

Abstract
Traditionally, multiple sclerosis (MS) has been categorized by distinct clinical descriptors — relapsing-remitting, secondary-progressive, and primary-progressive — for patient care, research, and regulatory approval of medications. Accumulating evidence suggests the clinical course of MS is better considered as a continuum, with contributions from concurrent pathophysiologies that vary across individuals and over time. The apparent evolution to a progressive course reflects a partial shift from predominantly localized acute injury to widespread inflammation and neurodegeneration coupled with failure of compensatory mechanisms, such as neuroplasticity and remyelination. Aging increases neural susceptibility to injury and decreases resiliency. These observations encourage a new consideration of the course of MS as a spectrum defined by the relative contributions of overlapping pathological and reparative/compensatory processes. New understanding of key mechanisms underlying progression and measures to quantify progressive pathology will potentially have important and beneficial implications for clinical care, treatment targets, and regulatory decision-making.
Introduction
Multiple sclerosis (MS) is an immune-mediated disease of the CNS. The heritability of MS risk is approximately 25%, with the remainder of susceptibility attributed to environmental, epigenetic, and gene-gene or gene-environment interactions.1 The International Advisory Committee on Clinical Trials in MS (Supplementary Materials) categorized clinical course descriptors (commonly referred to as the Lublin-Reingold classification) in 1996, with revision in 2013.2,3 They defined three clinical courses: relapsing-remitting (RRMS) (acute attacks followed by recovery), primary progressive (PPMS) (gradual worsening from onset), and secondary progressive (SPMS) (relapsing-remitting at onset but gradual worsening later in the disease course. The descriptors provided consistency in defining patient groups for natural history studies, enhanced homogeneity in clinical trials, and greatly improved communication between clinicians and patients.2 In the 2013 revision, clinico-radiological disease activity and progression were introduced as modifiers of the basic clinical courses to better reflect treatment-relevant aspects of the disease, such as relapses.3 These refinements were incorporated into trials that led to the first approvals of drugs for progressive MS (for example, the approval of siponimod for “active” SPMS).4,5
It seems clear now that disability progression is neither dichotomous nor genetically determined.6 Rather, accumulating data suggest that MS patients share qualitatively similar (but quantitatively different) pathology features independent of clinical course, including inflammation and neurodegeneration, both of which are already present at disease onset.7–10 In line with this observation, in relapsing-onset MS, a substantial proportion of disability progression is independent from relapses.11,12 Phenotypic differences in disease expression may be driven by patient-specific factors, including sex, age, social and environmental exposures, genetic factors, and disease duration.13,14
Since the introduction of the Lublin-Reingold descriptors, there have been calls for development of a disease classification more rooted in the biological mechanisms of MS. As a first step in this direction, the International Advisory Committee on Clinical Trials of MS focused on clarifying the 1996 and 2013 clinical course descriptors.15 The committee has since undertaken an effort to more comprehensively examine the current clinical course descriptors with the goal of determining an approach to development of a new paradigm for describing the disease.16–18 Herein, we present concepts and results relevant to the pathophysiology of injury and compensatory mechanisms in MS and summarize the tools that can be used in clinical practice, trials, and research to identify the spectrum of MS pathology and clinical progression. We consider knowledge gaps in identifying injury and failure of compensatory mechanisms and indicate how these gaps should be addressed. We suggest that clinical characterization and treatment selection should be guided by identification of disease-driving pathophysiological mechanisms rather than the traditional clinical descriptors. This approach lays the groundwork for a future consensus-based classification that would transform drug discovery and improve patient care.
Mechanisms of Injury
Nonresolving inflammation
Focal inflammatory demyelination in the white matter is a relatively stereotyped process characterized by perivenular inflammation involving both adaptive and innate immune cells, parenchymal astrocytic and microglial reaction, blood-brain-barrier opening, a rapid wave of demyelination manifested over the course of days to weeks (sometimes corresponding to clinical relapse), and a phase of tissue repair that typically lasts weeks to months.19 Focal inflammation can be observed as gadolinium enhancement on MRI, which allows identification of “active” disease (Fig. 1). The perivenular topography of focal inflammatory lesions can be detected using susceptibility-based MRI.20 In approximately one quarter of lesions, inflammation may “burn out” despite the absence of adequate repair, leaving behind an astroglial scar.21 Residua of these processes can be detected in vivo using T2-weighted hyperintensity on MRI; T1-weighted hypointensity ensues in the case of loss of neuropil (“black holes”) (Table 1). Abrogation of new MRI lesions is a cornerstone for assessing response to treatments aiming to block MS relapses but has limited value in predicting the benefit of therapy on slowing of clinical progression, although, as discussed below, the residua of focal inflammatory demyelination have emerged as key drivers of that progression.12,22,23
Figure 1. Mechanisms of injury and compensation and associated measures in MS.
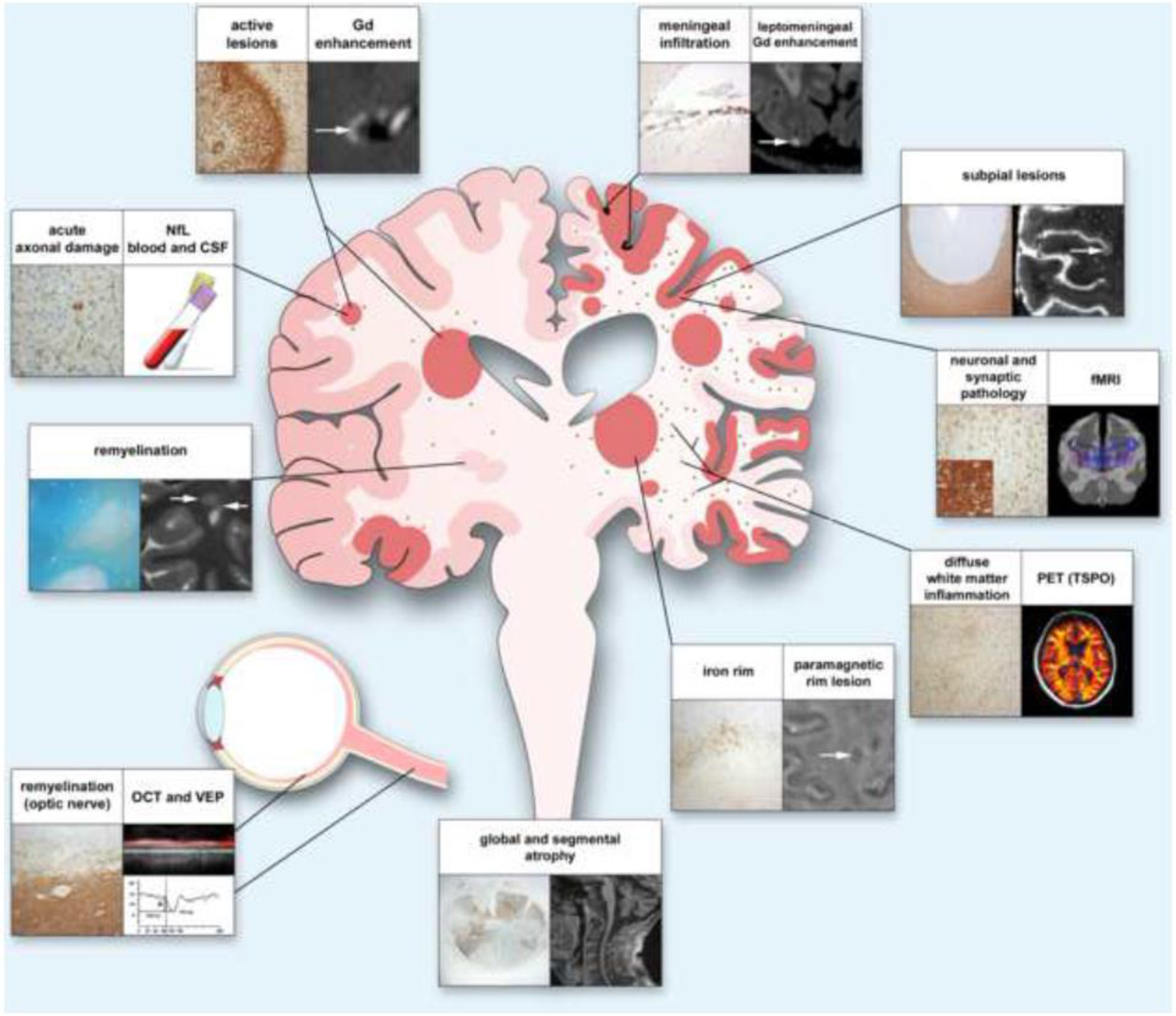
Early in the disease (left half of the figure), injury caused by focal lesions and associated axon damage can be compensated by mechanisms such as remyelination. Over time, lesions in grey and white matter, as well as axon damage, accumulate, and meningeal inflammation, diffuse microglial activation in the extralesional white matter, and slow expansion of existing lesions become more prominent (right side). Progression is further driven by decreased remyelination capacity and damage to neuronal networks mediated by loss of neurons and synapses. Ongoing low-level inflammation and loss of compensatory mechanisms result in segmental and global atrophy. In the figure, headings explain the content of each panel. The histological panel depicting the optic nerve shows axon neurofilaments, whereas the inset shows CD68-positive myeloid cells. The VEP trace depicts delayed latency, indicating slow conduction related to demyelination. Neuronal and synaptic pathology can be detected by NeuN, a marker for neurons (panel), and synaptophysin, a marker for synapses (insert); the blue lines in the radiological correlate symbolize neuronal connectivity. White arrows indicate radiological correlates of histopathological findings. Abbreviations: Gd, gadolinium; NfL, neurofilament light chain; CSF, cerebrospinal fluid; fMRI, functional magnetic resonance imaging; PET, positron emission tomography; TSPO, translocator protein 18 kilodaltons; OCT, optical coherence tomography; VEP, visual evoked potentials.
Table. Pathological Mechanisms of MS Progression and Current Approaches How to Measure Them.
Table shows pathological mechanisms of MS; tools that are implemented for their assessment in clinical practice (green), clinical trials (yellow), or clinical research (red); and relevant clinical correlates.
| Inflammation | ||||
| White matter inflammation | MRI for lesion volume/count (T2-FLAIR, Gd+) | Relapse (count, time to, annualized) | Clinical Practice | Filippi et al.80 |
| MRI for central vein sign (T2*) | Relapse | Clinical Practice | Al-Louzi et al.103 | |
| MRI for paramagnetic (iron) rim lesions (T2* phase, QSM) | Clinical progression | Clinical Practice | Filippi et al.102 | |
| Gray matter inflammation | MRI for lesion count/volume (T2, STIR, PSIR, PD, MPRAGE) | Relapse Clinical progression | Clinical Practice | Moccia et al.104 |
| Spinal cord inflammation | MRI for lesion count/volume (T2, STIR, PSIR, PD, MPRAGE) | Relapse Clinical progression | Clinical Practice | Moccia et al81 |
| Optic nerve inflammation | MRI for lesion count/volume (STIR) | Optic neuritis Changes in visual acuity | Clinical Practice | Kolappan et al.105 |
| OCT (pRNFL) | Changes in visual acuity | Clinical Practice | Sotirchos et al.50 | |
| Leptomeningeal inflammation | MRI (post-gadolinium 3D T2-FLAIR) | Clinical progression | Clinical Trials | Choi et al.106 |
| Microglia and astrocytes | PET (TSPO, acetate) | Clinical progression | Clinical Research | Moccia et al.48 |
| Neurodegeneration | ||||
| Neuro-axonal damage | Blood/CSF (neurofilament light chain levels) | Relapse Clinical progression | Clinical Trials | Khalil et al.41 |
| MRI (AD and FA DTI, ODI/NDI) | Clinical progression | Clinical Trials | Bagnato et al.107 | |
| MRS (GABA, choline) | Unknown | Clinical Research | Moccia et al.48 | |
| PET (GABA, choline) | Unknown | Clinical Research | Moccia et al.48 | |
| Neuro-axonal loss | MRI for intralesional axonal loss (T1 black holes) | Clinical progression | Clinical Practice | Filippi et al.102 |
| MRI for global and regional brain atrophy (3DT1) | Clinical progression | Clinical Trials | Eshaghi et al.108 | |
| MRI for spinal cord atrophy (3DT1) | Clinical progression | Clinical Trials | Moccia et al.104 | |
| OCT for optic nerve atrophy (GCL, pRNFL) | Low contrast visual acuity | Clinical Trials | Sotirchos et al.50 | |
| PET for synapse loss | Unknown | Clinical Research | Moccia et al.48 | |
| Molecular mechanisms of injury: Oxidative stress and mitochondrial dysfunction | ||||
| Energy failure | MRS (NAA, creatine, phosphocreatine) | Clinical progression | Clinical Research | Moccia M, et al.48 |
| Metabolic imbalance | Sodium imaging | Clinical progression | Clinical Research | Eisele et al.45 |
| MRS (glutamate, glutamine, glutathione) | Clinical progression | Clinical Research | Choi et al.106 | |
| Blood/CSF (oxidation products) | Clinical progression | Clinical Research | Pegoretti et al.109 | |
| Failure of compensatory mechanisms | ||||
| Demyelination and remyelination | Visual evoked potentials (VEP) | Changes in visual acuity | Clinical Practice | Green et al.75 |
| MRI (MT, MWF, RD DTI, MP2RAGE) | Clinical progression | Clinical Trials | Bagnato et al.107 | |
| PET (amyloid) | Clinical progression | Clinical Research | Moccia et al.48 | |
| Neuroplasticity | fMRI (BOLD) | Clinical progression | Clinical Trials | Loitfelder et al.110 |
The existence of an ongoing intrathecal immune response is usually demonstrated at the time of diagnosis by the presence of CNS-specific oligoclonal bands.24 In the acute phase, activation of microglia and infiltrates of macrophages and lymphocytes accompany demyelination and plaque formation.25,26 However, these inflammatory mechanisms fail to resolve in approximately 20% of lesions.19 Inflammation becomes more organized, with tissue-resident CD8+ memory cells and monocyte populations, fostering inflammatory changes in brain-resident cells (astrocytes and microglia), and ultimately resulting in chronic tissue remodelling and damage.25,27,28 These characteristics are especially prominent in “mixed active and inactive lesions”, a recent term that subsumes previous descriptions of “chronic active,” “smouldering,” and “slowly expanding” lesions, which in many (but not all) cases are identifiable on high-field MRI because of iron-laden phagocytes at the lesion’s white matter-bordering edge (the so-called “paramagnetic rim sign”) (Fig. 1).19,29 In vivo MRI studies confirm speculations based on autopsy studies that inflammatory changes within paramagnetic rim lesions can enlarge slowly into previously healthy perilesional tissue, accompanied by low-grade demyelination and transection of axons passing through or near lesions.29,30 Axon transection results in retro- and anterograde axon degeneration, with potentially detrimental effects on separate but anatomically connected areas of the brain. Therefore, it is not surprising that a high burden of these lesions is associated with more rapid disability accumulation.30 Recent data demonstrate that the paramagnetic rim sign may disappear over a period of years, raising the possibility that chronic focal white matter inflammation may be susceptible to therapeutic modulation.31,32 Changes in paramagnetic rim lesions are currently included as outcome measures in ongoing and newly designed MS clinical trials as potential correlates or predictors of MS progression. A separate MRI approach combines data from the entire time course of a clinical trial to capture the slow enlargement of MS lesions (so-called “slowly evolving lesions”), but whether and how these changes are related to chronic inflammation remains uncertain.33
Another important site of chronic inflammation is the leptomeninges (Fig. 1), where innate and adaptive immune cells may aggregate and occasionally organize into tertiary lymphoid structures.34 Many (but not all) autopsy studies have shown a spatial correspondence between leptomeningeal inflammatory aggregates, which are more prevalent in cases of clinically progressive MS, and demyelination of the underlying subpial cortex.35 Despite the advent of MRI-based approaches that can identify some current or previous areas of leptomeningeal inflammation due to accompanying blood-meningeal barrier abnormalities, such techniques are not sufficiently robust to quantify accumulation of leptomeningeal inflammation over time.
Finally, diffuse microglial activation and multifocal microglial nodules in the extralesional white matter have been reported in MS autopsies, especially in cases of progressive MS (Fig. 1).10,36 The causes and consequences of this diffuse (and occasionally profound) microglial activation are poorly understood. Similarly, whether microglial nodules represent areas of incipient but aborted focal demyelination, reaction to local tissue perturbation, or something else, remains unclear.37 Positron emission tomography (PET) studies using radioligands that bind to activated microglia and astrocytes have provided some in vivo evidence for widespread microglial involvement, although data generated by these scans are often noisy, spatial localization is poor, and cellular specificity is imperfect (Table 1).38 These same PET radioligands may identify some mixed active and inactive white matter lesions and have been used for this purpose in clinical trials.39–41 Given the new appreciation of massive glial and neuronal heterogeneity in the CNS, an important research goal is to improve the cellular specificity of molecular imaging techniques.
Nonresolving inflammation not only drives injury but may also prevent repair. An open and critical question is whether inflammation needs to resolve before tissue repair can commence. The development of sensitive and specific, noninvasive imaging markers that detect such inflammation, such as the paramagnetic rim sign, along with future development of robust CSF and blood biomarkers of the same processes, might allow this question to be answered. Similar approaches could elucidate the importance of nonresolving inflammation, and any potential group or individual effect on that inflammation of existing or future disease-modifying therapies for MS clinical progression.
Neurodegeneration
Inflammation is closely linked to axon and neuron injury in MS. Axon damage is already prominent at the earliest lesion stages, whereas neuronal loss may start early but becomes more obvious in tissue samples from patients with progressive disease (Fig. 1)42,43. As a consequence of primarily axon damage, neurofilament light chain (NfL), a cytoskeletal protein, is released into the interstitial space and subsequently enters CSF and peripheral blood (Table 1).44 NfL concentration has been directly associated with relapses and clinical progression, is now routinely included in clinical trials as an outcome measure, and is moving closer to clinical practice. NfL will likely be important as a prognostic biomarker to monitor MS patients for progression, disease activity, and treatment efficacy.45 At the molecular level, demyelination leads to dysfunction and anomalous distribution of ion channels along the axons. One consequence of aberrant function of ion channels is accumulation of intra-axonal calcium, which may stimulate catabolism and trigger intra-axonal proteolytic degradation.46–48 Altered ion channel distribution is difficult to detect in clinical practice, but a few MRI studies in MS patients have demonstrated that the tissue sodium concentrations is elevated in acute and chronic lesions compared to are-as of extralesional white matter, suggesting widespread or focal ion imbalance.49,50
At the metabolic level, myelin contributes to axon and neuron survival.51 In addition, astrocytes transfer metabolites to oligodendrocytes, which in turn support neuroaxonal metabolism.52 These metabolic changes can be studied using MR spectroscopy and PET, though their applications are currently limited to small samples in proof-of-concept studies; broader use would require standardization in acquisition and processing and substantial improvements in signal-to-noise ratio.53
While cellular, molecular, and metabolic mechanisms of neuroaxonal damage are still difficult to measure, the resulting global and regional brain atrophy — detectable from early in the disease course — has been associated with a higher risk of progressive disability accumulation. In particular, accelerated brain atrophy has been associated with long-term disability progression independent of relapse activity (so-called “silent progression”).54 Atrophy indices have been utilized as primary outcome measures in phase 2 clinical trials for progressive MS. Brain and spinal cord volume measurements are beginning to be available for clinical practice and will benefit from standardized acquisition protocols and analysis methods (Table 1).55 Axon loss, mostly from inflammatory demyelination in the optic nerve, is reflected in thinning of the retinal nerve fibre and ganglion cell layers on optical coherence tomography (OCT), which is in turn correlated with brain atrophy and disability accumulation (Fig. 1).56
Molecular mechanisms of injury: Oxidative stress and mitochondrial dysfunction
Oxidative stress and mitochondrial dysfunction contributing to glial and neuronal injury, axonal energy failure, and loss of neuronal network function may be key molecular mechanisms driving disease progression. High levels of oxidative stress in the CNS, as determined by lipid peroxides, their breakdown aldehydes, and oxidized DNA, can induce axon, neuron, dendrite, and oligodendroglia injury in MS lesions.57–59 Excessive iron deposition in CNS parenchyma has been hypothesized to be a source of oxidative stress in MS, and iron has been noted to accumulate in deep grey matter nuclei by susceptibility-based MRI as well as in macrophages and microglia in the rim of mixed active and inactive lesions.60 The pro-oxidative environment is ag-gravated by relative deficiency of protective brain glutathione in progressive MS, as potentially detected in vivo by glutathione spectroscopy.61
Mitochondria are also perturbed in MS. Following demyelination, mitochondria move from the cell soma to the demyelinated axon; however, the peak of this potentially beneficial mitochondrial response is only reached after axonal degeneration has been begun.62 Chronic demyelination, iron accumulation, and oxidative injury may further produce dysfunctional mitochondria, which accumulate over the disease course.63 Dysfunctional kinesins (motor proteins responsible for axonal transport of mitochondria) also impair export of mitochondria from the soma into the axon, further contributing to axonal energy failure and injury. In autopsies of progressive MS cases, the density of respiratory complex IV-deficient neurons is elevated throughout the grey matter, and there are multiple deletions of mitochondrial DNA in individual neurons resembling those seen with aging. Dysfunctional mitochondria may not complete oxidative phosphorylation, leading to energy failure, a state of “virtual hypoxia,” and amplification of oxidative injury through electron leakage in axons and neurons, which may contribute to neuronal network failure and disease progression.64
Energy failure can in principle be assayed in vivo using MR spectroscopy, but a combination of laboratory and imaging techniques that can reliably assess ongoing oxidative injury and mitochondrial dysfunction in lesions is needed (Table 1). As such, evidence of associations between molecular mechanisms of injury and MS progression mostly comes from small proof-of-concept studies, and standardization of methods will be necessary for implementation in clinical trials and practice.
Failure of Compensatory Mechanisms
Remyelination
Myelin is required for saltatory conduction of action potentials, supplying trophic factors for axons and protecting them against the inflammatory milieu. Remyelination is a spontaneous repair process in which new myelin sheaths are formed after a demyelinating event (Fig. 1).21,65 Repaired compared to native myelin is characterized by shorter and thinner myelin sheaths, resulting in slower action potential conduction.66,67 The extent of remyelination varies across and within individuals and may be influenced by lesion location, extent and composition of inflammation, age, genetic factors, disease duration, and potentially other factors to be identified.68,69 A high proportion of remyelinated lesions is associated with slower disease progression.37,70 MRI studies suggest that remyelination starts quickly after the onset of demyelination and continues over approximately six months.71 Whether remyelination continues beyond six months is uncertain but of tremendous interest.
In animal models of demyelination, proliferation and migration of oligodendrocyte progenitor cells (OPC) and their differentiation into mature myelinating oligodendrocytes are required for successful remyelination. In inactive as well as mixed active and inactive lesions OPC remain present, albeit in reduced numbers and uneven distribution, whereas mature oligodendrocytes are almost completely lost.72,73 These findings suggest that impaired oligodendrocyte differentiation contributes to remyelination failure in progressive MS.66,67 Recent studies suggest that not only OPC, but also mature oligodendrocytes, may contribute to successful lesion remyelination and that the reasons for remyelination failure in MS may be diverse and dependent on disease duration, lesion stage, and lesion location.31,65,74,75
Several methods can assess remyelination clinically though are not routinely used in clinical practice (Table 1). Longitudinal voxel-based magnetization transfer MRI has been used to quantify remyelination in several clinical trials; however, inflammation, oedema, and axon loss may also influence the measurement.76 T1 mapping at 7-tesla MRI allows partial differentiation of demyelinated and remyelinated white matter lesions (Fig. 1).77 Myelin water fraction imaging is another technique currently used to identify myelin changes in the human brain.78 Radiotracers that label amyloid (e.g., [11C]PIB) are sensitive to myelin, and longitudinal data raise the possibility that this method allows detection of both demyelination and remyelination in MS.70,79 Visual evoked potentials (VEP) have been extensively used to assess demyelination and remyelination, both in clinical practice and as a primary outcome in proof-of-concept clinical trials evaluating the potential of remyelination-promoting compounds.80
Neuroplasticity
Neuroplasticity and functional reorganization in response to damage are intrinsic properties of the CNS. Mechanisms include molecular changes, synaptogenesis, alteration of synaptic function, and dendrite and axon sprouting. Reorganization of neural networks can be demonstrated in persons with MS by task-oriented and resting state functional MRI (fMRI) (Fig. 1, Table 1). Motor, sensory, visual, and particularly cognitive functions (processing speed and efficiency, attention, memory, and executive function) are associated with widespread and bilateral brain activation in MS, especially with longer disease duration and more severe disability, compared to healthy controls.81 Acute and chronic inflammation not only cause CNS damage, which stimulates reorganization, but also probably interferes with the processes required for functional reorganization.42 Preservation of functional connectivity also depends on cognitive reserve, despite accumulation of structural damage, suggesting that such reserve can directly affect neuroplasticity potential.82 The magnitude of functional reorganization correlates with extent of lesional and extralesional damage. In patients with preserved motor function, greater lesion volume and microstructural damage are associated with widespread activation of brain areas, suggesting that reorganization is compensatory. However, the degree of recovery relates to the specific pattern of functional changes, indicating that compensation might in some instances be maladaptive.42
The severity of MS-related CNS damage as assessed clinically and by MRI is an important factor affecting quantitative and qualitative aspects of functional reorganization, interacting with age at disease onset, disease duration, and disease-modifying therapy.83 Other important factors, such as age, sex, comorbidities, and health behaviours (e.g., smoking and exercise) influence the capacity for compensatory reorganization.84–86
One explanation for the emergence of progressive disability worsening in MS is the accumulation of irreversible damage exceeding the capacity of the CNS to compensate. Future longitudinal studies should integrate fMRI findings with clinical and neuropsychological measures and other methods that assess neural networks structurally (e.g., diffusion tensor imaging and ultrahigh-field anatomic imaging) and functionally (e.g., magnetoencephalography). Aspects of fMRI acquisition parameters and analysis need to be refined and standardized. Additionally, a better understanding of the network characteristics that are most clinically relevant is required.42 Most importantly, among the changes associated with motor and cognitive disability worsening, it is critical to distinguish those that are clinically irrelevant, those that are appropriate but inadequate to compensate for accumulating CNS damage, and those that are maladaptive.
The Role of Aging in MS
Older chronological age is robustly associated with non-relapse related progression. Progressive MS is very rare in children, and progression from onset occurs in <1% of children vs. ~10% of adults diagnosed with MS.87 In adults, older age at diagnosis is associated with faster accumulation of ambulatory disability, a defining feature of progressive MS as currently described, as well as greater cognitive impairment.88,89
The prototypical biological marker of aging is telomere attrition. Leukocyte telomere length is a reliable marker of telomere length from different cell types throughout the body.90 In a cohort of over 500 MS cases, leukocyte telomere length attrition was associated with higher disability in both cross-sectional and longitudinal analyses independent of disease duration and chronological age.91 While current linkage of telomere shortening to MS subtype is only associative, there is strong biological plausibility that processes downstream of telomere attrition including the DNA damage response and cellular senescence contribute to disability progression. Intriguingly, immunosenescence of lymphocyte subsets has been linked to MS pathology.92–94
Senescence of different CNS cell subtypes, which might be accelerated due to the disease itself, may also impact progression. Senescent microglia may both promote chronic secretion of inflammatory cytokines and contribute to an inhibitory environment for remyelination due to their decreased phagocytic activity. Senescent astrocytes are detrimental to synaptic plasticity, blood-brain-barrier function, and the metabolic balance of neighbouring neurons.95,96 Overall, aging has been associated with declining neural plasticity and less capacity for functional recovery from inflammatory injury. By contrast, better physical outcome from MS attacks in children has been attributed to high functional reserve and capacity for plasticity.97
Systemic aging also leads to increased burden of comorbid illnesses, including vascular disease, which may further hasten development of MS ambulatory disability.98 While the mechanisms by which vascular disease worsens progression are not fully elucidated, injury to brain white matter is a likely contributor.99
Reproductive aging may also affect MS progression. Whereas women are at increased risk for developing MS, men with MS may have earlier and faster disability development.88,89 Several studies suggest that progressive MS pathology and disability accelerate in the perimenopausal period.100 Potential mechanisms for an association of ovarian functional decline with progressive MS pathology include the loss of neuroprotective effects of oestrogens and immune changes in the perimenopausal period. Loss of sex-specific steroid production may explain the phenomenon of women appearing to catch up in disability to men in later decades of life.
Conclusions and future directions
Despite substantial gains in knowledge of MS pathophysiology and the proliferation of treatments to forestall MS relapses, halting and reversing disease progression remain unmet needs. To address these needs, it is critical to move from clinically to biologically based definitions of MS progression and to develop and validate tools that can reliably assess and track relevant disease biology in clinical settings. Data suggest that disability progression is not caused by one uniform disease mechanism but instead results from a combination of several mechanisms, which play out variably across patients and within individual patients over time (Fig. 2). Indeed, over time, mechanisms of injury such as those discussed in this paper (nonresolving inflammation, neurodegeneration, oxidative stress, and mitochondrial dysfunction) can occur separately or in various combinations in the same individual, and together with failure of compensatory mechanisms (e.g., remyelination and neuroplasticity), all interacting with aging, define the clinical picture across the disease course. The field must develop methods to identify and quantify these mechanisms, minimally invasively and on the patient level, and incorporate the relevant measures into both clinical trials and clinical practice. Achieving this goal will require correlative clinical-radiological-pathological studies of people with fast versus slow disease progression independent of relapses and active lesions on MRI, as well as longitudinal studies correlating imaging and other paraclinical tools with disease progression as measured using state-of-the-art techniques (e.g., clinical, cognitive, and digital tools, as well as blood and CSF biomarkers).101–104
Figure 2. Assessments relevant to a mechanism-driven framework for MS progression.
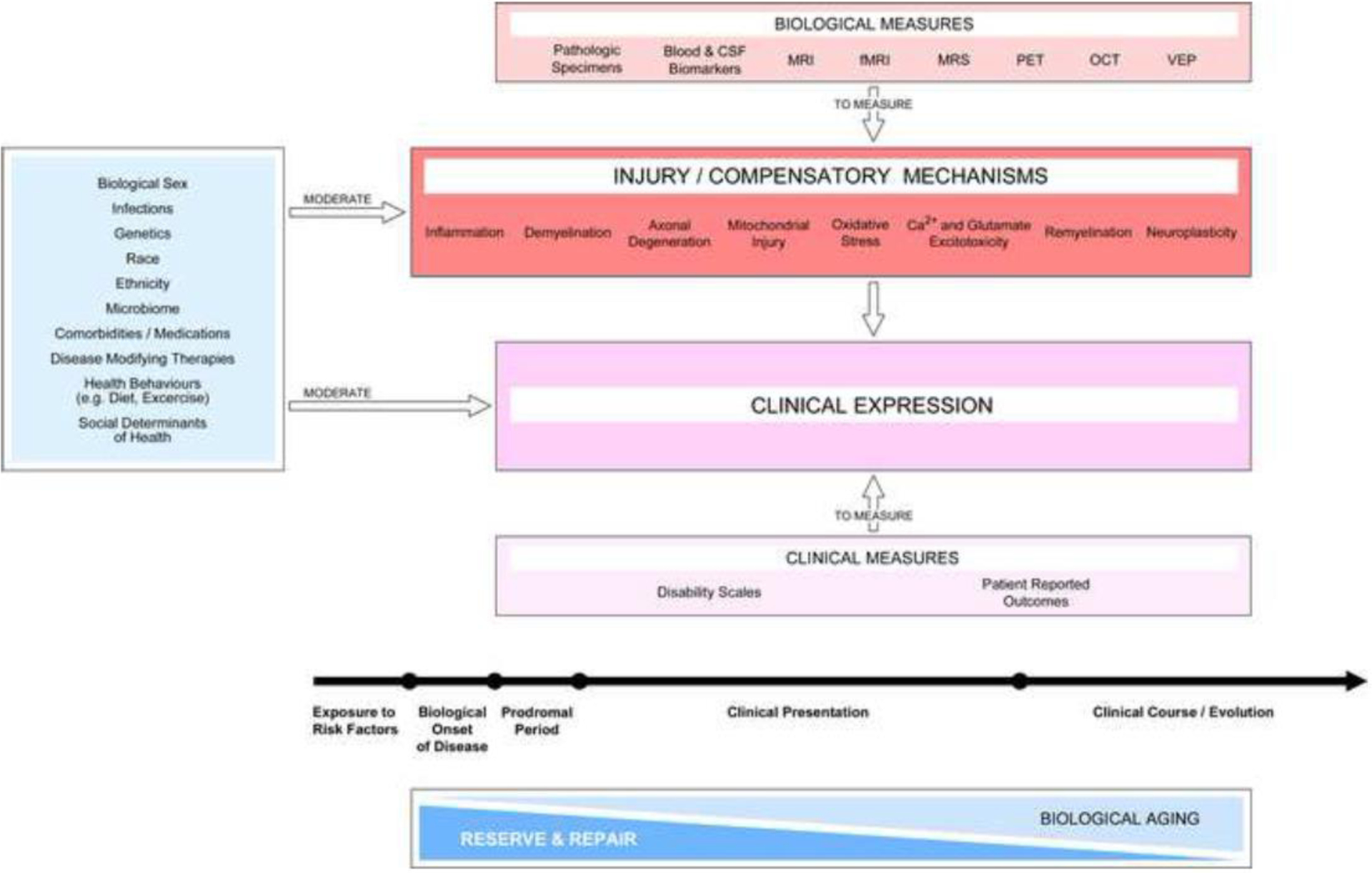
MS progression reflects a combination of mechanisms of injury and compensation (red box) that exist contemporaneously and contribute to clinical expression. The activation of these mechanisms marks the biological onset of the disease and initiate the prodromal period. The balance of such mechanisms, together with tissue repair, jointly determine clinical expression during the whole disease course. The age-associated decrease in reserve and repair capacity also influences clinical progression. Development of clinical and biological measures with high sensitivity and specificity is required to continuously monitor the clinical presentation of the disease and identify relevant injury and compensatory mechanisms in individuals. Potential mediators (light blue box on the left) exert positive and negative influences on injury and compensatory mechanisms and thus impact clinical expression over the whole disease course. The list of mediators is illustrative rather than comprehensive. Abbreviations: CSF, cerebrospinal fluid; MRI, magnetic resonance imaging, fMRI, functional MRI; MRS, magnetic resonance spectroscopy; PET, positron emission tomography; OCT, optical coherence tomography; VEP, visual evoked potentials.
In keeping with current trends throughout medicine, we envision a future where clinical benefit accrues directly from biomarker-based, biologically informed treatment decisions. The concepts described in this paper are a first step towards a new framework that eliminates the current phenomenological classification of patients into RR, SP, and PP descriptors.3 However, until a deeper understanding of underlying mechanisms and how they interact to drive progression is achieved, we expect that any new framework will require additional modification over time. Adoption of biologically based definitions of MS progression will be operationally challenging, as the existing descriptors are deeply embedded in clinical research and healthcare ecosystems. Patients rely on the current descriptors to understand their disease journey and inform healthcare decisions. In addition, regulatory authorities have integrated the descriptors, albeit with complicated and differing interpretations, in approval documents for MS treatments. As such, ensuring a smooth transition from the current state to a future framework is nontrivial but critical given its importance for patients.
The authors of this paper are cognizant that a new framework, albeit necessary for developing biologically based treatment approaches and algorithms, would require validation in clinical and research settings. Coordinated efforts of stakeholders (e.g., researchers, funders, health authorities, and patient organizations) will be key. Focused efforts will then be needed to integrate the new framework into clinical trials and practice and to transition away from the legacy framework used by regulatory agencies and health authorities for drug approvals. Comprehensive patient education efforts will also be required. As such, development of any roadmap for implementation will be a key future focus of the International Advisory Committee on Clinical Trials in MS.
Search strategy and selection criteria
References for this Review were identified by searches of English articles in PubMed between 01.01.2012 and 01.04.2022 and references from relevant articles. The search terms “multiple sclerosis”, “inflammation”, “neurodegeneration”, “mitochondrial dysfunction”, “oxidative stress”, and “remyelination”, “neuronal networks”, “neural plasticity”, “aging”, “imaging”, and “OCT” were used. The final reference list was generated on the basis of relevance to the topics covered in this Review.
Supplementary Material
Acknowledgment
This study was supported by the German Research Foundation (SFB128 B07, Ku1477/11-1) to TK and the Intramural Research Program of NINDS/NIH to DSR. We thank Antonio Carotenuto (Federico II University of Naples, Italy) for help with the figures. AJT acknowledges support from the UCL/UCLH NIHR Biomedical Research Centre. The work of the International Advisory Committee on Clinical Trials in MS is jointly sponsored by the European Committee for Treatments and Research in MS and the National Multiple Sclerosis Society (USA).
Declaration of interests
TK received research funding from the German Research Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Münster, National MS Society, European Leukodystrophy Association, Progressive MS Alliance, European Commission (H2020-MSCA-ITN-2018) and Novartis. She received compensation for serving on scientific advisory boards (Novartis) and speaker honoraria from Novartis and Roche.
MM has received research grants from MAGNIMS-ECTRIMS, UK MS Society, and Merck; consulting fees from Ipsen, BMS Celgene, Biogen, Sanofi-Genzyme, Roche, and Merck; honoraria for lectures from Merck, Roche, and Sanofi-Genzyme; support for attending meetings from Merck, Biogen, and Sanofi-Genzyme.
TC is an employee of one of the sponsors (National Multiple Sclerosis Society) of the International Advisory Committee on Clinical Trials in MS.
J.Coh has received personal compensation for consulting for Biogen, Bristol-Myers Squibb, Convelo, Genentech, Janssen, NervGen, Novartis, and PSI; speaking for H3 Communications; and serving as an Editor of Multiple Sclerosis Journal.
J. Cor. has received grants or contracts from Biogen, Merck, UC San Francisco, payments or honoraria for lectures, speaker bureaus or presentations from Biogen, Merck, Sanofi Genzyme, Novartis, Bristol-Myers and Roche, participation on data safety Monitoring Boards or Advisory Board from Novartis, Merck, Sanofi Genzyme and Biogen.
JG over the past year has received grant/contract research support from the National MS Society, Biogen, and Octave Biosciences. She serves on a steering committee for a trial supported by Novartis. She has received speaker fees from Alexion and BMS and served on an advisory board for Genentech.
XM received speaking honoraria and travel expenses for participation in scientific meetings, has been a steering committee member of clinical trials or participated in advisory boards of clinical trials in the past years with Abbvie, Actelion, Alexion, Bayer, Biogen, Bristol-Myers Squibb/Celgene, EMD Serono, Genzyme, Hoffmann-La Roche, Immunic, Janssen Pharmaceuticals, Medday, Merck, Mylan, Nervgen, Novartis, Sanofi-Genzyme, Teva Pharmaceutical, TG Therapeutics, Excemed, MSIF and NMSS.
RAM receives research funding from Biogen Idec and Roche. She is the chair of the Medical Advisory Committee of the MS Society of Canada.
VWY is funded by research grants from the MS Society of Canada, the Canadian Institutes of Health Research, Canadian Cancer Society, and Genentech. He has received speaker honoraria from Biogen, EMD Serono, Novartis, Roche and Sanofi-Genzyme. He is the recipient of unrestricted educational grants from Biogen, EMD Serono, Novartis, Roche, Sanofi-Genzyme and Teva Canada to support educational activities of the Alberta MS Network, which he directs. AJT reports personal fees as an editorial board member for The Lancet Neurology receiving a free subscription; is Editor-in- Chief for Multiple Sclerosis Journal receiving an honorarium from SAGE Publications; receive support from the UCL/UCLH NIHR Biomedical Research Centre; and receives support for travel as Chair, Scientific Advisory Committee, International Progressive MS Alliance, from the National MS Society (USA) as member, NMSS Research Programs Advisory Committee and as a Board member of the European Charcot Foundation. In the last 36 months, AJT has received payment (paid to the institution) from Eisai Ltd and from the German Aerospace Center, Health Research (ERA-NET NEURON). He has received fees or support for travel from Hoffman La Roche, Novartis and CanProCo SAB; and honoraria or support for travel from EXCEMED and Almirall. In November 2019 he received support for travel to PACTRIMS and in December 2019, he received support for travel to the MS Society of Canada. His unpaid roles include as a Guarantor of BRAIN; Trustee of the National Brain Appeal (National Hospital for Neurology & Neurosurgery); and as Chair of the Scientific Ambassadors, ‘STOP MS’ Appeal Board (UK MS Society).
DSR reports personal fees from Bounds Law Group, LLC, grants from Vertex, grants from Sanofi-Genzyme, grants from Abata Therapeutics, outside the submitted work; In addition, Dr. Reich has a patent System and method of automatically detecting tissue abnormalities (US Patent 9,607,392) issued, a patent Method of analyzing multi-sequence MRI data for analyzing brain abnormalities in a subject (US Patent 9,888,876) issued, a patent Automatic identification of subjects at risk of multiple sclerosis (US Patent application 16/254,710) issued, and a patent High-resolution cerebrospinal fluid-suppressed T2*-weighted magnetic resonance imaging of cortical lesions (US Patent application 62/838,578) pending.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol 2017; 13(1): 25–36. [DOI] [PubMed] [Google Scholar]
- 2.Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996; 46(4): 907–11. [DOI] [PubMed] [Google Scholar]
- 3.Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014; 83(3): 278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med 2017; 376(3): 209–20. [DOI] [PubMed] [Google Scholar]
- 5.Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 2018; 391(10127): 1263–73. [DOI] [PubMed] [Google Scholar]
- 6.Fitzgerald KC, Kim K, Smith MD, et al. Early complement genes are associated with visual system degeneration in multiple sclerosis. Brain 2019; 142: 2722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magliozzi R, Howell OW, Nicholas R, et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann Neurol 2018; 83(4): 739–55. [DOI] [PubMed] [Google Scholar]
- 8.Moccia M, van de Pavert S, Eshaghi A, et al. Pathologic correlates of the magnetization transfer ratio in multiple sclerosis. Neurology 2020; 95(22): e2965–e76. [DOI] [PubMed] [Google Scholar]
- 9.Absinta M, Maric D, Gharagozloo M, et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yong HYF, Yong VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nature reviews Neurology 2022; 18(1): 40–55. [DOI] [PubMed] [Google Scholar]
- 11.Lublin FD, Haring DA, Ganjgahi H, et al. How patients with multiple sclerosis acquire disability. Brain 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kappos L, Wolinsky JS, Giovannoni G, et al. Contribution of Relapse-Independent Progression vs Relapse-Associated Worsening to Overall Confirmed Disability Accumulation in Typical Relapsing Multiple Sclerosis in a Pooled Analysis of 2 Randomized Clinical Trials. JAMA Neurol 2020; 77(9): 1132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fransen NL, Crusius JBA, Smolders J, et al. Post-mortem multiple sclerosis lesion pathology is influenced by single nucleotide polymorphisms. Brain Pathol 2020; 30(1): 106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vandebergh M, Andlauer TFM, Zhou Y, et al. Genetic Variation in WNT9B Increases Relapse Hazard in Multiple Sclerosis. Annals of neurology 2021; 89(5): 884–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lublin FD, Coetzee T, Cohen JA, Marrie RA, Thompson AJ, MS IACoCTi. The 2013 clinical course descriptors for multiple sclerosis: A clarification. Neurology 2020; 94(24): 108892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eshaghi A, Young AL, Wijeratne PA, et al. Identifying multiple sclerosis subtypes using unsupervised machine learning and MRI data. Nat Commun 2021; 12(1): 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Meo E, Portaccio E, Giorgio A, et al. Identifying the Distinct Cognitive Phenotypes in Multiple Sclerosis. JAMA Neurol 2021; 78(4): 414–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsagkas C, Parmar K, Pezold S, et al. Classification of multiple sclerosis based on patterns of CNS regional atrophy covariance. Hum Brain Mapp 2021; 42(8): 2399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Absinta M, Sati P, Reich DS. Advanced MRI and staging of multiple sclerosis lesions. Nat Rev Neurol 2016; 12(6): 358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solomon AJ. Progress towards a diagnostic biomarker for MS: Central vein sign. Multiple sclerosis (Houndmills, Basingstoke, England) 2020; 26(4): 394–6. [DOI] [PubMed] [Google Scholar]
- 21.Frischer JM, Weigand SD, Guo Y, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015; 78(5): 710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodini B, Chard D, Altmann DR, et al. White and gray matter damage in primary progressive MS: The chicken or the egg? Neurology 2016; 86(2): 170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016; 387(10023): 1075–84. [DOI] [PubMed] [Google Scholar]
- 24.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018; 17(2): 162–73. [DOI] [PubMed] [Google Scholar]
- 25.Fransen NL, Hsiao CC, van der Poel M, et al. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain 2020; 143(6): 1714–30. [DOI] [PubMed] [Google Scholar]
- 26.Schirmer L, Velmeshev D, Holmqvist S, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019; 573(7772): 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Absinta M, Maric D, Gharagozloo M, et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021; 597(7878): 709–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wheeler MA, Clark IC, Tjon EC, et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020; 578(7796): 593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dal-Bianco A, Grabner G, Kronnerwetter C, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol 2017; 133(1): 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Absinta M, Sati P, Masuzzo F, et al. Association of Chronic Active Multiple Sclerosis Lesions With Disability In Vivo. JAMA Neurol 2019; 76(12): 1474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dal-Bianco A, Grabner G, Kronnerwetter C, et al. Long-term evolution of multiple sclerosis iron rim lesions in 7 T MRI. Brain 2021; 144(3): 833–47. [DOI] [PubMed] [Google Scholar]
- 32.Zinger N, Ponath G, Sweeney E, et al. Dimethyl Fumarate Reduces Inflammation in Chronic Active Multiple Sclerosis Lesions. Neurol Neuroimmunol Neuroinflamm 2022; 9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elliott C, Wolinsky JS, Hauser SL, et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult Scler 2019; 25(14): 1915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lassmann H. Multiple Sclerosis Pathology. Cold Spring Harb Perspect Med 2018; 8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011; 134(Pt 9): 2755–71. [DOI] [PubMed] [Google Scholar]
- 36.Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005; 128(Pt 11): 2705–12. [DOI] [PubMed] [Google Scholar]
- 37.Luchetti S, Fransen NL, van Eden CG, Ramaglia V, Mason M, Huitinga I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol 2018; 135(4): 511-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Högel H, Rissanen E, Vuorimaa A, Airas L. Positron emission tomography imaging in evaluation of MS pathology in vivo. Mult Scler 2018; 24(11): 1399–412. [DOI] [PubMed] [Google Scholar]
- 39.Kaunzner UW, Kang Y, Zhang S, et al. Quantitative susceptibility mapping identifies inflammation in a subset of chronic multiple sclerosis lesions. Brain 2019; 142(1): 133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaunzner UW, Kang Y, Monohan E, et al. Reduction of PK11195 uptake observed in multiple sclerosis lesions after natalizumab initiation. Mult Scler Relat Disord 2017; 15: 27–33. [DOI] [PubMed] [Google Scholar]
- 41.Sucksdorff M, Rissanen E, Tuisku J, et al. Evaluation of the Effect of Fingolimod Treatment on Microglial Activation Using Serial PET Imaging in Multiple Sclerosis. J Nucl Med 2017; 58(10): 1646–51. [DOI] [PubMed] [Google Scholar]
- 42.Chard DT, Alahmadi AAS, Audoin B, et al. Mind the gap: from neurons to networks to outcomes in multiple sclerosis. Nat Rev Neurol 2021; 17(3): 173–84. [DOI] [PubMed] [Google Scholar]
- 43.Zoupi L, Booker SA, Eigel D, et al. Selective vulnerability of inhibitory networks in multiple sclerosis. Acta Neuropathol 2021; 141(3): 415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018; 14(10): 577–89. [DOI] [PubMed] [Google Scholar]
- 45.Bittner S, Oh J, Havrdova EK, Tintore M, Zipp F. The potential of serum neurofilament as biomarker for multiple sclerosis. Brain 2021; 144(10): 2954–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci 2005; 233(1–2): 3–13. [DOI] [PubMed] [Google Scholar]
- 47.Witte ME, Schumacher AM, Mahler CF, et al. Calcium Influx through Plasma-Membrane Nanoruptures Drives Axon Degeneration in a Model of Multiple Sclerosis. Neuron 2019; 101(4): 615–24 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Estacion M, Vohra BP, Liu S, et al. Ca2+ toxicity due to reverse Na+/Ca2+ exchange contributes to degeneration of neurites of DRG neurons induced by a neuropathy-associated Nav1.7 mutation. J Neurophysiol 2015; 114(3): 1554–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eisele P, Kraemer M, Dabringhaus A, et al. Characterization of chronic active multiple sclerosis lesions with sodium ((23) Na) magnetic resonance imaging-preliminary observations. Eur J Neurol 2021; 28(7): 2392–5. [DOI] [PubMed] [Google Scholar]
- 50.Inglese M, Madelin G, Oesingmann N, et al. Brain tissue sodium concentration in multiple sclerosis: a sodium imaging study at 3 tesla. Brain 2010; 133(Pt 3): 847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Funfschilling U, Supplie LM, Mahad D, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012; 485(7399): 517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown AM, Baltan Tekkök S, Ransom BR. Energy transfer from astrocytes to axons: the role of CNS glycogen. Neurochem Int 2004; 45(4): 529–36. [DOI] [PubMed] [Google Scholar]
- 53.Moccia M, Ciccarelli O. Molecular and Metabolic Imaging in Multiple Sclerosis. Neuroimaging Clin N Am 2017; 27(2): 343–56. [DOI] [PubMed] [Google Scholar]
- 54.Cree BAC, Hollenbach JA, Bove R, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol 2019; 85(5): 653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wattjes MP, Ciccarelli O, Reich DS, et al. 2021 MAGNIMS-CMSC-NAIMS consensus recommendations on the use of MRI in patients with multiple sclerosis. Lancet Neurol 2021; 20(8): 653–70. [DOI] [PubMed] [Google Scholar]
- 56.Sotirchos ES, Gonzalez Caldito N, Filippatou A, et al. Progressive Multiple Sclerosis Is Associated with Faster and Specific Retinal Layer Atrophy. Ann Neurol 2020; 87(6): 885–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lassmann H, van Horssen J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim Biophys Acta 2016; 1862(3): 506–10. [DOI] [PubMed] [Google Scholar]
- 58.Haider L, Zrzavy T, Hametner S, et al. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016; 139(Pt 3): 807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haider L, Simeonidou C, Steinberger G, et al. Multiple sclerosis deep grey matter: the relation between demyelination, neurodegeneration, inflammation and iron. J Neurol Neurosurg Psychiatry 2014; 85(12): 1386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Filippi M, Bruck W, Chard D, et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol 2019; 18(2): 198–210. [DOI] [PubMed] [Google Scholar]
- 61.Choi IY, Lee P, Adany P, et al. In vivo evidence of oxidative stress in brains of patients with progressive multiple sclerosis. Mult Scler 2018; 24(8): 1029–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Licht-Mayer S, Campbell GR, Canizares M, et al. Enhanced axonal response of mitochondria to demyelination offers neuroprotection: implications for multiple sclerosis. Acta Neuropathol 2020; 140(2): 143–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campbell G, Mahad DJ. Mitochondrial dysfunction and axon degeneration in progressive multiple sclerosis. FEBS Lett 2018; 592(7): 1113–21. [DOI] [PubMed] [Google Scholar]
- 64.Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 2015; 14(2): 183–93. [DOI] [PubMed] [Google Scholar]
- 65.Heß K, Starost L, Kieran NW, et al. Lesion stage-dependent causes for impaired remyelination in MS. Acta Neuropathol 2020; 140(3): 359–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stangel M, Kuhlmann T, Matthews PM, Kilpatrick TJ. Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat Rev Neurol 2017; 13(12): 742–54. [DOI] [PubMed] [Google Scholar]
- 67.Franklin RJM, Ffrench-Constant C. Regenerating CNS myelin - from mechanisms to experimental medicines. Nat Rev Neurosci 2017; 18(12): 753–69. [DOI] [PubMed] [Google Scholar]
- 68.Goldschmidt T, Antel J, Konig FB, Brück W, Kuhlmann T. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 2009; 72(22): 1914–21. [DOI] [PubMed] [Google Scholar]
- 69.Chang A, Staugaitis SM, Dutta R, et al. Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol 2012; 72(6): 918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bodini B, Veronese M, Garcia-Lorenzo D, et al. Dynamic imaging of individual remyelination profiles in multiple sclerosis. Ann Neurol 2016; 79: 726–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen JT, Collins DL, Atkins HL, Freedman MS, Arnold DL. Magnetization transfer ratio evolution with demyelination and remyelination in multiple sclerosis lesions. Ann Neurol 2008; 63(2): 254–62. [DOI] [PubMed] [Google Scholar]
- 72.Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. New Engl J Med 2002; 346: 165–73. [DOI] [PubMed] [Google Scholar]
- 73.Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Brück W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008; 131: 1749–58. [DOI] [PubMed] [Google Scholar]
- 74.Yeung M, Djelloul M, Steiner E, et al. Dynamics of oligodendrocyte generation in multiple sclerosis. Nature 2019; 566(7745): 538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jakel S, Agirre E, Mendanha Falcao A, et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019; 566(7745): 543–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cadavid D, Mellion M, Hupperts R, et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2019; 18(9): 845–56. [DOI] [PubMed] [Google Scholar]
- 77.Kolb H, Absinta M, Beck ES, et al. 7T MRI Differetiates Remyelinated from Demyelinated Multiple Sclerosis Lesions. Ann Neurol 2021; 90: 612–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elkady AM, Wu Z, Leppert IR, Arnold DL, Narayanan S, Rudko DA. Assessing the differential sensitivities of wave-CAIPI ViSTa myelin water fraction and magnetization transfer saturation for efficiently quantifying tissue damage in MS. Mult Scler Relat Disord 2021; 56: 103309. [DOI] [PubMed] [Google Scholar]
- 79.Auvity S, Tonietto M, Caillé F, et al. Repurposing radiotracers for myelin imaging: a study comparing 18F-florbetaben, 18F-florbetapir, 18F-flutemetamol,11C-MeDAS, and 11C-PiB. Eur J Nucl Med Mol Imaging 2020; 47(2): 490–501. [DOI] [PubMed] [Google Scholar]
- 80.Green AJ, Gelfand JM, Cree BA, et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet 2017; 390(10111): 2481–9. [DOI] [PubMed] [Google Scholar]
- 81.Rimkus CM, Schoonheim MM, Steenwijk MD, et al. Gray matter networks and cognitive impairment in multiple sclerosis. Multiple sclerosis (Houndmills, Basingstoke, England) 2019; 25(3): 382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fuchs TA, Benedict RHB, Bartnik A, et al. Preserved network functional connectivity underlies cognitive reserve in multiple sclerosis. Hum Brain Mapp 2019; 40(18): 5231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rocca MA, Colombo B, Falini A, et al. Cortical adaptation in patients with MS: a cross-sectional functional MRI study of disease phenotypes. Lancet Neurol 2005; 4(10): 618–26. [DOI] [PubMed] [Google Scholar]
- 84.Schoonheim MM, Hulst HE, Landi D, et al. Gender-related differences in functional connectivity in multiple sclerosis. Mult Scler 2012; 18(2): 164–73. [DOI] [PubMed] [Google Scholar]
- 85.Sumowski JF, Wylie GR, Deluca J, Chiaravalloti N. Intellectual enrichment is linked to cerebral efficiency in multiple sclerosis: functional magnetic resonance imaging evidence for cognitive reserve. Brain 2010; 133(Pt 2): 362–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sandroff BM, Jones CD, Baird JF, Motl RW. Systematic Review on Exercise Training as a Neuroplasticity-Inducing Behavior in Multiple Sclerosis. Neurorehabil Neural Repair 2020; 34(7): 575–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harding KE, Liang K, Cossburn MD, et al. Long-term outcome of paediatric-onset multiple sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 2013; 84(2): 141–7. [DOI] [PubMed] [Google Scholar]
- 88.Kalincik T, Vivek V, Jokubaitis V, et al. Sex as a determinant of relapse incidence and progressive course of multiple sclerosis. Brain 2013; 136(Pt 12): 3609–17. [DOI] [PubMed] [Google Scholar]
- 89.Confavreux C, Vukusic S. Age at disability milestones in multiple sclerosis. Brain 2006; 129(Pt 3): 595–605. [DOI] [PubMed] [Google Scholar]
- 90.Demanelis K, Jasmine F, Chen LS, et al. Determinants of telomere length across human tissues. Science 2020; 369(6509). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krysko KM, Henry RG, Cree BAC, et al. Telomere Length Is Associated with Disability Progression in Multiple Sclerosis. Ann Neurol 2019; 86(5): 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Claes N, Fraussen J, Vanheusden M, et al. Age-Associated B Cells with Proinflammatory Characteristics Are Expanded in a Proportion of Multiple Sclerosis Patients. J Immunol 2016; 197(12): 4576–83. [DOI] [PubMed] [Google Scholar]
- 93.Thewissen M, Linsen L, Somers V, Geusens P, Raus J, Stinissen P. Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann N Y Acad Sci 2005; 1051: 255–62. [DOI] [PubMed] [Google Scholar]
- 94.Thewissen M, Somers V, Venken K, et al. Analyses of immunosenescent markers in patients with autoimmune disease. Clin Immunol 2007; 123(2): 209–18. [DOI] [PubMed] [Google Scholar]
- 95.Correale J, Farez MF. The Role of Astrocytes in Multiple Sclerosis Progression. Front Neurol 2015; 6: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oost W, Talma N, Meilof JF, Laman JD. Targeting senescence to delay progression of multiple sclerosis. J Mol Med (Berl) 2018; 96(11): 1153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rocca MA, Absinta M, Ghezzi A, Moiola L, Comi G, Filippi M. Is a preserved functional reserve a mechanism limiting clinical impairment in pediatric MS patients? Hum Brain Mapp 2009; 30(9): 2844–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marrie RA, Rudick R, Horwitz R, et al. Vascular comorbidity is associated with more rapid disability progression in multiple sclerosis. Neurology 2010; 74(13): 1041–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Geraldes R, Esiri MM, DeLuca GC, Palace J. Age-related small vessel disease: a potential contributor to neurodegeneration in multiple sclerosis. Brain Pathol 2017; 27(6): 707-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Graves JS, Henry RG, Cree BAC, et al. Ovarian aging is associated with gray matter volume and disability in women with MS. Neurology 2018; 90(3): e254–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McKay KA, Bedri SK, Manouchehrinia A, et al. Reduction in Cognitive Processing Speed Surrounding Multiple Sclerosis Relapse. Ann Neurol 2022; 91(3): 417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moccia M, Lanzillo R, Palladino R, et al. Cognitive impairment at diagnosis predicts 10-year multiple sclerosis progression. Multiple sclerosis (Houndmills, Basingstoke, England) 2016; 22(5): 659–67. [DOI] [PubMed] [Google Scholar]
- 103.Dillenseger A, Weidemann ML, Trentzsch K, et al. Digital Biomarkers in Multiple Sclerosis. Brain Sci 2021; 11(11): 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.De Angelis M, Lavorgna L, Carotenuto A, et al. Digital Technology in Clinical Trials for Multiple Sclerosis: Systematic Review. J Clin Med 2021; 10(11): 2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.