

Abstract
Background
We previously reported activity of pelareorep, pembrolizumab and chemotherapy. Patients developed new T-cell clones and increased peripheral T-cell clonality, leading to an inflamed tumour. To evaluate a chemotherapy-free regimen, this study assesses if pelareorep and pembrolizumab has efficacy by inducing anti-tumour immunological changes (NCT03723915).
Methods
PDAC patients who progressed after first-line therapy, received iv pelareorep induction with pembrolizumab every 21-days. Primary objective is overall response rate. Secondary objectives included evaluation of immunological changes within tumour and blood.
Results
Clinical benefit rate (CBR) was 42% amongst 12 patients. One patient achieved partial response (PR) and four stable disease (SD). Seven progressed, deemed non-responders (NR). VDAC1 expression in peripheral CD8+ T cells was higher at baseline in CBR than NR but decreased in CBR upon treatment. On-treatment peripheral CD4+ Treg levels decreased in CBR but not in NR. Analysis of tumour demonstrated PD-L1+ cells touching CD8+ T cells, and NK cells were more abundant post-treatment vs. baseline. A higher intensity of PD-L1 in tumour infiltrates at baseline, particularly in CBR vs. NR. Finally, higher levels of soluble (s)IDO, sLag3, sPD-1 observed at baseline among NR vs. CBR.
Conclusion
Pelareorep and pembrolizumab showed modest efficacy in unselected patients, although potential immune and metabolic biomarkers were identified to warrant further evaluation.
Subject terms: Cancer immunotherapy, Translational research
Introduction
Pancreatic cancer remains a leading cause of cancer-related deaths in the United States, with incidences continuing to increase yearly at around 1% for both sexes. The 5-year relative survival rate of all stages combined is only 10% [1, 2]. Since early disease is usually asymptomatic, PDAC is most often detected only when the tumour is no longer surgically resectable and the treatment options are limited [3, 4].Therefore, new drugs and therapies are urgently needed.
Pelareorep is an intravenously administered, non-genetically modified reovirus derived from the Type 3 Dearing strain that selectively infects and replicates in tumour cells [5]. Pelareorep activates both innate and adaptive immune responses against the tumour and promotes an inflamed tumour phenotype including increased CD8+ T-cell infiltration, and PD-L1 expression [6, 7]. Pelareorep replicates preferentially in cancer cells with activated or mutated RAS signalling pathways, which is also the main genetic event in PDAC patients [8–11]. The efficacy of pelareorep in combination with other therapies including gemcitabine has been shown in several pre-clinical models [12, 13].
A phase 1, open-label, dose-escalation study demonstrated that pelareorep combined with gemcitabine had an acceptable safety profile in patients with advanced malignancies. A subsequent phase 2 clinical trial evaluated the combination of pelareorep and gemcitabine in 34 chemotherapy naïve patients with advanced PDAC reported modest clinical activity and good tolerability [14]. On-treatment biopsies revealed pelareorep replication in, and apoptosis of, tumour cells along with increased levels of PD-L1 expression in the tumour microenvironment (TME) [14].
Given the observed increase in PD-L1 expression, a subsequent phase 1b clinical trial in PDAC patients who had progressed after first-line therapy was conducted in which the anti-PD-1 checkpoint inhibitor pembrolizumab was combined with pelareorep and chemotherapy [15]. The triple-drug combination of pelareorep, pembrolizumab and single agent chemotherapy (gemcitabine, irinotecan or 5-flurouracil) was again well-tolerated and modest clinical benefit in selected patients. On-treatment biopsies showed viral replication in tumours with positive staining of viral dsRNA and reovirus capsid protein, similar to previous studies with pelareorep [5, 15–17]. In addition, increased intratumoral CD8+ T cells was seen along with an increase in LILRA4 and ICOS expression, both of which genes are involved in IFN production in response to reovirus infection. Blood levels of several IFN-inducible chemokines were also increased in the first treatment cycle, which has previously been observed during the first 48 h following pelareorep administration [18]. Although T-cell clone expansion was also seen with pembrolizumab, it was the expansion of early and durable clones associated with pelareorep (and not pembrolizumab), which correlated with improved long-term outcomes in patients, suggesting T-cell clonality as a potential biomarker in reovirus combination therapies.
The role of chemotherapy combinations in oncolytic viral therapy studies was to attenuate development of neutralising antireoviral antibodies (NARA) and to enhance the systemic delivery of virus to tumour tissues [19]. Conversely, chemotherapy immunosuppression may alter expected viral induced anti-tumour responses. To evaluate if the anti-tumour immune responses may be enhanced further through chemotherapy omission and assess potential biomarkers of oncolytic viral immunomodulation, we present the results of a phase 2 study (NCT03723915) in which pelareorep and pembrolizumab were administered as a chemo-free second-line therapy to patients with advanced PDAC.
Methods
Trial patient’s inclusion/exclusion criteria
To be eligible, patients were required to have histologically confirmed advanced (unresectable or metastatic) pancreatic adenocarcinoma with documented radiographic progression or intolerance of first-line therapy (systemic chemotherapy). Patients were required to have an ECOG Performance Score ≤ 1, measurable disease as defined by RECIST v 1.1, adequate organ function, and a life expectancy of more than 6 months. Patients who had chemotherapy/radiotherapy within 4 weeks prior to day 1 of study drug and those who had not recovered from adverse events due to agents administered more than 4 weeks before cycle 1, day 1 (C1D1) were excluded from the study. Other key exclusion criteria included an active central nervous (CNS) metastasis and/or carcinomatous meningitis, a diagnosis of immunodeficiency, autoimmune disease, history of human immunodeficiency virus (HIV) infection and an active hepatitis B or C diagnosis.
Patients must have signed an informed consent prior to enrollment. The study protocol was approved by the ethics and scientific committees of Northwestern University, Chicago, IL on 5/02/2018 (IRB number: STU00207577), and conducted according to the guidelines on good clinical practice (GCP) and the ethical principles outlined in the Declaration of Helsinki.
Trial design, treatment, and procedures
This study was a Phase 2 single-arm, open-label, Simon two-stage study conducted at Northwestern University. Each cycle lasted for 21 days, with 200 mg pembrolizumab administered intravenously, over 30 min on day 1 of each cycle. In addition, given the omission of chemotherapy, additional dosing of pelareorep was administered with cycle 1 compared to the previous Phase 1b study, with pelareorep given at 4.5 × 1010TCID50 (50% tissue culture infective dose) intravenously on days 1 (after pembrolizumab infusion was completed), 2, 3 and 8 of the first cycle and on days 1 and 8 from cycle 2 onwards (Fig. 1a). Dose modifications for either pembrolizumab or pelareorep were not permitted throughout the study. Treatment was discontinued if consent was withdrawn, unacceptable adverse events (AEs) occurred or if radiographic disease progression was observed. The safety and tolerability of pembrolizumab and pelareorep combination was assessed using the Common Terminology Criteria for Adverse Evens (CTCAE) version 4.03. Tumour assessment and disease progression were assessed by CT or MRI following the RECIST guidelines (version 1.1) performed at baseline (on C1D1 or up to 7 days before study treatment initiation) and every 3 weeks thereafter. Tumour biopsies were collected prior to treatment (or archival biopsies were used) and any time between C1D4 and C1D15 (optional), and blood samples were collected on C1D1, C1D8, C2D1, C3D1, C4D1 and at the End of Study (EoST).
Fig. 1. Study design and clinical efficacy analysis.

a Study design: patients were given pembrolizumab on day 1 of each cycle, and pelareorep on days 1, 2,3 and 8 of cycle 1 and days 1 and 8 of cycle 2 onwards. Blood and tissue biopsies were collected (as indicated) for molecular response analysis to treatment. b Swimmer plot of best responses of the 13 patients on the trial. c Progression Free Survival and d Overall Survival.
Trial objectives
The primary objective of the trial was to determine the overall response rate (ORR) defined as the proportion of patients with complete (CR) or partial (PR) response to treatment with pembrolizumab and pelareorep by RECIST v 1.1 criteria. The secondary objectives were to determine: (i) progression free survival (PFS), 1-year, 2-year, median overall survival (mOS), and clinical benefit rate (CBR) defined as the proportion of patients with ORR or stable disease (SD). (ii) safety and tolerability as determined by NCI CTCAE v 4.03, and (iii) to elucidate the effects of pembrolizumab and pelareorep combination therapy on immune responses by analysing pre-and post-treatment biopsies and blood-based immune markers. Exploratory objectives include ORR and PFS using iRECIST criteria.
Multiplex immunohistochemistry (mIHC)
The formalin-fixed and paraffin-embedded (FFPE) tumour biopsies from the pre- (baseline) and post-treated patients were collected for mIHC staining of reoviral capsid protein, CD8, CD56, FoxP3, PD-L1, VDAC1 and DAPI followed the manual of the Opal 7-Colour IHC kit (AKOYA Biosciences) as described previously [20]. The five-μm thick biopsies were baked at 65 °C for 1 h, deparaffinized, re-hydrolysed and re-fixed in 10% neutral buffered formalin prior to antigen retrieve with heated AR9 retrieval buffer (AKOYA Biosciences) in a high-pressure antigen retrieval oven. The biopsy sections were subjected to six rounds of staining procedures, including non-specific blocking, incubation of primary antibodies, HRP-labelled second antibodies and visualised with the assigned Opal fluorophore. Each round of staining was finalised with a heating retrieval using AR6 retrieval buffer to release the bounded primary and secondary antibodies without disturbing the developed Opal fluorophore signal. Afterwards, the slides were counterstained with DAPI (AKOYA biosciences) and mounted with Diamond anti-fade fluorescence mounting media (Thermo Fisher Scientific). A spectral library was generated from seven single fluorophore staining including six Opal dyes and DAPI for the “spectral unmixing process”, and the unstained slide served as the background control. The antibodies and corresponding Opal fluorophores are listed in Supplementary Table 1. The antibody to reovirus capsid protein was a gift from Dr. Matt Coffey (Oncolytics Biotech, Inc.).
Acquirement of multispectral images (MSI) and data analysis
The stained biopsy sections and the single Opal fluorophore staining were imaged at high magnification (20x) using the Vectra 3 Automated Quantitative Pathology Imaging System (Perkin Elmer), which is equipped with five emission spectral filters including DAPI, FITC, Cy3, Texas Red and Cy5. The images were proceeded with spectral unmixing into seven individual fluorophores based on the unique emitting spectrum pattern of each single staining fluorophore using InForm Advanced Image Analysis software (AKOYA Biosciences). Subsequently, the unmixed images underwent cell segmentation based on DAPI, membrane and cytoplasm specific markers, subsequently cell phenotyping based on specific cellular markers. The data containing composite images, cell segmentation and cell phenotyping from InForm were further evaluated by quantitative analyses of cellular densities (Count of phenotypic cell/DAPI count) and protein intensities using R-based phenoptrReports & phenoptr (AKOYA biosciences).
Spectral flow cytometry (CyTEK)
The peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by Ficoll-Paque Plus (GE Healthcare) density gradient centrifugation and cryopreserved for subsequent testing. For CyTEK analysis, PBMCs were incubated in Fc blocker for 10 min at room temperature, followed by viability staining for 15 min at room temperature. Cells were washed and incubated with surface staining antibody cocktail for 30 min at 4 °C. Cells were washed with 1x Permeabilization/Wash buffer (eBioscienceTM, 00-8333-56), and fixed/permeabilized with 1x Fixation/Permeabilization reagent for 20 min at room temperature (eBioscienceTM, 00-5223-56). Cells were washed with 1x Permeabilization/Wash buffer twice. Intracellular staining (ICS) was performed in 100 µL 1x Permeabilization/Wash buffer with ICS antibody cocktail (Supplementary Table 2) for 45 min at room temperature. Cells were washed and resuspended in phosphate buffer saline (PBS) for acquisition on a 3 laser Cytek Aurora spectral flow cytometer. FCS files were analysed using Flowjo v10 software and OMIQ. Clustering analyses were performed using the viSNE algorithm [21] within the Cytobank and OMIQ web applications per the developers’ instructions. All events were sampled with a minimum estimated cluster size of 1% (~1000 events). The Significance Analysis of Microarrays (SAM) association model [22] was also used for the differential analyses of individual clusters.
Measurement of plasma soluble checkpoints
The levels of plasma soluble checkpoints were measured by Luminex using The Human Immuno-Oncology Checkpoint 14-Plex ProcartaPlex Panel (ThermoFisher) according to the manufacturer’s instructions. All standards and samples were measured in duplicates.
Statistical methods
This Phase 2 study followed a Simon’s two-stage optimum design to evaluate the efficacy of pembrolizumab in combination with Pelareorep. The “Go/No Go” criteria in Phase 2 (Stage 1) was defined using null response rate, alternative response rate, power, and alpha. In Stage 1, with null rate 10%, alternative rate 35%, power 90%, and 1-sided alpha 0.025, the Simon two-stage "optimum" design specified a stage 1 sample size of 11, stopping for futility if < = 1 response out of 11 in stage 1, and adding 19 additional patients if > = 2 responses at stage 1. In Stage 2, at least 7 responses out of 30 were required to reject a null rate of <10%. Comparison of values was performed using two-sided t-tests, ANOVA test or Mann–Whitney U-test for unpaired data, and Wilcoxon matched-pairs signed rank test for paired data via GraphPad Prism. Additional statistical methods are described where appropriate.
Results
Patient characteristics
Fifteen patients had signed consent for the study, 2 screen failed (one for declining performance status and one for a concurrent second malignancy). The first patient enrolled on 11/13/2018 and the last patient was entered on 10/18/2019. A total of 13 histologically confirmed advanced (unresectable or metastatic) pancreatic adenocarcinoma patients with documented objective radiographic progression on first-line therapy systemic chemotherapy for advanced PDAC were enrolled in this study. One patient who was deemed ineligible for efficacy analysis developed ischaemic cerebrovascular accident (CVA) in C1, unrelated to study medication and did not continue on study. Table 1 summarises the patients’ demographic and clinical factors. Most of the patients were female (n = 9, 69.2%), Caucasian (n = 9, 69.2%) and non-Hispanic (n = 12, 92.3%) with the overall median age of 64 years (range: 50–71). The median CA 19-9 at baseline was 240 U/ml, ranging from 7-20881 U/ml (normal range healthy individuals 0–37 U/ml). Two patients had previous resection of their primary tumour, three were treated with radiotherapy and all received chemotherapy prior to this trial. Two patients had an ECOG performance status of 0 and 11 had an ECOG performance status of 1.
Table 1.
Demographic details.
| Overall (N = 13) | |
|---|---|
| Age (years) | |
| Mean (SD) | 61.3 (6.98) |
| Median [min, max] | 64.0 [50.0, 71.0] |
| Sex | |
| Female | 9 (69.2%) |
| Male | 4 (30.8%) |
| Race | |
| Asian | 1 (7.7%) |
| Black | 1 (7.7%) |
| Not reported/refused | 2 (15.4%) |
| White | 9 (69.2%) |
| Ethnicity | |
| Non-hispanic | 12 (92.3%) |
| Not reported/refused | 1 (7.7%) |
| ECOG PS | |
| 0 | 2 (15.4%) |
| 1 | 11 (84.6%) |
Patient’s demographic and clinical factors.
Safety
All 13 patients who received study medications were evaluable for safety. Overall, the combination of pembrolizumab with pelareorep was tolerable with manageable adverse events. The most frequent AEs are listed in Table 2. The most common AEs were chills (85%), fever (77%), vomiting (69%) and nausea (54%). Most AEs were grade 1 or 2. No grade 4 AEs were reported, and the only grade 3 AEs that occurred with a frequency >10% were fatigue, decreased lymphocyte count and hyponatremia, each of which was observed in 15% of patients (2/13).
Table 2.
Adverse events summary.
| AE | All grades | Grade 1 | Grade 2 | Grade 3 |
|---|---|---|---|---|
| Chills | 11 (84.62%) | 10 (76.92%) | 1 (7.69%) | 0 (0%) |
| Fever | 10 (76.92%) | 7 (53.85%) | 2 (15.38%) | 1 (7.69%) |
| Vomiting | 9 (69.23%) | 8 (61.54%) | 0 (0%) | 1 (7.69%) |
| Nausea | 7 (53.85%) | 3 (23.08%) | 3 (23.08%) | 1 (7.69%) |
| Fatigue | 6 (46.15%) | 4 (30.77%) | 0 (0%) | 2 (15.38%) |
| Diarrhoea | 5 (38.46%) | 5 (38.46%) | 0 (0%) | 0 (0%) |
| Hyperglycaemia | 5 (38.46%) | 0 (0%) | 5 (38.46%) | 0 (0%) |
| Anaemia | 4 (30.77%) | 3 (23.08%) | 0 (0%) | 1 (7.69%) |
| Headache | 4 (30.77%) | 4 (30.77%) | 0 (0%) | 0 (0%) |
| White blood cell decreased | 4 (30.77%) | 2 (15.38%) | 1 (7.69%) | 1 (7.69%) |
| Abdominal pain | 3 (23.08%) | 2 (15.38%) | 1 (7.69%) | 0 (0%) |
| Alkaline phosphatase increased | 3 (23.08%) | 1 (7.69%) | 1 (7.69%) | 1 (7.69%) |
| Anorexia | 3 (23.08%) | 3 (23.08%) | 0 (0%) | 0 (0%) |
| Aspartate aminotransferase increased | 3 (23.08%) | 3 (23.08%) | 0 (0%) | 0 (0%) |
| Blood bilirubin increased | 3 (23.08%) | 3 (23.08%) | 0 (0%) | 0 (0%) |
| Lymphocyte count decreased | 3 (23.08%) | 0 (0%) | 1 (7.69%) | 2 (15.38%) |
| Neutrophil count decreased | 3 (23.08%) | 0 (0%) | 2 (15.38%) | 1 (7.69%) |
| Platelet count decreased | 3 (23.08%) | 2 (15.38%) | 1 (7.69%) | 0 (0%) |
| Proteinuria | 3 (23.08%) | 2 (15.38%) | 1 (7.69%) | 0 (0%) |
| Hyponatremia | 2 (15.38%) | 0 (0%) | 0 (0%) | 2 (15.38%) |
| Hypothyroidism | 2 (15.38%) | 2 (15.38%) | 0 (0%) | 0 (0%) |
| Musculoskeletal and connective tissue disorder—other, specify | 2 (15.38%) | 2 (15.38%) | 0 (0%) | 0 (0%) |
| Myalgia | 2 (15.38%) | 2 (15.38%) | 0 (0%) | 0 (0%) |
| Nasal congestion | 2 (15.38%) | 2 (15.38%) | 0 (0%) | 0 (0%) |
| Alanine aminotransferase increased | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Arthralgia | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Back pain | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Chronic kidney disease | 1 (7.69%) | 0 (0%) | 1 (7.69%) | 0 (0%) |
| Cough | 1 (7.69%) | 0 (0%) | 1 (7.69%) | 0 (0%) |
| Dry mouth | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Dyspnoea | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Generalised muscle weakness | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| GGT increased | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Hyperhidrosis | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Hypertension | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Hypoalbuminemia | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Lymphocyte count increased | 1 (7.69%) | 0 (0%) | 1 (7.69%) | 0 (0%) |
| Pleural effusion | 1 (7.69%) | 0 (0%) | 1 (7.69%) | 0 (0%) |
| Postnasal drip | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Pruritus | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Rash acneiform | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Rash maculo-papular | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Sinus tachycardia | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Urinary tract infection | 1 (7.69%) | 0 (0%) | 1 (7.69%) | 0 (0%) |
| Urine discoloration | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
| Vaginal dryness | 1 (7.69%) | 1 (7.69%) | 0 (0%) | 0 (0%) |
The safety and tolerability of pembrolizumab and pelareorep combination are evaluated by the frequencies and percentages of AEs for all grades, determined by NCI CTCAE v 4.03. The AEs are listed in a descending order of frequencies of AEs >10 percent for all grades.
Antitumor activity—clinical efficacy
Twelve of the 13 enrolled patients were evaluable for efficacy based on tumour response per RECIST v1.1 and iRECIST. Based on both the iRECIST and RECIST version 1.1, no patients had a CR, 1 had a PR, 4 had SD and 7 had progressive disease (PD) as their best response (Fig. 1b). The swimmer’s plot shows the distribution of time to progression or death, differentiating between best response types by colour and duration of response for individual patients. Disease control or Clinical Benefit Rate (CBR), defined as PR + SD, was achieved in 5/12 (42%) of the patients. The overall median PFS and OS was 1.9 months and 6.3 months, respectively (Fig. 1c, d). The 6, 12- and 24-month OS rates were 58.8%, 8.3% and 8.3%, respectively. Amongst patients with CBR the median OS was 7.3 vs. 6.2 months in non-responders (NR) who had imaging proven PD on first evaluation.
Immune profiles associated with response
The peripheral blood mononuclear cells (PBMCs) from a cohort of 11 patients before (C1D1) and after treatment (C2D1) were characterised using a high-dimensional spectral flow cytometry (CyTEK) panel that incorporated hallmark surface markers for all major lymphocyte populations. The dimensionality reduction tool viSNE was employed to compare CBR vs. NR. Live intact single cells gated from the PBMC could be clearly grouped into distinct subsets (Fig. 2a), including CD4+ T cells (CD3+CD4+), CD8+ T cells (CD3+CD8+), Treg (CD3+CD4+ Foxp3+CD25+), non-Treg CD4+ T cells (CD3+CD4+ Foxp3−), B cells (CD19+ CD3−), NK (CD3−CD56+), NKT (CD3+CD56+). The frequencies of all baseline major lymphocyte subsets using conventional supervised gating on FlowJo, did not show significant differences between CBR and NR (Suppl. Fig. S1), although there was a trend for an increase in B cell levels but a decrease in NKT cell levels at C2D1 in comparison to C1D1 in both CBR and NR (Fig. 2b). Notably, a significant reduction in the ratio of CD4+ Treg levels at C2D1 over C1D1 was observed in CBR but not in NR (Fig. 2c). Given the key role of T cells in response to anti-PD-1 immune checkpoint blockade therapy, we focused on this cell type. Both CD4+ and CD8+ T-cell populations were examined by using the combination of CD45RA and CCR7 (CD197) cell surface markers to define naive (CD197+ CD45RA+), central memory (CM, CD197+ CD45RA−), effector memory (EM, CD197− CD45RA−), or effector memory RA+ (EMRA, CD197−CD45RA+) subsets. We found no significant differences in these T-cell memory subsets at C1D1 and C2D1 between CBR and NR (Suppl. Fig. S2). The gating strategy is shown in Suppl. Fig. S3.
Fig. 2. Identification of differences in immune cell populations between CBR and NR using spectral flow cytometry (CyTEK).

a Exemplified tSNE visualisation of merged events from PBMCs of NR (n = 6) and CBR (n = 5), before (C1D1) and after therapy (C2D1). b The summarised immune cell composition of PBMCs of NR and CBR, before (C1D1) and after therapy (C2D1). c Fold changes in Treg abundance in response to therapy between NR and CBR. Error bars represent mean with SEM. *p < 0.05. The p-value was calculated by Wilcoxon rank-sum test.
Likewise, we observed no differences in expression of the activation or exhaustion markers CD69, KLRG1, PD-1, or HLA-DR (not shown) among different T-cell subsets. In contrast, peripheral CD8+ T cells in CBR displayed higher expression levels of mitochondrial marks Voltage Dependent Anion Channel-1 (VDAC1), Hexokinase II (HK2) and the epigenetic mark H3K27me3 with a higher proliferative capacity (Ki67 expression) than those in NR at C1D1 (Fig. 3a). Particularly, CD8 EM cells had significantly increased expression of VDAC1, while CD8 naïve cells had significantly increased expression of Ki67 in CBR than those in NR at C1D1 (Fig. 3b). Furthermore, the VDAC1 expression in CD8+ T cells was significantly increased in NR but decreased in CBR after therapy (Fig. 3c). Taken together, these results suggest that distinct populations of CD8+ T-cell with active epigenetic and mitochondrial programs likely possess a superior ability of response to immunotherapy. Nevertheless, more work needs to be done to prove these points.
Fig. 3. Distinct features of peripheral CD8+ T cells between CBR and NR.

a The heat map and quantifications represent the relative median expression for metabolic markers VDAC1 and HK2, the epigenetic mark H3K27me3, and the proliferation marker Ki67 the exhausting markers PD-1 and KLRG1 within CD8+ T cells from NR (n = 6) and CBR (n = 5) at C1D1. b Relative median expression of Ki67 in naïve CD8+ T cells, and the VDAC1 expression in EM CD8+ T cells were significantly increased in CBR compared to NR at C1D1 by SAM analysis. c Relative expression levels of VDAC1 in total CD8+ T cells at C1D1 and C2D1 between CBR and NR. Error bars represent mean with SEM. *p < 0.05, **p < 0.01, ***p < 0.001. All p-values were calculated using two-sided t-tests and were corrected for the multiple comparison using the Benjamini–Hochberg adjustment.
To explore potential mechanisms of therapeutic immune response, we examined pre- and post-treated tumour biopsy sections for immune infiltration by mIHC using Opal staining, which allowed for the simultaneous assessment of seven markers in a single FFPE tissue section. We stained the following set of markers: CD8, Foxp3, CD56, VDAC1, PD-L1, reoviral capsid protein and DAPI (nuclear stain) (Fig. 4a). All the 7 post-treatment biopsies were positive for reoviral capsid. Through the spectral unmixing algorithm, we found no significant differences in density of intratumoral CD8+ T cells, CD56+ NK cells, FoxP3+ Treg cells, PD-L1+ cells and VDAC1+ cells between baseline and post-treatment specimens (Fig. 4b). However, the post-treatment samples tended to display a greater expression level (intensity) of PD-L1 in CD8+ T cells, CD56+ NK cells, as well as in FoxP3+ Treg cells, and VDAC1+ cells than the baseline samples in the total analysed areas (Fig. 4c), although statistical significance was not reached. Similarly, there was a trend for a decrease in VDAC1 intensity in CD8+ T cells, CD56+ NK cells, as well as in FoxP3+ Treg cells, and PD-L1+ cells upon treatment (Fig. 4d). The subsequent spatial analysis revealed that the PD-L1+ cells touching CD8+ T cells, CD56+ NK cells, as well as likely FoxP3+ Treg cells, were more abundant in the post-treatment samples than in the baseline samples (Fig. 4e). More interestingly, we found a significantly higher intensity of PD-L1 in CD56+ NK cells, FoxP3+ Treg cells, VDAC1+ cells as well as likely CD8+ T cells, particularly in CBR than NR (Fig. 4f) in the baseline samples. The CBR also tended to display higher intensities of VDAC1 in CD8+ T cells, CD56+ NK cells, and FoxP3+ Treg cells than NR in the total analysed area (Fig. 4g).
Fig. 4. Distinct features of tumour-infiltrating leucocytes (TILs) between CBR and NR.

a Representative image of TIL profiling including cytotoxic T cells (CD8+), NK cells (CD56+), Treg cells (CD8-FoxP3+), and reovirus-infected tumour cells (Capsid+) with functional markers PD-L1 and VDAC1 in a responder patient after treatment measured by mIHC. Original magnification, ×20. b Density of intratumoral CD8+ T cells, CD56+ NK cells, FoxP3+ Treg cells, PD-L1+ cells and VDAC1+ cells (c) from C1D1 (n = 7) and C2D1 (n = 7). C VDAC1 intensity of intratumoral CD8+ T cells, CD56+ NK cells, FoxP3+ Treg cells, PD-L1+ cells and Capsid+ cells from C1D1 (n = 7) and C2D1 (n = 7). d PD-L1 intensity of intratumoral CD8+ T cells, CD56+ NK cells, FoxP3+ Treg cells, VDAC1+ cells and Capsid+ cells from C1D1 (n = 7) and C2D1 (n = 7). e Representative image file written using Phenoptr to calculate touching pairs of phenotyped one infiltrating cell population (yellow) with the other phenotyped one (red). Touching events among different cellular components as indicated from paired C1D1 (n = 6) and C2D1 (n = 6). f VDAC1 intensity of intratumoral CD8+ T cells, CD56+ NK cells, FoxP3+ Treg cells, and PD-L1+ cells at C1D1 from CBR (n = 4) and NR (n = 3). g PD-L1 intensity of intratumoral CD8+ T cells, CD56+ NK cells, FoxP3+ Treg cells, and VDAC1+ cells at C1D1 from CBR (n = 4) and NR (n = 3). Box and whiskers represent mean ± SD, and each point represents one patient, *p < 0.05. The p-values were calculated by a two-way ANOVA analysis.
Lastly, expression levels of soluble immune checkpoints were examined from plasma samples at C1D1, C2D1 and C3D1 (Fig. 5). Notably, there were significantly higher levels of soluble (s)IDO, sLag3, sPD-1 in the baseline samples from NR than those from CBR, indicating the possible predictive role of sIDO, sLag3, sPD-1 in unfavourable immune checkpoint inhibitor (ICI) response in PDAC patients.
Fig. 5. Plasma levels of soluble immune checkpoints between NR and CBR.
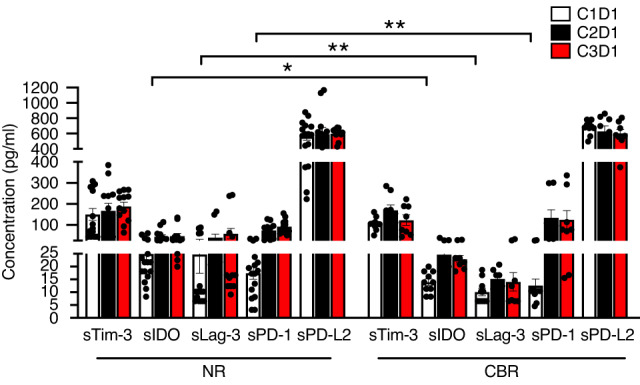
The levels of plasma soluble checkpoints as indicated were measured from NR (n = 8) and CBR (n = 5) by Luminex using The Human Immuno-Oncology Checkpoint 14-Plex ProcartaPlex Panel. Error bars represent mean with SEM. *p < 0.05, **p < 0.01. The p-values were calculated using two-sided t-tests.
Discussion
Building upon the pre-clinical and clinical evidence of immunomodulation with pelareorep in PDAC, we designed a series of combinatory studies evaluating the role of pelareorep in combination with an immune checkpoint inhibitor (ICI) and/or chemotherapy in patients with advanced PDAC who had progressed on first-line chemotherapy. We previously reported the triple-drug combination of pelareorep, pembrolizumab and single agent chemotherapy (gemcitabine, irinotecan or 5-flurouracil) was well-tolerated and no immune-related adverse events were noted. Of the 10 evaluable patients, three showed prolonged clinical benefit: one had a PR lasting for 17.4 months, and two had SD lasting 4 and 9.2 months. On treatment tumour analysis confirmed reoviral replication and increased intertumoral CD8+ T cells. The sequential administration of pelareorep/chemotherapy on C1D1 followed a week later (C1D8) with pembrolizumab allowed us to characterise emerging and expanding T-cell clones from each constituent of the combination strategy. We found that, while clonal expansion was more pronounced after pembrolizumab, it was the early clonal expansion achieved with pelareorep priming as well as the durable clonal expansion that appeared to be associated with improved long-term outcomes [6].
The previous use of chemotherapy was hypothesised to improve clinical efficacy by attenuating development of NARA therefore enhancing systemic delivery of reovirus to tumour tissue [19]. Conversely, in the era of combinatory therapy with immune-modulating therapies, such as oncolytic viruses, the resulting immune suppression from chemotherapy may result in inferior anti-tumour immune response when combined with ICI. We sought to investigate this further through this chemotherapy-free regimen. Given the long half-life of pembrolizumab, we opted incorporate both agents simultaneously in the current study. Finally, to allow for a more robust immune induction we gave pelareorep intravenously more frequently resulting in a higher total dose with cycle 1 to augment viral induced tumour immune cell infiltration. This dose was deemed to be safe in previous studies [15]. The combination was well-tolerated with grade 1 or 2 flu-like symptoms reported as the most common treatment related AE. No viral enhanced immune-related toxicity was also noted compared to single agent pembrolizumab.
In terms of clinical efficacy, the primary endpoint was to determine the overall response rate (ORR) by RECIST v 1.1. One patient had a PR, 4 had SD and 7 had PD as their best response. None of these patients had MSI-H disease. Our study did not meet the pre-specified threshold of two or more responses in stage I to allow for study expansion into stage II. The lack of clinical activity suggests omission of chemotherapy alone to improve viral immunomodulation had no impact in enhancing tumour response in this unselected PDAC patient population. Amongst the 12 evaluable patients in stage I, five patients had PR or SD or clinical benefit rate (CBR) of 42%. An additional 7 patients had PD on first scan evaluation or were deemed NR. This grouping allowed for tumour and blood correlative analysis to help predict or enhance future immunotherapeutic strategies in PDAC patients.
We used clustering to determine peripheral immune cell populations that contribute to anti-tumour response. Different immunosuppressive cell populations (e.g., Treg and MDSCs) have been investigated as potential predictive or prognostic biomarkers in cancer patients receiving ICIs. Interestingly, we found that peripheral Treg abundance was significantly decreased in CBR but not NR after pelareorep pembrolizumab treatment, indicating that relative changes in circulating Tregs during combinatorial immunotherapy could be an indicative of response to ICI treatment. This is consistent with a recent report [23], showing that a decline of PD‐1+ Tregs in peripheral blood of melanoma patients after the first treatment cycle of nivolumab or pembrolizumab was significantly associated with more favourable outcome. However, the biomarker results with regards to peripheral Tregs are overly controversial [24]. Thus, the clinical implication of Treg abundance and functionality particularly in PDAC patients treated with ICIs requires further investigation.
In an attempt to explore the mechanism of therapeutic response, we devised a specialised cytometric CyTEK panel to interrogate metabolic programs within peripheral lymphocytes at a single cell level given the importance of metabolic reprogramming for immune cell differentiation and function [25–27]. Our data have revealed elevated expression levels of mitochondrial marks VDAC1 and HK2, and the epigenetic mark H3K27me3 in baseline peripheral CD8+T cells in responders compared to that in non-responders. An emerging study indicates the presence of peripheral H3K27me3hiVDAC1hi T-cell population in COVID-19 patients, and increased expression of VDAC1 in these cells, are linked to mitochondrial dysfunction and apoptosis [28]. Thus, we infer that reduced VDAC1 expression could contribute to a protective immunity or improve the functionality of pre-existing effector T-cell immunity in response to ICIs. Indeed, we found that the VDAC1 expression in CD8+ T cells was significantly decreased in patients with CBR with treatment, while it increased in NR. Increased survival in TCR-stimulated CD8+ T cells was also observed from CBR after therapy when compared to NR (data not shown). Similar to peripheral counterparts, intratumoral CD8+ T cells (together with NK cells) tended to display lower intensities of VDAC1 upon treatment. Taken together, our data suggest that increased VDAC1 expression in effector immune cells may drive dysregulated antitumor immunity, but possibly represents potentially potent biomarkers to predict and track ICI response.
PD-1/PD-L1 inhibitors demonstrated no therapeutic benefit in PDAC, except those patients with MSI-H/dMMR [29]. Comprehensive IHC analysis of PD-L1 showed scarce expression mainly on the surface of stromal and immune cells in PDAC [30–33], possibly accounting for the ineffectiveness of anti-PD-1/PD-L1 antibodies. We have recently reported a PD-L1 upregulation in PDAC after oncolytic virotherapy in combination with gemcitabine [14]. Similarly, in the present study PD-L1 expression tended to increase particularly in different immune cell populations after pelareorep+ pembrolizumab treatment, and more abundance of the CD8+ T cells and NK cells touching PD-L1+ cells was observed in post-treatment samples. This is likely the result of an active immune response to IFN-γ release by effector T cells through a negative feedback loop that sustains PD-L1 expression [34]. These results are consistent with a recent study that has shown that PD-L1 on tumour-infiltrating immune cells (TIIC) in addition to cancer cells is important for predicting best response to atezolizumab in non-small cell lung cancer [35]. Several other studies have also indicated that high PD-L1 expression on TIIC, but not on cancer cells, is a favourable prognostic factor in multiple types of cancer [36–38]. In addition, we found significantly greater expression levels of PD-L1 in CBR than NR from baseline samples, indicator of pre-existing immunity, supporting the value of PD-L1 expression level as a predictor of response to combinatorial immunotherapy in PDAC. These findings together underscore the importance of comprehensive assessment of PD-L1 expression on both compartment of TIIC and cancer cells in PDAC especially in response to PD-1/PD-L1 inhibitors.
Besides peripheral and tumour immune cell characterisation, we measured the plasma levels of soluble forms of both stimulatory and inhibitory factors that regulate T-cell expansion and function involved in the cancer-immunity cycle [39]. While the fluctuation of these proteins in the NR and CBR groups showed no difference after therapy, significantly higher levels of sIDO, sLag3, sPD-1 were observed in the baseline samples from NR than those from CBR. Although soluble inhibitory molecules do not always exert negative immune effects, we speculate that the pre-existing immune systems could be exhausted by their potential immunomodulatory activity, leading to unfavourable ICI response in PDAC patients, consistent with recent findings that these circulating soluble checkpoints could predict shorter survival and reduced clinical benefit from PD-1/PD-L1 checkpoint blockade in other solid tumours [40–44]. Nevertheless, the regulation and immunologic function of these proteins still remain elusive.
Conclusion
Pelareorep in combination with pembrolizumab in patients with advanced PDAC progressing on first-line systemic chemotherapy, had limited activity in an unselected PDAC patient population. By extending our previous studies [14], in the current research we observed comprehensive immunomodulatory effects of pelareorep in combination with pembrolizumab when analysing on-treatment tumour biopsies including viral replication with associated infiltration of CD8+ T cells and NK cells interacting with PD-L1+ cells, and reduction of peripheral Tregs in patients responding to therapy through integration of tumour mIHC analysis and circulating immune cell characterisation using spectral cytometric CyTEK. Lower expression levels of PD-L1 in on-treatment tumour biopsies but higher soluble inhibitory molecules such as sIDO, sLag3 and sPD-1 in plasma are observed at baseline amongst NR than CBR. Furthermore, increased VDAC1 expression in effector cells may drive dysregulated antitumor immunity and may predict future immunomodulation strategies with ICI. The findings in our study need to be validated in a larger patient cohort, a limitation of our study. There is an ongoing phase I/II clinical study in Europe exploring pelareorep in combination with atezolizumab in multiple gastrointestinal cancer treatment combination (GOBLET) [45, 46], including PDAC patients in the first-line setting with gemcitabine and nab-paclitaxel, that will provide further pooled clinical and correlative biomarker data to enhance the current limitation of immunotherapy in PDAC patients.
Supplementary information
Acknowledgements
The study received Pembrolizumab from Merck (MISP 55500). The study received Reolysin and research funding from Oncolytics Biotech.
Author contributions
Conceptualisation and design: DM, BZ. Data acquisition: DM, SC, PX, AK, SC, VC, MM, AB, BZ. Data analysis: DM, SC, PX, HL, TH, AK, SC, IBH, XM, MC, MM, AB, BZ. Manuscript writing: DM, SC, PX, HL, TH, BZ. Manuscript revisions and approval of final version: All authors. Accountability of work: all authors.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
DM received research funding Oncolytics Biotech and drug support from Merck. HL, TH and MC are employees of Oncolytics Biotech.
Ethics approval and consent to participate
This study was conducted upon approval of the Institutional Review Board (IRB) at Northwestern University, Chicago, IL on 5/02/2018 (IRB number: STU00207577) and in accordance with current U.S. Food and Drug Administration (FDA) regulations, the International Conference on Harmonisation (ICH), Good Clinical Practices (GCPs), the Declaration of Helsinki, and local ethical and legal requirements. All patients signed a written informed consent before the conduct of any study procedures and after a full explanation of the study to the patient by the study investigator.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Siqi Chen, Ping Xie.
Contributor Information
Devalingam Mahalingam, Email: mahalingam@northwestern.edu.
Bin Zhang, Email: bin.zhang@northwestern.edu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-023-02344-5.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Tempero MA, Malafa MP, Al-Hawary M, Behrman SW, Benson AB, Cardin DB, et al. Pancreatic adenocarcinoma, version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021;19:439–57.. doi: 10.6004/jnccn.2021.0017. [DOI] [PubMed] [Google Scholar]
- 3.Garg SK, Chari ST. Early detection of pancreatic cancer. Curr Opin Gastroenterol. 2020;36:456–61.. doi: 10.1097/MOG.0000000000000663. [DOI] [PubMed] [Google Scholar]
- 4.Pereira SP, Oldfield L, Ney A, Hart PA, Keane MG, Pandol SJ, et al. Early detection of pancreatic cancer. Lancet Gastroenterol Hepatol. 2020;5:698–710. doi: 10.1016/S2468-1253(19)30416-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10:eaam7577. doi: 10.1126/scitranslmed.aam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahalingam D, Kalyan A, Kircher SM, Maurer V, Kocherginsky M, Xu J, et al. Pembrolizumab in combination with the oncolytic virus pelareorep in patients progressing on systemic chemotherapy for advanced pancreatic adenocarcinoma: A phase II study. J Clin Oncol. 2020;38:2020. doi: 10.1200/JCO.2020.38.15_suppl.e16789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fountzilas C, Patel S, Mahalingam D. Review: Oncolytic virotherapy, updates and future directions. Oncotarget. 2017;8:102617–39.. doi: 10.18632/oncotarget.18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Twigger K, Roulstone V, Kyula J, Karapanagiotou EM, Syrigos KN, Morgan R, et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the EGFR pathway. BMC Cancer. 2012;12:368. doi: 10.1186/1471-2407-12-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roulstone V, Pedersen M, Kyula J, Mansfield D, Khan AA, McEntee G, et al. BRAF- and MEK-targeted small molecule inhibitors exert enhanced antimelanoma effects in combination with oncolytic reovirus through ER stress. Mol Ther. 2015;23:931–42.. doi: 10.1038/mt.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–62. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17:153–68.. doi: 10.1038/s41575-019-0245-4. [DOI] [PubMed] [Google Scholar]
- 12.Groeneveldt C, Kinderman P, van den Wollenberg DJ, van den Oever RL, Middelburg J, Mustafa DA, et al. Preconditioning of the tumor microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J Immunother Cancer. 2020;8:e001191. doi: 10.1136/jitc-2020-001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gujar SA, Clements D, Dielschneider R, Helson E, Marcato P, Lee PW. Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br J Cancer. 2014;110:83–93. doi: 10.1038/bjc.2013.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahalingam D, Goel S, Aparo S, Patel Arora S, Noronha N, Tran H, et al. A phase II study of pelareorep (REOLYSIN((R))) in combination with gemcitabine for patients with advanced pancreatic adenocarcinoma. Cancers (Basel) 2018;10:160. doi: 10.3390/cancers10060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahalingam D, Wilkinson GA, Eng KH, Fields P, Raber P, Moseley JL, et al. Pembrolizumab in combination with the oncolytic virus pelareorep and chemotherapy in patients with advanced pancreatic adenocarcinoma: a phase Ib Study. Clin Cancer Res. 2020;26:71–81. doi: 10.1158/1078-0432.CCR-19-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sborov DW, Nuovo GJ, Stiff A, Mace T, Lesinski GB, Benson DM, Jr., et al. A phase I trial of single-agent reolysin in patients with relapsed multiple myeloma. Clin Cancer Res. 2014;20:5946–55. doi: 10.1158/1078-0432.CCR-14-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adair RA, Roulstone V, Scott KJ, Morgan R, Nuovo GJ, Fuller M, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4:138ra77. doi: 10.1126/scitranslmed.3003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Sherbiny YM, Holmes TD, Wetherill LF, Black EV, Wilson EB, Phillips SL, et al. Controlled infection with a therapeutic virus defines the activation kinetics of human natural killer cells in vivo. Clin Exp Immunol. 2015;180:98–107. doi: 10.1111/cei.12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roulstone V, Mansfield D, Harris RJ, Twigger K, White C, de Bono J, et al. Antiviral antibody responses to systemic administration of an oncolytic RNA virus: the impact of standard concomitant anticancer chemotherapies. J Immunother Cancer. 2021;9:320. doi: 10.1136/jitc-2021-002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poropatich K, Dominguez D, Chan WC, Andrade J, Zha Y, Wray B, et al. OX40+ plasmacytoid dendritic cells in the tumor microenvironment promote antitumor immunity. J Clin Invest. 2020;130:3528–42.. doi: 10.1172/JCI131992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–52. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gambichler T, Schroter U, Hoxtermann S, Susok L, Stockfleth E, Becker JC. Decline of programmed death-1-positive circulating T regulatory cells predicts more favourable clinical outcome of patients with melanoma under immune checkpoint blockade. Br J Dermatol. 2020;182:1214–20.. doi: 10.1111/bjd.18379. [DOI] [PubMed] [Google Scholar]
- 24.An HJ, Chon HJ, Kim C. Peripheral blood-based biomarkers for immune checkpoint inhibitors. Int J Mol Sci. 2021;22:9414. doi: 10.3390/ijms22179414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DePeaux K, Delgoffe GM. Metabolic barriers to cancer immunotherapy. Nat Rev Immunol. 2021;21:785–97.. doi: 10.1038/s41577-021-00541-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. 2019;16:425–41.. doi: 10.1038/s41571-019-0203-7. [DOI] [PubMed] [Google Scholar]
- 27.Madden MZ, Rathmell JC. The complex integration of T-cell metabolism and immunotherapy. Cancer Discov. 2021;11:1636–43.. doi: 10.1158/2159-8290.CD-20-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson EA, Cascino K, Ordonez AA, Zhou W, Vaghasia A, Hamacher-Brady A, et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. 2021;34:108863. doi: 10.1016/j.celrep.2021.108863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rahn S, Kruger S, Mennrich R, Goebel L, Wesch D, Oberg HH, et al. POLE Score: a comprehensive profiling of programmed death 1 ligand 1 expression in pancreatic ductal adenocarcinoma. Oncotarget. 2019;10:1572–88.. doi: 10.18632/oncotarget.26705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Ma Q, Chen X, Guo K, Li J, Zhang M. Clinical significance of B7-H1 and B7-1 expressions in pancreatic carcinoma. World J Surg. 2010;34:1059–65. doi: 10.1007/s00268-010-0448-x. [DOI] [PubMed] [Google Scholar]
- 32.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–31. doi: 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38:1–11. doi: 10.1097/CJI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to pd-1 blockade in melanoma. N Engl J Med. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowanetz M, Zou W, Gettinger SN, Koeppen H, Kockx M, Schmid P, et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1) Proc Natl Acad Sci USA. 2018;115:E10119–E26.. doi: 10.1073/pnas.1802166115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim HR, Ha SJ, Hong MH, Heo SJ, Koh YW, Choi EC, et al. PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for head and neck cancer patients. Sci Rep. 2016;6:36956. doi: 10.1038/srep36956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong Q, Shou J, Ying J, Ling Y, Yu Y, Shen Z, et al. High PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor in urothelial carcinoma. Future Oncol. 2021;17:2893–905.. doi: 10.2217/fon-2021-0092. [DOI] [PubMed] [Google Scholar]
- 38.Kuo YT, Liao CK, Chen TC, Lai CC, Chiang SF, Chiang JM. A high density of PD-L1-expressing immune cells is significantly correlated with favorable disease free survival in nonmetastatic colorectal cancer. Med (Baltim) 2022;101:e28573. doi: 10.1097/MD.0000000000028573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 40.Botticelli A, Cerbelli B, Lionetto L, Zizzari I, Salati M, Pisano A, et al. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J Transl Med. 2018;16:219. doi: 10.1186/s12967-018-1595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Botticelli A, Zizzari IG, Scagnoli S, Pomati G, Strigari L, Cirillo A, et al. The Role of soluble LAG3 and soluble immune checkpoints profile in advanced head and neck cancer: a pilot study. J Pers Med. 2021;11:651. doi: 10.3390/jpm11070651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agullo-Ortuno MT, Gomez-Martin O, Ponce S, Iglesias L, Ojeda L, Ferrer I, et al. Blood predictive biomarkers for patients with non-small-cell lung cancer associated with clinical response to nivolumab. Clin Lung Cancer. 2020;21:75–85. doi: 10.1016/j.cllc.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Ugurel S, Schadendorf D, Horny K, Sucker A, Schramm S, Utikal J, et al. Elevated baseline serum PD-1 or PD-L1 predicts poor outcome of PD-1 inhibition therapy in metastatic melanoma. Ann Oncol. 2020;31:144–52.. doi: 10.1016/j.annonc.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Tiako Meyo M, Jouinot A, Giroux-Leprieur E, Fabre E, Wislez M, Alifano M, et al. Predictive value of soluble PD-1, PD-L1, VEGFA, CD40 ligand and CD44 for nivolumab therapy in advanced non-small cell lung cancer: a case-control study. Cancers (Basel) 2020;12:473. doi: 10.3390/cancers12020473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collienne M, Arnold D, Stein A, Goekkurt E, Martens U, Loghmani H, et al. P-49 GOBLET: a phase 1/2 multiple indication signal finding and biomarker study in advanced gastrointestinal cancers treated with pelareorep and atezolizumab–safety and preliminary response results. Ann Oncol. 2022;33:SUPPLEMENT 4, S264.
- 46.Collienne M, Loghmani H, Heineman TC, Arnold D. GOBLET: a phase I/II study of pelareorep and atezolizumab +/− chemo in advanced or metastatic gastrointestinal cancers. Future Oncol. 2022;18:2871–8. doi: 10.2217/fon-2022-0453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.