

Abstract
The need for accurate and efficient force fields for modeling 3D structures of macrobiomolecules and in particular intrinsically disordered proteins (IDPs) has increased with recent findings to associate IDPs and human diseases. However, most conventional protein force fields and recent IDP-specific force fields are limited in reproducing accurate structural features of IDPs. Here, we present an environmental specific precise force field (ESFF1) based on CMAP corrections of 71 different sequence environments to improve the accuracy and efficiency of MD simulation for both IDPs and folded proteins. MD simulations of 84 different short peptides, IDPs, and structured proteins show that ESFF1 can accurately reproduce spectroscopic properties for different peptides and proteins whether they are disordered or ordered. The successful ab initio folding of five fast-folding proteins further supports the reliability of ESFF1. The extensive analysis documented here shows that ESFF1 is able to achieve a reasonable balance between ordered and disordered states in protein simulations.
Graphical Abstract
INTRODUCTION
Intrinsically disordered proteins (IDPs) including proteins with disordered regions (consecutive disordered segments ≥40 in length) are characterized by lack of well-defined tertiary structures. IDPs exist as an ensemble of flexible conformers. At the same time, those various flexible conformers allow IDPs to play different essential roles at various conditions as IDPs are able to adopt different conformers upon binding to diverse partners such as ligands, proteins, DNA, and RNA. As a result, plenty of IDPs are associated with various human diseases, such as Tau protein, IAPP and β-amyloid peptide in neurodegenerative disease, p53 protein in cancer, and α-synuclein in Parkinson’s disease. With the motivation of exploring the structure–function relationship of IDPs and designing drugs that targeting IDPs, tremendous efforts have been devoted to studying IDPs. However, due to the flexibility of IDP conformers, conventional experimental tools are limited in investigating the IDP structures.1,2 Molecular dynamics (MD) simulations are in an ideal position to compliment experimental studies of IDPs due to their detailed conformer sampling ability.
Recent improvements in computer software and hardware allow molecular dynamics simulations to sample IDPs in relevant time scales. However, the accuracy of force fields severely limited the quality of IDP simulations. Most of current protein force fields were designed to reproduce the highly structured 3D structures, leading to excessive stability of secondary structures, especially α-helix, in MD simulations. Although many efforts have been invested to correct the improper preference of backbone potential, resulting in balanced propensity among secondary structures in structured proteins, accurate modeling of secondary structures in IDPs is still a tremendous challenge. Very recently, great efforts were devoted to improve the modeling of IDPs, and tremendous progress was made in this direction. These improvements were mainly focused on solvent models and main-chain torsional preferences. On the one hand, upon the observation that traditional force fields are limited in modeling thermodynamic properties of water, the TIP4P-Ew model was proposed to improve the modeling of water,3 and the combination of ff99SB/TIP4P-Ew showed reasonable protein–water interactions in simulations of IDP Ab21–30 and Ab42 peptides.4,5 Furthermore, ff03 variants, ff03 * and ff03w, in combination of the TIP4P/20056 model were also found to improve the conformers sampling of IDPs.7 In addition, the TIP4P-D model, introduced by modifying parameters in the TIP4P model to correct for the deficiencies in water dispersion interactions, was found to improve the radii of gyration of some IDPs.8 On the other hand, to reduce the biases toward the structural proteins in conventional force fields, ff99IDPs9 was first developed by Chen and co-workers to improve IDP sampling by transplanting residue-specific CMAP corrections of eight disordered promoting residues (A, E, G, K, P, Q, R, and S) to the ff99SBildn10 force field.11 On the basis of the same idea, ff14IDPSFF was also proposed by extending these CMAP corrections to all 20 residues, which was found to dramatically improve the quality in reproducing secondary chemical shifts of multiple short disordered proteins.12 Most recently the CHARMM36m13 and a99SB-disp14 force fields were also proposed to model both IDPs and structured proteins.
Obviously, in these latest force fields, the main-chain torsional terms become increasingly sophisticated. On the other hand, to reduce the overstabled α-helix in ff99SBildn and ff14SB simulations, we adjusted the PPII and right helix regions in the ff14IDPSFF force field but found the strategy underestimated the stability of α-helix.12,15 CHARMM36m was also reported not to be able to fold back the all-β structure peptide GTT.14 Thus, additional efforts are still needed to improve the current force fields to reproduce experimentally observed propensities of secondary structures.
As reported in Baldwin’s study, J-coupling constants for GGXGG peptides are quite different from dipeptide values for some polar residues such as X = D, N, T, and Y due to probable side-chain–backbone interactions in GGXGG peptides.16 In addition, Shi et al.’s work on J-coupling constants of Ac-LXP-NH2 provides direct experimental evidence for the efforts of the neighboring residues. Shi et al. proposed that the neighboring residues play important roles in determining the stabilities of secondary structures of short peptides by influencing their hydration environments.17 However, our investigation of widely used protein force fields, such as ff14IDPSFF, ff14SB, a99SB-disp, ff03ws, and CHARMM36m shows that MD simulations were not accurate enough to reproduce the reported neighboring-residue effects for the Ac-LXP-NH2 peptides (Figure S1).
Therefore, we intend to take into consideration the chemical environments of neighboring residues in our refinement of protein force fields. Indeed, our benchmark database investigation of neighboring residues on phi/psi distributions of central residues suggests that neighbors’ chemical properties (such as polar versus nonpolar) are important (Figures S2 and S3), whose efforts are yet to be fully captured by standard protein force fields in MD simulations, as shown below. Our hypothesis is that incorporation of neighboring chemical environments into protein force fields would increase the accuracy and predictive power of MD simulations. These efforts would lead to environment-specific force fields. Here, we document our very first effort in developing such a force field, which is termed as ESFF1 throughout this Article.
As discussed in detail in the Supporting Information, ESFF1 was developed by transplanting 71 sophisticated backbone CMAP energy terms into ff14SB. In MD simulations, a CMAP term is used to correct each residue’s specific phi/psi dihedral energy based on its neighboring residues at both the N-terminus and C-terminus. To assess the performance of ESFF1, a total of 247.4 μs trajectories was simulated for 84 peptides and proteins and compared with experiments (Table S1).
MATERIALS AND METHODS
Benchmark Construction and Environment Definition.
A database of 24,236 protein structures with a sequence identity less than 50% and R factor less than 0.25 was constructed from the RCSB protein structure databank. The DSSP18 program was utilized to assign the secondary structures and dihedral angles for these proteins. Residues without any secondary structures are defined in the “coil” state. State “S” is used to denote high-curvature structures, for which the angle between vector Cαi–Cαi+2 and vector Cαi-2–Cαi is at least 70 degrees. Our preliminary analysis shows that blending S residues into a database of pure coil residues can enhance its helicity, which was found to be a limitation of our previous databases. Finally, a total of 2,611,450 coil and 1,195,531 S residues was, respectively, collected from the protein structure database.
To define a benchmark phi/psi database, all coil residues and 25% of randomly selected S residues were mixed together, with their phi/psi angle, respectively, classified based on the N-terminal and C-terminal chemical environments of the central residue. As illustrated in Figure S4, for residue Ala in the middle of tripeptide Ser-Ala-Leu, the N-terminal residue Ser was defined as the polar environment (P), and the C-terminal residue Leu was defined as the nonpolar environment (NP). Therefore, the phi/psi angle of Ala in Ser-Ala-Leu (133.9, −164.5) is classified as the P-Ala-NP class. For the 20 naturally occurring amino acids, Gly, Ser, Tyr, Cys, Asn, Gln, Thr, His, Glu, Asp, Arg, and Lys were defined as polar environments, and Met, Trp, Phe, Val, Leu, Ile, Pro, and Ala as nonpolar environments. As a result, residue “X” would exist in four different environments of NP-X-NP, NP-X-P, P-X-NP, and P-X-P. Thus, for all 20 amino acids, 80 environmental combinations corresponding to 80 different phi/psi distributions were collected, with the number of samples of each environmental combination shown in Figure S5.
CMAP Training and Force Field Integration.
Two-dimensional grid-based potential energy correction maps (termed CMAP)19 were utilized to minimize the difference of the dihedral distribution between MD simulations and benchmark data for each of the 80 environmental combinations. This method is previously utilized and described in the development of ff99IDPs, ff14IDPs, ff14IDPSFF, ff03CMAP, OPLSIDPSFF, and CHARMM36IDPSFF.9,12,20-23
The dipeptide models (Ace-X-Nme, Ace means acetyl, X represents a given amino acid, and Nme means aminomethyl) were utilized during the CMAP optimization via explicit solvent MD simulations. The training of the CMAP parameter is also described in our previous works.9,12,20,21 It is worth mentioning that CMAP parameters were optimized iteratively until the phi/psi distributions acquired from MD simulations match those from benchmarks for each environmental combination within a given tolerance, set to be less than 0.10%.
To improve the agreement between the backbone dihedral angles preferences of the dipeptide models and those from the database benchmark, an iterative optimization of 10 rounds was conducted to obtain the CMAP parameters of each environmental combination. The root-mean-square deviations of populations (RMSp) of these optimizations are shown in Figure S6, which shows that the RMSp for four types of environmental combinations of 20 amino acids between CMAP0 (ff14SB) and the database benchmark are higher than 0.22%. After the optimization, the RMSp for 17 amino acids’ environmental combinations between the best CMAP parameter and the benchmark decreases to 0.073%. For residues Cys, Met, and Trp, all four environmental combinations were combined together without considering their environments due to insufficient sampling.
After optimization, the CMAP parameter with the lowest RMSp among all 10 training rounds was selected for each specific environmental combination. As a result, a total of 71 CMAP parameters corresponding to 71 environmental combinations are organized into a text database file to be integrated into the topology files of the ff14SB force field, including three residue-specific parameters for Cys, Met, and Trp due to insufficient sampling. More specifically, a perl script was developed to help to choose suitable CMAP and generate an ESFF1 topology file. After that, standard MD simulations can be executed without extra operations.
Molecular Dynamics Simulations.
Initial dipeptide structures for 20 amino acids were built in an all-trans conformation with the LEaP module in the Amber 14 suit.24 Counterions (Na+/Cl−) were added to neutralize the systems, which were then solvated in a truncated octahedron box of TIP3P water molecules with a buffer of 10 Å. In order to verify the improvement of TIP4P-D8 in Rg reproduction, corresponding simulations at the same conditions for RS1,25 Histatin5,26 ab40,27 drkN SH3,28 and ACTR29 were conducted with ESFF1 and TIP4P-D. The CUDA version of PMEMD was used to accelerate the MD simulations.30 All systems were relaxed for 20,000 steps with the steepest descent minimization, then were heated for 20 ps and equilibrated for 20 ps in the NPT ensemble at the corresponding temperature listed in Table S1. During the simulation, the covalent bonds with hydrogen atoms were constrained using the SHAKE algorithm.31 The electrostatic interactions were calculated using the particle mesh Ewald (PME) method32 with a cutoff of 8 Å. Lennard-jones interactions were truncated at 8 Å. Temperature coupling was controlled using a Berendsen thermostat, and pressure coupling was controlled using a Berendsen barostat.33
To verify the environmental effects, multiple widely studied short peptides were simulated with three force fields ESFF1, ff14IDPSFF, and ff14SB. To compare the performances of these force fields in IDP modeling, five independent trajectories per force field were simulated for RS1,25 Histatin5,26 c-Myb,34 RevARM,35 MevN,36 rIAPP,37 ab40,27 ab42,27 drkN SH3,28 KID,38 ACTR,29 IA3,39 p53N,40 tauF4,41 and α-synuclein.42 To compare the influence of the thermostat method, a Langevin thermostat with a collision frequency of 2 ps−1 was also used for the extra simulations of c-Myb. For ACTR, an extra five trajectories were also simulated with ESFF1 and ff14SB but started from the well-folded state. To investigate the performances of ESFF1 in modeling structured proteins, 14 typical structured proteins were simulated, including TC5b,43 villin HP,44 crambin,45 LysM,46 IgGbG,47 BPTI,48 CSEv5,48 FAS1,49 CspB,50 Ccc2a,51 DMAP1, ubiquitin,52 FKBP12,53 and LUSH.54 Furthermore, to validate whether ESFF1 is able to simulate order–disorder transition in protein–protein interactions, two complexes ACTR-CBP29 and p53TAD-CBP55 were also simulated with ff14SB and ESFF1. For REMD simulations of six fast-folding proteins CLN025, TC5b, GB1, Trpzip2, WW GTT, and villin HP, the replica exchange interval was set to 1 ps, and the exchange acceptance ratio was around 10%. The totally extended unfolded REMD initial structures of CLN025, TC5b, GB1, Trpzip2, and WW GTT were obtained from 10 ns NVT high-temperature MD at 500 K. A separate 24 μs (1.2 μs × 20 replicas) REMD for villin HP was also started from the folded PDB structure 1VII44 to check the sampling issue. Detailed conditions of these simulations are summarized in Table S1.
Data Analysis.
CPPTRAJ56 was used to process all MD trajectories and calculate RMSD, RMSF, radii of gyration, dihedral angles, distances, and hydrogen bonds. Conformer clustering was performed with Kclust.57 The secondary structures were assigned with the DSSP program.18 The experimental data of the secondary chemical shift, J-coupling, SAXS, radius gyration, melting curve, and fraction of the specific secondary structure for all tested systems were downloaded from the BRMB database58 or related literatures summarized in Table S1. Secondary chemical shifts of MD simulations were calculated with ShiftX2,59 and J-coupling constants were calculated with the Karplus equation with latest parameters.60 All these analyses were calculated using the production portions of the MD trajectories listed in Table S1.
RESULTS AND DISCUSSION
ESFF1 Reproduces Local Environmental Effects in Tetrapeptides.
As listed in Table S1, tetrapeptides Ac-LXP-NH2 (X = A, C, D, E, F, H, I, K, L, M, N, Q, R, S, T, V, W, and Y) were studied by Shi et al. with NMR spectroscopy.17 The finding that the J-coupling constant of Leu depends on its neighboring residue (X) clearly shows the influence of local sequence environments on the structural preference of Leu, as expected. This set of tetrapeptides thus provides a nice initial test to study whether existing force fields can reproduce the effects of local environments in MD simulations. All simulated J-coupling constants are shown in Figure 1 along with experimental values. The P-values of the paired Wilcoxon signed rank test between ESFF1, ff14IDPSFF, ff14SB, ff03ws, a99SB-disp, and CHARMM36m simulations with NMR experiments are 0.181, 0.043, 7.63 × 10−6, 7.63 × 10−6, 7.63 × 10−6, and 1.53 × 10−5, respectively, and RMSD values are 0.183, 0.205, 0.793, 0.633, 0.943, and 0.424 Hz, respectively. Obviously, the simulations with ESFF1 agree the best with the experiment with the highest P-value and the lowest deviation, demonstrating the ability of ESFF1 in reproducing local sequence environments.
Figure 1.

Agreement of computed and measured 3J(HN,Hα) for residue Leu in the Ace-LXP-NH2 peptides. Computed data from ESFF1, ff14IDPSFF, ff14SB, ff03ws, a99SB-disp, and CHARMM36m simulations. P-value less than 0.05 means rejecting the none hypothesis of “there is no significant difference between simulation and experiment”. Red data points represent nonpolar residues X = A, F, I, L, M, V, and W in Ace-LXP-NH2. Blues data points represent polar residues X = C, D, E, H, K, N, Q, R, S, T, and Y in Ace-LXP-NH2.
ESFF1 Reproduces NMR Observables of Short Peptides.
We next tested ff14SB, ff14IDPSFF, and ESFF1 in MD simulations of 46 short peptides (3–19 residues). Two NMR observables, Δδ Cα chemical shift and 3J(HN,Hα), were monitored to assess the quality of MD simulations. Table S2 lists the RMSDs between MD simulated and NMR measured values. For the simpler single-residue repeats of GLY3, VAL3, ALA3, ALA5, and ALA7, although the RMSDs for Δδ Cα chemical shifts are similar among all three simulations, the RMSDs for 3J(HN,Hα) are significantly lower in the simulations with ESFF1 than those with ff14SB and ff14IDPSFF. These results indicate that ESFF1 can better reproduce monitored NMR observables of these simple peptides.
Another measurement worth pointing out is the temperature dependence of 3J(HN,Hα) of the central residue in ALA3 as reported by Schwalbe et al.61 Figure S7 shows the comparison of this interesting temperature-dependent observable in all three simulations with that in experiment. Overall, the trends with ESFF1 are in qualitative agreement with experiments. This is clearly different from those simulations with ff14SB and ff14IDPSFF, indicating that the ESFF1 force field also improves the temperature dependence of these observables in MD simulations.
Other NMR observables such as 3J(HN,Hα), 3J(HN,C′), and 3J(HN,Cβ) were also monitored for short peptides with the motif of GXG (X = A, E, F, K, L, M, S, and V). The agreement between MD and NMR values are listed in Table S3. It is clear that ESFF1 and ff14IDPSFF perform better than ff14SB in reproducing these NMR observables as well.
With increasing length and complexity, the different performances among the three tested force fields becomes more noticeable. An analysis of MD simulations for 14 peptides pepX (EGAAXAASS, X = D, E, G, H, I, K, L, N, P, Q, T, V, W, and Y) is shown in Table S2. Among these simulations ESFF1 was found to have consistent low RMSD values of less than 0.24 ppm for the monitored chemical shifts. In contrast, all RMSD values with ff14SB are consistently higher than 0.45 ppm. In addition the RMSD values for the monitored J-coupling constants are also significantly lower in simulations with ESFF1 and ff14IDPSFF than those with ff14SB.
Finally, Table S2 also presents a comparison for the simulations of peptide HEWL19. Consistent with the above simulations, use of ESFF1 leads to the lowest RMSD values in Δδ Cα. It is clear that with ESFF1’s focus to correct subtle main-chain torsional biases, the quality of the sampled conformers in MD simulations was noticeably improved for the tested short peptides.
ESFF1 Reproduces NMR Observables and Conformational Distributions of IDPs.
To assess the performance of ESFF1 in modeling IDPs, we simulated 15 intrinsically disordered proteins (Table S1) for which NMR observables are available. Conformational sampling for these IDPs is apparently more challenging, and care has been taken to make sure the sampling is sufficient as shown in Figure S8.
A summary of the RMSD values between experimental and simulated ΔδCα chemical shifts for these IDPs is shown in Table 1. In the simulations with ESFF1, the RMSD values are less than 0.5 ppm for nine out of 15 IDPs, and the rest are between 0.5 and 0.75 ppm. When comparing with our previous residue-specific force field ff14IDPSFF with only correct phi/psi distributions, we also observed good improvement in NMR prediction for some partially disordered proteins such as c-Myb, RevARM, MevN, and KID. On the contrary, the RMSD values are mostly greater than 1.0 ppm in simulations with ff14SB. As chemical shifts Δδ Cα are closely related to protein secondary structures, these data suggest that ESFF1 is able to represent the structural preferences of the tested IDPs better, supporting our strategy to focus on correcting subtle biases in conventional protein force fields.
Table 1.
RMSD for Secondary Δδ Cα and 3J(HN,Hα) between MD Simulations and NMR Experiments
| RMSD ofΔδ Cα (ppm) | RMSD of 3J(HN,Hα) (Hz) | |||||
|---|---|---|---|---|---|---|
| System | ff14SB | ff14IDPSFF | ESFF1 | ff14SB | ff14IDPSFF | ESFF1 |
| RS1 | 0.775 | 0.221 | 0.298 | 1.190 | 0.626 | 0.667 |
| Histatin5 | – | – | – | 1.838 | 1.153 | 0.977 |
| RevARM | 1.335 | 0.645 | 0.480 | 1.481 | 0.742 | 0.766 |
| ab40 | 1.115 | 0.409 | 0.310 | 1.481 | 0.569 | 0.577 |
| ab42 | 1.472 | – | 0.371 | 1.631 | – | 0.606 |
| drkN SH3 | 0.887 | 0.687 | 0.487 | 0.989 | 1.170 | 0.999 |
| α-synuclein | 1.016 | 0.619 | 0.636 | 1.384 | 0.546 | 0.669 |
| MevN | 1.082 | 0.657 | 0.450 | – | – | – |
| c-Myb | 1.092 | 0.847 | 0.337 | – | – | – |
| tauF4 | 1.169 | 0.750 | 0.616 | – | – | – |
| p53N | 1.178 | 0.601 | 0.721 | – | – | – |
| IA3 | 1.433 | 0.569 | 0.448 | – | – | – |
| KID | 0.678 | 0.561 | 0.483 | – | – | – |
| rIAPP | 1.183 | 0.442 | 0.509 | – | – | – |
| ACTR (PDB) | 1.288 | – | 0.719 | – | – | – |
| ACTR (extended) | 0.743 | – | 0.520 | – | – | – |
Three-bond J-coupling constants are related to backbone torsion angles by the Karplus equation.62 In particular, 3J-(HN,Hα) values are extensively used to evaluate structural preferences, with the 3J(HN,Hα) average around 5.2 Hz in helical regions and the average around 8.5 Hz in β-sheet/extended conformers.63 Therefore, we calculated J-couplings for these simulations and compared them with corresponding available experimental data (Table 1). As shown in the right part of Table 1, almost all RMSD values for ff14SB simulations are greater than 1 Hz except for drkN SH3. However, ff14IDPSFF was found to significantly reduce the RMSD values. Comparing with ff14IDPSFF, ESFF1 further improves the J-coupling constants of IDPs with all RMSD values less than 1 Hz. Obviously J-coupling constants were poorly reproduced in ff14SB simulations of IDPs, while ff14IDPSFF and ESFF1 alleviate the limitation in all tested IDPs. More interestingly, as shown in Shaw group’s analysis of six traditional force fields and their new force field a99SB-disp, the lowest RMSD value was found to be from CHARMM36m at 0.59 Hz for ab42,14 which is still higher than the values from ff14IDPSFF and ESFF1. In summary, in simulations for ab42, Histatin5, RevARM, and RS1, ESFF1 results in more reasonable J-coupling constants than many other force fields tested in the literatures.13,64,65
Comparing with our IDP-specific force field ff14IDPSFF, ESFF1 was found to achieve a better balance between ordered and disordered states in MD simulations. As shown in Figures S9-S12, simulated Δδ Cα values of c-Myb, RevARM, MevN, and KID underestimated the Δδ Cα chemical shift in those partially folded regions when ff14IDPSFF is used, while ESFF1 corrected the biases by importing condign “S” into the phi/psi benchmark database to balance the probability of PPII, sheet, and helix. According to all non-Gly, for non-Pro residues’ phi/psi distributions in MD simulations for c-Myb, RevARM, MevN, and KID shown in Figure 2 and Figures S13-S15, ff14SB always dominated the α-right region the same as the crystal structures retrieved from PDB, and ff14IDPSFF prefers the PPII and β regions rather than the α-right region. Only ESFF1 shows a balanced fraction among different secondary structure regions. As a result, the helicity of local fragments is also reproduced well in the ESFF1 simulations of c-Myb, MevN, and KID, while that in the ff14IDPSFF simulations is significantly lower and that in the ff14SB simulations was overestimated for the same proteins (Figures S16-S18). The detailed information on secondary chemical shifts, J-coupling constants, SAXS, secondary structure populations, and conformational clusters under different force fields for the tested proteins are shown in Figures S9-S29 and Tables S-S18. In addition, the extra simulations of c-Myb with a Langevin thermostat were performed and compared with a Berendsen thermostat. The results show that different thermostats have no significant influence on the conformer sampling of c-Myb with an ESFF1 force field (shown in Figure S30). However, the Langevin thermostat should be widely tested in future simulations.
Figure 2.

Main-chain torsional distributions in MD simulations of c-Myb. (A) Ramachandran plots of all non-Pro and non-Gly residues in PDB structures and in simulations with ff14SB, ff14IDPSFF, and ESFF1 force fields. (B) Fractions of secondary structure regions in ff14SB, ff14IDPSFF, and ESFF1 simulations.
ESFF1 Reproduces the Conformers and Dynamics of Structured Proteins.
To evaluate the performance of ESFF1 in modeling structured proteins, 14 typical proteins covering common folds from previous studies13,66 were simulated for 1000 ns per trajectory. Backbone structures were found to be stable throughout the production trajectories (Figure S31). The Cα RMSD profiles show that the structured proteins stay stable without the tendency for unfolding. The dominant MD conformers are also consistent with the crystal structures (Figure 3). These findings show that the ESFF1 force field is able to maintain the stability of both secondary and tertiary structures in tested structured proteins.
Figure 3.

Superposition of crystal structure (gray) and dominated structure (green) in 1000 ns ESFF1 simulations of 14 structured proteins. The dominated structure is the representative conformer of the top cluster in conformation clustering analysis by Kclust software with the RMSD cutoff set to 4 A. The RMSD between the dominated structures and the corresponding crystal structures and the percent occupancies of dominated structures are labeled.
In addition, with the available experimental side-chain J-coupling for ubiquitin, we compared the simulated 3J(C′,Cγ) and 3J(N,Cγ) coupling constants and experimental data (Table S19). The results show that ESFF1 can also well reproduce the side-chain sampling for ubiquitin. We also compared the energy landscape of eight IDPs and four folded proteins, as presented in Figure S32. The results show that the energy landscape of IDPs is more rugged than that of the folded proteins.
ESFF1 Reproduces Conformational Preferences of IDPs in Bound and Free States.
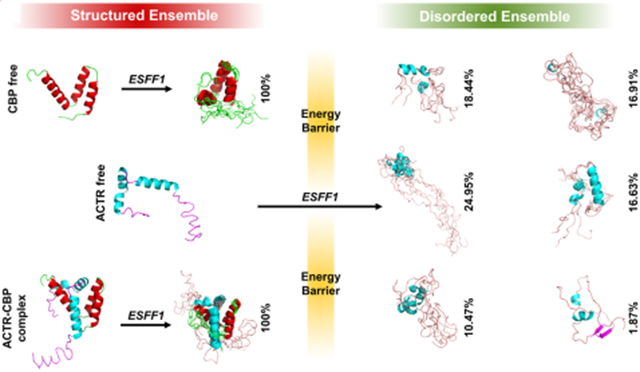
IDPs often play functional roles upon binding to receptors such as proteins, DNA, or RNA. Thus, the performance of force fields to reproduce ordered conformers in the bound state is also crucial in force field evaluations. Here, we tested two IDP complexes ACTR-CBP and p53TAD-CBP in both bound and free states. The NMR observables of ACTR, p53TAD, and CBP calculated from MD simulations with ESFF1 correlate well with experimental data in both bound and free states (Figure 4 and Tables S20-S21). More importantly, NMR experiments29,55 have shown that IDP ACTR is in the highly structured state when bound to CBP, while another IDP p53TAD only contains a few helical structures and many random coils when bound to the same receptor CBP. This interesting phenomenon was also reproduced correctly in the ESFF1 simulations (Figures S33 and S34). These data show that the ESFF1 is able to distinguish ordered and disordered states effectively and samples the most reasonable conformational ensembles.
Figure 4.

Performance of ESFF1 in modeling order–disorder complex ACTR-CBP in both free and bound states. (A) Conformers sampling of CBP, ACTR, and ACTR-CBP complex in MD simulations run by ESFF1. (B) Secondary Cα chemical shifts of ACTR and CBP in free or bound state under ff14SB, ESFF1, and NMR experiments.
ESFF1 Reproduces Melting Thermodynamics of Fast Folding Proteins.
We also studied ab initio folding simulations for six fast-folding proteins CLN025, TC5b, GB1, Trpzip2, WW GTT, and villin HP via replica exchange molecular dynamics (REMD) (Table S1). The dominant conformers from REMD simulations and corresponding experimental structures are shown in Figure 5. It is interesting to note that ESFF1 is able to fold five proteins CLN025, TC5b, GB1, Trpzip2, and WW GTT to the native state from fully extended conformers in REMD simulations. Although ESFF1 failed to sample the native state for villin HP in an extensive REMD simulation (1 μs × 20 replicas), another REMD simulation starting from the folded state (1.2 μs × 20 replicas) shows a reasonable agreement with experimental melting curve. This indicates that the sampling is still not very effective for the larger villin HP in our REMD simulations. It is also interesting to note that the melting curves for four out of five proteins (except TC5b) also agree well with experiments (Figure 6). The detailed values are listed in Table S22. The stability of TC5b is much lower than the experimental value, which is also similar to the performance of CHARMM22*/TIP3P, ff99SBildn/TIP4P-D, ff99disp, and CHARMM36m force fields in the study by the Shaw group.14 These data suggest that ESFF1 performs respectfully among all recent protein force field revisions. It is also worth pointing out that ESFF1 may underestimate the stability of some helical structures.
Figure 5.

Ab initio folding of fast-folding proteins with ESFF1. (A) Initial conformers of CLN025, Trpzip-2, GB1 hairpin, TC5b, and WW GTT for REMD. (B) Dominated conformers (magenta) from ESFF1 simulations compared with corresponding experimental structures (cyan) from the PDB database. The representative structures were calculated from the replica at the lowest temperature by clustering analysis. The centroid conformer of the Top 1 cluster for each protein is aligned with its PDB structure with corresponding Cα RMSD listed below the image. The percentage of this cluster is also listed in parentheses.
Figure 6.

Melting curves of CLN025, Trpzip-2, GB1 hairpin, TC5b, WW GTT, and villin for REMD with ESFF1 compared with corresponding experimental data.43,67-71
Usage of TIP4P-D Further Improves Simulations of IDPs.
Solvent modeling is another key factor that limits the accuracy of MD simulations of IDPs. As the Shaw group pointed out, typical solvent models such as TIP3P significantly underestimate the London dispersion interactions, leading to underestimation of the radius of gyration (Rg) and unreasonable compact conformers in MD simulations of IDPs.8 Thus, we analyzed the Rg of every tested protein in every tested force field and compared them to the corresponding experimental Rg values (Table 2). We also observed significant underestimation of Rg for both ff14SB and ESFF1 when combined with TIP3P. Despite the observation that ff14IDPSFF slightly improves Rg values, the simulated Rg values hardly agree with experimental values. Therefore, MD simulations at the same conditions for RS1, Histatin5, ab40, drkN SH3, and ACTR were repeated with the TIP4P-D solvent model. The simulated Rg values in both TIP4P-D and TIP3P solvents are listed in Table 2, and the simulated NMR properties are shown in Tables S8, S9, S11, S13, and S14, respectively. These data suggest that TIP4P-D dramatically improves the simulated Rg values. Even more interesting, it also improves the accuracy of simulated NMR properties. Finally, we also observed that simulated Rg values in TIP4P-D are often higher than experimental values, which indicates that TIP4P-D overestimates the flexibility of IDPs in MD simulations which is consistent with the findings in the literature.72
Table 2.
Rg Comparison between Simulation and Experiment
| Calculated Rg (Å) in TIP3P | Calculated Rg (Å) in TIP4P-D | ||||||
|---|---|---|---|---|---|---|---|
| System | Length | Experimental Rg (Å) | ff14SB | ff14IDPSFF | ESFF1 | ff14SB | ESFF1 |
| RS1 | 24 | 12.6 | 10.17 | 11.71 | 9.96 | 13.92 | 15.41 |
| Histatin5 | 24 | 13.8 | 10.15 | 10.38 | 10.51 | 13.97 | 14.92 |
| ab40 | 40 | 12 | 11.62 | 15.34 | 12.25 | 17.57 | 19.22 |
| ab42 | 42 | 12.4 | 11.02 | – | 11.27 | – | – |
| drkN SH3 | 59 | 16.7 | 13.22 | 19.03 | 14.97 | 22.55 | 25.59 |
| ACTR | 71 | 25 | 17.37 | – | 17.2 | 20.98 | 27.35 |
| α-synuclein | 140 | 31 | 22.95 | 28.99 | 27.69 | – | – |
CONCLUSIONS
We developed an environment-specific force field ESFF1 based on CMAP correlation of 71 types of environmental combinations. A comprehensive analysis of 61 different short peptides and IDPs shows that that ESFF1 simulations can accurately reproduce NMR properties for different peptides and proteins. With the sophisticated CMAP correlation for every specific environmental combination, ESFF1 is able to achieve a reasonable balance between ordered and disordered populations in MD simulations. Thus, ESFF1 performs excellently in conformer sampling of IDPs, suggesting that ESFF1 might be helpful for structural prediction of IDPs. In addition, long time simulations of 14 fully folded proteins show that ESFF1 is also suitable for structured proteins. More interestingly, MD simulations for ACTR-CBP and p53TAD-CBP complexes illustrate the best applications for ESFF1, where both ordered and disordered states are reproduced well. In addition, the successful ab initio folding of five fast-folding proteins also lends support in the reliability of ESFF1 in simulating structured proteins. Finally, MD simulations with ESFF1/TIP4P-D suggest TIP4P-D may further improve the performance of ESFF1, though limitations are also observed. Therefore, developing more reasonable solvent models for ESFF1 are necessary next steps. The ESFF1 force field parameters are deposited at https://github.com/chaohao2010/ADD-CMAP.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Center for HPC at Shanghai Jiao Tong University, the National Natural Science Foundation of China (31770771, 21977068, and 31620103901), the National Key Research and Development Program of China (2018YFC0310803), the Medical Engineering Cross Fund of Shanghai Jiao Tong University (YG2017MS08), and the National Institutes of Health/NIGMS (GM093040 and GM079383).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c00059.
Figures S1–S3: Initial investigation of environmental effects. Figures S4–S6: Sequence environment, environmental combination, and CMAP parameters. Figures S7 and S9–S26: Detailed comparison between the experimental data and simulated results from different force fields for disordered peptides and proteins. Figure S8: Simulation convergence of RS1. Figures S27–S28 showed the secondary structure of ab40 in ESFF1 and ff14SB simulation. Figure S29 showed the conformer cluster for ab42. Figure S30 showed the comparison of simulated results using ESFF1 force field with different thermostats. Figure S31: Cα RMSD of 14 folded proteins. Figure S32: Energy landscape of eight IDPs and four structured proteins. Figures S33 and S34: Chemical shifts and conformer clusters in simulations of p53TAD and CBP complex with ESFF1 and ff14SB force fields. Table S1: Simulation conditions used in this study. Tables S2 and S3: Comparison between experimental data and simulated results from different force fields for short peptides. Tables S4–S18: Rg and secondary chemical shift comparisons for IDPs. Tables S19 and S20: RMSDs of secondary chemical shifts between MD simulations and NMR experiments for IDP complex. (PDF)
The authors declare no competing financial interest.
Contributor Information
Dong Song, State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic & Developmental Sciences, Department of Bioinformatics and Biostatistics, National Experimental Teaching Center for Life Sciences and Biotechnology, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Hao Liu, State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic & Developmental Sciences, Department of Bioinformatics and Biostatistics, National Experimental Teaching Center for Life Sciences and Biotechnology, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Ray Luo, Departments of Molecular Biology and Biochemistry, Chemical and Molecular Engineering, Materials Science and Engineering, and Biomedical Engineering, University of California, Irvine, California 92697-3900, United States.
Hai-Feng Chen, State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic & Developmental Sciences, Department of Bioinformatics and Biostatistics, National Experimental Teaching Center for Life Sciences and Biotechnology, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China; Shanghai Center for Bioinformation Technology, Shanghai 200235, China.
REFERENCES
- (1).Brucale M; Schuler B; Samori B Single-Molecule Studies of Intrinsically Disordered Proteins. Chem. Rev 2014, 114, 3281–3317. [DOI] [PubMed] [Google Scholar]
- (2).Jensen MR; Zweckstetter M; Huang JR; Blackledge M Exploring Free-Energy Landscapes of Intrinsically Disordered Proteins at Atomic Resolution Using NMR Spectroscopy. Chem. Rev 2014, 114, 6632–6660. [DOI] [PubMed] [Google Scholar]
- (3).Horn HW; Swope WC; Pitera JW; Madura JD; Dick TJ; Hura GL; Head-Gordon T Development of an Improved Four-Site Water Model for Biomolecular Simulations: TIP4P-Ew. J. Chem. Phys 2004, 120, 9665–9678. [DOI] [PubMed] [Google Scholar]
- (4).Chong SH; Ham S Assessing the Influence of Solvation Models on Structural Characteristics of Intrinsically Disordered Protein. Comput. Theor. Chem 2013, 1017, 194–199. [Google Scholar]
- (5).Fawzi NL; Phillips AH; Ruscio JZ; Doucleff M; Wemmer DE; Head-Gordon T Structure and Dynamics of the Abeta(21–30) Peptide from the Interplay of NMR Experiments and Molecular Simulations. J. Am. Chem. Soc 2008, 130, 6145–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Abascal JL; Vega C A General Purpose Model for the Condensed Phases of Water: Tip4p/2005. J. Chem. Phys 2005, 123, 234505. [DOI] [PubMed] [Google Scholar]
- (7).Best RB; Mittal J Protein Simulations with an Optimized Water Model: Cooperative Helix Formation and Temperature-Induced Unfolded State Collapse. J. Phys. Chem. B 2010, 114, 14916–14923. [DOI] [PubMed] [Google Scholar]
- (8).Piana S; Donchev AG; Robustelli P; Shaw DE Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [DOI] [PubMed] [Google Scholar]
- (9).Wang W; Ye W; Jiang C; Luo R; Chen HF New Force Field on Modeling Intrinsically Disordered Proteins. Chem. Biol. Drug Des 2014, 84, 253–269. [DOI] [PubMed] [Google Scholar]
- (10).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved Side-Chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins: Struct., Fund, Genet 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ye W; Ji D; Wang W; Luo R; Chen HF Test and Evaluation of ff99IDPs Force Field for Intrinsically Disordered Proteins. J. Chem. Inf. Model 2015, 55, 1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Song D; Luo R; Chen HF The IDP-Specific Force Field ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model 2017, 57, 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Robustelli P; Piana S; Shaw DE Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E4758–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Duong VT; Chen Z; Thapa MT; Luo R Computational Studies of Intrinsically Disordered Proteins. J. Phys. Chem. B 2018, 122, 10455–10469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Avbelj F; Grdadolnik SG; Grdadolnik J; Baldwin RL Intrinsic Backbone Preferences Are Fully Present in Blocked Amino Acids. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang Y; Zhou Y; He L; Fu Y; Zhang W; Hu J; Shi Z Hydration Effects on Leu’s Polyproline II Population in AcLXPNH2. Chem. Commun. (Cambridge, U. K.) 2018, 54, 5764–5767. [DOI] [PubMed] [Google Scholar]
- (18).Kabsch W; Sander C Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- (19).MacKerell AD Jr.; Feig M; Brooks CL 3rd. Improved Treatment of the Protein Backbone in Empirical Force Fields. J. Am. Chem. Soc 2004, 126, 698–699. [DOI] [PubMed] [Google Scholar]
- (20).Song D; Wang W; Ye W; Ji D; Luo R; Chen HF ff14IDPs Force Field Improving the Conformation Sampling of Intrinsically Disordered Proteins. Chem. Biol. Drug Des 2017, 89, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu H; Song D; Lu H; Luo R; Chen HF Intrinsically Disordered Protein-Specific Force Field CHARMM36IDPSFF. Chem. Biol. Drug Des 2018, 92, 1722–1735. [DOI] [PubMed] [Google Scholar]
- (22).Zhang Y; Liu H; Yang S; Luo R; Chen H-F Well-Balanced Force Field ff03CMAP for Folded and Disordered Proteins. J. Chem. Theory Comput 2019, 15, 6769–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yang S; Liu H; Zhang Y; Lu H; Chen H Residue-Specific Force Field Improving the Sample of Intrinsically Disordered Proteins and Folded Proteins. J. Chem. Inf. Model 2019, 59, 4793–4805. [DOI] [PubMed] [Google Scholar]
- (24).Case DA; Babin V; Berryman JT; Betz RM; Cai Q; Cerutti DS; Cheatham TE III; Darden TA; Duke RE; Gohlke H; Goetz AW; Gusarov S; Homeyer N; Janowski P; Kaus J; Kolossvary I; Kovalenko A; Lee TS; LeGrand S; Li P; Luchko T; Luo R; Madej B; Merz KM; Paesani F; Roe DR; Roitberg A; Sagui C; Salomon-Ferrer R; Seabra G; Simmerling CL; Smith W; Swails J; Walker RC; Wang J; Wolf RM; Wu X; Kollman PA AMBER 2014; University of California: San Francisco, 2014. [Google Scholar]
- (25).Xiang S; Gapsys V; Kim HY; Bessonov S; Hsiao HH; Mohlmann S; Klaukien V; Ficner R; Becker S; Urlaub H; Luhrmann R; de Groot B; Zweckstetter M Phosphorylation Drives a Dynamic Switch in Serine/Arginine-Rich Proteins. Structure 2013, 21, 2162–2174. [DOI] [PubMed] [Google Scholar]
- (26).Raj PA; Marcus E; Sukumaran DK Structure of Human Salivary Histatin 5 in Aqueous and Nonaqueous Solutions. Biopolymers 1998, 45, 51–67. [DOI] [PubMed] [Google Scholar]
- (27).Hou L; Shao H; Zhang Y; Li H; Menon NK; Neuhaus EB; Brewer JM; Byeon IJ; Ray DG; Vitek MP; Iwashita T; Makula RA; Przybyla AB; Zagorski MG Solution NMR Studies of the a Beta(1–40) and a Beta(1–42) Peptides Establish That the Met35 Oxidation State Affects the Mechanism of Amyloid Formation. J. Am. Chem. Soc 2004, 126, 1992–2005. [DOI] [PubMed] [Google Scholar]
- (28).Marsh JA; Forman-Kay JD Structure and Disorder in an Unfolded State under Nondenaturing Conditions from Ensemble Models Consistent with a Large Number of Experimental Restraints. J. Mol. Biol 2009, 391, 359–374. [DOI] [PubMed] [Google Scholar]
- (29).Ebert MO; Bae SH; Dyson HJ; Wright PE NMR Relaxation Study of the Complex Formed between CBP and the Activation Domain of the Nuclear Hormone Receptor Coactivator ACTR. Biochemistry 2008, 47, 1299–1308. [DOI] [PubMed] [Google Scholar]
- (30).Gotz AW; Williamson MJ; Xu D; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (32).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A Smooth Particle Mesh Ewald Method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- (33).Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- (34).Zor T; De Guzman RN; Dyson HJ; Wright PE Solution Structure of the KIX Domain of CBP Bound to the Transactivation Domain of C-Myb. J. Mol. Biol 2004, 337, 521–534. [DOI] [PubMed] [Google Scholar]
- (35).Casu F; Duggan BM; Hennig M The Arginine-Rich RNA-Binding Motif of HIV-1 Rev Is Intrinsically Disordered and Folds Upon Rre Binding. Biophys. J 2013, 105, 1004–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kingston RL; Hamel DJ; Gay LS; Dahlquist FW; Matthews BW Structural Basis for the Attachment of a Paramyxoviral Polymerase to Its Template. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 8301–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Williamson JA; Miranker AD Direct Detection of Transient Alpha-Helical States in Islet Amyloid Polypeptide. Protein Sci. 2007, 16, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Radhakrishnan I; Perez-Alvarado GC; Dyson HJ; Wright PE Conformational Preferences in the Ser(133)-Phosphorylated and Non-Phosphorylated Forms of the Kinase Inducible Transactivation Domain of CREB. FEBS Lett. 1998, 430, 317–322. [DOI] [PubMed] [Google Scholar]
- (39).Green TB; Ganesh O; Perry K; Smith L; Phylip LH; Logan TM; Hagen SJ; Dunn BM; Edison AS IA3, an Aspartic Proteinase Inhibitor from Saccharomyces Cerevisiae, Is Intrinsically Unstructured in Solution. Biochemistry 2004, 43, 4071–4081. [DOI] [PubMed] [Google Scholar]
- (40).Wong TS; Rajagopalan S; Freund SM; Rutherford TJ; Andreeva A; Townsley FM; Petrovich M; Fersht AR Biophysical Characterizations of Human Mitochondrial Transcription Factor a and Its Binding to Tumor Suppressor p53. Nucleic Acids Res. 2009, 37, 6765–6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sibille N; Huvent I; Fauquant C; Verdegem D; Amniai L; Leroy A; Wieruszeski JM; Lippens G; Landrieu I Structural Characterization by Nuclear Magnetic Resonance of the Impact of Phosphorylation in the Proline-Rich Region of the Disordered Tau Protein. Proteins: Struct., Funct., Genet 2012, 80, 454–462. [DOI] [PubMed] [Google Scholar]
- (42).Bermel W; Bertini I; Felli IC; Lee YM; Luchinat C; Pierattelli R Protonless NMR Experiments for Sequence-Specific Assignment of Backbone Nuclei in Unfolded Proteins. J. Am. Chem. Soc 2006, 128, 3918–3919. [DOI] [PubMed] [Google Scholar]
- (43).Neidigh JW; Fesinmeyer RM; Andersen NH Designing a 20-Residue Protein. Nat. Struct. Biol 2002, 9, 425. [DOI] [PubMed] [Google Scholar]
- (44).McKnight CJ; Matsudaira PT; Kim PS NMR Structure of the 35-Residue Villin Headpiece Subdomain. Nat. Struct. Biol 1997, 4, 180–184. [DOI] [PubMed] [Google Scholar]
- (45).Jelsch C; Teeter MM; Lamzin V; Pichon-Pesme V; Blessing RH; Lecomte C Accurate Protein Crystallography at Ultra-High Resolution: Valence Electron Distribution in Crambin. Proc. Natl. Acad. Sci U. S. A 2000, 97, 3171–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bateman A; Bycroft M The Structure of a Lysm Domain from E. Coli Membrane-Bound Lytic Murein Transglycosylase D (MltD). J. Mol. Biol 2000, 299, 1113–1119. [DOI] [PubMed] [Google Scholar]
- (47).Ross SA; Sarisky CA; Su A; Mayo SL Designed Protein G Core Variants Fold to Native-Like Structures: Sequence Selection by ORBIT Tolerates Variation in Backbone Specification. Protein Sci. 2001, 10, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wlodawer A; Walter J; Huber R; Sjolin L Structure of Bovine Pancreatic Trypsin Inhibitor: Results of Joint Neutron and X-Ray Refinement of Crystal Form II. J. Mol. Biol 1984, 180, 301–329. [DOI] [PubMed] [Google Scholar]
- (49).Le Du M; Marchot P; Bougis P; Fontecilla-Camps JC 1.9-Å Resolution Structure of Fasciculin 1, an Anti-Acetylcholinesterase Toxin from Green Mamba Snake Venom. J. Biol. Chem 1992, 267, 22122–22130. [DOI] [PubMed] [Google Scholar]
- (50).Schindelin H; Marahiel MA; Heinemann U Universal Nucleic Acid-Binding Domain Revealed by Crystal Structure of the B. Subtilis Major Cold-Shock Protein. Nature 1993, 364, 164. [DOI] [PubMed] [Google Scholar]
- (51).Banci L; Bertini I; Ciofi-Baffoni S; Huffman DL; O’Halloran TV Solution Structure of the Yeast Copper Transporter Domain Ccc2a in the Apo and Cu (I)-Loaded States. J. Biol. Chem 2001, 276, 8415–8426. [DOI] [PubMed] [Google Scholar]
- (52).Vijay-Kumar S; Bugg CE; Cook WJ Structure of Ubiquitin Refined at 1.8 Å resolution. J. Mol. Biol 1987, 194, 531–544. [DOI] [PubMed] [Google Scholar]
- (53).Szep S; Park S; Boder ET; Van Duyne GD; Saven JG Structural Coupling between Fkbp12 and Buried Water. Proteins: Struct., Funct., Genet 2009, 74, 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kruse SW; Zhao R; Smith DP; Jones DN Structure of a Specific Alcohol-Binding Site Defined by the Odorant Binding Protein Lush from Drosophila Melanogaster. Nat. Struct. Mol. Biol 2003, 10, 694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lee CW; Martinez-Yamout MA; Dyson HJ; Wright PE Structure of the p53 Transactivation Domain in Complex with the Nuclear Receptor Coactivator Binding Domain of CREB Binding Protein. Biochemistry 2010, 49, 9964–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Roe DR; Cheatham TE 3rd. Ptraj and Cpptraj: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (57).Feig M; Karanicolas J; Brooks CL 3rd. MMTSB Tool Set: Enhanced Sampling and Multiscale Modeling Methods for Applications in Structural Biology. J. Mol. Graphics Modell 2004, 22, 377–395. [DOI] [PubMed] [Google Scholar]
- (58).Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z; Nakatani E; Schulte CF; Tolmie DE; Kent Wenger R; Yao H; Markley JL Biomagresbank. Nucleic Acids Res. 2007, 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Han B; Liu Y; Ginzinger SW; Wishart DS SHIFTX2: Significantly Improved Protein Chemical Shift Prediction. J. Biomol. NMR 2011, 50, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Vogeli B; Ying J; Grishaev A; Bax A Limits on Variations in Protein Backbone Dynamics from Precise Measurements of Scalar Couplings. J. Am. Chem. Soc 2007, 129, 9377–9385. [DOI] [PubMed] [Google Scholar]
- (61).Graf J; Nguyen PH; Stock G; Schwalbe H Structure and Dynamics of the Homologous Series of Alanine Peptides: A Joint Molecular Dynamics/Nmr Study. J.Am. Chem. Soc 2007, 129, 1179–1189. [DOI] [PubMed] [Google Scholar]
- (62).Karplus M Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J. Chem. Phys 1959, 30, 11–15. [Google Scholar]
- (63).Smith LJ; Bolin KA; Schwalbe H; MacArthur MW; Thornton JM; Dobson CM Analysis of Main Chain Torsion Angles in Proteins: Prediction of NMR Coupling Constants for Native and Random Coil Conformations. J. Mol. Biol 1996, 255, 494–506. [DOI] [PubMed] [Google Scholar]
- (64).Kang W; Jiang F; Wu YD Universal Implementation of a Residue-Specific Force Field Based on Cmap Potentials and Free Energy Decomposition. J. Chem. Theory Comput 2018, 14, 4474–4486. [DOI] [PubMed] [Google Scholar]
- (65).Carballo-Pacheco M; Strodel B Comparison of Force Fields for Alzheimer’s a Beta42: A Case Study for Intrinsically Disordered Proteins. Protein Sci. 2017, 26, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Emperador A; Orozco M Discrete Molecular Dynamics Approach to the Study of Disordered and Aggregating Proteins. J. Chem. Theory Comput 2017, 13, 1454–1461. [DOI] [PubMed] [Google Scholar]
- (67).Honda S; Akiba T; Kato YS; Sawada Y; Sekijima M; Ishimura M; Ooishi A; Watanabe H; Odahara T; Harata K Crystal Structure of a Ten-Amino Acid Protein. J. Am. Chem. Soc 2008, 130, 15327–15331. [DOI] [PubMed] [Google Scholar]
- (68).Cochran AG; Skelton NJ; Starovasnik MA Tryptophan Zippers: Stable, Monomeric Beta-Hairpins. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Munoz V; Thompson PA; Hofrichter J; Eaton WA Folding Dynamics and Mechanism of Beta-Hairpin Formation. Nature 1997, 390, 196–199. [DOI] [PubMed] [Google Scholar]
- (70).Piana S; Sarkar K; Lindorff-Larsen K; Guo M; Gruebele M; Shaw DE Computational Design and Experimental Testing of the Fastest-Folding Beta-Sheet Protein. J. Mol. Biol 2011, 405, 43–48. [DOI] [PubMed] [Google Scholar]
- (71).Kubelka J; Chiu TK; Davies DR; Eaton WA; Hofrichter J Sub-Microsecond Protein Folding. J. Mol. Biol 2006, 359, 546–553. [DOI] [PubMed] [Google Scholar]
- (72).Wu HN; Jiang F; Wu YD Significantly Improved Protein Folding Thermodynamics Using a Dispersion-Corrected Water Model and a New Residue-Specific Force Field. J. Phys. Chem. Lett 2017, 8, 3199–3205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.