

SUMMARY
Carboxylic acids are valuable building blocks for pharmaceutical discovery because of their chemical stability, commercial availability, and structural diversity. Decarboxylative coupling reactions enable versatile functionalization of these feedstock chemicals, but many of the most general methods require prefunctionalization of carboxylic acids with redox-active moieties. These internal oxidants can be costly, their installation impedes rapid library synthesis, and their use results in environmentally problematic organic byproducts. We report herein a method for the direct decarboxylative cross-coupling of native carboxylic acids with nucleophilic coupling partners mediated by inexpensive, terrestrially abundant, and nontoxic Fe(III) salts. This method involves an initial photochemical decarboxylation followed by radical-polar crossover, which enables the construction of diverse carbon–carbon, carbon–oxygen, and carbon–nitrogen bonds with remarkable generality.
Graphical Abstract
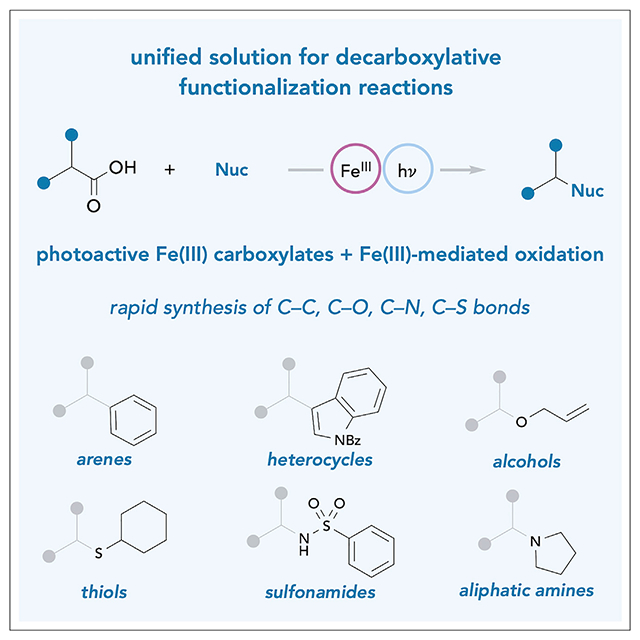
We present a photochemical reaction that enables the direct coupling of carboxylic acids with structurally diverse amines, alcohols, and arenes. This method takes advantage of the ready availability of these building blocks from commercial libraries, the photoactivity of terrestrially abundant iron salts, and the energy of visible light. The result is a versatile oxidative coupling reaction with the potential to streamline rapid synthesis of compound libraries for pharmaceutical discovery.
INTRODUCTION
Modern medicinal chemistry relies heavily on robust cross-coupling methods that enable the rapid assembly of large libraries of structurally diverse drug candidates. Increasingly, the commercial accessibility of large building block sets has become recognized as an important consideration; methods that couple functionalities with greater commercial availability can more efficiently span diverse chemical space and accelerate discovery.1 For example, the development of cross-electrophile coupling methods was inspired in part by the recognition that electrophilic organohalides have much greater commercial availability than the nucleophilic organometallic reagents used in traditional cross-coupling methods.2 The resulting reductive coupling methods have thus been developed into an indispensable tool for contemporary pharmaceutical discovery. An analysis of the commercial availability of functionalized organic molecules, however, reveals that alcohols, amines, and carboxylic acids are dramatically better represented in building block libraries (Figure 1A).3 These functional groups are intrinsically nucleophilic, and cross-nucleophile coupling methods that exploit the native reactivity of these abundant feed-stocks, therefore, are net-oxidative in nature. As new methods for oxidative decarboxylative coupling have emerged in the past several years, these reactions have become increasingly valued in pharmaceutical discovery.4,5
Figure 1. Development of a modular decarboxylative cross-nucleophile coupling.
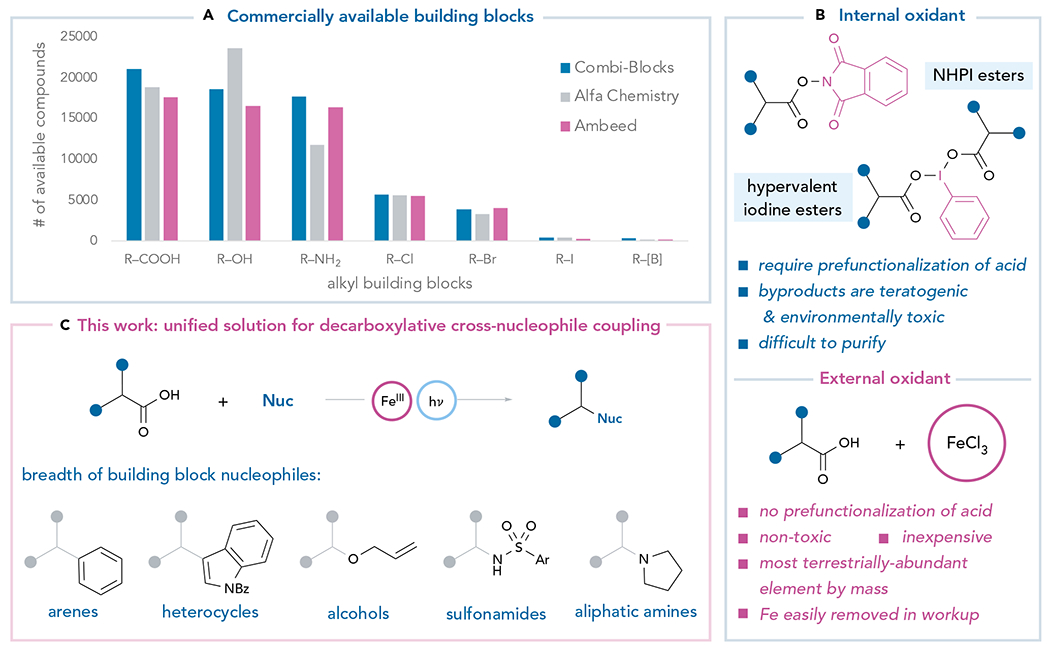
(A) Analysis of the fragments available for purchase at three commercial vendors.
(B) Cross-nucleophile coupling requires an oxidant.
(C) This work: decarboxylative cross-nucleophile coupling enabled by Fe(III) salts and visible light.
Existing methods for oxidative decarboxylative coupling, however, commonly face two important challenges that remain to be fully resolved. First, the majority of these reactions are transition metal-catalyzed cross-coupling methods that involve reductive elimination from a discrete organometallic intermediate as the key bond-forming step.6–9 Because the electronic properties of the nucleophilic ligands exert a significant perturbation on the reactivity of these complexes, reoptimization of the metal catalyst and reaction conditions is often required for the formation of electronically dissimilar C–C, C–O, and C–N bonds. Second, the terminal oxidants most frequently used in these coupling reactions have unattractive characteristics.10 The most common strategies prefunctionalize the carboxylic acid partner with a redox-active moiety as an internal oxidant, which increases step count, compromises atom economy, and hinders applications in rapid compound library synthesis (Figure 1B).11–22 The precursors of these internal organic oxidants can be costly on a per-mole basis, and the most common byproducts of their use are aromatic organics such as phthalimide and iodobenzene that are believed to be toxic or teratogenic.23,24 Baran has recently reported a remarkable electrochemical decarboxylative coupling reaction of unfunctionalized carboxylic acids;25,26 this method, however, requires a stoichiometric silver salt as the terminal oxidant. The development of a unified platform for cross-nucleophile coupling of native carboxylic acid feedstocks with diverse nucleophilic partners, ideally using inexpensive and non-toxic terminal oxidants, therefore, remains an unsolved challenge.
Our group recently reported a method for copper-mediated decarboxylative couplings of carboxylic acid feedstocks that proved to be general for a wide range of alcohol and protected nitrogen nucleophiles.27 Our strategy combined the propensity of Cu(II) salts to self-assemble into photoactive carboxylate complexes with their ability to rapidly oxidize photogenerated organoradical intermediates. This method, however, was not a complete solution to the challenges outlined above. First, although the reaction works well for the coupling of structurally diverse alcohols, sulfonamides, and other protected nitrogen nucleophiles, the scope with respect to carbon nucleophiles was limited to a small number of very electron-rich arenes. This limitation is unfortunate because the diarylethane unit is a common pharmacophore found in small-molecule natural products, agrochemicals, therapeutic agents, and other bioactive compounds.28 Second, although the cost and toxicity of Cu salts are relatively low, they are not negligible, and as Cu(II) is also the terminal oxidant in these reactions, it must be used stoichiometrically.
We hypothesized that a more complete solution to both problems might arise from an investigation of other earth-abundant base metals for their ability to mediate oxidative decarboxylative coupling reactions. These studies have resulted in a method for oxidative decarboxylative coupling of native carboxylic acid feedstocks with a remarkably broad range of carbon, oxygen, and nitrogen nucleophiles that is mediated by simple Fe(III) salts (Figure 1C). The propensity of Fe(III) carboxylate complexes to undergo oxidative photodecarboxylation has been extensively studied, perhaps most importantly in the photochemical decomposition of the canonical ferrioxalate chemical actinometer.29,30 The applications of Fe(III) carboxylate photochemistry in organic synthesis, however, have been limited until quite recently,31–35 despite significant contemporary interest in Fe complexes as earth-abundant, nontoxic, and sustainable chemical reagents.30 Here, we show that the photoreactivity of Fe(III) carboxylate complexes assembled in situ can be productively merged with a unique mechanism for oxidative radical substitution with remarkable generality for diverse carbon, oxygen, and nitrogen nucleophiles. Moreover, Fe(III) salts are ideal terminal oxidants. Iron is the most abundant element on Earth by mass.36 Its salts are generally inexpensive (FeCl3, $3/mol), particularly in comparison with the most common organic internal oxidants utilized in decarboxylative couplings (NHPI, $51/mol; PhI(OAc)2, $667/mol). Its toxicity is minimal and is of negligible concern in the pharmaceutical industry,37 and consequently, the development of new methods utilizing the chemistry of iron coordination complexes is an area of active investigation for synthetic chemists interested in sustainable chemistry.38 Thus, this protocol improves significantly on previous methods for oxidative decarboxylative coupling in terms of generality, substrate compatibility, cost, and sustainability.
RESULTS AND DISCUSSION
Our preliminary studies began with an investigation of the decarboxylative coupling of acid 1 with carbon nucleophile 2 to produce 1,1-diarylalkane 3 (Figure 2). Despite significant attempts at optimization using various Cu(II) complexes, the yield and chemoselectivity remained poor, favoring a C–O dimer formed from the nucleophilic attack of a second equivalent of the carboxylic acid (Tables S1 and S2). We thus expanded our survey to other first-row metals that participate in photoinduced ligand-to-metal charge transfer (LMCT) processes (Table S2).35,39 Ce(III), which is known to engage carboxylate40,41 and chloride42,43 ligands in LMCT transitions, gives no conversion. Likewise, NiCl2, which has recently been identified as an LMCT catalyst, provides no desired reactivity.44,45 Other metals, including Cr(III) and Mn(III), similarly afford little to no product formation. However, we found that Fe(III) salts uniquely provide high yields of the desired cross-coupling product with good regioselectivity (Figure 2, entries 2 and 3). Routine investigation of reaction parameters identified optimal reaction conditions using FeCl3 as an inexpensive chromophore and terminal oxidant (entries 4–7). Importantly, control reactions verified that the Fe(III) salt and visible light are both necessary for the coupling to proceed (entries 8 and 9).
Figure 2. Optimization of carbon–carbon bond formation.
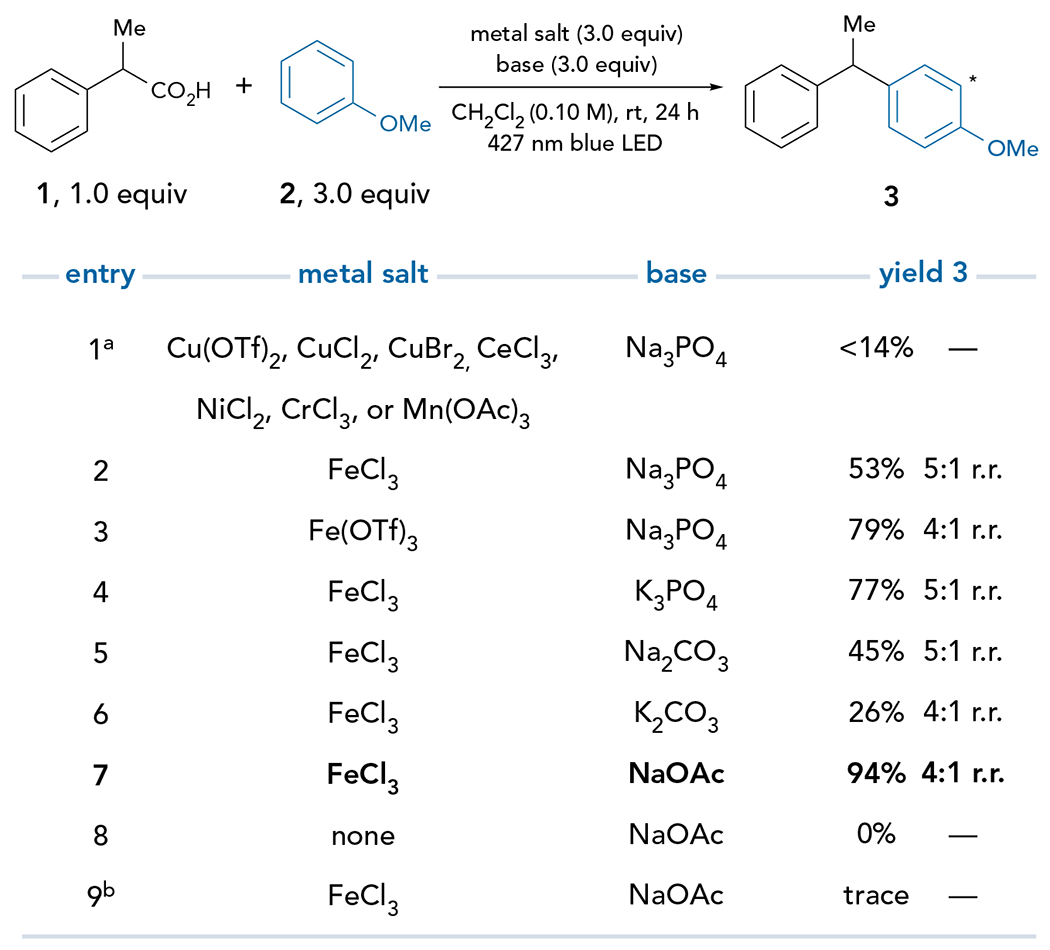
Reaction conducted using metal salt (3.0 equiv), base (3.0 equiv), nucleophile 2 (3.0 equiv), and carboxylic acid 1 (0.1 mmol) in CH2Cl2 (0.10 M) setup under inert atmosphere and irradiated with a 427 nm blue Kessil Lamp at RT for 24 h. Yields were determined by 1H NMR analysis of the crude reaction mixture using 1-methylnaphthalene as an internal standard. aSee supplemental information for full reaction details. bReaction vessel was wrapped in aluminum foil.
Our examination of reaction scope commenced by applying these optimized conditions to construct a variety of 1,1-diarylalkanes (Figure 3; additional examples in supplemental information). A survey of aryl nucleophiles revealed reactivity trends that are consistent with standard electrophilic aromatic substitution reactions. Yields are highest with electron-rich arenes such as anisole (3), toluene (4), and biphenyl (5). Benzene (6) and fluorobenzene (7) are both competent nucleophiles, although the yield diminishes significantly with chlorobenzene (8). Additionally, arene and polyarene nucleophiles containing both electron-donating and electron-withdrawing substituents are competent nucleophiles affording 20 and 21 in high yields. A variety of five- and six-membered heterocycles are found to be excellent reaction partners, including furans (25), pyrroles (26), and an electron-rich pyridine (29). Benzo-fused heterocyclic partners are also viable nucleophiles, including N-protected indoles (22), benzothiophenes (23), and benzofurans (24).
Figure 3. Scope of decarboxylative carbon–carbon bond formation.
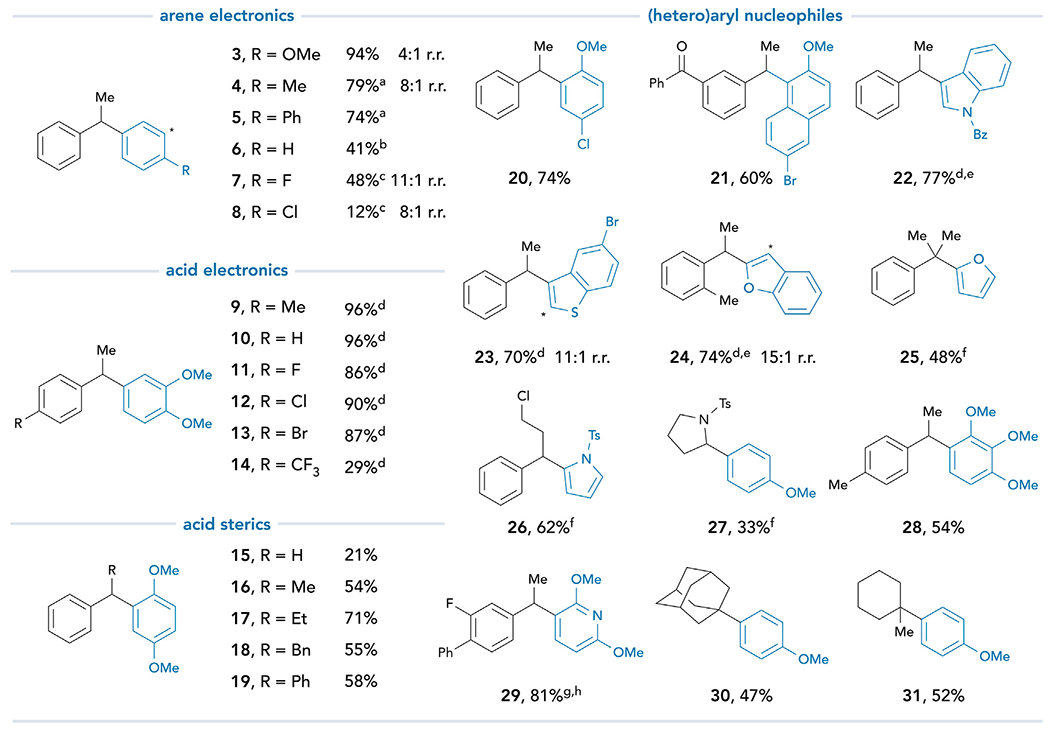
Reaction conditions: FeCl3 (3.0 equiv), NaOAc (3.0 equiv), nucleophile (3.0 equiv), carboxylic acid (1.0 equiv), and CH2Cl2 (0.1 0 M). See supplemental information for experimental details. All yields are isolated yields. a10 equiv of the nucleophile. b20 equiv of the nucleophile. c50 equiv of the nucleophile. dNa2CO3 used as the base. eFe(OTf)3 used as the Fe(III) salt. f5 equiv of the nucleophile. gNa3PO4 used as the base. hAfter irradiation, the reaction was heated to 80°C for 16 h.
We next evaluated the scope with respect to the carboxylic acid, which revealed that activated and halogenated substrates react smoothly (9–13), whereas the yield decreases with strongly deactivating substituents such as a trifluoromethyl group (14). Although primary carboxylic acids couple less rapidly (15), increasing the steric bulk at the α-carbon of secondary derivatives does not inhibit reactivity (16–19). Similarly, tertiary acids readily participate in the desired cross-coupling reaction, affording valuable, fully substituted products (25, 30, and 31). A pendant chloride is tolerated (26), which could serve as a handle to access the corresponding alkyl amine derivatives commonly found in bioactive γ-amino diarylalkane scaffolds.26 Proline derivative 27 highlights the application of this method to α-heteroatom-bearing carboxylic acids. We also synthesized isoerianin analog 28, belonging to a class of 1,1-diarylethanes with known cytotoxic activity.46 Furthermore, the common nonsteroidal anti-inflammatory drug (NSAID) flurbiprofen successfully undergoes cross-coupling with a pyridine derivative to afford 29 in 81% yield. Finally, tertiary aliphatic carboxylic acids are viable coupling partners in this chemistry, favoring nucleophilic substitution over rapid decarboxylative elimination (30 and 31).
We next probed the modularity of the system toward C–O and C–N bond formation (Figure 4). Under the same reaction conditions optimized for C–C coupling, the pairing of 2-phenylethanol with α,4-dimethylphenylacetic acid results in an NMR yield of 32% (entry 1). Optimization of base identity allows 32 to be isolated in 80% yield (entry 2, for full optimization, see supplemental information). Additionally, Fe(OTf)3 is an equally competent iron salt (entry 3) and provides generally cleaner conversion to the ether products compared with FeCl3 (vide infra). Indeed, a broad scope of the cross-coupling between secondary and tertiary carboxylic acids with differing electronic and steric properties is successful with 2-phenylethanol (33–38). Primary alcohols bearing pendant functionalities react smoothly, including a phthalimide (39) and an olefin (45). Although secondary alcohols are viable substrates (47), the yield decreases with sterically hindered tertiary derivatives (see supplemental information). Etherification of 3-oxo-1-indancarboxylic acid provides rapid access to a functionalized cyclic indanone core (40). Benzylic alcohols bearing aryl halides are well tolerated, providing avenues for further functionalization of the products through orthogonal transition metal-mediated methods (41–43 and 48). The successful cross-coupling of an α-oxo acid to furnish mixed acetal 41 demonstrates the viability of non-benzylic acids to participate in this coupling. Moreover, this method can affect the direct etherification of several NSAIDs in high yields (43–45 and 47–48). Finally, this protocol is directly applicable to thioetherification; sulfide 46 is obtained in 53% yield when cyclohexanethiol is used as the coupling partner, without a noticeable formation of sulfoxide or sulfone byproducts.
Figure 4. Scope of decarboxylative carbon–heteroatom bond formation.
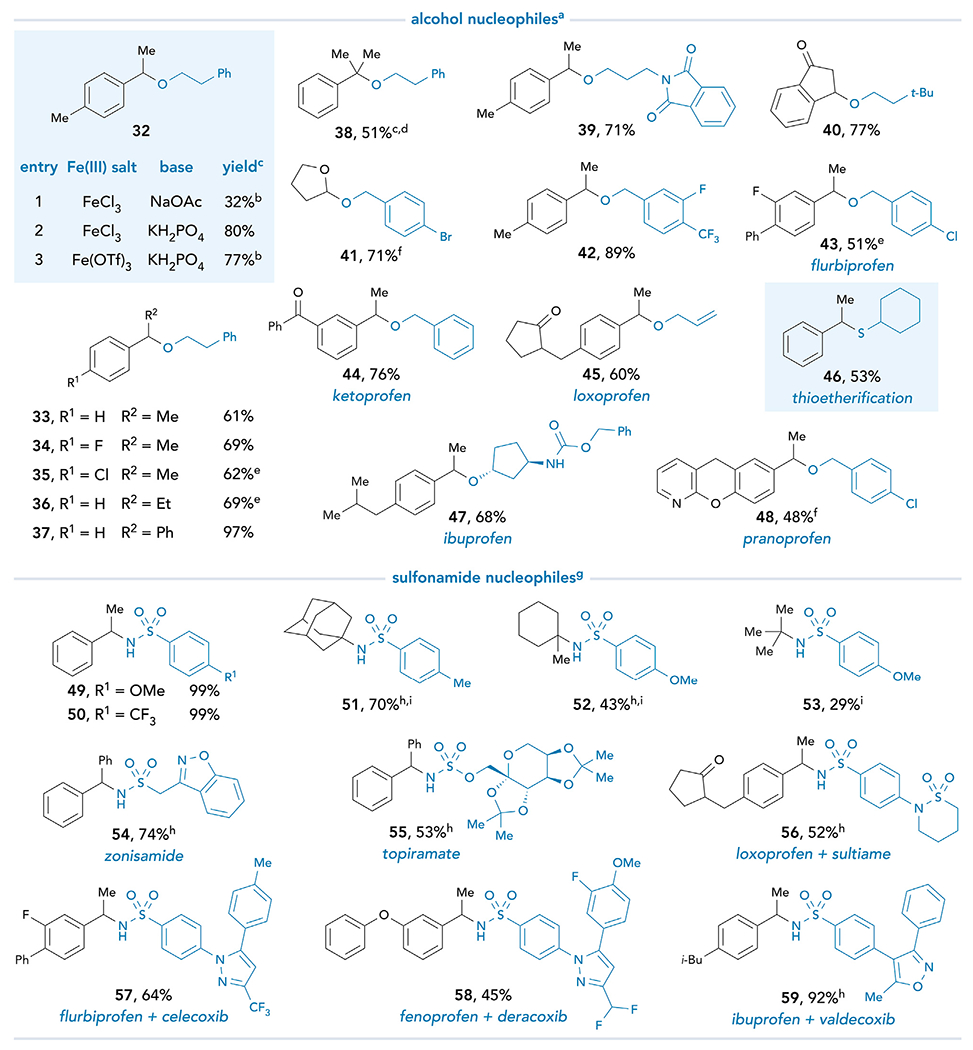
See supplemental information for experimental details. All yields are isolated unless otherwise noted. aReaction conditions for O- and S-nucleophiles: Fe(OTf)3 (3.0 equiv), K2HPO4 (5.0 equiv), nucleophile (3.0 equiv), carboxylic acid (1.0 equiv), and CH2O2 (0.10 M). bYield was determined by 1H NMR analysis of the crude reaction mixture using 1-methylnaphthalene as an internal standard. c1,2-DCE used as the solvent. dFeCl3 used as iron salt. e48 h. fPyridine (3 equiv) used as the base. gReaction conditions for N- nucleophiles: FeCl3 (3.0 equiv), Na3PO4 (3.0 equiv), nucleophile (3.0 equiv), carboxylic acid (1.0 equiv), and CH2O2 (0.10 M). hFe(OTf)3 used as the iron salt. iNaOAc used as the base.
We also examined the Fe-promoted decarboxylative coupling with sulfonamides as nitrogen nucleophiles. Electron-rich (49) and electron-deficient (50) sulfonamides undergo the desired transformation in quantitative yields. Tertiary aliphatic acids couple readily with sulfonamide nucleophiles (51–53). To showcase the potential relevance of this method to medicinal chemistry, we studied cross-coupling reactions involving several drug molecules. Zonisamide, topiramate, and sultiame, all medications used to treat epilepsy, react readily to afford derivatives 54–56 in good yields. Additionally, we successfully coupled a series of NSAIDs with COX-2-inhibitor sulfa drugs to access conjugates 57–59. These examples highlight the tolerance for a wide range of valuable functional groups, including protected sugars, benzisoxazoles, isoxazoles, pyrazoles, aliphatic ketones, diaryl ethers, and halogens.
The ease of reaction setup is notable; throughout our investigations, the reactions were set up under an inert atmosphere for easy handling of hygroscopic Fe(III) salts. After setup, however, the reactions can be conducted under air (Table S16). Additionally, we observed only a modest loss of yield when all reagents are added on the benchtop, with only a sparge with nitrogen prior to irradiation as a precaution (Table S17). Although adjustments were made to maximize yields for each nucleophile class, a single set of conditions (FeCl3 and Na3PO4) provides synthetically useful yields across the major nucleophile families investigated (53% for 3, 64% for 33, and 99% for 49), demonstrating the exceptional modularity of this reaction platform (Table S15).
Additionally, we consistently found that the generality of this reaction was superior to that of our previously reported Cu(II) protocol (Figure 5A). This was particularly notable with modestly electron-rich arene nucleophiles, which gave unsatisfactory yields using the Cu(II) conditions but were excellent reaction partners using FeCl3. Moreover, although tertiary aliphatic carboxylic acids exclusively undergo oxidative elimination to alkenes under Cu(II) conditions (Tables S19 and S20), they were adequate reaction partners using the present protocol. Interestingly, the superior reactivity of Fe(III) over Cu(II) for C–C bond formation implies that different reactive intermediates are likely involved.
Figure 5. Fe(III) salts expand the breadth of cross-nucleophile coupling reactions.
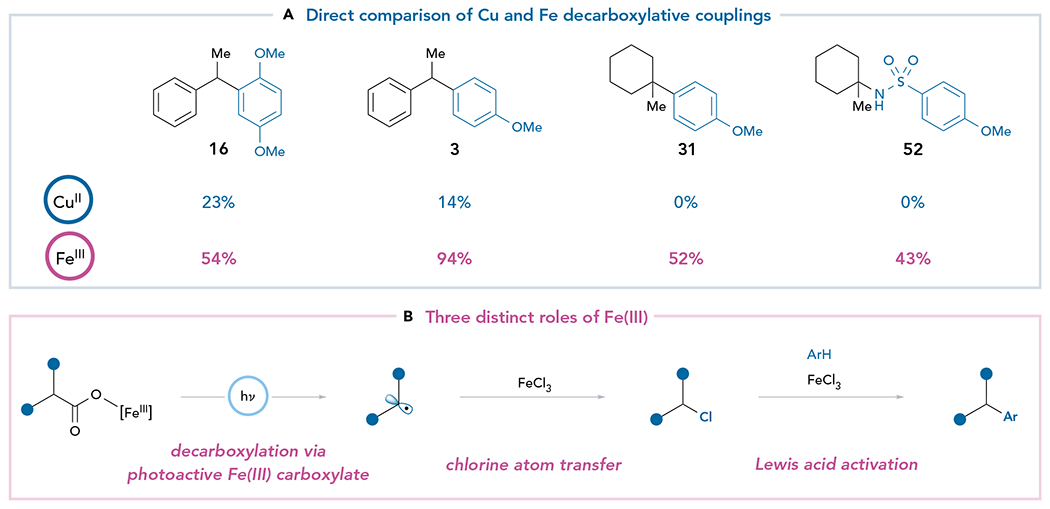
(A) Comparison of reactivity using Cu(II) and Fe(III) salts. Reaction conditions for Cu(II) mediated couplings: Cu(OTf)2 (2.5 equiv), Na3PO4 (3.0 equiv), i-PrCN (5.5 equiv), nucleophile (3.0 equiv), and carboxylic acid 1 (0.1 mmol) in CH2Cl2 (0.10M) setup under inert atmosphere and irradiated with a 427 nm blue Kessil Lamp at RT for 24 h. Yields were determined by 1H NMR analysis of the crude reaction mixture using 1-methylnaphthalene as an internal standard. Reaction conditions for Fe(III)-mediated couplings are reported in Figures 2 and 3.
(B) Proposed roles of Fe(III) in the C–C bond-forming reaction.
Although the mechanisms of reactions involving iron complexes are notoriously difficult to deconvolute,47 one clue arose from the observation that reactions stopped before completion often produced the corresponding alkyl chloride byproduct. We hypothesized this alkyl chloride resulted from rapid chlorination of the decarboxylated radical by FeCl3 prior to functionalization by the nucleophile. Indeed, in experiments where the nucleophile is omitted, quantitative yields of the decarboxylative chlorination product can be obtained (Tables S21 and S22). Moreover, Fe(III) salts can activate chloroalkanes toward nucleophilic substitution and are common Lewis acid catalysts for Friedel-Crafts alkylation (Table S23).48 Importantly, CuCl2 does not mediate an analogous photochemical decarboxylative chlorination, and it is ineffective in catalyzing nucleophilic substitution of benzylic chlorides (Tables S25 and S26). Thus, we propose that this coupling reaction involves a unique mechanism in which FeCl3 can play three mechanistically different roles (Figure 5B). First, we hypothesize that Fe(III) produces the key photogenerated carboxy radical intermediate, either via direct LMCT of a Fe(III) carboxylate assembled in situ or through the intermediacy of a photogenerated Cl radical.49 Second, FeCl3 produces a chemically stable alkyl chloride by trapping the decarboxylated organoradical intermediate via halogen atom transfer (XAT).50 Finally, it promotes nucleophilic substitution by Lewis acid activation of the alkyl chloride intermediate.
Simple aliphatic amines, unfortunately, are not compatible with this method, possibly due to the strong binding affinity of the amines to the Lewis acidic Fe(III) center (Table S27). This is a significant constraint because modern drug discovery relies heavily on methods for the introduction of basic amine fragments into substrate molecules.51 However, we recognized that the chloride intermediate provides an opportunity to overcome this limitation. Formal decarboxylative amination can be readily achieved by adding the nucleophile to the reaction directly after initial irradiation of the acid in the presence of FeCl3 (Figure 6). Employing this telescoped strategy, we realized the cross-coupling of flurbiprofen with the most common nitrogen-containing fragments in pharmaceutical candidates, including pyrrolidines (60), piperidines (61), piperazines (62), morpholines (63), alkylamines (64 and 65), heterocycles (66), and arylamines (67).46 Furthermore, the versatility of the reported platform encouraged us to further expand the coupling reaction to nucleophiles that were incompatible with the one-step conditions. When sodium p-toluenesulfinate is employed as the nucleophile, formal decarboxylative sulfinylation affords sulfone 68 in 73% yield via the telescoped two-step protocol. Finally, the ability to efficiently generate benzylic chlorides suggests that the synthetic scope of Fe-mediated photodecarboxylation could be further expanded by leveraging a diverse range of modern transition metal-catalyzed reactions. As a preliminary example of this versatility, we directly subjected the photogenerated alkyl chloride to known Cu(I)-catalyzed borylation conditions enabling the formal decarboxylative borylation of flurbiprofen in 51% yield (69).52
Figure 6. The intermediary of an alkyl chloride enables further diversification of carboxylic acid feedstocks.
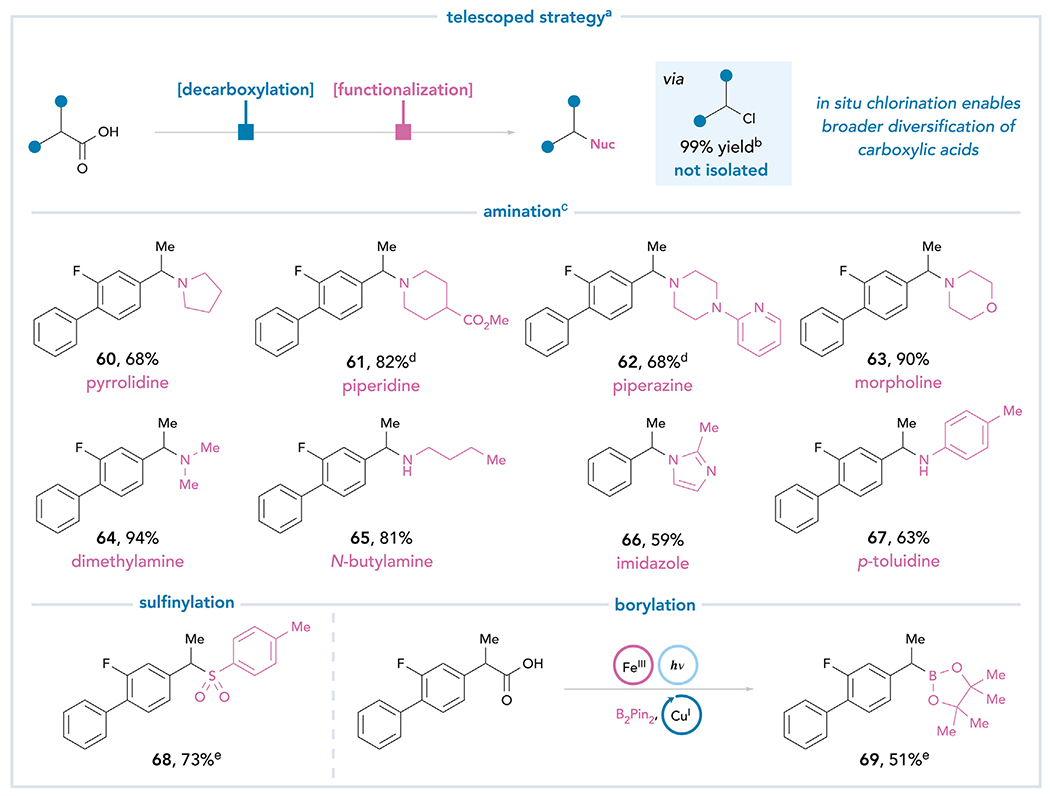
All yields are isolated unless otherwise noted. aReaction conditions: FeCl3 (3.0 equiv), Na3PO4 (3.0 equiv), carboxylic acid (1.0 equiv), and MeCN (0.10 M), 24 h, rt, 427 nm blue LED. bYield was determined by 1H NMR analysis of the crude reaction mixture using 1-methylnaphthalene as an internal standard. cAfter irradiation, amine (10 equiv) and KI (1.5 equiv) were added directly to the reaction mixture and heated to 70°C overnight. d7.0 equiv of the amine. eSee supplemental information for experimental details.
Conclusions
In conclusion, we have developed a robust protocol for decarboxylative cross-coupling of native carboxylic acids with diverse carbon-, oxygen-, and nitrogen-centered nucleophilic partners mediated by simple iron salts. Because Fe(III) serves as both the chromophore in this reaction as well as the terminal oxidant, it improves on conventional methods for decarboxylative coupling in several ways. The reaction involves a unique mechanism for oxidative radical coupling that is operational with a remarkably broad scope of electronically dissimilar nucleophilic reaction partners. The use of Fe(III) as a terminal oxidant in place of a redox-active internal organic oxidant dramatically improves the practicality and environmental impact of the method, and the ability to conduct the coupling without prefunctionalization of either reaction partner should accelerate the preparation of compound libraries and the structural optimization of lead drug candidates. Finally, the reaction setup is straightforward and requires little specialized equipment other than a blue LED light source. These results provide a further demonstration of the synthetic utility of the photochemistry of base metal coordination complexes, and we hope that they stimulate new ideas in the design of photochemical coupling reactions.
EXPERIMENTAL PROCEDURES
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Tehshik Yoon (tyoon@chem.wisc.edu).
Materials availability
Full experimental details are available in the supplemental information. This includes experimental procedures, optimization investigations, mechanistic studies, and characterization of unique compounds (1H NMR, 13C NMR, 19F NMR, 11B NMR, HRMS, and melting point).
Data and code availability
All data is available in the supplemental information.
General experimental procedure
An oven-dried 6-mL vial equipped with a stir bar is brought into a nitrogen-filled glovebox and charged with Fe(III) salt (3.0 equiv), base (3.0 equiv), nucleophile (3.0 equiv), the carboxylic acid (1.0 equiv), and methylene chloride (0.10 M). The vial is sealed with a screwcap bearing a Teflon septum, removed from the glovebox, and placed on a stir plate. The vial is irradiated at 427 nm with two 40 W Kessil Lamp PR160 lamps at a distance of 10 cm with stirring at 800 rpm. A fan is used to maintain the vial at room temperature. After 24 h, the crude reaction mixture is diluted with 1.5 mL EtOAc and absorbed directly on diatomaceous earth (Celite). The product is purified by flash chromatography.
Supplementary Material
Highlights.
Unified solution to decarboxylative cross-nucleophile coupling reactions
Photoactive Fe(III) carboxylates + Fe(III)-mediated oxidation
Fe(III) salts are non-toxic, earth abundant, and low cost
Direct formation of C–C, C–O, C–S, and C–N bonds without prefunctionalization
THE BIGGER PICTURE.
Carboxylic acids are valuable chemical building blocks because they are widely available with significant structural diversity. Decarboxylative coupling reactions are increasingly utilized in modern drug discovery, but many of the most general methods require the preinstallation of redox-active organic moieties as internal oxidants. Here, we report a photochemical method for the direct decarboxylative functionalization of carboxylic acids with a wide range of simple carbon-, oxygen-, and nitrogen-centered nucleophiles. Key to the success of this process is the diverse reactivity of simple iron salts, which serve both as the light-absorbing center in this reaction and as a sustainable terminal oxidant.
ACKNOWLEDGMENTS
Funding for this work was provided by the NIH (R35 GM144129, T.P.Y.), an ACS GCI Pharmaceutical Roundtable Research Grant (T.P.Y.), and Pfizer (T.P.Y.). S.N.G. thanks the NIH for a fellowship grant (F32GM139373), and G.A.L. is the recipient of a 3M Science & Technology Fellowship. Analytical facilities at UW–Madison are funded by the NIH (S10OD012245), NSF (CHE-9304546), and a generous gift from the Paul J. and Margaret M. Bender Fund.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.chempr.2023.04.008.
DECLARATION OF INTERESTS
M.W.B. and S.W.B. are employees and shareholders of Pfizer, Inc.
REFERENCES
- 1.Kutchukian PS, Dropinski JF, Dykstra KD, Li B, DiRocco DA, Streckfuss EC, Campeau L-C, Cernak T, Vachal T, Davies IW, et al. (2016). Chemistry informer libraries: a chemoinformatics enabled approach to evaluate and advance synthetic methods. Chem. Sci 7, 2604–2613. 10.1039/C5SC04751J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biswas S, and Weix DJ (2013). Mechanism and selectivity in nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides. J. Am. Chem. Soc 135, 16192–16197. 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reaxys. https://reaxys.com (accessed 12/8/2022); see supplemental information for details.
- 4.Beil SB, Chen TQ, Intermaggio NE, and MacMillan DWC (2022). Carboxylic acids as adaptive functional groups in Metallaphotoredox catalysis. Acc. Chem. Res 55, 3481–3494. 10.1021/acs.accounts.2c00607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laudadio G, Palkowitz MD, El-Hayek Ewing T, and Baran PS (2022). Decarboxylative cross-coupling: a radical tool in medicinal chemistry. ACS Med. Chem. Lett 13, 1413–1420. 10.1021/acsmedchemlett.2c00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong D, Moon PJ, Bsharat O, and Lundgren RJ (2020). Direct catalytic decarboxylative amination of aryl acetic acids. Angew. Chem. Int. Ed. Engl 59, 1313–1319. 10.1002/anie.201912518. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen VT, Nguyen VD, Haug GC, Vuong NTH, Dang HT, Arman HD, and Larionov OV (2020). Visible-light-enabled direct decarboxylative N-alkylation. Angew. Chem. Int. Ed. Engl 59, 7921–7927. 10.1002/anie.201916710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dow NW, Pedersen PS, Chen TQ, Blakemore DC, Dechert-Schmitt AM, Knauber T, and MacMillan DWC (2022). Decarboxylative borylation and cross-coupling of (hetero)aryl acids enabled by copper charge transfer catalysis. J. Am. Chem. Soc 144, 6163–6172. 10.1021/jacs.2c01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drapeau MP, Bahri J, Lichte D, and Gooβen LJ (2019). Decarboxylative ipso amination of activated benzoic acids. Angew. Chem. Int. Ed. Engl 58, 892–896. 10.1002/anie.201812068. [DOI] [PubMed] [Google Scholar]
- 10.Li P, Zbieg JR, and Terrett JA (2021). The direct decarboxylative N-alkylation of azoles, sulfonamides, ureas, and carbamates with carboxylic acids via photoredox catalysis. Org. Lett 23, 9563–9568. 10.1021/acs.orglett.1c03761. [DOI] [PubMed] [Google Scholar]
- 11.Liang Y, Zhang X, and MacMillan DWC (2018). Decarboxylative sp3 C–N coupling via dual copper and photoredox catalysis. Nature 559, 83–88. 10.1038/s41586-018-0234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mao R, Frey A, Balon J, and Hu X (2018). Decarboxylative C(sp3)–N cross-coupling via synergetic photoredox and copper catalysis. Nat. Catal 1,120–126. 10.1038/s41929-017-0023-z. [DOI] [Google Scholar]
- 13.Zhao W, Wurz RP, Peters JC, and Fu GC (2017). Photoinduced, copper-catalyzed decarboxylative C–N coupling to generate protected amines: an alternative to the Curtius rearrangement. J. Am. Chem. Soc 139, 12153–12156. 10.1021/jacs.7b07546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, and Zhang D (2020). Cuphotoredox-catalyzed C(sp)-C(sp3) coupling of redox-active esters with terminal alkynes. Org. Biomol. Chem 18, 4479–4483. 10.1039/D00B00835D. [DOI] [PubMed] [Google Scholar]
- 15.Li P, Zbieg JR, and Terrett JA (2021). A platform for decarboxylative couplings via photoredox catalysis: direct access to carbocations from carboxylic acids for carbon–oxygen bond formation. ACS Catal. 11, 10997–11004. 10.1021/acscatal.1c03251. [DOI] [Google Scholar]
- 16.Shibutani S, Kodo T, Takeda M, Nagao K, Tokunaga N, Sasaki Y, and Ohmiya H (2020). Organophotoredox-catalyzed decarboxylative C(sp3)–O bond formation. J. Am. Chem. Soc 142, 1211–1216. 10.1021/jacs.9b12335. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa M, Nagao K, Ikeda Z, Reynolds M, Ibáñez I, Wang J, Tokunaga N, Sasaki Y, and Ohmiya H (2021). Organophotoredox-catalyzed decarboxylative N-alkylation of sulfonamides. ChemCatChem 13, 3930–3933. 10.1002/cctc.202100803. [DOI] [Google Scholar]
- 18.Guo G, Yuan Y, Bao X, Cao X, Sang T, Wang J, and Huo C (2021). Photocatalytic redox-neutral approach to diarylmethanes. Org. Lett 23, 6936–6940. 10.1021/acs.orglett.1c02523. [DOI] [PubMed] [Google Scholar]
- 19.Webb EW, Park JB, Cole EL, Donnelly DJ, Bonacorsi SJ, Ewing WR, and Doyle AG (2020). Nucleophilic (radio)fluorination of redox-active esters via radical-polar crossover enabled by photoredox catalysis. J. Am. Chem. Soc 142, 9493–9500. 10.1021/jacs.0c03125. [DOI] [PubMed] [Google Scholar]
- 20.Murarka S (2018). N-(acyloxy)phthalimides as redox-active esters in cross-coupling reactions. Adv. Synth. Catal 360, 1735–1753. 10.1002/adsc.201701615. [DOI] [Google Scholar]
- 21.Chen KQ, Wang ZX, and Chen XY (2020). Photochemical decarboxylative C(sp3)–X coupling facilitated by weak interaction of N-heterocyclic carbene. Org. Lett 22, 8059–8064. 10.1021/acs.orglett.0c03006. [DOI] [PubMed] [Google Scholar]
- 22.Maeda B, Sakakibara Y, Murakami K, and Itami K (2021). Photoredox-catalyzed benzylic esterification via radical-polar crossover. Org. Lett 23, 5113–5117. 10.1021/acs.orglett.1c01645. [DOI] [PubMed] [Google Scholar]
- 23.Sigma-Aldrich (2022). Safety data sheet. Phthalimide. CAS RN: 85-41-6; 24023 (Sigma-Aldrich, MilliporeSigma; ). https://www.sigmaaldrich.com/US/en/sds/aldrich/240230. [Google Scholar]
- 24.Sigma-Aldrich (2021). Safety data sheet. Iodobenzene for synthesis. CAS RN: 591-50-4; 8.20730; (Sigma-Aldrich, MilliporeSigma; ). https://www.sigmaaldrich.Com/US/en/sds/mm/8.20730. [Google Scholar]
- 25.Xiang J, Shang M, Kawamata Y, Lundberg H, Reisberg SH, Chen M, Mykhailiuk P, Beutner G, Collins MR, Davies A, et al. (2019). Hindered dialkyl ether synthesis with electrogenerated carbocations. Nature 573, 398–402. 10.1038/s41586-019-1539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheng T, Zhang HJ, Shang M, He C, Vantourout JC, and Baran PS (2020). Electrochemical decarboxylative N-alkylation of heterocycles. Org. Lett 22, 7594–7598. 10.1021/acs.orglett.0c02799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li QY, Gockel SN, Lutovsky GA, DeGlopper KS, Baldwin NJ, Bundesmann MW, Tucker JW, Bagley SW, and Yoon TP (2022). Decarboxylative cross-nucleophile coupling via ligand-to-metal charge transfer photoexcitation of Cu(II) carboxylates. Nat. Chem 14, 94–99. 10.1038/s41557-021-00834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ameen D, and Snape TJ (2013). Chiral 1,1-diaryl compounds as important pharmacophores. Med. Chem. Commun 4, 893–907. 10.1039/C3MD00088E. [DOI] [Google Scholar]
- 29.Parker CA (1953). A new sensitive chemical actinometer. I. Some trials with potassium ferrioxalate. Proc. R. Soc. Lond A 220,104–116. 10.1098/rspa.1953.0175. [DOI] [Google Scholar]
- 30.Hatchard CG, and Parker CA (1956). A new sensitive chemical actinometer. II. Potassium ferrioxalate as a standard chemical actinometer. Proc. R. Soc. Lond. A 235, 518–536. 10.1098/rspa.1956.0102. [DOI] [Google Scholar]
- 31.Vernia JE, Warmin MR, Krause JA, Tierney DL, and Baldwin MJ (2017). Photochemistry and anion-controlled structure of Fe(III) complexes with an α-hydroxy acid-containing tripodal amine chelate. Inorg. Chem 56, 13029–13034. 10.1021/acs.inorgchem.7b01799. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Wang X, Xia S, and Jin J (2019). Ligand-accelerated iron photocatalysis enabling decarboxylative alkylation of heteroarenes. Org. Lett 21, 4259–4265. 10.1021/acs.orglett.9b01439. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Qian J, Wang M, Huang Y, and Hu P (2022). Visible-light-induced decarboxylative fluorination of aliphatic carboxylic acids catalyzed by iron. Org. Lett 24, 5972–5976. 10.1021/acs.orglett.2c02242. [DOI] [PubMed] [Google Scholar]
- 34.Wang S, Li T, Gu C, Han J, Zhao CG, Zhu C, Tan H, and Xie J (2022). Decarboxylative tandem C-N coupling with nitroarenes via SH2 mechanism. Nat. Commun 13, 2432. 10.1038/s41467-022-30176-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abderrazak Y, Bhattacharyya A, and Reiser O (2021). Visible-light-induced homolysis of earth-abundant metal-substrate complexes: A complementary activation strategy in photoredox catalysis. Angew. Chem. Int. Ed. Engl 60, 21100–21115. 10.1002/anie.202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frey PA, and Reed GH (2012). The ubiquity of iron. ACS Chem. Biol 7, 1477–1481. 10.1021/cb300323q. [DOI] [PubMed] [Google Scholar]
- 37.European Medicines Agency (2008). Guideline on the specification limits for residues of metal catalysts or metal reagents. EMEA/CHMP/SWP/4446/2000. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-specification-limits-residues-metal-catalysts-metal-reagents_en.pdf.
- 38.Guðmundsson A, and Bäckvall JE (2020). On the use of iron in organic chemistry. Molecules 25. 10.3390/molecules25061349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juliá F (2022). Ligand-to-metal charge transfer (LMCT) photochemistry at 3d-metal complexes: an emerging tool for sustainable organic synthesis. ChemCatChem 14. e202200916. 10.1002/cctc.202200916. [DOI] [Google Scholar]
- 40.Sheldon RA, and Kochi JK (1968). Photochemical and thermal reduction of cerium(IV) carboxylates. Formation and oxidation of alkyl radicals. J. Am. Chem. Soc 90, 6688–6698. 10.1021/ja01026a022. [DOI] [Google Scholar]
- 41.Shirase S, Tamaki S, Shinohara K, Hirosawa K, Tsurugi H, Satoh T, and Mashima K (2020). Cerium(IV) carboxylate photocatalyst for catalytic radical formation from carboxylic acids: decarboxylative oxygenation of aliphatic carboxylic acids and lactonization of aromatic carboxylic acids. J. Am. Chem. Soc 142, 5668–5675. 10.1021/jacs.9b12918. [DOI] [PubMed] [Google Scholar]
- 42.Yang Q, Wang YH, Qiao Y, Gau M, Carroll PJ, Walsh PJ, and Schelter EJ (2021). Photocatalytic C–H activation and the subtle role of chlorine radical complexation in reactivity. Science 372, 847–852. 10.1126/science.abd8408. [DOI] [PubMed] [Google Scholar]
- 43.Yatham VR, Bellotti P, and König B (2019). Decarboxylative hydrazination of unactivated carboxylic acids by cerium photocatalysis. Chem. Commun. (Camb) 55, 3489–3492. 10.1039/C9CC00492K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heitz DR, Tellis JC, and Molander GA (2016). Photochemical nickel-catalyzed C–H arylation: synthetic scope and mechanistic investigations. J. Am. Chem. Soc 138, 12715–12718. 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shields BJ, and Doyle AG (2016). Direct C(sp3)–H cross coupling enabled by catalytic generation of chlorine radicals. J. Am. Chem. Soc 138, 12719–12722. 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Messaoudi S, Hamze A, Provot O, Tréguier B, Rodrigo De Losada JR, Bignon J, Liu JM, Wdzieczak-Bakala J, Thoret S, Dubois J, et al. (2011). Discovery of isoerianin analogues as promising anticancer agents. ChemMedChem 6, 488–497. 10.1002/cmdc.201000456. [DOI] [PubMed] [Google Scholar]
- 47.Neidig ML, Carpenter SH, Curran DJ, DeMuth JC, Fleischauer VE, Iannuzzi TE, Neate PGN, Sears JD, and Wolford NJ (2019). Development and evolution of mechanistic understanding in iron-catalyzed cross-coupling. Acc. Chem. Res 52, 140–150. 10.1021/acs.accounts.8b00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horne DA (1983). The synthesis of 4,4′-ditertbutyl biphenyl: a sophomore organic chemistry experiment. J. Chem. Educ 60, 246. 10.1021/ed060p246. [DOI] [Google Scholar]
- 49.The addition of carboxylic acid substrate 1 and base to FeCl3 results in a stronger UV-vis absorption than FeCl3 alone. We observe a similar increase in absorptivity upon addition of 1 and Base to Fe(OTf)3, but these spectra are not identical. It follows that the light-absorbing species in this reaction is an Fe(III) carboxylate, but that different complexes are formed under these conditions. See Figures S9–S11.
- 50.We surmise that decarboxylative couplings using Fe(OTf)3 proceed through an analogous mechanism. Although the instability of benzylic triflates preclude their isolation, evidence of their formation can be obtained by GC-MS analysis. See Figures S5–S8.
- 51.Roughley SD, and Jordan AM (2011). The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem 54, 3451–3479. 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 52.Ito H, and Kubota K (2012). Copper(I)-catalyzed boryl substitution of unactivated alkyl halides. Org. Lett 14, 890–893. 10.1021/ol203413w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the supplemental information.