

Abstract
Introduction:
NOD-like receptor family pyrin domain containing 3 (NLRP3) can sense a plethora of exogenous and endogenous dangers. Upon activation, a multimeric protein complex, the NLRP3 inflammasome, is formed to initiate the innate immune responses. Emerging studies have implicated the pathophysiological roles of this protein complex in human disorders, highlighting that it represents a druggable target for therapeutics development.
Areas covered:
The current review summarizes the functional facets of the NLRP3 inflammasome, its association with autoimmune diseases, and recent patents on the development of NLRP3 inhibitors. Literature search was conducted using SciFinder and Google Patents with the key word NLRP3 and NLRP3 inhibitors.
Expert opinion:
Although significant advances have been made in understanding the NLRP3 inflammasome, more studies are still needed to elucidate the molecular mechanisms underlying its roles in autoimmune diseases. A number of NLRP3 inhibitors have been patented, however, none of them have been approved for clinical use. Due to the complex nature of the NLRP3 inflammasome, novel screening assays along with target engagement methods could benefit the drug discovery and clinical translation. In addition, clinical trials on NLRP3 inhibitors are still in their early stages, and continuous investigations are needed to fully assess their safety and effectiveness.
Keywords: NLRP3 inflammasome, autoimmune diseases, small molecule inhibitors, SAR
1. Introduction
The innate immunity and adaptive immunity constitute the immune system and are responsible for surveilling the host from endogenous and exogenous dangers [1]. The innate immune system serves as the first line of defense and relies on pattern-recognition receptors (PRRs) to recognize pathogens and danger signaling through pathogen-associated molecular patterns (PAMPs), and/or damage-associated molecular patterns (DAMPs), and to initiate inflammatory responses [2–4]. PRRs can be classified into membrane-bound PRRs, e.g., Toll like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic PRRs including NOD-like receptors (NLRs) and RIG-I-like receptors (RLRs) [5,6]. Several cytoplasmic PRRs are able to form a multimeric protein complex known as “inflammasome” to activate the proinflammatory caspases and lead to the production of inflammatory cytokines [7–10]. Among the known inflammasomes, the NLR family pyrin domain containing 1 (NLRP1), NLRP3, NLRP6, and NLR family card domain containing 4 (NLRC4) inflammasomes are subsets of the NLR family. Absent in melanoma 2 (AIM2) and interferon (IFN)-inducible protein 16 (IFI16) inflammasomes belong to the pyrin and HIN (hematopoietic expression, interferon-inducible nature, and nuclear localization) domain-containing (PYHIN) family [11–13]. Recent studies have highlighted the pathological roles of inflammasome dysregulation in certain human diseases, and the NLRP3 inflammasome has attracted much attention as a viable drug target for therapeutic interventions. Herein, we review the NLRP3 inflammasome activation process, its association with autoimmune diseases, and summarize the small molecule inhibitors developed by targeting the inflammasomes from patents filed recently and their potential application in autoimmune diseases.
2. NLPR3 inflammasome and its activation
Assembly of the NLRP3 inflammasome requires a sensor protein, NLRP3 (containing Leucine rich repeat-NACHT-PYD domains), an adaptor apoptosis-associated speck-like protein (ASC, containing PYD and CARD domains), and an effector pro-caspase 1 (containing the CARD domain) [14,15]. Under canonical conditions, two steps are typically needed to assemble and activate the NLRP3 inflammasome (Fig. 1). The priming is to upregulate the expression of NLRP3 protein upon the signaling of PAMPs or DAMPs, e.g., microbial products or endogenous cytokines such as tumor necrosis factor (TNF-α) or interleukin (IL)-1β through the activation of the NF-κB (nuclear factor kappa light chain enhancer of activated B cells) pathway [16–18]. Additionally, the priming step also induces posttranslational modifications (PTMs) of NLRP3, including ubiquitylation, phosphorylation, and sumoylation, which leads to the stabilization of inactive NLRP3 [19]. A plethora of molecular and cellular events have been implicated to trigger NLRP3 inflammasome activation, including K+ efflux [20–22], Ca2+ flux [23], Cl− efflux [24,25], lysosomal disruption [26], mitochondrial dysfunction [27], metabolic changes [28,29], and trans-Golgi disassembly [30], among others. Upon activation, the NIMA-related kinase 7 (NEK7) binds to NLRP3, causing structural rearrangement that allows for NLRP3 oligomerization through its NACHT domain. This is followed by the recruitment of ASC via the PYD-PYD interactions between NLRP3 and ASC to form the “speck” [31]. Then, pro-caspase 1 is recruited to form the complex through CARD–CARD interactions with ASC [32,33]. Upon assembly of the NLRP3 inflammasome complex, self-cleavage converts pro-caspase 1 to its active form, caspase-1, followed by the proteolytic cleavage of pro-IL-1β and pro-IL-18 into proinflammatory cytokines, IL-1β and IL-18. This ultimately leads to pyroptosis, a highly regulated and programmed mechanism of cell death [34]. Another critical component involved in the pyroptotic process, gasdermin D (GSDMD), is also cleaved by the active caspase-1 to produce an N-terminal fragment (NT-GSDMD) by which a pore structure will be formed in the cell membranes to release IL-1β and IL-18 [35].
Figure 1.
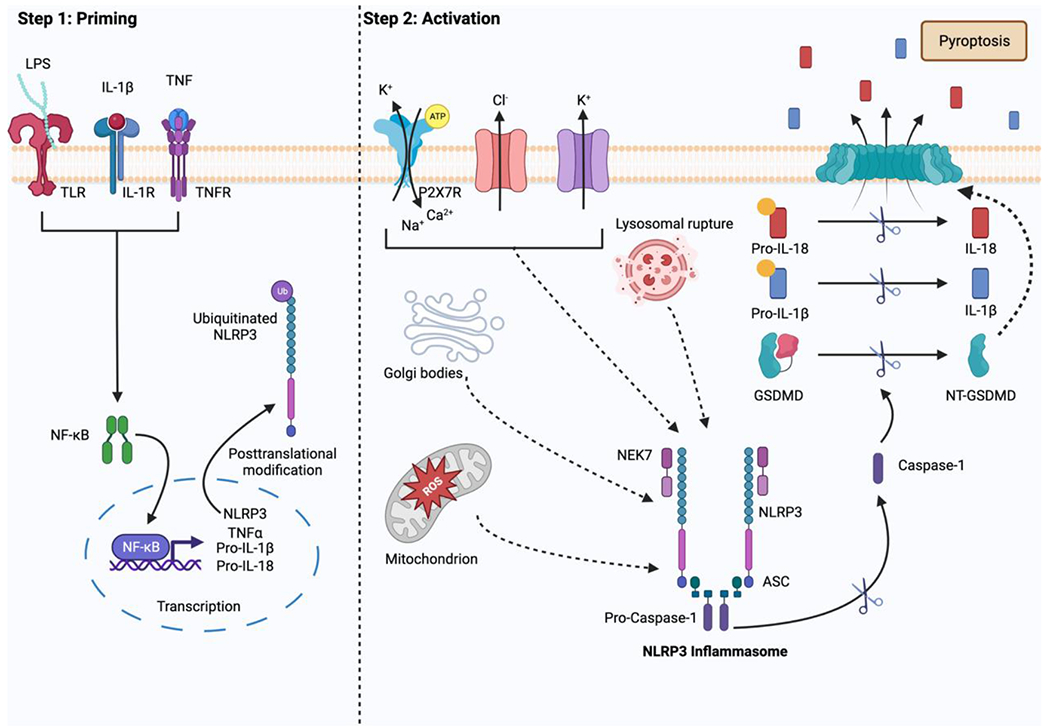
Canonical activation of the NLRP3 Inflammasome (created with BioRender.com). Step 1 illustrates the priming step in the process of NLRP3 inflammasome activation. Upon the activation of the NF-κB pathway, the expression of NLRP3, pro-IL-1β, and pro-IL18 are increased. Step 2 shows the activation process of the NLRP3 inflammasome. The activation triggers, such as K+ efflux and lysosomal rupture, lead to the assembly of the inflammasome complex. This is followed by the proteolytic cleavage of pro-caspase-1 to active caspase-1, ultimately leading to the release of IL-1β and IL-18.
The non-canonical activation of the NLRP3 inflammasome involves the recognition of cytosolic lipopolysaccharide (LPS) by human caspase-4, caspase-5, or mouse caspase-11 during gram-negative bacterial infection (Fig. 2) [36]. The activation of these caspases results in the proteolytic cleavage of GSDMD and pyroptosis without IL-1β and IL-18 release [37,38]. Notably, pannexin-1 (a membrane channel for adenosine triphosphate (ATP)) can be cleaved by the activated caspase-11 to induce an efflux event of ATP and K+, which in turn activates the NLRP3 inflammasome and leads to IL-1β and IL-18 secretion [39]. This reflects the interconnection between the non-canonical and canonical pathways.
Figure 2.
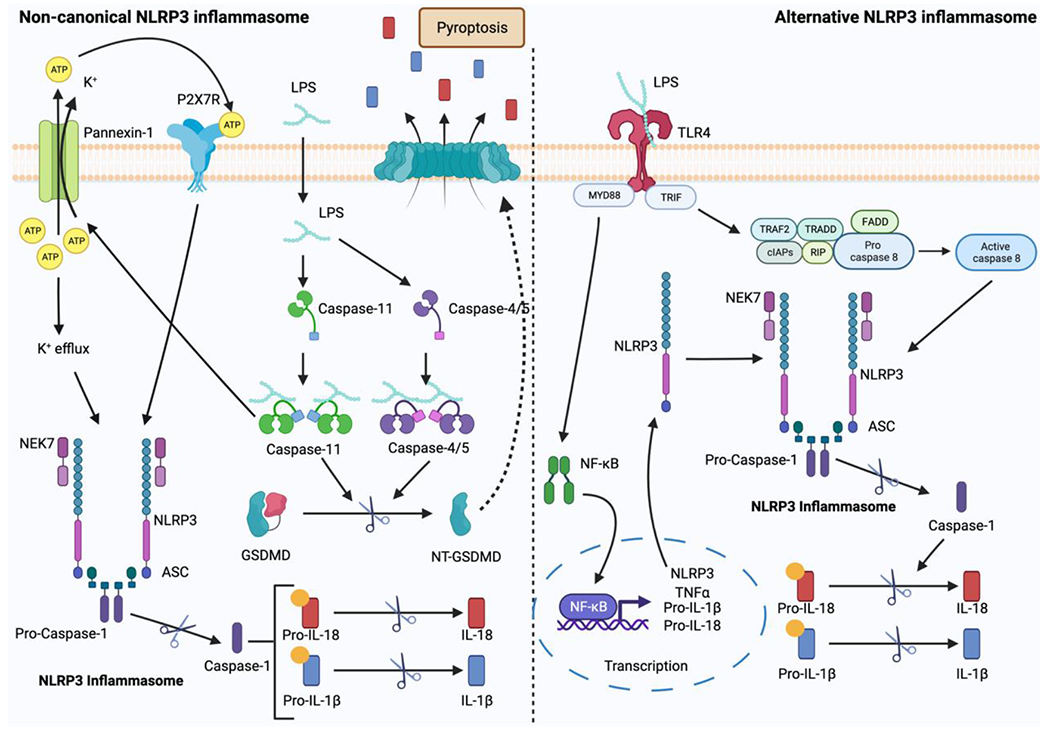
The noncanonical and alternative activations of the NLRP3 inflammasome (created with BioRender.com). Under the non-canonical activation pathway, caspase-4/5 or caspase-11 is activated to cleave GSDMD and lead to pyroptosis. Under the alternative activation pathway, caspase-8 is activated and this subsequently leads to the activation of the NLRP3 inflammasome.
The activation of the NLRP3 inflammasome via the alternative pathway is observed in human and porcine monocytes but not in murine monocytes (Fig. 2) [40]. Upon stimulation with LPS, TLR4 activates myeloid differentiation primary response 88 (MyD88) and Toll/IL-1R (TIR)-domain-containing adapter-inducing interferon-β (TRIF), leading to the activation of the NF-κB pathway, and the formation of a complex comprising receptor-interacting serine/threonine protein kinase 1 (RIPK1) and FAS-associated death domain protein (FADD). This complex then triggers the activation of caspase-8, ultimately leading to the activation of the NLRP3 inflammasome. In the TRIF-RIPK1-FADD-caspase 8 cascade, the events observed under canonical NLRP3 inflammasome activation, such as ASC speck formation, K+ efflux, GSDMD cleavage, and pyroptosis, are typically dispensable [40].
3. Dysregulation of the NLRP3 inflammasome in autoimmune diseases
As a crucial component of the innate immune system, the NLRP3 inflammasome is widely expressed in a variety of immune cells, including monocytes, macrophages [41], osteoblasts [42], epithelial cells [43], fibroblasts [44], T cells, and B cells [45]. The T-helper (Th)17 cells, a subpopulation of the CD4+ T cells, have been implicated to play roles in autoimmune and inflammatory disorders, e.g., multiple sclerosis (MS), rheumatoid arthritis (RA), and psoriasis [46]. Regulatory Th17 cells (Treg17) modulate the inflammatory responses via Th17/Th1 signaling [47,48]. Dysregulation of Th17/Treg has been shown to have pathophysiological roles in autoimmune diseases [49–52]. Studies have shown that IL-1β prolongs the survival of T cells and induces their differentiation and polarization, especially towards the Th17 population. In addition, IL-1β stimulates the proliferation of B cells leading to enhanced antibody production [53]. Furthermore, studies demonstrated that IL-18 enhances the proliferation of Th1 cells and leads to the production of IFN-γ [53–55]. Collectively, the NLRP3/IL-1 axis plays an essential role in promoting the differentiation and amplification of Th17 and Th1 cells, highlighting the importance of the NLRP3 inflammasome in autoimmune diseases.
3.1. Role of the NLRP3 inflammasome in Systemic Lupus Erythematosus (SLE)
SLE is a chronic autoimmune illness that damages several organs as a result of impaired innate and adaptive immunity, which leads to the generation and buildup of nuclear autoantibody complexes in response to inadequate auto-antigen recognition. Although the exact cause of SLE is undetermined, excessive activation of the NLRP3 inflammasome is thought to be a major factor in the disease’s pathogenesis. Nearly 70% of SLE patients suffer from lupus nephritis (LN) [56,57]. Studies have shown that patients with SLE, especially those suffering with LN, more significantly expressed a gain-of-function mutation on the NLRP3 gene than other groups, resulting in increased activity of the NLRP3 inflammasome in patients with SLE [58]. Additionally, increased levels of NLRP3 gene expression, as well as other genes in the NLRP3 inflammasome pathway, have been shown in both cells isolated from SLE patients and in lupus-like mouse models, such as MLR/lpr mice [59–61]. Moreover, over activation of the NLRP3 inflammasome was observed in samples derived from SLE patients [59,60]. The accumulation of DAMPs, such as reactive oxygen species (ROS), intracellular ATP, nucleic acids, and others, as a result of an elevated rate of apoptosis, decreased clearance of neutrophil extracellular traps (NETs), formed by activated low density granulocytes (LDGs), and abnormal autophagy are thought to be major factors influencing the hyperactivation of NLRP3, which results in the aberrant release of pro-inflammatory cytokines that ultimately cause tissue damage. Inhibition of the NLRP3 inflammasome has exhibited marked improvement of the disease state both in bone marrow-derived mesenchymal stem cells from SLE patients (SLE-BMSCs) and MRL/lpr mice, showing decreased inflammation, significant improvement of renal pathology, reduced autoantibody production, and reduced incidence of mortality compared to untreated mice [59,62]. Furthermore, assessment of wild-type and caspase-1−/− Balb/c and C57BL/6 mice treated with pristane to induce lupus showed the importance of the NLRP3 inflammasome in autoantibody production and the development of LN [63]. These studies suggested the importance of the NLRP3 inflammasome in the progression of SLE and highlight the need for NLRP3 inhibitors as potential treatments for SLE. However, there are also reports that the decreased level of NLRP3 results in the progression of disease in SLE patients as the NLRP3 pathway is associated with programmed cell death, which aids in the maintenance of homeostasis by removal of overactivated lymphocytes [64]. These studies also indicated decreased levels of NLRP3 and its inflammasome components in patients with SLE, which is contradictory to the information published by others [64,65].
3.2. Role of the NLRP3 inflammasome in Systemic Sclerosis (SSc)
Characterized by autoimmunity, vascular injury, and aggressive tissue fibrosis, SSc is an autoimmune disease mainly powered by genetic and environmental factors including exposure to viruses, silica dust, organic solvents, and chemicals. Immune dysregulation and uncontrolled inflammation play a pivotal role in the synthesis of extracellular matrix components (EMCs), e.g., collagen, whose over-accumulation leads to hardening, scarring, and deformation of tissue which ultimately leads to organ damage [66]. The overproduction of collagen in SSc has been linked to inflammasome activation, and studies have shown the increased level of NLRP3 and its downstream cytokines in SSc patients. Comparing the multiple skin layers of healthy people versus SSc patients, increased level of NLRP3 can be observed in SSc patients [67]. Additionally, studies have shown a ~7-fold increase of both NLRP3 and IL-1β gene expression levels in SSc patient-derived versus normal dermal fibroblast cell lines. Furthermore, studies have shown that pharmacological suppression of the NLRP3 inflammasome by caspase-1 inhibitor Z-YVAD(OMe)-FMK results in a decrease in the amount of collagen and stress fibers [68]. Regulated by the NLRP3-IL1 signaling, the skewing of Th1/Th2 towards Th2 plays an important role in the pathogenesis of SSc due to an increased production of pro-fibrotic cytokines in comparison to anti-fibrotic IFN-γ by Th1 cells [69]. NLRP3-mediated activation of B cells results in fibrosis and production of autoantibodies and pro-fibrotic cytokines [70,71]. This causal relationship between NLRP3 activation and the pathogenesis of SSc has also been demonstrated in mouse models treated with bleomycin to induce dermal fibrosis associated with SSc. The skin thickness in the NLRP3−/− and ASC−/− mice was significantly less than the wild-type controls and is comparable to their untreated knockout counterparts [68].
3.3. Role of the NLRP3 inflammasome in Irritable Bowel Disease (IBD)
Resulting from chronic and recurring inflammation of the gastrointestinal (GI) tract, Crohn’s disease (CD) and ulcerative colitis are the two main clinical manifestations of IBD. Development of IBD might be due to the dysfunction of the mucosal immune system of the GI tract caused by the disruption of the intestinal mucosa that keeps gut micro-organisms separated from the immune cells [72]. The permeability of the tight junctions of the mucosal membrane has been shown to be impaired under increased levels of proinflammatory cytokines, such as IL-1β and TNF-α [73], and these increased levels are attributed to the severity of inflammation in IBD [74–76]. Caspase-1 activation and maturation of IL-1β and IL-18 are thought to occur as a result of abnormal NLRP3 activation caused by NF-κB in IBD [77]. Genetic analysis of CD patients associated a single nucleotide polymorphism of the NLRP3 gene to a higher susceptibility of developing the disease [78]. However, the exact role of NLRP3 in IBD pathogenesis is not completely understood, both NLRP3’s protective and pathogenic functions have been proposed. In a study employing a dextran sodium sulfate (DSS)-induced colitis mouse model, the NLRP3−/− mice exhibited far lower clinical and histological scores associated with colitis as well as lower proinflammatory cytokine levels in the colon. Additionally, these lowered levels and scores were comparable to those of the NLRP3+/+ mice treated with a caspase-1 inhibitor, suggesting that the NLRP3 inflammasome plays a critical role in the pathology of IBD [79].
3.4. Role of the NLRP3 inflammasome in RA
Driven by pro-inflammatory cytokines, RA is a systemic polygenic autoimmune disease that causes synovial inflammation of joints resulting in cartilage and bone destruction. NLRP3, as the major regulator of the maturation of IL-1β and IL-18, has been implicated an active role in the pathogenesis of RA [80,81]. NF-κB, a critical transcriptional modulator of NLRP3 and proinflammatory cytokine expression, is constitutively activated in the synovial tissue of RA patients [82,83]. Consequently, gene expression of NLRP3 in PBMCs of RA patients, as well as other inflammasome components, has been shown to be significantly higher than in the healthy control population [84,85]. Additionally, single nucleotide polymorphisms associated with NLRP3 have been shown to modulate the susceptibility of an individual to develop RA and response to therapy [84]. Furthermore, it was determined that not only do RA patients exhibit increased NLRP3 inflammasome activity, but that upon exogenous stimulation, caspase-1 contributes most significantly to IL-1β release compared to other caspases [85]. Studies have also highlighted the role of IL-1β following pyroptosis in stimulating the priming step of the NLRP3 inflammasome formation in RA tissues, thus perpetuating the inflammatory cycle [83]. Through IL-1β, NLRP3 in synovial tissue promotes Th17 cell differentiation in CD4+ T cells which further aggravates RA by the production of TNF-α and IL-17a. Through NLRP3 activation, anti-citrullinated protein antibodies (ACPA) increase the production of IL-1 in RA patients. Additionally, genetic knock out studies in mice also suggested the crucial roles of the NLRP3 inflammasome in the pathology of arthritis [86].
4. Small molecule inhibitors of the NLRP3 inflammasome
Given the growing evidence of the NLRP3 inflammasome’s involvement in multiple pathological disorders, efforts have been dedicated to the development of small molecule inhibitors of this protein platform to achieve disease interventions. Recently, patents have been filed from both industry and academia to cover a variety of chemical scaffolds and their use. In this review, we summarized the compounds that are reported as NLRP3 inhibitors from patents published from 2016 to present. We conducted a thorough search using SciFinder and Google Patents. NLRP3 and NLRP3 inhibitors were used as the key words for this search. The list of references identified was then checked individually for patent publications. In total, forty-four patents were identified. The institutions and their related patents numbers are shown in Table 1. Additionally, the summarized NLRP3 inhibitors in preclinical or early clinical stages reported from recently filed patents and articles are illustrated in Table 2.
Table 1.
Summary of the institutions and their related patents numbers.
| Company/Institution Name | Numbers of patents |
|---|---|
| Inflazome Ltd. | 11 |
| IFM Therapeutics, Inc. | 6 |
| Janssen Pharmaceuticals | 5 |
| NodThera Ltd. | 4 |
| Novartis AG | 4 |
| The University Of Queensland | 3 |
| AC Immune SA | 1 |
| Cadila Healthcare Ltd. | 1 |
| Galderma Research and Development SNC. | 1 |
| Icahn School of Medicine at Mount Sinai. | 1 |
| Innate Tumor Immunity, Inc. | 1 |
| Jecure Therapeutics, Inc. | 1 |
| Pfizer Inc. | 1 |
| The University Of Manchester | 1 |
| University of Turin | 1 |
| Ventus Therapeutics, Inc. | 1 |
| Virginia Commonwealth University | 1 |
Table 2.
Representative NLRP3 inhibitors are in preclinical or early clinical stages reported from recently filed patents and articles.
| Inhibitor | MOAs | IC50 (IL-1β release)# | Preclinical disease models* | Development Stage |
|---|---|---|---|---|
CY-09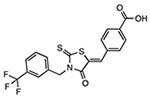
|
Binds to Walker A site in NACHT domain Inhibits ATPase activity |
6 μM | T2DM, diabetic retinopathy, diabetic liver injury, NAFLD, peritonitis, Muckle-Wells syndrome, epilepsy | Preclinical |
Tranilast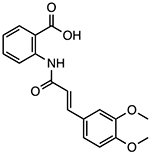
|
Disrupts NLRP3-NLRP3 oligomerization through NACHT domain binding | <25 μM | T2DM, NASH, colitis, peritonitis, Muckle-Wells syndrome, atherosclerosis | Phase II clinical trials to treat cryopin associated periodic syndrome (CAPS) (NCT03923140) Phase IV trial for percutaneous coronary intervention (NCT05130892) |
Oridonin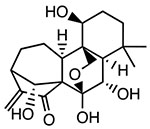
|
Covalently binds to Cys279 of NACHT domain and reduces NLRP3-NEK7 interaction | <0.5 μM | T2DM, liver fibrosis, IBD, peritonitis, hearing loss, pleurisy | Phase IV trial for percutaneous coronary intervention (NCT05130892) |
RRx-001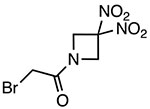
|
Covalently binds to Cys409 of NACHT domain and reduces NLRP3-NEK7 interaction | 117 nM | EAE, DSS colitis | Phase III clinical trials adjuvant treatment of small cell lung cancer with platinum (NCT03699956) |
Bay-11-7082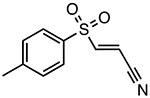
|
Inhibits ATPase activity Unknown binding site |
~3 μM | T2DM, IBD, psoriasis, lupus nephritis | Preclinical |
BOT-4-one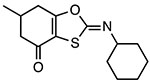
|
Inhibits ATPase activity Unknown binding site |
1.28 μM (LPS/ATP) 0.67 μM (LPS/nigericin) |
Peritonitis, dermatitis, psoriasis, arthritis | Preclinical |
OLT1177 (Dapansutrile)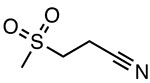
|
Inhibits ATPase activity Unknown binding site |
ND | AD, EAE, colitis, arthritis | Phase II clinical trials to treat acute gouty arthritis (EudraCT 2016-000943-14) |
INF39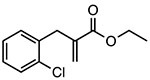
|
Covalent inhibitor of ATPase activity Unknown binding site |
~10 μM | IBD, colitis, pancreatitis | Preclinical |
JC-171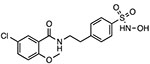
|
Reduces NLRP3-ASC interaction Unknown binding site |
8.45 μM | EAE | Preclinical |
| VTX2735 | Peripherally restricted NLRP3 inhibitor | 4 nM | N/A | Phase II clinical trial to treat cryopyrin associated periodic syndrome (NTC05812781) |
Emlenoflast (Inzomelid; MCC7840; IZD174)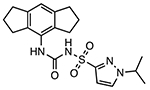
|
NLRP3 inflammasome inhibitor Unknown binding site |
4.7 nM | N/A | Phase I clinical trials for safety and tolerability (NCT04015076) Phase IIb clinical trials to treat cryopyrin associated periodic syndrome (CAPS) (EudraCT2020-000489-40) |
Selnoflast (RG6418; RO7486967)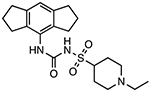
|
NLRP3 inflammasome inhibitor Unknown binding site |
≤ 1 μM (THP-1 cells) | N/A | Phase Ib clinical trials for safety and tolerability in ulcerative colitis patients (BP43099) Phase Ib clinical trials for safety and tolerability in patients with early idiopathic Parkinson’s disease (BP43176, CPMS 50823, IRAS 307220) |
IC50 values for IL-1β release inhibition in murine macrophage cells.
Abbreviations: AD: Alzheimer’s disease, PD: Parkinson’s disease, EAE: Experimental autoimmune encephalomyelitis, EAT: Experimental autoimmune thyroiditis, T2DM: Type 2 diabetes mellitus, NAFLD: Non-alcoholic fatty liver disease, NASH: Non-alcoholic steatohepatitis, IBD: Irritable bowel disease, DSS: Dextran sulfate sodium. ND: not determined.
4.1. CRID3/MCC950-based sulfonylurea, sulfonamide, and urea derivatives
Early studies from Perregaux et al. reported the inhibitory effects of glyburide, a sulfonylurea type of FDA approved antidiabetic agent, and a series of biaryl sulfonylurea compounds on the release of IL-1β induced by stimuli in human monocytes and murine macrophages [87]. These biaryl sulfonylurea compounds were termed as cytokine release inhibitory drugs (CRIDs). Studies from Genentech demonstrated inhibition of the NLRP3 inflammasome as the mechanism of action (MOA) for glyburide’s anti-inflammatory effects [88]. However, further studies of the CRIDs initially suggested glutathione S-transferase omega-1 (GSTO1) as the molecular target for these compounds. CRID3 (also known as CP-456773 and MCC950) was used as a competitor for the affinity chromatography experiments in this study from which the GSTO1 was eluted. Although further studies by Coll et al. suggested that CRID3 targets the NLRP3 and AIM2 inflammasomes, later studies from the same group demonstrated CRID3 as a potent NLRP3 inhibitor and this compound was re-termed as MCC950 [89–90]. To credit the original inventor and to reflect the current term use in most of the literature, we use CRID3/MCC950 for this compound in the rest of the manuscript. Recently, mechanistic studies from two groups demonstrated that CRID3/MCC950 functions as a direct NLRP3 inhibitor by binding to the NACHT domain of NLRP3 [91–93]. Recent structural biology studies supported this conclusion by revealing that CRID3/MCC950 interacts with multiple sites within the NACHT domain, including the nucleotide binding domain (NBD), helical domain 1 (HD1), winged-helix domain (WHD), and helical domain 2 (HD2). The unique binding by CRID3/MCC950 locks NLRP3 in an inactive state, therefore preventing the assembly of the inflammasome [94]. Since the discovery of CRID3/MCC950 as a potent NLRP3 inhibitor, this compound has been extensively tested in multiple disease models including autoimmune diseases. CRID3/MCC950 showed good skin penetration and tolerance for the treatment and/or prevention of an inflammatory skin disorder, such as atopic dermatitis and psoriasis [95]. In EAE (experimental autoimmune encephalomyelitis) mice, an experimental mouse model of MS, treatment with CRID3/MCC950 alleviated cognitive deficits, hippocampal pathology and synaptic loss [96]. CRID3/MCC950 has also been shown to significantly reduce infiltrating T cells and proinflammatory cytokines, e.g., IL-17a, in experimental autoimmune thyroiditis (EAT) mice [97]. Furthermore, pharmacological suppression of the NLRP3 inflammasome by CRID3/MCC950 led to significant improvement in cognitive function and insulin sensitivity in the diabetic encephalopathy mice [98]. Given the promising results obtained from studies using CRID3/MCC950 as a tool compound, several patents based on the CRID3/MCC950 scaffold have been reported to cover a series of analogs. The structure of CRID3/MCC950 consists of a central sulfonylurea moiety flanked with a dimethyl-3-furanmethanol moiety and a 1,2,3,5,6,7-hexahydro-s-indacene moiety (Fig. 3). The derivatives of CRID3/MCC950 covered in these patents are compounds with modifications on these three moieties of CRID3/MCC950.
Figure 3.
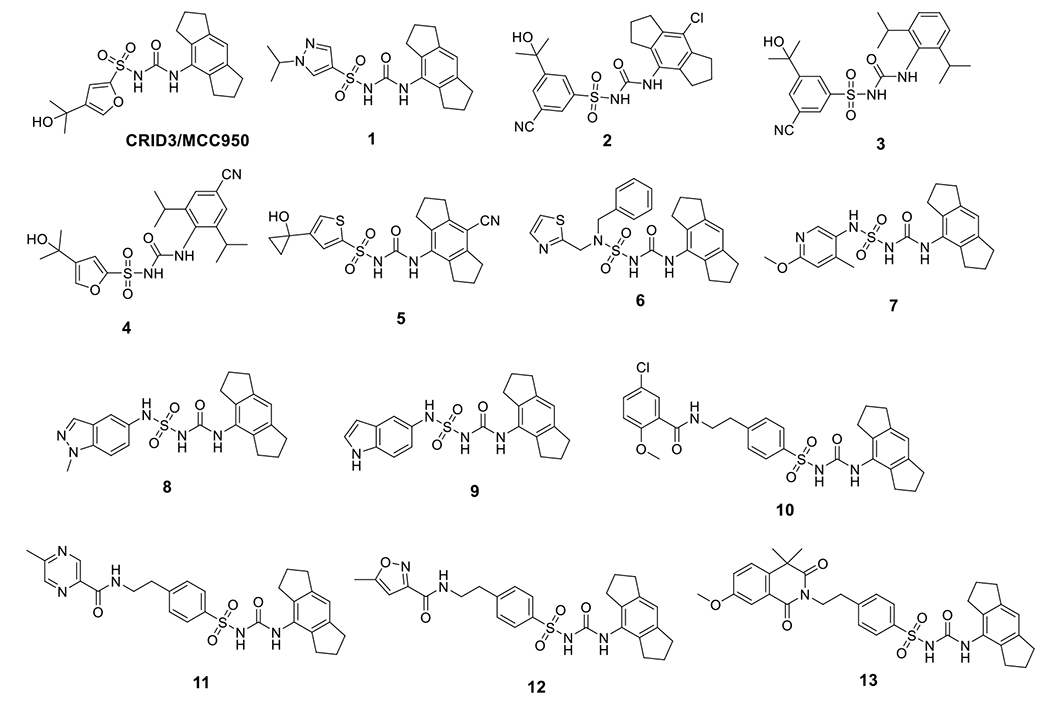
Chemical structures of CRID3/MCC950 and sulfonylurea derivatives.
4.1.1. Sulfonylurea derivatives
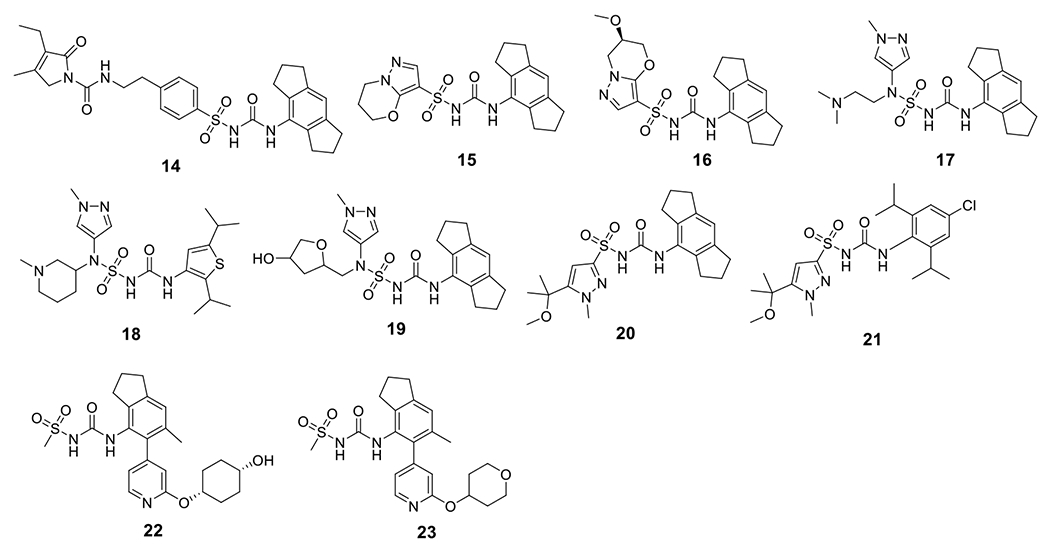
Similar to CRID3/MCC950, compound 1 showed potent inhibition on IL-1β release with an IC50 of 11 nM in human monocyte-derived macrophages (HMDM) [99]. A structurally similar compound, emlenoflast (also known as MCC7840, IZD174, or inzomelid) (Table 2), was reported in the same patent with an IC50 of 13 nM in HMDM cells [99] and an IC50 of 4.7 nM in murine microglia under LPS/ATP stimulation conditions [100]. Emlenoflast was originally developed by the University of Queensland and licensed to Inflamzome Ltd.. This compound has entered Phase IIb clinical trials to treat cryopyrin associated periodic syndrome (EudraCT2020-000489-40) [101–103]. Inflazome Ltd., merged with F. Hoffman-La Roche Ltd. in 2020 [104], also developed selnoflast (also known as RG6418 or RO7486967) with an IC50 of ≤ 1 μM for IL-1β inhibition in THP-1 cells [105], and has since entered Phase Ib clinical trials to treat patients with ulcerative colitis [106], as well as patients with early idiopathic Parkinson’s disease [107]. IFM Therapeutics Inc. designed a series compounds as exemplified by 2-5 showing an IC50 < 1 μM on inhibition of IL-1β release in THP-1 cells [108,109]. Pyroptosis assays using human THP-1 cells demonstrated the protective effects of compounds 6-9 with IC50 < 1 μM [110]. Structural extension delivered compounds 10-14 that exhibited similar potency with IC50 < 1 μM to inhibit the production of IL-1β in BMDM cells under the stimulation of LPS/ATP [111]. Jecure Therapeutics Inc., acquired by F. Hoffman-La Roche Ltd. in 2018 [112], reported a series of pyrazolo-oxazine compounds (15 and 16, Fig. 3) and showed inhibitory effects by these compounds on the production of IL-1β and pyroptosis in human Kupffer cells (KC). In a C57BL/6 mouse model, a single oral dose of compound 15 at 1.5 mg/kg, and compound 16 at 1 mg/kg reduced the release of IL-1β induced by LPS/ATP by > 98% [113]. Replacement of the furan moiety and the 1,2,3,5,6,7-hexahydro-s-indacene in CRID3/MCC950 with heterocycles yielded compounds 17-19 that retained the inhibitory effects on IL-1β production [114,115]. Inflazome Ltd. also developed pyrazole-containing compounds, exemplified by compounds 20 and 21, to show inhibitory effects on the NLRP3 inflammasome with improved oral bioavailability [116]. Removal of the furan moiety and replacement of the 1,2,3,5,6,7-hexahydro-s-indacene moiety with a bi-aryl ring system in CRID3/MCC950 led to compounds 22 and 23 that showed potent inhibition of the NLRP3 inflammasome [117]. The change of the tricyclic indacene moiety to a bi-aryl ring system might be intended to explore the chemical space within NLRP3 where CRID3/MCC950 binds to.
4.1.2. Acyl sulfonamide and Acyl sulfoximine compounds
Novartis reported a series of compounds that contain an acyl sulfonamide moiety, among which compounds 24-26 showed potent inhibition on IL-1β release with an IC50 < 1 μM upon stimulation by gramicidin in PMA-differentiated THP-1 cells (Fig. 4) [118]. IFM Therapeutics, Inc., acquired by Bristol-Myers Squibb in 2017 [119], developed compounds 27-30 with comparable inhibitory potency under the same assay conditions [120]. Further modifications led to compounds 31-33, and the biological results suggested that the aryl sulfonamide moiety is dispensable to retain the activities as NLRP3 inhibitors [121]. However, as evidenced by compounds 34 and 35 in a following study, the aryl sulfonamide moiety might be important to provide favorable pharmacokinetic (PK) properties [122]. Change of the sulfonylurea to a sulfoximine led to improved inhibitory potency on the NLRP3 inflammasome as reflected by 36-39 [123]. Similarly, compounds 40-43 also exhibited single digit nanomolar potency to inhibit the NLRP3 inflammasome [124]. Combination of urea and sulfoximine into a sulfonimidamide moiety also led to active compounds as NLRP3 inflammasome inhibitors (44-49) [125]. A potential covalent alkenyl sulfonylurea derivative, 50, developed by Zydus Lifesciences Ltd. (originally known as Candila Healthcare Ltd.) [126], demonstrated promising potency both in vitro and in vivo [127]. Interestingly, Inflazome Ltd. designed and reported macrocyclic 1,3,4-triazole and 3-pyrrazole sulfonamide, sulfonylurea and acyl sulfonamide analogs as evidenced by 51-59 as NLRP3 inhibitors. The compounds seem to be weaker inhibitors with sub-micromolar potency (Fig. 5) [128–130]. Although the rationale behind such design was not disclosed, it might be intended to explore the effects of conformational constrain on the biological activities of these scaffolds.
Figure 4.
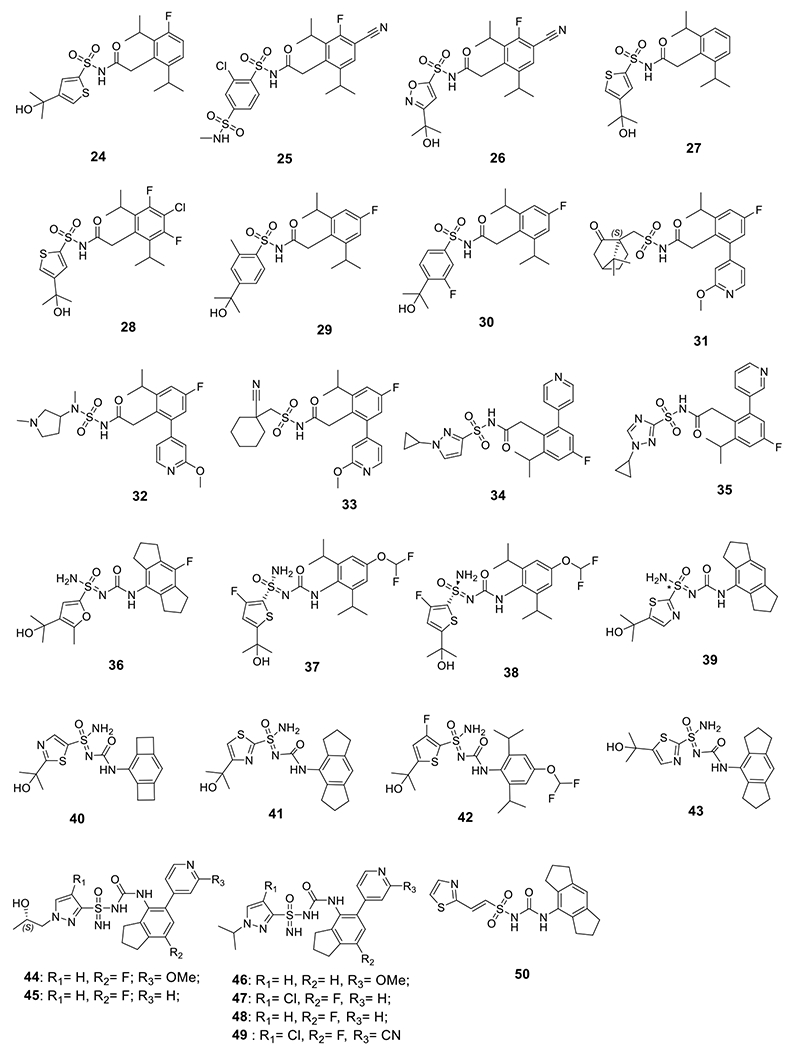
Chemical structures of sulfonamide, sulfoximine, sulfonimidamide, and alkenyl sulfonylurea compounds.
Figure 5.
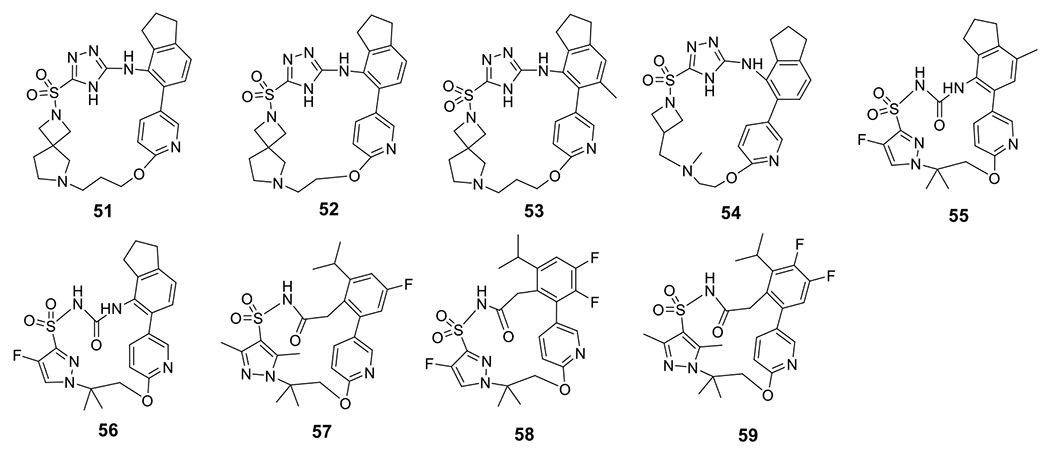
Chemical structures of macrocyclic sulfonyl triazole and sulfonamide compounds.
4.1.3. Replacement of sulfonylurea moiety with a heterocyclic ring
NodThera Ltd. reported small molecule NLRP3 inhibitors based on a thiazole scaffold (Fig. 6). Among them, 60 was found to be active with an IC50 < 1 μM for the inhibition of IL-1β release in PBMC cells under stimulation by LPS/ATP or LPS/nigericin [131]. Replacement of the thiazole moiety with a 1,3,4-triazole, as observed in the patent by Inflazome Ltd., led to compounds 61 and 62 with IC50 ≤ 2 μM under stimulation by LPS/nigericin in THP-1 cells [132]. Among the inhibitors reported by AC Immune SA, compounds 63-65 are the most potent inhibitors with an IC50 < 1 μM. These compounds were also demonstrated with in vivo efficacy to suppress IL-1β by 99%, 73%, >99%, respectively, in an LPS/ATP induced peritonitis mouse model [133].
Figure 6.
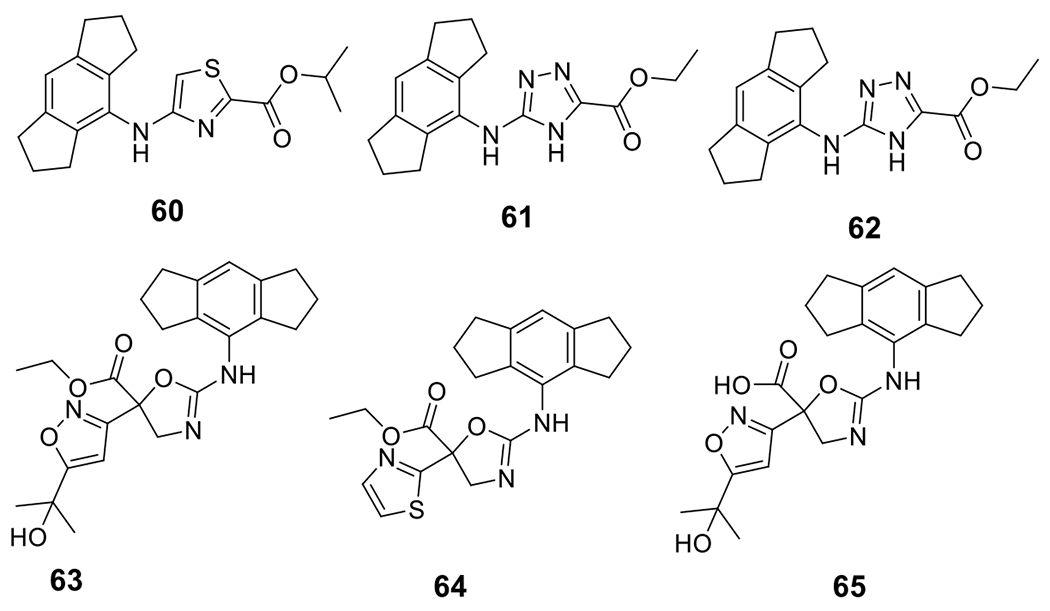
Chemical structures of thiazole, triazole, and dihydroxazole compounds.
5. Non-MCC950-based NLRP3 inflammasome inhibitors
Janssen Pharmaceuticals reported SAR studies based on a pyridazine scaffold and compounds 66-73 were identified as active NLRP3 inhibitors (Fig. 7). While maintaining potent inhibition on the IL-1β release (IC50 < 0.03 μM) induced by LPS, they also demonstrated good selectivity with no significant inhibition on TNF-α production (IC50 > 10 μM) [134–138]. Novartis AG disclosed novel tricyclic thienopyrrolotriazinacetamide compounds 74 and 75 with an IC50 of 0.4 nM and 0.7 nM, respectively, against IL-1β release induced by nigericin in THP-1 cells. Notably, no inhibition on TNF-α was observed even at much higher concentrations (> 100 μM) [139].
Figure 7.
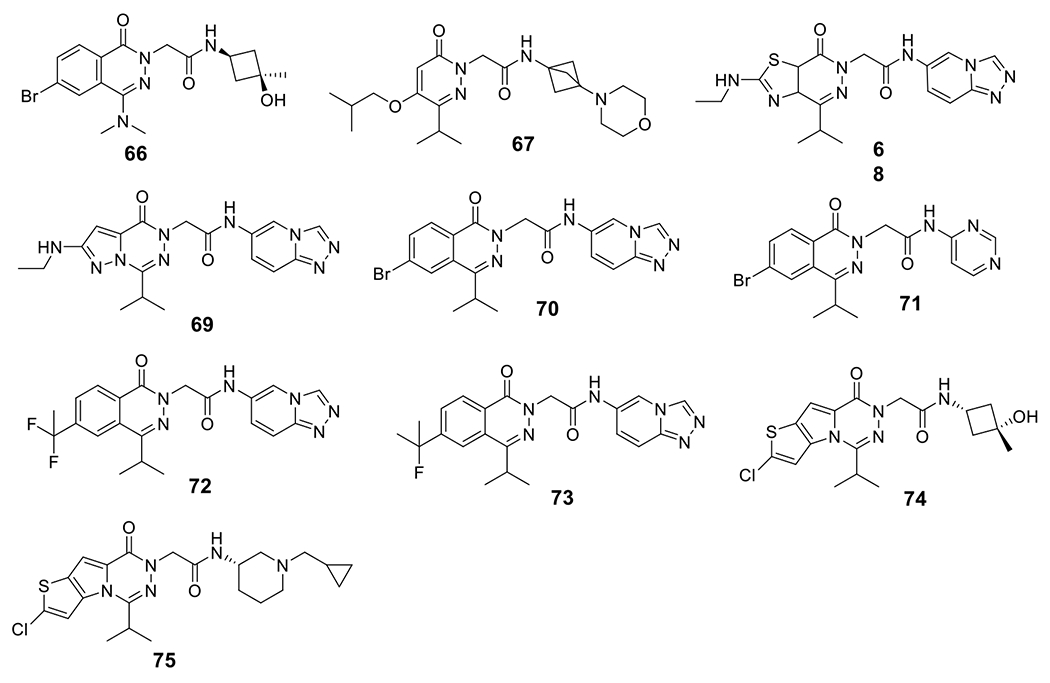
Chemical structures of pyridazine and thienopyrrolotriazinacetamide compounds.
Compounds based on an imidazo-quinoline scaffold were disclosed by IFM Therapeutics, among those compounds, 76-85 showed activity as hNLRP3 agonists with an EC50 ≤ 1 μM in PMA-differentiated THP-1 cells. Notably, these compounds showed no activity on the activation of TLR7 and TLR8 with an EC50 > 100 μM [140,141]. Interestingly, compounds 86 and 87 showed potent activation of the NLRP3 inflammasome with an EC50 of 0.19 μM and 0.11 μM, respectively [142]. (Fig. 8) This clearly suggests the versatility of this chemical scaffold to provide novel NLRP3 agonists.
Figure 8.
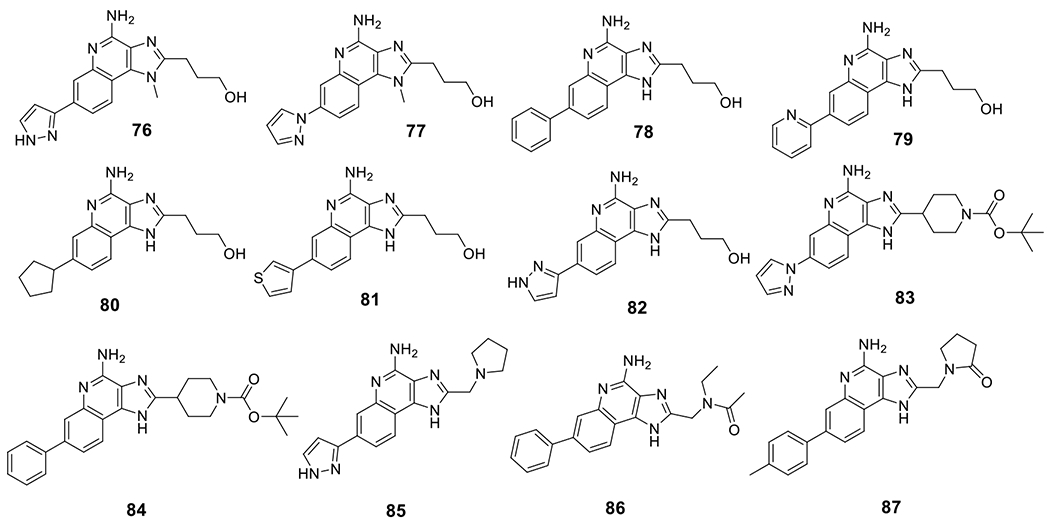
Chemical structures of NLRP3 inflammasome agonists with imidazo-quinoline scaffold.
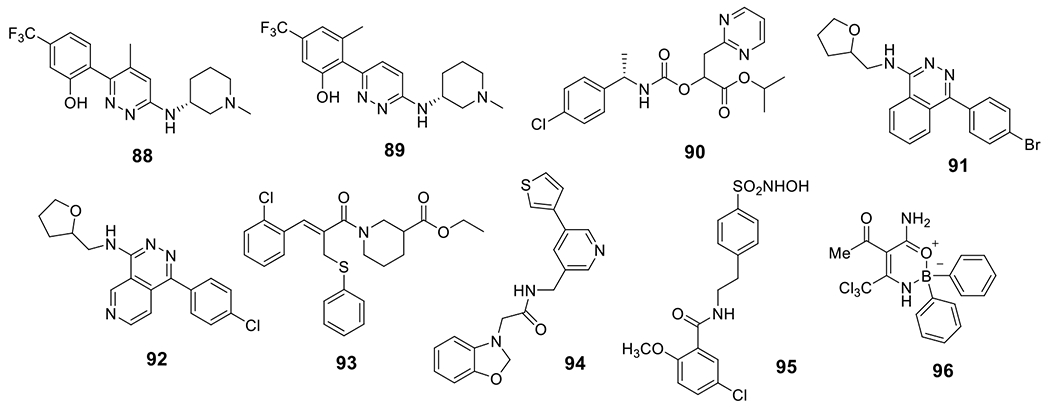
Novartis AG disclosed novel pyridazin-3-yl phenol compounds as NLRP3 inhibitors (Fig. 9). Among the reported compounds, 88 and 89 exhibited potent inhibition on the production of IL-1β in PMA-differentiated THP-1 cells with an IC50 of 4 nM and 6 nM, respectively, while no inhibition on TNF-α secretion was observed (IC50 > 100 μM) [143]. Carbamate containing compounds, exemplified by compound 90, were also disclosed by NodThera Ltd. as active NLRP3 inhibitors [144]. Ventus Therapeutics recently discovered bicyclic pyridazine analogs as NLRP3 inhibitors, and among them, compounds 91 and 92 showed moderate inhibitory potency [145]. Sulfide containing compounds developed by the University of Turin were also reported. Compound 93 was demonstrated to show in vivo efficacy in a DSS-induced colitis animal model to reduce intestinal inflammation [146]. Mount Sinai reported benzoxazolone-containing compounds, and 94 is the most potent within this series and notably exhibited dual inhibition on the NLRP3 and NLRC4 inflammasomes. Mechanistically, this compound may compete with ATP for the ATP binding site within the NLRP3 and block ASC oligomerization. Compound 94 was also demonstrated with in vivo activity to reduce IL-1β in the brains of LPS-treated mice [147]. Our lab recently developed sulfonamide analogs from the structure of glyburide and demonstrated their activity as NLRP3 inhibitors. Among the compounds reported, compound 95 dose dependently inhibited IL-1β release with an IC50 of 8.45 μM in J774A.1 cells under the stimulation by LPS/ATP. Notably, this compound selectively engaged the inflammasome in vivo and exhibited comparable efficacy as CRID3/MCC950 in an EAE mouse model to improve the clinical score and to suppress the pathology [148,149]. A series of cyclic diarylboron derivatives were disclosed by the University of Manchester, among which compound 96 was identified as the most potent one with an IC50 of 0.54 μM to inhibit the production of IL-1β under stimulation conditions by LPS/nigericin in THP-1 cells [150].
Figure 9.
Chemical structures of pyridazine-3-yl phenol, benzoxazolone, and other associated compounds.
Conclusion
Dysregulation of the NLRP3 inflammasome has been linked to the development of inflammatory and autoimmune disorders, such as SSc, SLE, IBD, and RA. As a result, small molecule inhibitors to target the NLRP3 inflammasome have been actively pursued to achieve disease interventions. Since the discovery of CRID3/MCC950 as a direct NLRP3 inhibitor and the demonstrated efficacy of this compound in a variety of preclinical disease models, many NLRP3 inflammasome inhibitors have been developed and patented recently.
Overall, the majority of the reported compounds are based on the sulfonylurea skeleton found in CRID3/MCC950. Notably, most of the derivatives are able to inhibit the release of IL-1β upon activation of the NLRP3 inflammasome under various experimental conditions. Interestingly, structural modifications of the sulfonylurea moiety led to compounds with a wide range of potency from high micromolar to single digit nanomolar potency, highlighting the importance of the sulfonylurea moiety in interacting with the NLRP3 protein, consistent with the structural biology study results.
New chemical scaffolds that are completely free of the sulfonylurea moiety, evidenced by the pyridazine and carbamate analogs, were also reported as NLRP3 inflammasome inhibitors with significantly improved potency. It is worth noting that most of the patented compounds exhibit selectivity for inhibiting the production of IL-1β, without affecting the levels of TNF-α.
Expert opinion
Small molecule inhibitors of the NLRP3 inflammasome have gained growing attention as potential therapeutics. Meanwhile, accumulating evidence indicates the vital role of NLRP3 inflammasome in the pathogenesis and development of several autoimmune diseases, such as SSc, SLE, IBD, and RA. A review of the patent landscape in this area reveals a notable increase in the number of patents filed as NLRP3 inhibitors in recent years. Some of these compounds are currently in clinical trials. CRID3/MCC950, a widely used tool compound, has shown efficacy in a variety of preclinical models of autoimmune diseases. Treatment with CRID3/MCC950 has been shown to reduce the severity of the disease in animal models of gout and MS. However, CRID3/MCC950 displayed significant hepatotoxicity in phase II clinical trials as RA treatment [151,152]. Over the past few years, numerous compounds based on the CRID3/MCC950 scaffold have been developed and tested as NLRP3 inhibitors. In most of the cases, inhibition of activation and subsequent release of downstream cytokines, e.g., IL-1β, was reported. Combination therapy using NLRP3 inhibitors together with TNF-α inhibitors or IL-1 inhibitors has been the subject of several patent applications.
With the availability of structural biology information for the inactive and active states of NLRP3, as well as the cryo-EM structure of NLRP3 in complex with small molecule inhibitors, the development of novel NLRP3 inhibitors will continue to evolve. It is also hoped that, in the near future, the availability of small molecule NLRP3 inhibitors with different MOAs will complement the molecular and genetic studies to help elucidate the pathological roles of the NLRP3 inflammasome in human diseases including the autoimmune diseases. In addition, the recent interests in developing inhibitors of ASC, one of the components of the NLRP3 inflammasome, will be expanding the avenue to facilitate drug discovery efforts by targeting the inflammasome.
Despite the promising preclinical results, clinical trials of small molecule NLRP3 inhibitors for autoimmune diseases are still in their early stages, and further investigations are needed to fully assess their safety and validate their efficacy in human beings. There are several challenges that need to be considered in the future development of NLRP3 inhibitors. Firstly, possible immune suppression by the NLRP3 inhibitors may increase the chance of infections. Although the clinical success of IL-1β based treatments may alleviate the concerns, clinical evidence is still needed to prove the safety of such inhibitors. Secondly, the complexity of this protein platform and its activation under a plethora of conditions warrant biomarker and target engagement confirmation to facilitate clinical validation of NLRP3 inhibitors. In this regard, it is promising to see the recent development of imaging tools based on some of the NLRP3 inhibitors. Lastly, it is possible that not all patients with autoimmune disorders may benefit from NLRP3 suppression by the inhibitors due to the complex interplay between the innate and adaptive immune responses. Studies in understanding the cellular and molecular mechanisms underlying the association of the NLRP3 inflammasome with autoimmune diseases are still needed to help the drug discovery efforts. Although challenges exist down the road in developing NLRP3 inhibitors as effective treatments for autoimmune diseases, the results and data from preclinical and clinical studies of NLRP3 inhibitors are encouraging, and we may see breakthroughs in the near future to validate NLRP3 as a druggable target and successful trials with expected outcomes.
Article highlights.
The review provided a comprehensive examination of the NLRP3 inflammasome, including its architecture, activation, and role in autoimmune diseases.
The review provided a coverage of the patents on NLRP3 inflammasome inhibitors from 2016-present. Their efficiencies in preclinical disease models are also discussed.
We comprehensively reviewed the inhibitors based on different chemical scaffolds and their SARs from published patents.
The potential applications and mechanism of action of MCC950 in treating autoimmune diseases are discussed in detail.
Funding
The work was supported in part by the NIA of the NIH under award number U01AG076481 and RF1AG076912 (SZ), Commonwealth of Virginia’s Alzheimer’s and Related Disease Research Award Fund administered by the Center on Aging, School of Allied Health Professions, Virginia Commonwealth University (SZ).
Footnotes
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010; 464:1367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140:821–32. [DOI] [PubMed] [Google Scholar]
- 3.Fullard N, O Reilly S. Role of innate immune system in systemic sclerosis. Semin Immunopathol 2015; 37:511–7. [DOI] [PubMed] [Google Scholar]
- 4.Alexandre YO, Cocita CMD, Ghilas S, Dalod M. Deciphering the role of DC subsets in MCMV infection to better understand immune protection against viral infections. Front Microbiol 2014; 5:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourgeois C, Kuchler K. Fungal pathogens-a sweet and sour treat for toll-like receptors. Front Cell Infect Microbiol 2012; 2:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dzopalic T, Rajkovic I, Dragicevic A, Colic M. The response of human dendritic cells to co-ligation of pattern-recognition receptors. Immunol Res 2012; 52:20–33. [DOI] [PubMed] [Google Scholar]
- 7.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002; 10:417–26. [DOI] [PubMed] [Google Scholar]
- 8.Gentile LF, Cuenca AL, Cuenca AG, Nacionales DC, Ungaro R, Efron PA, et al. Improved emergency myelopoiesis and survival in neonatal sepsis by caspase-1/11 ablation. Immunology 2015; 145:300–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev 2015; 265:130–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanders MG, Parsons MJ, Howard AGA, Liu J, Fassio SR, Martinez JA, et al. Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death Dis 2015; 6:e1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res 2015; 8:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue M, Shinohara ML. The role of interferon-β in the treatment of multiple sclerosis and experimental autoimmune encephalomyelitis - in the perspective of inflammasomes. Immunology 2013; 139:11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eigenbrod T, Dalpke AH. Bacterial RNA: an underestimated stimulus for innate immune responses. J Immunol 2015; 195:411–8. [DOI] [PubMed] [Google Scholar]
- 14.Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun 2015; 6:7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inoue M, Shinohara ML. NLRP3 inflammasome and MS/EAE. Autoimmune Dis 2013; 2013:859145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183:787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franchi L, Eigenbrod T, Núñez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 2009; 183:792–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, Mcgettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013; 496:238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zangiabadi S, Abdul-Sater AA. Regulation of the NLRP3 Inflammasome by Posttranslational Modifications. J Immunol 2022; 208:286–292. [DOI] [PubMed] [Google Scholar]
- 20.Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 1994; 269: 15195–203. [PubMed] [Google Scholar]
- 21.Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996; 272:735–8. [DOI] [PubMed] [Google Scholar]
- 22.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013; 38:1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murakami T, Ockinger J, Yu J, Byles V, Mccoll A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci 2012; 109:11282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Domingo-Fernández R, Coll RC, Kearney J, Breit S, O’Neill LAJ. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1β transcription and activate the NLRP3 inflammasome. J Biol Chem 2017; 292:12077–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. Clics-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun 2017; 8:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221–5. [DOI] [PubMed] [Google Scholar]
- 28.Sanman LE, Qian Y, Eisele NA, Ng TM, van der Linden WA, Monack DM, et al. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 2016; 5:e13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011; 12:408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J, Chen ZJ. Ptdins4p on dispersed trans-golgi network mediates NLRP3 inflammasome activation. Nature 2018; 564:71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, Chen ZJ. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014; 156: 1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schröder GF, Fitzgerald KA, Wu H, Egelman EH. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014; 156:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidt FI, Lu A, Chen JW, Ruan J, Tang C, Wu H, Ploegh HL. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J Exp Med 2016; 213:771–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med 2018; 215:827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019; 19:477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514:187–92. [DOI] [PubMed] [Google Scholar]
- 37.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016; 535:111–6. [DOI] [PubMed] [Google Scholar]
- 38.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016; 535:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the Pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 2015; 43:923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity 2016; 44:833–46. [DOI] [PubMed] [Google Scholar]
- 41.Shalhoub J, Falck-Hansen MA, Davies AH, Monaco C. Innate immunity and monocyte-macrophage activation in atherosclerosis. J Inflamm (Lond) 2011; 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mccall SH, Sahraei M, Young AB, Worley CS, Duncan JA, Ting JP, et al. Osteoblasts express nlrp3, a nucleotide-binding domain and leucine-rich repeat region containing receptor implicated in bacterially induced cell death. J Bone Miner Res 2008; 23:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem 2007; 55:443–52. [DOI] [PubMed] [Google Scholar]
- 44.Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum 2011; 63:3563–74. [DOI] [PubMed] [Google Scholar]
- 45.Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol 2011; 186:5738–48. [DOI] [PubMed] [Google Scholar]
- 46.Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-Mccrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol 2018; 15:458–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Artlett CM, Thacker JD. Molecular activation of the NLRP3 inflammasome in fibrosis: common threads linking divergent fibrogenic diseases. Antioxid Redox Signal 2015; 22:1162–75. [DOI] [PubMed] [Google Scholar]
- 48.La Cava A Tregs are regulated by cytokines: implications for autoimmunity. Autoimmun Rev 2008; 8:83–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A 2009; 106:7119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009; 30:576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol 2011; 187:6143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29:301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009; 27:519–50.. [DOI] [PubMed] [Google Scholar]
- 54.Okamura H, Nagata K, Komatsu T, Tanimoto T, Nukata Y, Tanabe F, Akita K, Torigoe K, Okura T, Fukuda S, et al. A novel costimulatory factor for gamma interferon induction found in the livers of mice causes endotoxic shock. Infect Immun 1995; 63:3966–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev 2001; 12:53–72. [DOI] [PubMed] [Google Scholar]
- 56.Oliveira CB, Lima CAD, Vajgel G, Sandrin-Garcia P. The role of NLRP3 inflammasome in lupus nephritis. Int J Mol Sci 2021; 22:12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen H, Yang Y, Meng X, Luo X, Li X, Shuai Z, et al. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun Rev 2018; 17:694–702. [DOI] [PubMed] [Google Scholar]
- 58.Da Cruz HLA, Cavalcanti CAJ, de Azêvedo Silva J, de Lima CAD, Fragoso TS, Barbosa AD, et al. Differential expression of the inflammasome complex genes in systemic lupus erythematosus. Immunogenetics 2020; 72:217–24. [DOI] [PubMed] [Google Scholar]
- 59.Tan W, Gu Z, Leng J, Zou X, Chen H, Min F, et al. Let-7f-5p ameliorates inflammation by targeting NLRP3 in bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Biomed Pharmacother 2019; 118:109313. [DOI] [PubMed] [Google Scholar]
- 60.Yang CA, Huang ST, Chiang BL. Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatology (Oxford) 2015; 54:324–31. [DOI] [PubMed] [Google Scholar]
- 61.Honarpisheh M, Desai J, Marschner JA, Weidenbusch M, Lech M, Vielhauer V, et al. Regulated necrosis-related molecule mRNA expression in humans and mice and in murine acute tissue injury and systemic autoimmunity leading to progressive organ damage, and progressive fibrosis. Biosci Rep 2016; 36:e00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao J, Zhang H, Huang Y, Wang H, Wang S, Zhao C, et al. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-κB activation. Int Immunopharmacol 2013; 17:116–22. [DOI] [PubMed] [Google Scholar]
- 63.Kahlenberg JM, Yalavarthi S, Zhao W, Hodgin JB, Reed TJ, Tsuji NM, et al. An essential role of caspase 1 in the induction of murine lupus and its associated vascular damage. Arthritis Rheumatol 2014; 66:152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang Q, Yu C, Yang Z, Wei Q, Mu K, Zhang Y, Zhao W, Wang X, Huai W, Han L. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematosus. J Rheumatol 2014; 41:444–52. [DOI] [PubMed] [Google Scholar]
- 65.Ma ZZ, Sun HS, Lv JC, Guo L, Yang QR. Expression and clinical significance of the NEK7-NLRP3 inflammasome signaling pathway in patients with systemic lupus erythematosus. J Inflamm (Lond). 2018; 15:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008; 214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martínez-Godínez MA, Cruz-Domínguez MP, Jara LJ, Domínguez-López A, Jarillo-Luna RA, Vera-Lastra O, Montes-Cortes DH, Campos-Rodríguez R, López-Sánchez DM, Mejía-Barradas CM, Castelán-Chávez EE, Miliar-García A. Expression of NLRP3 inflammasome, cytokines and vascular mediators in the skin of systemic sclerosis patients. Isr Med Assoc J 2015; 17:5–10. [PubMed] [Google Scholar]
- 68.Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum 2011; 63:3563–74. [DOI] [PubMed] [Google Scholar]
- 69.Birnhuber A, Crnkovic S, Biasin V, Marsh LM, Odler B, Sahu-Osen A, et al. IL-1 receptor blockade skews inflammation towards Th2 in a mouse model of systemic sclerosis. Eur Respir J 2019; 54:1900154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Worrell JC, O’Reilly S. Bi-directional communication: conversations between fibroblasts and immune cells in systemic sclerosis. J Autoimmun 2020; 113:102526. [DOI] [PubMed] [Google Scholar]
- 71.Lin C, Jiang Z, Cao L, Zou H, Zhu X. Role of NLRP3 inflammasome in systemic sclerosis. Arthritis Res Ther 2022; 24:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhen Y, Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front Immunol 2019;10:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol 2007; 178:4641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishiguro Y Mucosal proinflammatory cytokine production correlates with endoscopic activity of ulcerative colitis. J Gastroenterol 1999; 34:66–74. [DOI] [PubMed] [Google Scholar]
- 75.Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R, et al. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol 1999; 163:143–7. [PubMed] [Google Scholar]
- 76.Sartor RB. Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology 1994; 106:533–9. [DOI] [PubMed] [Google Scholar]
- 77.Liu L, Li X. NLRP3 inflammasome in inflammatory bowel disease: friend or foe? Dig Dis Sci 2017; 62:2211–4. [DOI] [PubMed] [Google Scholar]
- 78.Villani A, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to crohn’s disease susceptibility. Nat Genet 2009; 41:71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (dss) is mediated by the NLRP3 inflammasome. Gut 2010; 59:1192–9. [DOI] [PubMed] [Google Scholar]
- 80.Makkar R, Behl T, Bungau S, Kumar A, Arora S. Understanding the role of inflammasomes in rheumatoid arthritis. Inflammation 2020; 43:2033–47. [DOI] [PubMed] [Google Scholar]
- 81.Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol 2018; 194:231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Handel ML, McMorrow LB, Gravallese EM. Nuclear factor-kappa B in rheumatoid synovium. Localization of p50 and p65. Arthritis Rheum 1995; 38:1762–70. [DOI] [PubMed] [Google Scholar]
- 83.Georganas C, Liu H, Perlman H, Hoffmann A, Thimmapaya B, Pope RM. Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: the dominant role for NF-kappa B but not C/EBP beta or c-Jun. J Immunol 2000; 165:7199–206. [DOI] [PubMed] [Google Scholar]
- 84.Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Eyre S, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis 2014; 73:1202–10. [DOI] [PubMed] [Google Scholar]
- 85.Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, et al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther 2015; 17:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014; 512:69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pfizer Inc. Efficient synthesis of furan sulfonamide compounds useful in the synthesis of new IL-1 inhibitors. US6022984A. 2000.
- 88.Perregaux DG, Mcniff P, Laliberte R, Hawryluk N, Peurano H, Stam E, et al. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J Pharmacol Exp Ther 2001; 299:187–97. [PubMed] [Google Scholar]
- 89.Laliberte RE, Perregaux DG, Hoth LR, Rosner PJ, Jordan CK, Peese KM, et al. Glutathione s-transferase omega 1-1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1beta posttranslational processing. J Biol Chem 2003; 278:16567–78. [DOI] [PubMed] [Google Scholar]
- 90.Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, de Torre-Minguela C, Cerón-Carrasco JP, Pérez-Sánchez H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol 2019; 15:560–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, de Torre-Minguela C, Cerón-Carrasco JP, Pérez-Sánchez H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol 2019; 15:560–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 2019; 15:556–9. [DOI] [PubMed] [Google Scholar]
- 94.Hochheiser IV, Pilsl M, Hagelueken G, Moecking J, Marleaux M, Brinkschulte R, et al. Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature 2022; 604:184–9. [DOI] [PubMed] [Google Scholar]
- 95.Galderma Research and Development SNC. NLRP3 inhibitors for the treatment of inflammatory skin disorders. US20190192478A1. 2019. [Google Scholar]
- 96.Hou B, Zhang Y, Liang P, He Y, Peng B, Liu W, et al. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis 2020; 11:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ding H, Sun X, Xu H. Pharmacological suppression of NLRP3 inflammasome attenuated the development of autoimmune thyroiditis. Cell Immunol 2023; 384:104659. [DOI] [PubMed] [Google Scholar]
- 98.Zhai Y, Meng X, Ye T, Xie W, Sun G, Sun X. Inhibiting the NLRP3 inflammasome activation with MCC950 ameliorates diabetic encephalopathy in db/db mice. Molecules 2018; 23:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.The University Of Queensland. Sulfonylureas and related compounds and use of same. WO2016131098A1. 2016.
- 100.Inflazome Ltd. Treatment or prevention of psychiatric brain disorders using the NLRP3 inhibitor n-((1,2,3,5,6,7-hexahydro-s-indacen-4-yl)carbamoyl)-1-isopropyl-1 h-pyrazole-3-sulfonamide WO2021089781A1. 2021.
- 101. Available from: https://www.alzforum.org/therapeutics/inzomelid.
- 102. Available from: https://clinicaltrials.gov/ct2/show/NCT04015076.
- 103. Available from: https://www.clinicaltrialsregister.eu/ctr-search/trial/2020-000489-40/GB#P.
- 104. Available from: https://cen.acs.org/business/mergers-&-acquisitions/Roche-buys-Inflazome-445-million/98/i37.
- 105.Inflazome Ltd. Novel sulfonamide carboxamide compounds. WO2019008025A1. 2019.
- 106. Available from: https://www.isrctn.com/ISRCTN16847938.
- 107. Available from: https://www.isrctn.com/ISRCTN85338453.
- 108.IFM Therapeutics, Inc. Compounds and compositions for treating conditions associated with nlrp activity. WO 2017/184623 A1. 2017. [Google Scholar]
- 109.IFM Therapeutics, Inc. Compounds and compositions for treating conditions associated with nlrp activity. WO 2017/184624 A1. 2017.
- 110.The University Of Queensland. Sulfonylureas and related compounds and use of same. WO 2017/140778 A1. 2017.
- 111.The University Of Queensland. Novel compounds and uses. WO 2018/215818 A1. 2018.
- 112. Available from: https://www.businesswire.com/news/home/20181127005232/en/Genentech-Acquires-Jecure-Therapeutics.
- 113.Jecure Therapeutics Inc. Chemical compounds as inhibitors of interleukin-1 activity. WO2018136890A1. 2018.
- 114.Nodthera Ltd. Sulfonylurea derivatives and uses thereof. WO2020249667A1. 2020. [Google Scholar]
- 115.Nodthera Ltd. Sulphamoyl urea derivatives containing alkyl-oxacycloalkyl moiety and uses thereof. WO2022051582A1. 2022.
- 116.Inflazome Ltd. NLRP3 inhibitors. WO2020104657A1. 2020.
- 117.Inflazome Ltd. Novel sulfoneurea compounds. WO2020035466A1. 2020.
- 118.Novartis inflammasome research Inc. Compounds and compositions for treating conditions associated with nlrp activity. WO2020086728A1. 2020.
- 119.Available from: https://news.bms.com/news/details/2017/Bristol-Myers-Squibb-Completes-Previously-Announced-Acquisition-of-IFM-Therapeutics/default.aspx.
- 120.IFM Therapeutics, Inc. Compounds and compositions for treating conditions associated with nlrp activity. WO 2017/184604 A1. 2017.
- 121.Inflazome Ltd. Sulfonamide derivates as NLRP3 inhibitors. WO2019166633A1. 2019.
- 122.Inflazome Ltd. Novel compounds. WO2019166632A1. 2019.
- 123.Novartis inflammasome research Inc. Compounds and compositions for treating conditions associated with NLRP3 activity. WO2020102576A1. 2020.
- 124.IFM Therapeutics, Inc. Compounds and compositions for treating conditions associated with NLRP activity. WO2019023147A1. 2019.
- 125.Inflazome Ltd. Novel Compounds. WO2019068772A1. 2019.
- 126.Available from: https://zyduslife.com/zyduslife.
- 127.Cadila Healthcare Ltd. Novel substituted sulfonylurea derivatives. WO2019043610A1. 2019.
- 128.Inflazome ltd. Compounds. WO2021165245A1. 2021.
- 129.Inflazome Ltd. macrocyclic sulfonylurea derivatives useful as NLRP3 inhibitors. WO2021032591A1. 2021.
- 130.Inflazome Ltd. Macrocyclic sulfonylamide derivatives useful as NLRP3 inhibitors. WO2021032588A1. 2021.
- 131.Nodthera Ltd. Amino heterocyclic compounds and uses thereof. WO2020157069A1. 2020.
- 132.Ltd Inflazome. NLRP3 Inhibitors. WO2021043966A1. 2021.
- 133.AC Immune SA. Dihydrooxazole and thiourea derivatives modulating the NLRP3 inflammasome pathway. WO2021255279A1. 2021.
- 134.Janssen Pharmaceuticals NV. Compounds. WO2021239885A1. 2021.
- 135.Janssen Pharmaceuticals NV. Phthalazinone derivatives as NLRP3 inflammasome inhibitors. WO2022229315A1. 2022.
- 136.Janssen Pharmaceuticals NV. 4-alkoxy-6-oxo-pyridazine derivatives modulating NLRP3. WO2022184842A1. 2022.
- 137.Janssen Pharmaceuticals NV. New compounds. WO2022063876A1. 2022.
- 138.Janssen Pharmaceuticals NV. New compounds. WO2022063896A1. 2022.
- 139.Novartis AG. NLRP3 inflammasome inhibitors. WO2020021447A1. 2020.
- 140.IFM Therapeutics, Inc. NLRP3 modulators. WO 2017/184746 A1. 2017.
- 141.IFM Therapeutics, Inc. NLRP3 modulators. WO 2017/184735 A1. 2017.
- 142.Innate Tumor Immunity, Inc. Substituted imidazo-quinolines as NLRP3 modulators. WO 2018/152396 A1. 2018.
- 143.Novartis AG. NLRP3 inflammasome inhibitors. US20200361898A1. 2020.
- 144.Nodthera Ltd. Carbamate derivatives and uses thereof. WO2020152361A1. 2020.
- 145.Ventus Therapeutics U.S., Inc. Pyridazine compounds for inhibiting NLRP3. WO2022216971A1. 2022.
- 146.University of Turin. NLRP3 inflammasome-inhibiting compounds and the use thereof. WO2022234447A1. 2022.
- 147.Icahn School of Medicine at Mount Sinai. Benzoxazolone inhibitors of inflammasomes. WO2022187804A1. 2022.
- 148.Guo C, Fulp JW, Jiang Y, Li X, Chojnacki JE, Wu J, et al. Development and characterization of a hydroxyl-sulfonamide analogue, 5-chloro-n-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci 2017; 8:2194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Virginia Commonwealth University. Cryopyrin inhibitors for preventing and treating inflammation. US10343985B2. 2019.
- 150.The University Of Manchester. Cyclic diarylboron derivatives as NLRP3 inflammasome inhibitors. WO 2017/017469 A1. 2017.
- 151.Available from: 10.1021/cen-09807-cover. [DOI]
- 152.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 2018; 17:588–606. [DOI] [PubMed] [Google Scholar]