

ABSTRACT
Diabetes mellitus is still expanding globally and is epidemic in developing countries. The combat of this plague has caused enormous economic and social burdens related to a lowered quality of life in people with diabetes. Despite recent significant improvements of life expectancy in patients with diabetes, there is still a need for efforts to elucidate the complexities and mechanisms of the disease processes to overcome this difficult disorder. To this end, the use of appropriate animal models in diabetes studies is invaluable for translation to humans and for the development of effective treatment. In this review, a variety of animal models of diabetes with spontaneous onset in particular will be introduced and discussed for their implication in diabetes research.
Keywords: animal model, diabetes mellitus, pathophysiology
Animal models of diabetes and application to human studies

INTRODUCTION
For a better understanding of the disease mechanisms and for the development of effective treatments, animal models have contributed enormously to the knowledge gained in diabetes research 1 , 2 . The results obtained have translated well to humans in daily practice. On September 3, 2020, Professor Yoshio Goto, a world renowned diabetologist and a lifelong researcher in experimental diabetes and its complications (Figure 1), passed away at the age of 94. He was a founder of the Japan Society of Experimental Diabetes and Obesity the meeting of which is held annually, now in its 36th year. He also established the global study group of animal diabetes, holding the Lessons from Animal Diabetes (LAD) workshop together with Professors Arnold Renold in Geneva, Dan Porte in Seattle, USA and Eleazar Shafrir in Jerusalem, Israel. His work is well remembered for the development of the Goto Kakizaki rat (GK rat), an animal model of spontaneous onset type 2 diabetes 3 , 4 . Currently, GK rats are distributed worldwide for studies on diabetes. The advent of GK rats promoted discoveries of many models of spontaneous onset diabetes, in particular with the selective breeding of animals. It is fortunate that we are now able to choose suitable animal models for our studies depending on the study design 5 . This review, with a tribute to the establishment of the GK rat by Goto and Kakizaki, introduces mainly rodent models of diabetes and their characteristic features. Some larger mammalian models as well as non‐mammalians will also be described briefly.
Figure 1.

Late Professor Yoshio Goto, Founder of the Japan Society of Experimental Diabetes and Obesity, Emeritus Professor of Tohoku University.
RATIONALE FOR THE USE OF ANIMAL MODELS
Pandy and Dvorakova 6 explained the rationale of the use of animal models for medicine. It is mainly based on the intention to understand biological function in humans, being designated according to their use such as: (1) Exploratory models, aimed at understanding the mechanism of action in biological systems and could be related to fundamental or basic research on biological systems or a mechanism associated with disease or abnormal biological function. (2) Explanatory models, aimed at understanding the complex biological problem. They should not necessarily be reliant only on animal usage, but could be a physical, bioinformatics, or mathematical model system developed to unravel complex mechanisms. They are used to develop a scientific hypothesis and to discover fundamental laws. (3) Predictive models are the most important animal models and are generally used for preclinical or applied research. They are unique animal models, which are aimed at assessing a possible effect in humans 7 . They are also used to discover and quantify the impact of the treatment, in addition to the evaluation of therapy such as pharmacokinetic/pharmacodynamics and toxicity of drugs 8 . Translation of the results to humans may be an ultimate goal for benefits to human health care and improvement of quality of life.
DIAGNOSTIC CRITERIA OF DIABETES IN ANIMAL MODELS
There is no consensus for the diagnosis of diabetes applicable to animal models. This situation is different to the status of human diabetes. The Study Group of the Japan Society of Experimental Diabetes and Obesity (JSEDO) provided the guideline for the use of animal diabetes and the definition of diabetes in animals 9 . The criterion of human diabetes as an elevated fasting blood glucose (>126 mg/dL; 7.0 mmoL/L) is based on significant correlation with the probabilities of the presence of retinal complications, either in the American Diabetes Association or in the Japan Diabetic Society. There are no data, however, on the relationship between blood glucose levels and microvascular complications in animal models. In addition, blood glucose dynamics should be variable among animal species. To obtain some guidance, if any, for markers of diabetes, the Japan Society of Animal Diabetes and Obesity sent out a questionnaire on the diagnosis of animal diabetes to investigators who were involved in studies on diabetic animals. Most commonly the investigators used levels of casual blood glucose for the designation of diabetes. For convenience, animals with blood glucose levels more than 300 mg/dL (16.7 mmoL/L) are regarded as animals with overt diabetes. When fasting blood glucose levels were used as indices of elevation, values above 150 (8.3 mmoL/L) or 200 mg/dL (11 mmoL/L) were arbitrarily adopted as a sign of hyperglycemia depending on the purpose of their studies. On the other hand, the continuous presence of sugar in the urine is regarded as a sign of diabetes in NOD mice when repeated blood withdrawal is not feasible. In contrast to models of type 1 diabetes, glucose tolerance tests with an oral or intraperitoneal load of glucose are commonly applied for models of type 2 diabetes. In either case, comparison with a non‐diabetic healthy control group is essential. The study group advocated the use of animal models for diabetes research under a sufficient rationale following ethical guidelines (Table 1).
Table 1.
Rationale and recommendation for the use of animal models in diabetes research
|
ANIMAL MODELS FOR TYPE 1 DIABETES
Bio Breeding (BB) rats and NOD mice are the most popular models for type 1 diabetes. The clinical features of human type 1 diabetes are characterized by the acute onset of polyuria, polydipsia, hyperglycemia, and insulin deficiency. Autoimmune processes to the islet β‐cells serve as the pathologic basis in these animals. The exact pathogenesis of type 1 diabetes in humans, however, remains unclear. Consequently, the data obtained from these models should be interpreted carefully. In contrast to islet damage, the presence of long‐term hyperglycemia (‘glucotoxicity’) can be used to explore the pathogenesis of diabetic complications of the target organs, but the animals should survive long enough to show tissue damage to this end (Table 2).
Table 2.
List of major rodent animal models for type 1 diabetes mellitus
| Strain | Founder(s)/place or institution | Gender (M vs F) | DM onset | Immune basis | Major islet pathology | Application and complication | Others |
|---|---|---|---|---|---|---|---|
| Type 1 | |||||||
| NOD mice (non‐obese diabetes) |
Makino, 1980 17 (Shiga, Japan) Jcl: ICR origin (cataract) |
F > M DMpop; 90% of F 60% of M |
Insulitis 4–5 weeks β‐cell loss 10–14 weeks DM onset ~40 weeks Autoab+, GAD+ |
Autoimmune basis Analogous to human type 1 diabetes Islet contains CD4,8+ & cytokines |
Insulitis β‐cell loss |
Useful for intervention study DM onset promoted by cyclophosphamide Nephropathy onset Neuropathy (mainly autonomic) |
NON as control |
| BB rat (Bio Breeding) |
Nakhooda, 1974 10 Ottawa, Canada, and Worcestor, MA, USA |
F > M 90% of F 60% of M |
Insulitis 4–5 weeks DM ~12 weeks of age after puberty 8–16 weeks Autoab+, GAD+ |
Autoimmune basis Analogous to human type 1 diabetes, weight loss T‐lymphopenia (ART2+) |
Insulitis β‐cell loss MHC‐II CD4↓Mφ |
Useful for intervention study Polyendocrinopathy (thyroid, salivary & lacrimal gland) Nephropathy Neuropathy |
BBDR (diabetes resistant strain) |
| LEW/AR1/‐IDDM |
Lenzen, 2001 35 Hannover, Germany (congenic) |
M ≒ F 20–60% |
20–60% DM onset; around puberty (8–9 weeks) |
Autoimmune basis T, Mφ, NK infiltration MHC‐II upregulation No lymphopenia |
Insulitis β‐cell loss |
Suitable for complication study No other organ abnormality Polyendocrinopathy |
|
| LETL |
Kawano, 1980 42 Tokushima, Japan |
M ≒ F |
Low DM onset ca 20% Autoab |
No lymphopenia Autoimmune basis |
Insulitis β‐cell loss | Low incidence of DM | LETO as a control |
| KDP |
Komeda, 1988 43 Tokyo, Japan |
M ≒ F | 80% | Autoimmune basis |
Insulitis+ β‐cell loss |
KND as a control | |
| Chemically & virally induced STZ rat and mice |
Rakieten, STZ, 1963 4 Indiana, USA Lukens, Alloxan, 1948 46 Like & Rossini 1986 53 Worcestor, MA, USA Virus (EMC) Craighead, 1968 55 |
M ≒ F | Any time but susceptible around puberty | β‐cell toxicity | Massive β cell loss |
Over 90% success for STZ but care for survival (10% show recovery) STZ adverse action of tumor induction (liver, kidney, soft tissues) Direct cytotoxicity of virus (EMC‐D variant) |
|
In contrast to spontaneous diabetes, animals with diabetes induced by chemicals are much more commonly used as a model of insulin‐deficient diabetes animals because of the convenience and lower expenses. Streptozotocin and alloxan are frequently used as diabetogenic agents. Such chemically induced diabetic animals are not equivalent to those in human diabetes, but are well studied for the evaluation of the effects of long‐term hyperglycemia on target tissues with diabetic complications.
Spontaneous onset type 1 diabetes model
BB (Bio Breeding) rat
The BB rats were discovered in 1974 in a colony of outbred Wistar rats at Bio Breeding Laboratories in Ottawa, Canada. They manifested a sudden onset of polyuria, hyperglycemia, insulin deficiency, and weight loss after puberty 10 . Without insulin treatment, BB rats develop severe ketoacidosis sufficient to be fatal. The clinical features appear to be consistent with human type 1 diabetes. Shortly after the discovery, the incidence of diabetes onset was only 10%. By transfer to a suitable environment, nearly 90% of rats developed diabetes between 8 and 16 weeks of age, but 6–8 months later in some substrains. The frequency of the overt diabetic phenotype is comparable in males and females. When diabetes starts early, maturation is impaired 11 . All substrains have been derived from two founder colonies of inbred [dp (diabetes‐prone) BB/Wor (Worcestor)] and outbred (BBdp) 11 . BB rats resistant to diabetes (BBDR) have also been bred to serve as controls.
The pathologic basis of diabetes is characterized by inflamed islets with infiltration of T cells, B cells, NK cells, and macrophages, consistent with the feature of so‐called insulitis encountered in human type 1 diabetes. The progressive decline of islet β cells is in parallel with insulin deficiency. Distinct from human type 1 diabetes, blood analysis of BB rats disclosed lymphopenia with a severe reduction in CD4+ T cells and a near absence of CD8+ T cells 11 . This model has contributed substantially to elucidating the role of genetics in type 1 diabetes 12 , 13 . BB rats have been used in detailed studies of diabetic neuropathy 14 . Systemic observation in BB rats disclosed robust lymphocytic inflammation in the thyroid gland and gastric mucosa, in which polyendocrine autoimmune processes are exerted 15 .
Recently, a new substrain named BB/O(ttawa)K(arlsburg) (BB/OK) rats was further cross‐bred with SHR (spontaneously hypertensive rats) to obtain diabetic rats with vascular complications named BB.SHR 16 . BB.SHR rats are not lymphopenic nor diabetic, but obese and hypertensive, typical of metabolic disorder. This congenic BB.SHR strain is now used in studies on the effects of hypertension and diabetes on cardiovascular systems 16 .
NOD mouse
The NOD mouse was found at the Shionogi Research Laboratories in Osaka, Japan in 1974 17 , 18 . This model develops marked inflammatory changes of pancreatic islets at 3–4 weeks of age. In the prediabetic stage, the islets are infiltrated mainly by CD4+ and CD8+ lymphocytes with a lesser population of B cells and NK cells 19 , 20 . The insulitis is accompanied by β cell destruction, but the onset of overt diabetes is not apparent until approximately 90% of pancreatic insulin is lost at around 10–14 weeks of age. Diabetes is more prevalent in females ranging from 60% to 90%, whereas the incidence in males ranges from 10% to 30% 20 . When the mice become overtly diabetic, they rapidly lose weight and require insulin treatment for survival. The MHC class 2 in NOD mice shares structural similarities to those in humans, which may confer resistance or susceptibility to the disease in both NOD mice and humans 21 . Genes that influence the onset of diabetes are mapped on chromosome 3, 11, and outside MHC 22 .
The NOD mice are suitable for exploring therapies in which modulation of the autoimmune response is being targeted 23 , 24 . Caution should be taken, however, that they are kept in pathogen‐free (SPF) conditions to maintain the incidence of diabetes because microbial exposure suppresses the development of diabetes 25 . The NOD mouse has helped to identify many of the genetic and signaling pathways that can lead to type 1 diabetes, as it does represent many aspects of human disease. In fact, the onset of diabetes was significantly inhibited by cyclosporin treatment 26 , whereas cyclophosphamide enhanced the onset and severity of diabetes 27 , 28 . Despite the fact that there are a number of drugs that are effective in NOD mice, they were found to be ineffective in humans. One of the major reasons for this discrepancy may be ascribed to the time point of intervention. The lack of suitable biomarkers and translating dosing from the NOD mouse to humans add problems to effective treatment. NOD mice can also be used in a recurrent autoimmunity model, where syngeneic islets from young non‐diabetic NOD mice are transplanted into diabetic NOD mice 29 . The graft is rapidly destroyed by autoimmune mechanisms.
Extrapancreatic tissue damage is found in NOD mice. Destruction of islet sympathetic nerves concomitant with marked degenerative, dystrophic changes of neurites are salient features of the peripheral nerves, reminiscent of the structure encountered in experimental and human diabetic autonomic neuropathy 30 . Genetic control is implicated in the manifestation of autoimmune neuropathy in this model 31 . There are substrains of NOD mice with features of autoimmune neuropathy comparable to chronic inflammatory demyelinating polyneuropathy (CIDP) 32 . There have been some investigations on kidney changes, such as renal hypertrophy and glomerular sclerosis. These alterations are accounted for by activated immune and inflammatory reactions, but the precise mechanisms are largely unknown. A variety of compounds have been used in an experimental attempt to prevent or restore the renal changes 33 . Complete recovery was not obtained because of the lack of complete inhibition of the autoimmune processes 34 .
LEW.1AR1/‐IDDM rat
LEW rats are derived from congenic Lewis rats and represent a model of spontaneous autoimmune type 1 diabetes with a defined MHC haplotype. This model was established at the Institute of Laboratory Animal Science of Hannover Medical School in Germany 35 . Original LEW rats manifested diabetes at a rate of ~20% at puberty (between around 8 and 9 weeks of age without any gender differences between male and female). With further inbreeding of LEW diabetic rats, the incidence of diabetes increased to ~60% with a comparable incidence in both genders. Pancreatic islets contained apoptotic β cells infiltrated with macrophages and lymphocytes about a week before the animals became hyperglycemic 36 . The diabetic phenotype of LEW rats is characterized by hyperglycemia, glycosuria, ketonuria, and polyuria, but without lymphopenia in which T cells are normally populated. Proinflammatory cytokines are upregulated to exert damage to the islets 37 . In contrast to robust inflamed islets, LEW rats usually survive well without insulin supplementation 36 , and thus are suitable for studies on the progression of diabetic complications. The slow progression of autoimmune diabetes resembles latent autoimmune diabetes in adults (LADA) 38 , alternatively slowly progressive insulin dependent diabetes mellitus (SPIDDM) reported from Japan 39 .
The treatment approach for type 1 diabetes may start with initial monotherapy followed by combination therapy to target proinflammatory cytokines 40 . So far in this model, most studies have been investigating the mechanisms involved in the development of diabetes. Among these, Fingolimod (FTY720) treatment protected islet infiltration of immune cells in the prediabetic and early diabetic phase in this model, but not in humans 41 . The rats with diabetic syndrome exhibit an autosomal recessive mode of inheritance with an incomplete penetrance of the mutant phenotype. Other endocrine and exocrine organs such as adrenals, thyroid, and salivary glands as well as lacrimal glands are unaffected in this model.
LETL rat and KDP rat
The Long‐Evans Tokushima Lean (LETL) rat is an inbred strain with type 1 diabetes based on autoimmune destruction of islet β cells without lymphopenia 42 . This strain was established in 1989 from an outbred colony of Long‐Evans rats in Otsuka Pharm Co., Tokushima, Japan. These rats develop sudden onset of diabetes with polyuria, polyphagia, hyperglycemia, and weight loss. Islets are marked with lymphocytic infiltration which is also detected in the salivary and lacrimal glands. Two recessive genes were suggested to be involved in the pathogenesis of insulitis, which is closely linked with RT1u. There is no gender difference in the incidence of diabetes. The incidence of diabetes is low, ranging at about ~20%. This drawback of a low incidence was overcome by the efforts of Komeda and his group who established substrains of diabetes‐prone and non‐diabetic rats (Komeda non‐diabetes rats, KND) and diabetes prone diabetic rats (Komeda Diabetes Prone rats, KDP) 43 . The KDP rats show a high incidence of diabetes (overall ~70%) and 100% development of mild to severe insulitis at 120–220 days of age. Different from other type 1 diabetes models of NOD, BB, and LEW rats in which islets are infiltrated with IL‐1β and TNFα, the major cytokines in KDP are IFNγ and TNFα 44 . There is a complete absence of diabetes incidence in the KND substrain. Among 165 SSLP marker loci throughout all the rat chromosomes, there was no loci of variation in either KDP, KND, or their parenteral LETL rats, indicating that the genetic background of these substrains appears to be uniform except for the major gene(s), responsible for diabetes 45 .
Chemically induced type 1 diabetes and virus‐induced type 1 diabetes
The most potent and popular diabetogenic chemicals are alloxan and streptozotocin (STZ) [2‐deoxy‐2‐(3‐(methyl‐3‐nitrosoureido)‐D‐glucopyranose)], the latter of which is synthesized by Streptomycetes chromogenes 46 , 47 . The diabetogenic effects are different among the species and also in individuals 48 , 49 . These chemicals are cytotoxic glucose analogs, which accumulate in β‐cells via glucose transporter 2 (GLUT2) 50 , 51 . A higher dosage such as 50 mg/kg STZ and above can produce irreversible diabetes in rats, whereas a dose of 150 mg/kg is required to develop diabetes in other species (e.g. Cynomologus monkey, pigs). Partial correction of hyperglycemia may occur in pigs 4 weeks after STZ injection 50 . The mechanism of STZ action depends on poly(ADP)‐ribose activation (the DNA alkylating activity) by its methyl‐nitrosourea moiety 52 . The methyl group from STZ is transferred to the DNA molecule and causes damage along with other defined events, which leads to DNA fragmentation. In chemically induced diabetes, the majority of the endogenous β‐cells are destroyed, resulting in little endogenous insulin production. Diabetes is usually induced 5–7 days after treatment with streptozotocin or alloxan. Due to their similarity in structure to glucose, glucose can compete with alloxan and streptozotocin, and thus, fasting animals tend to be more susceptible. Both alloxan and streptozotocin are relatively unstable, and the solutions should ideally be made immediately prior to injection. Repeated injection of a low dose of streptozotocin to neonates of rat and mice was used for the production of a non‐insulin‐dependent diabetes model in which β‐cell destruction is not marked enough to end with fatal hyperglycemia 53 , 54 . It is problematic, however, that streptozotocin is a potent carcinogenic compound yielding epithelial and mesenchymal tumors in the kidney, liver, and soft tissues over long‐term observations.
Similar to streptozotocin, viruses have a tropism for the islet to elicit β‐cell damage resulting in type 1 diabetes. Among these, Coxackie B virus, Encephalomyocarditis (EMC) virus, Kilham rat virus, and LCMV under an insulin promoter have been found to be diabetogenic inducing insulitis, hypoinsulinemia, and hyperglycemia 55 , 56 . Classic studies on virus‐induced diabetes disclosed a clear difference from human type 1 diabetes 57 . However, the exact role of viruses in human type 1 diabetes such as fulminant type 1 diabetes, LADA (latent autoimmune diabetes in adult), or SPIDDM (slowly progressive insulin‐dependent diabetes mellitus) is yet to be elucidated, and the relationship of viruses with islet inflammation and exocrine pancreatic changes are largely unknown, still worthy of critical studies.
ANIMAL MODELS FOR TYPE 2 DIABETES
Type 2 diabetes is the most common type of diabetes in humans (Table 3). The clinical features are variable but usually silent for symptoms until various vascular and neurological complications become manifest. The underlying mechanisms of this disorder are ascribed to impaired insulin secretion (insulin deficit) and ineffective insulin action (insulin resistance), resulting in glucose intolerance and overt hyperglycemia 58 . Obesity causes insulin resistance with resultant hyperglycemia in animals and contributes to the onset of diabetes. The C57BL6/J mouse develops hyperphagia‐induced obesity subserving as a model for human obesity 59 . The induction of obesity by a high fat diet (HFD) appears to depend on epigenetic differences, reminiscent of the diverse range of susceptibility to obesity among humans 60 . Not all animals with diabetes are obese and different mechanisms may operate in obesity and diabetes. In this section, spontaneously occurring diabetic animals with polygenic and with monogenic involvement will be discussed.
Table 3.
List of major rodent animal models for type 2 diabetes mellitus
| Founder(s)/place or institution | Gender (M vs F) | DM onset | Metabolic basis | Major islet pathology | Complication | Others | |
|---|---|---|---|---|---|---|---|
| Type 2 diabetes polygenic | |||||||
| GK rat |
Goto, Sendai & Hirosaki, 1975 3 , 4 , Japan Portha, 2009 62 Ostensson, 1993 61 |
M > F |
Around 8–12 weeks Age‐related decline of β‐cell function and structure Type 2 diabetes mellitus Selectively bred |
Impaired insulin secretion Insulin resistance Inherent decrease in β‐cell mass |
Progressive β‐cell loss Fibrosis with irregular contour |
Abnormal OGTT Vasculopathy; thickened basal lamina Neuropathy; distal axonal changes Polycystic ovary syndrome model 71 |
Control Wistar |
| SDT rat |
Shinohara, 2000 72 , Chiba, Japan Sasase, 2013 74 Osaka |
M > F Male |
20–24 weeks Non‐obese type 2 diabetes mellitus |
Ins deficiency Islet pathology Hyperglycemia Hypoinsulinemia |
β‐cell loss Islet hemorrhage |
Proliferative retinopathy, cataract, glaucoma Nephropathy, mesangial growth Nodular lesion Neuropathy |
Non‐obese Wistar |
|
Sand rat Desert gerbil |
Marquie, 1984 78 Shafrir, 1992 79 Israel |
M ≒ F |
80–90% on high energy diet ~8 month age |
Obesity Ins resist |
Islet hyperplasia Hyperinsulinemia |
Hyperglycemia Diet‐induced obesity No hyperphagia |
|
| UCD‐T2DM rat |
Cummings, 2008 163 , UCSF, USA Cummings, 2011 166 , Davis, CA, USA |
M > F M 92% F 43% |
Polygenic HFD‐obesity Hybrid of SD&ZDF |
Insulin resistance Type 2 diabetes mellitus Dyslipidemia |
Islet atrophy hypoplasia β‐cell loss |
GLP‐1 and leptin responsive Impaired insulin secretion |
LSD rat |
| NZO mice |
Bielschowsky, 1953 85 Bielschowsky, 1970 86 |
M ≫ F |
Type 2 diabetes, polygenic HFD‐induced obesity |
Insulin resistance Type 2 diabetes, lipidemia |
Islet hypertrophy |
Metabolic syndrome model Hyperinsulinemia, leptin resist Hyperglycemia |
F: infertile Kept in Dusseldorf |
| KK & KK‐Ay mice |
Nakamura, 1967 90 Michaud, 1994 93 |
Agouti mutated |
Type 2 diabetes, polygenic Obesity Yellow fur |
Insulin resistance Obesity |
Islet hypertrophy |
Hyperinsulinemia Hyperglycemia, hyperleptinemia |
|
| NSY mice |
Shibata, 1980 96 Nagoya, Japan STZ origin, JCl:ICR |
M > F M 98% F 31% |
HNFa‐mut Selectively bred Polygenic |
Type 2 diabetes |
No distinct Islet pathology |
Hyperglycemia, fatty liver Hyper → hypoinsulinemia Amyloid in kidney & other organs |
|
| ON mice |
Tokyo, Japan |
M > F |
Selectively bred Polygenic HFD‐induced |
Mildly obese Modest type 2 diabetes |
Mild islet changes Insulin resistance |
Atherosclerosis Hyperglycemia, hypoinsulinemia |
|
| TallyHO mice | Kim, 2001 109 , 2006 111 Theiler Original colony, W Virginia, USA |
Type 2 diabetes mellitus Male only |
polygenic |
Obesity both M&F Dyslipidemia Insulin resistance |
Islet changes Islet hyperplasia Degranulated β‐cell |
Hyperinsulinemia Hyperglycemia lipidemia |
|
| TSOD mice | Suzuki, 1992 118 (Tsumura Co, Ibaraki, Japan) Slc:ddY origin |
Type 2 diabetes mellitus M ≫ F 100% |
Type 2 diabetes mellitus, polygenic Metabolic syndrome |
Obesity with marked insulin resistance |
Lipidemia Hyperglycemia |
Fatty liver, hepatoma Hyper → hypoinsulinemia |
|
| Monogenic | |||||||
| OLETF |
Kawano, 1992 130 Otsuka Long Evans Tokushima Fatty rat Tokushima, Japan |
CCK‐R Type A mutated |
Obesity Type 2 diabetes |
Obesity Lipidemia Type 2 diabetes |
Islet atrophy β‐cell loss Lipid deposition |
Fatty liver Type 2 diabetes; chronic hyperglycemia Insulin resistance Lipotoxicity |
LETO |
| ZF |
Zucker fatty 1972 141 Zucker and Zucker Cross Merck M‐strain and Sherman |
Male Leptin−/− (missense mut) |
Fa/fa |
Obesity at 4 weeks Hyperphagia Mets, lipidemia Hyperinsulinemia Not DM but IGT |
Islet hyperplasia |
Marked insulin resistance Hyperinsulinemia Lipotoxicity Infertility |
|
| ZDF |
Peterson, 1990 147 Indianapolis, IN, USA Genetic Models Inc. |
Male Leptin‐R(−/−) (mutated) |
ZDF diabetes‐prone (dp) 7–10 weeks DM Selectively bred Hyperglycemic rats |
Type 2 diabetes with obesity Insulin resistance Lipidemia Female nonDM, but become DM by HFD |
Islet degeneration Progressive β‐cell loss with apoptosis Lipid droplets in β‐cell |
Infertility Lipotoxicity Initial hypo‐ to hyperinsulinemia Premature death Mild retinal microvessel changes Neuropathy changes |
|
| Koletskey (SHROB) rat |
Kolestky (SHROB), 1973 160 , Cleveland, OH, USA Obese SHR |
M > F |
Mets 5w obese Hybrid SHR&ZF |
Model of Mets Insulin resistance Hypertension Lipidemia |
Lipidemia (type 4) |
Vasculopathy Membranous glomerulonephritis Inflamed large vessels (mesenteric arteries) |
|
| ob/ob mice |
Jackson lab (USA) Ingalls, 1950 171 |
> F Leptin (−) |
Obese, nonDM Defect of leptin |
Insulin resistance Hyperleptinemia Hyperinsulinemia |
Lipidemia, hyperphagia Islet hypertrophy |
Transient hyperglycemia | B6‐ +/+ mice |
| db/db mice |
Jackson lab (USA) Coleman, 1969 183 LeptinR(−/−) |
M > F Mutated Lepr |
Type 2 diabetes mellitus 6–8 weeks Mutated leptin receptor (defect of lepr) |
Initial hyperphagia Non‐obese |
Hyper‐to hypoinsulinemia, islet atrophy and β‐cell loss |
Premature death Microangiopathy (retina, kidney) Cardiomyopathy Neuropathy |
BKS‐ +/+ mice |
| Akita mice (Mody2) |
Yoshioka et al., 1997 194 Kayo et al., 1998 195 |
Ins2‐mutated ER tress |
C57Bl/6NSlc origin Male only Mimic type 1 |
Onset 3–4 weeks Insulin deficiency Hyperglycemia Polyuria |
Hypoinsulinemia Islet atrophy β‐cell apoptosis |
Premature death ~12 weeks of age Neuropathy Nephropathy, albuminuria Microangiopathy |
|
| Unique | Pancreatic diabetes | ||||||
| WBN/Kob rat |
Wistar‐Bonn Kobori Tsuchitani, 1985 203 |
Insulin def M > F Pancreatic DM |
Exocrine inflame Age‐related Hyperglycemia, hemorrhage |
Endocrine Exocrine insufficiency Non‐obese Lean |
Islet fibrosis Hemorrhage Chronic pancreatitis Hemosiderin deposit |
Motor neuropathy Exocrine insufficiency Vascular complication Sclerotic changes CIDP model |
|
Polygenic models
GK (Goto‐Kakizaki) rat
This model was developed in Hirosaki and Sendai, Japan by Professor Yoshio Goto and his group 3 , 4 , 61 , 62 . They first selected normal Wistar rats with mild glucose intolerance and repeated selective breeding of rats with hyperglycemia. After the 6th–7th generation, all offspring developed overt hyperglycemia fulfilling the criteria of diabetes. They do not present obesity. Both insulin resistance and impaired insulin secretion are present. Inheritance is polygenic. The pancreatic islets became mildly inflamed with macrophage infiltration together with distortion and fibrosis with robust reduction of β‐cells 63 , 64 . Amyloid deposition is not apparent, possibly due to genetic differences in the amyloid peptide of rodents 65 . This model is characterized by lean animals, impaired insulin secretion, moderate hyperglycemia, and insulin resistance. At the age of 6–8 weeks, there is a gradual increase in postprandial hyperglycemia, and impaired glucose tolerance. Studies on this model disclosed an early emergence of microangiopathic changes in the kidney, peripheral nerves, validating this model for type 2 diabetes 66 , 67 , 68 . The GK rat is used widely in USA, Europe, UK, and Japan. Genetic analysis identified the genetic foci related to diabetes onset 69 , 70 . More recently, this model has exhibited a resemblance to the ovarian structure and hormonal perturbations of polycystic ovary syndrome 71 .
SDT (spontaneously diabetic Torii) rat
The spontaneously diabetic Torii rat is an inbred strain of spontaneously onset non‐obese diabetic rat of the type 2 diabetes phenotype established in 1992 at Torii Pharmaceutical Laboratory in Japan 72 . Diabetes develops in male rats after 20 weeks of age to nearly 100% at 40 weeks of age. Diabetes manifests with severe hyperglycemia, polydipsia, and polyuria, impaired insulin secretion and hypoinsulinemia. Pancreatic islets undergo atrophy with a depletion of β‐cells, mixed with changes of hemorrhage and fibrosis. In this model, it is noteworthy that ocular complications of diabetes characterized by cataract, proliferative retinopathy, and retinal detachment develop with the longer duration of diabetes 73 . Vascular complications also accompany peripheral neuropathy 74 . They develop diabetes with a polygenic basis, regulated by at least three quantitative trait loci (QTLs) each located on rat chromosome 1, 2, and X 75 . These investigators further developed a new, obese type animal model with diabetes, incorporating Lepr (fatty) gene by breeding with Zucker fatty rat, to explore whether the specific complications of diabetes become worse 76 . They found aggravated signs of somatic neuropathy in this model, but still await further confirmation of the neuropathy phenotype 77 .
Sand rat (Psammomys obesus)
The sand rat, a diurnal gerbil (Psammomys obesus), is an outbred polygenic model of nutrition‐dependent obesity and diabetes 78 . In its native desert habitat, with access to a low‐calorie plant‐based diet, the sand rat is lean and normoglycemic. In the laboratory environment, however, with free access to an energy rich laboratory diet, the sand rat becomes obese and rapidly develops hyperglycemia. Diabetes is characterized by severe hypoinsulinemia, hyperlipidemia, and ketosis, often observed within 3–4 weeks of birth 79 . Similar to the progression of type 2 diabetes in humans, the diabetic sand rat suffers from β‐cell degradation, retinopathy, nephropathy, body weight loss, and premature death. Induction of glucotoxicity elicited by excessive diet accelerates the aggravation of β‐cell loss 80 , 81 . Suppressed expression of PDX1 is associated with progressive β‐cell reduction and diabetes onset 81 . On a cholesterol‐rich diet, the sand rat also develops non‐alcoholic steatohepatitis with morphological and functional consequences that are similar to the human pathology 82 . Through selective breeding, researchers in Israel have created diabetes‐prone and diabetes‐resistant sand rat groups in order to study the interaction between diet and genes in the development of diabetes and obesity 83 . Careful investigations in the chronically observed sand rat disclosed the presence of retinal changes comparable to those in humans with diabetes 84 .
NZO (New Zealand Obese) mouse
The NZO mouse is a model of type 2 diabetes with a polygenic nature exhibiting classic characteristics such as insulin resistance and impaired glucose‐mediated insulin secretion. This model was developed from a mixed colony at the University of Otago Medical School in Dunedin, New Zealand 85 , 86 . Initially, three different coat colors were found in this colony, namely agouti, tan, and chocolate. Inbred animals with agouti color commonly manifested obesity and were established as a new strain of obese and agouti‐colored mice termed NZO.B1. The other line of mice developed from agouti was the New Zealand Black (NZB/B1). Inbreeding of the tan colored mice gave rise to the NZY/B1 line and inbreeding of chocolate‐colored mice the NZC line, respectively 86 . The lack of a suitable control strain has been a problem, because the NZO mouse is an inbred model. A number of lean mice were used as controls including the C57BL/6, Balb C, and the albino ICR mouse. Some groups attempted to use the lean NZC mouse as a control since it was derived from the same population 87 . To raise the inbreeding efficiency, treatment with a β3 adrenergic receptor agonist was adopted. Diet restriction to reduce body weight was also effective. This model typically exhibits hyperphagia, inactivity, and obesity 88 . Blood analysis demonstrates lipidemia, hyperglycemia, and hyperinsulinemia in particular in the fed state and elevated levels of leptin which do not respond to its receptor 88 . Islet pathology depicts enlarged islets composed mostly of β cells and lack of peripheral mixed composition of α‐ and β‐cells 89 .
KK (Kondo Kasukabe) and KKAy (KK‐Agouti‐yellow) mice
Nakamura and Yamada 90 introduced a new mouse model of diabetes named KK mice after the founder's name (Kondo) and the location (Kasukabe in Saitama, Japan). The features are modest obesity (30–35 g), polyphagia, polyuria, glucosuria, glucose intolerance, moderate hyperglycemia, lipidemia, insulin resistance, hyperinsulinemia, and histological changes in the pancreas 91 , 92 . Because of the modest diabetes and obesity, Nishimura, a co‐worker of Kondo, transferred the yellow gene (Ay) into KK mice by repeated crossing of yellow obese mice and KK mice. The Ay allele (dominant allele at the mouse agouti locus) is associated phenotypically with yellow fur, hyperphagia, and obesity 93 . This congenic strain of KK mice was named yellow KK or KKAy mice. Yellow KKAy mice are easily distinguished by the black color of the fur from KK mice at weaning. The diabetic phenotype manifests in KKAy mice at young ages (6–8 weeks) but reverts to normal after 40 weeks of age. This may be due to a reduction in food intake. They have a latent prediabetic state of glucose intolerance and insulin resistance before the development of hyperglycemia and glucosuria. Treatment with an α‐glucosidase inhibitor effectively suppressed hyperglycemia more potently in KK‐Ay mice than in normal control mice, mediated by intestinal sucrase activities 94 . The KK mice develop diabetes based on polygenic inheritance. A retinoid X receptor antagonist successfully improved leptin resistance without affecting plasma leptin levels in KK‐Ay mice 95 .
NSY (Nagoya Shibata Yasuda) mice
As with the KK mice, this model was developed by selective inbreeding using a laboratory strain of mouse termed Jcl:ICR, originally chosen from hyperglycemic mice susceptible to STZ 96 . NSY mice spontaneously develop diabetes in an age‐dependent manner. The key features include the type 2 diabetes phenotype exemplified by impaired insulin secretion and mild insulin resistance. Obesity is not a major feature and there is a marked gender difference with almost all males developing hyperglycemia, but less than a third of females, being affected.
The NSY mouse is particularly useful when considering age‐related phenotypes (e.g. decline of β‐cell function). A genome‐wide linkage scan identified a sequence variation in the hepatic nuclear factor 1β gene, a gene also implicated in human MODY syndrome 97 , 98 , 99 . Thus, it showed the polygenic transmission of diabetes genes 100 showing impaired insulin secretion in response to glucose as well as to arginine. Ikegami and his group uncovered the genetic loci of NSY mice responsible for the diabetes phenotype 99 , 101 . It is interesting to note that environmental factors interrelate with this genetic background for the onset of diabetes in which enhanced autonomic nerve activity is characteristic around the endocrine pancreas 102 . A substrain of obese type of NSY mice with yellow fur was obtained by cross breeding with agouti Ay mice 102 . This strain has a good fertility and does not display inter‐male aggression, making them useful as a new model for type 2 diabetes with early onset and persistent hyperglycemia 103 .
ON (Oikawa‐Nagao) mice
A new mouse model of type 2 diabetes exhibiting the clinical phenotype of diabetes was established by repeated selective inbreeding of mouse lines of hyperglycemia fed with a high fat diet (HFD) 104 . Inbred breeding started using hybrid mice of C57BL/6J, C3H/HeJ, and AKR/N backgrounds. After 5 weeks of HFD feeding, mice with both high and low 2 h blood glucose levels in an oral glucose tolerance test were separately and repeatedly selected over 14 generations. Two lines of mice prone and resistant to diet‐induced glucose intolerance designated ON‐DP (Oikawa‐Nagao diabetes prone) and ON‐DR (Oikawa‐Nagao diabetes resistant) mice were obtained 105 , 106 . The ON‐DP mice exhibit mild diabetes and obesity. These mice likely carry the polygenic and heritable nature of type 2 diabetes, showing impaired insulin secretion and reduced insulin sensitivity (insulin resistance). They are mildly obese with impaired glucose tolerance, hyperinsulinemia and hyperglycemia. Pancreatic islets were slightly hyperplastic with an increased β‐cell area 105 . The clinical phenotype of glucose intolerance and hyperphagia was likely associated with impaired glucose uptake and methylation of leptin gene promoter in adipose tissue in this model 107 . This model may also be applicable to the exploration of the role of lipid in the genesis of macroangiopathy and therapeutic effects on atherosclerotic lesions 108 .
TH (TALLYHO/Jng) mice
This model is a newly developed model of type 2 diabetes with mild obesity named the TALLYHO/Jng (TH) mouse of an inbred polygenic nature 109 . The TH mice develop overt diabetes rather late (~40 weeks of age) and exhibit reduced insulin‐stimulated glucose uptake in adipose tissue and skeletal muscle and abnormal pancreatic morphology and function. The clinical and pathologic features, in many aspects, resemble polygenic human type 2 diabetes 110 in which leptin has a role in impaired insulin secretion in response to glucose. Despite hyperinsulinemia and obesity, the female TH mice remain euglycemic. The molecular basis of the diabetic phenotype appeared to relate to lower levels of insulin receptor substrate 1 (IRS‐1) and impaired GLUT4 mobilization in the adipose tissue 111 . This model also manifests cerebrovascular dysfunction 112 , and male TH mice present drastically increased plasma triglyceride levels. The obesity is related to leptin resistance 113 . Finally, in addition to the obese diabetic phenotype 114 , the TH mouse displays reduced bone mineral density, making it an interesting model for studying the interplay between type 2 diabetes and the skeleton 115 and cognitive decline in diabetes 116 . Dysregulated mitochondria are invoked in the complicated diabetic phenotype 117 .
TSOD (Tsumura, Suzuki, Obese diabetic) mice
The TSOD mouse is a polygenic type 2 diabetes model with obesity, hyperglycemia, and hyperlipidemia. This model was developed 1999 in Tsumura Central Laboratories, in Ibaraki, Japan by Suzuki et al. (1999) and named after Tsumura, Suzuki, obesity, and diabetes 118 , 119 . It is derived from the selective breeding of obese male mice of the ddY strain using indices of heavy weight and the appearance of urinary glucose. The male TSOD mice constantly showed signs of obesity and urine glucose with increased intake of food and water. With a longer duration of diabetes, this model was found to develop characteristic complications of diabetes 120 . Alterations of autonomic activity were accompanied by metabolic aberrations 121 . Concurrently, increased blood glucose, insulin, and lipid levels are found in males, with some involved with steatohepatitis 118 , 120 . They are similar to ob/ob mice showing obesity and hyperinsulinemia, polydipsia, polyuria, and hyperglycemia. Pancreatic islets are hypertrophic without any signs of insulitis, or fibrosis. Although this model is alluded to accompany diabetes‐like complications, the presence of nephropathy and neuropathy is not well characterized. More recently, associated fatty liver, steatohepatitis, as well as the development of hepatic neoplasm have been well studied to constitute the current topics of this model 122 , 123 .
Chemically induced type 2 diabetes model
Inflammatory as well as environmental insults to the islet are known to cause functional and structural damage to islet endocrine cells. If the injury is sufficient to destroy a considerable population of more than 70% of the islets, the resultant loss of β‐cells induces hyperglycemia and diabetes onset. If it is modest or not sufficient to cause diabetes, the minor injury still provides a sufficient basis for susceptibility to diabetes to cause diabetes by subsequent environmental influences on the islets. Based on this presumption, Like and Rossini succeeded in the establishment of an animal model of type 2 diabetes, first in mice and then in rats 53 , 124 . They applied repeated injections of a small dose of streptozotocin to neonatal mice or rats for the production of rodent models of type 2 diabetes 125 , 126 . The diabetes occurred with a male predominance. They found lymphocytic infiltration in the islets reminiscent of insulitis encountered in a virus induced model of type 1 diabetes. However, hyperglycemia is modest in these mice injected with small dose of streptozotocin, and rather mimics type 2 diabetes 127 , 128 . The presence of type C viral particles may also underlie the potential additional injurious effects on the islet 53 , 126 .
After birth the animals are usually fed with a high fat diet (HFD) to accelerate the incidence of diabetes 127 , 128 . Concurrent with the onset of hyperglycemia, polyuria and polydipsia manifest. Islets in these animals show hyperplasia and hypertrophy, but low insulin secretion. With maturation, they show a modest weight gain but not to the extent of obesity. Low insulin secretion and insulin resistance are two characteristics in this model. More recently, a distinct phenotype of neuropathic complications was found in this model 129 .
Monogenic/multigenic models and related type 2 diabetes model
Monogenic animal models are extremely useful for understanding the function of the specific target genes in metabolic diseases and monogenic forms of diabetes and obesity. They have contributed as indispensable research tools to the development of effective drug discovery (Table 3). The obese diabetic rats named Otsuka Long Evans Tokushima Fatty (OLETF) rats, Zucker fatty (ZF) rats, Zucker Diabetic Fatty (ZDF) rats are based on the mutated cholecystokinin receptor gene, leptin gene, and leptin receptor gene, respectively 1 , 5 . On the other hand, the ob/ob mouse model with a C57BL/6J background has been used frequently to assess the potency of novel anti‐obesity medications to overcome a strong hyperphagia‐driven obesity 5 , 60 . Furthermore, this model was also used for studies investigating whether leptin action is required as a cofactor for the efficacy of specific therapies. The db/db mouse on the C57BLKS/J background is often used in studies investigating the efficacy of anti‐diabetic drugs. Similar to rat models, the leptin gene and leptin receptor gene are responsible for the phenotype of ob/ob mouse and db/db mouse, respectively.
OLETF (Otsuka Long Evans Tokushima Fatty) rat
The OLETF rat derived from the closed colony of Long‐Evans rat is a model of type 2 diabetes characterized by polydipsia, polyphagia, obesity, and diabetes 130 , 131 . In this model, diabetes develops rather late around 25 to 60 weeks of age. Male OLETF rats show a high probability (nearly all) of the development of diabetes in contrast to less frequent and later onset of diabetes in female rats 130 . The OLETF rats show higher levels of plasma insulin after 8 weeks of age, and after 60 weeks of age, the plasma insulin levels are lower than that of LETO, terminating in hypoinsulinemia 132 . After 12 weeks of age, this model shows impaired insulin secretion and insulin resistance in peripheral tissues. With the progression of insulin deficiency, there appears a marked hyperplasia of the islet and ongoing β cell demise. Concomitantly, the islets undergo compromised vascular architecture which is associated with marked insulin resistance in overtly diabetic animals 133 . Together with robust hyperlipidemia, a marked deposition of fat droplets was noted in β‐cells and contributed to the development of diabetes 134 . A lower frequency of diabetes onset in female rats is ascribed to ovarian hormones providing compensatory β‐cell hypertrophy in this model 135 . Diet restriction or exercise in juvenile stages or shortly after the onset of diabetes postpones the onset of diabetes or an improvement of diabetes. It is of note that atypical hyperplasia of bile and pancreatic ducts is often encountered in this model which may provide some clues to the increased frequencies of atypical growth of ductal neoplasms 136 .
The OLETF rats lack the cholecystokinin‐receptor (CCK‐R) type A, contributing to their phenotype 137 . CCK is a gut derived peptide hormone that functions as a peripheral satiety signal, and the OLETF rat has been useful in studying the function of CCK in metabolism. The pancreatic growth exerted in troglitazone‐treated OLETF rats is suggested for the involvement of CCK‐R in the maturation of exocrine and endocrine pancreas 138 . Multiple genetic components play a role in the development of diabetes in OLETF rats 139 . Among these, odb1 is mapped on the X‐chromosome, and odb2 on chromosome 14 which is a candidate gene for CCK‐R type A. These genes cooperate mutually and result in a marked increase in plasma glucose concentrations. This model is also used to test the validity of effective compounds on vascular complications of diabetes 140 .
ZF (Zucker fatty) rat and ZDF (Zucker diabetic fatty) rat
Among the healthy rat colonies bred in Massachusetts, obese hyperphagic rats were found in the Massachusetts Harriet GB Memorial laboratory by Zucker 141 , 142 . This strain of outbred breeding was established from a mutation named Zucker, i.e. Zucker fatty rat (mutated leptin gene). It was then transferred to the laboratory of Dr Walter Shaw at Eli Lilly Research Laboratories in Indianapolis, IN and Dr J Clark in Indiana University in 1974–75. The rats were not originally diabetic, but only showed obesity with marked insulin resistance in muscle and fatty tissues 143 , 144 . The pancreatic islets were age‐dependently hyperplastic with an increased β‐cell population 145 . Degenerative changes or cell death were not documented. Among the transferred Zucker rats, several offspring of the animals were identified as hyperglycemic from which a substrain was rederived. Inbreeding of selected pairs from this rederivation yielded nonobese but a robust diabetic phenotype (type 2 diabetes) and were named Zucker diabetic rats (mutated leptin receptor) 146 . Subsequently, two colonies of Zucker diabetic fatty (ZDF) rats were established in two laboratories of Dr Richard Peterson at Indiana University Medical School (IUMS) 147 and Dr Roger Unger at Texas Southwestern Medical Center, Dallas, USA. Although direct comparisons were not made, the phenotype of the two lines appears to be more or less similar. In these rats, a progressive depletion of β‐cells resulting from apoptosis affected by hyperglycemia as well as lipotoxic effects are the main feature of the islet pathology in these rats 148 . Accumulation of fatty acid in β‐cells with subsequent apoptotic cell death was noted 149 , 150 . Suppression of glucose transporters (GLUT) is also implicated in the onset of diabetic hyperglycemia 151 . Unger and his associates introduced the significant role of leptin in the regulation of intracellular triglycerides for the protection of lipotoxicity 152 . Genetic analysis of the ZDF rat disclosed a mutant leptin receptor (lepr −/− ) responsible for obesity and insulin resistance in ZDF rat (obese fa/fa) while the control ZDF (lean fa/‐) rats exhibited wild type expression 147 . Forced overexpression of normal leptin receptor (OB‐Rb) in ZDF islets by perfusing ZDF pancreata with a vector containing the OB‐Rb cDNA restored the insulin secretion and inhibited lipid storage 153 . The coat color of ZDF is black hooded with a black stripe down the length of the back. The ZDF rats carry an autosomal recessive defect in the β‐cell transcription machinery that is inherited independently from the mutation in Lepr. The ZDF rats are also used in the exploration of effective compounds for specific vascular complications of diabetes including neuropathy and micro‐ and macrovascular complications 154 , 155 , 156 .
Efforts were extended to establish an obese type 2 diabetes substrain of the Zucker Diabetic Sprague–Dawley (ZDSD) rat. This model was established by repeated crossing of lean ZDF rats with polygenic obese Sprague–Dawley rats to eliminate the influence of the mutated leptin pathway 157 . The ZDSD rat colony is maintained currently in Charles River Co. 158 . Exploration of the pathogenesis of diabetes complications and treatment regimens have been conducted using this model 159 .
The Koletsky rat, also known as the spontaneously hypertensive obese (SHROB) rat 160 , 161 , lacks a functional leptin receptor because of a nonsense point mutation of Lepr gene. This model was originally based on a cross between spontaneously hypertensive rats and obese Sprague Dawley rats 160 . They are hyperphagic, morbidly obese, and hypertensive with reduced energy expenditure, impaired glucose tolerance, impaired insulin sensitivity, and stunted linear growth owing to lower activity. Female and male rats that are homozygous for the mutation are infertile. In addition, and in contrast to the ZDF rat, the Koletsky rat develops hypertension starting at an age of 30 days 162 . The Koletsky rat is not prone to developing diabetes mellitus but exhibits extensive sclerotic vascular lesions.
University of California Davis‐T2DM (UCD‐T2DM) rat
The UCD‐T2DM rat was obtained from crossing obese and insulin‐resistant Sprague–Dawley rats with lean ZDF rats which were wild‐type for the leptin receptor. The inbred breeding of the rats exhibited an overt type 2 diabetes phenotype with conspicuous β‐cell loss and marked hyperglycemia, as well as impaired glucose‐stimulated insulin secretion 163 , 164 . These animals develop type 2 diabetes on a low‐fat, low‐sugar diet, and both sexes are affected. However, the onset of diabetes is earlier in males (~4–8 months of age) than in females (~8–12 months of age) 163 . Unlike ZDF rats, UCD‐T2DM rats are fertile and responsive to chronic leptin administration showing an improvement of hyperglycemia and a high HbA1c 165 . Leptin administration results in a reduction of endoplasmic reticulum stress in the liver, muscle, adipose tissue, and pancreas 166 , 167 . Impairment of glucose sensing in the gut and vagal afferent nerve activity contributes to the onset of the diabetes phenotype 168 . The UCD‐T2DM rats develop a number of complications of type 2 diabetes, including albuminuria, defects in vertebral disc and bone structure and function, and defects in brain and/or neuronal plasticity and metabolism 169 . Overall, the UCD‐T2DM rat is a versatile rodent model that can be used in the development and evaluation of novel strategies for the therapeutic management and prevention of type 2 diabetes.
ob/ob mice (lep −/− mice)
Also known as B6/ob, these animals are homozygous for the obese spontaneous mutation, Lepob (often referred to as ob or ob/ob), show obesity, hyperphagia, transient hyperglycemia, glucose intolerance, and elevated plasma insulin 170 . ob/ob mice are also hypometabolic, hypothermic, and often subfertile 171 , 172 . Hormone production from both pituitary and adrenal glands is increased 173 , and wound healing is impaired 174 . The obesity is characterized by a growth in the number and size of adipocytes 175 . Brain neuropeptide β‐endorphin is altered and related to polyphagic behavior in this model 176 . Although hyperphagia furthers the obesity, limited access to food did not influence the basal hyperglycemia nor surplus fat deposition in this model, indicating that hyperphagia alone is not responsible for the maintenance of many of the characteristic features in obesity 177 . This model is sterile but treatment with the human recombinant leptin successfully corrected this defect 178 . The physiology of obese‐hyperglycemic mice (ob/ob mice) is well summarized as an early phase of type 2 diabetes and obesity 179 , 180 .
db/db mice (lepr −/− mice)
db/db mice are used as a model of type 2 diabetes and obesity 181 , 182 . Affected mice are polyphagic, polydipsic, and polyuric. Obesity begins at 3–4 weeks of age. Mice that are homozygous for the spontaneous mutation (Lepr db ) demonstrate morbid obesity, chronic hyperglycemia, pancreatic β‐cell atrophy, and hypoinsulinemia at a later stage. Genetic and environmental factors are implicated in the phenotype expressions, as a parabiosis study demonstrated 183 , 184 . The connection of diabetes onset with obesity is complicated 185 . Islet pathology is characterized by a robust β‐cell depletion and a modest increase in α‐cells 186 . The onset of diabetes is influenced by diet behavior implicating the importance of diet composition underlying the pathologic basis of diabetes 187 and autonomic nerve regulation 188 . Compared with the original strain, crossbreeding of the mice with a BK background yielded the more severe diabetes phenotype black tan‐brachyury (BT‐BR) ob/ob mouse; an uncontrolled rise in blood sugar, a severe depletion of insulin‐producing β‐cells of the pancreatic islets, nephropathy, neuropathy, retinopathy, and myocardial disease and death by 10 months 189 , 190 , 191 , 192 . This strain was transferred to Charles River Co. from the Jackson laboratory in May 2002. Obviously, the leptin receptor gene is mutated, responsible for the onset of diabetes 180 , 193 .
OTHER ANIMAL MODELS WITH MONOGENIC DIABETES OR PANCREATIC DIABETES
Akita mouse (insulin 2 gene)
The Akita mouse was first discovered by Koizumi and his associates in Akita University, Japan 194 , 195 . The model is derived from the C57BL/6NSlc mouse with a spontaneous mutation of insulin 2 gene (Ins2+/C96Y mutation) along with incorrect proinsulin processing. The mutation causes the aggregation of misfolded proteins, resulting in the production of endoplasmic reticulum process (ER stress) 196 . Excessive ER stress exerts progressive β‐cell death, and finally results in severe insulin deficiency starting from 3 to 4 weeks of age. The clinical and metabolic symptoms are characterized by hypoinsulinemia, hyperglycemia, polydispsia, and polyuria. Untreated diabetic mice rarely survive longer than 12 weeks. Although originally considered to be a model of type 2 diabetes, the rapid and total depletion of β‐cell mass in the Akita mouse renders it an alternative to a model of type 1 diabetes, comparable to STZ‐induced diabetes in transplantation studies 197 . Brain orexin‐related peptide is involved in the phenotype of hyperphagia and anorexia in this model 198 .
This Akita mouse is used for the study of macrovascular and neuropathic disease in type 1 diabetes. This model can also be considered as a valid animal model of diabetic sympathetic autonomic neuropathy, which closely resembles the pathology of humans as well as other diabetic rodent models 199 . More recently, the mechanism of the altered pain sensation elicited by hyperglycemia was explored in this model 200 . High levels of albuminuria and systematic renal pathological symptoms have been observed in the Akita mice 201 . However, the renal structural changes in this model need clear validation as to whether these changes in fact result from diabetes or other mechanisms 202 . The Akita mice have advantages for developing nephropathy over the chemically induced STZ animal model of diabetes in which streptozotocin is toxic to the renal tissues.
WBN/Kob rat
This strain was originally developed for an investigation of stomach cancer at the University of Bonn in Germany. Dr Oichiro Kobori, a surgeon, and one of the Japanese investigators who worked in the laboratory brought this strain back to Japan. Among these animals, rats with the diabetic phenotype were found and named WBN/Kob (Slc Co., Hamamatsu, Japan). Only male rats that developed hyperglycemia, weight loss, cataract, and motor neuropathy were found 203 . In the beginning of the characterization of this model, this strain was represented as a model of type 2 diabetes. Further exploration disclosed that the exocrine pancreas underwent fibrosis and atrophy suggesting the presence of exocrine dysfunction, recapitulating the features of chronic pancreatitis 204 , 205 . Thus, this model was regarded as a model of pancreatic diabetes. Progressive pancreatic fibrosis invades both endocrine and exocrine pancreatic tissues, eliciting depletion of endocrine cells and acinar atrophy. The preclinical application of trypsin inhibitors successfully prevented the impaired exocrine pancreatic function 206 , 207 . In a similar manner, angiotensin converting enzyme inhibitor attenuated pancreatic inflammation and fibrosis 208 . Genetic analysis (QTLs) demonstrated that chromosome 7 and X; PRSS1. CFTR, and SPINK1 were increased in rats with pancreatitis in WBN rats 209 . The findings indicated that chronic pancreatitis in WBN/Kob rats is controlled by multiple genes and a genetic analysis in WBN/Kob rats might be useful for gene targeting for human chronic pancreatitis. Of interest this model exhibits characteristic motor neuropathy recapitulating chronic immune‐mediated demyelinating neuropathy 210 , as suggested by the presence of the autoimmune involvement in salivary, as well as other endocrine, tissues 211 . Recently, incorporation of a modified fatty gene into WBN rat was attempted by mating with the ZDF strain carrying the gene for Lepr +/+ obesity 212 . This model exhibits enhanced pancreatic fibrosis, indicating the role of insulin resistance in the augmented pathology.
OTHER NON‐HUMAN MAMMALIAN SPECIES
Dog, cat, minipig, rabbit, and monkey
Models of diabetes and obesity are needed for an experimental treatment approach for transplantation of tissues and organs, cannulation of vessels, and access to anatomic regions not feasible in humans (e.g. the hepatic portal vein, hepatic and renal veins, or cardio‐ and cerebro‐vascular systems). Historically, dogs (canine model) were used for studies on metabolic changes, such as the discovery of insulin and its action in the brain. Originally, extirpation of the whole pancreas was applied to obtain a diabetic condition 213 . In particular, a beagle dog named Marjorie is the most well known for the discovery of insulin by Banting and Best 214 . Nowadays, mongrel hound dogs likely serve as a convenient model because of the consistency of the development of overweight and/or obesity, prediabetes or low dose STZ‐induced diabetes mellitus within a reasonable period of time 215 . Duct ligation is also applied for the induction of diabetes in the dog, causing atrophy and marked fibrosis in the pancreas 216 .
The cat is most likely to suffer from diabetes which is commonly found by veterinarians. In common with human diabetes, the pancreatic islets undergo marked amyloid deposition 217 . The nucleic acid composition of islet amyloid polypeptide (amylin) is analogous to that of type 2 diabetes in humans 218 . Clinically, obesity is accompanied by polydipsia, polyphagia, polyuria, and loss of hair in these animals. With progression of the disease, they lose weight 219 .
The pig is another large animal model used for translational studies in research on obesity and diabetes 220 . In practice, the minipig (a small pig) is commonly used for convenience and ease of handling. Depending on the place of breeding, minipigs are called Ossabaw, Yucatan, or Gőttingen minipigs. Obesity in pigs is induced routinely by high‐energy high‐fat and/or high carbohydrate diet. Gőttingen minipigs are used most widely because they can be reared to adulthood at reasonable cost. Initially this model was considered to be a model of type 1 diabetes, occasionally requiring additional streptozotocin or alloxan injection to increase the reduction of the β‐cell mass 221 . Subsequent exploration disclosed the correlation of the β‐cell mass and the extent of insulin deficiency, providing a model of type 2 diabetes 222 . Inconsistency of hyperglycemia is ascribed to compensatory β‐cell activation in the remaining β‐cells 223 . The large size of animals allowed the sequence of perturbed insulin secretion and its association with islet pathology to be shown 224 .
There is a long history of studying non‐human primates for translational research in metabolic diseases (Figure 2). Commonly used species include rhesus macaques (Macaca mulatta), cynomologus monkeys (Macaca fascicularis), baboons (Papio species), African Green Monkeys (Chlorocebus species), and common marmocets (Callithrix jacchus) 225 , 226 . There are clear differences in the metabolic regulation of lipogenesis, thermogenesis as well as insulin‐mediated glucose utilization between monkeys and humans. Notwithstanding, studies in non‐human primates have led directly to translational studies confirming the same mechanism in humans. In particular, it is of note that the presence of islet amyloidosis with marked β‐cell loss characteristic of type 2 diabetes is common in humans and diabetic monkeys, implicating a similar etiology of islet lesions in monkeys and humans 227 . An important advantage of the use of non‐human primate models includes their close genetic relationship to humans and physiological similarity to humans. The relatively larger size of the animal is well suited to imaging studies of islets or transplantation studies 228 . The development of vascular complications of diabetes in the model is useful for studies on the treatment effects of drug compounds 229 .
Figure 2.
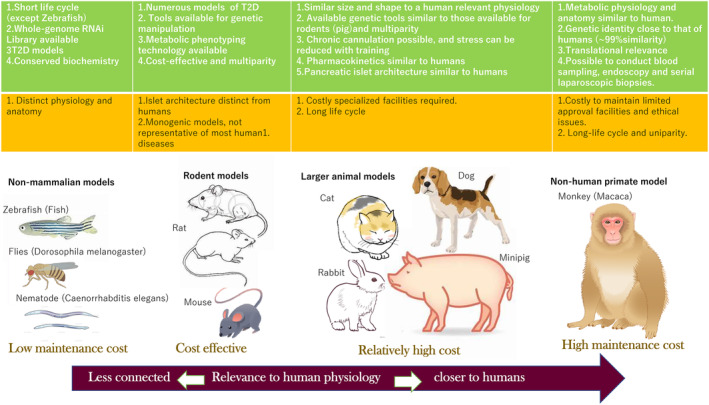
Characteristic features of different species of animal models for diabetes mellitus. There are a variety of animal models for diabetes mellitus research which have distinct advantages (green square) and shortcomings (red square). As shown in the figure, non‐mammalian models have the advantages of a low maintenance cost and a short life cycle. By contrast, the physiology of large animals, especially that of non‐human primates, closely resembles human physiology. However, these species have high maintenance costs and long life cycles. As such, these models are unfeasible for large scale, time efficient experiments. Small rodents represent a good combination of throughput and translational physiology (modified from Kleinert et al. 5 ).
NON‐MAMMALIAN SPECIES FOR MASS AND SHORT TERM STUDIES (FISH, FLIES, AND NEMATODE)
Fish (carp and zebrafish)
Classically, fish are known to suffer from high blood sugar when exposed to high osmolality. The affected fish, represented by carp, given too much food gradually become skinny and thinner. Ultrastructural investigation disclosed the degranulation of islet β‐cells in diseased fish 230 . Since such affected fish show thinned skin losing scales, and their eyes developing cataract, this condition was named “Sekoke disease (fish with falling off scales)”, comparable to the condition of human diabetes. Dr Yokote found a marked expansion of the mesangial area of glomeruli and microaneurysmal swelling of retinal microvessels in the diseased carp 231 , 232 . Recent studies suggested that the scale loss reflects skeletal fragility in the fish with the type 2 diabetes phenotype, reminiscent of impaired bone mineral composition 233 .
More recently, investigators have used zebrafish (Danio rerio) to uncover the molecular mechanisms of the metabolic alterations occurring in diabetes, with the advantage of their short lifespan and related genetic analogy to mammalians 234 , 235 . Further support for the convenience of the zebrafish is based on the evidence that they have mammalian‐like ghrelin and leptin activity 236 , 237 in addition to the concrete presence of insulin‐producing cells 238 , 239 . Similar to what occurs in mammals, β‐cell differentiation from common precursors in zebrafish requires both Notch and pancreas transcription factor (Ptf1) signaling 240 , 241 . Ablation of β‐cells in zebrafish causes hyperglycemia 242 . Other metabolic pathways will also be explored using Zebrafish to target the discovery of new mechanisms in diabetes. Zebrafish provides advantages of time‐saving analyses replacing the prolonged and transgenerational needs to explore the effects of obesity and diabetes. In addition, the availability of adequate tissues allows high‐throughput analyses such as whole‐genome RNA interference (RNAi) libraries. It may be exemplified by time‐efficient Zebrafish studies on the relationship between hyperglycemia and dementia, compared with a much longer time for humans 243 .
Nematodes (Caenorhabditis elegans) and flies (Drosophila melanogaster)
Small organisms such as the nematode Caenorhabditis elegans (C. elegans) 244 , 245 and the fruit‐fly Drosophila melanogaster 246 , 247 are also used for molecular studies on lifespan and disease. They have also served as a good model to explore the genetic and environmental factors involved in longevity 248 . Indeed, a high glucose diet shortened their lifespan 249 , whereas diet restriction of glucose successfully prolonged their life 250 . Although the metabolic pathways involved in the extended lifespan are not clear, involvement of autophagy 251 or mitochondria‐driven oxidative stress 250 is suggested to play a role in nematode studies. Metformin exposure of C. elegans successfully delayed the process of aging which was associated with altered folate and methionine metabolism 252 . In this setting, activation of AMP‐Kinase may be relevant to the extended lifespan of subjects under diet restriction 253 .
The mechanisms of diet‐induced obesity are well studied in the model of the fruitfly D. melanogaster 254 . Dietary composition specifies consumption, obesity, and lifespan in this species 255 . Interestingly, although diet restriction was confirmed to extend their lifespan, its effects depended on the amount of glucose but not of yeast which provided more potent dietary effects on the lifespan 256 . Extensive evidence suggests the presence of orthologous insulin and insulin‐like growth factor I (IGF‐I)‐like signaling peptides in Drosophila 257 . Several different cell types including neurons and hypodermic cells produce insulin and IGF‐I‐like signaling peptides 258 . Ablation of insulin producing cells induced the diabetes phenotype in Drosophila. This was not the case in C. elegans, however, in which it did not induce a comparable diabetes‐like state. Insulin resistance can be induced in Drosophila by a high‐calorie diet 259 . Collectively, non‐mammalian species possess multiple pathways of body mass control and energy balance found in mammals. They exhibit conserved features of insulin‐like signaling and secretion, develop diabetes‐like states when exposed to genetic or environmental (particularly nutritive) perturbations. After all, they serve as valuable tools to provide a conceptual basis for more detailed analyses in higher organisms.
CAUTION FOR THE USE OF ANIMAL MODELS
Ethical issues are of particular concern for animal experimentation. Minimal numbers of animals should be employed and special care may be needed for the appropriate use of animals. Unnecessary painful conditions, malnutrition, or an excessive supply of foods are all negative factors or an uncomfortable environment for the animals, which may pose stressful conditions to the animals and may influence the results. Water supply and a clean wood‐chip floor should be prepared for the animals. Investigators should pay special attention to such environmental conditions.
CONCLUDING REMARKS
In this review, I have summarized an updated list of animal models for diabetes and their characteristics with an introduction of the experimental use of non‐mammalian organisms. Investigators should be aware of the fundamental differences between humans and animals but extrapolate the translation of experimental results from animal models towards to human diabetes. There may be many other promising animal models for diabetes which are not referred to in this review, in part due to the limitation of space. I also avoided referring to the use of transgenic or knockout animal models which is an expanding area in diabetes research and should be discussed in future publications. I recommend the readers of this review to look into the related references, and only emphasize the enormous efforts of our predecessors in establishing models for human diseases.
DISCLOSURE
There is nothing to declare for the publication of this review. There is no financial support, or specific relationship to the companies for lectures or publications.
Approval of the research protocol: N/A.
Informed consent: N/A.
Registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
ACKNOWLEDGMENT
The author acknowledges the long‐term support of his mentors and collaborators.
REFERENCES
- 1. Rees DA, Alcolado JC. Animal models of diabetes mellitus. Diabetic Med 2005; 22: 359–370. [DOI] [PubMed] [Google Scholar]
- 2. King AJF. The use of animal models in diabetes research. Br J Pharmacol 2012; 166: 877–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goto Y, Kakizaki M, Kaseda N. Spontaneous diabetes produced by selective breeding of normal Wistar rats. Proc Jpn Acad 1975; 51: 80–85. [Google Scholar]
- 4. Goto Y, Kakizaki M, Masaki N. Production of spontaneous diabetic rats by repetition of selective breeding. Tohoku J Exp Med 1976; 119: 85–90. [DOI] [PubMed] [Google Scholar]
- 5. Kleinert M, Clemmensen C, Hofmann SM, et al. Animal models of obesity and diabetes mellitus. Nat Rev 2018; 14: 140–152. [DOI] [PubMed] [Google Scholar]
- 6. Pandy S, Dvorakova MC. Future perspective of diabetic animal models. Endocr Metab Immune Disord Drug Targets 2020; 20: 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 2001; 358: 221–229. [DOI] [PubMed] [Google Scholar]
- 8. McGonigle P, Ruggeri B. Animal models of human disease: challenging in enabling translation. Biochem Pharmacol 2014; 87: 162–171. [DOI] [PubMed] [Google Scholar]
- 9. Report of the Japan Society of Experimental Diabetes and Obesity Working Group (in Japanese) , Yagihashi S, Terauchi Y, et al. Diagnostic criteria of diabetes in experimental animal models. J Jpn Soc Diabetes 2010; 53: 379–384. [Google Scholar]
- 10. Nakhooda AF, Like AA, Chappel CI, et al. The spontaneously diabetic Wistar rat: metabolic and morphologic studies. Diabetes 1977; 26: 100–112. [DOI] [PubMed] [Google Scholar]
- 11. Mordes JP, Bortell R, Groen H, et al. Autoimmune diabetes mellitus in the BB rat. In: Sima AAF, Shafrir E (eds). Animal Models of Diabetes. A Primer, Vol. 2. Amsterdam: Harwood Academic Publishers, Frontiers in Animal Diabetes Research, 2001; 1–41. [Google Scholar]
- 12. Jacob HJ, Petterson A, Wilson L, et al. Genetic dissection of autoimmune type 1 diabetes in the BB rat. Nat Genet 1992; 2: 56–60. [DOI] [PubMed] [Google Scholar]
- 13. Wallis RH, Wang K, Marandi L, et al. Type 1 diabetes in the BB rat: a polygenic disease. Diabetes 2009; 58: 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sima AAF, Brismar T, Yagihashi S. Neuropathies encountered in the spontaneously diabetic BB Witar rat. In: Dyck PJ, Thomas PK, Asbury AK, et al. (eds). Diabetic Neuropathy. Philadelphia, PA: WB Sounders Co., 1987; 253–265. [Google Scholar]
- 15. Wallis RH, Wang K, Dabrowski D, et al. A novel susceptibility locus on rat chromosome 8 affects spontaneous but not experimentally induced type 1 diabetes. Diabetes 2007; 56: 1731–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klöting I, Kovacs P, van den Brandt J. BB/OK rat, BB/SHR: congenic BB.SHR (D4Mit6‐Npy‐Spr) rats: a new aid to dissect the genetics of obesity. Obes Res 2002; 10: 1074–1077. [DOI] [PubMed] [Google Scholar]
- 17. Makino S, Kunimoto K, Muraoka Y, et al. Breeding of non‐obese, diabetic strain of mice. Exp Anim 1980; 29: 1–13. [DOI] [PubMed] [Google Scholar]
- 18. Makino S, Muraoka Y, Kishimoto Y, et al. Genetic analysis for insulitis in NOD mice. Exp Anim 1985; 34: 425–432. [DOI] [PubMed] [Google Scholar]
- 19. Yoon JW, Yoon CS, Lim HW, et al. Control of autoimmune diabetes in NOD mice by GAD expression or suppression in beta cells. Science 1999; 284: 1183–1187. [DOI] [PubMed] [Google Scholar]
- 20. Jun HS, Yoon CS, Zbytnuik L, et al. The role of macrophages in T cell‐mediated autoimmune diabetes in nonobese diabetic mice. J Exp Med 1999; 189: 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hattori M, Buse J, Jackson R, et al. The NOD mouse: recessive diabetogenic gene in the major histocompatibility complex. Science 1986; 231: 733–735. [DOI] [PubMed] [Google Scholar]
- 22. Todd JA, Aitman T, Conall R, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature 1991; 351: 542–547. [DOI] [PubMed] [Google Scholar]
- 23. Yang Y, Santamaria P. Lessons on autoimmune diabetes from animal models. Clin Sci (Lond) 2006; 110: 627–639. [DOI] [PubMed] [Google Scholar]
- 24. von Herrath M, Nepom GT. Animal models of human type 1 diabetes. Nat Immunol 2009; 10: 129–132. [DOI] [PubMed] [Google Scholar]
- 25. Coleman DL, Kuzuva JE, Leiter EH. Effect of diet on incidence of diabetes in nonobese diabetic mice. Diabetes 1990; 39: 432–436. [DOI] [PubMed] [Google Scholar]
- 26. Mori Y, Suko M, Okudaira H, et al. Preventive effects of cyclosporin on diabetes in NOD mice. Diabetologia 1986; 29: 244–247. [DOI] [PubMed] [Google Scholar]
- 27. Atlan‐Gepner C, Bouabdallah R, Valero R, et al. A cyclophosphamide‐induced autoimmune diabetes. Lancet 1998; 352: 373–374. [DOI] [PubMed] [Google Scholar]
- 28. Caquard M, Ferret‐Bernard S, Haurogné K, et al. Diabetes acceleration by cyclophosphamide in the non‐obese diabetic mouse is associated with differentiation of immunosuppressive monocytes into immunostimulatory cells. Immunol Lett 2010; 129: 85–93. [DOI] [PubMed] [Google Scholar]
- 29. Aldrich VR, Hernandez‐Rovira BB, Chandwani A, et al. NOD mice‐good model for T1D but not without limitations. Cell Transplant 2020; 29: 963689720939127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidt RE, Dorsey DA, Beaudet LN, et al. Non‐obese diabetic mice rapidly develop dramatic sympathetic neuritic dystrophy: a new experimental model of diabetic autonomic neuropathy. Am J Pathol 2003; 163: 2077–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bour‐Jordan H, Thompson HL, Giampaolo JR, et al. Distinct genetic control of autoimmune neuropathy and diabetes in the non‐obese diabetic background. J Autoimmn 2013; 45: 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soliven B. Immune mechanisms in spontaneously occurring CIDP in NOD mice. J Periph Nerv Syst 2011; 16(Suppl 1): 56–59. [DOI] [PubMed] [Google Scholar]
- 33. Segev Y, Landau D, Marbach M, et al. Renal hypertrophy in hyperglycemin non‐obese diabetic mice is associated with persistent renal accumulation of insulin‐like growth factor‐1. J Am Soc Nephrol 1997; 8: 436–444. [DOI] [PubMed] [Google Scholar]
- 34. Xiao X, Ma B, Dong B, et al. Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice. J Autoimmun 2009; 32: 85–93. [DOI] [PubMed] [Google Scholar]
- 35. Lenzen S, Tiedge M, Elsner M, et al. The LEW.1AR1/Ztm‐iddm rat: a new model of spontaneous insulin‐dependent diabetes mellitus. Diabetologia 2001; 44: 1189–1196. [DOI] [PubMed] [Google Scholar]
- 36. Jörns A, Kubat B, Tiedge M, et al. Pathology of the pancreas and other organs in the diabetic LEW.1AR1/Ztm‐iddm rat, a new model of spontaneous insulin‐dependent diabetes mellitus. Virchows Arch 2004; 444: 183–189. [DOI] [PubMed] [Google Scholar]
- 37. Jörns A, Günther A, Hedrich HJ, et al. Immune cell infiltration, cytokine expression, and beta‐cell apoptosis during the development of type 1 diabetes in the spontaneously diabetic LEW.1AR1/Ztm‐iddm rat. Diabetes 2005; 54: 2041–2052. [DOI] [PubMed] [Google Scholar]
- 38. Jörns A, Wedekind D, Jähne J, et al. Pancreas pathology of latent autoimmune diabetes in adults (LADA) in patients and in a LADA rat model compared with type 1 diabetes. Diabetes 2020; 69: 624–633. [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi T, Tanaka S, Harii N, et al. Immunopathological and genetic features in slowly progressive insulin‐dependent diabetes mellitus and latent autoimmune diabetes in adults. Ann N Y Acad Sci 2006; 1079: 60–66. [DOI] [PubMed] [Google Scholar]
- 40. Lenzen S. Animal models of human type 1 diabetes for evaluating combination therapies and successful translation to the patient with type 1 diabetes. Diabetes Metab Res Rev 2017; 33: e2915. [DOI] [PubMed] [Google Scholar]
- 41. Jörns A, Rath KJ, Terbish T, et al. Diabetes prevention by immunomodulatory FTY720 treatment in the LEW.1AR1‐iddm rat despite immune cell activation. Endocrinology 2010; 151: 3555–3565. [DOI] [PubMed] [Google Scholar]
- 42. Kawano K, Hirashima T, Mori S, et al. New inbred strain of long‐evans Tokushima lean rats with IDDM without lymphopenia. Diabetes 1991; 40: 1375–1381. [DOI] [PubMed] [Google Scholar]
- 43. Komeda K, Noda M, Terao K, et al. Establishment of two substrains, diabetes‐prone and non‐diabetic, from long‐evans Tokushima lean (LETL) rats. Endocr J 1998; 45: 737–744. [DOI] [PubMed] [Google Scholar]
- 44. Jörns A, Arndt T, Meyer zu Vilsendorf A, et al. Islet infiltration, cytokine expression and beta cell death in the NOD mouse, BB rat, Komeda rat, LEW.1AR1‐iddm rat and humans with type 1 diabetes. Diabetologia 2014; 57: 512–521. [DOI] [PubMed] [Google Scholar]
- 45. Yokoi N, Kanazawa M, Kitada K, et al. A non‐MHC locus essential for autoimmune type I diabetes in the Komeda diabetes‐prone rat. J Clin Invest 1997; 100: 2015–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lukens FD. Alloxan diabetes. Physiol Rev 1948; 28: 304–330. [DOI] [PubMed] [Google Scholar]
- 47. Rakieten N, Rakieten ML, Nadkarni MR. Studies on the diabetogenic action of streptozotocin (NSC‐37917). Cancer Chemother Rep 1963; 29: 91–98. [PubMed] [Google Scholar]
- 48. Eizirik DL, Pipeleers DG, Ling Z, et al. Major species differences between humans and rodents in the susceptibility to pancreatic beta cell injury. Proc Natl Acad Sci USA 1994; 91: 9253–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lenzen S. The mechanisms of alloxan‐ and streptozotocin‐induced diabetes. Diabetologia 2008; 51: 216–226. [DOI] [PubMed] [Google Scholar]
- 50. Dufrane D, van Steenberghe M, Guiot Y, et al. Streptpzotocin‐induced diabetes in large animals (pigs/primates): role of GLUT2 transporter and beta cell plasticity. Transplantation 2006; 81: 36–45. [DOI] [PubMed] [Google Scholar]
- 51. Tyrberg B, Andersson A, Borg LA. Species differences in susceptibility of transplanted and cultured pancreatic islets to the beta‐cell toxin alloxan. Gen Comp Endocrinol 2001; 122: 238–251. [DOI] [PubMed] [Google Scholar]
- 52. Lee JH, Yag SH, Oh JM, et al. Pharmacokinetics of drugs in rats with diabetes mellitus induced by alloxan or streptozocin: comparison with those in patients with type 1 diabetes mellitus. J Pharm Pharmacol 2010; 62: 1–23. [DOI] [PubMed] [Google Scholar]
- 53. Like AA, Rossini AA. Streptozotocin‐induced pancreatic insulitis: new model of diabetes mellitus. Science 1986; 193: 415–417. [DOI] [PubMed] [Google Scholar]
- 54. Wang Z, Gleichmann H. GLUT2 in pancreatic islets: crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice. Diabetes 1998; 47: 50–56. [DOI] [PubMed] [Google Scholar]
- 55. Craighead JE, Kanich RE, Kessler JB. Lesions of the islets of Langerhans in encephalomyocarditis virus‐infected mice with diabetes mellitus‐like disease. I. Acute lesions. Am J Pathol 1974; 74: 287–300. [PMC free article] [PubMed] [Google Scholar]
- 56. D'Andrea BJ, Wilson GL, Craighead JE. Effect of genetic obesity in mice on the induction of diabetes by encephalomyocarditis virus. Diabetes 1981; 30: 451–454. [DOI] [PubMed] [Google Scholar]
- 57. Yoon JW, McClintock PR, Bachurski CJ, et al. Virus‐induced diabetes mellitus. No evidence for immune mechanisms in the destruction of beta‐cells by the D‐variant of encephalomyocarditis virus. Diabetes 1985; 34: 922–925. [DOI] [PubMed] [Google Scholar]
- 58. Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol Med 2010; 16: 407–416. [DOI] [PubMed] [Google Scholar]
- 59. Attané C, Peyot ML, Lussier R, et al. Differential insulin secretion of high‐fat diet‐fed C57BL/6NN and C57BL/6NJ mice: implications of mixed genetic background in metabolic studies. PLoS One 2016; 11: e0159165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fontane DA, Davis DB. Attention to background strain is essential for metabolic research: C57BL/6 and the international knockout mouse consortium. Diabetes 2016; 65: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ostenson CG, Khan A, Abdel‐Halim SM, et al. Abnormal insulin secretion and glucose metabolism in pancreatic islets from the spontaneously diabetic GK rat. Diabetologia 1993; 36: 3–8. [DOI] [PubMed] [Google Scholar]
- 62. Portha B, Lacraz G, Kergoat M, et al. The GK rat beta‐cell: a prototype for the diseased human beta‐cell in type 2 diabetes? Mol Cell Endocrinol 2009; 297: 73–85. [DOI] [PubMed] [Google Scholar]
- 63. Koyama M, Wada R, Sakuraba H, et al. Accelerated loss of islet beta cells in sucrose‐fed Goto‐Kakizaki rats, a genetic model of non‐insulin‐dependent diabetes mellitus. Am J Pathol 1998; 153: 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Homo‐Delarche F, Calderari S, Irminger JC, et al. Islet inflammation and fibrosis in a spontaneous model of type 2 diabetes, the GK rat. Diabetes 2006; 55: 1625–1633. [DOI] [PubMed] [Google Scholar]
- 65. Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid and diabetes mellitus. Physiol Rev 2011; 91: 795–826. [DOI] [PubMed] [Google Scholar]
- 66. Yagihashi S, Goto Y, Kakizaki M, et al. Thickening of glomerular basement membrane in spontaneously diabetic rats. Diabetologia 1978; 15: 309–312. [DOI] [PubMed] [Google Scholar]
- 67. Wada R, Koyama M, Mizukami H, et al. Effect of long‐term treatment with a‐glucosidase inhibitor on the peripheral nerve function and structure in Goto‐Kakizaki rats: a genetic model for type 2 diabetes. Diabetes Metab Res Rev 1999; 15: 332–337. [DOI] [PubMed] [Google Scholar]
- 68. Williams J, Floege J. Association of prolonged hyperglycemia with glomerular hypertrophy and renal basement membrane thickening in the Goto Kakizaki model of non‐insulin‐dependent diabetes mellitus. Am J Kidney Dis 2001; 37: 400–410. [DOI] [PubMed] [Google Scholar]
- 69. Gauguier D, Froguel P, Parent V, et al. Chromosomal mapping of genetic loci associated with non‐insulin dependent diabetes in the GK rat. Nat Genet 1996; 12: 38–43. [DOI] [PubMed] [Google Scholar]
- 70. Galli J, Fakhrai‐Rad H, Kamel A, et al. Pathophysiological and genetic characterization of the major diabetes locus in GK rats. Diabetes 1999; 48: 2463–2470. [DOI] [PubMed] [Google Scholar]
- 71. Bourgneuf C, Bailbé D, Lamazière A, et al. The Goto‐Kakizaki rat is a spontaneous prototypical rodent model of polycystic ovary syndrome. Nat Commun 2021; 12: 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shinohara M, Masuyama T, Shoda T, et al. A new spontaneously diabetic non‐obese Torii rat strain with severe ocular complications. Int J Exp Diabetes Res 2000; 1: 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kakehashi A, Saito Y, Mori K, et al. Characteristics of diabetic retinopathy in SDT rats. Diabetes Metab Res Rev 2006; 22: 455–461. [DOI] [PubMed] [Google Scholar]
- 74. Sasase T, Ohta T, Masuyama T, et al. The spontaneously diabetic torii rat: an animal model of nonobese type 2 diabetes with severe diabetic complications. J Diabetes Res 2013; 2013: 976209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Masuyama T, Fuse M, Yokoi N, et al. Genetic analysis for diabetes in a new rat model of nonobese type 2 diabetes, Spontaneously Diabetic Torii rat. Biochem Biophys Res Commun 2003; 25: 196–206. [DOI] [PubMed] [Google Scholar]
- 76. Masuyama T, Katsuda Y, Shinohara M. A novel model of obesity‐related diabetes: introgression of the Lepr(fa) allele of the Zucker fatty rat into nonobese Spontaneously Diabetic Torii (SDT) rats. Exp Anim 2005; 54: 13–20. [DOI] [PubMed] [Google Scholar]
- 77. Maekawa T, Tadaki H, Sasase T, et al. Pathophysiological profiles of SDT fatty rats, a potential new diabetic peripheral neuropathy model. J Pharmacol Toxicol Methods 2017; 88: 160–166. [DOI] [PubMed] [Google Scholar]
- 78. Marquié G, Duhault J, Jacotot B. Diabetes mellitus in sand rats (Psammomys obesus). Metabolic pattern during development of the diabetic syndrome. Diabetes 1984; 33: 438–443. [DOI] [PubMed] [Google Scholar]
- 79. Shafrir E. Animal models of non‐insulin‐dependent diabetes. Diabetes Metab Rev 1992; 8: 179–208. [DOI] [PubMed] [Google Scholar]
- 80. Leibowitz G, Yuli M, Donath MY, et al. β‐cell glucotoxicity in the Psammomys obesus model of type 2 diabetes. Diabetes 2001; 50(Suppl 1): S113–S117. [DOI] [PubMed] [Google Scholar]
- 81. Leibowitz G, Ferber S, Apelqvist A, et al. IPF1/PDX1 deficiency and β‐cell dysfunction in Psammomys obesus, an animal with type 2 diabetes. Diabetes 2001; 50: 1799–1806. [DOI] [PubMed] [Google Scholar]
- 82. Kaiser N, Yuli M, Uçkaya G, et al. Dynamic changes in β‐cell mass and pancreatic insulin during the evolution of nutrition‐dependent diabetes in psammomys obesus: impact of glycemic control. Diabetes 2005; 54: 138–145. [DOI] [PubMed] [Google Scholar]
- 83. Kaiser N, Nesher R, Donath MY, et al. Psammomys obesus, a model for environment‐gene interactions in type 2 diabetes. Diabetes 2005; 54(Suppl 2): S137–S144. [DOI] [PubMed] [Google Scholar]
- 84. Saïdi T, Mbarek S, Omri S, et al. The sand rat, Psammomys obesus, develops type 2 diabetic retinopathy similar to humans. Invest Ophthalmol Vis Sci 2011; 52: 8993–9004. [DOI] [PubMed] [Google Scholar]
- 85. Bielschowsky M, Bielschowsky F. A new strain of mice with hereditary obesity. Proc Univ Otago Med School 1953; 31: 29–31. [Google Scholar]
- 86. Bielschowsky M, Goodall CM. Origin of inbred NZ mouse strains. Cancer Res 1970; 30: 834–836. [PubMed] [Google Scholar]
- 87. Harrison LC, Itin A. A possible mechanism for insulin resistance and hyperglycemia in NZO mice. Nature 1979; 279: 334–336. [DOI] [PubMed] [Google Scholar]
- 88. Igel M, Becker W, Herberg L, et al. Hyperleptinemia, leptin resistance, and polymorphic leptin receptor in the New Zealand obese mouse. Endocrinology 1997; 138: 4234–4239. [DOI] [PubMed] [Google Scholar]
- 89. Andrikopoulos S, Thorburn AW, Proietto J. The New Zealand obese mouse: a polygenic model of type 2 dabetes. In: Sima AAF, Shafrir E (eds). Animal Models of Diabetes. A Primer, Vol. 2. Amsterdam: Harwood Academic Publishers, Frontiers in Animal Diabetes Research, 2001; 171–183. [Google Scholar]
- 90. Nakamura M, Yamada K. Studies on a diabetic (KK) strain of the mouse. Diabetologia 1967; 3: 212–221. [DOI] [PubMed] [Google Scholar]
- 91. Iwatsuka H, Shino A. Studies on diabetogenic action of obesity in mice congenital insulin resistance of KK mice. Endocrinol Jpn 1970; 17: 535–540. [DOI] [PubMed] [Google Scholar]
- 92. Iwatsuka H, Shino A, Taketomi S. Streptozotocin resistance of the genetically diabetic KK mouse. Diabetes 1974; 23: 856–857. [DOI] [PubMed] [Google Scholar]
- 93. Michaud EJ, Bultman SJ, Klebig ML, et al. A molecular model for the genetic and phenotypic characteristics of the mouse lethal yellow (ay) mutation. Proc Natl Acad Sci USA 1994; 91: 2562–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Miura T, Koide T, Ohichi R, et al. Effect of acarbose (α‐glucosidase inhibitor) on disaccharase activity in small intestine in KK‐ay and ddY mice. J Nutr Sci Vitaminol (Tokyo) 1998; 44: 371–379. [DOI] [PubMed] [Google Scholar]
- 95. Yotsumoto T, Naitoh T, Kanaki T, et al. A retinoid X receptor antagonist, HX531, improves leptin resistance without increasing plasma leptin level in KK‐Aay mice under normal dietary conditions. Metabolism 2005; 54: 573–578. [DOI] [PubMed] [Google Scholar]
- 96. Shibata M, Yasuda B. New experimental congenital diabetic mice (N.S.Y. mice). Tohoku J Exp Med 1980; 130: 139–142. [DOI] [PubMed] [Google Scholar]
- 97. Ueda H, Ikegami H, Kawaguchi Y, et al. Genetic analysis of late‐onset type 2 diabetes in a mouse model of human complex trait. Diabetes 1999; 48: 1168–1174. [DOI] [PubMed] [Google Scholar]
- 98. Ueda H, Ikegami H, Kawaguchi Y, et al. Age‐dependent changes in phenotypes and candidate gene analysis in a polygenic animal model of type II diabetes mellitus; NSY mouse. Diabetologia 2000; 43: 932–948. [DOI] [PubMed] [Google Scholar]
- 99. Ikegami H, Fujisawa T, Ogihara T. Mouse models of type 1 and type 2 diabetes derived from the same closed colony: genetic susceptibility shared between two types of diabetes. ILAR J 2004; 45: 268–277. [DOI] [PubMed] [Google Scholar]
- 100. Hamada Y, Ikegami H, Ueda H, et al. Insulin secretion to glucose as well as nonglucose stimuli is impaired in spontaneously diabetic Nagoya‐Shibata‐Yasuda mice. Metabolism 2001; 50: 1282–1285. [DOI] [PubMed] [Google Scholar]
- 101. Babaya N, Fujisawa T, Nojima K, et al. Direct evidence for susceptibility genes for type 2 diabetes on mouse chromosomes 11 and 14. Diabetologia 2010; 53: 1362–1371. [DOI] [PubMed] [Google Scholar]
- 102. Kaneko K, Chikamoto A, Hsu JC, et al. Effects of environmental enrichment on autonomic nervous activity in NSY mice. Exp Anim 2020; 69: 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ohno T, Miyasaka Y, Yoshida K, et al. A novel model mouse for type 2 diabetes mellitus with early onset and persistent hyperglycemia. Exp Anim 2022; 71: 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nagao M, Asai A, Kawahara M, et al. Selective breeding of mice for different susceptibilities to high fat diet‐induced glucose intolerance: development of two novel mouse lines, selectively bred diet‐induced glucose intolerance‐prone and ‐resistant. J Diabetes Investig 2012; 3: 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nagao M, Asai A, Inaba W, et al. Characterization of pancreatic islets in two selectively bred mouse lines with different susceptibilities to high‐fat diet‐induced glucose intolerance. PLoS One 2014; 9: e84725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Nagao M, Esguerra JLS, Wendt A, et al. Selectively bred diabetes models: GK rats, NSY mice, and ON mice. Methods Mol Biol 2020; 2128: 25–54. [DOI] [PubMed] [Google Scholar]
- 107. Asai A, Nagao M, Hayakawa K, et al. Leptin production capacity determines food intake and susceptibility to obesity‐induced diabetes in Oikawa‐Nagao diabetes‐prone and diabetes‐resistant mice. Diabetologia 2020; 63: 1836–1846. [DOI] [PubMed] [Google Scholar]
- 108. Asai A, Shuto Y, Nagao M, et al. Metformin attenuates early‐stage atherosclerosis in mildly hyperglycemic Oikawa‐Nagao mice. J Atheroscler Thromb 2019; 26: 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kim JH, Sen S, Avery CS, et al. Genetic analysis of a new mouse model for non‐insulin‐dependent diabetes. Genomics 2001; 74: 273–286. [DOI] [PubMed] [Google Scholar]
- 110. Sung YY, Lee YS, Jung WH, et al. Glucose intolerance in young TallyHo mice is induced by leptin‐mediated inhibition of insulin secretion. Biochem Biophys Res Commun 2005; 338: 1779–1787. [DOI] [PubMed] [Google Scholar]
- 111. Kim JH, Stewart TP, Soltani‐Bejnood M, et al. Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. J Endocrinol 2006; 191: 437–446. [DOI] [PubMed] [Google Scholar]
- 112. Didion SP, Lynch CM, Faraci FM. Cerebral vascular dysfunction in TallyHo mice: a new model of type II diabetes. Am J Physiol Heart Circ Physiol 2007; 292: H1579–H1583. [DOI] [PubMed] [Google Scholar]
- 113. Rhee SD, Sung YY, Lee YS, et al. Obesity of TallyHO/JngJ mouse is due to increased food intake with early development of leptin resistance. Exp Clin Endocrinol Diabetes 2011; 119: 243–251. [DOI] [PubMed] [Google Scholar]
- 114. Kim JH, Saxton AM. The TALLYHO mouse as a model of human type 2 diabetes. Methods Mol Biol 2012; 933: 75–87. [DOI] [PubMed] [Google Scholar]
- 115. Devlin MJ, Van Vliet M, Motyl K, et al. Early‐onset type 2 diabetes impairs skeletal acquisition in the male TALLYHO/JngJ mouse. Endocrinology 2014; 155: 3806–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ramasubramanian B, Reddy PH. Are TallyHo mice a true mouse model for type 2 diabetes and Alzheimer's disease? J Alzheimers Dis 2019; 72(s1): S81–S93. [DOI] [PubMed] [Google Scholar]
- 117. Bhatti JS, Thamarai K, Kandimalla R, et al. Mitochondria‐targeted small peptide, SS31 ameliorates diabetes induced mitochondrial dynamics in male TallyHO/JngJmMice. Mol Neurobiol 2021; 58: 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Suzuki W, Iizuka S, Tabuchi M, et al. A new mouse model of spontaneous diabetes derived from ddY strain. Exp Anim 1999; 48: 181–189. [DOI] [PubMed] [Google Scholar]
- 119. Hirayama I, Yi Z, Izumi S, et al. Genetic analysis of obese diabetes in the TSOD mouse. Diabetes 1999; 48: 1183–1191. [DOI] [PubMed] [Google Scholar]
- 120. Iizuka S, Suzuki W, Tabuchi M, et al. Diabetic complications in a new animal model (TSOD mouse) of spontaneous NIDDM with obesity. Exp Anim 2005; 54: 71–83. [DOI] [PubMed] [Google Scholar]
- 121. Takahashi A, Tabuchi M, Suzuki W, et al. Insulin resistance and low sympathetic nerve activity in the Tsumura Suzuki obese diabetic mouse: a new model of spontaneous type 2 diabetes mellitus and obesity. Metabolism 2006; 55: 1664–1669. [DOI] [PubMed] [Google Scholar]
- 122. Nishida T, Tsuneyama K, Fujimoto M, et al. Spontaneous onset of nonalcoholic steatohepatitis and hepatocellular carcinoma in a mouse model of metabolic syndrome. Lab Invest 2013; 93: 230–241. [DOI] [PubMed] [Google Scholar]
- 123. Tsuneyama K, Nishitsuji K, Matsumoto M, et al. Animal models for analyzing metabolic syndrome‐associated liver diseases. Pathol Int 2017; 67: 539–546. [DOI] [PubMed] [Google Scholar]
- 124. Rossini AA, Like AA, Chick WL, et al. Studies of streptozotocin‐induced insulitis and diabetes. Proc Natl Acad Sci USA 1977; 74: 2485–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Rossini AA, Appel MC, Williams RM, et al. Genetic influence of the streptozotocin‐induced insulitis and hyperglycemia. Diabetes 1977; 26: 916–920. [DOI] [PubMed] [Google Scholar]
- 126. Like AA, Appel MC, Williams RM, et al. Streptozotocin‐induced pancreatic insulitis in mice. Morphologic and physiologic studies. Lab Invest 1978; 38: 470–486. [PubMed] [Google Scholar]
- 127. Reed MJ, Meszaros K, Entes LJ, et al. A new rat model of type 2 diabetes: the fat‐fed, streptozotocin‐treated rat. Metabolism 2000; 49: 1390–1394. [DOI] [PubMed] [Google Scholar]
- 128. Srinivasan K, Viswanad B, Asrat L, et al. Combination of high‐fat diet‐fed and low‐dose streptozotocin treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res 2005; 52: 313–320. [DOI] [PubMed] [Google Scholar]
- 129. Yorek MA. Alternatives to the streptozotocin‐diabetic rodent. Int Rev Neurobiol 2016; 127: 89–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kawano K, Hirashima T, Mori S, et al. Spontaneous long‐term hyperglycemic rat with diabetic complications. OLETF (Otsuka Long‐Evans Tokushima Fatty) strain. Diabetes 1992; 41: 1422–1428. [DOI] [PubMed] [Google Scholar]
- 131. Kawano K, Hirashima T, Mori S, et al. OLETF (Otsuka Long‐Evans Tokushima Fatty) rat: a new NIDDM rat strain. Diab Res Clin Pract 1994; 24(Suppl): S317–S320. [DOI] [PubMed] [Google Scholar]
- 132. Zhu M, Noma Y, Mizuno A, et al. Poor capacity for proliferation of pancreatic beta‐cells in Otsuka‐Long‐Evans‐Tokushima fatty rat: a model of spontaneous NIDDM. Diabetes 1996; 45: 941–946. [DOI] [PubMed] [Google Scholar]
- 133. Mizuno A, Noma Y, Kuwajima M, et al. Changes in islet capillary angioarchitecture coincide with impaired B‐cell function but not with insulin resistance in male Otsuka‐long‐Evans‐Tokushima fatty rats: dimorphism of the diabetic phenotype at an advanced age. Metabolism 1999; 48: 477–483. [DOI] [PubMed] [Google Scholar]
- 134. Man ZW, Zhu M, Noma Y, et al. Impaired β‐cell function and deposition of fat droplets in the pancreas as a consequence of hypertriglyceridemia in OLETF rat, a model of spontaneous NIDDM. Diabetes 1997; 46: 1718–1724. [DOI] [PubMed] [Google Scholar]
- 135. Zhu M, Mizuno A, Kuwajima M, et al. Ovarian hormone‐induced beta‐cell hypertrophy contributes to the homeostatic control of beta‐cell mass in OLETF female rat, a model of type II diabetes. Diabetologia 1998; 41: 799–805. [DOI] [PubMed] [Google Scholar]
- 136. Kondo M, Kanemoto N, Taniguchi Y, et al. Atypical hyperplasia of choledocho‐pancreatic duct epithelium in an Otsuka Long Evans Tokushima fatty strain of rats. Pathol Int 2000; 50: 126–135. [DOI] [PubMed] [Google Scholar]
- 137. Funakoshi A, Miyasaka K, Jimi A, et al. Little or no expression of the cholecystokinin‐A receptor gene in the pancreas of diabetic rats (Otsuka Long‐Evans Tokushima Fatty = OLETF rats). Biochem Biophys Res Commun 1994; 199: 482–488. [DOI] [PubMed] [Google Scholar]
- 138. Jia DM, Fukumitsu KI, Tabaru A, et al. Troglitazone stimulates pancreatic growth in congenitally CCK‐A receptor‐deficient OLETF rats. Am J Physiol Regul Integr Comp Physiol 2001; 280: R1332–R1340. [DOI] [PubMed] [Google Scholar]
- 139. Hirashima T, Kawano K, Mori S, et al. A diabetogenic gene (ODB‐1) assigned to the X‐chromosome in OLETF rats. Diabetes Res Clin Pract 1995; 27: 91–96. [DOI] [PubMed] [Google Scholar]
- 140. Nakamura S, Makita Z, Ishikawa S, et al. Progression of nephropathy in spontaneous diabetic rats is prevented by OPB‐9195, a novel inhibitor of advanced glycation. Diabetes 1997; 46: 895–899. [DOI] [PubMed] [Google Scholar]
- 141. Zucker LM, Antoniades HN. Insulin and obesity in the Zucker genetically obese rat “fatty”. Endocrinology 1972; 90: 1320–1330. [DOI] [PubMed] [Google Scholar]
- 142. Zucker LM. Efficiency of energy utilization by the Zucker hereditarily obese rat “fatty” (38569). Proc Soc Exp Biol Med 1975; 148: 498–500. [DOI] [PubMed] [Google Scholar]
- 143. Czech MP, Richardson DK, Becker SG, et al. Insulin response in skeletal muscle and fat cells of the genetically obese Zucker rat. Metabolism 1978; 27(12 Suppl 2): 1967–1981. [DOI] [PubMed] [Google Scholar]
- 144. Clark JB, Keen S, Clark CM Jr. Studies on the regulation of insulin binding by liver plasma membranes from Zucker fatty rats. Diabetes 1982; 31: 867–873. [DOI] [PubMed] [Google Scholar]
- 145. Larsson LI, Boder GB, Shaw WN. Changes in the islets of langerhans in the obese Zucker rat. Lab Invest 1977; 36: 593–598. [PubMed] [Google Scholar]
- 146. Clark JB, Palmer CJ, Shaw WN. The diabetic Zucker fatty rat. Proc Soc Exp Biol Med 1983; 173: 68–75. [DOI] [PubMed] [Google Scholar]
- 147. Peterson RG. The Zucker diabetic fatty (ZDF) rat. In: Sima AAF, Shafrir E (eds). Animal Models of Diabetes. A Primer. Amsterdam: Frontiers in Animal Diabetes Research, Harwood Academic Publishers, 2001; 109–128. [Google Scholar]
- 148. Pick A, Clark J, Kubstrup C, et al. Role of apoptosis in failure of β‐cell mass compensation for insulin resistance and β‐cell defects in the male Zucker diabetic fatty rat. Diabetes 1998; 47: 358–364. [DOI] [PubMed] [Google Scholar]
- 149. Lee Y, Hirose H, Ohneda M, et al. Beta‐cell lipotoxicity in the pathogenesis of non‐insulin‐dependent diabetes mellitus of obese rats: impairment in adipocyte‐beta‐cell relationships. Proc Natl Acad Sci USA 1994; 91: 10878–10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Shimabukuro M, Zhou YT, Levi M, et al. Fatty acid‐induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA 1998; 95: 2498–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Orci L, Ravazzola M, Baetens D, et al. Evidence that down‐regulation of β‐cell glucose transporters in non‐insulin‐dependent diabetes may be the cause of diabetic hyperglycemia. Proc Natl Acad Sci USA 1990; 87: 9953–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Unger RH, Zhou YT, Orci L. Regulation of fatty acid homeostasis in cells: novel role of leptin. Proc Natl Acad Sci USA 1999; 96: 2327–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wang MY, Koyama K, Shimabukuro M, et al. OB‐Rb gene transfer to leptin‐resistant islets reverses diabetogenic phenotype. Proc Natl Acad Sci USA 1998; 95: 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Corsetti JP, Sparks JD, Peterson RG, et al. Effect of dietary fat on the development of non‐insulin dependent diabetes mellitus in obese Zucker diabetic fatty male and female rats. Atherosclerosis 2000; 148: 231–241. [DOI] [PubMed] [Google Scholar]
- 155. Lu X, Guo X, Karathanasis SK, et al. Rosiglitazone reverses endothelial dysfunction but not remodeling of femoral artery in Zucker diabetic fatty rats. Cardiovasc Diabetol 2010; 9: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Schmidt RE, Dorsey DA, Beaudet LN, et al. Analysis of the Zucker Diabetic Fatty (ZDF) type 2 diabetic rat model suggests a neurotrophic role for insulin/IGF‐I in diabetic autonomic neuropathy. Am J Pathol 2003; 163: 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Peterson RG, Jackson CV, Zimmerman K, et al. Characterization of the ZDSD rat: a translational model for the study of metabolic syndrome and type 2 diabetes. J Diabetes Res 2015; 2015: 487816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wang AN, Carlos J, Fraser GM, et al. Zucker diabetic‐Sprague Dawley (ZDSD) rat: type 2 diabetes translational research model. Exp Physiol 2022; 107: 265–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Peterson RG, Jackson CV, Zimmerman KM. The ZDSD rat: a novel model of diabetic nephropathy. Am J Transl Res 2017; 9: 4236–4249. [PMC free article] [PubMed] [Google Scholar]
- 160. Koletsky S. Obese spontaneously hypertensive rats – a model for study of atherosclerosis. Exp Mol Pathol 1973; 19: 53–60. [DOI] [PubMed] [Google Scholar]
- 161. Koletsky S. Animal model: obese hypertensive rat. Am J Pathol 1975; 81: 463–466. [PMC free article] [PubMed] [Google Scholar]
- 162. Koletsky S. Pathologic findings and laboratory data in a new strain of obese hypertensive rats. Am J Pathol 1975; 80: 129–142. [PMC free article] [PubMed] [Google Scholar]
- 163. Cummings BP, Digitale EK, Stanhope KL, et al. Development and characterization of a novel rat model of type 2 diabetes mellitus: the UC Davis type 2 diabetes mellitus UCD‐T2DM rat. Am J Physiol Regul Integr Comp Physiol 2008; 295: R1782–R1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Rountree AM, Reed BJ, Cummings BP, et al. Loss of coupling between calcium influx, energy consumption and insulin secretion associated with development of hyperglycaemia in the UCD‐T2DM rat model of type 2 diabetes. Diabetologia 2013; 56: 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Cummings BP, Stanhope KL, Graham JL, et al. Chronic administration of the glucagon‐like peptide‐1 analog, liraglutide, delays the onset of diabetes and lowers triglycerides in UCD‐T2DM rats. Diabetes 2010; 59: 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Cummings BP, Bettaieb A, Graham JL, et al. Subcutaneous administration of leptin normalizes fasting plasma glucose in obese type 2 diabetic UCD‐T2DM rats. Proc Natl Acad Sci USA 2011; 108: 14670–14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Cummings BP. Leptin therapy in type 2 diabetes. Diabetes Obes Metab 2013; 15: 607–612. [DOI] [PubMed] [Google Scholar]
- 168. Lee J, Cummings BP, Martin E, et al. Glucose sensing by gut endocrine cells and activation of the vagal afferent pathway is impaired in a rodent model of type 2 diabetes mellitus. Am J Physiol Regul Integr Comp Physiol 2012; 302: R657–R666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Agrawal R, Zhuang Y, Cummings BP, et al. Deterioration of plasticity and metabolic homeostasis in the brain of the UCD‐T2DM rat model of naturally occurring type‐2 diabetes. Biochim Biophys Acta 2014; 1842: 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Lindstrom P. The physiology of obese‐hyperglycemic mice [Ob/Ob mice]. Sci World J 2007; 7: 666–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ingalls AM, Dickie MM, Snell G. Obese, a new mutation in the mouse. J Hered 1950; 41: 318. [DOI] [PubMed] [Google Scholar]
- 172. Lindström P. β‐Cell function in obese‐hyperglycemic mice [Ob/Ob mice]. Adv Exp Med Biol 2010; 654: 463–477. [DOI] [PubMed] [Google Scholar]
- 173. Edwardson JA, Hough CA. The pituitary‐adrenal system of the genetically obese (Ob/Ob) mouse. J Endocrinol 1975; 65: 99–107. [DOI] [PubMed] [Google Scholar]
- 174. Seitz O, Schürmann C, Hermes N, et al. Wound healing in mice with high‐fat diet‐ or Ob gene‐induced diabetes‐obesity syndromes: a comparative study. Exp Diabetes Res 2010; 2010: 476969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Loten EG, Rabinovitch A, Jeanrenaud B. In vivo studies on lipogenesis in obese hyperglycaemic (Ob‐Ob) mice: possible role of hyperinsulinaemia. Diabetologia 1974; 10: 45–52. [DOI] [PubMed] [Google Scholar]
- 176. Margules DL, Moisset B, Lewis MJ, et al. β‐Endorphin is associated with overeating in genetically obese mice (Ob/Ob) and rats (fa/fa). Science 1978; 202: 988–991. [DOI] [PubMed] [Google Scholar]
- 177. Dubuc PU. Effects of limited food intake on the obese‐hyperglycemic syndrome. Am J Physiol 1976; 230: 1474–1479. [DOI] [PubMed] [Google Scholar]
- 178. Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet 1996; 12: 318–320. [DOI] [PubMed] [Google Scholar]
- 179. Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432. [DOI] [PubMed] [Google Scholar]
- 180. Wang B, Chandrasekera PC, Pippin JJ. Leptin‐ and leptin receptor‐deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev 2014; 10: 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science 1966; 153: 1127–1128. [DOI] [PubMed] [Google Scholar]
- 182. Coleman DL, Hummel KP. Studies with the mutation, diabetes, in the mouse. Diabetologia 1967; 3: 238–248. [DOI] [PubMed] [Google Scholar]
- 183. Coleman DL, Hummel KP. Effects of parabiosis of normal with genetically diabetic mice. Am J Physiol 1969; 217: 1298–1304. [DOI] [PubMed] [Google Scholar]
- 184. Hunt CE, Lindsey JR, Walkley SU. Animal models of diabetes and obesity, including the PBB/Ld mouse. Fed Proc 1976; 35: 1206–1217. [PubMed] [Google Scholar]
- 185. Herberg L, Coleman DL. Laboratory animals exhibiting obesity and diabetes syndromes. Metabolism 1977; 26: 59–99. [DOI] [PubMed] [Google Scholar]
- 186. Baetens D, Stefan Y, Ravazzola M, et al. Alteration of islet cell populations in spontaneously diabetic mice. Diabetes 1978; 27: 1–7. [DOI] [PubMed] [Google Scholar]
- 187. Leiter EH, Coleman DL, Eisenstein AE, et al. Dietary control of pathogenesis in C57BL/KsJ db/db mice. Metabolism 1981; 30: 554–562. [DOI] [PubMed] [Google Scholar]
- 188. Brown MR, Dyck PJ, McClearn GE, et al. Central and peripheral nervous system complications. Diabetes 1982; 31(Suppl 1 Pt 2): 65–70. [DOI] [PubMed] [Google Scholar]
- 189. Li Y, Hu Q, Li C, et al. PTEN‐induced partial epithelial‐mesenchymal transition drives diabetic kidney disease. J Clin Invest 2019; 129: 1129–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. O'Brien PD, Hur J, Hayes JM, et al. BTBR Ob/Ob mice as a novel diabetic neuropathy model: neurological characterization and gene expression analyses. Neurobiol Dis 2015; 73: 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Lee VK, Hosking BM, Holeniewska J, et al. BTBR Ob/Ob mouse model of type 2 diabetes exhibits early loss of retinal function and retinal inflammation followed by late vascular changes. Diabetologia 2018; 61: 2422–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Ye Y, Bajaj M, Yang HC, et al. SGLT‐2 inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther 2017; 31: 119–132. [DOI] [PubMed] [Google Scholar]
- 193. Chen H, Charlat O, Tartaglia LA, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996; 84: 491–495. [DOI] [PubMed] [Google Scholar]
- 194. Yoshioka M, Kayo T, Ikeda T, et al. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early‐onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 1997; 46: 887–894. [DOI] [PubMed] [Google Scholar]
- 195. Kayo T, Koizumi A. Mapping of murine diabetogenic gene mody on chromosome 7 at D7Mit258 and its involvement in pancreatic islet and beta cell development during the perinatal period. J Clin Invest 1998; 101: 2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the chop gene delays endoplasmic reticulum stress‐mediated diabetes. J Clin Invest 2002; 109: 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Bishop NH, Nelsen MK, Beard KS, et al. Differential impact of chronic hyperglycemia on humoral versus cellular primary alloimmunity. Diabetes 2017; 66: 981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Toyoshima M, Asakawa A, Fujimiya M, et al. Dimorphic gene expression patterns of anorexigenic and orexigenic peptides in hypothalamus account male and female hyperphagia in Akita type 1 diabetic mice. Biochem Biophys Res Commun 2007; 352: 703–708. [DOI] [PubMed] [Google Scholar]
- 199. Schmidt RE, Green KG, Snipes LL, et al. Neuritic dystrophy and neuropathy in Akita (Ins2(Akita)) diabetic mouse. Exp Neurol 2009; 216: 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Vastani N, Guenther F, Gentry C, et al. Impaired nociceptor in the diabetic (Ins2+/Akita) mouse. Diabetes 2018; 67: 1650–1662. [DOI] [PubMed] [Google Scholar]
- 201. Gurley SB, Mach CL, Stegbauer J, et al. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am J Physiol Renal Physiol 2010; 298: F788–F795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Harlan SM, Heinz‐Taheny KM, Sullivan JM, et al. Progressive renal disease established by renin‐coding adeno‐associated virus‐driven hypertension in diverse diabetic models. J Am Soc Nephrol 2018; 29: 477–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Tsuchitani M, Saegusa T, Narama I, et al. A new diabetic strain of rat (WBN/Kob). Lab Anim 1985; 19: 200–207. [DOI] [PubMed] [Google Scholar]
- 204. Ohashi K, Kim JH, Hara H, et al. WBN/Kob rats. A new spontaneously occurring model of chronic pancreatitis. Int J Pancreatol 1990; 6: 231–247. [PubMed] [Google Scholar]
- 205. Mori Y, Yokoyama J, Nishimura M, et al. Diabetic strain (WBN/Kob) of rat characterized by endocrine‐exocrine pancreatic impairment due to distinct fibrosis. Pancreas 1990; 5: 452–459. [DOI] [PubMed] [Google Scholar]
- 206. Shimoda I, Koizumi M, Shimosegawa T, et al. Physiological characteristics of spontaneously developed diabetes in male WBN/Kob rat and prevention of development of diabetes by chronic oral administration of synthetic trypsin inhibitor (FOY‐305). Pancreas 1993; 8: 196–203. [DOI] [PubMed] [Google Scholar]
- 207. Graf R, Schiesser M, Bimmler D. Increased secretion of the pancreatic secretory trypsin inhibitor (PSTI‐I, monitor peptide) during development of chronic pancreatitis in the WBN/Kob rat. Pancreatology 2002; 2: 108–115. [DOI] [PubMed] [Google Scholar]
- 208. Kuno A, Yamada T, Masuda K, et al. Angiotensin‐converting enzyme inhibitor attenuates pancreatic inflammation and fibrosis in male Wistar Bonn/Kobori rats. Gastroenterology 2003; 124: 1010–1019. [DOI] [PubMed] [Google Scholar]
- 209. Tshuji A, Nishikawa T, Mori M, et al. Quantitative trait locus analysis for chronic pancreatitis and diabetes mellitus in the WBN/Kob rat. Genomics 2001; 73: 365–369. [DOI] [PubMed] [Google Scholar]
- 210. Yagihashi S, Wada R, Kamijo M, et al. Peripheral neuropathy in the WBN/Kob rat with chronic pancreatitis and spontaneous diabetes. Lab Invest 1993; 68: 296–307. [PubMed] [Google Scholar]
- 211. Sakaguchi Y, Inaba M, Tsuda M, et al. The Wistar Bonn Kobori rat, a unique animal model for autoimmune pancreatitis with extrapancreatic exocrinopathy. Clin Exp Immunol 2008; 152: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Akimoto T, Nakama K, Katsuta Y, et al. Characterization of a novel congenic strain of diabetic fatty (WBN/Kob‐Lepr(fa)) rat. Biochem Biophys Res Commun 2008; 366: 556–562. [DOI] [PubMed] [Google Scholar]
- 213. von Mehring J, Minkowski O. Diabetes mellitus nach Pankreasexstirpation. Arch Exp Path Pharmacol 1889; 26: 371–387. [Google Scholar]
- 214. Banting FG, Best CH. The internal secretion of the pancreas. J Lab Clin Med 1922; 7: 256–271. [Google Scholar]
- 215. Yu EN, Winnick JJ, Edgerton DS, et al. Hepatic and whole‐body insulin metabolism during proestrus and estrus in mongrel dogs. Comp Med 2016; 66: 235–240. [PMC free article] [PubMed] [Google Scholar]
- 216. Traverso LW, Bockman DE, Pleis SK, et al. Nesidioblastosis as a mechanism to prevent fibrosis‐induced diabetes after pancreatic duct obstruction. Pancreas 1993; 8: 325–329. [DOI] [PubMed] [Google Scholar]
- 217. Johnson KH, Stevens JB. Light and electron microscopic studies of islet amyloid in diabetic cats. Diabetes 1973; 22: 81–90. [DOI] [PubMed] [Google Scholar]
- 218. Betsholtz C, Christmanson L, Engström U, et al. Structure of cat islet amyloid polypeptide and identification of amino acid residues of potential significance for islet amyloid formation. Diabetes 1990; 39: 118–122. [DOI] [PubMed] [Google Scholar]
- 219. Nelson RW, Reusch CE. Animal models of disease: classification and etiology of diabetes in dogs and cats. J Endocrinol 2014; 222: T1–T9. [DOI] [PubMed] [Google Scholar]
- 220. Renner S, Blutke A, Dobenecker B, et al. Metabolic syndrome and extensive adipose tissue inflammation in morbidly obese Göttingen minipigs. Mol Metab 2018; 16: 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Larsen MO, Rolin B. Use of the Göttingen minipig as a model of diabetes, with special focus on type 1 diabetes research. ILAR J 2004; 45: 303–313. [DOI] [PubMed] [Google Scholar]
- 222. Larsen MO, Rolin B, Raun K, et al. Evaluation of beta‐cell mass and function in the Göttingen minipig. Diabetes Obes Metab 2007; 9(Suppl 2): 170–179. [DOI] [PubMed] [Google Scholar]
- 223. Larsen MO, Rolin B, Gotfredsen CF, et al. Reduction of beta cell mass: partial insulin secretory compensation from the residual beta cell population in the nicotinamide‐streptozotocin Göttingen minipig after oral glucose in vivo and in the perfused pancreas. Diabetologia 2004; 47: 1873–1878. [DOI] [PubMed] [Google Scholar]
- 224. Larsen MO, Juhl CB, Pørksen N, et al. Beta‐cell function and islet morphology in normal, obese, and obese beta‐cell mass‐reduced Göttingen minipigs. Am J Physiol Endocrinol Metab 2005; 8: E412–E421. [DOI] [PubMed] [Google Scholar]
- 225. Hansen BC. Obesity and diabetes in monkeys. In: Bjorntorp P, Brodoff BN (eds). Obesity. New York: JB Lippincott, Williams & Wilkins Co., 1992; 256–265. [Google Scholar]
- 226. Bodkin NL. The rhesus monkey (Macaca mulatta): a unique and valuable model for the study of spontaneous diabetes mellitus ad associated conditions. In: Sima AAF, Shafrir E (eds). Animal Models of Diabetes. A Premier, Vol. 2. Amsterdam: Frontiers in Animal Diabetes Research, Harwood Academic Publishers, 2001; 309–325. [Google Scholar]
- 227. O'Brien TD, Wagner JD, Litwak KN, et al. Islet amyloid and islet amyloid polypeptide in cynomolgus macaques (Macaca fascicularis): an animal model of human non‐insulin‐dependent diabetes mellitus. Vet Pathol 1996; 33: 479–485. [DOI] [PubMed] [Google Scholar]
- 228. Eriksson O, Johnström P, Cselenyi Z, et al. In vivo visualization of β‐cells by targeting of GPR44. Diabetes 2018; 67: 182–192. [DOI] [PubMed] [Google Scholar]
- 229. Tso MO, Kurosawa A, Benhamou E, et al. Microangiopathic retinopathy in experimental diabetic monkeys. Trans Am Ophthalmol Soc 1988; 86: 389–421. [PMC free article] [PubMed] [Google Scholar]
- 230. Nakamura M, Yamada K, Yokote M. Ultrastructural aspects of the pancreatic islets in carps of spontaneous diabetes mellitus. Experientia 1971; 27: 75–76. [DOI] [PubMed] [Google Scholar]
- 231. Yokote M. Retinal and renal microangiopathy in carp with spontaneous diabetes mellitus. Adv Metab Disord 1973; 2(Suppl 2): 299–230. [DOI] [PubMed] [Google Scholar]
- 232. Wolf S. Animal model of human diseases. Diabetes mellitus. Animal. Model: sekoke: diabetes of nutritional origin in carp. Am J Pathol 1976; 85: 805–808. [PMC free article] [PubMed] [Google Scholar]
- 233. Suzuki N, Kitamura KI, Hattori A. Fish scale is a suitable model for analyzing determinants of skeletal fragility in type 2 diabetes. Endocrine 2016; 54: 575–577. [DOI] [PubMed] [Google Scholar]
- 234. Oka T, Nishimura Y, Zang L, et al. Diet‐induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC Physiol 2010; 10: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Mansfeld J, Urban N, Priebe S, et al. Branched‐chain amino acid catabolism is a conserved regulator of physiological ageing. Nat Commun 2015; 6: 10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Cruz SA, Tseng YC, Kaiya H, et al. Ghrelin affects carbohydrate‐glycogen metabolism via inhibition and glucagon stimulation in the zebrafish (Danio rerio) brain. Comp Biochem Physiol A Mol Integr Physiol 2010; 156: 190–200. [DOI] [PubMed] [Google Scholar]
- 237. Gorissen M, Bernier NJ, Nabuurs SB, et al. Two divergent leptin paralogues in zebrafish (Danio rerio) that originate early in teleostean evolution. J Endocrinol 2009; 201: 329–339. [DOI] [PubMed] [Google Scholar]
- 238. Argenton F, Zecchin E, Bortolussi M. Early appearance of pancreatic hormone‐expressing cells in the zebrafish embryo. Mech Dev 1999; 87: 217–221. [DOI] [PubMed] [Google Scholar]
- 239. Polakof S, Panserat S, Soenga JL, et al. Glucose metabolism in fish: a review. J Comp Physiol B 2012; 182: 1015–1045. [DOI] [PubMed] [Google Scholar]
- 240. Parsons MJ, Pisharath H, Yusuff S, et al. Notch‐responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev 2009; 126: 898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Lin JW, Biankin AV, Horb ME, et al. Differential requirement for ptf1a in endocrine and exocrine lineages of developing zebrafish pancreas. Dev Biol 2004; 270: 474–486. [DOI] [PubMed] [Google Scholar]
- 242. Papasani MR, Robison BD, Hardy RW, et al. Early developmental expression of two insulins in zebrafish (Danio rerio). Physiol Genom 2006; 27: 79–85. [DOI] [PubMed] [Google Scholar]
- 243. Capiotti KM, De Moraes DA, Menezes FP, et al. Hyperglycemia induces memory impairment linked to increased acetylcholinesterase activity in zebrafish (Danio rerio). Behav Brain Res 2014; 274: 319–325. [DOI] [PubMed] [Google Scholar]
- 244. Ashrafi K, Chang FY, Watts JL, et al. Genome‐wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 2003; 421: 268–272. [DOI] [PubMed] [Google Scholar]
- 245. Watts JL. Fat synthesis and adiposity regulation in Caenorhabditis elegans . Trends Endocrinol Metab 2009; 20: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Leopold P, Perrimon N. Drosophila and the genetics of the internal milieu. Nature 2007; 450: 186–188. [DOI] [PubMed] [Google Scholar]
- 247. Pospisilik JA, Schramek D, Schnidar H, et al. Drosophila genome‐wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell 2010; 140: 148–160. [DOI] [PubMed] [Google Scholar]
- 248. Fontana L, Partridge L, Longo VD. Extending healthy life span – from yeast to humans. Science 2010; 328: 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Schlotterer A, Kukudov G, Bozorgmehr F, et al. C. elegans as model for the study of high glucose‐mediated life span reduction. Diabetes 2009; 58: 2450–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Schulz TJ, Zarse K, Voigt A, et al. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 2007; 6: 280–293. [DOI] [PubMed] [Google Scholar]
- 251. Mörck C, Pilon M. Caloric restriction and autophagy in Caenorhabditis elegans . Autophagy 2007; 3: 51–53. [DOI] [PubMed] [Google Scholar]
- 252. Weir HJ, Yao P, Huynh FK, et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab 2017; 26: 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Cabreiro F, Au C, Leung KY, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 2013; 153: 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Trinh I, Boulianne GL. Modeling obesity and its associated disorders in Drosophila . Phys Ther 2013; 28: 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Skorupa DA, Dervisefendic A, Zwiener J, et al. Dietary composition specifies consumption, obesity and lifespan in Drosophila melanogaster . Aging Cell 2008; 7: 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Mair W, Piper MDW, Partridge L. Calories do not explain extension of life span by dietary restriction in drosophila. PLoS Biol 2005; 3: 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Brogiolo W, Stocker H, Ikeya T, et al. An evolutionarily conserved function of the drosophila insulin receptor and insulin‐like peptides in growth control. Curr Biol 2001; 11: 213–221. [DOI] [PubMed] [Google Scholar]
- 258. Rulifson EJ, Kim SK, Nusse R. Ablation of insulin‐producing neurons in flies: growth and diabetic phenotypes. Science 2002; 296: 1118–1120. [DOI] [PubMed] [Google Scholar]
- 259. Birse RT, Choi J, Reardon K, et al. High fat diet‐induced obesity and heart dysfunction is regulated by the TOR pathway in drosophila. Cell Metab 2010; 12: 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]