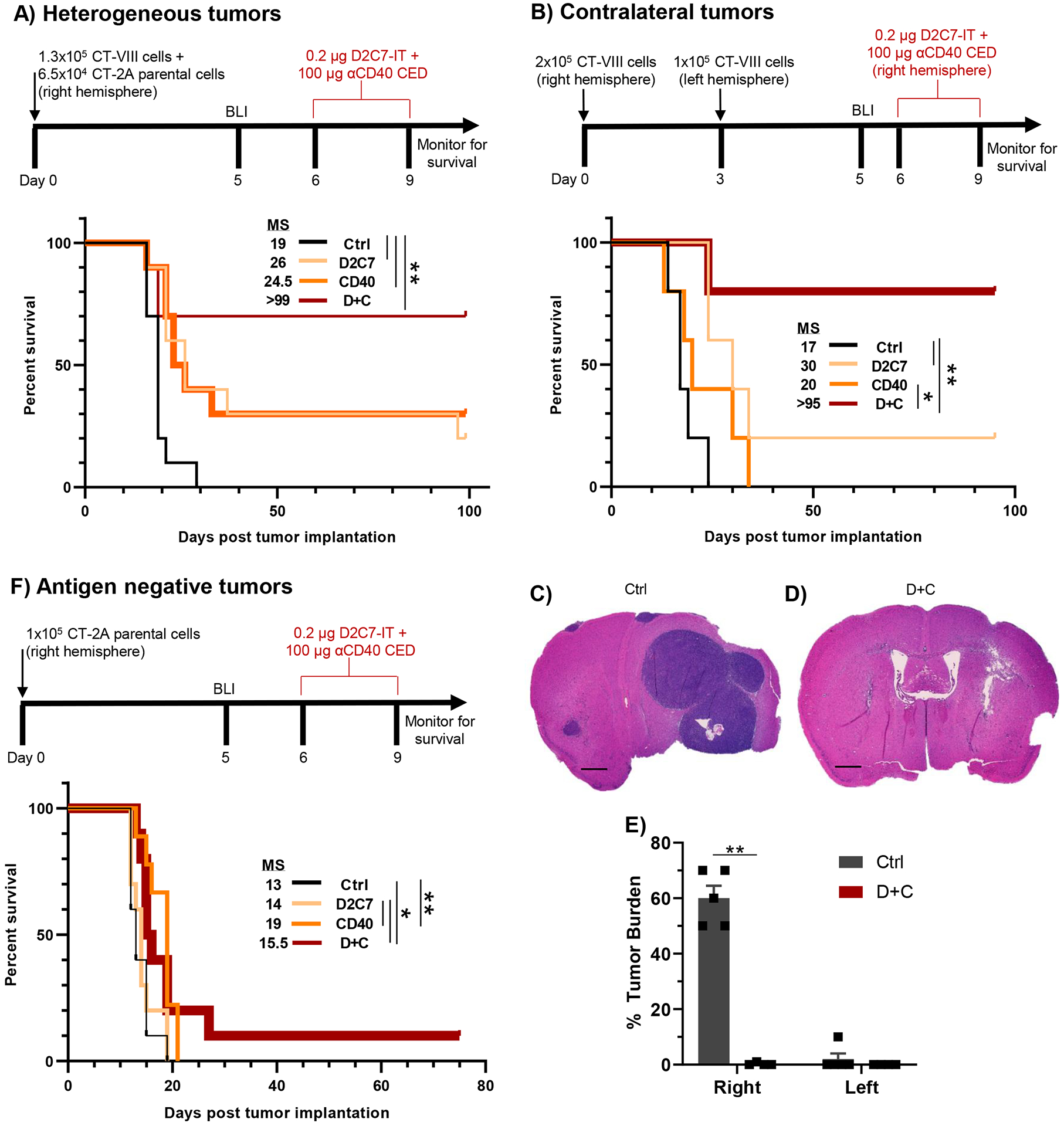
(A) Survival of CT-VIII+CT-2A tumor-bearing mice (n=10/group) treated with Ctrl, D2C7, αCD40, or D+C as indicated. (B) Survival of CT-VIII tumor-bearing mice (n=5/group) treated with Ctrl, D2C7, αCD40, or D+C as indicated in a model of metastatic cancer. (C-D) Representative H&E images of tumor sections from CT-VIII contralateral tumors (N=4–5/group), treated with control (C) or D+C (D) harvested 7-d post-therapy. Scale bar: 600 μm. (E) Percentage composition of tumor cells of entire tumor section cohort from C and D. Data are mean ± SEM. (F) Survival of parental CT-2A tumor-bearing mice (n=9–10/group) treated with Ctrl, D2C7, αCD40, or D+C as indicated. *P<0.05, **P<0.01.
The high degree of tumor cell infiltration into the surrounding brain parenchyma is a major challenge for the clinical management of GBM (36). Therefore, we evaluated the efficacy of D+C against micrometastatic tumor cells in a CT-VIII tumor model reflective of this behavior as outlined (Fig. 2B). All mice in control and αCD40 groups developed tumors, with a MS of 17 days and 20 days, respectively (Fig. 2B). Compared to control (MS=17 days), we observed significant increases in survival in the D2C7 (MS=30 days, P<0.01) and D+C (MS>95 days, P<0.01) groups. Histological examination of coronal sections taken at the tumor injection site in CT-VIII tumor brains at 7 days post-treatment demonstrated large and small tumors in the right and left hemispheres, 50–70% and 0–10% tumor burden, respectively, in the control group (Fig. 2C and 2E). There was almost complete tumor eradication, 1 and 0% tumor burden in the right and left hemispheres, respectively, in the D+C group (Fig. 2D and 2E), confirming the efficacy of the combination therapy against both treated and untreated distant tumors.
We also examined the efficacy of D+C in the antigen-negative CT-2A model to determine the contribution of D2C7-mediated killing of dmEGFRvIII antigen-expressing tumors in the generation of antitumor responses (Fig. 2F). Compared to control (MS=13 days), αCD40 therapy showed an increase in MS to 19 days, whereas D2C7 or D+C therapy failed to increase MS beyond those generated by αCD40 (Fig. 2F). These data indicate that D2C7-mediated tumor cell killing is required for the D+C therapy-mediated antitumor response.
D2C7 and αCD40 combination therapy induces an activated proinflammatory phenotype in tumor-associated myeloid cells
Given the functional role of CD40 on diverse immune cells (18), we next performed intratumoral immune profiling following treatment with D2C7 and αCD40 therapies using flow cytometry (FC). Analysis of CT-VIII tumors six days following therapy demonstrated an increase in the relative frequency of neutrophils (3.3%) and microglia (41.3%) in the D2C7 group (Fig. 3A and 3C) and a decrease in the relative frequency of natural killer (NK) cells (0.69–0.92%) (Fig. 3B) and macrophages (19.8–22.2%) (Fig. 3D) in the mono and combination therapies compared to the control (1.9% and 42.4%, respectively). Treatment with αCD40 or D+C resulted in similar amounts of neutrophils, NK cells, microglia, and macrophages (Fig. 3A–D). We next investigated the activation phenotype of tumor-associated microglia and macrophages by examining CD80 and major histocompatibility complex II (MHCII) expression in treated tumors. High upregulation of CD80 and MHCII on microglia was present only in the αCD40 and D+C groups (Fig. 3E and 3F). In contrast, marginally higher upregulation of CD80 and MHCII on macrophages compared to the control and D2C7 groups were noted in the αCD40 and D+C groups (Fig. 3G and 3H).
Fig. 3. D+C therapy promotes antitumor phenotype in intratumoral microglia and macrophages.
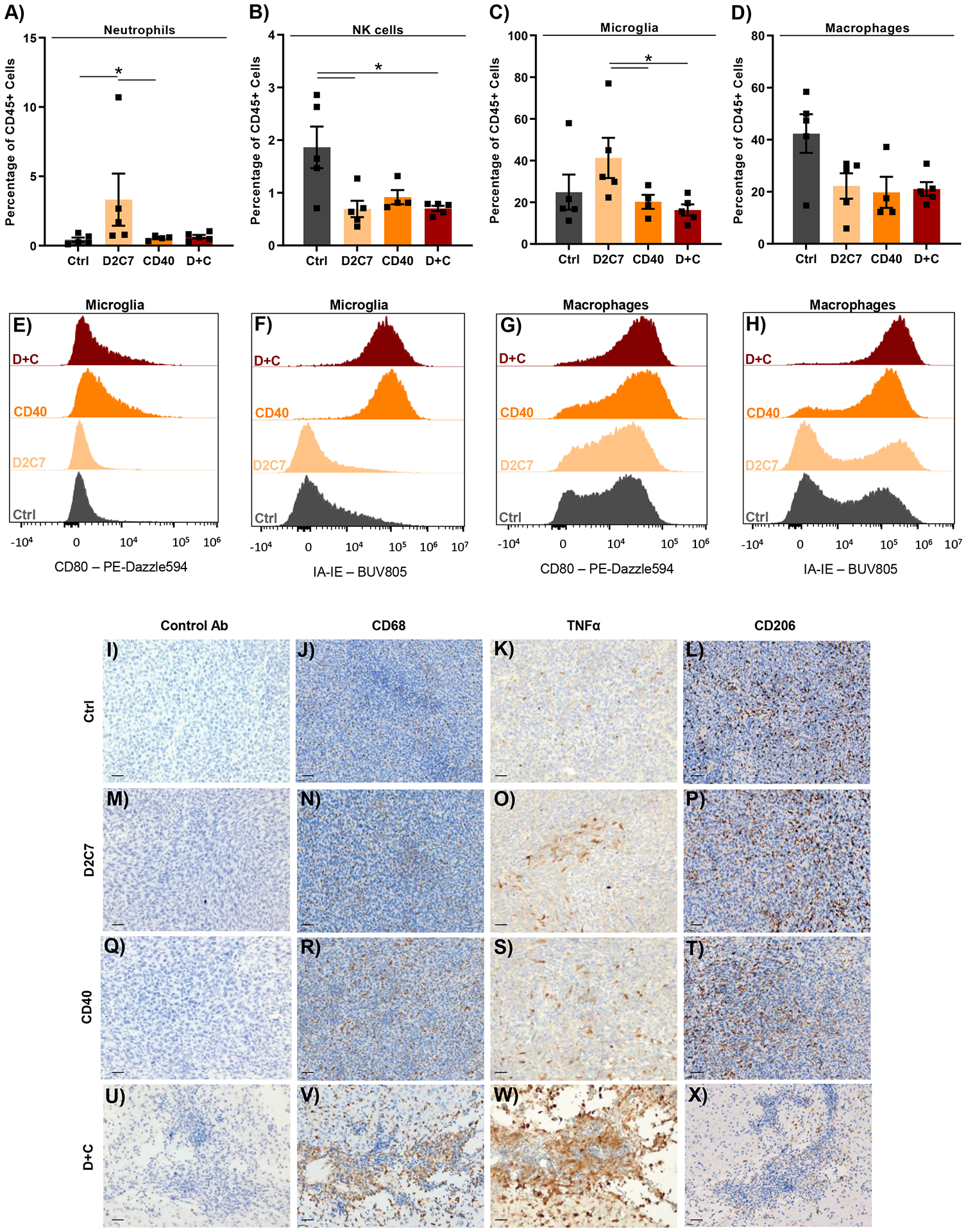
CT-VIII tumors were treated from days 6–9 after implantation by CED with Ctrl, D2C7, αCD40, and D+C therapies (n=4–5/group). 6-d post treatment, tumor hemispheres were harvested and analyzed by flow cytometry (A-H) or whole brains were collected for IHC staining (I-X). (A-D) Relative frequencies of neutrophils (A), natural killer cells (B), microglia (C), and macrophages (D) are presented. Data are mean ± SEM. (E-H) Expression of CD80 and MHCII (IA-IE) on microglia (E and F) and macrophages (G and H) are presented. (I-X) Representative IHC images of Control (I-L), D2C7 (M-P), αCD40 (Q-T), and D+C (U-X) stained with control, CD68, TNFα, or CD206 Abs. Scale bar: 30 μm.
*P<0.05
Having observed the upregulation of activation markers, we further analyzed the post-treatment status of TAMs using immunohistochemistry (IHC). Six days post-therapy, coronal sections of CT-VIII-bearing brains were taken at the tumor injection site and stained appropriately. In accordance with the FC data, compared to control (Fig. 3J) and D2C7 (Fig. 3N), a higher macrophage activation was evidenced by higher CD68 staining in mice treated with αCD40 and D+C therapies (Fig. 3R and 3V). However, compared to the control (Fig. 3K and 3L) and monotherapies (Fig. 3O, 3S, 3P, and 3T), the production of the proinflammatory cytokine TNFα was increased, and anti-inflammatory macrophage marker CD206 was decreased in the D+C group (Fig. 3W and 3X). In line with the IHC data, similar trends in CD206 expression on macrophages (decreased) (fig. S3A) and intracellular expression of TNFα in microglia (increased) (fig. S3B) were noted by FC analysis in the D+C group. The above data indicate that the antitumor effect of D+C therapy is, in part, due to an increase in the proinflammatory activity of microglia and TAMs.
Cytotoxic D2C7 plus agonistic αCD40 therapy increases intratumoral CD8+ T cells which controls glioma growth and improves survival
Given the role of CD8+ tumor-infiltrating lymphocytes (TILs) in αCD40 therapy-mediated tumor response (34, 37), we examined the post-treatment frequency of CD4+ T cells, CD8+ T cells, and B cells in the CT-VIII and GL-VIII glioma models. FC analysis of CT-VIII tumors 6 days post-therapy demonstrated an increase in CD4+ TILs (11.9%), CD8+ TILs (22.7%), and B cells (18.7%) in the combination compared to the control (~5%, all cell types) (Fig. 4A). Compared to the control, an increase in CD4+ (13.5%) and CD8+ (20.8%) TILs were noted in the D2C7 and αCD40 groups, respectively (Fig. 4A). Similarly, analysis of GL-VIII tumors 6 days post-therapy demonstrated an increase in CD4+ TILs (13.9%), CD8+ TILs (14.1%), and B cells (7.2%), only in D+C compared to control (1–5%, all cell types) (fig. S4A).
Fig. 4. CD8+ T cells mediate D+C antitumor response in gliomas.
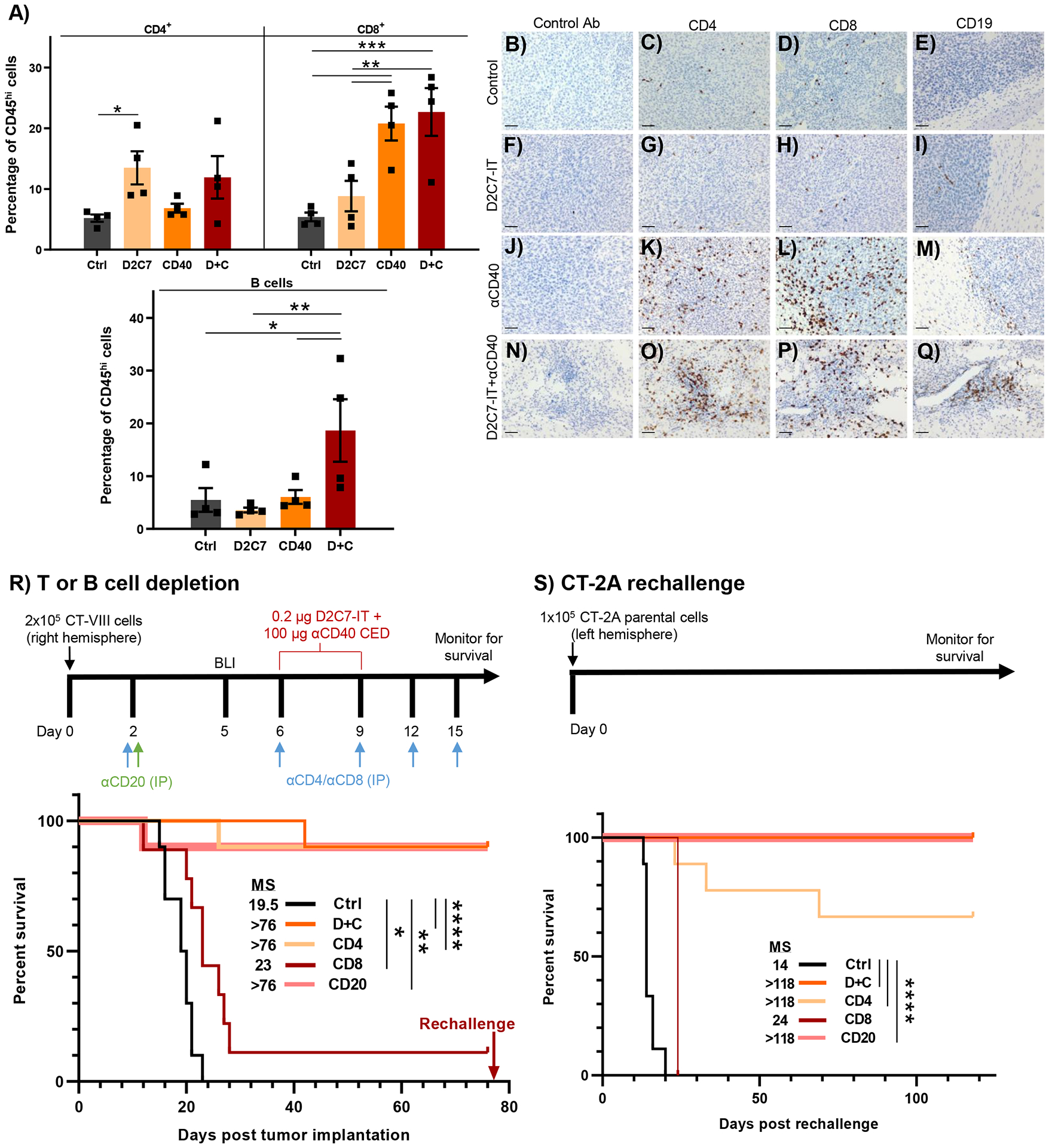
(A) FC analysis of CD4+, CD8+ T cells and B cells in CT-VIII tumor-bearing mice, 6-days post Ctrl, D2C7, αCD40, and D+C therapies (n=4/group). Data are mean ± SEM. (B-Q) Representative IHC images from CT-VIII tumors 7-d post-therapy, Control (B-E), D2C7 (F-I), αCD40 (J-M), and D+C (N-Q), stained with control, CD4, CD8, or CD19 Abs. Scale bar: 50 μm. (R) Survival of CT-VIII tumor-bearing mice (n=9–10/group) treated with Ctrl or D+C therapy with or without CD4, CD8, or B cell depletion beginning at day 2 post implantation. (S) Treated mice from (R) that experienced complete tumor regression for >75-d, were rechallenged with the CT-2A tumor cells. Tumor naïve mice (n=9) served as controls. The single CD8-depleted mouse was excluded from comparative analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Post-treatment frequency of T and B cells in the coronal sections of CT-VIII and GL-VIII tumor brains from the tumor injection site were analyzed using IHC. In agreement with the FC data, CD4+ TILs increased at the tumor cell low injection site (Fig. 1H and 1M) in the D+C group (Fig. 4O and S4O). Similarly, there was an increase in CD8+ TILs distributed throughout the tumors in the αCD40 group (Fig. 4L and S4L) or the tumor cell low injection site (Fig. 1H and 1M) in the D+C group (Fig. 4P and S4P). Again, an increase in B cells was noted in the D+C-treated brain (Fig. 4Q and S4Q).
To determine the function of T and B cells in D+C antitumor response, we depleted these lymphocytes in CT-VIII tumor-bearing mice (Fig. 4R). The D+C therapy without lymphocyte depletion resulted in 9/10 survivors with >76 days MS (Fig. 4R). An identical number of survivors and increase in MS were observed upon CD4+ T cell and B cell depletion (αCD20) (Fig. 4R). The efficacy of D+C was almost completely abolished upon CD8+ T cell depletion (1/9 survivors) (Fig. 4R), establishing their role as effectors of the adaptive immune response generated by the combination. Contralateral tumor rechallenge with CT-2A cells resulted in complete tumor uptake and euthanasia in all mice from the control group and in one mouse from the CD8+ T cell-depleted group (Fig. 4S). Six of nine mice from the CD4+ T cell-depleted group, and all mice from the groups without lymphocyte depletion and with B cell depletion, survived tumor rechallenge and remained tumor-free for >6 months after D+C therapy (Fig. 4S), establishing that the CD8+ T cells are critical for both the primary and long-term D+C antitumor immune response.
D2C7 and αCD40 treatment promotes progenitor cell states and prevents terminal exhaustion phenotype in CD8+ TILs in gliomas
Given that CD8+ TILs are needed for D+C antitumor efficacy, we investigated their phenotype further. FC analysis of CT-VIII (Fig. 5A) and GL-VIII (Fig. 5B) tumors 6 days post-therapy demonstrated that the majority of CD8+ TILs in all groups exhibited an effector memory (CD8+ Tem) phenotype (CD3+CD8+CD62L−CD44+), with significant increases in the αCD40 (P<0.01) and D+C (P<0.001) groups.
Fig. 5. D2C7+αCD40 therapy prevents CD8+ T cell exhaustion in gliomas.
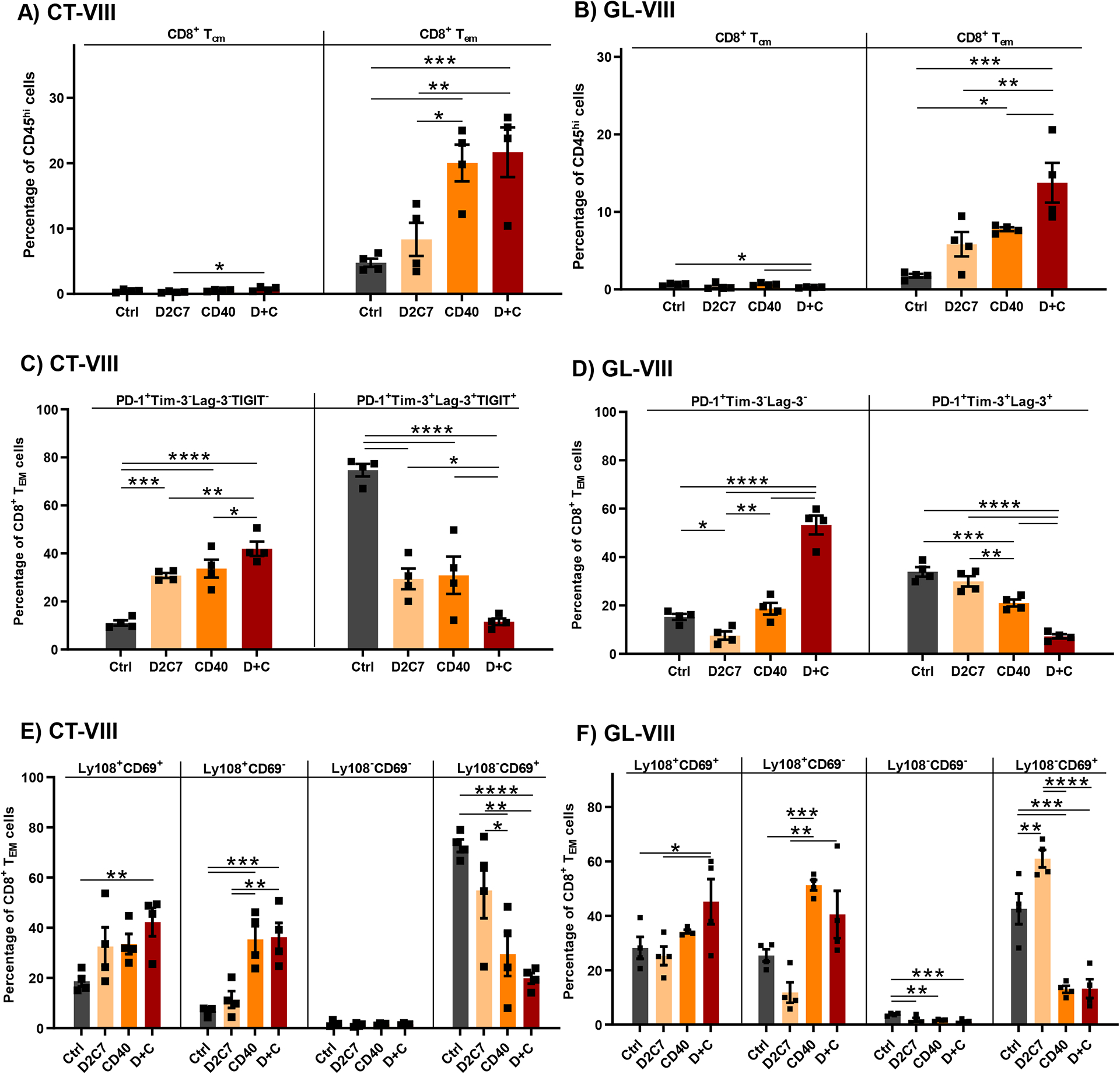
CT-VIII (A, C, E) or GL-VIII (B, D, F) tumors were treated from days 6–9 after implantation by CED with Ctrl, D2C7, αCD40, and D+C therapies (n=4/group). Tumor hemispheres harvested 6-d post treatment were analyzed by flow cytometry. Data are mean ± SEM. (A-B) Frequencies of CD8+ T cells with central memory (Tcm: CD3+CD8+CD62L+CD44+) or effector memory (Tem: CD3+CD8+CD62L−CD44+) phenotype are presented. (C-D) Frequencies of CD8+ Tem cells expressing PD-1 only or multiple exhaustion markers (PD-1/TIGIT/Tim-3/Lag-3) are presented. (E-F) Frequencies of CD8+ Tem cells classified into specific exhaustion subsets based on Ly108+ and CD69+ expression are presented. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Although T cell dysfunction through expression of multiple inhibitory receptors is a hallmark of GBM (12), αCD40 agonism reverses CD8+ TIL exhaustion and improves therapy response in various tumor models (37, 38). Hence, we examined CD8+ TIL exhaustion by examining PD-1, Tim-3, Lag-3, and TIGIT expression, post-D2C7 and αCD40 treatment in CT-VIII (Fig. 5C) and GL-VIII (Fig. 5D) gliomas. Highest frequency of CD8+PD-1+ TILs, a marker of early activation and exhaustion, and a lowest frequency CD8+PD-1+Tim-3+Lag-3+TIGIT+ TILs, a marker of terminal exhaustion, (39) was observed in the D+C group, followed by the next highest and lowest frequencies of activation and terminal exhaustion markers in the two monotherapy groups, in both CT-VIII and GL-VIII models. The terminal exhaustion phenotype was highest in the control group (~34–75% CD8+ TILs) (Fig. 5C and 5D). Similarly, median fluorescence intensity was higher for PD-1, TIGIT, Tim-3, Lag-3, and CD39 in the control group, followed by D2C7 and αCD40 groups, with the least expression in the D+C group in CT-VIII gliomas (fig. S5A and S5E). We validated our FC manual gating analysis with the Astrolabe Cytometry Platform (40) using unsupervised clustering for cell subset labeling and differential expression (DE) analysis to examine inhibitory receptor expression. In agreement with the manual analysis, the Astrolabe platform demonstrated a higher frequency of CD8+ TILs with a memory phenotype in the αCD40 (15.2%) and D+C (16.9%) groups in CT-VIII tumors (fig. S5F). DE analysis demonstrated PD-1, TIGIT, Tim-3, Lag-3, and CD39 expression was highest in control, lowest in D+C, and intermediate in D2C7 and αCD40 groups (fig. S5G and S5K).
Given that CD8+ TILs are terminally exhausted in glioma models (Fig. 5C and 5D), and since the inhibition of T cell exhaustion is required for successful antitumor response, we next examined CD8+ TIL activation potential in gliomas. Activation of exhausted CD8+ T cells (Tex) is defined by Ly108, also known as Slamf6, a surrogate for T cell factor family member TCF1 (Tcf7) transcription factor, and CD69 expression. Thus the following types of expression represent the following state of activation in CD8+ TILs: Ly108+CD69+ (progenitor 1; Texprog1), Ly108+CD69− (progenitor 2; Texprog2), Ly108−CD69− (intermediate; Texint), and Ly108−CD69+ (terminal; Texterm) (41). CD8+ TILs in the Texprog1 and Texprog2 exhibit high proliferative (Ki-67+) capacity whereas Texint and Texterm subsets exhibit minimal proliferation, with activation of Texprog subsets achievable by αPD-1 therapy (41). The FC analysis of CT-VIII and GL-VIII tumors demonstrated a higher frequency of Texprog1 and Texprog2 and a lower frequency of Texterm subsets in the D+C (CT-VIII: 42%, 36%, 20% and GL-VIII: 45%, 41%, 13%, respectively) and αCD40 (CT-VIII: 34%, 35%, 30% and GL-VIII: 34%, 51% and 13%, respectively) groups (Fig. 5E and 5F). In agreement with the terminal exhaustion defined by PD-1+TIGIT+Tim-3+Lag-3+ expression (Fig. 5C), the frequency of Texterm was highest in control (CT-VIII: 73%) and lowest in the D+C group. The Texint subset was minimal in all four groups (Fig. 5E and 5F). These data demonstrate that D+C treatment inhibits terminal exhaustion and maintains CD8+ TILs in an active Tex progenitor state.
Intratumoral CD8+ T cells are sufficient for tumor control and improve survival post D+C therapy in gliomas
Since CD8+ TILs are the primary effectors of the D+C antitumor response (Fig. 4R), we investigated the contribution of intratumoral versus peripheral CD8+ T cells to treatment efficacy in the CT-VIII model. We used FTY720 (FTY), a sphingosine 1-phosphate receptor-1 inhibitor, to prevent T cell egress from lymphoid organs, and thus blocked the influx of new T cells into the tumor during therapy. This forced the D+C-mediated immune response to be carried out by pre-treatment lymphocytes in the brain (42) as outlined (Fig. 6A). All control mice in both DMSO (Ctrl) and FTY (Ctrl+F) groups required euthanasia, with the FTY (MS=16 days) mice performing significantly (P<0.01) worse than the DMSO (MS=19 days) mice. The D+C therapy with DMSO (D+C) or FTY (D+C+F) resulted in 8/10 and 7/10 survivors, respectively, and increased MS to >98 days (Fig. 6A). The difference between the D+C or D+C+F groups were not statistically significant (P=0.5183). The above data indicate that the antitumor activity of D+C in CT-VIII gliomas is mediated by T cells which infiltrated the tumors before therapy.
Fig. 6. Pre-existing intratumoral T cells facilitate D+C antitumor response against gliomas.
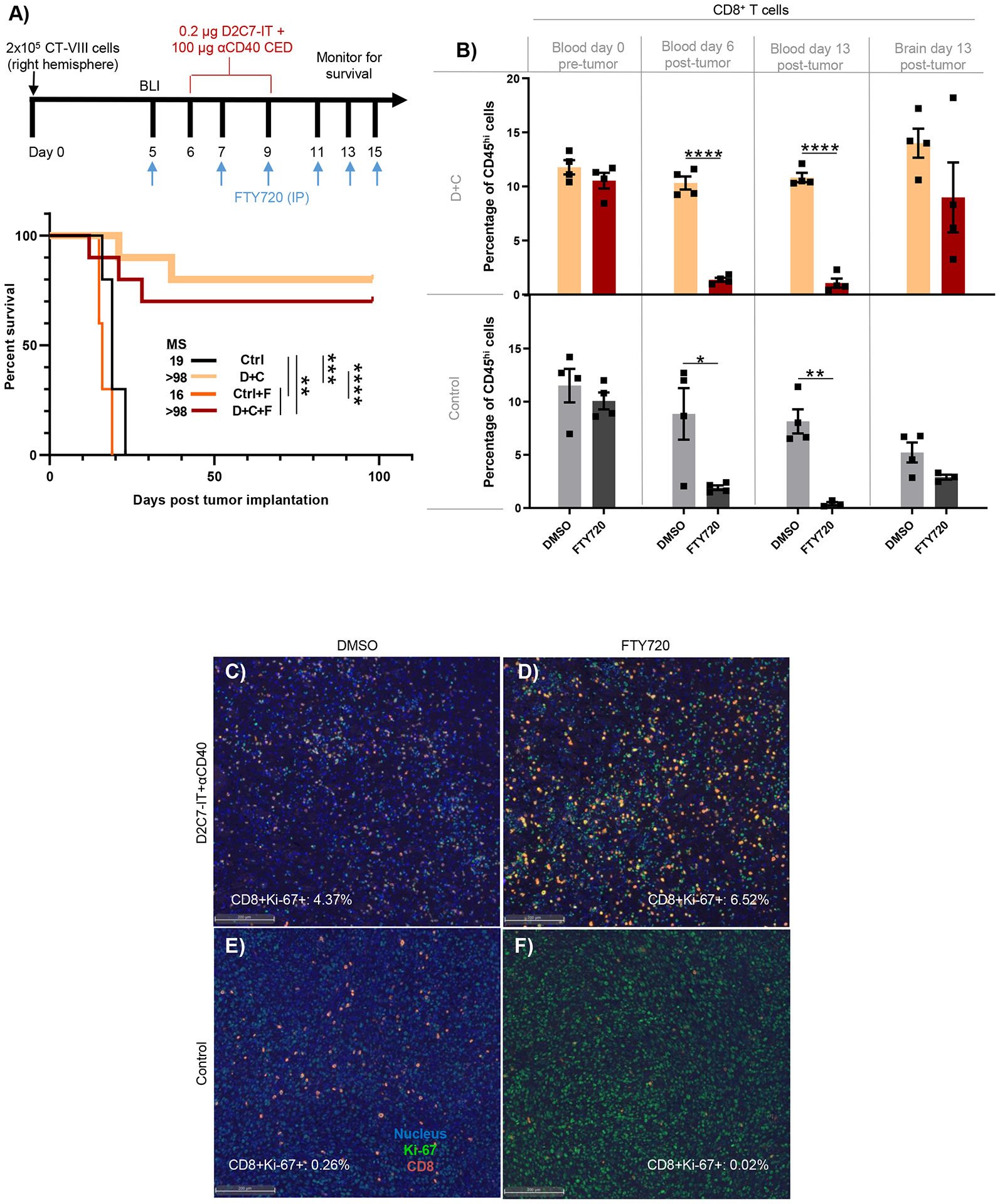
(A) Survival of CT-VIII tumor mice (n=10/group) treated with control or D+C therapy with or without FTY as indicated. (B) FC analysis of CD8+ T cells in day 0 blood (pre-tumor), day 6 blood (post-tumor), day 13 blood and brain (post-tumor) from CT-VIII tumor-bearing mice (n=4/group) treated as in 5A. Data are mean ± SEM. (C-F) Representative IF images of tumor sections from CT-VIII tumors (n=4/group), treated as in 5A and harvested 7-d post-therapy, stained for DAPI (nucleus, blue), CD8+ T cells (red), and Ki-67 (green) proliferation marker. The CD8+Ki-67+ cell frequency by QMIF analysis post indicated treatment is presented. Scale bar: 200 μm. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
We performed FC analysis to gain mechanistic insights into D+C+F-induced antitumor responses. Analysis of day 0 blood, before CT-VIII implantation, demonstrated similar amounts of CD8+ T cells in all groups (10–12%) (Fig. 6B). On day 6, before the start of the D+C CED, the abundance of CD8+ T cells in blood was reduced by ~80–90% in FTY-treated animals from both control (DMSO:8.9% versus FTY:1.9%) and D+C groups (DMSO:10.3% versus FTY:1.4%) (Fig. 6B), confirming the blockade of CD8+ T cell exit from lymphoid organs before treatment. On day 13, at the time of euthanasia, CD8+ T cell reduction in FTY-treated blood was maintained at >90% in both control (DMSO:8.2% versus FTY:0.4%) and D+C groups (DMSO:10.8% versus FTY:1.1%) (Fig. 6B). Additionally, on day 13, tumors were harvested and the frequencies of CD8+ TILs were examined. Compared to control+FTY (2.9%), a 3.1-fold increase in the frequency of CD8+ TILs in the brain was noted in the D+C+FTY group (9%) (Fig. 6B). Similarly, a ~2.7-fold increase in CD8+ TILs was observed between the control (5.2%) and D+C (14%) treatments with DMSO. There was no statistical difference in CD8+ TILs between D+C groups with and without FTY (Fig. 6B).
The increased frequency of CD8+ TILs post-D+C+FTY indicated proliferation and self-renewal of CD8+ TILs within the brain. To understand the dynamics of CD8+ TILs in the TME, we used quantitative multiplex immunofluorescence (QMIF) analysis to determine CD8+ TIL proliferation using Ki-67 co-expression in brains treated as in Fig. 6B and harvested on day 13. Overall, compared to control (Fig. 6E and 6F) and D+C+DMSO (CD8+Ki-67+; 4.37%) (Fig. 6C), an increased frequency of proliferating CD8+ TILs (CD8+Ki-67+; 6.52%) was associated with D+C+FTY (Fig. 6D). The above data support the hypothesis that the frequency of cytotoxic TILs post-D+C therapy is driven by local proliferation, resulting in higher cancer cell killing and cures.
D2C7 and αCD40 therapy enhances proliferating functional and tumor antigen-specific CD8+ TILs in gliomas
Given the observed intratumoral changes in CD8+ T cell subsets post-D+C, we next evaluated their effector functions and proliferation potential. CD8+ TILs from CT-VIII gliomas were assessed for intracellular cytokine expression and proliferation (Ki-67). Treatment with D+C resulted in significant increases (14.7%, P<0.05) in the proportion of CD8+ TILs positive for Ki-67 and producing multiple cytokines (IFNγ+TNFα+) compared to control (3.7%) or monotherapies (D2C7:8% and αCD40:6.6%) (Fig. 7A). This increase in polyfunctional CD8+ TILs in the D+C treatment cohort coincided with an increase in the amounts of tumor antigen (Trp2)-specific T cells within tumors, as determined by IFNγ ELISpot assay (Fig. 7B and 7C). D2C7 monotherapy had no effect on the proportion of IFNγ+ Trp2-specific TILs, compared to controls, whereas αCD40 monotherapy increased IFNγ+ Trp2-specific TIL concentrations ~5-fold. D+C therapy resulted in a ~10-fold enhancement in the proportion of IFNγ+ Trp2-specific TILs compared to vehicle controls (Fig. 7B). Together, the data indicate that the combination of D2C7 tumor cell killing and αCD40 immune-stimulation increases the proliferation, function, and tumor antigen-specificity of CD8+ TILs in gliomas.
Fig. 7. D2C7+αCD40 therapy increases functional and tumor antigen-specific CD8+ TILs in WT mice and loses antitumor efficacy in cDC1-lacking Batf3−/− mice bearing gliomas.
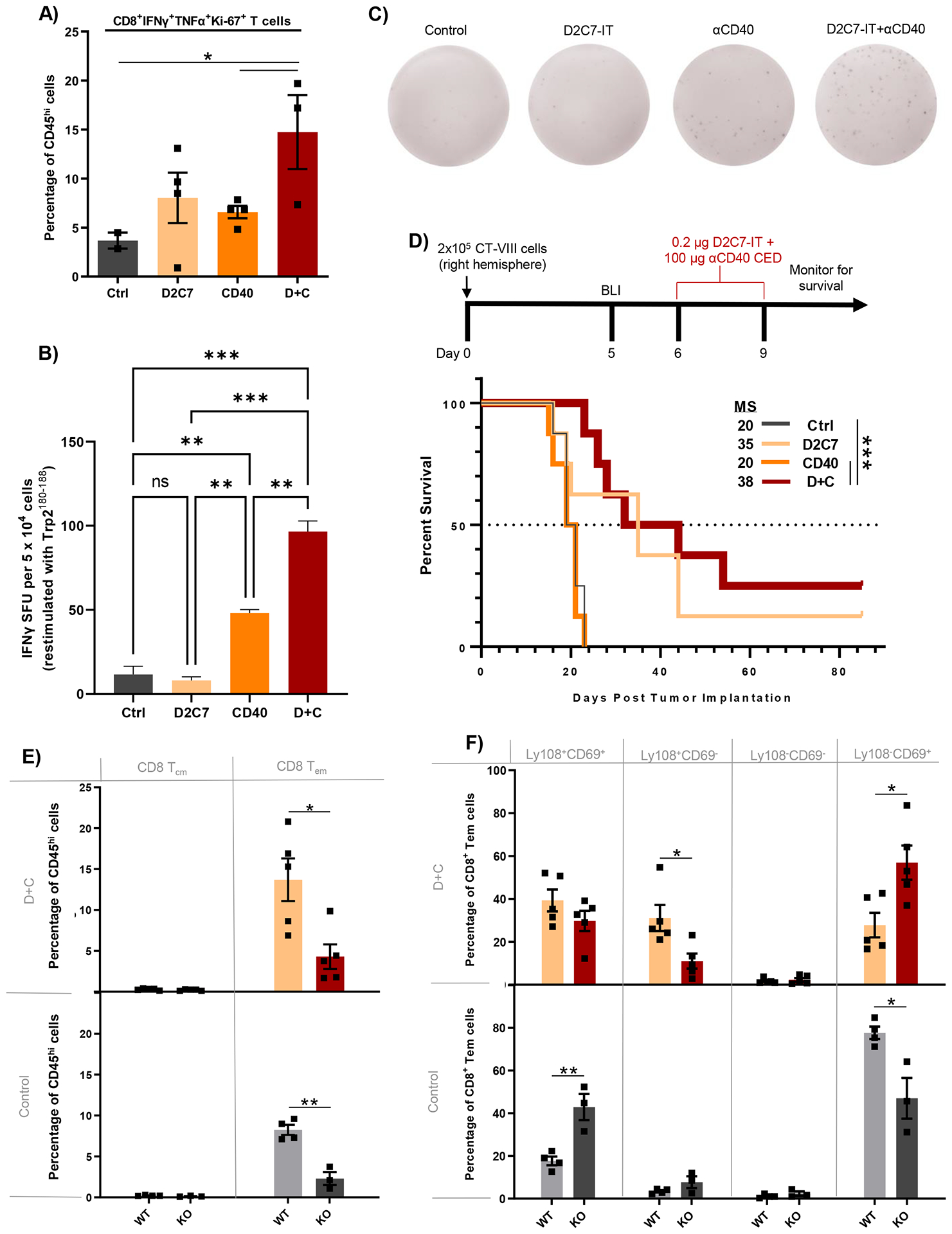
(A) FC analysis of CD8+IFNγ+TNFα+Ki-67+ T cells in CT-VIII tumor-bearing C57BL/6J WT mice, 6-days post Ctrl, D2C7, αCD40, and D+C therapies (n=2–4/group). (B) IFNγ ELISpot analysis performed on TILs harvested from CT-VIII-Trp2 tumors from C57BL/6J WT mice 6-days post-Ctrl, D2C7, CD40, and D+C therapies (n=3–6/group) stimulated with Trp2180–188 peptide. Background response from irrelevant peptide stimulation (< ~4 IFNγ spot forming units [SFUs]) subtracted from all plotted results. (C) Representative images of wells containing leukocytes isolated from Ctrl, D2C7, CD40, and D+C treated brains from C57BL/6J WT mice, and the IFNγ SFU observed following stimulation with Trp2180–188. (D) Survival of Batf3−/− (KO) mice implanted with CT-VIII (n=10/group) cells and treated with Ctrl, D2C7, αCD40, or D+C as indicated. (E-F) Frequencies of CD8+ T cells with central memory (Tcm) or effector memory (Tem) phenotype (E) and CD8+ Tem cells classified into specific exhaustion subset based on Ly108+ and CD69+ expression (F) in CT-VIII tumor hemispheres harvested 6-days post control or D+C therapy in WT and KO mice by FC analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are mean ± SEM.
Batf3 deficiency eliminates D+C-induced survival improvements and promotes Texterm CD8+ TIL phenotype in gliomas
The recruitment, priming, and expansion of tumor-specific stem-like Tcf1+Slamf6+CD8+ TILs following CD40 costimulation is mediated by cDC1s (conventional dendritic cells type 1) (43, 44). Thus, we examined the role of cDC1s in D+C induced antitumor responses by utilizing Batf3−/− knockout (KO) mice lacking cDC1s (45). Control and αCD40 groups generated similar survival (MS=20 days) in Batf3 KO mice in the absence of cures in mice(Fig. 7D). Contrarily, D2C7 and D+C groups extended MS to 35 and 38 days, respectively (Fig. 7D), indicating that D2C7-mediated tumor cell killing generated improved survival in Batf3 KO mice. However, D2C7 (1/8 survivors) and D+C (2/8 survivors) treatments produced fewer survivors in Batf3 KO CT-VIII tumor-bearing mice than Batf3 WT mice, highlighting the importance of cDC1-CD8+ T cell crosstalk for robust D+C mediated antitumor response. Additionally, Batf3 deficiency has been shown to alter CD8+ T cell survival, proliferation, and memory responses (46). Thus, we investigated the effect of Batf3 deficiency on CD8+ TILs in CT-VIII tumors post-D+C therapy. Compared to WT mice, we found a significant decrease in intratumoral CD8+ Tem numbers from both control (8.3% vs. 2.3%, P<0.01) and D+C (13.7% vs. 4.3%, P<0.05) groups in Batf3 KO CT-VIII tumor-bearing mice (Fig. 7E). We next tested the Tex phenotype of CD8+ TILs post-therapy in WT and Batf3 KO mice with CT-VIII tumors. Consistent with our findings above, D+C promoted a higher frequency of Texprog1 and Texprog2 CD8+ TILs in WT mice whereas the majority of CD8+ TILs were Texterm subset in Batf3 KO mice (Fig. 7F). Collectively, these data indicate that the cDC1-CD8+ T cell crosstalk facilitates D+C antitumor response in gliomas.
D2C7-IT reprograms glioma gene expression, upregulates CD40, and induces pattern recognition receptor (PRR) pathway transcriptome expression
Next, we investigated molecular drivers impacting D2C7 and αCD40 therapies by assessing gene expression differences between treatments at the end of 72 hr CED (post-therapy) and 3-d post-therapy in CT-VIII gliomas. Bulk RNA sequencing (RNA-seq) analysis of tumors revealed ~2- to 4-fold (post-therapy) and ~17-fold increases (3-d post-therapy) in differentially expressed (DE) genes in the D2C7 and D+C groups compared to αCD40 group (Fig. 8A and fig. S6A), establishing that D2C7 cytotoxicity compared to the αCD40 immune modulation induces intratumoral gene expression changes. There was minimal overlap between the genes modulated by D2C7 and αCD40, demonstrating distinct mechanisms of action of the two therapeutics. The highest overlap in DE genes at both time points was noted between D2C7 and D+C treatments (Fig. 8A and fig. S6A). The top genes upregulated by D2C7 (Fig. 8B and fig. S6B) and D+C (Fig. 8D and fig. S6D) were involved in pleiotropic functions, including apoptosis (Gadd45b), chemoattraction and inflammatory response (Cxcl1, Ccl7, Steap4), and intracellular movements (Selp, Myo15). The top genes upregulated by αCD40 (Fig. 8C and fig. S6C) primarily mediated immune effects, including T cell trafficking (Ccl22), immune response (Adgre4), and cytokine signaling (Gbp4). CD40 upregulation post-therapy was observed with D2C7 (Fig. 8B) and D+C (Fig. 8D), but not with αCD40 (Fig. 8C). These unique transcriptomic changes highlight the complementary activity of the D+C therapy.
Fig. 8. D2C7 and D+C therapies instigate proinflammatory transcriptional and cytokine changes in the tumor microenvironment in the CT-VIII glioma model.
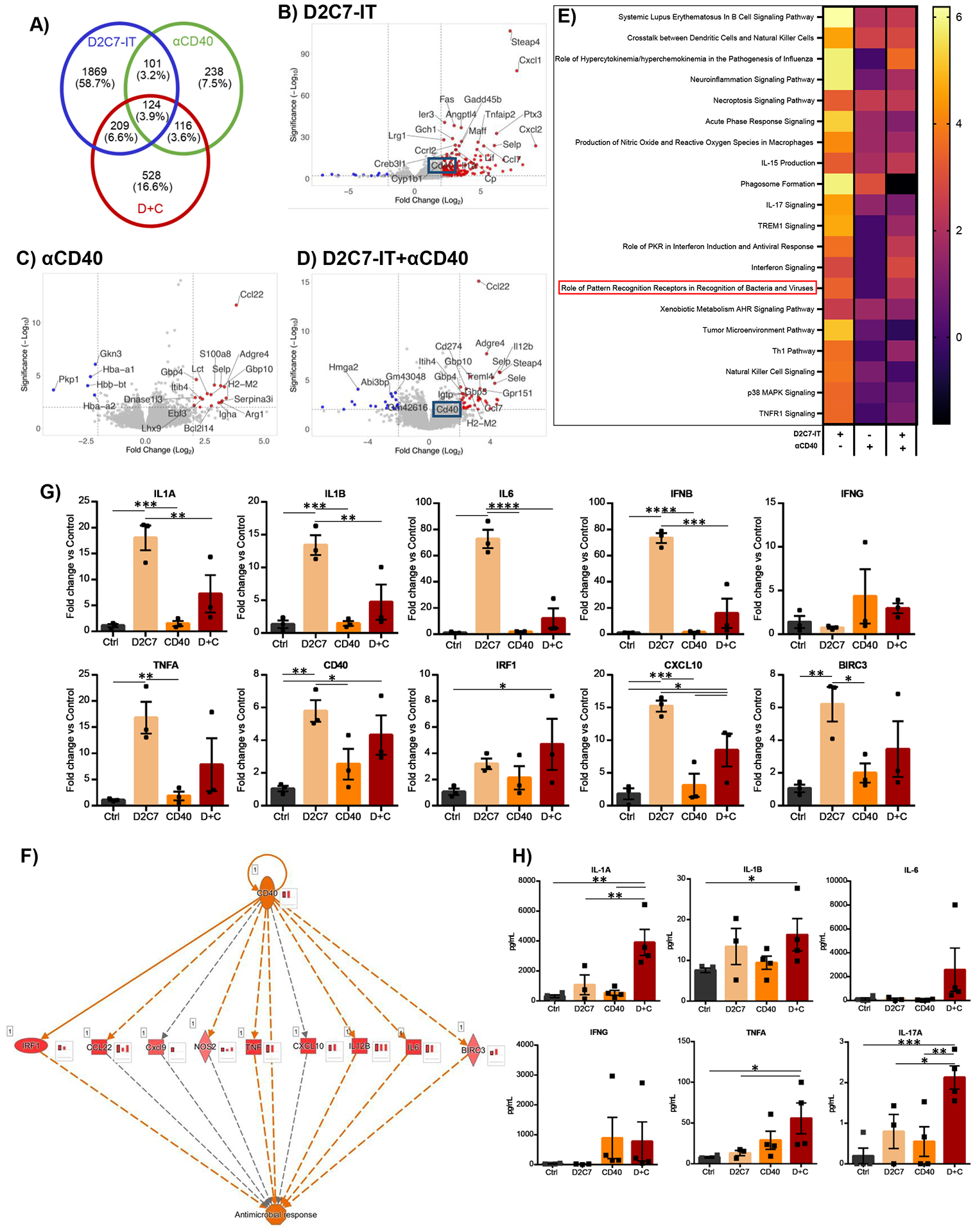
(A) Venn diagram of differentially expressed genes (up- and down-regulated compared to control group) in D2C7, αCD40, and D+C groups by RNA-Seq analysis of CT-VIII gliomas post 72 hr CED (n=3/group). (B-D) Volcano plots depicting the log2(fold change) in gene expression in D2C7 (B), αCD40 (C), and D+C (D) treated versus control tumors post 72 hr CED. Top 25 differentially expressed with P<0.05 are shown in red (upregulated) and blue (down-regulated). (E-F) Heatmap of z-scores of Top 20 IPA Canonical Pathway (E) and IPA CD40 regulatory network (F) gene transcripts in D2C7, αCD40, and D+C treated versus control tumors post 72 hr CED. The red bar charts in insets next to each gene represent the amount of activation in D+C (left), D2C7(middle), and αCD40 (right) treated versus control tumors post 72 hr CED. (G) The qPCR gene expression analysis of D2C7, αCD40, and D+C treated versus Ctrl tumors post 72 hr CED (n=3/group) in CT-VIII model. Data indicate the mean fold change over control after normalization to the average of ACTB housekeeping gene.
(H) Cytokine concentrations in tumor lysates of D2C7, αCD40, and D+C treated versus Ctrl tumors post 72 hr CED (n=3–4/group) in CT-VIII model. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are mean ± SEM.
We next investigated molecular interactions influencing the D2C7, αCD40, and D+C antitumor responses using Ingenuity Pathway Analysis (IPA). Based on the z-score the top 20 canonical pathways activated post-therapy include crosstalk between DCs and NK cells, necroptosis, neuroinflammation, acute phase response, production of nitric oxide and reactive oxygen species, interferon, and pattern recognition receptor (PRR) recognition of bacteria and viruses (Fig. 8E, Table S1). Considering that D2C7 is derived from Pseudomonas exotoxin and PRR pathway activation occurs exclusively with D2C7 and D+C, we investigated the genes activated in the PRR pathway post-therapy. Compared to αCD40, an increased upregulation of extracellular (complement C3), membrane-bound (TLR), and cytoplasmic (RIG-1, MDA-5, NALP3) PRR genes were noted in the D2C7 and D+C groups (fig. S6E and S7, Table S1). To understand if the PRR pathway activation is specific to D2C7, we compared our data with published RNA-Seq data of MMTV-PyMT breast tumors treated with αVEGFA+αAng2+αCD40 (GSE94920) (47). As before, PRR pathway activation was noted only in D2C7 and D+C groups and not in the diverse cell types, such as F4/80+ TAMs, CD4+, and CD8+ T cells, and CD31+ endothelial cells, from tumors treated with αVEGFA+αAng2+αCD40, confirming that this is a distinct function of D2C7 (fig. S6F, Table S1). Next, given the unique upregulation of CD40 by D2C7 (Fig. 8B) and D+C (Fig. 8D), we utilized the regulator effects analytical tool in IPA for predicting CD40 transcriptomic and functional outcomes downstream. The analysis identified a potential role for CD40 in antimicrobial response through activation of transcription regulator IRF1, cytokines TNF and IL-6, chemokine CXCL10, and BIRC3 (Fig. 8F). Finally, 3 days post-therapy, pathways associated with cell death through senescence and tumor growth suppression, such asinhibition of matrix metalloproteases, were upregulated by D2C7 (fig. S6G–S6I, Table S2). In contrast, immune response pathways, such as theTh1 pathway and regulation of cytokine production by IL17A and IL17F, were upregulated by αCD40 (fig. S6G–S6I, Table S2).; However, both cell death and immune response pathways were activated by D+C (fig. S6G–S6I, Table S2).
We further investigated the expression of the PRR pathway proinflammatory and CD40 antimicrobial response genes, including IL-1A, IL-1B, IL-6, IFNB, IFNG, TNFA, CD40, IRF1, CXCL10, and BIRC3, in bulk CT-VIII tumors post-therapy by qPCR. All genes except IFNG displayed the highest induction following D2C7 and D+C, whereas αCD40 had minimal effects on most of the genes (Fig. 8G). We next determined D2C7 and αCD40 antitumor effects by assessing cytokine expression in tumor tissue lysate post-therapy (Fig. 8H). Consistent with the optimal antitumor response observed in the D+C group, the highest concentration of IL-1A, IL-1B, IL-6, IFNG, TNFA, and IL-17A were specifically observed in the D+C group, with varying amounts observed in the control and monotherapies (Fig. 8H). These data indicate the contribution of D2C7 in tumor cell killing and induction of a potent proinflammatory milieu in the TME that supports a robust αCD40-induced immune response against the tumor.
Single-cell proteomics identify polyfunctional CD8+ TIL activation post D+C treatment in gliomas
In addition to CD8+IFNγ+TNFα+ TILs identified above, we investigated whether the D+C proinflammatory and immune-stimulatory milieu generated a polyfunctional T cell response in CT-VIII gliomas. A 28-plex Ab array single-cell proteomic analysis of CD8+ TILs from CT-VIII tumors 6-d post-treatment identified upregulation of a small number of polyfunctional CD8+ TILs secreting 5+ cytokines only in the D+C group (fig. S8A). The polyfunctional strength index (PSI), an indicator of the frequency of cells secreting multiple cytokines, showed the highest value in D2C7, followed by the αCD40 and D+C groups (fig. S8B). Whereas effector and chemoattractive cytokines were the predominant types secreted by αCD40, both D2C7 and D+C had a broader range, including stimulatory, regulatory, inflammatory, and other cytokine types (fig. S8B). A heatmap of secreted cytokines demonstrated antitumor-associated Granzyme B and IFNγ, and inflammatory MIP-1α to be highly secreted across D2C7, αCD40, and D+C therapies, and the specific subsets of polyfunctional CD8+ TILs generated by D2C7 and αCD40 therapies (fig. S8C). For example, MIP-1α+IFNγ+GZMB+, MIP-1α+GZMB+, MIP-1α+IFNγ+, GZMB+RANTES+ were some common CD8+ TIL subsets that were noted in D2C7 and αCD40, whereas additional CD8+ TIL subsets unique to individual treatments were also present. As depicted in fig. S8A (D+C: 5+ cytokines), CD8+ TILs secreting ten different cytokines were only present in the D+C group (fig. S8C). Post-therapy RNA-Seq cytokine expression data showed activation of cytokines, such as IFNγ, TNFα, IL-2, IL-6, IL-17A, IL-21, IL-18, IL-12, and IL-15, identical to the single-cell secretome analysis, predominantly in the D2C7 and D+C groups (fig. S8D, Table S1). Together, these data demonstrate that the D+C therapy promoted an antitumoral polyfunctional CD8+ TIL phenotype conducive to tumor rejection in mice. Collectively, our data indicate that the D+C cytotoxic immunotherapy combination promotes the generation of a proinflammatory and immune-stimulatory TME, which results in complementary antitumoral effects against gliomas.
CD40 expression in GBMs
To support the clinical translation of the D+C therapy for patients with GBM, we next performed CD40 IHC on 30 GBM specimens derived from human biopsies. The strongest localization of CD40 staining was found in proliferative endothelial cells in which 2–3+ intensity was identified in 27/30 cases, where 3 cases were negative for any reactivity (fig. S9A). Also noted in 20/27 reactive cases was 1–2+ intensity of CD40 staining in non-proliferative endothelial cells (fig. S9B). Finally, 1–2+ CD40 staining was noted in mononuclear inflammatory cells, in 7/27 reactive cases, predominantly around areas of necrosis (fig. S9C and S9D). We did not observe reactivity with isotype-matched non-specific IgG control. The CD40 expression analysis in GBM patient specimens validates our hypothesis that αCD40 immune stimulation could augment D2C7-IT antitumor response and potentially improve survival in patients with GBM.
DISCUSSION
Tumor heterogeneity and immune dysfunction generated by local and systemic immunosuppression have hampered survival improvements following tumor-targeted or immunotherapy single-agent trials in patients with GBM (48, 49). Hence, we aimed to overcome GBM immunosuppression using a cytotoxic-immunotherapy combination approach. Here we have demonstrated in two orthotopic murine tumor models that D2C7 cytotoxicity and CD40 agonism work together to eradicate gliomas and produce long-term survival when either monotherapy is ineffective in extending survival. The D+C immunotherapeutic regimen described herein utilizes a unique mechanism for activating microglia and TAMs and converting poorly T cell-infiltrated gliomas into a heavily infiltrated TME resulting in robust CD8+ T cell-mediated tumor regression.
There is conflicting evidence in gliomas about CD40 expression on tumor cells and its association with survival outcomes (29, 30, 50). In preclinical glioma models, agonistic αCD40 combinations only generated modest survival improvements and no cures (29, 34, 35, 51, 52). The contrasting preclinical efficacy of αCD40-based combination immunotherapies in syngeneic glioma models could be due to differences in dosing (single versus multiple), route of delivery (IP versus intravenous), and drug combinations. Alternatively, intratumoral CD40 costimulation to activate antigen-presenting cells (APCs) and increase T cell infiltration (37) could be leveraged to enhance the antitumor efficacy of D2C7 and improve the clinical outcomes in GBM, as well as overcoming the adverse events observed in the clinic with high dose systemic delivery of agonistic αCD40 (31, 53).
Microglia in naïve brain exhibit little or no MHCII expression. Upon activation in neurological disorders, microglia upregulate MHCII, CD80, and CD86 expression (54, 55). Similarly, macrophages upregulate MHCII, CD80, and CD86 upon activation (56). The FC analysis established microglia and macrophage activation (CD80hiMHCIIhi) following αCD40 and D+C therapies, whereas IHC and FC analysis established a robust induction of a proinflammatory myeloid milieu (CD68hi, TNFαhi, CD206low) specifically by the D+C in the CT-VIII gliomas.
Both FC and IHC analyses showed that D+C increased tumor-infiltrating CD4+ T cells, CD8+ T cells, and B cells (Fig. 4 and fig. S4). However, lymphocyte depletion studies showed that CD8+, but not CD4+ T cell or B cell loss, impaired D+C therapy-induced tumor inhibition, demonstrating the prominent role of CD8+ T cells in the treatment-derived immune response. Consistent with findings in other tumor models, analysis of CD8+ TILs in D+C-treated gliomas revealed an increase in the CD8+ Tem subset displaying an activated (CD8+PD-1+ TILs) or a progenitor phenotype (CD8+Slamf6+; Texprog) and a decrease in CD8+ Tem subset with a terminal exhaustion phenotype (CD8+PD-1+Tim-3+Lag-3+TIGIT+ TILs) (37, 38), resulting in tumor control (44, 57, 58). Together, our data demonstrate that CD8+ TIL phenotype, rather than their presence, dictates tumor response to immunotherapies. Our experiments in the CT-VIII glioma model with FTY revealed that D+C-mediated antitumor efficacy was associated with proliferating pre-existing T cells rather than newly activated T cells. Therefore, we hypothesize that in minimally T cell–infiltrated gliomas, D+C treatment inhibits T cell exhaustion and increases progenitor T cells, thus leading to tumor-resident T cell activation, proliferation, cytotoxicity, and tumor control.
Studies in multiple tumors, including gliomas, demonstrated that Batf3-dependent cDCs are critical for antigen cross-presentation and priming tumor-specific T cell immunity (45, 59, 60). Accordingly, Batf3−/− KO mice demonstrated impaired rejection of CT-VIII gliomas in response to D+C therapy. Additionally, CD8+ TIL phenotyping of CT-VIII gliomas in Batf3−/− KO and WT mice demonstrated a decrease in the CD8+ Tem subset in Batf3−/− KO mice. The reduction in CD8+ T cell accumulation within the TME following D+C in Batf3−/− KO mice could be due to the absence of Batf3-lineage cDC1s. However, our FC analysis did not show a significant increase in cDC1s intratumorally following control (0.28%±0.04%) and D+C (0.22%±0.03%) therapy.
The innate immune system senses exogenous pathogens (pathogen-associated molecular patterns [PAMPs]) and endogenous alarmins (damage-associated molecular patterns [DAMPs]) through PRRs (61). Lipopolysaccharide, phospholipids, and proteins on the bacterial cell wall are common PAMPs that activate diverse PRRs (62). Although derived from the gram-negative Pseudomonas aeruginosa exotoxin A, D2C7 lacks a bacterial cell wall or nucleic acid PAMPs for PRR activation, and therefore we are unable to demonstrate a role for PAMPs in D2C7- and D+C-mediated antitumor response. On the contrary, immunogenic cell death (DAMPs) has been established as a mode of action for immunotoxins in mesothelioma (63). Here, we have identified additional DAMPs, including IL-1a, ROS, and NLRP3, that are upregulated following D2C7 therapy, which could potentially activate PRRs and create a proinflammatory TME in gliomas. Further, through transcriptome profiling, we demonstrate global gene expression changes, including CD40 upregulation and PRR pathway activation specifically by D2C7 and D+C therapies but not by αCD40 therapy. Thus, we have uncovered a unique role for D2C7 in exerting pleiotropic proinflammatory effects on the TME potentially through the Pseudomonas exotoxin mediated cytotoxicity, which, when combined with αCD40, improves tumor clearance and animal survival.
CD40 activation on endothelial cells enhances the expression of adhesion molecules, such as E-selectin, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1, increasing leukocyte recruitment and inflammatory response during ischemic stroke, atherosclerosis, and intestinal inflammation (64, 65). Herein, we hypothesize that CD40 expressed on human brain endothelial cells could enhance the adhesion and migration of immune cells across the blood-brain barrier and improve the antitumor response in the brain following D+C therapy.
Our study has some limitations, such as delineating the role of peripheral T cells, myeloid cells, and endothelial cells in D+C therapy-mediated antitumor response. Future studies using transgenic mouse models will focus on confirming the role of microglia (Tmem119CreERT2:ROSA-DTA and Tmem119CreERT2:CD40fl/fl) and macrophages (LysMCre:ROSA-DTA and LysMCre:CD40fl/fl) as mediators of innate immune responses during D+C mediated tumor rejection. We have not examined the role of cDC1s in tumor-draining lymph nodes. Given our findings in the Batf3−/−KO mice, characterization of cDC1 function in D+C antitumor immunity in gliomas in tumor-draining lymph nodes in WT mice and using Xcr1Cre:ROSA-DTA and Xcr1Cre:CD40fl/fl mice is warranted (66). Finally future studies using transgenic mouse models will also investigate the role of endothelial cells (Tie2-Cre (Tek-Cre):CD40fl/fl) in D+C mediated antitumor response.
In conclusion, D+C cytotoxic immunotherapy combination activates microglia and TAMs, creates a proinflammatory TME, inhibits CD8+ TIL exhaustion, increases Texprog, polyfunctional, and tumor antigen-specific CD8+ TILs, extends survival, and generates long-term antitumor immune responses against malignant gliomas in mice (fig. S11). Thus, D2C7-IT+αCD40 is a promising combinatorial intervention for patients with malignant gliomas. A phase 1 clinical trial of D2C7-IT+αhumanCD40 (2141-V11) administered intratumorally using CED in patients with recurrent malignant glioma (NCT04547777) is in progress.
MATERIALS AND METHODS
Study Design
The primary objective of this study was to determine the antitumor activity and gain mechanistic insights into the D2C7-IT and αCD40 combination therapy in orthotopic glioma models. Primary outcomes for in vivo therapy studies included weight loss measurements and survival, and toxicity studies included complete blood count, ALT, and AST measurement.In vitro mechanistic studies utilized IHC, immunofluorescence, flow cytometric, ELISpot, gene expression, and single-cell proteomics analysis to determine phenotypic and functional changes in myeloid and T cells and glioma microenvironment. In adherence with the requirement for species-matched antibody-receptor systems to precisely model the activity of therapeutic reagents, these studies used syngeneic glioma cell lines overexpressing mouse EGFRvIII and C57BL/6J WT or transgenic Batf3 KO mice to evaluate the effects of D2C7-IT and murine αCD40 combination. The role of T cells in D2C7-IT and αCD40 combination therapy antitumor response was studied by blocking T cell egress from lymphoid organs by pharmacologic inhibition with FTY720. Since CT-2A and GL261 cell lines were established from brain tumors from C57BL/6 female mice, we used female mice in all our studies. Tumor-bearing mice were randomized into different treatment groups using an open-access randomization software, Research Randomizer, Version 4.0. The sample size was determined based on bootstrapping simulations of preliminary in vivo therapy studies showing that sample sizes must be ten per group for ~80% power (α=0.05). For all experiments, the number of animals in each group and p values are reported in the figure legends, and no outliers or other data points were excluded. All survival experiments were repeated 1–3 times, all flow cytometry or IHC experiments were repeated 2–3 times, and ELISpot, gene expression, LegendPlex cytokine measurement, Isoplexis single-cell proteomics studies were performed once. The reported replicates refer to biological replicates. Weight loss measurements and survival monitoring were blinded.
Cell Lines and culture conditions
Mouse brain tumor cell lines CT-2A (12) and GL261 (National Cancer Institute) were passaged at confluence with Accutase Cell Detachment Solution (BD Biosciences). They were cultured in an incubator maintained at 37°C with 5% carbon dioxide. Both cell lines were cultured in DMEM-high glucose media (Thermo Fisher Scientific) supplemented with 10% FBS (Thermo Fisher Scientific). The cell lines were tested for rodent pathogens and authenticated by whole-exome sequencing. The cell culture supernatants were tested for mycoplasma infection. After thawing, all cell lines were maintained in culture for >10 passages.
Patient tumor tissues
De-identified archival GBM tissue samples (n=30) were obtained with IRB approval from the Preston Robert Tisch Brain Tumor Center Biorepository at Duke University Medical Center (DUMC). Cases were selected from archived Formalin-Fixed, Paraffin-Embedded (FFPE) blocks stemming from resected tissue. Tissue blocks were selected by a DUMC neuropathologist as ~70–100% viable tumor and >1 cm2 of tissue by light microscopic examination of Hematoxylin and Eosin (H&E) stained sections. Serial unstained sections (5 μm thickness) were cut from these FFPE blocks and stained for CD40 by IHC.
Establishment of D2C7-IT target cell lines
The chimeric DNA fragment designated D2C7 (d)-mouse (m)-EGFRvIII (dmEGFRvIII) was ligated into the pLXIN retroviral vector (Clontech Laboratories). Mouse astrocytoma cell lines CT-2A and GL261 were transduced with the dmEGFRvIII retroviral particles and luciferase (pLVX-IRES-FFLuc, Clontech Laboratories) lentiviral particles, and stable cell lines (CT-2A-dmEGFRvIII-FFLuc [CT-VIII] and GL261-dmEGFRvIII-FFLuc [GL-VIII]) were established upon G418 and hygromycin selection. The mouse Trp2 gene (Origene), was cloned into pLenti-6.3-V5 lentiviral vector (Thermo Fisher Scientific). Mouse astrocytoma cell line CT-2A-dmEGFRvIII-FFLuc was transduced with the Trp2 lentiviral particles, and CT-2A-dmEGFRvIII-FFLuc-Trp2 stable cell line was established upon blasticidin selection.
Preparation of recombinant immunotoxins
D2C7-IT (D2C7) was expressed in E. coli BL21 (λ DE3) (Agilent), under the control of T7 promoter. D2C7 from inclusion bodies was reduced, refolded, and further purified as monomers by ion exchange and size-exclusion chromatography as described previously (15). Finally, endotoxin removal from the purified immunotoxin was achieved with ActiClean Etox resin (Sterogene). The purity of the final D2C7 preparation was >95% by SDS-PAGE and size-exclusion chromatography (HPLC).
Mice
All experiments were done in accordance with the Institutional Animal Care and Use Committee of Duke University Medical Center (A082-21-04). Animals were maintained in a barrier facility, under pathogen-free conditions according to NIH guidelines. Female C57BL/6J mice or Batf3−/− were purchased from The Jackson Laboratory. All mice used were 7–8 weeks old and weighed 16–20 gm at the start of the study.
Intracranial tumor implantation
For tumor implantation, mice were anesthetized and mounted onto a stereotactic frame, and a 0.5-inch midline incision was made to expose the skull. A cordless rotary tool (Dremel) with a carbide bur operative friction grip 1/4 (Henry Schein) was used to create a pilot hole at 0.5 mm anterior and 2.0 mm lateral to bregma. An automatic stereotaxic injector holding a 25 μl Hamilton syringe (Hamilton Company) with a 27 gauge needle was inserted vertically into the pilot hole. It was lowered down until the tip of the needle reached a depth of 3.3 mm below the skull. A total of 2×105 CT-VIII or 1×105 GL-VIII mouse glioma cells were injected in 5 μl of PBS containing 3% methylcellulose (Sigma) at a rate of 3.33 μl per min. Medical Bone wax (DLAR Pharmacy) was used to close the drill hole, and veterinary surgical adhesive glue (DLAR Pharmacy) was used to close the incision site. For tumor rechallenge studies, a total of 1×105 antigen-negative CT-2A (denoted as CT-2A parental cells) or 7.5×104 antigen-negative GL261 cells were injected at the same coordinates in the contralateral hemisphere.
Staggered contralateral tumor implantation
Each mouse underwent two intracranial tumor implantations with 2×105 CT-VIII (day 0, right hemisphere) and 1×105 CT-VIII (day 3, left hemisphere) cells, as described above.
Bioluminescence imagining (BLI) and randomization
For the D2C7 and αCD40 mono or combination therapy studies, 5 d post-tumor implantation, mice were anesthetized, injected with 3 mg D-Luciferin – K+ Salt (Perkin Elmer) dissolved in 100 μl of PBS, and imaged for tumor presence using a Perkin Elmer IVIS Lumina III In Vivo Imaging System. Mice were then randomized into different treatment groups (n=5–10 per group for antitumor efficacy studies and 3–5/group for flow cytometry, immunohistochemistry, immunofluorescence, RNA-Seq, Real-time PCR, LegendPlex, and Single Cell Secretome studies) by total flux using an open-access randomization software (67)). Research Randomizer (Version 4.0) [Computer software], randomizer.org).
For the CD8+ T cell, CD4+ T cell, and B cell depletion studies, mice were randomized into different treatment groups (n=10 mice/group) based on their weight measurements before tumor implantation using the same randomization software. Five doses (250 μg/dose) of rat IgG2b isotype control antibody (Clone LTF-2, Bio-X-Cell), αCD4 antibody (Clone GK1.5, Bio-X-Cell), αCD8 antibody (Clone 2.43, Bio-X-Cell), or αCD20 antibody (clone MB20–11, kindly provided by Dr. Thomas Tedder) (68) were administered IP post-implantation of CT-VIII cells (Isotype/αCD4/αCD8: days 2, 6, 9, 12, and 15 and αCD20: day 2) on day 0.
Convection-enhanced delivery (CED) treatment
On day 6 post-tumor implantation, mice were treated with D2C7 (0.2 μg) and αCD40 (10–300 μg; Clone FGK4.5/FGK45 Bio-X-Cell) mono or combination therapies by convection-enhanced delivery using Alzet Micro-Osmotic Pumps (Model 1007D) (Durect Corporation) at a rate of 0.5 μl/hr for 72 hr. Control groups were treated with 2% MSA-PBS with 100 μg rat IgG2a Ab (Bio-X-Cell InVivoPlus Clone 2A3). On day 9 at the end of 72 hr CED, the Alzet Micro-Osmotic Pumps were removed and animals were monitored accordingly.
Tumor rechallenge studies
Mice from efficacy studies surviving symptom-free for >70 d post-tumor implantation and age-matched naïve control mice were rechallenged on days 75–90, and were monitored for survival.
FTY720 Administration
Fingolimod Hydrochloride (FTY720-FTY) (Sigma) was dissolved in 100% dimethylsulfoxide (DMSO) (Sigma) at a concentration of 5 mg/ml. The FTY stock solution was then diluted in 1x phosphate-buffered saline (PBS) to achieve a working concentration of 0.125 mg/ml immediately before treatment, and 200 μl (25 μg total dose) was administered to the study mice using intraperitoneal injections for a total of six doses on days 5, 7, 9, 11, 13, and 15.
Statistical Analysis
Pairwise differences of survival curves were assessed using the generalized Wilcoxon test. Pairwise differences of histology, flow cytometry, AST, ALT, and Platelets data were assessed using the Mann-Whitney U test. The statistical significance of qPCR, LEGENDPlex, and ELISpot data was assessed using a one-way analysis of variance (ANOVA). Data indicate mean values ± standard error of the mean (SEM).
Supplementary Material
Acknowledgments:
We thank O. Alzate and C. Pirozzi for their critical reading of the manuscript and valuable comments. We thank S. Clayton for his help with cell culture, A. Nguyen and H. Jackson for assistance with intracranial surgeries and tumor tissue processing for flow cytometry and IHC analyses, E. Thomas, M. Burnett, A. Byrnes, and D. Satterfield for support with FFPE sections and H&E staining. We thank H. Beggs (BioLegend) for helpful discussions on LEGENDplex assay and analysis. We thank A. Danisi for his technical assistance and helpful discussions on Single-Cell Secretome assay and analysis. Supplementary Figure 11 was created with BioRender.com.
Funding:
The work was supported, in part, by Silvian Foundation (to V.C.), Tisch family through the Jewish Communal Fund (to D.D.B.), a National Institutes of Health grant R01NS119183 (to V.C.), a National Institutes of Health grant R35CA197264 (to D.D.B.), a National Cancer Institute grant K08CA248966 (to D.A.K.), and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (to I.H.P.).
Competing interests:
P.D. is an employee of Cytek Biosciences, Inc. V.C., D.D.B., and I.H.P. are inventors on patent application (DU6879US; Immunotherapy with combination therapy comprising an Immunotoxin) licensed to Vimana. D.D.B. serves as a consultant for Istari Oncology and Vimana. L.S.P. is currently an employee of Tune Therapeutics, Inc. A.B. is presently an employee of Xilis. A.M.S. is currently an employee of Immorna. T.F.T. is the founder of Cellective Biotherapy, Inc. and Antigenomycs, Inc., not related to the present work. A.D. serves as an advisory board member for Orbus Therapeutics and Midatech Pharma, served on the advisory board of Istari Oncology within the last two years, receives clinical research support (to the institution) from Orbus Therapeutics and Midatech Pharma, and has stock options with Istari Oncology. D.M.A. serves as an advisory board member for Immunogenesis, MAIA Biotechnology, and Diverse Biotech and a consultant for Jackson Laboratories. M.D.G. serves as an advisory board member for Myeloid Therapeutics. D.A.K. serves as an expert consultant for Shoreline Biosciences, not related to the present work. S.K.N. is a member of the Pfizer mRNA advisory panel, an inventor on patents licensed to Istari Oncology and Vimana, and a co-founder of NanoVenari Technologies, LLC. The other authors declare that they have no competing interests.
Data and materials availability:
The datasets generated and/or analyzed in the current study is available in the NCBI Gene Expression Omnibus (GSE221376). Mouse tumor cell lines generated in this study will be made available to the broader scientific community upon request to the lead contact under a material transfer agreement.
References
- 1.Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS, CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol 22, iv1–iv96 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, European Organisation for R, Treatment T of Cancer Brain, G. Radiotherapy, G. National Cancer Institute of Canada Clinical Trials, Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Fernandes C, Costa A, Osorio L, Lago RC, Linhares P, Carvalho B, Caeiro C, in Glioblastoma, De Vleeschouwer S, Ed. (Codon Publications, 2017). [PubMed] [Google Scholar]
- 4.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, Kawaguchi ES, Du L, Li G, Yong WH, Gaffey SC, Cohen AL, Mellinghoff IK, Lee EQ, Reardon DA, O’Brien BJ, Butowski NA, Nghiemphu PL, Clarke JL, Arrillaga-Romany IC, Colman H, Kaley TJ, de Groot JF, Liau LM, Wen PY, Prins RM, Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 25, 477–486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, Friedman HS, McSherry F, Muscat AM, Nair S, Peters KB, Randazzo D, Sampson JH, Vlahovic G, Harrison WT, McLendon RE, Ashley D, Bigner DD, Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med 379, 150–161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, Huse JT, de Groot J, Li S, Overwijk WW, Spetzler D, Heimberger AB, Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 19, 1047–1057 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE, Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zagzag D, Salnikow K, Chiriboga L, Yee H, Lan L, Ali MA, Garcia R, Demaria S, Newcomb EW, Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Invest 85, 328–341 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, Woroniecka K, Elsamadicy AA, Dechant CA, Kemeny HR, Sanchez-Perez L, Cheema TA, Souders NC, Herndon JE, Coumans JV, Everitt JI, Nahed BV, Sampson JH, Gunn MD, Martuza RL, Dranoff G, Curry WT, Fecci PE, Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24, 1459–1468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunn GP, Dunn IF, Curry WT, Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in human glioma. Cancer Immun 7, 12 (2007). [PMC free article] [PubMed] [Google Scholar]
- 11.Gutmann DH, Kettenmann H, Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 104, 442–449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, Elsamadicy AA, Cui X, Koyama S, Jackson C, Hansen LJ, Johanns TM, Sanchez-Perez L, Chandramohan V, Yu YA, Bigner DD, Giles A, Healy P, Dranoff G, Weinhold KJ, Dunn GP, Fecci PE, T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res 24, 4175–4186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang JH, Desjardins A, Convection-enhanced delivery for high-grade glioma. Neurooncol Pract 9, 24–34 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, Network TR, The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandramohan V, Bao X, Keir ST, Pegram CN, Szafranski SE, Piao H, Wikstrand CJ, McLendon RE, Kuan CT, Pastan IH, Bigner DD, Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin Cancer Res 19, 4717–4727 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandramohan V, Bao X, Yu X, Parker S, McDowall C, Yu YR, Healy P, Desjardins A, Gunn MD, Gromeier M, Nair SK, Pastan IH, Bigner DD, Improved efficacy against malignant brain tumors with EGFRwt/EGFRvIII targeting immunotoxin and checkpoint inhibitor combinations. J Immunother Cancer 7, 142 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desjardins A RD, Chandramohan V, Peters K, Johnson M, Landi D, Khasraw M, Threatt S, Bullock C, Herndon JE II, Lipp E, Sampson J, Friedman A, Friedman H, Ashley D, and Bigner DD, in Neuro-Oncology. (2020), vol. 22, pp. ii38. [Google Scholar]
- 18.van Kooten C, Banchereau J, CD40-CD40 ligand. J Leukoc Biol 67, 2–17 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Beatty GL, Li Y, Long KB, Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther 17, 175–186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR, Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393, 478–480 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Ridge JP, Di Rosa F, Matzinger P, A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393, 474–478 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ, T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393, 480–483 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, Offringa R, Toes RE, CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med 5, 774–779 (1999). [DOI] [PubMed] [Google Scholar]
- 24.French RR, Chan HT, Tutt AL, Glennie MJ, CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med 5, 548–553 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, Fein S, Schoenberger S, Levitsky HI, Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med 5, 780–787 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH, CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL, CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol 174, 6013–6022 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Rakhmilevich AL, Buhtoiarov IN, Malkovsky M, Sondel PM, CD40 ligation in vivo can induce T cell independent antitumor effects even against immunogenic tumors. Cancer Immunol Immunother 57, 1151–1160 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chonan M, Saito R, Shoji T, Shibahara I, Kanamori M, Sonoda Y, Watanabe M, Kikuchi T, Ishii N, Tominaga T, CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol 17, 1453–1462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Werner JM, Kuhl S, Ulrich K, Krischek B, Stavrinou P, Goldbrunner R, Timmer M, Expression of CD40 Correlates Negatively with Overall and Progression-Free Survival of Low- and High-Grade Gliomas. World Neurosurg 130, e17–e25 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Li DK, Wang W, Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol Lett 20, 176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medina-Echeverz J, Ma C, Duffy AG, Eggert T, Hawk N, Kleiner DE, Korangy F, Greten TF, Systemic Agonistic Anti-CD40 Treatment of Tumor-Bearing Mice Modulates Hepatic Myeloid-Suppressive Cells and Causes Immune-Mediated Liver Damage. Cancer Immunol Res 3, 557–566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazzaccara C, Labruna G, Cito G, Scarfo M, De Felice M, Pastore L, Sacchetti L, Age-Related Reference Intervals of the Main Biochemical and Hematological Parameters in C57BL/6J, 129SV/EV and C3H/HeJ Mouse Strains. PLoS One 3, e3772 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kosaka A, Ohkuri T, Okada H, Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol Immunother 63, 847–857 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Hooren L, Vaccaro A, Ramachandran M, Vazaios K, Libard S, van de Walle T, Georganaki M, Huang H, Pietila I, Lau J, Ulvmar MH, Karlsson MCI, Zetterling M, Mangsbo SM, Jakola AS, Olsson Bontell T, Smits A, Essand M, Dimberg A, Agonistic CD40 therapy induces tertiary lymphoid structures but impairs responses to checkpoint blockade in glioma. Nat Commun 12, 4127 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fabian C, Han M, Bjerkvig R, Niclou SP, Novel facets of glioma invasion. Int Rev Cell Mol Biol 360, 33–64 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Garris CS, Wong JL, Ravetch JV, Knorr DA, Dendritic cell targeting with Fc-enhanced CD40 antibody agonists induces durable antitumor immunity in humanized mouse models of bladder cancer. Sci Transl Med 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ngiow SF, Young A, Blake SJ, Hill GR, Yagita H, Teng MW, Korman AJ, Smyth MJ, Agonistic CD40 mAb-Driven IL12 Reverses Resistance to Anti-PD1 in a T-cell-Rich Tumor. Cancer Res 76, 6266–6277 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ, Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 10, 29–37 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amir ED, Lee B, Badoual P, Gordon M, Guo XV, Merad M, Rahman AH, Development of a Comprehensive Antibody Staining Database Using a Standardized Analytics Pipeline. Front Immunol 10, 1315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, Yan P, Nzingha K, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Huang AC, Wherry EJ, Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841 e828 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H, Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296, 346–349 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Oba T, Hoki T, Yamauchi T, Keler T, Marsh HC, Cao X, Ito F, A Critical Role of CD40 and CD70 Signaling in Conventional Type 1 Dendritic Cells in Expansion and Antitumor Efficacy of Adoptively Transferred Tumor-Specific T Cells. J Immunol 205, 1867–1877 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oba T, Long MD, Keler T, Marsh HC, Minderman H, Abrams SI, Liu S, Ito F, Overcoming primary and acquired resistance to anti-PD-L1 therapy by induction and activation of tumor-residing cDC1s. Nat Commun 11, 5415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM, Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ataide MA, Komander K, Knopper K, Peters AE, Wu H, Eickhoff S, Gogishvili T, Weber J, Grafen A, Kallies A, Garbi N, Einsele H, Hudecek M, Gasteiger G, Holzel M, Vaeth M, Kastenmuller W, BATF3 programs CD8(+) T cell memory. Nat Immunol 21, 1397–1407 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Kashyap AS, Schmittnaegel M, Rigamonti N, Pais-Ferreira D, Mueller P, Buchi M, Ooi CH, Kreuzaler M, Hirschmann P, Guichard A, Rieder N, Bill R, Herting F, Kienast Y, Dirnhofer S, Klein C, Hoves S, Ries CH, Corse E, De Palma M, Zippelius A, Optimized antiangiogenic reprogramming of the tumor microenvironment potentiates CD40 immunotherapy. Proc Natl Acad Sci U S A 117, 541–551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chuntova P, Chow F, Watchmaker PB, Galvez M, Heimberger AB, Newell EW, Diaz A, DePinho RA, Li MO, Wherry EJ, Mitchell D, Terabe M, Wainwright DA, Berzofsky JA, Herold-Mende C, Heath JR, Lim M, Margolin KA, Chiocca EA, Kasahara N, Ellingson BM, Brown CE, Chen Y, Fecci PE, Reardon DA, Dunn GP, Liau LM, Costello JF, Wick W, Cloughesy T, Timmer WC, Wen PY, Prins RM, Platten M, Okada H, Unique challenges for glioblastoma immunotherapy-discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro Oncol 23, 356–375 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, Wen P, Reifenberger G, Weller M, Molecular targeted therapy of glioblastoma. Cancer Treat Rev 80, 101896 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Xie F, Shi Q, Wang Q, Ge Y, Chen Y, Zuo J, Gu Y, Deng H, Mao H, Hu Z, Zhou Y, Zhang X, CD40 is a regulator for vascular endothelial growth factor in the tumor microenvironment of glioma. J Neuroimmunol 222, 62–69 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Shoji T, Saito R, Chonan M, Shibahara I, Sato A, Kanamori M, Sonoda Y, Kondo T, Ishii N, Tominaga T, Local convection-enhanced delivery of an anti-CD40 agonistic monoclonal antibody induces antitumor effects in mouse glioma models. Neuro Oncol 18, 1120–1128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang F, He Z, Duan H, Zhang D, Li J, Yang H, Dorsey JF, Zou W, Nabavizadeh SA, Bagley SJ, Abdullah K, Brem S, Zhang L, Xu X, Byrne KT, Vonderheide RH, Gong Y, Fan Y, Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat Commun 12, 3424 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vonderheide RH, CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med 71, 47–58 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Ponomarev ED, Shriver LP, Dittel BN, CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J Immunol 176, 1402–1410 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Yang I, Han SJ, Kaur G, Crane C, Parsa AT, The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci 17, 6–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suttles J, Stout RD, Macrophage CD40 signaling: a pivotal regulator of disease protection and pathogenesis. Semin Immunol 21, 257–264 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, Haining WN, Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W, Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF, Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yan J, Zhao Q, Gabrusiewicz K, Kong LY, Xia X, Wang J, Ott M, Xu J, Davis RE, Huo L, Rao G, Sun SC, Watowich SS, Heimberger AB, Li S, FGL2 promotes tumor progression in the CNS by suppressing CD103(+) dendritic cell differentiation. Nat Commun 10, 448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bianchi ME, DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81, 1–5 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Mogensen TH, Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22, 240–273, Table of Contents (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leshem Y, King EM, Mazor R, Reiter Y, Pastan I, SS1P Immunotoxin Induces Markers of Immunogenic Cell Death and Enhances the Effect of the CTLA-4 Blockade in AE17M Mouse Mesothelioma Tumors. Toxins (Basel) 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chatzigeorgiou A, Lyberi M, Chatzilymperis G, Nezos A, Kamper E, CD40/CD40L signaling and its implication in health and disease. Biofactors 35, 474–483 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Ishikawa M, Vowinkel T, Stokes KY, Arumugam TV, Yilmaz G, Nanda A, Granger DN, CD40/CD40 ligand signaling in mouse cerebral microvasculature after focal ischemia/reperfusion. Circulation 111, 1690–1696 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, Davidson J. T. t., Bagadia P, Liu T, Briseno CG, Li L, Gillanders WE, Wu GF, Yokoyama WM, Murphy TL, Schreiber RD, Murphy KM, cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 584, 624–629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Urbaniak GC PS, Research Randomizer (Version 4.0) (Computer sofware), 2013. Available at https://www.randomizer.org..
- 68.Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF, The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med 199, 1659–1669 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu YA, O’Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER. Que L, Gunn MD, A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLoS One 11, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gassen SV, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y, FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–45 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Amir ED, Lee B, Badoual P, Gordon M, Guo XV, Merad M, Rahman AH, Development of a Comprehensive Antibody Staining Database Using a Standardized Analytics Pipeline. Front Immunol 10, 1315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Finak G, Langweiler M, Jaimes M, Malek M, Taghiyar J, Korin Y, Raddassi K, Devine L, Obermoser G, Pekalski ML, Pontikos N, Diaz A, Heck S, Villanova F, Terrazzini N, Kern F, Qian Y, Stanton R, Wang K, Brandes A, Ramey J, Aghaeepour N, Mosmann T, Scheuermann RH, Reed E, Palucka K, Pascual V, Blomberg BB, Nestle F, Nussenblatt RB, Brinkman RR, Gottardo R, Maecker H, McCoy JP, Standardizing Flow Cytometry Immunophenotyping Analysis from the Human ImmunoPhenotyping Consortium. Sci Rep 6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maecker HT, McCoy JP, Nussenblatt R, Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol 12, 191–200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Swartz AM, Reap E, Norberg P, Schmittling R, Janetzki S, Sanchez-Perez L, Sampson JH, A simple and enzyme-free method for processing infiltrating lymphocytes from small mouse tumors for ELISpot analysis. J Immunol Methods 459, 90–93 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goedhart J, Luijsterburg MS, VolcaNoseR is a web app for creating, exploring, labelling, and sharing volcano plots. Sci Rep 10, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed in the current study is available in the NCBI Gene Expression Omnibus (GSE221376). Mouse tumor cell lines generated in this study will be made available to the broader scientific community upon request to the lead contact under a material transfer agreement.