

Abstract
An abnormal chromosome number, or aneuploidy, underlies developmental disorders and is a common feature of cancer, with different cancer types exhibiting distinct patterns of chromosomal gains and losses. To understand how specific aneuploidies emerge in certain tissues and how they contribute to disease development, various methods have been developed to alter the karyotype of mammalian cells and mice. In this review, we provide an overview of both classic and novel strategies for inducing or selecting specific chromosomal gains and losses in human and murine cell systems. We highlight how these customized aneuploidy models helped expanding our knowledge of the consequences of specific aneuploidies to (cancer) cell physiology.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10577-023-09735-7.
Keywords: Aneuploidy, CIN, chromosome, cancer, CRISPR/Cas9
Introduction
Aneuploidy, defined as a number of chromosomes that deviates from a multiple of the haploid genome, is a prominent feature of spontaneous pregnancy loss, congenital disorders such as Down syndrome, and of cancer (Hassold et al. 1980; Hassold and Jacobs 1984; Sahoo et al. 2017; Ben-David and Amon 2019; Gruhn and Hoffmann 2022). This karyotype aberration is one of the consequences of erroneous chromosome segregation during meiosis and mitosis (referred to as chromosomal instability, or CIN) and includes gains and losses of whole chromosomes or chromosomal arms. Long before the first genomic alterations in proto-oncogenes were discovered in human cancers (Der et al. 1982; Parada et al. 1982; Dalla-Favera et al. 1982), aneuploidy was already described as a distinct feature of cancer by Boveri (Boveri 2008, translated from the original 1914 article). In fact, abnormal karyotypes are observed in ~80% of all solid tumors and in ~60% of hematopoietic cancers (Weaver and Cleveland 2006; Duijf et al. 2013; Taylor et al. 2018). While the role of CIN in cancer initiation and metastasis is becoming increasingly evident (Bakhoum et al. 2018, reviewed in van Jaarsveld and Kops 2016; Sansregret et al. 2018; Tijhuis et al. 2019), the exact contribution of whole chromosome or chromosomal arm gains and losses to disease development has remained less clear (Ben-David and Amon 2019). This knowledge gap is in part due to the challenges associated with generating mammalian models with specific aneuploidies. In this review, we focus on various strategies developed over the years to manipulate the karyotypes of human and mouse cells and animals.
Of note, Saccharomyces cerevisiae (budding yeast) has been a valuable organism for modeling aneuploidies, thanks to its short doubling time, the possibility of karyotype manipulations through mating, and genetic tools readily applicable for this model organism. These yeast-specific methods have been reviewed elsewhere (Mulla et al. 2014; Gilchrist and Stelkens 2019), but it is worth highlighting that they have produced valuable collections of stable isogenic aneuploid strains. These strains have been used to uncover generic cellular responses to aneuploidy (Torres et al. 2007; Pavelka et al. 2010; Beach et al. 2017; Ravichandran et al. 2018). Subsequent analyses of human and mouse cell lines carrying specific trisomies or monosomies have revealed very similar effects on cell physiology as seen in yeast, including impaired cellular fitness and proliferative potential due to the proteotoxic, metabolic and replication stresses associated with chromosomal gains, or impaired ribosomal biogenesis linked to chromosomal losses (Williams et al. 2008; Stingele et al. 2012; Nicholson et al. 2015; Meena et al. 2015; Ohashi et al. 2015; Santaguida et al. 2015; Passerini et al. 2016; Passerini et al. 2016; Chunduri et al. 2021).
While the adverse effects of aneuploidy may explain its detrimental consequences for the developing embryo, it also raises the “aneuploidy paradox” in cancer. Why does a disease characterized by proliferation frequently exhibit aneuploid karyotypes? Furthermore, different tumor types display distinct aneuploidy patterns. For instance, colorectal tumors commonly have recurrent gains of chr7, 13, 20 and loss of chr18, while clear cell renal tumors commonly lose chr3p and gain chr5q (Knouse et al. 2017; Mitchell et al. 2018). How these cancer-specific aneuploidy patterns are established, and to what extent they contribute to disease progression remains a major puzzle in the field. Chromosome loss may support the loss of heterozygosity (LOH) of tumor suppressor genes, while chromosome gain could drive the amplification of one or more oncogenes (Nowak et al. 2002; Rajagopalan et al. 2003). Consistent with this idea, the presence of oncogenes or tumor suppressor genes on certain chromosomes correlates with their recurrent gain or loss in cancer (Davoli et al. 2013). Still, accomplishing LOH or oncogene amplification by losing or gaining an entire chromosome appears challenging because of the collateral deregulation of hundreds of other genes on the affected chromosome. An alternative hypothesis is that the gain or loss of specific chromosomes is tolerated in a tissue-specific manner. Chromosomes harboring genes that are highly expressed in certain tissues may be more easily tolerated as trisomy in those tissues because such gains would cause a milder gene expression imbalance (Sack et al. 2018; Patkar et al. 2021). While attractive, these hypotheses have never been formally tested, partly because it requires the modeling of specific aneuploidies across different tissue types. Finally, whether tumors are addicted to recurrent aneuploidies is another question currently addressed in the field (Girish et al. 2023). While certain cancers are dependent on an activated oncogene to maintain their malignant properties (Weinstein and Joe 2008), it is less clear how the elimination of specific recurrent aneuploidies from cancer cells affects cancer maintenance (Girish et al. 2023).
To understand the contribution of specific chromosomal gains or losses to developmental disorders and cancer, there is a clear need to create models with customized aneuploidies. We will discuss the current successful strategies to generate chromosome-specific gains and losses in mouse and human model systems. Each method offers unique opportunities and distinct hurdles in their experimental application. We will highlight the general principles of these approaches, focus on the insights into cancer biology thus far gathered through their application, and discuss future developments and prospects for the field.
Strategies to introduce specific chromosomal gains
Many of the present mammalian cell lines and mouse strains that possess a single chromosome gain, or a corrected chromosomal loss have been generated using microcell-mediated chromosome transfer (MMCT) or by crossing mice with Robertsonian translocations. The models generated by these techniques provided significant insights into the effects of aneuploidy on non-transformed and cancer cell physiology, as well as developmental disorders like Down syndrome.
Microcell-mediated chromosome transfer
MMCT was originally developed by Fournier and Ruddle in the 1970s as a method for introducing murine chromosomes into mouse, Chinese hamster, and human cells (Fournier and Ruddle 1977). The process involves two steps: first, the induction of micronucleation in donor cells via irradation or prolonged treatment with colcemid (i.e., a drug that depolymerizes microtubules), followed by a rate-limiting second step, in which the micronuclei are fused to recipient cells of choice (Fig. 1a). To facilitate subsequent positive selection of recipient cell clones with the specific chromosomal gain, an antibiotic resistance gene is usually integrated in the chromosome of interest from the donor cell line. The most commonly used donor cells for MMCT are mouse A9 fibroblasts and Chinese hamster ovary (CHO) cells, as these cells easily form micronuclei and can survive prolonged exposure to colcemid (Nakayama et al. 2015). The application of MMCT to human cell systems has been greatly improved by the generation and commercialization of libraries of CHO/human and A9/human hybrid donor cell lines, each containing a selectable single human autosome or chromosome X (Kugoh et al. 1999; Tanabe et al. 2000).
Fig. 1.
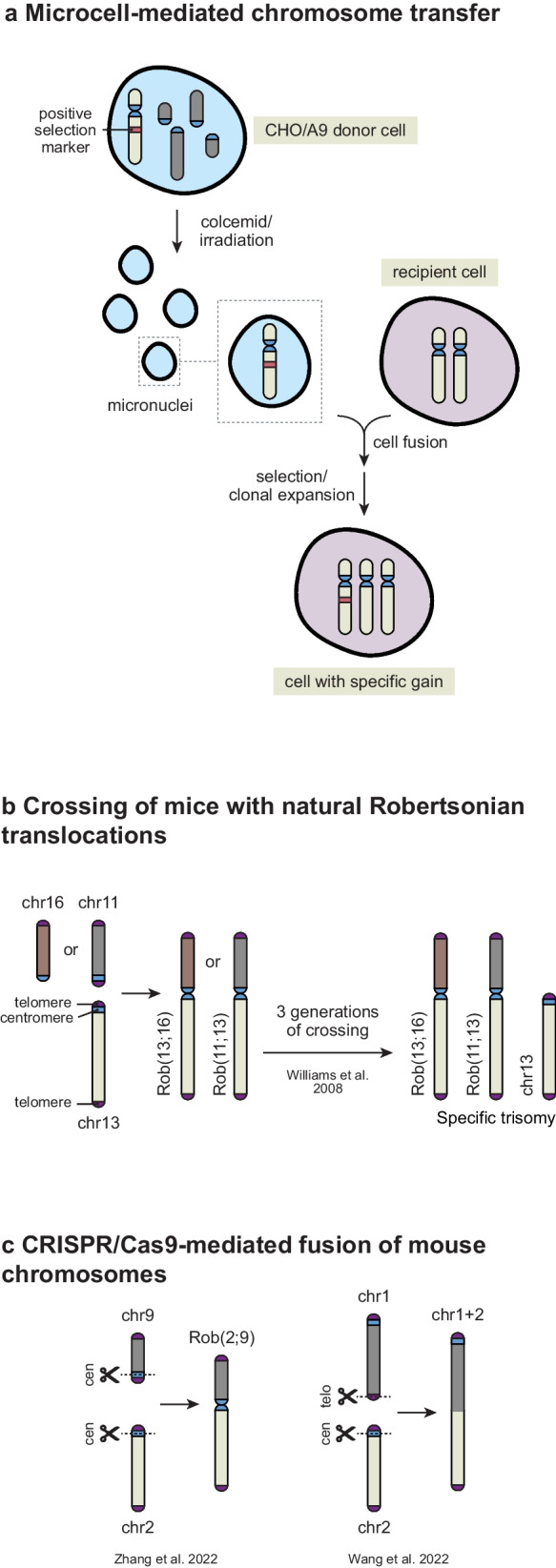
Strategies to introduce an additional chromosome into mammalian cells and mice. a Microcell-mediated chromosome transfer consists of 2 steps: (1) micronuclei formation in donor cells by colcemid treatment or irradiation; and (2) fusion of micronuclei to recipient cells of choice. Hybrid Chinese hamster ovary (CHO) or mouse A9 cells containing a single human chromosome are mostly used as donor cells (Tanabe et al. 2000). A positive selection marker, (often puromycin or neomycin resistance genes), is usually integrated into the chromosome of interest to facilitate the recovery of recipient cells with specific chromosomal gains. b Crossings of mice carrying Robertsonian chromosomes can generate mice and mouse ESCs with specific trisomies (Williams et al. 2008). As starting point, two mouse strains are crossed each carrying a different Robertsonian translocation involving the chromosome of interest (e.g., Rob (13;16) and Rob (11;13)). c CRISPR/Cas9-induced targeted chromosome fusions can generate either Robertsonian-like metacentric chromosomes (Zhang et al. 2022) or a large telocentric fused chromosome (Wang et al. 2022) (Supplemental Table 2). This could be applied to create parental mouse strains carrying specific (viable) chromosome fusions for crossings that eventually generate specific trisomies
MMCT was originally used for biological applications ranging from gene mapping to gene imprinting analysis (Saxon et al. 1986; Kugoh et al. 1999; reviewed in Yoshida et al. 2000; Meaburn et al. 2005) and to model embryonic aneuploidies leading to congenital disorders (reviewed in Akutsu et al. 2020). These included the generation of mouse models for Down syndrome by transferring either a large part of human chr21 (Shinohara et al. 2001; O’Doherty et al. 2005), or a mouse artificial chomosome containing the q-arm of the human chr21, into mouse embryonic stem cells (ESCs) (Kazuki et al. 2020). Moreover, early studies in the 90s utilized MMCT to map tumor growth suppression and anti-metastatic activities on human chromosomes by reverting a chromosomal loss or inducing a specific chromosomal gain in various cancer cell lines (Oshimura et al. 1989; Yamada et al. 1990; Kugoh et al. 1990; Ogata et al. 1993; Tanaka et al. 1998). As a result, a considerable number of human, mouse, and rat cell lines that harbor an extra single human chromosome were generated (reviewed by Kugoh et al. 2016). Functional studies on these cell lines led to the hypothesis that loss of a specific chromosome or chromosome arm could be a strategy to silence tumor-suppressing genes located on that chromosome. For example, MMCT of human chr3 into 3p monosomic renal carcinoma cells induced senescence in vitro (Ohmura et al. 1995; Tanabe et al. 2000). Similarly, induction of trisomy for chr3p or the whole chr3 in oral squamous carcinoma cells inhibited cellular growth and suppressed tumor formation in athymic mice upon subcutaneous injection (Uzawa et al. 1995; Nishio et al. 2015). These anti-tumorigenic effects of chr3 gain were subsequently attributed to multiple tumor-suppressing genes on chr3p, including several telomerase repressors (Uzawa et al. 1998; Abe et al. 2010; Nishio et al. 2015). Furthermore, transferring either whole chromosomes or parts of human chromosomes 1-8, 10-13, 16-20, 22, X, or Y into different cancer or non-transformed immortalized cell lines mainly inhibited the proliferative, tumorigenic and/or metastatic potential of the recipient cell line (reviewed in Yoshida et al. 2000; Meaburn et al. 2005; and Kugoh et al. 2016). However, only in a few cases could the tumor-suppressing effects of a specific chromosome transfer be narrowed down to a single gene or gene cluster (Dong et al. 1995; Yoshida et al. 1999; Seraj et al. 2000; Goldberg et al. 2003).
More recent studies focused on the generic and specific effects of chromosomal gains on cell physiology. Analyses of transformed and non-transformed human cell lines with a single trisomic chromosome (Supplemental table 1) revealed that a chromosome gain often leads to increased CIN, replication stress, as well as global transcriptomic and proteomic changes (Phillips et al. 2001; Phillips et al. 2001; Nawata et al. 2011; Stingele et al. 2012; Dürrbaum et al. 2014; Nicholson et al. 2015; Passerini et al. 2016). Furthermore, in line with earlier observations (Yoshida et al. 2000; Meaburn et al. 2005; Kugoh et al. 2016), these recent studies also showed that nearly all single chromosomal gains negatively impact cellular transformation and metastasis formation. However, this generally appears not to be due to expression of tumor-suppressing genes on the gained chromosome, but rather due to the stresses associated with the gene expression imbalances (Vasudevan et al. 2020). A few studies reported chromosome-specific effects on human cell physiology. For instance, MMCT of chr2, but not of chromosomes 3, 8, 7, 11, or 12, induced senescence in human cervical cancer cells (Uejima et al. 1995). In contrast, introduction of an extra copy of chr7, but not of chromosomes 1, 2, 6, 9, or 11, was found to have tumor-suppressive effects in human choriocarcinoma cells CC1 (Matsuda et al. 1997). Moreover, trisomy of chromosome 8, 16, 17, or 19 suppressed metastatic behavior of human colorectal cancer HCT116 cancer cells, whereas trisomy of chr5 enhanced their metastatic potential (Vasudevan et al. 2020). Meanwhile, trisomy of chr7 and chr13 conferred a growth advantage to colorectal cancer DLD-1 cells when cultured under challenging conditions such as hypoxia or low serum, although these aneuploid cells proliferated slower than their euploid counterparts in standard culture conditions (Rutledge et al. 2016). Interestingly, trisomy 13, but not trisomy 7, was found to cause cytokinesis failure and elevated CIN in human colorectal cancer DLD-1 cells due to SPG20 overexpression (Nicholson et al. 2015). Despite these significant findings, their clinical relevance and the molecular mechanisms by which these aneuploidies potentially drive cancer progression and metastasis require further investigation.
Robertsonian translocations
The second approach to generate specific trisomies takes advantage of Robertsonian fusions, which naturally occur in various races of the house mouse (M.m. domesticus). A translocation or fusion is considered Robertsonian when two different chromosomes are attached to each other at their centromeres (Fig. 1b; Robertson 1916). In mice, which have telocentric chromosomes, Robertsonian translocations result in the fusion of two entire chromosomes into one large metacentric chromosome. Although over a hundred races of M.m. domesticus with at least one Robertsonian metacentric chromosome have been identified in the wild, not all of these are used as laboratory mouse strains (Capanna et al. 1976; Garagna et al. 2014). By crossing mice according to a complex protocol, trisomic embryos for chr1, 13, 16, or 19 have been obtained within three generations (Supplemental table 1, Fig. 1b, Williams et al. 2008). However, none of these trisomic embryos survived to term, except for those with an extra copy of chr19, which lived for a short time after birth. All trisomic embryos exhibited developmental abnormalities such as growth restriction and nuchal edema, demonstrating the overall detrimental effects of a single chromosomal gain on mouse embryonic development. Additionally, studies using mouse embryonic fibroblasts (MEFs) derived from these trisomic embryos showed that having an extra chromosome led to metabolic changes, inhibited cell proliferation, and impeded oncogene-induced transformation (Williams et al. 2008; Sheltzer et al. 2017). While powerful, this method is only applicable to mice and cannot be used to study the contribution of a particular trisomy to cancer development, due to the limited lifespan of the trisomic animals and the inherent incompatibility of the method with conditional and tissue-specific control over the event that generates the trisomy. Moreover, it is difficult to apply to other chromosomes since the combinations of Robertsonian chromosome fusions in laboratory mice are limited. However, this limitation may be overcome by recent advances that allow for targeted whole-chromosome fusions via CRISPR/Cas9-induced DNA double stranded breaks (DSBs) in telomere and centromere regions of different mouse chromosomes (Fig. 1c, Supplemental table 2). Thus far, this impressive tour de force has delivered viable homozygous pups carrying engineered fusion of chr4-5, chr1-13, chr2-11, and chr5-17 (Supplemental table 2; Wang et al. 2022; Zhang et al. 2022). In theory, these mice could be used to create mouse strains with different specific chromosomal gains using similar crossing protocols as those used for mice with natural Robertsonian fusions (Williams et al. 2008, Fig. 1b).
Methods to eliminate specific chromosomes
A quarter of a century ago, Lewandoski and Martin pioneered the field of targeted chromosome elimination by generating male mice that lacked an entire copy of the Y chromosome (X0) (Lewandoski and Martin 1997) through the use of Cre recombinase and a male transgenic mouse line that had accidentally integrated inverted loxP sites into the Y chromosome. With the introduction of targetable nucleases such as CRISPR/Cas9 and TALEN, the toolbox for targeted chromosome elimination has greatly expanded, allowing for more efficient manipulation of karyotypes.
Cre/loxP-mediated chromosome loss
Cre recognizes and catalyzes the recombination of loxP sites (Sternberg and Hamilton 1981; Sauer and Henderson 1988) and particularly inverted loxP sites integrated into a single chromosome (i.e., in cis) give rise to acentric and dicentric chromosome fragments when the loxP sites of the duplicated sister chromatids recombine (Lewandoski and Martin 1997). The acentric fragments are lost during successive rounds of cell division following their formation (Ly et al. 2017). The dicentric chromosome fragments, on the other hand, often undergo multiple breakage-fusion-bridge (BFB) cycles before they are lost (Thomas et al. 2018, Fig. 2a). Although not intensively investigated, depending on the chromosome and site of loxP integration, these BFB cycles as well as the acentric fragments could in theory also generate oncogene amplifying recombination products, such as extrachromosomal (ec) DNA (Thomas et al. 2018; Warecki and Sullivan 2020; Shoshani et al. 2021b). This potential side-effect needs to be considered when studying the oncogenic potential of Cre/loxP generated monosomies (Thomas et al. 2018). Yet, since both the acentric and dicentric chromosome fragments tend to get lost over time, Cre/loxP has been widely used to induce targeted whole and partial chromosome loss in vitro and in vivo (Fig. 2a, Supplemental table 3).
Fig. 2.

Methods to eliminate specific chromosomes. a Two inverted LoxP sites are integrated into a chromosome arm of interest. Upon expression of Cre recombinase after S phase, the two sister chromatids can recombine, generating dicentric and acentric chromosomes that are eventually lost after one or multiple mitoses. An antibiotic resistance gene such as pac (puromycin resistance) or neo (neomycin resistance) is usually inserted between the two loxP sites to facilitate selection of cells harboring loxP integration. To efficiently recover cells with the targeted chromosomal loss, a number of transgenes can be inserted, including ones encoding for fluorescent proteins (FP), cell-surface proteins such as human (h)CD2, or a suicide gene such as herpes simplex virus thymidine kinase (HSV-tk), allowing for FACS sorting or Ganciclovir (GCV)-induced negative selection, respectively. b (i) With one or multiple chromosome-specific sgRNAs, one or multiple DNA double stranded breaks (DSBs) are induced either in the arm or in the (peri-)centromere of the targeted chromosome by CRISPR/Cas9, leading to either whole or partial loss of the targeted chromosome. Integration of a suicide gene (i.e., HSV-tk) in the arm of the targeted chromosome can facilitate selection of cells that have lost the targeted chromosome (arm). Alternatively, CRISPR/Cas9 or TALEN can be used to induce two DSBs flanking the chromosomal region to be deleted. This will lead to ligation of the endogenous telomere to the centromere-proximal break site, leading to specific segmental arm loss. (ii) Telomere-mediated chromosome truncation: CRISPR/Cas9 induces a single DSB near the centromere, and the break is repaired using a repair template containing a positive selection marker (pac or L-histidinol dihydrochloride, hisD), a human telomere sequence (telo), and frequently homology arms (HA) overlapping the break site. The incorporation of a synthetic S. cerevisiae cytosine deaminase-uracil phosphoribosyl transferase fusion gene (Fcy::Fur) outside the HA, can be used to eliminate cells with an off-target integration by 5-fluorocytosine
LoxP sites are frequently co-integrated with a gene encoding a fluorescent protein to facilitate FACS-based enrichment of cells with the desired karyotype (Matsumura et al. 2007; Otsuji et al. 2008; Zhu et al. 2010; Sato et al. 2017; Thomas et al. 2018; Wakita et al. 2022). Alternatively, co-integration of loxP with, for instance, the herpes simplex virus thymidine kinase gene (HSV-tk) allows for the negative selection of cells with an integration in the chromosome of interest (Sato et al. 2017; Wakita et al. 2022). Mammalian cells expressing HSV-tk are killed by the anti-viral drug Ganciclovir (GCV) (Borrelli et al. 1988), and thus only cells that have eliminated the chromosome with the integrated HSV-tk gene can survive this treatment. In fact, it even supports the recovery of cells that have spontaneously lost the HSV-tk-bearing chromosome in the absence of Cre-induced recombination, a strategy successfully applied, albeit at very low efficiency, to restore a disomic state of chr21 in Down syndrome induced pluripotent stem cells (iPSCs) (Li et al. 2012). Of note, GCV was recently shown to be mutagenic; thus, cells recovered after GCV selection have likely acquired additional GCV-associated mutations (de Kanter et al. 2021).
Cre/LoxP has been extensively used to eliminate stem cell-derived chromosomes from tetraploid hybrids generated by cell fusion of mouse ESCs and differentiated mouse cells. Mouse ESC-somatic cell fusions result in the reprogramming of somatic nuclei to a pluripotent state. By eliminating individual ESC-derived chromosomes from the hybrid, their role in maintaining the pluripotent state was studied (Matsumura et al. 2007; Otsuji et al. 2008). To facilitate this type of stem cell research, a panel of mouse ESC lines with inverted loxP sites across 13 different autosomes (mouse chromosomes 1-6, 10-13, 17, 19) and the Y chromosome was generated (Tada et al. 2009). Importantly, Cre/loxP was further exploited to generate mice with tissue-specific monosomies via crosses between mice carrying inverted loxP sites in a chromosome of interest and mice expressing Cre in a defined cell lineage or tissue. Using this strategy, a copy of chromosome 4, 9, 10, 11, or 14 was eliminated from mouse lymphocytes (Zhu et al. 2010), and a copy of chr2 from the limb buds of mouse embryos (Grégoire and Kmita 2008). Both studies reported cell death in the Cre-expressing tissue, albeit to different extents, likely reflecting chromosome- and tissue-specific responses. Of note, since Cre recombination efficiency varies across tissues partly due to differences in Cre expression levels (Duchon et al. 2008), the induced aneuploidies in mouse tissues are mostly mosaic, meaning that only a fraction of the cells display the intended chromosomal loss (Hérault et al. 2010). Aneuploid mosaicism is observed in both non-transformed and transformed human tissues (Riccardi and Crandall 1978; Hasle et al. 1995; Taylor et al. 2014; Forsberg et al. 2014; Knouse et al. 2014; Fragouli et al. 2017), indicating these mouse models mimic a physiologically relevant condition.
More recently, Cre/loxP was used to eliminate one of the three copies of chr21 in HeLa cells, a human hypertriploid cervical cancer line. Interestingly, HeLa cells disomic for chr21 displayed impaired growth compared to the parental trisomic cells (Sato et al. 2017). In marked contrast, Cre/LoxP-induced elimination, or transcriptional silencing of the extra copy of chr21 in Down syndrome iPSCs alleviated the proliferative defects linked to trisomy 21 (Li et al. 2012; Jiang et al. 2013). Furthermore, to address the consequences of chromosome loss in the context of cancer, Thomas et al. derived four different tetraploid immortalized MEF cell lines lacking one copy of chromosomes 9, 10, 12, or 14 (Thomas et al. 2018). Except for the line with a loss of chr12, tetraploid MEFs with a chromosome loss displayed enhanced tumorigenic potential compared to isogenic controls. Transformation was associated with ongoing genomic instability in the MEFs with targeted chromosome loss. Importantly, chromosome elimination in diploid cells did not induce transformation, suggesting that a tetraploid background supports tumorigenesis after chromosome loss.
Chromosome elimination by targetable nucleases
The most simplistic yet versatile way to eliminate a specific chromosome is by inducing multiple DSBs along the arms of a chromosome of interest by CRISPR/Cas9 (Fig. 2b, i). This type of chromosome fragmentation likely produces unrepaired acentric fragments that form micronuclei during cell division and eventually lead to the loss of the targeted chromosome or (segments of) the chromosomal arm (Supplemental table 3) (Leibowitz et al. 2021; Papathanasiou et al. 2021). Multiple chromosome-specific DSBs can be accomplished by one sgRNA targeting chromosome-specific repeats, or by a cocktail of sgRNAs, each targeting a unique sequence. For instance, sex chromosome elimination in mouse ESCs was achieved with relatively high efficiency by directing a single sgRNA towards chromosome-specific repeats in an arm of chrX or chrY (Adikusuma et al. 2017; Zuo et al. 2017). Likewise, chromosome fragmentation effectively achieved loss of chr21 from human Down syndrome iPSCs and of chr7 from the colorectal cancer cell line HT-29 (Zuo et al. 2017).
Alternatively, fragmenting the (peri)centromere seems even more effective (Fig. 2b, i, Supplemental table 3), as inducing multiple breaks in the (peri)centromeric region of chrY induced whole chrY and Y arm loss more efficiently than fragmentation of the long arm of chrY (Adikusuma et al. 2017). In fact, this strategy effectively eliminated mouse chrY from bone marrow cells derived from Cas9 knock-in mice (Sano et al. 2022). When injected into irradiated wild-type recipient mice, the chimeric mice displayed a mosaic loss of chrY in their hematopoietic cells, a condition frequently observed in older males (Forsberg et al. 2014; Dumanski et al. 2016). Strikingly, these mice displayed shorter life spans and developed age-associated cardiomyopathies (incl. myocardial fibrosis) earlier in life than control mice, attributed to chrY-deficient macrophages in the heart that somehow overactivated a pro-fibrotic signaling network (Sano et al. 2022).
While the identification of centromere-specific sgRNAs was a challenge in the past, the recent publication of the T2T genome assembly now enables researchers to excavate targetable centromeric repeats, thereby expanding the range of chromosomes that can be eliminated with this strategy (Altemose et al. 2022; Nurk et al. 2022; Bosco et al. 2023). Furthermore, inducing two DSBs flanking the targeted chromosome arm (one DSB proximal and one distal to the centromere) by CRISPR/Cas9 or TALEN was shown to efficiently delete large parts of chromosomal arms, most likely because the distal chromosome fragment harboring the telomere recombined and capped the truncated chromosome (Fig 2b, i). This approach was used to eliminate segments of chr8p from non-malignant MCF10A mammary epithelial cells (Cai et al. 2016), and of chr11q and chr6q from human neuroblastoma SKNSH and NMB cells (Eleveld et al. 2021, Supplemental table 2).
Finally, several recent reports detected specific chromosome loss as an on-target, undesirable side-effect of a single DSB induced by CRISPR/Cas9-mediated gene editing (Zuccaro et al. 2020; Leibowitz et al. 2021; Papathanasiou et al. 2021; Turocy et al. 2022; Nahmad et al. 2022, Supplemental table 3). In fact, CRISPR/Cas9 targeting of single genes caused both whole-chromosome and segmental loss of chromosomes 2, 5, 6, and X in hTERT-RPE1 cells (Leibowitz et al. 2021); chr2 and chr17 in mouse embryos (Papathanasiou et al. 2021); segmental loss of chr7 and either gain or loss of whole chr14 in human primary T cells (Nahmad et al. 2022). Specific losses were detected at low frequencies shortly after the first cell division cycles following CRISPR/Cas9 and sgRNA expression. Interestingly, a relatively high incidence (over 35%) of whole or partial losses of chromosomes 6, 16, 17, or X could be achieved in human embryos after inducing a single DSB near the centromere of these chromosomes in either pre-fertilized oocytes or 2-cell stage zygotes (Zuccaro et al. 2020; Turocy et al. 2022). Thus, targeted single DSBs, especially the ones near or in the centromere, can be leveraged to either generate specific monosomies, or correct specific trisomies in mammalian cancerous and developmental models. It is important to realize, however, that additional rearrangements involving the targeted chromosome take place or precede the chromosomal loss (Leibowitz et al. 2021; Papathanasiou et al. 2021; Turocy et al. 2022). Moreover, how well recently generated monosomies can be stably maintained depends on the cell type and genetic background in which these losses are generated. Both human and mouse embryos seem to exhibit greater tolerance for monosomies compared to cultured ESCs, as evidenced by the detection of both arm-level and whole-chromosome losses in trophectoderm biopsies of blastomeres, and the unsuccessful attempts to derive monosomic ESCs from these early embryos (Biancotti et al. 2012; Zuccaro et al. 2020). Furthermore, the loss or inactivation of TP53 is an important factor to recover somatic human cell lines with stable monosomies (Chunduri et al. 2021).
An advantage of the CRISPR/Cas9 based approaches is that they do not require prior engineering of the chromosome of interest. This makes them in principle versatile and translatable to many different cell types, provided that these cells can be efficiently transfected or transduced with CRISPR/Cas9 and expanded from single cell cultures. However, this is often a problem and hence complementary strategies allowing for enrichment of cells with the engineered loss have been developed, such as telomere-mediated arm truncation (Fig. 2b, ii). Here, a centromere-proximal DSB is induced, and cells are provided with a repair template harboring an artificial telomere (Uno et al. 2017; Taylor et al. 2018). This repair template contains 100–1000 bp of human telomeric seed sequence, a homologous sequence to the targeted region, and a positive selection marker (often a puromycin resistance gene) to facilitate the recovery of cells with neo-telomere incorporation. Additionally, a negative selection marker incorporated into the repair template outside the homology arm can be used to eliminate cells with random integration of the artificial telomere (Uno et al. 2017). Using such an approach, Taylor et al. deleted chr3p from squamous lung cancer cells to model its recurrent loss in this cancer type and found that the mere loss of chr3p was not sufficient to induce transformation (Taylor et al. 2018). In addition, this strategy was used to delete chr8p from immortalized lung epithelial cells to validate the BISCUT algorithm prediction that non-homozygous loss of the DNA helicase WRN contributes to the positive selection of chr8p loss in cancer (Shih et al. 2023). Alternatively, a negative selection marker such as HSV-tk can be integrated into the chromosome (arm) of interest prior to induction of a peri-centromeric DSB (Fig. 2b, i). Although this requires additional chromosome engineering, it allows for selection of cells that have lost the chromosome (arm) of interest, for example by GCV treatment (discussed above). This strategy allowed Girish et al. to correct trisomy 1q in several cancer cell lines including A2058 (melanoma), AGS (gastric cancer), and A2780 (ovarian cancer). Restoring the chr1q disomic state in these cell lines reduced their anchorage-independent growth ability in vitro and in vivo (Girish et al. 2023). This was partly attributed to over-expression of the TP53 inhibitor MDM4 and the Wnt/β-catenin effector BCL9 in trisomic 1q cells, making these cells addicted to this specific aneuploidy in an oncogene-like manner. Moreover, the isogenic trisomic-disomic 1q cell lines provided a platform to investigate potential therapeutic vulnerabilities of cells with a chr1q gain. Over-expression of the pyrimidine salvage kinase UCK2 caused by the 1q trisomy, rendered these cells especially sensitive to the nucleotide analogs RX-3117 and 3-deazauridine compared to their disomic counterparts (Girish et al. 2023).
Inducing, detecting, and isolating cells with specific chromosomal gains and losses
Most of the strategies described above require (prior) genetic engineering of the chromosome of interest, and/or extensive clonal expansion of cells with the intended karyotype change. Alternative approaches involve the isolation and analysis of cells after experimentally inducing chromosome segregation errors (CIN, Fig. 3). In cultured mammalian cells, CIN is most frequently induced or enhanced by disturbing the chromosome segregation machinery. This disruption can be achieved by exposing cells to compounds that interfere with microtubule dynamics (e.g., paclitaxel, nocodazole, and Aurora B kinase inhibitors), prevent the formation of a bipolar mitotic spindle (e.g., Eg5 inhibitors like monastrol) or impede the mitotic checkpoint, such as MPS1 kinase inhibitors (Mayer et al. 1999; Cimini et al. 2001; Ditchfield et al. 2003; Hauf et al. 2003; Lampson et al. 2004; Santaguida et al. 2010; Weaver 2014; Maia et al. 2018). The mitotic checkpoint ensures that anaphase only begins after kinetochores (i.e., multi protein structures that assemble on centromeres and function as microtubule binding sites of the chromosomes) have properly attached to the mitotic spindle (Musacchio and Salmon 2007). Checkpoint inactivation is sometimes combined with inhibition of the kinetochore-localized kinesin CENP-E, to diminish chromosome congression and favor whole chromosome mis-segregations (Weaver et al. 2003; Soto et al. 2017). Additionally, several CIN-induced aneuploidy mouse models have been generated through overexpression, heterozygous deletion, or mutation of mitotic checkpoint proteins, or by overexpression of Polo-like-kinase 4 (PLK4) to induce centrosome amplification (Michel et al. 2001; Sotillo et al. 2007; Weaver et al. 2007; Iwanaga et al. 2007; Li et al. 2009; Baker et al. 2009; Foijer et al. 2014; Levine et al. 2017; Hoevenaar et al. 2020). Collectively, these and other in vivo models demonstrated that depending on the level of CIN, tissue context and genetic background, CIN can either suppress or promote carcinogenesis (Schvartzman et al. 2010; Simon et al. 2015). More recently, some of these mouse models were refined to more tightly control the level and duration of CIN in adult mouse tissues (Foijer et al. 2017; Trakala et al. 2021; Shoshani et al. 2021a). Only these latest mouse models are discussed below.
Fig. 3.

Approaches to induce, detect, and isolate cells with specific aneuploidies. Non-transformed or transformed near-diploid cells with or without functional TP53 can acquire heterogeneous aneuploid karyotypes after transient or chronic induction of CIN by compounds that disrupt the chromosome segregation machinery. In addition, the mere knockout (KO) or knockdown (KD) of TP53 in hTERT-RPE1 cells or sequential mutation of APC, TP53, KRAS and SMAD4 (APKS) in colorectal organoids is sufficient to increase CIN. FACS-based single-cell sorting followed by WG DNA or RNA sequencing shortly after CIN induction permits assessment of karyotype heterogeneity in the initial aneuploidy landscape thereby revealing potential mis-segregation biases. At the same time, single cell culture after CIN induction can generate monoclonal lines harboring specific monosomies or trisomies. These can be subjected to bulk WG DNA or RNA sequencing to reveal their (altered) karyotypes. Finally, the initially heterogeneous and mosaic aneuploid population can be further cultured under standard conditions or under certain challenging conditions such as anti-cancer drugs. Aneuploidy patterns that eventually emerge can be detected by single-cell WG DNA or RNA sequencing. Of note, WG RNA sequencing, carried out either shortly after CIN induction or after prolonged culture, also allows for analyses of cellular responses to specific chromosomal gains and losses. i, inhibitor. This figure was partially created with Biorender
Evidently, these in vitro and in vivo CIN-inducing strategies do not permit precise control over the identity of the chromosome that mis-segregates. However, they generate a population of cells with heterogenous aneuploid karyotypes. Via single-cell whole genome (WG) DNA or RNA sequencing (Bakker et al. 2016; Zhao et al. 2019; Kashima et al. 2020), or by chromosome fluorescence in situ hybridization (FISH) in combination with image-based flow cytometry (Image-Stream, Worrall et al. 2018), the aneuploidy landscapes and cellular responses have been analyzed shortly after the mis-segregation events or after a period of adaptation in either standard or challenging in vitro culture conditions, or in vivo environments (Fig. 3).
Conditions resulting in chromosome mis-segregation biases
Single-cell whole genome DNA sequencing (scWGS) and high-throughput chromosome FISH applied to cultured human cells shortly after CIN induction showed that certain chromosomes have a higher tendency to mis-segregate than others. In fact, it supported earlier conclusions based on FISH with a limited set of chromosome-specific probes that distinct chromosome mis-segregation biases are seen depending on cell type and the mode of CIN induction (Drpic et al. 2018; reviewed in Klaasen and Kops 2022). For example, temporary depolymerization of microtubules (MT) by nocodazole treatment and washout elevated the mis-segregation rates of chr1 and 2 more than that of other chromosomes in non-transformed hTERT-RPE1 and BJ-hTERT cells (Worrall et al. 2018). These large chromosomes appeared to be prone to cohesion fatigue, the gradual failure to maintain sister-centromere cohesion during a mitotic delay (Daum et al. 2011), in this set-up caused by the nocodazole-induced spindle disruption and consequent activation of the mitotic checkpoint (Worrall et al. 2018).
Second, inducible degradation of the centromere-specific histone CENP-A was shown to specifically perturb kinetochore function and faithful segregation of the human Y chromosome in the male DLD1 colorectal cancer cell line. This is because the human chromosome Y centromere harbors alpha satellite DNA repeats that cannot bind CENP-B, and CENP-B temporarily maintains kinetochores in the absence of CENP-A (Fachinetti et al. 2015). This unique property of the Y chromosome centromere may account for its slightly higher mis-segregation rate compared to chromosome X or 4, even in the presence of CENP-A (Fachinetti et al. 2015). Furthermore, it may contribute to the observed mosaic loss of chrY in peripheral blood lymphocytes of ~20% of the male population (Thompson et al. 2019; Lau 2020), and to the frequent loss of chrY in a variety of tumor types in males (Qi et al. 2023). Interestingly, centromeres of the other chromosomes vary in their number of CENP-B binding sites (Earnshaw et al. 1989; Dumont et al. 2020). In the absence of CENP-A, this is reflected in differences in “kinetochore strengths” per chromosome. Accordingly, large chromosomes with the least CENP-B binding sites and hence weakest kinetochores (i.e., chr3, 6, and X) were found to mis-segregate more often in female hTERT-RPE1 cells lacking CENP-A. Conversely, small chromosomes with large centromeres (i.e., chr 17–20) mis-segregated the least under these conditions (Dumont et al. 2020). Whether these mis-segregation biases caused an equal increase in both losses and gains of these specific chromosomes in the population is currently not clear.
Finally, by combining scWGS with individual chromosome tracking and manipulation, Klaasen et al. showed that human chromosomes occupying the periphery of the interphase nucleus have a higher probability to mis-segregate in hTERT-RPE1 cells when MPS1 is inhibited (Klaasen et al. 2022). These include the larger chromosomes (1–5), but also the smaller chr18. Peripheral chromosomes are thought to more frequently end up near or behind the spindle poles at the beginning of mitosis, and therefore take longer to congress and bi-orient on the mitotic spindle (Klaasen et al. 2022). In line with this, larger chromosomes were also more often stalled near the spindle poles after inhibition of CENP-E, the MT plus-end directed kinesin that facilitates chromosome transport from the spindle pole to the equator (Kapoor et al. 2006; Tovini and McClelland 2019).
It is currently not clear whether these chromosome mis-segregation biases contribute to the establishment of cancer-associated aneuploidy patterns. Since mis-segregating chromosomes can also form micronuclei that can trigger chromothripsis (Stephens et al. 2011; Crasta et al. 2012; Zhang et al. 2015; Ly et al. 2017; Umbreit et al. 2020), chromosomes that mis-segregate more frequently than others may instead be over-represented in chromothriptic recombinations observed in certain cancers (Cortés-Ciriano et al. 2020; Klaasen et al. 2022).
Specific aneuploidies evolving after CIN induction
As mentioned, the (transient) induction of CIN generates a population of cells with a variety of aneuploid karyotypes that can serve as substrates for (adaptive) evolution. WGS of a significant number of single cells sampled from the population at various time points during adaptation will subsequently reveal the number and type of aneuploidies that evolve and dominate the culture during adaptation and selection (Fig. 3). Because cells with and without aneuploid genomes co-exist in the initial population, the dominant aneuploidies are most likely selected because they confer a fitness advantage. Such an approach demonstrated that segmental aneuploidies, a consequence of chromosome breakage, are almost exclusively tolerated and propagated in cells that lack functional TP53 (Santaguida et al. 2017; Soto et al. 2017). Moreover, it uncovered that acquired resistance to a variety of chemotherapeutic drugs is associated with the selection of cells harboring specific chromosome gains and/or losses (Ippolito et al. 2021; Lukow et al. 2021). For example, resistance to paclitaxel of hTERT-RPE1 cells correlated with a stable gain of chr11 or loss of chr10 (Ippolito et al. 2021; Lukow et al. 2021). In contrast, resistance to the BRAF inhibitor vemurafenib recurrently selected for a gain of chr7 in the BRAF mutant colorectal cancer cell line Colo205, but for recurrent gains of chr11 and 18 in A375, a BRAF mutant melanoma cell line (Lukow et al. 2021).
Of note, the mutation or knock-down of TP53 in the hTERT-RPE1 cell line supports clonal outgrowth of cells with specific karyotypes, either with or without experimentally increasing chromosome mis-segregation rates (Chunduri et al. 2021; Hintzen et al. 2022; Adell et al. 2023) (Fig. 3). Monoclonal cell lines with a single monosomy for chr10, 13, 19p, X, or double monosomies for chr10;18 or chr7;10 could be derived after single-cell culture of hTERT-RPE1 cells in which TP53 was inactivated, albeit at low frequency. Re-expression of TP53 in these cell lines suppressed their viability and proliferative capacity, suggesting that TP53 acts as an important barrier against the proliferation of cells with chromosome loss (Chunduri et al. 2021). Similarly, sequential mutation of APC, TP53, KRAS and SMAD4 (APKS) by CRISPR/Cas9 in colorectal organoids derived from healthy human tissues also supported the evolution of specific aneuploidies. Particularly, aneuploidies frequently observed in colorectal cancers, such as monosomies of chr18, 8p, and 4, emerged and dominated after long-term culture of the APKS organoids (Kester et al. 2022).
Furthermore, in adult mouse tissues, heterogeneous and mosaic aneuploidies were observed three months after CIN induction by bypassing the mitotic checkpoint via inducible expression of a mutant form of the APC/C activator CDC20 that cannot bind MAD2 (CDC20AAA) (Trakala et al. 2021). However, the T cell lymphomas that developed in these animals later in life displayed characteristic and recurrent gains of chr14 and 15, together with less frequent gains and losses of other chromosomes (Trakala et al. 2021). Importantly, the frequent gain of chr15 was attributed to the presence of c-MYC on this chromosome, and expressing this oncogene from chr6 was sufficient to select for trisomy of chr6 instead of chr15 (Trakala et al. 2021). Similarly, T cell lymphomas that developed in TP53+/− or TP53−/− mice also exhibited recurrent gains of chr15, yet often accompanied by gains of chr4, 5, and 14. These aneuploidies arose after either transient induction of CIN via temporary PLK4 overexpression (Shoshani et al. 2021a), or after chronic CIN induced in the T cell compartment (Foijer et al. 2017; Shoshani et al. 2021a). Collectively, these examples show that under certain in vitro and in vivo selection pressures, CIN-induced heterogeneous and mosaic aneuploidies can evolve towards more homogeneous, cell-type specific aneuploidies.
Taken together, scWGS and bulk WGS of individual cell clones are powerful technologies to assess the karyotype of cell populations or monoclonal cell lines, respectively. By revealing the level of aneuploidy per individual chromosome and the level of aneuploid mosaicism in a cell population following various CIN-inducing treatments in different cell lines (Bakker et al. 2016), these analyses provide a starting point for understanding how cancer type-specific aneuploidy patterns arise and evolve, and how specific aneuploidies may contribute to drug resistance and tumor formation. Importantly, ongoing developments in computational methods to analyze single-cell sequencing data have made it possible to reliably deduce chromosome copy number states from single-cell RNA sequencing data (Patel et al. 2014; Bosco et al. 2023). This development enables the linking of cellular states and responses to specific chromosomal gains and losses in cell lines, organoids, and cancer tissues (De Falco et al. 2023; Gao et al. 2023).
dCas9-based approaches to mis-segregate specific human chromosomes
Instead of inducing multiple, mostly random chromosome mis-segregations per division and retrospectively assessing the identity of the aneuploid chromosomes in the progeny, very recent approaches have attempted to prospectively mis-segregate a single specific chromosome for inducing its respective gain and loss in the daughter cells (Bosco et al. 2023; Tovini et al. 2023; Truong et al. 2023). The common principle of these approaches is the use of a nuclease-dead Cas9 (dCas9) and a sgRNA to tether a certain protein to a chromosome of interest. This protein then interferes with the faithful segregation of that chromosome during cell division (Fig. 4a–d). dCas9 is directed by the sgRNA to a chromosome-specific repetitive DNA sequence, repeats that are present in both homologs and predominantly found near the telomere, the pericentromere, or underlying the centromere (Bosco et al. 2023; Tovini et al. 2023). As the complementary sgRNA sequence is sometimes present in the repeat over 1000 times, many dCas9 molecules can accumulate on the repeat using one unique sgRNA. Thus far, the following strategies have been tested.
Fig. 4.

dCas9-based methods for mis-segregating specific human chromosomes. a–c dCas9 fused to either P. patens Kinesin14VIb (Kin14VIb) (b), or human CENP-T1-243 (c) has been used in combination with a sgRNA targeting chromosome-specific DNA repeats in chr1 and 9 for inducing chromosome-specific mis-segregation and aneuploidy. d A third strategy targets chromosome-specific higher order repeats (HOR) of centromeres with unique sgRNAs to recruit dCas9 fused to either KNL11-86/S24A;S60, KNL11-86/RVSF/AAAA, or NDC801-207/6xS/T>A. We propose that dCas9-KNL1-86/RVSF/AAAA and dCas9-NDC801-207/6xST>A together with a centromere-specific sgRNA create an ectopic microtubule binding site on the centromere, which can lead to (pseudo-) merotelic attachment of the targeted chromosome during mitosis. Whole chromosome and chromosomal arm aneuploidies for chr6, 7, 8, 9, 12, 16, 18, and X could be generated using this approach. Number of predicted sgRNA binding sites (b.s.) based on the T2T genome assembly are indicated in a, d. AurB, Aurora B
Counteracting chromosome congression
Through tethering of a MT minus-end directed motor protein onto a chromosome of interest, Truong and Cané-Gasull et al. aimed to counteract the forces that drive chromosome congression and to cause a selected chromosome to misalign and mis-segregate during metaphase and anaphase, respectively (Truong et al. 2023, Fig. 4b). The motor and stalk domain of Kinesin 14VIb (Kin14VIb) from the land moss Physcomitrella patens (Jonsson et al. 2015; Nijenhuis et al. 2020) were indirectly bound to dCas9 via rapalog-induced FRB-FKBP12 heterodimerization. dCas9 and kinesin were directed by specific sgRNAs to either a subtelomeric repeat of chr1p or a large pericentromeric repeat of chr9q in hTERT-RPE1 cells with functional TP53. Despite poleward transport of the Kin14VIb-bound locus during (pro)metaphase, the kinetochores of the kinesin-bound sister chromatids acquired bi-oriented MT attachments that silenced the mitotic checkpoint. The counteracting pulling forces caused by Kin14VIb motors walking towards one spindle pole and kinetochore MTs attached to the opposite spindle pole caused typical morphological changes of the targeted chromosomes. In the case of chr1, the 1p arm was heavily stretched in anaphase, while in the case of chr9, the Kin14VIb-bound pericentromere and 9q arm became separated from the kinetochore and 9p arm during metaphase and anaphase (Fig. 4b). This eventually led to arm-level aneuploidies of either 1p or 9q after a single cell division (Truong et al. 2023). While the 9q aneuploidies included tetrasomies, nullisomies, and monosomies, the number of cells with trisomies was relatively low, for reasons not yet well understood.
Ectopic kinetochore assembly
Inspired by prior LacO-LacI tethering studies demonstrating that the N-terminus of Centromere Protein-T (CENP-T1-243) is sufficient to assemble a functional kinetochore on an integrated LacO repeat (Gascoigne et al. 2011; Gascoigne and Cheeseman 2013), Tovini et al. fused CENP-T1-243 to dCas9 to create an extra kinetochore on a chromosome of interest (Fig. 4c, Tovini et al. 2023). Indeed, transfection of dCas9-CENP-T in HEK293T and HCT116 cells together with the same sgRNAs for chr1 and chr9 as described above recruited high levels of the kinetochore proteins NDC80/HEC1 and KNL1 near the telomere of chr1p and to the pericentromere of chr9q, respectively. These ectopic kinetochores attached to spindle MTs and activated the mitotic checkpoint, and MPS1 was therefore inhibited to promote anaphase onset and the mis-segregation of the targeted chromosome. Single-cell sequencing of HEK293T cells shortly after expressing dCas9-CENP-T revealed an increase in a range of large copy number alterations for chr1p and chr9q, compared to cells expressing dCas9 that was not fused to CENP-T (Tovini et al. 2023). Thus, assembly of an extra kinetochore either close to the telomere or nearby the native kinetochore generates segmental aneuploidies of the targeted chromosome.
Centromere targeting
Bosco et al. developed an elaborate computational pipeline to analyze the T2T human genome assembly and identify sgRNAs targeting chromosome-specific alpha-satellite centromeric repeats (Altemose et al. 2022; Nurk et al. 2022; Bosco et al. 2023). This analysis delivered sgRNAs targeting the centromeres of 15 different chromosomes (2, 4, 5-13, 16, 18, 19, X). sgRNAs selected based on their ability to recruit dCas9 at centromeres by imaging was used to dock either the N-terminal MT binding domain (aa 1-207) of NDC80/HEC1 (DeLuca et al. 2006), or the N-terminus (aa 1-86) of KNL1 onto centromeres via direct fusion to dCas9 (Bosco et al. 2023) (Fig. 4d). While all dCas9 fusion proteins appeared effective, dCas9-KNL11-86/RVSF/AAAA was studied most extensively. Its expression in hTERT-RPE1-CDKN1A/RB1 knock-down or hCEC-TP53KO cells together with sgRNAs for chr7 or chr18 induced the mis-segregation of these chromosomes during mitosis and their aneuploidies in a significant fraction of the cells. Impressively, with their strategy, Bosco et al. successfully generated not only segmental, but also whole-chromosome gains and losses of various other specific chromosomes (chr6, 8, 9, 12, 16, and X)(Bosco et al. 2023). Although successful, how centromere-docking of dCas9-KNL1-86/RVSF/AAAA induces chromosome mis-segregation remains unclear. KNL11-86 can bind to MTs and to protein phosphatase 1 (PP1) via its SSILK and RVSF motifs (Liu et al. 2010; Bajaj et al. 2018). PP1 recruitment by KNL1 supports mitotic checkpoint silencing and presumably the stabilization of kinetochore MT attachments by dephosphorylating MPS1 and Aurora B substrates within the kinetochore (Liu et al. 2010; Zhang et al. 2014; Nijenhuis et al. 2014). Phosphorylation of the SSILK and RVSF motifs of KNL1 by Aurora B, on the other hand, counteracts PP1 recruitment, thereby reinforcing mitotic checkpoint signaling and potentially kinetochore MT destabilization in early mitosis (Liu et al. 2010; Nijenhuis et al. 2014; Nasa et al. 2018). Bosco et al. propose that recruiting a small N-terminal KNL1 fragment with a mutated RVSF motif to centromeres disrupts the Aurora B:PP1 balance, such that Aurora B decreases the stability of kinetochore MT attachments on the chromosome of interest (Bosco et al. 2023). It remains, however, difficult to envision how a small part of KNL1, unable to bind PP1, and tethered to the centromere, can interfere with endogenous KNL1-PP1 at the kinetochore. Since KNL11-86 also binds MTs, especially in the absence of PP1 (Espeut et al. 2012; Bajaj et al. 2018), a more likely scenario may be that dCas9-KNL1-86/RVSF/AAAA, in combination with centromere-specific sgRNAs, creates an ectopic MT binding site on the centromere similar to dCas9-NDC801-207. This extra MT binding site on the centromere may increase the risk of acquiring a (pseudo) merotelic attachment (i.e., a single chromatid bound by MTs emanating from opposing spindle poles), that causes the targeted chromosome to lag and mis-segregate during anaphase. Alternatively, dCas9 binding is known to act as a roadblock during replication, especially on repetitive DNA sequences (Whinn et al. 2019; Doi et al. 2021), and it might as well be that the mere binding of dCas9-KNL11-86/RVSF/AAAA causes replication or transcriptional problems that result in incomplete replication or inactivation of the centromere. Irrespective of the mechanism, dCas9-based targeting of centromeric repeats appears to work very efficiently to induce both whole and partial specific chromosomal gains and losses (Bosco et al. 2023).
The advantages of the dCas9-based approaches are that they do not require prior engineering of the chromosome of interest, are applicable to many different cell types, and in principle can generate segmental as well as whole chromosome gains and losses, depending on the chromosomal region targeted. The main challenges lie in expressing sufficient levels of the dCas9-fusion proteins and in identifying sgRNAs that are not only chromosome-specific, but also able to bind to a chromosomal region at least 1000 times. Either way, this dCas9-based toolbox opens up new avenues to manipulate individual mitotic chromosomes and to systematically test the immediate and late cellular responses of various tissue types to the gain and loss of a single specific chromosome.
Conclusion and future perspectives
In this review, we aimed to provide a complete and updated overview of the current methods and technologies for inducing specific aneuploidies in mouse and human cell systems. The past decade has witnessed an explosion of novel and improved strategies for manipulating human and mouse chromosomes and karyotypes. With the development of various CRISPR/Cas9 and dCas9-based approaches, as well as state-of-the-art WG DNA and RNA-sequencing methods (i.e. single-cell and spatial transcriptomics, Erickson et al. 2022), a large toolbox is now available to generate, isolate, detect, and study specific aneuploidies in healthy and diseased mammalian tissues. Moreover, for many of the described approaches (MMCT, CENP-A loss, CRISPR/Cas9, and dCas9-based methods), chromosome gain or loss is accompanied by micronucleus formation of the targeted chromosome. Hence, these methods may also provide unique opportunities to investigate the faith of micronuclei with known chromosome content (Ly et al. 2017; Ly et al. 2019; Kneissig et al. 2019; Leibowitz et al. 2021; Papathanasiou et al. 2021; Bosco et al. 2023; Truong et al. 2023). In fact, recent findings suggests that the content of a micronucleus can determine if and when it will rupture in the following cell cycle (Mammel et al. 2022). Additionally, chromothripsis of specific micronucleated chromosomes can drive oncogenic amplification and drug resistance in cancer through ecDNA generation (Shoshani et al. 2021b).
These methods also offer the possibility to address whether certain tissues tolerate particular chromosomal gains or losses better than others by engineering a specific aneuploidy in a mouse tissue of interest via inducible and tissue-specific expression of either Cre recombinase, active Cas9, or nuclease-dead Cas9. Additionally, transplantation of mouse cancer cells with an engineered karyotype into isogenic immuno-competent mice allows assessment of how specific aneuploidies affect metastasis formation or remodel the tumor microenvironment. In fact, injection of a mouse bladder cancer cell line with a CRISPR/Cas9-engineered loss of chrY, into immunocompetent male C57BL/6 mice, revealed that tumors lacking chrY are more efficient in evading anti-tumor adaptive immunity because these tumors promote the dysfunction of CD8+ T cells in their tumor microenvironment (Abdel-Hafiz et al. 2023). Finally, introducing these chromosome manipulation methods into gastruloids or other embryonic models (Shahbazi et al. 2019; van den Brink and van Oudenaarden 2021; Oldak et al. 2023; Weatherbee et al. 2023) will enable studies on how (stem) cell fate decisions are affected by certain karyotype alterations during mouse and human development.
While the generation of models for specific aneuploidies forms an essential step to evaluate their impact on developmental and cancer biology, additional analyses and manipulations are required to pinpoint the underlying mechanisms by which specific chromosomal gains or losses contribute to cancer in particular tissue or genetic contexts. By analyzing differentially expressed genes in response to specific aneuploidies, researchers uncovered a link between chr1q gain and MDM4-mediated TP53 suppression in cancer cells (Girish et al. 2023) and validated that chr18q loss can drive TGF-β resistance in colon cancer (Bosco et al. 2023). Elaborate computational algorithms can be applied to identify candidate genes underlying the phenotypes driven by specific aneuploidies. For instance, the TUSON Explorer algorithm, which predicts cancer drivers based on their mutational patterns, can be combined with copy number analysis to identify potential driver genes on recurrently gained or lost chromosomes in cancer (Davoli et al. 2013). In addition, weighted correlation network analysis (aka WGCNA) can be used to identify overexpressing genes that correlate with a specific chromosomal gain (Su et al. 2021). Candidate genes can subsequently be validated by RNAi or CRISPRa/CRISPRi screens using libraries of shRNA, siRNAs, or sgRNAs targeting coding genes on the chromosome of interest (Xue et al. 2012; Gilbert et al. 2013; Gilbert et al. 2014; Bock et al. 2022), or by transduction of a library of bar-coded open reading frames (ORF) (Sack et al. 2018; Su et al. 2021). Through a combination of these approaches, Su et al. demonstrated that dose-sensitive overexpression of RAD21 caused by trisomy 8 helped mitigate the replication stress induced by the oncogenic EWS-FLI1 fusion in Ewing sarcoma (Su et al. 2021). Next to this, integration of XIST, the long non-coding RNA (lncRNA) that inactivates one of the X chromosomes in females (Boumil and Lee 2001; Engreitz et al. 2013; Simon et al. 2013), was found to fully inactivate a copy of chr21 (Jiang et al. 2013; Chiang et al. 2018; Czermiński and Lawrence 2020), and to silence parts of mouse chr1 and human chromosomes 1p, 3q, 4q, 7p, 7q, 8p, 12q, and 15q (Kelsey et al. 2015; Loda et al. 2017; Naciri et al. 2021). Although less specific than RNAi or CRISPRi, the epigenetic silencing potential of this lncRNA could be leveraged to pinpoint which part of the chromosome of interest is responsible for certain aneuploidy-related phenotypes.
With the current possibilities to generate customized karyotypes in various cell types in a dish or whole organism, exciting times lie ahead for the field. Together with functional genetic screens and elaborate computational pipelines to analyze large WG DNA and RNA-sequencing data sets of patient-derived cancer tissues it will fuel advancements in understanding how cancer cells tolerate and benefit from aneuploidy, and how specific aneuploidies impact development.
Supplementary information
(PDF 139 kb)
Abbreviations
- APC
adenomatous polyposis coli
- APC/C
anaphase promoting complex/cyclosome
- APKS
APC, TP53, KRAS and SMAD4
- AurB
Aurora B
- BCL9
B-cell lymphoma 9
- BFB
breakage-fusion-bridge
- BISCUT
breakpoint identification of significant cancer undiscovered targets
- BJ-hTERT
hTERT immortalized human BJ foreskin fibroblasts
- BRAF
rapidly accelerated fibrosarcoma homolog B
- CD
cluster of differentiation
- CDC20
cell division cycle protein 20
- CDKN1A
cyclin dependent kinase inhibitor 1A
- hCEC
human colonic epithelial cells
- CHO
Chinese hamster ovary
- CENP
centromere protein
- chr
chromosome
- CIN
chromosomal instability
- c-MYC
cellular myelocytomatosis oncogene
- Cre
causes/cyclization recombination
- CRISPR
clustered regularly interspaced short palindromic repeats
- CRISPRa
CRISPR activator
- CRISPRi
CRISPR interference
- dCas9
nuclease-dead Cas9
- Dox
doxycyclin
- DSB
DNA double-stranded break
- ecDNA
extra-chromosomal DNA
- ESC
embryonic stem cell
- EWS
Ewing sarcoma
- FACS
fluorescence activated cell sorting
- Fcy::Fur
S. cerevisiae cytosine deaminase-uracil phosphoribosyl transferase fusion gene
- FISH
fluorescence in situ hybridization
- FKBP12
FK506 binding protein 12
- FLI
friend leukemia virus integration 1
- FP
fluorescent protein
- FRB
FKBP12–rapamycin-binding
- GCV
ganciclovir
- HA
homology arm
- HEC1
highly expressed in cancer 1; mammalian homolog of NDC80
- HEK293T
human embryonic kidney 293T
- HisD
L-histidinol dihydrochloride
- HSV-tk
herpes symplex virus thymidine kinase
- hTERT
human telomerase reverse transcriptase
- hTERT-RPE1
hTERT immortalized retinal pigment epithelial cell line
- iPSC
induced pluripotent stem cell
- KD
knockdown
- Kin14VIb
physcomitrella patens kinesin14VIb
- KNL1
kinetochore scaffold 1
- KO
knockout
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- LacI
lac inhibitor
- LacO
lac operon
- lncRNA
long non-coding RNA
- LOH
loss of heterozygosity
- loxP
locus of crossing (x) over, P1
- MAD2
mitotic arrest deficient 2
- MDM4
murine double minute 4
- MEF
mouse embryonic fibroblast
- MMCT
microcell-mediated chromosome transfer
- M.m. domesticus
mus musculus domesticus
- MPS1
monopolar spindle 1
- MT
microtubule
- neo
neomycin resistance gene
- NDC80
nuclear division cycle 80
- ORF
open reading frame
- pac
puromycin resistance gene (puromycin N-acetyltransferase)
- PLK4
polo-like kinase 4
- PP1
protein phosphatase 1
- RB1
retinoblastoma protein 1
- Rob
Robertsonian chromosome
- rtTA
reverse-tet transactivator
- Seq
sequencing
- sgRNA
single guide RNA
- shRNA
small hairpin RNA
- siRNA
small interference RNA
- SMAD4
mothers against decapentaplegic homolog 4
- SPG20
spastic paraplegia 20
- T2T
telomere to telomere
- TALEN
transcription activator-like effector nuclease
- Telo
telomere
- TGF-β
transforming growth factor-β
- TP53
tumor protein 53
- TUSON
tumor suppressor and oncogene
- UCK2
uridine-cytidine kinase 2
- WGCNA
weighted gene co-expression network analysis
- WG
whole genome
- WGS
whole-genome sequencing
- WO
Washout
- WRN
Werner syndrome helicase
- XIST
X-inactive-specific transcript
Author contribution
S.M.A.L. conceived the review. M.A.T., P.C.G., and S.M.A.L. performed literature searches and drafted the manuscript. M.A.T. generated figure 1. P.C.G. generated figure 2. S.M.A.L. generated figures 3 and 4. M.A.T. and S.M.A.L. critically revised the work.
Funding
This work is part of Oncode Institute which is partly financed by the Dutch Cancer Society.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Declarations
Ethical approval
Not applicable: the study does not involve human participants/tissues or animals.
Consent to participate
Not applicable. The study does not involve human participants/tissues or animals.
Consent for publication
Not applicable: the study does not involve human participants/tissues or animals.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
My Anh Truong and Paula Cané-Gasull contributed equally to this work.
References
- Abdel-Hafiz HA, Schafer JM, Chen X, Xiao T, Gauntner TD, Li Z, Theodorescu D (2023) Y chromosome loss in cancer drives growth by evasion of adaptive immunity. Nature. 10.1038/s41586-023-06234-x [DOI] [PMC free article] [PubMed]
- Abe S, Tanaka H, Notsu T, Horike S, Fujisaki C, Qi D-L, Ohhira T, Gilley D, Oshimura M, Kugoh H. Localization of an hTERT repressor region on human chromosome 3p21.3 using chromosome engineering. Genome Integr. 2010;1:6. doi: 10.1186/2041-9414-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adell MAY, Klockner TC, Höfler R, Wallner L, Schmid J, Markovic A, Martyniak A, Campbell CS. Adaptation to spindle assembly checkpoint inhibition through the selection of specific aneuploidies. Genes Dev. 2023;37(5–6):171–190. doi: 10.1101/gad.350182.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adikusuma F, Williams N, Grutzner F, Hughes J, Thomas P. Targeted deletion of an entire chromosome using CRISPR/Cas9. Mol Ther. 2017;25(8):1736–1738. doi: 10.1016/j.ymthe.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akutsu SN, Fujita K, Tomioka K, Miyamoto T, Matsuura S. Applications of genome editing technology in research on chromosome aneuploidy disorders. Cells. 2020;9(1):239. doi: 10.3390/CELLS9010239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altemose N, Logsdon GA, Bzikadze AV, Sidhwani P, Langley SA, Caldas GV, Hoyt SJ, Uralsky L, Ryabov FD, Shew CJ, et al. Complete genomic and epigenetic maps of human centromeres. Science. 2022;376(6588):eabl4178. doi: 10.1126/science.abl4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj R, Bollen M, Peti W, Page R. KNL1 binding to PP1 and microtubules is mutually exclusive. Structure. 2018;26(10):1327–1336.e4. doi: 10.1016/j.str.2018.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell. 2009;16(6):475–486. doi: 10.1016/j.ccr.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553(7689):467–472. doi: 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker B, Taudt A, Belderbos ME, Porubsky D, Spierings DCJ, de Jong TV, Halsema N, Kazemier HG, Hoekstra-Wakker K, Bradley A, et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. 2016;17(1):1–15. doi: 10.1186/S13059-016-0971-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach RR, Ricci-Tam C, Brennan CM, Moomau CA, Hsu P, Hua B, Silberman RE, Springer M, Amon A. aneuploidy causes non-genetic individuality. Cell. 2017;169(2):229–242.e21. doi: 10.1016/J.CELL.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat Rev Genet. 2019;21(1):44–62. doi: 10.1038/s41576-019-0171-x. [DOI] [PubMed] [Google Scholar]
- Biancotti JC, Narwani K, Mandefro B, Golan-Lev T, Buehler N, Hill D, Svendsen CN, Benvenisty N. The in vitro survival of human monosomies and trisomies as embryonic stem cells. Stem Cell Res. 2012;9(3):218–224. doi: 10.1016/J.SCR.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Bock C, Datlinger P, Chardon F, Coelho MA, Dong MB, Lawson KA, Lu T, Maroc L, Norman TM, Song B, Stanley G, et al. High-content CRISPR screening. Nat Rev Methods Primer. 2022;2(1):1–23. doi: 10.1038/s43586-021-00093-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli E, Heyman R, Hsi M, Evans RM. Targeting of an inducible toxic phenotype in animal cells. Proc Natl Acad Sci. 1988;85(20):7572–7576. doi: 10.1073/pnas.85.20.7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco N, Goldberg A, Zhao X, Mays JC, Cheng P, Johnson AF, Bianchi JJ, Toscani C, Katsnelson L, Annuar D, et al. KaryoCreate: a CRISPR-based technology to study chromosome-specific aneuploidy by targeting human centromeres. Cell. 2023;186(9):1985–2001.e19. doi: 10.1016/j.cell.2023.03.02910.1101/2022.09.27.509580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumil R, Lee JT. Forty years of decoding the silence in X-chromosome inactivation. Hum Mol Genet. 2001;10(20):2225–2232. doi: 10.1093/hmg/10.20.2225. [DOI] [PubMed] [Google Scholar]
- Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(Supplement 1):1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- Cai Y, Crowther J, Pastor T, Abbasi Asbagh L, Baietti MF, De Troyer M, Vazquez I, Talebi A, Renzi F, Dehairs J, Swinnen JV, Sablina AA. Loss of chromosome 8p governs tumor progression and drug response by altering lipid metabolism. Cancer Cell. 2016;29(5):751–766. doi: 10.1016/j.ccell.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Capanna E, Gropp A, Winking H, Noack G, Civitelli M-V. Robertsonian metacentrics in the mouse. Chromosoma. 1976;58(4):341–353. doi: 10.1007/BF00292842. [DOI] [PubMed] [Google Scholar]
- Chiang J-C, Jiang J, Newburger PE, Lawrence JB. Trisomy silencing by XIST normalizes Down syndrome cell pathogenesis demonstrated for hematopoietic defects in vitro. Nat Commun. 2018;9(1):5180. doi: 10.1038/s41467-018-07630-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chunduri NK, Menges P, Zhang X, Wieland A, Gotsmann VL, Mardin BR, Buccitelli C, Korbel JO, Willmund F, Kschischo M, Raeschle M, Storchova Z. Systems approaches identify the consequences of monosomy in somatic human cells. Nat Commun. 2021;12(1):5576. doi: 10.1038/s41467-021-25288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol. 2001;153(3):517–528. doi: 10.1083/jcb.153.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés-Ciriano I, Lee JJ-K, Xi R, Jain D, Jung YL, Yang L, Gordenin D, Klimczak LJ, Zhang C-Z, Pellman DS, Park PJ. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet. 2020;52(3):331–341. doi: 10.1038/s41588-019-0576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482(7383):53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermiński JT, Lawrence JB. Silencing trisomy 21 with XIST in neural stem cells promotes neuronal differentiation. Dev Cell. 2020;52(3):294–308.e3. doi: 10.1016/j.devcel.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci. 1982;79(24):7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum JR, Potapova TA, Sivakumar S, Daniel JJ, Flynn JN, Rankin S, Gorbsky GJ. Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr Biol. 2011;21(12):1018–1024. doi: 10.1016/J.CUB.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155(4):948. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Falco A, Caruso F, Su X-D, Iavarone A, Ceccarelli M. A variational algorithm to detect the clonal copy number substructure of tumors from scRNA-seq data. Nat Commun. 2023;14(1):1074. doi: 10.1038/s41467-023-36790-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kanter JK, Peci F, Bertrums E, Rosendahl Huber A, van Leeuwen A, van Roosmalen MJ, Manders F, Verheul M, Oka R, Brandsma AM, Bierings M, Belderbos M, van Boxtel R. Antiviral treatment causes a unique mutational signature in cancers of transplantation recipients. Cell Stem Cell. 2021;28(10):1726–1739.e6. doi: 10.1016/j.stem.2021.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, Salmon ED. Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell. 2006;127(5):969–982. doi: 10.1016/j.cell.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci. 1982;79(11):3637–3640. doi: 10.1073/pnas.79.11.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161(2):267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi G, Okada S, Yasukawa T, Sugiyama Y, Bala S, Miyazaki S, Kang D, Ito T. Catalytically inactive Cas9 impairs DNA replication fork progression to induce focal genomic instability. Nucleic Acids Res. 2021;49(2):954–968. doi: 10.1093/NAR/GKAA1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J-T, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, Barrett JC. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science. 1995;268(5212):884–886. doi: 10.1126/science.7754374. [DOI] [PubMed] [Google Scholar]
- Drpic D, Almeida AC, Aguiar P, Renda F, Damas J, Lewin HA, Larkin DM, Khodjakov A, Maiato H. Chromosome segregation is biased by kinetochore size. Curr Biol. 2018;28(9):1344–1356.e5. doi: 10.1016/j.cub.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchon A, Besson V, Pereira PL, Magnol L, Hérault Y. Inducing segmental aneuploid mosaicism in the mouse through targeted asymmetric sister chromatid event of recombination. Genetics. 2008;180(1):51–59. doi: 10.1534/genetics.108.092312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duijf PHG, Schultz N, Benezra R. Cancer cells preferentially lose small chromosomes. Int J Cancer. 2013;132(10):2316–2326. doi: 10.1002/ijc.27924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanski JP, Lambert J-C, Rasi C, Giedraitis V, Davies H, Grenier-Boley B, Lindgren CM, Campion D, Dufouil C, Pasquier F, Amouyel P, Lannfelt L, Ingelsson M, Kilander L, Lind L, Forsberg LA. Mosaic loss of chromosome Y in blood is associated with Alzheimer disease. Am J Hum Genet. 2016;98(6):1208–1219. doi: 10.1016/j.ajhg.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont M, Gamba R, Gestraud P, Klaasen S, Worrall JT, Vries SGD, Boudreau V, Salinas-Luypaert C, Maddox PS, Lens SM, Kops GJ, McClelland SE, Miga KH, Fachinetti D. Human chromosome-specific aneuploidy is influenced by DNA-dependent centromeric features. EMBO J. 2020;39(2):e102924. doi: 10.15252/EMBJ.2019102924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürrbaum M, Kuznetsova A, Passerini V, Stingele S, Stoehr G, Storchová Z. Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genomics. 2014;15(1):139. doi: 10.1186/1471-2164-15-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Ratrie H, Stetten G. Visualization of centromere proteins CENP-B and CENP-C on a stable dicentric chromosome in cytological spreads. Chromosoma. 1989;98(1):1–12. doi: 10.1007/BF00293329. [DOI] [PubMed] [Google Scholar]
- Eleveld TF, Bakali C, Eijk PP, Stathi P, Vriend LE, Poddighe PJ, Ylstra B. Engineering large-scale chromosomal deletions by CRISPR-Cas9. Nucleic Acids Res. 2021;49(21):12007–12016. doi: 10.1093/nar/gkab557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, Plath K, Guttman M. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341(6147):1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson A, He M, Berglund E, Marklund M, Mirzazadeh R, Schultz N, Kvastad L, Andersson A, Bergenstråhle L, Bergenstråhle J, et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature. 2022;608(7922):360–367. doi: 10.1038/s41586-022-05023-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeut J, Cheerambathur DK, Krenning L, Oegema K, Desai A. Microtubule binding by KNL-1 contributes to spindle checkpoint silencing at the kinetochore. J Cell Biol. 2012;196(4):469–482. doi: 10.1083/jcb.201111107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, Cleveland DW. DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev Cell. 2015;33(3):314–327. doi: 10.1016/j.devcel.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foijer F, Albacker LA, Bakker B, Spierings DC, Yue Y, Xie SZ, Davis S, Lutum-Jehle A, Takemoto D, Hare B, et al. (2017) Deletion of the MAD2L1 spindle assembly checkpoint gene is tolerated in mouse models of acute T-cell lymphoma and hepatocellular carcinoma. eLife 6:e20873. 10.7554/eLife.20873 [DOI] [PMC free article] [PubMed]
- Foijer F, Xie SZ, Simon JE, Bakker PL, Conte N, Davis SH, Kregel E, Jonkers J, Bradley A, Sorger PK. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc Natl Acad Sci U S A. 2014;111(37):13427–13432. doi: 10.1073/pnas.1400892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Rasi C, Malmqvist N, Davies H, Pasupulati S, Pakalapati G, Sandgren J, Diaz de Ståhl T, Zaghlool A, et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat Genet. 2014;46(6):624–628. doi: 10.1038/ng.2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier RE, Ruddle FH. Microcell-mediated transfer of murine chromosomes into mouse, Chinese hamster, and human somatic cells. Proc Natl Acad Sci. 1977;74(1):319–323. doi: 10.1073/pnas.74.1.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragouli E, Alfarawati S, Spath K, Babariya D, Tarozzi N, Borini A, Wells D. Analysis of implantation and ongoing pregnancy rates following the transfer of mosaic diploid-aneuploid blastocysts. Hum Genet. 2017;136(7):805–819. doi: 10.1007/s00439-017-1797-4. [DOI] [PubMed] [Google Scholar]
- Gao T, Soldatov R, Sarkar H, Kurkiewicz A, Biederstedt E, Loh P-R, Kharchenko PV. Haplotype-aware analysis of somatic copy number variations from single-cell transcriptomes. Nat Biotechnol. 2023;41(3):417–426. doi: 10.1038/s41587-022-01468-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garagna S, Page J, Fernandez-Donoso R, Zuccotti M, Searle JB. The Robertsonian phenomenon in the house mouse: mutation, meiosis and speciation. Chromosoma. 2014;123(6):529–544. doi: 10.1007/s00412-014-0477-6. [DOI] [PubMed] [Google Scholar]
- Gascoigne KE, Cheeseman IM. Induced dicentric chromosome formation promotes genomic rearrangements and tumorigenesis. Chromosome Res. 2013;21(4):407–418. doi: 10.1007/s10577-013-9368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Takeuchi K, Suzuki A, Hori T, Fukagawa T, Cheeseman IM. Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell. 2011;145(3):410–422. doi: 10.1016/j.cell.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159(3):647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–451. doi: 10.1016/J.CELL.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist C, Stelkens R. Aneuploidy in yeast: segregation error or adaptation mechanism? Yeast. 2019;36(9):525–539. doi: 10.1002/yea.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girish V, Lakhani AA, Thompson SL, Scaduto CM, Brown LM, Hagenson RA, Sausville EL, Mendelson BE, Kandikuppa PK, Lukow DA et al (2023) Oncogene-like addiction to aneuploidy in human cancers. Science:eadg4521. 10.1126/science.adg4521 [DOI] [PMC free article] [PubMed]
- Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub C, Freedman LP, Welch DR. Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP1. Cancer Res. 2003;63(2):432–440. [PubMed] [Google Scholar]
- Grégoire D, Kmita M. Recombination between inverted loxP sites is cytotoxic for proliferating cells and provides a simple tool for conditional cell ablation. Proc Natl Acad Sci U S A. 2008;105(38):14492–14496. doi: 10.1073/pnas.0807484105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruhn JR, Hoffmann ER. Errors of the Egg: the establishment and progression of human aneuploidy research in the maternal germline. Annu Rev Genet. 2022;56(1):369–390. doi: 10.1146/annurev-genet-072820-033609. [DOI] [PubMed] [Google Scholar]
- Hasle H, Clausen N, Pedersen B, Bendix-Hansen K. Myelodysplastic syndrome in a child with constitutional trisomy 8 mosaicism and normal phenotype. Cancer Genet Cytogenet. 1995;79(1):79–81. doi: 10.1016/0165-4608(94)00099-w. [DOI] [PubMed] [Google Scholar]
- Hassold T, Chen N, Funkhouser J, Jooss T, Manuel B, Matsuura J, Matsuyama A, Wilson C, Yamane JA, Jacobs PA. A cytogenetic study of 1000 spontaneous abortions. Ann Hum Genet. 1980;44(2):151–164. doi: 10.1111/j.1469-1809.1980.tb00955.x. [DOI] [PubMed] [Google Scholar]
- Hassold TJ, Jacobs PA. Trisomy in Man. Annu Rev Genet. 1984;18(1):69–97. doi: 10.1146/annurev.ge.18.120184.000441. [DOI] [PubMed] [Google Scholar]
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, Van Meel J, Rieder CL, Peters JM. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161(2):281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hérault Y, Duchon A, Maréchal D, Raveau M, Pereira PL, Dalloneau E, Brault V. Controlled somatic and germline copy number variation in the mouse model. Curr Genomics. 2010;11(6):470–480. doi: 10.2174/138920210793176038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintzen DC, Soto M, Schubert M, Bakker B, Spierings DCJ, Szuhai K, Lansdorp PM, RJC K, Foijer F, Medema RH, Raaijmakers JA. The impact of monosomies, trisomies and segmental aneuploidies on chromosomal stability. PLoS One. 2022;17(7):e0268579. doi: 10.1371/JOURNAL.PONE.0268579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoevenaar WHM, Janssen A, Quirindongo AI, Ma H, Klaasen SJ, Teixeira A, van Gerwen B, Lansu N, Morsink FHM, Offerhaus GJA, Medema RH, Kops GJPL, Jelluma N. Degree and site of chromosomal instability define its oncogenic potential. Nat Commun. 2020;11(1):1501. doi: 10.1038/s41467-020-15279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito MR, Martis V, Martin S, Tijhuis AE, Hong C, Wardenaar R, Dumont M, Zerbib J, Spierings DCJ, Fachinetti D, Ben-David U, Foijer F, Santaguida S. Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev Cell. 2021;56(17):2440–2454.e6. doi: 10.1016/J.DEVCEL.2021.07.006. [DOI] [PubMed] [Google Scholar]
- Iwanaga Y, Chi Y-H, Miyazato A, Sheleg S, Haller K, Peloponese J-M, Jr, Li Y, Ward JM, Benezra R, Jeang K-T. Heterozygous deletion of mitotic arrest–deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 2007;67(1):160–166. doi: 10.1158/0008-5472.CAN-06-3326. [DOI] [PubMed] [Google Scholar]
- Jiang J, Jing Y, Cost GJ, Chiang JC, Kolpa HJ, Cotton AM, Carone DM, Carone BR, Shivak DA, Guschin DY, et al. Translating dosage compensation to trisomy 21. Nature. 2013;500(7462):296–300. doi: 10.1038/nature12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson E, Yamada M, Vale RD, Goshima G. Clustering of a kinesin-14 motor enables processive retrograde microtubule-based transport in plants. Nat Plants. 2015;1(7):15087. doi: 10.1038/nplants.2015.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Lampson MA, Hergert P, Cameron L, Cimini D, Salmon ED, McEwen BF, Khodjakov A. Chromosomes can congress to the metaphase plate before biorientation. Science. 2006;311(5759):388–391. doi: 10.1126/science.1122142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima Y, Sakamoto Y, Kaneko K, Seki M, Suzuki Y, Suzuki A. Single-cell sequencing techniques from individual to multiomics analyses. Exp Mol Med. 2020;52(9):1419–1427. doi: 10.1038/s12276-020-00499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, Gao FJ, Li Y, Moyer AJ, Devenney B, Hiramatsu K, Miyagawa-Tomita S, Abe S, Kazuki K, Kajitani N, et al. A non-mosaic transchromosomic mouse model of Down syndrome carrying the long arm of human chromosome 21. eLife. 2020;9:e56223. doi: 10.7554/eLife.56223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsey AD, Yang C, Leung D, Minks J, Dixon-McDougall T, Baldry SEL, Bogutz AB, Lefebvre L, Brown CJ. Impact of flanking chromosomal sequences on localization and silencing by the human non-coding RNA XIST. Genome Biol. 2015;16(1):208. doi: 10.1186/s13059-015-0774-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kester L, de Barbanson B, Lyubimova A, Chen L-T, van der Schrier V, Alemany A, Mooijman D, Peterson-Maduro J, Drost J, de Ridder J, van Oudenaarden A. Integration of multiple lineage measurements from the same cell reconstructs parallel tumor evolution. Cell Genomics. 2022;2(2):100096. doi: 10.1016/j.xgen.2022.100096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaasen SJ, Kops GJPL. Chromosome inequality: causes and consequences of non-random segregation errors in mitosis and meiosis. Cells. 2022;11(22):3564. doi: 10.3390/cells11223564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaasen SJ, Truong MA, van Jaarsveld RH, Koprivec I, Štimac V, de Vries SG, Risteski P, Kodba S, Vukušić K, de Luca KL, et al. Nuclear chromosome locations dictate segregation error frequencies. Nature. 2022;607(7919):604–609. doi: 10.1038/s41586-022-04938-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneissig M, Keuper K, de Pagter MS, van Roosmalen MJ, Martin J, Otto H, Passerini V, Campos Sparr A, Renkens I, Kropveld F et al (2019) Micronuclei-based model system reveals functional consequences of chromothripsis in human cells. eLife 8. 10.7554/eLife.50292 [DOI] [PMC free article] [PubMed]
- Knouse KA, Davoli T, Elledge SJ, Amon A. Aneuploidy in cancer: Seq-ing answers to old questions. Annu Rev Cancer Biol. 2017;1(1):335–354. doi: 10.1146/annurev-cancerbio-042616-072231. [DOI] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, Amon A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci. 2014;111(37):13409–13414. doi: 10.1073/pnas.1415287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugoh H, Mitsuya K, Meguro M, Shigenami K, Schulz TC, Oshimura M. Mouse A9 cells containing single human chromosomes for analysis of genomic imprinting. DNA Res. 1999;6(3):165–172. doi: 10.1093/dnares/6.3.165. [DOI] [PubMed] [Google Scholar]
- Kugoh H, Ohira T, Oshimura M. Studies of tumor suppressor genes via chromosome engineering. Cancers. 2016;8(1):4. doi: 10.3390/cancers8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugoh HM, Hashiba H, Shimizu M, Oshimura M. Suggestive evidence for functionally distinct, tumor-suppressor genes on chromosomes 1 and 11 for a human fibrosarcoma cell line, HT1080. Oncogene. 1990;5(11):1637–1644. [PubMed] [Google Scholar]
- Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM. Correcting improper chromosomes-spindle attachments during cell division. Nat Cell Biol. 2004;6(3):232–237. doi: 10.1038/ncb1102. [DOI] [PubMed] [Google Scholar]
- Lau Y-FC. Y chromosome in health and diseases. Cell Biosci. 2020;10(1):97. doi: 10.1186/s13578-020-00452-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz ML, Papathanasiou S, Doerfler PA, Blaine LJ, Sun L, Yao Y, Zhang CZ, Weiss MJ, Pellman D. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat Genet. 2021;53(6):895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D, Foijer F, Holland AJ. Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell. 2017;40(3):313–322.e5. doi: 10.1016/j.devcel.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M, Martin GR. Cre-mediated chromosome loss in mice. Nat Genet. 1997;17(2):223–225. doi: 10.1038/ng1097-223. [DOI] [PubMed] [Google Scholar]
- Li LB, Chang KH, Wang PR, Hirata RK, Papayannopoulou T, Russell DW. Trisomy correction in down syndrome induced pluripotent stem cells. Cell Stem Cell. 2012;11(5):615–619. doi: 10.1016/j.stem.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fang X, Wei Z, York JP, Zhang P. Loss of spindle assembly checkpoint–mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol. 2009;185(6):983–994. doi: 10.1083/jcb.200904020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Vleugel M, Backer CB, Hori T, Fukagawa T, Cheeseman IM, Lampson MA. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J Cell Biol. 2010;188(6):809–820. doi: 10.1083/jcb.201001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loda A, Brandsma JH, Vassilev I, Servant N, Loos F, Amirnasr A, Splinter E, Barillot E, Poot RA, Heard E, Gribnau J. Genetic and epigenetic features direct differential efficiency of Xist-mediated silencing at X-chromosomal and autosomal locations. Nat Commun. 2017;8(1):690. doi: 10.1038/s41467-017-00528-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukow DA, Sausville EL, Suri P, Chunduri NK, Wieland A, Leu J, Smith JC, Girish V, Kumar AA, Kendall J, Wang Z, Storchova Z, Sheltzer JM. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev Cell. 2021;56(17):2427–2439.e4. doi: 10.1016/J.DEVCEL.2021.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P, Brunner SF, Shoshani O, Kim DH, Lan W, Pyntikova T, Flanagan AM, Behjati S, Page DC, Campbell PJ, Cleveland DW. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat Genet. 2019;51(4):705–715. doi: 10.1038/s41588-019-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, Cleveland DW. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol. 2017;19(1):68–75. doi: 10.1038/ncb3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia ARR, Linder S, Song J-Y, Vaarting C, Boon U, Pritchard CEJ, Velds A, Huijbers IJ, van Tellingen O, Jonkers J, Medema RH. Mps1 inhibitors synergise with low doses of taxanes in promoting tumour cell death by enhancement of errors in cell division. Br J Cancer. 2018;118(12):1586–1595. doi: 10.1038/s41416-018-0081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammel AE, Huang HZ, Gunn AL, Choo E, Hatch EM. Chromosome length and gene density contribute to micronuclear membrane stability. Life Sci Alliance. 2022;5(2):e202101210. doi: 10.26508/lsa.202101210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Sasaki M, Kato H, Yamada H, Cohen M, Barrett JC, Oshimura M, Wake N. Human chromosome 7 carries a putative tumor suppressor gene(s) involved in choriocarcinoma. Oncogene. 1997;15(23):2773–2781. doi: 10.1038/sj.onc.1201461. [DOI] [PubMed] [Google Scholar]
- Matsumura H, Tada M, Otsuji T, Yasuchika K, Nakatsuji N, Surani A, Tada T. Targeted chromosome elimination from ES-somatic hybrid cells. Nat Methods. 2007;4(1):23–25. doi: 10.1038/nmeth973. [DOI] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286(5441):971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- Meaburn KJ, Parris CN, Bridger JM. The manipulation of chromosomes by mankind: the uses of microcell-mediated chromosome transfer. Chromosoma. 2005;114(4):263–274. doi: 10.1007/s00412-005-0014-8. [DOI] [PubMed] [Google Scholar]
- Meena JK, Cerutti A, Beichler C, Morita Y, Bruhn C, Kumar M, Kraus JM, Speicher MR, Wang Z-Q, Kestler HA, d’Adda di Fagagna F, Günes C, Rudolph KL. Telomerase abrogates aneuploidy-induced telomere replication stress, senescence and cell depletion. EMBO J. 2015;34(10):1371–1384. doi: 10.15252/embj.201490070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409(6818):355–359. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, Martincorena I, Tarpey P, Angelopoulos N, Yates LR, et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell. 2018;173(3):611–623.e17. doi: 10.1016/j.cell.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulla W, Zhu J, Li R. Yeast: A simple model system to study complex phenomena of aneuploidy. FEMS Microbiol Rev. 2014;38(2):201–212. doi: 10.1111/1574-6976.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8(5):379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Naciri I, Lin B, Webb C-H, Jiang S, Carmona S, Liu W, Mortazavi A, Sun S. Linking chromosomal silencing with Xist expression from autosomal integrated transgenes. Front Cell Dev Biol. 2021;9:693154. doi: 10.3389/fcell.2021.693154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahmad AD, Reuveni E, Goldschmidt E, Tenne T, Liberman M, Horovitz-Fried M, Khosravi R, Kobo H, Reinstein E, Madi A, Ben-David U, Barzel A. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat Biotechnol. 2022;40(12):1807–1813. doi: 10.1038/s41587-022-01377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama Y, Uno N, Uno K, Mizoguchi Y, Komoto S, Kazuki Y, Nanba E, Inoue T, Oshimura M. Recurrent micronucleation through cell cycle progression in the presence of microtubule inhibitors. Cell Struct Funct. 2015;40(1):51–59. doi: 10.1247/csf.14005. [DOI] [PubMed] [Google Scholar]
- Nasa I, Rusin SF, Kettenbach AN, Moorhead GB. Aurora B opposes PP1 function in mitosis by phosphorylating the conserved PP1-binding RVxF motif in PP1 regulatory proteins. Sci Signal. 2018;11(530):eaai8669. doi: 10.1126/scisignal.aai8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawata H, Kashino G, Tano K, Daino K, Shimada Y, Kugoh H, Oshimura M, Watanabe M. Dysregulation of gene expression in the artificial human trisomy cells of chromosome 8 associated with transformed cell phenotypes. PLoS One. 2011;6(9):e25319. doi: 10.1371/journal.pone.0025319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JM, Macedo JC, Mattingly AJ, Wangsa D, Camps J, Lima V, Gomes AM, Dória S, Ried T, Logarinho E, Cimini D. Chromosome mis-segregation and cytokinesis failure in trisomic human cells. eLife. 2015;4:1–23. doi: 10.7554/eLife.05068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijenhuis W, Vallardi G, Teixeira A, Kops GJPL, Saurin AT. Negative feedback at kinetochores underlies a responsive spindle checkpoint signal. Nat Cell Biol. 2014;16(12):1257–1264. doi: 10.1038/ncb3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijenhuis W, van Grinsven MMP, Kapitein LC (2020) An optimized toolbox for the optogenetic control of intracellular transport. J Cell Biol 219(4). 10.1083/jcb.201907149 [DOI] [PMC free article] [PubMed]
- Nishio S, Ohira T, Sunamura N, Oshimura M, Ryoke K, Kugoh H. Repression of hTERT transcription by the introduction of chromosome 3 into human oral squamous cell carcinoma. Biochem Biophys Res Commun. 2015;466(4):755–759. doi: 10.1016/j.bbrc.2015.09.119. [DOI] [PubMed] [Google Scholar]
- Nowak MA, Komarova NL, Sengupta A, Jallepalli PV, Shih I-M, Vogelstein B, Lengauer C. The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci. 2002;99(25):16226–16231. doi: 10.1073/pnas.202617399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. The complete sequence of a human genome. Science. 2022;376(6588):44–53. doi: 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D, Linehan JM, Sharpe PT, Brandner S, Bliss TVP, Henderson DJ, Nizetic D, Tybulewicz VLJ, Fisher EMC. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309(5743):2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Ayusawa D, Namba M, Takahashi E, Oshimura M, Oishi M. Chromosome 7 suppresses indefinite division of nontumorigenic immortalized human fibroblast cell lines KMST-6 and SUSM-1. Mol Cell Biol. 1993;13(10):6036–6043. doi: 10.1128/mcb.13.10.6036-6043.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi A, Ohori M, Iwai K, Nakayama Y, Nambu T, Morishita D, Kawamoto T, Miyamoto M, Hirayama T, Okaniwa M, Banno H, Ishikawa T, Kandori H, Iwata K. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat Commun. 2015;6(1):7668. doi: 10.1038/ncomms8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmura H, Tahara H, Suzuki M, Ide T, Shimizu M, Yoshida MA, Tahara E, Shay JW, Barrett JC, Oshimura M. Restoration of the cellular senescence program and repression of telomerase by human chromosome 3. Jpn J Cancer Res. 1995;86(10):899–904. doi: 10.1111/j.1349-7006.1995.tb02998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldak B, Wildschutz E, Bondarenko V, Aguilera-Castrejon A, Zhao C, Tarazi S, Comar M-Y, Ashouokhi S, Lokshtanov D, Roncato F (2023) Transgene-free Ex utero derivation of a human post-implantation embryo model solely from genetically unmodified naïve PSCs. BioRxiv. 10.1101/2023.06.14.544922
- Oshimura M, Kugoh HM, Shimizu M, Yamada H, Hashiba H, Horikawa I, Sasaki M. Multiple chromosomes carrying tumor suppressor activity, via microcell-mediated chromosome transfer, for various tumor cell lines. Princess Takamatsu Symp. 1989;20:249–257. [PubMed] [Google Scholar]
- Otsuji T, Matsumura H, Suzuki T, Nakatsuji N, Tada T, Tada M. Rapid induction of large chromosomal deletions by a Cre/inverted loxP system in mouse ES cell hybrids. J Mol Biol. 2008;378(2):328–336. doi: 10.1016/j.jmb.2008.01.065. [DOI] [PubMed] [Google Scholar]
- Papathanasiou S, Markoulaki S, Blaine LJ, Leibowitz ML, Zhang CZ, Jaenisch R, Pellman D. Whole chromosome loss and genomic instability in mouse embryos after CRISPR-Cas9 genome editing. Nat Commun. 2021;12(1):5855. doi: 10.1038/s41467-021-26097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982;297(5866):474–478. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]
- Passerini V, Ozeri-Galai E, de Pagter MS, Donnelly N, Schmalbrock S, Kloosterman WP, Kerem B, Storchová Z. The presence of extra chromosomes leads to genomic instability. Nat Commun. 2016;7:10754. doi: 10.1038/ncomms10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patkar S, Heselmeyer-Haddad K, Auslander N, Hirsch D, Camps J, Bronder D, Brown M, Chen W-D, Lokanga R, Wangsa D, et al. Hard wiring of normal tissue-specific chromosome-wide gene expression levels is an additional factor driving cancer type-specific aneuploidies. Genome Med. 2021;13(1):1–16. doi: 10.1186/S13073-021-00905-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468(7321):321–325. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JL, Hayward SW, Wang Y, Vasselli J, Pavlovich C, Padilla-Nash H, Pezullo JR, Ghadimi BM, Grossfeld GD, Rivera A, Linehan WM, Cunha GR, Ried T. The consequences of chromosomal aneuploidy on gene expression profiles in a cell line model for prostate carcinogenesis. Cancer Res. 2001;61(22):8143–8149. doi: 10.1158/0008-5472.can-04-0474. [DOI] [PubMed] [Google Scholar]
- Qi M, Pang J, Mitsiades I, Lane AA, Rheinbay E. Loss of chromosome Y in primary tumors. Cell. 2023;186(14):3125–3136.e11. doi: 10.1016/j.cell.2023.06.006. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3(9):695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- Ravichandran MC, Fink S, Clarke MN, Hofer FC, Campbell CS. Genetic interactions between specific chromosome copy number alterations dictate complex aneuploidy patterns. Genes Dev. 2018;32(23–24):1485–1498. doi: 10.1101/GAD.319400.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi VM, Crandall BF. Karyotype-phenotype correlation: mosaic trisomy 8 and partial trisomies of different segments of chromosome 8. Hum Genet. 1978;41(3):363–367. doi: 10.1007/BF00284772. [DOI] [PubMed] [Google Scholar]
- Robertson WMRB. Taxonomic relationships shown in the chromosomes of tettigidae and acrididae: V-shaped chromosomes and their significance in acrididae, locustidae, and gryllidae: chromosomes and variation. J Morphol. 1916;27(2):179–331. doi: 10.1002/jmor.1050270202. [DOI] [Google Scholar]
- Rutledge SD, Douglas TA, Nicholson JM, Vila-Casadesús M, Kantzler CL, Wangsa D, Barroso-Vilares M, Kale SD, Logarinho E, Cimini D. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci Rep. 2016;6(1):22828. doi: 10.1038/srep22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, Leng Y, Liang AC, Scorsone KA, Westbrook TF, Wong K-K, Elledge SJ. Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell. 2018;173(2):499–514.e23. doi: 10.1016/j.cell.2018.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo T, Dzidic N, Strecker MN, Commander S, Travis MK, Doherty C, Tyson RW, Mendoza AE, Stephenson M, Dise CA, Benito CW, Ziadie MS, Hovanes K. Comprehensive genetic analysis of pregnancy loss by chromosomal microarrays: outcomes, benefits, and challenges. Genet Med. 2017;19(1):83–89. doi: 10.1038/gim.2016.69. [DOI] [PubMed] [Google Scholar]
- Sano S, Horitani K, Ogawa H, Halvardson J, Chavkin NW, Wang Y, Sano M, Mattisson J, Hata A, Danielsson M, et al. Hematopoietic loss of Y chromosome leads to cardiac fibrosis and heart failure mortality. Science. 2022;377(6603):292–297. doi: 10.1126/science.abn3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansregret L, Vanhaesebroeck B, Swanton C. Determinants and clinical implications of chromosomal instability in cancer. Nat Rev Clin Oncol. 2018;15(3):139–150. doi: 10.1038/nrclinonc.2017.198. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Richardson A, Iyer DR, M’Saad O, Zasadil L, Knouse KA, Wong YL, Rhind N, Desai A, Amon A. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev Cell. 2017;41(6):638–651.e5. doi: 10.1016/J.DEVCEL.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D’Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010;190(1):73–87. doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Vasile E, White E, Amon A. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 2015;29(19):2010–2021. doi: 10.1101/gad.269118.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Kato H, Yamaza H, Masuda K, Nguyen HTN, Pham TTM, Han X, Hirofuji Y, Nonaka K. Engineering of systematic elimination of a targeted chromosome in human cells. BioMed Res Int. 2017;2017:6037159. doi: 10.1155/2017/6037159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci U S A. 1988;85(14):5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon PJ, Srivatsan ES, Stanbridge EJ. Introduction of human chromosome 11 via microcell transfer controls tumorigenic expression of HeLa cells. EMBO J. 1986;5(13):3461–3466. doi: 10.1002/j.1460-2075.1986.tb04670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman J-M, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10(2):102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q131. Cancer Res. 2000;60(11):2764–2769. [PubMed] [Google Scholar]
- Shahbazi MN, Siggia ED, Zernicka-Goetz M. Self-organization of stem cells into embryos: a window on early mammalian development. Science. 2019;364(6444):948–951. doi: 10.1126/science.aax0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Ko JH, Replogle JM, Habibe Burgos NC, Chung ES, Meehl CM, Sayles NM, Passerini V, Storchova Z, Amon A. Single-chromosome gains commonly function as tumor suppressors. Cancer Cell. 2017;31(2):240–255. doi: 10.1016/J.CCELL.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih J, Sarmashghi S, Zhakula-Kostadinova N, Zhang S, Georgis Y, Hoyt SH, Cuoco MS, Gao GF, Spurr LF, Berger AC, Ha G, Rendo V, Shen H, Meyerson M, Cherniack AD, Taylor AM, Beroukhim R (2023) Cancer aneuploidies are shaped primarily by effects on tumour fitness. Nature. 10.1038/s41586-023-06266-3 [DOI] [PMC free article] [PubMed]
- Shinohara T, Tomizuka K, Miyabara S, Takehara S, Kazuki Y, Inoue J, Katoh M, Nakane H, Iino A, Ohguma A, Ikegami S, Inokuchi K, Ishida I, Reeves RH, Oshimura M. Mice containing a human chromosome 21 model behavioral impairment and cardiac anomalies of Down’s syndrome. Hum Mol Genet. 2001;10(11):1163–1175. doi: 10.1093/hmg/10.11.1163. [DOI] [PubMed] [Google Scholar]
- Shoshani O, Bakker B, de Haan L, Tijhuis AE, Wang Y, Kim DH, Maldonado M, Demarest MA, Artates J, Zhengyu O, et al. Transient genomic instability drives tumorigenesis through accelerated clonal evolution. Genes Dev. 2021;35(15–16):1093–1108. doi: 10.1101/gad.348319.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, Fang R, Castillon GA, Yu M, Li JSZ, Sun Y, Ellisman MH, Ren B, Campbell PJ, Cleveland DW. Chromothripsis drives the evolution of gene amplification in cancer. Nature. 2021;591(7848):137–141. doi: 10.1038/s41586-020-03064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JE, Bakker B, Foijer F. CINcere modelling: what have mouse models for chromosome instability taught us? Recent Results Cancer Res. 2015;200:39–60. doi: 10.1007/978-3-319-20291-4_2. [DOI] [PubMed] [Google Scholar]
- Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg-Schoenberg M, Bowman SK, Kesner BA, Maier VK, Kingston RE, Lee JT. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature. 2013;504(7480):465–469. doi: 10.1038/nature12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11(1):9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto M, Raaijmakers JA, Bakker B, Spierings DCJ, Lansdorp PM, Foijer F, Medema RH. p53 prohibits propagation of chromosome segregation errors that produce structural aneuploidies. Cell Rep. 2017;19(12):2423–2431. doi: 10.1016/J.CELREP.2017.05.055. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg N, Hamilton D. Bacteriophage P1 site-specific recombination: I. Recombination between loxP sites. J Mol Biol. 1981;150(4):467–486. doi: 10.1016/0022-2836(81)90375-2. [DOI] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol. 2012;8(1):608. doi: 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su XA, Ma D, Parsons JV, Replogle JM, Amatruda JF, Whittaker CA, Stegmaier K, Amon A. RAD21 is a driver of chromosome 8 gain in Ewing sarcoma to mitigate replication stress. Genes Dev. 2021;35(78):556–572. doi: 10.1101/gad.345454.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M, Matsumura H, Kurose Y, Nakatsuji N, Tada T. Target chromosomes of inducible deletion by a Cre/inverted loxP system in mouse embryonic stem cells. Chromosome Res. 2009;17(4):443–450. doi: 10.1007/s10577-009-9035-0. [DOI] [PubMed] [Google Scholar]
- Tanabe H, Nakagawa Y, Minegishi D, Hashimoto K, Tanaka N, Oshimura M, Sofuni T, Mizusawa H. Human monochromosome hybrid cell panel characterized by FISH in the JCRB/HSRRB. Chromosome Res. 2000;8(4):319–334. doi: 10.1023/a:1009283529392. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shimizu M, Horikawa I, Kugoh H, Yokota J, Barrett JC, Oshimura M. Evidence for a putative telomerase repressor gene in the 3p14.2–p21.1 region. Genes Chromosomes Cancer. 1998;23(2):123–133. doi: 10.1002/(SICI)1098-2264(199810)23:2<123::AID-GCC5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018;33(4):676–689.e3. doi: 10.1016/J.CCELL.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TH, Gitlin SA, Patrick JL, Crain JL, Wilson JM, Griffin DK. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum Reprod Update. 2014;20(4):571–581. doi: 10.1093/humupd/dmu016. [DOI] [PubMed] [Google Scholar]
- Thomas R, Marks DH, Chin Y, Benezra R. Whole chromosome loss and associated breakage–fusion–bridge cycles transform mouse tetraploid cells. EMBO J. 2018;37(2):201–218. doi: 10.15252/embj.201797630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DJ, Genovese G, Halvardson J, Ulirsch JC, Wright DJ, Terao C, Davidsson OB, Day FR, Sulem P, Jiang Y, et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature. 2019;575(7784):652–657. doi: 10.1038/s41586-019-1765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijhuis AE, Johnson SC, McClelland SE. The emerging links between chromosomal instability (CIN), metastasis, inflammation and tumour immunity. Mol Cytogenet. 2019;12(1):1–21. doi: 10.1186/s13039-019-0429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317(5840):916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Tovini L, Johnson SC, Guscott MA, Andersen AM, Spierings DCJ, Wardenaar R, Foijer F, McClelland SE. Targeted assembly of ectopic kinetochores to induce chromosome-specific segmental aneuploidies. EMBO J. 2023;42:e111587. doi: 10.15252/embj.2022111587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovini L, McClelland SE. Impaired CENP-E function renders large chromosomes more vulnerable to congression failure. Biomol. 2019;9(2):44. doi: 10.3390/BIOM9020044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakala M, Aggarwal M, Sniffen C, Zasadil L, Carroll A, Ma D, Su XA, Wangsa D, Meyer A, Sieben CJ, et al. Clonal selection of stable aneuploidies in progenitor cells drives high-prevalence tumorigenesis. Genes Dev. 2021;35:1079–1092. doi: 10.1101/gad.348341.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong MA, Cané-Gasull P, de Vries SG, Nijenhuis W, Wardenaar R, Kapitein LC, Foijer F, Lens SMA. A kinesin-based approach for inducing chromosome-specific mis-segregation in human cells. EMBO J. 2023;42:e111559. doi: 10.15252/embj.2022111559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turocy J, Marin D, Xu S, Xu J, Robles A, Treff N, Egli D (2022) DNA double strand breaks cause chromosome loss through sister chromatid tethering in human embryos. bioRxiv 2022.03.10.483502
- Uejima H, Mitsuya K, Kugoh H, Horikawa I, Oshimura M. Normal human chromosome 2 induces cellular senescence in the human cervical carcinoma cell line SiHa. Genes Chromosomes Cancer. 1995;14(2):120–127. doi: 10.1002/gcc.2870140206. [DOI] [PubMed] [Google Scholar]
- Umbreit NT, Zhang CZ, Lynch LD, Blaine LJ, Cheng AM, Tourdot R, Sun L, Almubarak HF, Judge K, Mitchell TJ, Spektor A, Pellman D (2020) Mechanisms generating cancer genome complexity from a single cell division error. Science 368(6488). 10.1126/science.aba0712 [DOI] [PMC free article] [PubMed]
- Uno N, Hiramatsu K, Uno K, Komoto S, Kazuki Y, Oshimura M. CRISPR/Cas9-induced transgene insertion and telomere-associated truncation of a single human chromosome for chromosome engineering in CHO and A9 cells. Sci Rep. 2017;7(1):12739. doi: 10.1038/s41598-017-10418-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzawa N, Yoshida MA, Hosoe S, Oshimura M, Amagasa T, Ikeuchi T. Functional evidence for involvement of multiple putative tumor suppressor genes on the short arm of chromosome 3 in human oral squamous cell carcinogenesis. Cancer Genet Cytogenet. 1998;107(2):125–131. doi: 10.1016/S0165-4608(98)00097-1. [DOI] [PubMed] [Google Scholar]
- Uzawa N, Yoshida MA, Oshimura M, Ikeuchi T. Suppression of tumorigenicity in three different cell lines of human oral squamous cell carcinoma by introduction of chromosome 3p via microcell-mediated chromosome transfer. Oncogene. 1995;11(10):1997–2004. [PubMed] [Google Scholar]
- van den Brink SC, van Oudenaarden A. 3D gastruloids: a novel frontier in stem cell-based in vitro modeling of mammalian gastrulation. Trends Cell Biol. 2021;31(9):747–759. doi: 10.1016/j.tcb.2021.06.007. [DOI] [PubMed] [Google Scholar]
- van Jaarsveld RH, Kops GJPL. Difference makers: chromosomal instability versus aneuploidy in cancer. Trends Cancer. 2016;2(10):561–571. doi: 10.1016/j.trecan.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Vasudevan A, Baruah PS, Smith JC, Wang Z, Sayles NM, Andrews P, Kendall J, Leu J, Chunduri NK, Levy D, Wigler M, Storchová Z, Sheltzer JM. Single-chromosomal gains can function as metastasis suppressors and promoters in colon cancer. Dev Cell. 2020;52(4):413–428.e6. doi: 10.1016/j.devcel.2020.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita S, Hara M, Kitabatake Y, Kawatani K, Kurahashi H, Hashizume R. Experimental method for haplotype phasing across the entire length of chromosome 21 in trisomy 21 cells using a chromosome elimination technique. J Hum Genet. 2022;67(10):565–572. doi: 10.1038/s10038-022-01049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LB, Li ZK, Wang LY, Xu K, Ji TT, Mao YH, Ma SN, Liu T, Tu CF, Zhao Q, Fan XN, Liu C, Wang LY, Shu YJ, Yang N, Zhou Q, Li W. A sustainable mouse karyotype created by programmed chromosome fusion. Science. 2022;377(6609):967–975. doi: 10.1126/SCIENCE.ABM1964. [DOI] [PubMed] [Google Scholar]
- Warecki B, Sullivan W. Mechanisms driving acentric chromosome transmission. Chromosome Res. 2020;28(3):229–246. doi: 10.1007/s10577-020-09636-z. [DOI] [PubMed] [Google Scholar]
- Weatherbee BA, Gantner CW, Daza RM, Hamazaki N, Iwamoto-Stohl LK, Shendure J, Zernicka-Goetz M (2023) Transgene directed induction of a stem cell-derived human embryo model. Nature. 10.1038/s41586-023-06368-y
- Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681. doi: 10.1091/mbc.e14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol. 2006;18(6):658–667. doi: 10.1016/j.ceb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Weaver BAA, Bonday ZQ, Putkey FR, Kops GJPL, Silk AD, Cleveland DW. Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J Cell Biol. 2003;162(4):551–563. doi: 10.1083/jcb.200303167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BAA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11(1):25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–3080. doi: 10.1158/0008-5472.CAN-07-3293. [DOI] [PubMed] [Google Scholar]
- Whinn KS, Kaur G, Lewis JS, Schauer GD, Mueller SH, Jergic S, Maynard H, Gan ZY, Naganbabu M, Bruchez MP, O’Donnell ME, Dixon NE, van Oijen AM, Ghodke H. Nuclease dead Cas9 is a programmable roadblock for DNA replication. Sci Rep. 2019;9(1):1–9. doi: 10.1038/s41598-019-49837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322(5902):703–710. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrall JT, Tamura N, Mazzagatti A, Shaikh N, van Lingen T, Bakker B, Spierings DCJ, Vladimirou E, Foijer F, McClelland SE. Non-random mis-segregation of human chromosomes. Cell Rep. 2018;23(11):3366–3380. doi: 10.1016/J.CELREP.2018.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, Revill K, Weissmueller S, Rappaport AR, Simon J, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci U S A. 2012;109(21):8212–8217. doi: 10.1073/pnas.1206062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Wake N, Fujimoto S, Barrett JC, Oshimura M. Multiple chromosomes carrying tumor suppressor activity for a uterine endometrial carcinoma cell line identified by microcell-mediated chromosome transfer. Oncogene. 1990;5(8):1141–1147. [PubMed] [Google Scholar]
- Yoshida BA, Dubauskas Z, Chekmareva MA, Christiano TR, Stadler WM, Rinker-Schaeffer CW. Mitogen-activated protein kinase kinase 4/stress-activated protein/Erk kinase 1 (MKK4/SEK1), a prostate cancer metastasis suppressor gene encoded by human chromosome 171. Cancer Res. 1999;59(21):5483–5487. [PubMed] [Google Scholar]
- Yoshida BA, Sokoloff MM, Welch DR, Rinker-Schaeffer CW. Metastasis-suppressor genes: a review and perspective on an emerging field. JNCI J Natl Cancer Inst. 2000;92(21):1717–1730. doi: 10.1093/jnci/92.21.1717. [DOI] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522(7555):179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Lischetti T, Nilsson J. A minimal number of MELT repeats supports all the functions of KNL1 in chromosome segregation. J Cell Sci. 2014;127(4):871–884. doi: 10.1242/jcs.139725. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Yan M, Yang Z, Xiang H, Tang W, Cai X, Wu Q, Liu X, Pei G, Li J. Creation of artificial karyotypes in mice reveals robustness of genome organization. Cell Res. 2022;32(11):1026–1029. doi: 10.1038/s41422-022-00722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Carter R, Natarajan S, Varn FS, Compton DA, Gawad C, Cheng C, Godek KM. Single-cell RNA sequencing reveals the impact of chromosomal instability on glioblastoma cancer stem cells. BMC Med Genomics. 2019;12(1):79. doi: 10.1186/s12920-019-0532-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Kim YM, Li S, Zhuang Y. Generation and analysis of partially haploid cells with Cre-mediated chromosome deletion in the lymphoid system. J Biol Chem. 2010;285(34):26005–26012. doi: 10.1074/jbc.M110.139196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccaro MV, Xu J, Mitchell C, Marin D, Zimmerman R, Rana B, Weinstein E, King RT, Palmerola KL, Smith ME, Tsang SH, Goland R, Jasin M, Lobo R, Treff N, Egli D. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell. 2020;183(6):1650–1664.e15. doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
- Zuo E, Huo X, Yao X, Hu X, Sun Y, Yin J, He B, Wang X, Shi L, Ping J, Wei Y, Ying W, Wei W, Liu W, Tang C, Li Y, Hu J, Yang H. CRISPR/Cas9-mediated targeted chromosome elimination. Genome Biol. 2017;18(1):1–18. doi: 10.1186/s13059-017-1354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 139 kb)
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.