

Abstract
Circular RNA (circRNA) has various advantages over linear mRNA that is gaining success as a new vaccine and therapeutic agent. Thus, circRNA and its engineering methods have attracted attention recently. In this study, we developed a new in vitro circRNA engineering method by end-to-end self-targeting and splicing (STS) reaction using Tetrahymena group I intron ribozyme. We found that only the P1 helix structure of the group I intron was enough to generate circRNA by STS reaction. The efficacy of circRNA generation by STS reaction was comparable to the method using a permuted intron-exon (PIE) reaction. However, an end-to-end STS reaction does not introduce any extraneous fragments, such as an intronic scar that can be generated by PIE reaction and might trigger unwanted innate immune responses in cells, into circRNA sequences. Moreover, generated circRNA was efficiently purified by ion pair-reversed phase high-pressure liquid chromatography and used for cell-based analysis. Of note, efficient protein expression and stability with least innate immune induction by the circRNA with coxsackievirus B3 IRES were observed in cells. In conclusion, our new in vitro circRNA strategy can effectively generate highly useful circRNAs in vitro as an alternative circRNA engineering method.
Keywords: MT: non-coding RNAs, circular RNA, mRNA, self-circularization, Tetrahymena, group I intron, trans-splicing ribozyme, RNA replacement, RNA therapeutics, RNA vaccine
Graphical abstract
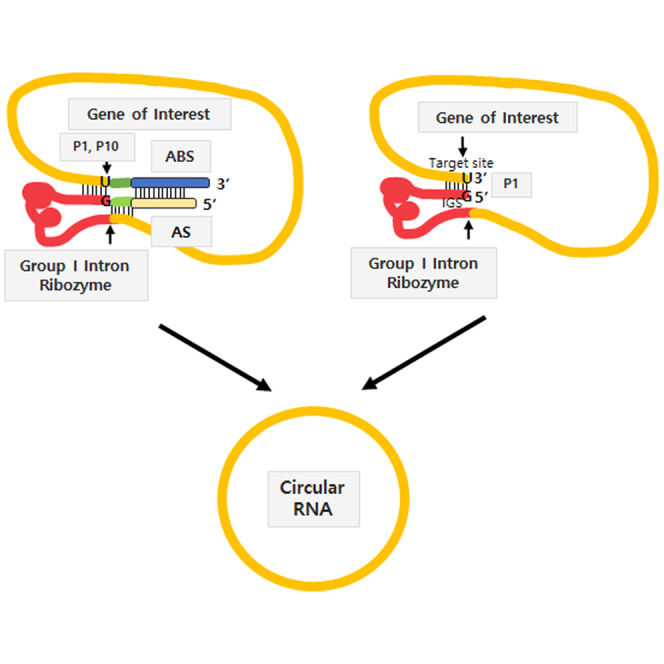
Lee et al. developed a new efficient in vitro circular RNA engineering method by end-to-end STS reaction using Tetrahymena group I intron ribozyme. Generated circRNAs did not harbor any extraneous intronic sequences. They showed efficient protein expression, stability, and least innate immune induction in cells.
Introduction
Circular RNA (circRNA) is considered as a new agent in RNA vaccine and therapeutics. It has advantages over linear RNAs, such as nuclease resistance and efficient ribosome recycling.1
Recent reports have suggested that circular mRNA platform is suitable as translatable RNA template for direct intratumoral administration for cancer therapy by encoding a mixture of cytokines for modulating anti-tumor immune responses,2 as well as for vaccine that elicits potent neutralizing antibodies and T cell responses by expressing antigenic protein.3 CircRNA can be useful as diverse biomedical tools, including adenosine deaminase acting on RNA (ADAR)-recruiting RNA with improved efficiency of targeted RNA editing because of the stability and resistance to degradation than linear ADAR-recruiting RNA,4 antisense RNA modulating target RNA stability, RNA aptamer to modulate various types of target molecules by direct binding with high affinity, and sponges of microRNAs or proteins that can regulate their activities by containing complementary microRNA binding sites or protein binding sequences.1 Moreover, circRNA preparation does not require the use of expensive modification for linear RNA such as 5′ capping and nucleotide modifications protected by intellectual property rights.5,6 Instead of 5′ capping, internal ribosome entry site (IRES) or IRES-like element is required for efficient translation initiation.7,8,9
To prepare circRNA in vitro, chemical-, enzyme-, and ribozyme-based methods have been used with their own pros and cons.10,11,12,13 In particular, the ribozyme-based method has advantages over other methods, including a relatively simple generation process, mass production, higher circularization efficiency for longer RNA, and less intermolecular interaction during circularization. However, recent reports have suggested that circRNAs with exogenous fragments have concerns,14 such as intronic scar that can trigger unwanted immune responses.15 Nevertheless, new circRNA preparation methods to overcome such drawback have not been intensively developed for decades.
In this study, we developed a new in vitro circRNA engineering method using the Tetrahymena group I intron ribozyme, which has been developed for various applications for RNA therapeutics16,17,18 and RNA imaging agents19 as a trans-splicing ribozyme.20
Group I intron-based trans-splicing ribozyme can replace target RNA sequences with desired RNA sequences tagged at the end of the ribozyme through two consecutive transesterification reactions (Figure 1A).20 Briefly, trans-splicing ribozyme can recognize its target site (5′-NNNNNU-3′) on the substrate RNA via specific complementary base parings including G·U wobble base pair with internal guide sequence (IGS) (5′-GN′N′N′N′N′-3′), forming a P1 helix structure (Figure 1A).21 P1 and P10 helixes define 5′ and 3′ splice sites, respectively (Figure 1A). In addition, extended base pairing between IGS and target site can increase the specificity and efficacy of ribozyme reaction. Approximately six to seven base pairs in the P10 helix can also increase 3′-splice site specificity.22,23 Moreover, antisense sequences that are complementary to the downstream region of the target site on the substrate RNA can be introduced into the upstream of group I intron ribozyme to achieve specificity and efficacy in cells and in vivo.24,25
Figure 1.

Self-circularization construct for end-to-end STS based on Tetrahymena group I intron
(A) Trans-splicing ribozyme reaction. (B and C) Self-circularization by end-to-end STS reaction in the presence (B) or absence of P10 and AS/ABS interaction (C). (D) Construct design of self-circularization RNA with extended P1, P10 helixes, and AS/ABS. Target site and IGS were underlined. Red part represents Tetrahymena group I intron ribozyme. Arrows indicate splicing sites.
For the first transesterification, the target site is attacked by the 3′ hydroxyl group (3′-OH) of exogenous guanosine, which can bind to a G-binding site within the ribozyme’s catalytic core and act as a nucleophile, resulting in the generation of a 3′-OH on the uridine at the 3′ end of the target site (Figure 1A). For the second transesterification, ωG at the 3′ end region of the group I intron can replace exogenous guanosine bound to the catalytic region of the ribozyme via a conformational change. After that, ωG is attacked by the 3′-OH of the cleaved target site, resulting in a ligation between the substrate RNA and the transgene RNA (Figure 1A).
Here, we rationally designed a Tetrahymena group I intron-mediated self-circularization construct to generate circRNA by an autocatalytical end-to-end self-targeting and splicing (STS) reaction within one RNA molecule instead of a trans-splicing reaction of two separate RNAs. Optimization and efficacy of circRNA generation and gene expression, stability, and innate immunity induction in cells by the circRNA were analyzed. This new in vitro circRNA engineering method is an alternative and more proper method than other methods, including ribozyme-based ones, to prepare circRNA for various applications in cells and in vivo without introducing extraneous fragments that might trigger unwanted innate immune responses in cells.15
Results
Design of self-circularization construct based on Tetrahymena group I intron ribozyme to generate circRNA in vitro
To generate circRNA using the Tetrahymena group I intron ribozyme, we placed target sequence for the ribozyme onto the 3′ end of a gene of interest (GOI) following group I intron sequence to allow end-to-end STS reaction through two consecutive transesterifications. The STS reaction cleavages the 3′ end of target site and consecutively links the 5′ end of the GOI to the cleaved 3′ end, similar to trans-targeting and splicing reaction for RNA replacement purposes.18 However, STS reaction in the circularization RNA construct does not link a separate transcript but generates a self-circularized RNA structure by joining both ends of the GOI region (Figures 1B and 1C). As extended P1, P10 helixes, and antisense sequence (AS)/antisense binding sequence (ABS) could affect the specificity and efficacy of splicing reaction,25 we included those when designing our self-circularization RNA construct in the beginning (Figures 1B and 1D). During optimization in this study, we identified that RNA construct with P1 helix only could efficiently perform self-circularization in vitro (Figure 1C). This will be explained later.
For the initial proof-of-concept experiments, target sequence, AS, and ABS (AS150: 150-nt complementary sequences) were selected based on trans-splicing ribozyme efficacy tested in our group (J.H.K., unpublished data). In addition, T7 polymerase promoter sequence was introduced for in vitro transcription (IVT) of self-circularization RNA.
Identification of circRNA generated by end-to-end STS reaction during IVT
To verify in vitro circRNA generation, IVT was carried out using a DNA template (GOI: EMCV IRES8-Gaussia luciferase) and T7 RNA polymerase. As expected, a band larger than an intact 1,874-nt-long RNA precursor appeared, probably due to changed migration by self-circularization (Figure 2A). Of note, the larger band was generated only through an IVT process without further procedures, such as chemical or enzyme treatment. Moreover, the band was resistant to RNase R digestion in denatured 4% polyacrylamide gel electrophoresis (PAGE) analysis after IVT and column purification. Other major bands are expected to be by-products from end-to-end STS reactions or self-cleaved fragments according to their respective sizes. The conjugated sequence of antisense and group I intron ribozyme and the sequence downstream of targeted site, both which are released after the self-circularization, would be by-products from end-to-end STS reaction. Self-cleaved fragments could be generated by cleavage at ωG position without first transesterification or by target site cleavage without the following second transesterification. We also observed nicked circRNA, as reported previously.8 We conducted additional end-to-end STS reactions to check if the circularization efficiency could be further increased as described previously.8 Conditions used for additional reactions were incubation at 37°C for 1 h and 3 h for second additional reaction after column purification or at 55°C for 15 min as the same condition with previous report8 in the presence of additional 2 mM guanosine triphosphate. No significant improvement of self-circularization was observed after first and second further end-to-end STS reactions (Figure 2A) or additional 55°C incubation (data not shown) compared with the IVT sample, suggesting that end-to-end STS reaction could be completed during IVT.
Figure 2.

Self-circularization during IVT by end-to-end STS
(A) Results of 4% denatured PAGE analysis of in vitro-transcribed self-circularization RNA construct (GOI: EMCV IRES-Gaussia Luciferase) with or without RNase R digestion. Additional end-to-end STS reactions were carried out for comparison. Sizes of other expected major bands besides circRNA are as follows: AS-group I intron ribozyme, 586 nt (193 nt + 393 nt); nicked circRNA, 1,092 nt; ωG cleavage, 1,291 nt; target site cleavage, 1,678 nt; intact RNA precursor, 1,874 nt (lane 1: IVT sample, lane 2: IVT sample with RNase R digestion, lane 3: IVT sample with first additional end-to-end STS reaction, lane 4: IVT sample with first additional end-to-end STS reaction and RNase R digestion, lane 5: IVT sample with second additional end-to-end STS reaction, lane 6: IVT sample with second additional end-to-end STS reaction and RNase R digestion). (B) Results of 1.5% agarose gel analysis of RT-PCR products using STS primers for circRNA detection of control linear RNA and circRNA IVT samples with or without RNase R digestion. Designed STS primers only amplify circRNA. NTC, no template control (lane 1, linear control RNA; lane 2, linear control RNA with RNase R digestion; lane 3, circRNA; lane 4, circRNA with RNase R digestion). (C) Sequencing analysis of RT-PCR band from circRNA sample of (B). Specific ligation junction region is indicated by arrow. (D) RNase H-based nicking assay. RNase H probe targeting the specific ligation junction allows RNase H digestion of circRNA, resulting in appearance of nicked circRNA band with monomeric size in 4% denatured PAGE (lane 1: HPLC-purified circRNA without RNase H digestion, lane 2: HPLC-purified circRNA with RNase H digestion). (E) Nonspecific nicking assay using magnesium ion and 65°C heating. Monomeric size of nicked circRNA band was observed in 4% denatured PAGE. In the presence of 2.5 mM Mg2+, circRNA band disappeared. However, the monomeric size of band was still observed as newly generated nicked circRNA without any multimeric size of new bands (lane 1, 0 mM; lane 2, 2.5 mM; lane 3, 5 mM of Mg2+).
The candidate RNA band was then extracted from the gel and subjected to RT-PCR reaction using specific primers and sequencing analysis to check if the band was a circRNA. Specific RT-PCR product was observed for the extracted RNA band using designed specific primers, which could amplify only circular form of RNA (Figure 2B), regardless of RNase R digestion. Control linear RNA, which was transcribed from the T7 DNA template without group I intron ribozyme sequence, was not able to generate a specific PCR band. A specific ligation junction of circRNA was confirmed by sequencing analysis (Figure 2C). More importantly, end-to-end STS reaction left only designed sequences in the generated circRNA without any extraneous sequences from group I intron ribozyme or other components.
Next, we checked whether the generated circRNA was monomeric or multimeric, as intermolecular targeting was possible. In both specific nicking by RNase H in the presence of the probe and nonspecific nicking by heating with Mg2+, amounts of circRNA band decreased, whereas the expected size of the monomeric linear band was increased or maintained, respectively (Figures 2D and 2E). These results suggest that an intermolecular reaction is unlikely, even in highly RNA precursor-concentrated condition in the reaction tube for IVT.
Optimization of self-circularization construct
To optimize the self-circularization construct, AS was added upstream of the IGS of the group I intron and tested with different lengths from 50 to 300 nucleotides (nt) with an interval of 50 nt. ABS is the reverse complementary sequence to the AS. It was added downstream of the target site. Relative band intensity (%) of circRNA was calculated to compare the relative in vitro self-circularization efficiency of each designed end-to-end STS RNA construct in denatured PAGE analysis (Figure S1). As a result, AS50, 100, and 150 showed no significant differences in relative band intensity of circRNA (6.4%, 6.8%, and 6%, respectively). In contrast, AS200, 250, and 300 showed reduced circularization efficiencies (4.3%, 4.2%, and 3.6% relative band intensity of circRNA, respectively). However, AS50 and AS100 showed 2.5 times and 20 times lower transcription efficiency, respectively, than others in the used T7 system. Thus, AS150 was selected for the self-circularization construct.
In addition, spacer (linker) sequence might be required between group I intron and IRES of the GOI region, as both ribozyme and IRES sequences are highly structured and the IRES could interfere with folding of the ribozyme structure or vice versa.8 Accordingly, we prepared and tested polyA spacers with 30 (A30) or 50 (A50) nt lengths (original construct has A10) between group I intron ribozyme and IRES as well as control linkers (control 1 and 2) designed to conserve secondary structures for ribozyme activity as reported previously (Table S1 and Figure S2).8 As a result, there were no significant differences in self-circularization efficiency among spacers with relative band intensities of 5.6%–6.7% (Figure S2). Therefore, an A10 or A30 spacer was used for further studies considering their appropriate structural flexibility and length.
P1 helix without P10 helix and AS/ABS in the self-circularizing RNA structure is an efficient minimum requirement for in vitro circRNA generation
Next, we designed and tested several constructs with or without important RNA helical structures for group I intron ribozyme activities such as P1, P10 helixes, and AS/ABS regions (Figure 3A).20 Unexpectedly, P1 helix without both P10 helix and AS/ABS in the self-circularization RNA construct (P1 RNA construct) was able to generate circRNA in vitro, whereas P1 and P10 helixes together without AS/ABS generated much lower amounts of circRNA in vitro (Figure 3B). Specific circRNA generated by P1 RNA construct was also confirmed by RT-PCR (Figure S3) and sequencing analysis (Figure 3C). These results suggest that only P1 helix for group I intron ribozyme interaction with the target site was enough to generate circRNA by end-to-end STS reaction in vitro without P10 helix or AS/ABS interaction. In addition, DNA template containing two 2′-O-methyl nucleosides at the 5′-termini of non-sense strand was prepared for IVT of P1 RNA construct, as such modification could dramatically decrease non-template extension at the 3′ end of transcribed RNAs.26 CircRNA was efficiently generated from the P1 RNA construct during IVT with a DNA template containing two 2′-O-methyl nucleosides, suggesting that the second transesterification only was enough to perform end-to-end STS reaction by P1 RNA construct (Figure 3D). This result indicates that the P1 RNA construct structure would be identical to the intermediate structure for the second transesterification (Figure 1A, middle) during STS reaction, which has a 3′-OH group at the uridine at the 3′ terminus.
Figure 3.

Self-circularization by P1 RNA construct without both P10 helix and AS/ABS
(A) Sequences around P1 and P10 helixes for the self-circularization RNA constructs (GOI: EMCV IRES-Gaussia Luciferase) composed of P1, P1 + P10, or P1+ P10 + 150-nt long AS (AS150)/ABS. (B) Results of 4% denatured PAGE analysis of in vitro-transcribed self-circularization RNA constructs with the selected structural components of group I intron ribozyme (lane 1, P1 RNA construct without P10 and AS; lane 2, P1 + P10 RNA construct without AS; lane 3, construct with all components). (C) Sequencing analysis of RT-PCR band from circRNA prepared by P1 RNA construct of (B). Specific ligation junction region is indicated by arrow. (D) Results of 4% denatured PAGE analysis of IVT samples of self-circularization RNA construct (GOI: EMCV IRES-Gaussia luciferase gene with AU-rich No. 16 target sequence) prepared using DNA templates with natural nucleosides (lane 1) or two 2′-O-methyl nucleosides at the 5′-termini of non-sense strand (lane 2).
AU-rich target sites are more efficient than other target sequences for end-to-end STS reaction of P1 RNA construct
We tested the efficacy and specificity of in vitro self-circularization of several variants of P1 RNA constructs with different IGS and target sequences in P1 helix for further optimization of the self-circularization RNA construct. Although it might be more efficient if the first transesterification step could be skipped for STS reaction, only 6-nuceotide-long P1 helix presence in the circularization RNA construct could raise the specificity concern.
We designed several P1 RNA constructs without including both P10 helix and AS/ABS structures based on a couple of validated IGS/target sites tested in our group (cases A, B, and C) (S.R.H. and J.H.K., unpublished data), as well as P1 constructs containing AU-rich or GC-rich P1 helix sequences (Figure 4A). P1 RNA constructs with two target sites in GOI or no complementary target sequence (i.e., no target site) to IGS in GOI were also designed (Figure 4A).
Figure 4.

Specificity and efficiency of self-circularization of various P1 RNA constructs
(A) Sequences around P1 helix for self-circularization of each P1 RNA construct case (GOI: EMCV-Gaussia Luciferase) with different target sequences. (B) Results of 4% denatured PAGE analysis of in vitro-transcribed self-circularization RNA constructs with different target sequences (lane 1, case A with P1 helix + P10 + AS150 components; lanes 2–8, each case with P1 helix only; lane 2, case A; lane 3, case B; lane 4, case C; lane 5, GC-rich; lane 6, AU-rich; lane 7, 2 target sites; lane 8, no target site). Relative band intensity of circRNA was shown as percentage of whole intensity of each lane. (C) Sequencing analysis of RT-PCR band from circRNA prepared with P1 RNA construct having AU-rich target sequence of (B). Specific ligation junction region is shown by arrow.
Surprisingly, a P1 RNA construct with AU-rich target sequences and IGS showed better circularization efficiency (with a relative band intensity of 12%) than constructs harboring all of P1, P10, and AS/ABS or other P1 RNA constructs in denatured PAGE analysis (Figure 4B). RT-PCR and sequencing analysis confirmed that P1 RNA construct with an AU-rich target sequence could generate specific circRNA with fidelity in vitro (Figure 4C). P1 RNA construct with GC-rich target/IGS interaction was less efficient with a relative band intensity of 5.4% than P1 RNA construct with AU-rich target/IGS interaction, suggesting that strong base pairing between the target site and IGS for the specific self-targeting might induce relatively lower efficiency in the second transesterification step.
A negligible amount of circRNA was detected by RNA construct without target/IGS interaction (no target site, Figure 4B). RT-PCR/sequencing reactions showed a specific ligation junction (but G instead of U at the ligation junction) (Figures S4A and S4B), as well as non-specific products, suggesting that the exposed 3′-OH group at the 3′ terminus of the RNA construct without complementary target site, which resembled the intermediate structure for the second transesterification, might be targeted by ωG by chance in vitro with a lower specificity. In addition, when two target sites existed in the self-circularization RNA construct, nonspecific products as well as specific circRNA sequences were observed in sequencing analysis (data not shown). Taken together, these results suggest that only a single existing target site should be selected from the GOI region to achieve specific end-to-end STS reaction for efficient circRNA generation.
Next, we tested all possible AU-rich target sequences to determine whether the efficiency could be further improved. This could also provide a reference data to initially identify an efficient target site at respective GOI sequences. Based on sequences of the target site (5′-NNNNNU-3′), a total 32 (25) possible AU-rich target sites were designed and tested (Table S2). As a result, most AU-rich target sequences showed self-circularization efficiencies comparable with the AU-rich No. 16 target sequence, which was tested in Figure 4 (Figures S5A and S5B).
Construct preparation of self-circularization RNA and high-pressure liquid chromatography purification of circRNA for protein translation and cell-based assay
For protein expression in cells, Coxsackievirus B3 (CVB3) IRES was used for efficient translation in circRNA for various cell types as reported previously.8 In addition, superfolder green fluorescent protein (sGFP)-coding sequences27 were placed downstream of CVB3 IRES to monitor the fluorescence intensity for checking protein expression levels in transfected cells directly. CircRNAs coding CVB3 IRES-derived sGFP were prepared by IVT with our method using end-to-end STS reaction. For comparison, circRNAs were generated with a permuted intron-exon (PIE) method using Anabaena pre-tRNA group I intron. The sGFP gene was introduced here instead of Gaussia luciferase gene in CVB3-GLuc-pAC that was constructed in the previous report.8 All sequences in the circularized RNA structure by STS reaction were the same as those by the PIE method, except that they had no intronic scar such as exon1/exon2 (E1/E2) fragments’ sequences essential for PIE reaction.
The efficacy of circularization by the end-to-end STS method was observed to be comparable with that of the PIE method based on an analysis of IVT RNAs in denatured PAGE (Figure S6A). E-gel electrophoresis (EX agarose gel) analysis of IVT RNAs also showed comparable circularization efficacy by the end-end STS reaction to the PIE method (Figure S6B). Of note, our denatured 4% PAGE condition provided better detection sensitivity and significant migration differences between linear RNAs and circRNAs compared with EX agarose gel analysis (Figure S6B). Thus, denatured PAGE analysis was able to detect more bands for PIE sample poorly detected in EX agarose gel analysis that was used previously (Figure S6).8 As a control, a linear sGFP RNA including short 5′ and 3′ UTRs was prepared with N1-methylpseudouridine (m1ψ), 5′ cap, and 3′ polyA. We changed Gaussia luciferase here to sGFP in a linear GLuc RNA that was tested in a previous study.8
Ion pair-reverse phase high-performance liquid chromatography (IP-RP HPLC) purification was carried out to purify CVB3-sGFP circRNA without any contaminants such as double-stranded RNA (dsRNA), which could induce unwanted innate immune responses.28 Initially, RNase R was not pre-treated for HPLC purification to decrease purification steps. Fractions of candidate peaks of circRNA were collected. Eluted circRNAs were then recovered by ethanol precipitation and analyzed by denatured PAGE (Figure 5A). When RNase R digestion was conducted before IP-RP HPLC purification, circRNAs were more readily separated (Figure 5B). Compared with size exclusion chromatography for circRNA purification reported previously,8,15 much clearer peak separation was observed for a control prepared with the PIE method (Figure S7A), suggesting that the IP-RP HPLC method could provide better peak resolution for circRNA purification. Linear m1ψ-modified control sGFP RNA with 5′ cap and 3′ polyA was also purified by IP-RP HPLC (Figure S7B).
Figure 5.

IP-RP HPLC purification of circRNAs prepared by end-to end STS reaction of P1 RNA construct
(A) HPLC chromatogram for IVT sample of P1 RNA construct (GOI: CVB3 IRES-sGFP) (top). Collected fractions were analyzed with 4% denatured PAGE (bottom). (B) HPLC chromatogram for IVT sample of P1 RNA construct after RNase R digestion (top). Collected major peak fraction was analyzed with 4% denatured PAGE (bottom).
Protein translation using circRNA and its stability and immunogenicity in cells
To check translation by circRNA in cells, CVB3 IRES-containing circRNAs prepared by PIE or end-to-end STS method and control m1ψ-modified linear RNA for sGFP expression were used to transfect HEK293A cells. Fluorescence intensities from transfected cells were then monitored for 7 days after transfection (Figure 6A). CircRNA prepared by PIE showed higher sGFP expression levels than m1ψ-modified linear RNA with 5′ cap and 3′ polyA, consistent with a previous report.8 CircRNA prepared by the end-to-end STS method also showed efficient sGFP expression in fluorescence intensity measurement. Its expression level was higher than that of circRNA prepared by the PIE method (Figure 6A). In quantitative RT-PCR (qRT-PCR) results, the quantity of circRNA prepared by the STS method in cells was also higher than that of circRNA prepared by the PIE method, with similar fold as shown in fluorescence intensity, suggesting comparable protein expression efficiency of circRNAs (Figure S8A). In the case of linear RNA, the fluorescence intensity was decreased from the 24 h after transfection, whereas those of circRNAs peaked at 2 days after transfection and maintained for much longer (Figure 6A), confirming higher stability of circRNA than linear RNA in cells, as reported previously.8,29 Higher fluorescence intensities by circRNAs compared with that of linear RNA were confirmed in fluorescence microscopy analysis at 24 h after transfection (Figure 6B).
Figure 6.

Efficient protein expression and stability with least innate immunity in cells induced by circRNA engineered through end-to-end STS reaction
(A) Efficient sGFP expression by circRNAs produced from P1 RNA construct (GOI: CVB3 IRES-sGFP) in HEK293A cells compared to linear RNA. Equimolar amounts of circRNA by PIE (circular PIE), circRNA by end-to-end STS (circular STS), or control m1ψ-modified linear RNA with 5′ cap/3′ polyA (linear m1ψ) were used for transfection. Fluorescence intensities of sGFP (Ex, 480 nm; Em, 510 nm) were then monitored. Data are presented as means ± SEMs (n = 8; #p < 0.01 for circular PIE vs. circular STS). (B) Fluorescence microscopy imaging (GFP filter) at 24 h after transfecting HEK293A cells with each RNA. (C) qRT-PCR analysis using specific primers for innate immunity markers with RNAs at 1 h or 6 h after transfecting A549 cells with each RNA. Poly(I:C) and linear RNA with m1ψ-modified base were used as controls. RNA levels were indicated relative to those in mock-transfected cells. Data are presented as means ± SEMs (n = 3; ∗p < 0.01 compared with Mock).
Since there are different observations about unwanted innate immune responses caused by circRNAs,6,14,15 we investigated innate immune responses by qRT-PCR analysis using specific primers for inflammatory cytokines including tumor necrosis factor (TNF)-α, IFN-β, IL-6, and RIG-I in HEK293A and A549 cells, as described previously.15 As a result, significant innate immune responses were not observed in HEK293A (Figure S8B) or A549 cells (Figure 6C) for all circRNAs purified by IP-RP HPLC. Therefore, the purity of circRNAs with HPLC might be a crucial factor affecting the innate immune responses. In our observation, IP-RP HPLC-purified circRNA with only natural nucleotides could trigger negligible innate immune responses in A549 cells.
Discussion
Currently, available major in vitro circRNA preparation methods have their own pros and cons.10 For example, with chemical- or protein enzyme-based ligation methods, further treatments of reaction products are needed after IVT for circularization and intermolecular interaction would be favored over an intramolecular interaction required for monomeric circRNA formation under condition of high RNA precursor concentration for circRNA preparation on a large scale. In contrast, a circRNA engineering method with ribozyme-based autocatalytic end-to-end STS reaction developed in this study showed that circularization occurred only during the IVT process without further treatment. Moreover, an intermolecular reaction was unlikely even under a high RNA precursor-concentrated condition in microtube for IVT. There are also distinct differences between ribozyme-based methods. The PIE method using group I intron introduces extraneous fragments in circRNA after the reaction. In contrast, the inverse splicing method, which is similar to PIE, using the group II intron does not introduce extraneous fragments in circRNA.30 However, group II intron-mediated formation of 2′,5′-phosphodiester linkage at the ligation/circularization site instead of natural 3′,5′-phosphodiester bond would be a concern.10,11,12,13 To the best of our knowledge, there are no reports to show the expression of GOI in mammalian cells with circRNAs prepared by group II intron in vitro. Noticeably, our method based on end-to-end STS reaction using the Tetrahymena group I intron did not introduce any extraneous fragments in circRNA together with efficient GOI translation. Extraneous fragments such as intronic scar would be a possible concern of the PIE method in terms of unwanted innate immune responses, probably caused by the generation of long E1-E2 junction15 or limited circRNA engineering optimization of sequences and structures.
In this study, self-circularization efficiency in vitro and protein expression in cells with circRNA by end-to-end STS reaction were comparable with that of PIE reaction for GOI with less than 2 K nt. Optimization of RNA construct for efficient self-circularization through the end-to-end STS method and HPLC purification for longer GOI of up to 11 K nt is currently underway. Noteworthy, in contrast with the PIE method having only limited options for circularization optimization including the spacer and AS, the end-to-end STS strategy can choose a target site from any regions in the GOI, suggesting that an efficient target site with greater self-circularization efficiency for longer GOI might be selected among many target sites in GOI, most probably due to conformation of the RNA construct which allows for an efficient target site-IGS interaction during end-to-end STS reaction.
For circRNA analysis in gel, we used a denatured PAGE system15 rather than an EX gel system used for circRNA analysis recently.8,31 In addition, compared with the denatured PAGE by others for circRNA analysis,15 we maintained the gel temperature at 50°C during electrophoresis using a temperature probe with the gel stained with SYBR gold nucleic acid gel stain instead of ethidium bromide for sensitive detection of by-products or contaminants. With this condition, we could check migration change for small size to more than 1,000 nt and observed that circRNAs migrated much more slowly, i.e., appearing larger. Hence, circRNAs and other bands were significantly more distinguished from the size of the linear precursor RNA compared with reported cases.31 Furthermore, gel size for longer running and polyacrylamide percentage for size separation could be easily optimized to achieve higher resolution. Our results suggest that a denatured PAGE method with some modifications would be a better option for circRNA analysis. In contrast, an EX gel system might be appropriate for extremely long RNAs, which are not ideal for denatured PAGE system. Unidentified bands with slower migration could be observed in denatured PAGE analysis, depending on target sequences and GOI. Nevertheless, those were negligible bands compared with other expected by-products and specific circRNA bands. They could be completely eliminated together with other by-products by the IP-RP HPLC purification procedure set up in this study.
The most interesting finding of this study was that self-circularization RNA construct harboring only P1 helix without both P10 helix and AS/ABS interaction could generate circRNA through end-to-end STS reaction in vitro with a high efficiency. Previous reports have suggested that P10 helix formation can increase the efficiency and specificity of group I intron-based trans-splicing riboyzme reactions.22,32,33,34 However, as self-circularization is an in vitro reaction and the target site can be exposed to the 3′ terminus of the RNA construct in the absence of P10 helix and AS/ABS structures, only 6-bp IGS and target interaction would sufficiently perform the specific and efficient reaction. The lower self-circularization efficiency by the P1 + P10 RNA construct was probably due to the context of P10 harboring 50% GC contents. It was reported that a strong P10 helix would be undesirable for group I intron-mediated splicing reaction probably because of premature docking of ωG into the G-binding site before the transesterification reaction step and poor substrate dissociation.35
Although any 5′-NNNNNU-3′ in GOI can be targeted with different self-circularization efficiencies, we observed that AU-rich target sequences in P1 RNA construct were most efficient for self-circularization when only one target sequence existed. A weak interaction between AU-rich target sequences and IGS in P1 helix could help with the efficient dissociation of ribozymes after the second transesterification, resulting in increased reaction kinetics. This suggests that an AU-rich sequence at any position in the GOI region might be selected as the target site of the P1 helix for an efficient self-circularization using end-to end STS reaction, if only one specific target sequence is present in the GOI. GOI sequences following the target site can be sent to the next position of ωG sequence of group I intron in the self-circularization RNA construct. As a final RNA product after self-circularization is circular, remaining GOI sequences can be reconnected with the other part of GOI in the circRNA structure, resulting in complete GOI sequences in circRNA (Figure 7).
Figure 7.

Self-circularization process through end-to-end STS reaction
Any position in GOI of self-circularization RNA construct can be selected as a target site for circularization through Tetrahymena group I intron-based end-to-end STS reaction. Split GOI parts can be reconnected in the correct order after circRNA formation by an end-to-end STS reaction.
CircRNAs without modified bases showed good translation efficiencies with negligible innate immune responses in cells, confirming that circRNA with natural nucleotides is applicable in cells and in vivo as reported previously.6 It is unclear exactly how exogenous circRNA with unmodified nucleotides provokes the least immune induction when it is introduced into cells. It has been reported that endogenous circRNAs tend to form 16–26 bp imperfect dsRNA that can act as inhibitors of innate immunity by dsRNA-activated protein kinase (PKR), indicating that the transfected exogenous circRNAs with unmodified nucleotides, which might form short dsRNA regions, could bind and inhibit PKR.15,36 Liu et al.15 have reported that synthesized circRNAs produced by T4 RNA ligase form short dsRNA regions and exhibit minimized immunogenicity through the efficient suppression of PKR activation, whereas circRNAs prepared by PIE can induce innate immunity, most probably due to complicated conformational changes rendered by introduced extraneous fragments. However, Wesselhoeft et al.6 have reported that intracellular transfection of circRNAs generated by PIE method with unmodified nucleotides could induce minimal innate immune response by bypassing cellular RNA sensors, such as RIG-I and Toll-like receptors. Our results showed insignificant innate immunity in A549 cells induced by circRNAs prepared with the PIE method and our end-to-end STS strategy when cells were transfected with circRNAs purified by IP-RP HPLC. The discrepancy between different groups in terms of innate immunity induction by circRNAs might be caused by differences in the sequence, length, and structure of GOI and/or the purification method of circRNAs.
Tetrahymena group I intron was engineered self to trans previously as shaping P1 helix through IGS and target sequence (5′-NNNNNU-3′) interaction is the only requirement for the splicing reaction.20,21,37 This simplicity enabled us to design end-to-end STS strategy and, consequently, we found out that P1 helix formation only was also enough for self-circularization in vitro. For the PIE method using a group I intron, Anabaena catalytic intron showed the better splicing efficiency compared with T4 phage thymidylate synthase catalytic intron.8 As each group I intron has quite a different structure with distinct structural elements that affect the minimum requirements for the reaction and kinetics,38,39 selecting and/or engineering an appropriate group I intron for each method would be helpful to improve self-circularization efficiency. Although an Anabaena group I intron can also form a P1 helix by base pairing between the intron sequence and the target sequence in the exon, additional interaction between exon sequences are required for the activity, as internal structural elements are missing in Anabaena group I intron compared with Tetrahymena group I intron.38 Therefore, for other group I introns to be used in an end-to-end STS design, the simplicity of the reaction seen in the Tetrahymena group I intron should be achieved through systematic engineering and/or artificial evolution.37
In summary, we developed a new in vitro circRNA engineering strategy by designing RNA construct to allow end-to-end STS reaction based on Tetrahymena group I intron. Importantly, circRNA produced through this method does not harbor any extraneous fragments such as intronic scar. Efficient translation of GOI in cells and negligible innate immune responses were also observed. Therefore, our new strategy could provide an alternative or more proper option than other methods, including other ribozyme-based ones for circRNA preparation in vitro for various applications.
Materials and methods
Design of DNA construct for circRNA and T7 DNA template preparation
DNAs encoding self-circularization RNA were synthesized by gBlocks service (Integrated DNA Technologies, Singapore) and inserted into pTOP vector using TOPcloner TA-Blunt kit (Enzynomics, Daejeon, South Korea) or an In-Fusion HD cloning kit (Takara Bio USA, Mountain View, CA) following the manufacturers’ protocols. When an A30 or A50 spacer sequence was inserted into the self-circularization construct between the group I intron and the IRES sequence, AatII restriction enzyme site was introduced at the 3′ end region of polyadenine sequences to easily switch the IRES part. GOI including IRES and transgene was introduced between AatII and AgeI sites (Enzynomics).
DNA templates for T7 IVT were prepared by standard PCR using a thermal cycler (Applied Biosystems, Waltham, MA) and a Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Fisher Scientific, Waltham, MA) or restriction enzyme reaction with plasmid DNA. Primers for PCR (Table S3) were synthesized by Cosmogenetech (Seoul, South Korea). Prepared DNA templates were purified using a LaboPass gel extraction kit (Cosmogenetech).
IVT, RNase R digestion, and denatured PAGE analysis
IVT was carried out using T7 DNA template and a HiScribe T7 high yield RNA synthesis kit following the manufacturer's protocols (New England Biolabs, Ipswich, MA). Resulting RNA transcripts or RNA samples after enzyme reactions were purified using a Monarch RNA cleanup kit (New England Biolabs). For control linear RNAs, m1ψ (100%) was incorporated during IVT using N1-methylpseudouridine-5′-triphosphate (TriLink Biotechnologies, San Diego, CA). For 5′ capping and 3′ polyA tailing of linear RNA, a vaccinia capping system and E. coli poly(A) polymerase (New England Biolabs) were used, respectively. RNase R (Biosearch Technologies, Hoddesdon, UK) was treated for selected IVT samples using the manufacturer’s protocol to identify circRNA bands.
For 4% polyacrylamide-7M urea gel electrophoresis (denatured PAGE, 20 × 20 cm gel size), samples were mixed with 10 M urea-bromophenol blue dye in 1× Tris-Borate-EDTA buffer and heated at 75°C for 5 min. Gel electrophoresis was carried out at 50°C using a temperature probe with 50 W condition (Bio-Rad Laboratories, Hercules, CA). After electrophoresis, gel was stained with SYBR gold nucleic acid gel stain (Invitrogen, Waltham, MA) and analyzed using an ImageQuant800 CCD imager (Cytiva, Marlborough, MA). As a size marker, 100 bp Plus DNA ladder (Bioneer, Daejeon, South Korea) or RiboRuler Low Range RNA ladder (Thermo Fisher Scientific) was loaded together with samples. The relative band intensity (%) of circRNA was calculated to compare the relative self-circularization efficiency ([Intensity of circRNA band/Intensity of lane] × 100) in vitro.
RT-PCR and sequencing
RT-PCR to identify end-to-end STS product as circRNA was carried out using specific forward and reverse primers (STS F and R) (Table S3). For reverse transcription, OneScript plus reverse transcriptase (Applied Biological Materials, Richmond, Canada) was used following the manufacturer’s protocol in 20-μL reactions with 100 ng RNAs. Resulting cDNAs were subjected to 35 cycles of standard PCR using an AccuPower Taq PCR premix (Bioneer).
The amplified specific band was extracted from agarose gel using a LaboPass gel extraction kit (Cosmogenetech). Sequencing was performed by Cosmogenetech after TA cloning using a TOPcloner TA-Blunt kit (Enzynomics).
Nicking assays
HPLC or gel-purified circRNAs were used for nicking assays. For RNase H-based nicking assay, 200 ng circRNA (target site: 5′-UUGGGU-3′) and 10-fold molar excess of RNase H probe targeting the ligation junction (5′-GCGTACTCGACCCAAACCGG-3′) were mixed. The sample was then heated at 65°C for 5 min. After annealing at room temperature for 10 min, RNase H (New England Biolabs) was treated following the manufacturer’s protocol. For nonspecific nicking assay, 100 ng circRNAs were incubated at 65°C for 30 min with 0, 2.5, or 5 mM of MgCl2. Samples were analyzed by 4% denatured PAGE.
HPLC purification of circRNAs
In vitro transcribed RNAs with or without RNase R digestion (≤160 μg) were used for HPLC purification. CircRNAs were purified by an IP-RP HPLC using a 1260 Infinity II bio-inert LC system (Agilent Technologies, Santa Clara, CA) and a 250 × 10 mm reverse-phase LC column with a particle size of 5 μm and a pore size of 4,000 Å (Agilent Technologies: PLRP-S 4000Å). RNA was heated at 65°C for 5 min and then placed at room temperature for 20 min. RNA was purified with a linear gradient using buffer A (100 mM hexylammonium acetate [HAA] in 10% acetonitrile [ACN]) and buffer B (100 mM HAA in 50% ACN; ADS Biotec, Omaha, NE) at a flow rate of 1.8 mL/min. Column oven temperature was set at 60°C. RNA was detected by UV absorbance at 260 nm. RNA fractions were recovered by ethanol precipitation method and then analyzed by 4% denatured PAGE.
Cell culture, cell-based assays, and qRT-PCR
Adenoviral E1-transformed human embryonic kidney cell line (HEK293A) was purchased from Invitrogen. A549 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). HEK293A and A549 cells were maintained in DMEM and RPMI-1640 (Invitrogen) supplemented with 10% fetal bovine serum (Cytiva), respectively.
For RNA transfection to analyze protein expression, 1 × 104 HEK293A cells/well were plated into 96-well black plates with a clear flat bottom (Corning, Kennebunk, ME). Equimolar amounts of each RNA were transfected with Lipofectamine MessengerMAX reagent (Invitrogen) following the manufacturer’s protocol. Fluorescence intensities of sGFP from transfected cells were measured at specific time points using a Synergy H1 microplate reader (BioTek, Winooski, VT) at excitation wavelength of 480 nm and emission wavelength of 510 nm.
For qRT-PCR assay to evaluate innate immune responses, RNAs were purified from cells at specific time points using an easy-spin total RNA extraction kit (Intron Biotechnology, Seongnam, South Korea) following the manufacturer’s protocol. cDNAs were synthesized with OneScript Plus reverse transcriptase (Applied Biological Materials) and hexanucleotides random primers. Real-time PCR analysis was performed in StepOnePlus equipment (Applied Biosystems) using SensiFAST SYBR Hi-ROX Kit (Meridian bioscience, Cincinnati, OH). Primer sequences listed in Table S3 were used for analyzing expression levels of inflammatory cytokine genes including IL-6, INF-β, RIG-I, and TNF-α.
For fluorescence microscopy analysis, HEK293A cells grown in DMEM medium were observed with a Leica DMi8 fluorescence microscope (Leica, Wetzlar, Germany). Images were acquired and quantified using Leica LAS X software.
Data and code availability
The data supporting the findings of this study are available within the article and its supplementary materials.
Acknowledgments
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF), funded by the Korean government (MSIT) (No. 2022M3E5F1017657).
Author contributions
K.H.L. and S.W.L. designed experiments; K.H.L., S.K., J.S., S.R.H., and J.H.K. conducted experiments; and K.H.L. and S.W.L. analyzed data and wrote the manuscript.
Declaration of interests
K.H.L., S.K., J.S., S.R.H., and J.H.K. are employee of Rznomics Inc. S.-W.L. is CEO of Rznomics Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.07.034.
Contributor Information
Kyung Hyun Lee, Email: khlee@rznomics.com.
Seong-Wook Lee, Email: swl0208@rznomics.com.
Supplemental information
References
- 1.Liu C.X., Chen L.L. Circular RNAs: characterization, cellular roles, and applications. Cell. 2022;185:2016–2034. doi: 10.1016/j.cell.2022.04.021. [DOI] [PubMed] [Google Scholar]
- 2.Yang J., Zhu J., Sun J., Chen Y., Du Y., Tan Y., Wu L., Zhai M., Wei L., Li N., et al. Intratumoral delivered novel circular mRNA encoding cytokines for immune modulation and cancer therapy. Mol. Ther. Nucleic Acids. 2022;30:184–197. doi: 10.1016/j.omtn.2022.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qu L., Yi Z., Shen Y., Lin L., Chen F., Xu Y., Wu Z., Tang H., Zhang X., Tian F., et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell. 2022;185:1728–1744.e16. doi: 10.1016/j.cell.2022.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi Z., Qu L., Tang H., Liu Z., Liu Y., Tian F., Wang C., Zhang X., Feng Z., Yu Y., et al. Engineering circular ADAR-recruiting RNAs increase the efficiency and fidelity of RNA editing in vitro and in vivo. Nat. Biotechnol. 2022;40:946–955. doi: 10.1038/s41587-021-01180-3. [DOI] [PubMed] [Google Scholar]
- 5.Gavira M., Kilic B. A network analysis of COVID-19 mRNA vaccine patents. Nat. Biotechnol. 2021;39:546–549. doi: 10.1038/s41587-021-00912-9. [DOI] [PubMed] [Google Scholar]
- 6.Wesselhoeft R.A., Kowalski P.S., Parker-Hale F.C., Huang Y., Bisaria N., Anderson D.G. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell. 2019;74:508–520.e4. doi: 10.1016/j.molcel.2019.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu C.X., Chen L.L. Expanded regulation of circular RNA translation. Mol. Cell. 2021;81:4111–4113. doi: 10.1016/j.molcel.2021.09.023. [DOI] [PubMed] [Google Scholar]
- 8.Wesselhoeft R.A., Kowalski P.S., Anderson D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 2018;9:2629. doi: 10.1038/s41467-018-05096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen R., Wang S.K., Belk J.A., Amaya L., Li Z., Cardenas A., Abe B.T., Chen C.K., Wender P.A., Chang H.Y. Engineering circular RNA for enhanced protein production. Nat. Biotechnol. 2023;41:262–272. doi: 10.1038/s41587-022-01393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee K.H., Kim S., Lee S.W. Pros and cons of in vitro methods for circular RNA preparation. Int. J. Mol. Sci. 2022;23:13247. doi: 10.3390/ijms232113247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X., Lu Y. Circular RNA: biosynthesis in vitro. Front. Bioeng. Biotechnol. 2021;9:787881. doi: 10.3389/fbioe.2021.787881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Müller S., Appel B. In vitro circularization of RNA. RNA Biol. 2017;14:1018–1027. doi: 10.1080/15476286.2016.1239009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petkovic S., Müller S. RNA circularization strategies in vivo and in vitro. Nucleic Acids Res. 2015;43:2454–2465. doi: 10.1093/nar/gkv045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisenstein M., Garber K., Landhuis E., DeFrancesco L. Nature Biotechnology’s academic spinouts 2021. Nat. Biotechnol. 2022;40:1551–1562. doi: 10.1038/s41587-022-01530-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C.X., Guo S.K., Nan F., Xu Y.F., Yang L., Chen L.L. RNA circles with minimized immunogenicity as potent PKR inhibitors. Mol. Cell. 2022;82:420–434.e6. doi: 10.1016/j.molcel.2021.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Sullenger B.A., Cech T.R. Ribozyme-mediated repair of defective mRNA by targeted, trans-splicing. Nature. 1994;371:619–622. doi: 10.1038/371619a0. [DOI] [PubMed] [Google Scholar]
- 17.Lan N., Howrey R.P., Lee S.W., Smith C.A., Sullenger B.A. Ribozyme-mediated repair of sickle beta-globin mRNAs in erythrocyte precursors. Science. 1998;280:1593–1596. doi: 10.1126/science.280.5369.1593. [DOI] [PubMed] [Google Scholar]
- 18.Han S.R., Lee C.H., Im J.Y., Kim J.H., Kim J.H., Kim S.J., Cho Y.W., Kim E., Kim Y., Ryu J.H., et al. Targeted suicide gene therapy for liver cancer based on ribozyme-mediated RNA replacement through post-transcriptional regulation. Mol. Ther. Nucleic Acids. 2021;23:154–168. doi: 10.1016/j.omtn.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.So M.K., Gowrishankar G., Hasegawa S., Chung J.K., Rao J. Imaging target mRNA and siRNA-mediated gene silencing in vivo with ribozyme-based reporters. Chembiochem. 2008;9:2682–2691. doi: 10.1002/cbic.200800370. [DOI] [PubMed] [Google Scholar]
- 20.Lee C.H., Han S.R., Lee S.W. Therapeutic applications of group I intron-based trans-splicing ribozymes. WIREs RNA. 2018;9:e1466. doi: 10.1002/wrna.1466. [DOI] [PubMed] [Google Scholar]
- 21.Been M.D., Cech T.R. One binding site determines sequence specificity of Tetrahymena pre-rRNA self-splicing, trans-splicing, and RNA enzyme activity. Cell. 1986;47:207–216. doi: 10.1016/0092-8674(86)90443-5. [DOI] [PubMed] [Google Scholar]
- 22.Köhler U., Ayre B.G., Goodman H.M., Haseloff J. Trans-splicing ribozymes for targeted gene delivery. J. Mol. Biol. 1999;285:1935–1950. doi: 10.1006/jmbi.1998.2447. [DOI] [PubMed] [Google Scholar]
- 23.Guo F., Cech T.R. In vivo selection of better self-splicing introns in Escherichia coli: the role of the P1 extension helix of the Tetrahymena intron. RNA. 2002;8:647–658. doi: 10.1017/s1355838202029011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byun J., Lan N., Long M., Sullenger B.A. Efficient and specific repair of sickle β-globin RNA by trans-splicing ribozymes. RNA. 2003;9:1254–1263. doi: 10.1261/rna.5450203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwon B.S., Jung H.S., Song M.S., Cho K.S., Kim S.C., Kimm K., Jeong J.S., Kim I.H., Lee S.W. Specific regression of human cancer cells by ribozyme-mediated targeted replacement of tumor-specific transcript. Mol. Ther. 2005;12:824–834. doi: 10.1016/j.ymthe.2005.06.096. [DOI] [PubMed] [Google Scholar]
- 26.Kao C., Rüdisser S., Zheng M. A simple and efficient method to transcribe RNAs with reduced 3′ heterogeneity. Methods. 2001;23:210. doi: 10.1006/meth.2000.1131. [DOI] [PubMed] [Google Scholar]
- 27.Pédelacq J.D., Cabantous S., Tran T., Terwilliger T.C., Waldo G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 28.Karikó K., Muramatsu H., Ludwig J., Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011;39:e142. doi: 10.1093/nar/gkr695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H.M., Ma X.L., Li H.G. Intriguing circles: conflicts and controversies in circular RNA research. WIREs RNA. 2019;10:e1538. doi: 10.1002/wrna.1538. [DOI] [PubMed] [Google Scholar]
- 30.Mikheeva S., Hakim-Zargar M., Carlson D., Jarrell K. Use of an engineered ribozyme to produce a circular human exon. Nucleic Acids Res. 1997;25:5085–5094. doi: 10.1093/nar/25.24.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abe B.T., Wesselhoeft R.A., Chen R., Anderson D.G., Chang H.Y. Circular RNA migration in agarose gel electrophoresis. Mol. Cell. 2022;82:1768–1777.e3. doi: 10.1016/j.molcel.2022.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suh E.R., Waring R.B. Base pairing between the 3′ exon and an internal guide sequence increases 3′ splice site specificity in the Tetrahymena self-splicing rRNA intron. Mol. Cell Biol. 1990;10:2960–2965. doi: 10.1128/mcb.10.6.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones J.T., Lee S.W., Sullenger B.A. Tagging ribozyme reaction sites to follow trans-splicing in mammalian cells. Nat. Med. 1996;2:643–648. doi: 10.1038/nm0696-643. [DOI] [PubMed] [Google Scholar]
- 34.Ayre B.G., Köhler U., Turgeon R., Haseloff J. Optimization of trans-splicing ribozyme efficiency and specificity by in vivo genetic selection. Nucleic Acids Res. 2002;30:e141. doi: 10.1093/nar/gnf141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell M.A., Sinha J., Johnson A.K., Testa S.M. Enhancing the second step of the trans excision-splicing reaction of a group I ribozyme by exploiting P9.0 and P10 for intermolecular recognition. Biochemistry. 2004;43:4323–4331. doi: 10.1021/bi035874n. [DOI] [PubMed] [Google Scholar]
- 36.Wilusz J.E. Circle the wagons: circular RNAs control innate immunity. Cell. 2019;177:797–799. doi: 10.1016/j.cell.2019.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller U.F. Design and experimental evolution of trans-splicing group I intron ribozymes. Molecules. 2017;22:75. doi: 10.3390/molecules22010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaug A.J., McEvoy M.M., Cech T.R. Self-splicing of the group I intron from Anabaena pre-tRNA: requirement for base-pairing of the exons in the anticodon stem. Biochemistry. 1993;32:7946–7953. doi: 10.1021/bi00082a016. [DOI] [PubMed] [Google Scholar]
- 39.Vicens Q., Cech T.R. Atomic level architecture of group I introns revealed. Trends Biochem. Sci. 2006;31:41–51. doi: 10.1016/j.tibs.2005.11.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and its supplementary materials.