

Abstract
Alzheimer's disease (AD) is characterized by the accumulation of pathological amyloid‐β (Aβ) and Tau proteins. According to the prion‐like hypothesis, both proteins can seed and disseminate through brain regions through neural connections and glial cells. The amygdaloid complex (AC) is involved early in the disease, and its widespread connections with other brain regions indicate that it is a hub for propagating pathology. To characterize changes in the AC as well as the involvement of neuronal and glial cells in AD, a combined stereological and proteomic analysis was performed in non‐Alzheimer's disease and AD human samples. The synaptic alterations identified by proteomic data analysis could be related to the volume reduction observed in AD by the Cavalieri probe without neuronal loss. The pathological markers appeared in a gradient pattern with the medial region (cortical nucleus, Co) being more affected than lateral regions, suggesting the relevance of connections in the distribution of the pathology among different brain regions. Generalized astrogliosis was observed in every AC nucleus, likely related to deposits of pathological proteins. Astrocytes might mediate phagocytic microglial activation, whereas microglia might play a dual role since protective and toxic phenotypes have been described. These results highlight the potential participation of the amygdala in the disease spreading from/to olfactory areas, the temporal lobe and beyond. Proteomic data are available via ProteomeXchange with identifier PXD038322.
Keywords: antioxidant protein 2 (AOP2), BM88 antigen (BM88), calpactin II, calpactin‐1 heavy chain (CAL1H), centaurin‐alpha‐1 (CENTA1), endonexin II (ENX2), nuclear chloride ion channel 27 (NCC27)
A combined stereological and proteomic analysis revealed amygdala volume reduction linked to synaptic dysfunction in Alzheimer''s disease samples. The pathological markers gradient suggested the relevance of connections in the distribution of the pathology among different brain regions. Extended astrogliosis, likely as response to pathologic deposits, could mediate phagocytic microglial activation. Microglia might play a dual role since protective and toxic phenotypes have been described. In conclusion, amygdala might promote the spreading of pathology from/to olfactory areas, the temporal lobe and beyond.
1. INTRODUCTION
Alzheimer's disease (AD) is characterized by executive dysfunction and memory impairment [1], with underlying accumulation of extracellular amyloid‐β (Aβ) and intracellular hyperphosphorylated Tau proteins. These two markers form aggregates in a predictable and sequential manner in the different brain regions established as Thal phases [2] and Braak stages [3, 4], respectively. According to the prion‐like hypothesis, both pathological markers can spread from cell to cell throughout brain regions [5, 6]. This premise is in consonance with Braak sequence stages since the affected areas are interconnected [7]. Nevertheless, growing evidence indicates that multiple pathological substrates could be linked to mild cognitive impairment and Alzheimer's clinical syndrome [8, 9]. Recently, limbic‐predominant age‐related TDP‐43 encephalopathy (LATE) has been described as new disease entity characterized by TDP‐43 proteinopathy and Alzheimer's type dementia, being the amygdala involved from early stages [10, 11]. In this sense, the amygdala constitutes a key hub that may contribute to the spread of pathologic molecules because of its vast connectivity with other brain regions [12].
Amygdala atrophy has been described in early stages of the disease [13], and it could be related to certain preclinical symptoms, such as olfactory deficits [14, 15] and/or emotional dysfunctions [16, 17, 18]. Moreover, amygdaloid complex (AC) volume reduction measured with magnetic resonance imaging (MRI) has been proposed as a diagnostic criterion for Alzheimer's disease (AD) [19]. A few histological studies have also confirmed amygdala atrophy [20] accompanied by neuronal and glial loss [21, 22]. However, neither neural nor glial‐specific markers have been employed. Furthermore, the diversity of nomenclature used to identify amygdaloid nuclei together with the lack of consistency in the studied nuclei make it difficult to understand how pathology can affect the AC.
Evidence for glial participation in Aβ and Tau aggregation [23] and propagation [24] has been increasing in recent decades, with special relevance of astrocyte involvement in Tau propagation [25]. Nonetheless, a dual role of glial cells has been postulated since glial‐mediated inflammation might cause damage (propagation) and beneficial effects (pathology clearance) in AD [26]. In this sense, multiple proteomic approaches are now booming with the aim of finding markers of interest. Unfortunately, proteomic analyses in the human amygdala are scarce; either limited to the study of healthy individuals [27] or focused on Aβ extracted from AD samples [28]. However, studies of complete AC in AD associated with the different cell populations are lacking.
Accordingly, the present study includes stereological quantification of volume, cellular populations, and pathology estimations in the AC. In addition, dia‐PASEF analysis of non‐Alzheimer's disease (non‐AD) and AD human amygdala samples was carried out. The aim was to characterize the involvement of neurons, microglia, and astrocytes in the amygdala in AD and to identify markers associated with the different cell populations.
2. MATERIALS AND METHODS
2.1. Human samples
Human brain samples and data were provided by Institut d'Investigacions Biomèdiques August Pi i Sunyer, Biobanco en Red de la Región de Murcia, Biobanco de Tejidos de la Fundación CIEN, Biobanco del Principado de Asturias and Biobanco Navarrabiomed (registration numbers: B.0000575, B.0000859, B.0000741, B.0000827, and B.0000735, respectively) integrated in the Spanish National Biobanks Network. The samples were processed following standard operating procedures with the appropriate approval of the Ethical and Scientific Committees. These protocols included obtaining written consent from the donors. All the experimental procedures carried out in the UCAI facilities of the Ciudad Real Medical School were approved by the Ethical Committee of Clinical Research of Ciudad Real University Hospital (SAF2016‐75768‐R and PID2019‐108659RB‐I00).
A total of 36 cases were selected for the study (Table 1): 18 cases were diagnosed as AD, and 18 cases were classified as non‐AD. Formalin‐fixed samples were employed for immunohistochemistry and stereological quantifications (N = 20, AD n = 10, non‐AD n = 10). Fresh‐frozen samples were used for dia‐PASEF analysis (N = 16, AD n = 8, non‐AD n = 8).
TABLE 1.
Human samples.
| Case | Sex | Age (y) | PMD (h) | Brain weight (g) | Cause of death | Braak stage | Braak syn | TDP‐43 | Treatment |
|---|---|---|---|---|---|---|---|---|---|
| AD cases (N = 18) | |||||||||
| 1 | M | 88 | 7:00 | 1150 | Sepsis | V | 0 | Negative | Formalin fixed |
| 2 | F | 92 | 4:00 | 1000 | Respiratory insufficiency | V | 0 | Positive | Formalin fixed |
| 3 | F | 62 | 9:00 | 900 | Cardiorespiratory arrest | V | NA | Negative | Formalin fixed |
| 4 | M | 59 | 6:00 | 1100 | Cardiorespiratory arrest | VI | NA | Negative | Formalin fixed |
| 5 | F | 91 | 7:00 | NA | Pulmonary thromboembolism | V | NA | Negative | Formalin fixed |
| 6 | F | 74 | 4:00 | 1042 | Cardiorespiratory arrest | V | NA | Negative | Formalin fixed |
| 7 | M | 77 | 6:00 | 1060 | Acute respiratory infection | VI | 0 | Negative | Formalin fixed |
| 8 | F | 71 | 10:00 | 1006 | NA | V | 0 | Negative | Formalin fixed |
| 9 | F | 68 | NA | 1100 | Gastric carcinoma | VI | 0 | Negative | Formalin fixed |
| 10 | F | 89 | NA | 910 | NA | V | 0 | NA | Formalin fixed |
| 11 | F | 91 | 8:00 | 1080 | Respiratory insufficiency | V | 0 | Negative | Fresh‐frozen |
| 12 | M | 78 | 5:00 | 1260 | Multiorganic arrest | V | 0 | Negative | Fresh‐frozen |
| 13 | M | 67 | 4:05 | 1100 | Acute respiratory insufficiency | VI | 0 | Positive | Fresh‐frozen |
| 14 | M | 85 | 3:15 | 1130 | Upper gastrointestinal bleeding | VI | 0 | Positive | Fresh‐frozen |
| 15 | F | 67 | 4:15 | 1160 | Bronchoaspirative pneumonia | VI | 0 | Negative | Fresh‐frozen |
| 16 | M | 69 | 2:25 | 900 | Multiorganic arrest | VI | 5 | Negative | Fresh‐frozen |
| 17 | F | 76 | 11:10 | 900 | Respiratory insufficiency | VI | 0 | Negative | Fresh‐frozen |
| 18 | F | 85 | 5:00 | 960 | Respiratory insufficiency | V | 0 | Negative | Fresh‐frozen |
| Non‐AD cases (N = 18) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 19 | M | 56 | 19:00 | 1400 | Cardiorespiratory arrest | I | NA | Negative | Formalin fixed |
| 20 | M | 84 | 3:00 | 1400 | Cardiorespiratory arrest | ‐ | NA | Negative | Formalin fixed |
| 21 | M | 74 | 7:00 | 1336 | Tumor of unknown origin | I | 0 | Negative | Formalin fixed |
| 22 | M | 88 | 3:00 | 1285 | NA | II | 0 | NA | Formalin fixed |
| 23 | F | 58 | 5:00 | 944 | Pneumonia | ‐ | 0 | Negative | Formalin fixed |
| 24 | F | 59 | 4:00 | 1200 | Respiratory insufficiency | ‐ | NA | Negative | Formalin fixed |
| 25 | M | 63 | 2:00 | 1400 | Cardiorespiratory arrest | I | NA | Negative | Formalin fixed |
| 26 | F | 62 | 2:00 | 1050 | Sepsis | ‐ | NA | Negative | Formalin fixed |
| 27 | F | 83 | 4:00 | 1152 | NA | II | 0 | Negative | Formalin fixed |
| 28 | M | 86 | 7:00 | 965 | Respiratory insufficiency | II | NA | Negative | Formalin fixed |
| 29 | F | 71 | 7:08 | 975 | Cardiorespiratory arrest | ‐ | 0 | Negative | Fresh‐frozen |
| 30 | M | 68 | 4:00 | 1220 | Cardiorespiratory arrest | ‐ | 0 | Negative | Fresh‐frozen |
| 31 | M | 68 | 4:10 | 1350 | Sepsis | ‐ | 0 | Negative | Fresh‐frozen |
| 32 | M | 77 | 10:31 | 1300 | Bronchoaspiration | ‐ | 0 | Negative | Fresh‐frozen |
| 33 | M | 72 | 2:55 | 1340 | Systemic vascular pathology | ‐ | NA | Negative | Fresh‐frozen |
| 34 | F | 68 | 16:30 | 1076 | Refractory asystolia | ‐ | NA | Negative | Fresh‐frozen |
| 35 | M | 81 | 5:00 | 1309 | Respiratory pathology | ‐ | NA | Negative | Fresh‐frozen |
| 36 | M | 72 | 9:00 | 1407 | ‐ | ‐ | NA | Negative | Fresh‐frozen |
Note: Detailed information about the samples employed in the study, including sex, age, postmortem delay, brain weight, cause of death, Braak stage, and treatment of the sample.
Abbreviations: F, female; M, male; NA, not available; PMD, postmortem delay; y, years.
Formalin‐fixed samples from different tissue banks were postfixed in fresh phosphate‐buffered 4% paraformaldehyde for 45 days. For cryoprotection, blocks were immersed for 48 h in a phosphate buffered (PB) solution of 2% dimethyl sulfoxide (DMSO) and 10% glycerol and for 48 h in a PB solution of 2% DMSO and 20% glycerol. A freezing sliding microtome was used to obtain 50‐μm‐thick coronal sections. Thirteen series were obtained for each block, and the distance between sections was 650 μm. The first series was used for Nissl staining. The remaining series were stored in 24‐well plates at −20°C in 30% ethylene glycol and 20% glycerol in 0.1 M PB (pH 7.4).
Frozen samples were homogenized following previously described procedures [29, 30, 31]. Briefly, tissue was homogenized in 0.4 mL of RIPA buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.1% Triton X‐100, 0.1% SDS, and 0.5% Na‐deoxycholate) containing a protease inhibitor cocktail (Sigma–Aldrich) and incubated for 2 h at 4°C. Protein extraction was performed by centrifugation at 12,000g for 5 min at 4°C, and the supernatant was collected.
2.2. Immunohistochemistry
Tissue epitopes were unmasked by boiling the tissue under pressure for 2 min in citrate buffer. The sections were immersed in formic acid for 3 min and rinsed in phosphate‐buffered saline (PBS). Endogenous peroxidase activity was inhibited by incubation in 1% H2O2 in PBS for 20 min. The sections were preincubated for 1 h (microtubule‐associated protein 2 [MAP2] and allograft inflammatory factor 1 [Iba‐1]) or 2 h (glial fibrillary acidic protein [GFAP], Tau and Aβ) with blocking buffer and overnight at 4°C with primary antibodies (MAP2, Iba‐1, GFAP, Tau, and Aβ) (for details, see Online Resource 1). The sections were then incubated in biotinylated anti‐rabbit secondary antibody (1:200; Vector Laboratories) for 2 h at room temperature and in avidin–biotin complex (ABC Standard; Vector Laboratories) and reacted with 0.025% 3.3′‐diaminobenzidine and 0.1% H2O2. The sections were mounted, counterstained with Nissl, dried, dehydrated, and coverslipped with DPX (Sigma–Aldrich).
2.3. Stereological quantifications
Human amygdala volume and neuronal, microglial and astroglial cell populations were quantified using a Zeiss Axio Imager M.2 microscope coupled to stereological software (StereoInvestigator, MBF Bioscience®). The amygdaloid nuclei were delimited with a 1× objective (Zeiss Plan‐Neofluar 1×/0.025, Ref. 420300‐9900), and quantification was performed under a 63× objective (Zeiss Plan‐Apochromat 63×/1,4 oil DIC, Ref. 420782‐9900).
Volume estimation was carried out using the Cavalieri estimator probe. The number of MAP2‐, Iba‐1‐, and GFAP‐expressing cells was quantified using the optical fractionator method. The dissector height (Z) was 9 μm, and the guard zones were 2 μm. The Tau‐ and Aβ‐positive areas were assessed with the area fraction fractionator (AFF) method under 40× (Zeiss Plan‐APOCHROMAT 40×/0.95, Ref. 420660‐9970) and 20× objectives (Zeiss Plan‐APOCHROMAT 20×/0.8, Ref. 420650‐9901), respectively.
2.4. Statistical analysis
For stereological quantifications, the normality of the data was assessed using the Shapiro Wilk test. The data are expressed as the mean ± SEM. For normal data, mean values were compared using either t tests or one‐way ANOVA, and the Mann–Whitney U test was used for non‐normal data. F tests were carried out to compare variables, and in the case of differences between variables, t tests with Welch's correction were performed. The ROUT method was employed for outlier identification. No data were removed for the analysis. A significance level of α = 0.05 was used. Statistical analyses were performed with the GraphPad Prism 8.0.2 software.
2.5. dia‐PASEF proteomic analysis
2.5.1. Sample preparation
Samples were precipitated using methanol/chloroform and resuspended in 100 μL of RapiGest SF (Waters). Total protein concentration was measured using the Qubit fluorimetric protein assay (Thermo Fisher Scientific). Twenty‐five micrograms of protein were digested using the iST kit (PreOmics). Peptides were diluted using LC–MS H2O 0.1% (v/v) formic acid to 10 ng/μL. Two hundred nanograms of peptides were loaded onto Evotips (Evosep) for purification. Pierce HeLa tryptic Digest Standard (Thermo Fisher Scientific) was also loaded for quality control.
2.5.2. LC–MS/MS
Liquid chromatography–tandem mass spectrometry (LC–MS/MS) was carried out using an Evosep One LC system (Evosep) coupled to a TIMS Q‐TOF instrument (timsTOF Pro, Bruker Daltonics) via a nanoelectrospray ion source (Captive Spray Source, Bruker Daltonics). An MS/MS peptide library was built from the peptides and proteins identified using data‐dependent acquisition (DDA) parallel accumulation‐serial fragmentation (PASEF) analyses of the samples. Each sample was analyzed using the same liquid chromatography–mass spectrometry (LC–MS) system and gradient as used for the previous DDA runs but using data independent acquisition (DIA) (for details, see Online Resource 2).
2.5.3. Protein identification
Peptide identification was performed using MSFragger. Databases of H. sapiens protein sequences (UP000005640) from UniProt (reviewed sequences only; Apr 2021) and common contaminating proteins, which contained 20,382 total sequences, were used. Inverted protein sequences were added to the original databases. The initial mass tolerance was set at 20 ppm for precursor and fragment ions. Trypsin was set as described above with a maximum of two missed cleavages. Methionine oxidation and N‐terminal acetylation were established as variable modifications, and carbamidomethylation was established as a fixed modification. Peptide lengths of 7–50 amino acids and peptide masses of 500–5000 Da were set. A maximum of three variable modifications per peptide was set. PeptideProphet was used to calculate the probability of correct identification of peptides for spectrum matching and to assemble peptides into proteins. Philosopher Filter was used to assign each identified peptide as a razor peptide to a single protein or protein group that had the greatest peptide evidence. The false discovery rate (FDR) was set to 1% for peptide spectrum match or ion/peptide and protein identification. EasyPQP was used for aligning peptides to a common indexed retention time scale and peptide ion mobility to that from one of the references runs automatically selected. The final spectral library was filtered at 1% FDR at the peptide and protein levels.
DIA‐NN 1.8 (https://github.com/vdemichev/DiaNN/releases/tag/1.8) was used for diaPASEF analysis and operated with maximum mass tolerances set to 15 ppm. The samples were analyzed with run‐to‐run pairing (match between ranks) enabled. Protein inference in DIA‐NN was configured to use the assembled proteins in the spectral library. Protein. The group column in the DIA‐NN report was used to identify the protein group and PG. MaxLFQ label‐free quantification was used to obtain the normalized amount. The DIA‐NN output was filtered at a q value <1% for precursors and proteins.
The FDR validation was filtered to include only unmodified peptides or peptides with carbamidomethylated cysteines, oxidized methionines, or excised N‐terminal methionines. The library was screened for precursors/proteins with a 2–4 charge range and a 100.0–1700.0 m/z mass range.
LC–MS/MS, protein identification and quantification were carried out at the Instituto Maimonides de Investigación Biomédica de Córdoba (IMIBIC) Proteomic Facility. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [32] partner repository with the dataset identifier PXD038322.
2.5.4. Proteomic data analysis
Perseus (1.6.15.0) was used to analyze identified proteins. After log2 transformation, data were normalized using the width adjustment method. Proteins with one razor peptide and missing values were removed. An unpaired two‐tailed t test was employed to estimate significant differences. The fold change (FC) cut off was established at 1.5, and a p value <0.05 was used to obtain differentially expressed proteins (DEPs). SynGo (dataset version: 20210225) and Metascape [33] were employed for functional analysis of synapses and processes. Lists of proteins that interact with pathological markers (APP and MAPT) were obtained with BioGRID4.4 [34]. Proteins expressed preferentially in each cellular type (neurons, microglia, and astrocytes [35]) were compared with DEPs and pathological marker interactomes using Venn diagrams.
2.5.5. Immunofluorescence
To validate proteomic data, tissue epitopes were unmasked, and sections were preincubated for 1 h with blocking buffer and overnight at 4°C with primary antibodies (for details, see Online Resource 1). Subsequently, the sections were incubated with Alexa Fluor 488‐conjugated anti‐rabbit, Alexa Fluor 594‐conjugated anti‐mouse or Alexa Fluor 647‐conjugated anti‐goat antibodies (1:200; Thermo Fisher) for 2 h and then with 0.05% DAPI for 10 min at room temperature. Sections were mounted and coverslipped with PVA‐DABCO.
2.5.6. Confocal analysis
Triple immunofluorescence staining of pathological proteins and proteins identified by dia‐PASEF analysis was analyzed with a Zeiss LSM 800 confocal microscope coupled to the Zen 2.3 software (Oberkochen, Germany). Spatial colocalization was analyzed in high magnification images obtained with a 63× objective (Zeiss Plan‐Apochromat 63×/1.4 Oil DIC M27‐oil, Ref. 420782‐9900‐799).
3. RESULTS
3.1. Volume reduction in the human amygdala
Nissl staining of human amygdala samples was employed for delimitation and volume estimation of the cortical nucleus (Co) and the basolateral complex (BLA), including its basomedial (BM), basolateral (BL), and lateral (La) nuclei (Figure 1A,B) (for nomenclature used, see [36]; in the present study, the Co plus BLA was referred to as the AC). The Cavalieri probe revealed a volume reduction in the AC (Mann–Whitney U = 5.000, p value = 0.0002) and particularly in the Co (unpaired t test t18 = 2.589, p value = 0.0185) and BLA (Mann–Whitney U = 5.000, p value = 0.002). When the different nuclei of the BLA were analyzed, a specific volume reduction in La was observed (unpaired t test t18 = 3.032, p value = 0.0072; Figure 1C; for detailed information on stereological data of volume estimations, see Online Resource 3).
FIGURE 1.
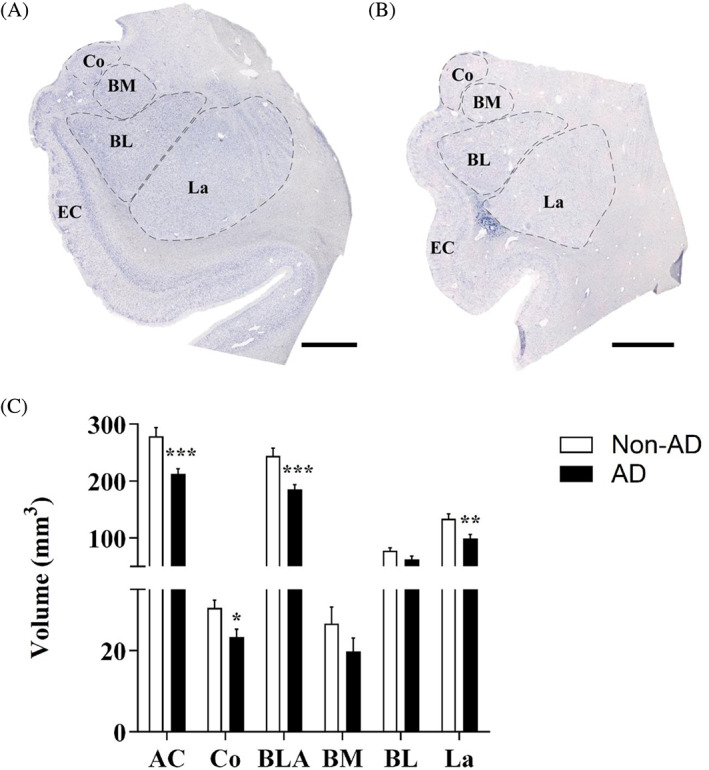
Amygdaloid volume reduction is specific to the Co and BLA, in particular the La. Nissl staining of the non‐AD (A) and AD (B) in the AC with delimitation of the amygdaloid nuclei studied. The global AC volume (C) and volume of the Co and BLA were significantly reduced in AD. In the BLA, volume was reduced specifically in the La (the graphs show the volume mean ± SEM, **p value <0.01, ***p value <0.001). AC, amygdaloid complex (Co, BLA); Co, cortical nucleus; BLA, basolateral complex (BM, BL, La); BM, basomedial nucleus; BL, basolateral nucleus; La, lateral nucleus. Scale bar = 1000 μm.
3.2. Cell population analysis revealed generalized astrogliosis in the AC in AD
Quantification of MAP2 (Figure 2A,B) and Iba‐1 (Figure 2D,E) positive cells revealed no differences in the number of neurons (Figure 2C) or microglia (Figure 2F). Microglial morphology was largely different in the non‐AD group (Figure 2D) compared with the AD group (Figure 2E), suggesting possible microglial activation in response to pathology. Regarding GFAP quantification (Figure 2G,H), a significant increase in the number of GFAP‐positive cells in the AC (unpaired t test t18 = 2.673, p value = 0.0155) as well as in every analyzed nucleus was reported (Co: Mann–Whitney U = 18.00, p value = 0.0279; BLA: Mann–Whitney U = 17.00, p value = 0.0115; BM: Mann–Whitney U = 19.00, p value = 0.0185; BL: Mann–Whitney U = 18.00, p value = 0.0147; La: Mann–Whitney U = 18.00, p value = 0.0147; Figure 2I).
FIGURE 2.
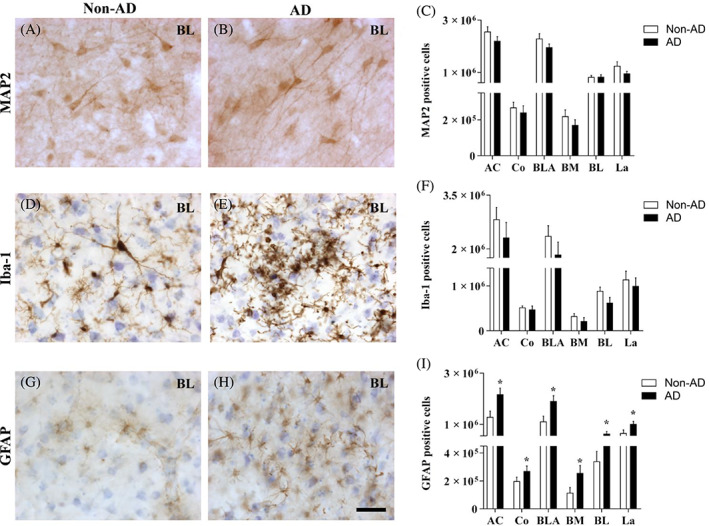
Generalized astrogliosis in the amygdaloid nuclei in AD. Immunohistochemical staining for MAP2 (A,B), Iba‐1 (D,E), and GFAP (G,H) in the BL in non‐AD and AD samples represents neurons, microglia, and astrocytes, respectively. The number of MAP2‐positive cells (C), Iba‐1‐positive cells (F), and GFAP‐positive cells (I) in the global AC and in the different nuclei are shown (the graphs show the mean ± SEM, *p value <0.05). Note that neither the number of neurons nor microglia was altered, and the number of astrocytes was increased in the whole AC. AC, amygdaloid complex (Co, BLA); Co, cortical nucleus; BLA, basolateral complex (BM, BL, La); BM, basomedial nucleus; BL, basolateral nucleus; La, lateral nucleus. Scale bar = 50 μm.
Concerning cell densities, neither neurons nor microglia showed changes (Online Resource 4). However, GFAP‐positive cell density was increased in the AC (unpaired t test t18 = 4.019, p value = 0.0008) and its different nuclei as well (Co: Mann–Whitney U = 14.00, p value = 0.0101; BLA: unpaired t test t18 = 3.905, p value = 0.001; BM: Mann–Whitney U = 7.00, p value = 0.0005; BL: unpaired t test t18 = 3.560, p value = 0.0022; La: unpaired t test t18 = 4.004, p value = 0.0008; Online Resource 4; for detailed information on stereological data of MAP2, Iba‐1 and GFAP estimations, see [Link], [Link], and 7, respectively).
3.3. Cortical and basal regions are the most affected by pathology in AD
The analysis of the area fraction occupied by pathological markers revealed a strong difference between the cortical and basal regions (BA; corresponding to the BM and BL) compared with La (Figure 3A,B). The area fraction occupied by Aβ was larger in the Co and BM than in the La (Figure 3C, one‐way ANOVA F (3, 36) = 5.726, p value = 0.0026), and the Tau area fraction was larger in the Co, BM, and BL than in the La (Figure 3D, one‐way ANOVA F (3, 36) = 10.74, p value <0.0001). Despite the differences in the staining pattern (Figure 3A,B), both Aβ and Tau appeared as a gradient with higher levels in medial (Co) regions (Figure 3A',B') than in lateral regions (Figure 3A'',B''; for detailed information on Aβ and Tau stereological data, see Online Resources 8 and 9, respectively).
FIGURE 3.
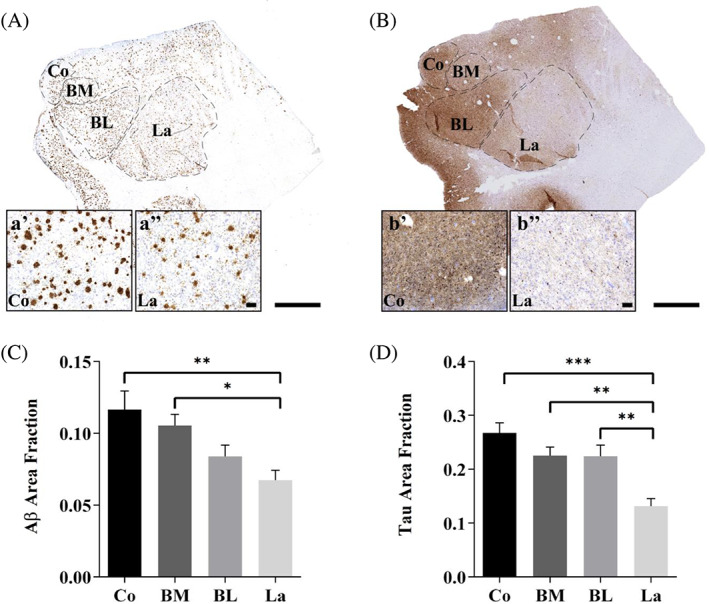
The cortical region is the most affected by pathology in AD. Aβ (A) and Tau (B) immunohistochemical staining of AD samples. Detail of Aβ (A', A'') and Tau (B', B'') staining pattern observed in Co and La, respectively. The area fractions of Aβ (C) and Tau (D) in the global AC and the different nuclei are shown (the graphs show the mean ± SEM, *p value <0.05, **p value <0.01, ***p value <0.001). Note that both Aβ and Tau appeared as a gradient with higher levels in medial (Co) regions than in lateral regions. Co: Cortical nucleus, BM: Basomedial nucleus, BL: Basolateral nucleus, La: Lateral nucleus. Scale bar = 1000 μm in (A,B); and 100 μm in (A',A''; B',B'').
In addition, because of the relevance of the amygdala regarding TDP‐43 related pathology, we performed an immunohistochemistry against phosphorylated TDP‐43 (TDP‐43‐P) selecting one of the positive cases provided by the biobanks (Table 1) (Online Resource 10). Interestingly, the staining of TDP‐43‐P was distributed in a similar manner as observed in Aβ and Tau labeling (Online Resource 10A). Whereas numerous intracellular accumulations of TDP‐43‐P were presented in Co (Online Resource 10B), clusters of TDP‐43‐P were common in BM and BL (Online Resource 10C,D, respectively). In contrast, scarce TDP‐43‐P deposits were found in La (Online Resource 10E).
3.4. Proteomic analysis revealed synaptic alteration and cellular responses to stress, with potential participation of astroglia and microglia
After restricted conditions of FC > 1.5 and p value <0.05 were applied to the 2153 quantified proteins by dia‐PASEF, a total of 178 proteins were considered DEPs in the proteomic analysis. From the 178 DEPs, 108 were considered up‐ and 70 were downregulated in AD (Table 2).
TABLE 2.
Differentially expressed proteins in AD amygdala.
| Protein IDs | Protein names | Genes | Protein description | FC | p value |
|---|---|---|---|---|---|
| Upregulated proteins | |||||
| P05362 | ICAM1_HUMAN | ICAM1 | Intercellular adhesion molecule 1 | 4.95 | 0.0001 |
| Q8IV08 | PLD3_HUMAN | PLD3 | 5′‐3′ exonuclease PLD3 | 4.21 | 0.0496 |
| P16070 | CD44_HUMAN | CD44 | CD44 antigen | 3.91 | 0.0020 |
| P02766 | TTHY_HUMAN | TTR | Transthyretin | 3.89 | 0.0049 |
| P22392 | NDKB_HUMAN | NME2 | Nucleoside diphosphate kinase B | 3.70 | 0.0013 |
| P13726 | TF_HUMAN | F3 | Tissue factor | 3.70 | 0.0137 |
| P10606 | COX5B_HUMAN | COX5B | Cytochrome c oxidase subunit 5B, mitochondrial | 3.20 | 0.0488 |
| P04083 | ANXA1_HUMAN | ANXA1 | Annexin A1 | 3.04 | 0.0014 |
| Q14019 | COTL1_HUMAN | COTL1 | Coactosin‐like protein | 2.90 | 0.0070 |
| P07355 | ANXA2_HUMAN | ANXA2 | Annexin A2 | 2.84 | 0.0012 |
| P49840 | GSK3A_HUMAN | GSK3A | Glycogen synthase kinase‐3 alpha | 2.72 | 0.0439 |
| Q15847 | ADIRF_HUMAN | ADIRF | Adipogenesis regulatory factor | 2.63 | 0.0045 |
| P00403 | COX2_HUMAN | MT‐CO2 | Cytochrome c oxidase subunit 2 | 2.59 | 0.0006 |
| P27105 | STOM_HUMAN | STOM | Stomatin | 2.41 | 0.0086 |
| P15531 | NDKA_HUMAN | NME1 | Nucleoside diphosphate kinase A | 2.41 | 0.0015 |
| P40429 | RL13A_HUMAN | RPL13A | 60S ribosomal protein L13a | 2.37 | 0.0311 |
| Q9H444 | CHM4B_HUMAN | CHMP4B | Charged multivesicular body protein 4b | 2.37 | 0.0407 |
| P35232 | PHB_HUMAN | PHB | Prohibitin | 2.37 | 0.0011 |
| Q92688 | AN32B_HUMAN | ANP32B | Acidic leucine‐rich nuclear phosphoprotein 32 family member B | 2.36 | 0.0203 |
| P31949 | S10AB_HUMAN | S100A11 | Protein S100‐A11 | 2.35 | 0.0192 |
| Q13907 | IDI1_HUMAN | IDI1 | Isopentenyl‐diphosphate Delta‐isomerase 1 | 2.28 | 0.0390 |
| P05387 | RLA2_HUMAN | RPLP2 | 60S acidic ribosomal protein P2 | 2.27 | 0.0328 |
| O76041 | NEBL_HUMAN | NEBL | Nebulette | 2.25 | 0.0219 |
| O75131 | CPNE3_HUMAN | CPNE3 | Copine‐3 | 2.24 | 0.0182 |
| O75828 | CBR3_HUMAN | CBR3 | Carbonyl reductase [NADPH] 3 | 2.22 | 0.0045 |
| O00299 | CLIC1_HUMAN | CLIC1 | Chloride intracellular channel protein 1 | 2.19 | 0.0008 |
| Q96HN2 | SAHH3_HUMAN | AHCYL2 | Adenosylhomocysteinase 3 | 2.18 | 0.0217 |
| P53367 | ARFP1_HUMAN | ARFIP1 | Arfaptin‐1 | 2.16 | 0.0484 |
| P45880 | VDAC2_HUMAN | VDAC2 | Voltage‐dependent anion‐selective channel protein 2 | 2.16 | 0.0205 |
| P10644 | KAP0_HUMAN | PRKAR1A | cAMP‐dependent protein kinase type I‐alpha regulatory subunit | 2.14 | 0.0180 |
| P0C0L5 | CO4B_HUMAN | C4B | Complement C4‐B | 2.10 | 0.0006 |
| Q8NBX0 | SCPDL_HUMAN | SCCPDH | Saccharopine dehydrogenase‐like oxidoreductase | 2.08 | 0.0202 |
| P01011 | AACT_HUMAN | SERPINA3 | Alpha‐1‐antichymotrypsin | 2.07 | 0.0168 |
| P26038 | MOES_HUMAN | MSN | Moesin | 2.07 | 0.0002 |
| P15259 | PGAM2_HUMAN | PGAM2 | Phosphoglycerate mutase 2 | 2.04 | 0.0355 |
| P10909 | CLUS_HUMAN | CLU | Clusterin | 2.03 | 0.0245 |
| Q07020 | RL18_HUMAN | RPL18 | 60S ribosomal protein L18 | 2.02 | 0.0081 |
| P50995 | ANX11_HUMAN | ANXA11 | Annexin A11 | 2.02 | 0.0046 |
| Q09666 | AHNK_HUMAN | AHNAK | Neuroblast differentiation‐associated protein AHNAK | 1.98 | 0.0012 |
| P48681 | NEST_HUMAN | NES | Nestin | 1.98 | 0.0093 |
| Q13938 | CAYP1_HUMAN | CAPS | Calcyphosin | 1.97 | 0.0084 |
| P21796 | VDAC1_HUMAN | VDAC1 | Voltage‐dependent anion‐selective channel protein 1 | 1.97 | 0.0197 |
| P04179 | SODM_HUMAN | SOD2 | Superoxide dismutase [Mn], mitochondrial | 1.95 | 0.0008 |
| P40121 | CAPG_HUMAN | CAPG | Macrophage‐capping protein | 1.94 | 0.0124 |
| P62277 | RS13_HUMAN | RPS13 | 40S ribosomal protein S13 | 1.94 | 0.0497 |
| Q14254 | FLOT2_HUMAN | FLOT2 | Flotillin‐2 | 1.94 | 0.0164 |
| Q09028 | RBBP4_HUMAN | RBBP4 | Histone‐binding protein RBBP4 | 1.93 | 0.0081 |
| Q9ULC3 | RAB23_HUMAN | RAB23 | Ras‐related protein Rab‐23 | 1.92 | 0.0379 |
| P13796 | PLSL_HUMAN | LCP1 | Plastin‐2 | 1.92 | 0.0130 |
| P13073 | COX41_HUMAN | COX4I1 | Cytochrome c oxidase subunit 4 isoform 1, mitochondrial | 1.90 | 0.0054 |
| P30047 | GFRP_HUMAN | GCHFR | GTP cyclohydrolase 1 feedback regulatory protein | 1.90 | 0.0375 |
| P15311 | EZRI_HUMAN | EZR | Ezrin | 1.89 | 0.0004 |
| O15488 | GLYG2_HUMAN | GYG2 | Glycogenin‐2 | 1.86 | 0.0216 |
| Q15417 | CNN3_HUMAN | CNN3 | Calponin‐3 | 1.84 | 0.0398 |
| P61421 | VA0D1_HUMAN | ATP6V0D1 | V‐type proton ATPase subunit d 1 | 1.83 | 0.0356 |
| Q01995 | TAGL_HUMAN | TAGLN | Transgelin | 1.82 | 0.0454 |
| Q9Y3E1 | HDGR3_HUMAN | HDGFL3 | Hepatoma‐derived growth factor‐related protein 3 | 1.82 | 0.0157 |
| Q96C23 | GALM_HUMAN | GALM | Galactose mutarotase | 1.82 | 0.0352 |
| P50897 | PPT1_HUMAN | PPT1 | Palmitoyl‐protein thioesterase 1 | 1.80 | 0.0180 |
| P08758 | ANXA5_HUMAN | ANXA5 | Annexin A5 | 1.79 | 0.0123 |
| P25788 | PSA3_HUMAN | PSMA3 | Proteasome subunit alpha type‐3 | 1.77 | 0.0043 |
| P08133 | ANXA6_HUMAN | ANXA6 | Annexin A6 | 1.76 | 0.0001 |
| Q96DG6 | CMBL_HUMAN | CMBL | Carboxymethylenebutenolidase homolog | 1.76 | 0.0429 |
| Q96G03 | PGM2_HUMAN | PGM2 | Phosphoglucomutase‐2 | 1.75 | 0.0067 |
| Q9NPH2 | INO1_HUMAN | ISYNA1 | Inositol‐3‐phosphate synthase 1 | 1.75 | 0.0059 |
| P04080 | CYTB_HUMAN | CSTB | Cystatin‐B | 1.75 | 0.0051 |
| P62266 | RS23_HUMAN | RPS23 | 40S ribosomal protein S23 | 1.75 | 0.0194 |
| Q8TC26 | TM163_HUMAN | TMEM163 | Transmembrane protein 163 | 1.75 | 0.0261 |
| P30041 | PRDX6_HUMAN | PRDX6 | Peroxiredoxin‐6 | 1.74 | 0.0018 |
| Q3KQU3 | MA7D1_HUMAN | MAP7D1 | MAP7 domain‐containing protein 1 | 1.72 | 0.0421 |
| Q8NBF2 | NHLC2_HUMAN | NHLRC2 | NHL repeat‐containing protein 2 | 1.72 | 0.0062 |
| Q96AQ6 | PBIP1_HUMAN | PBXIP1 | Pre‐B‐cell leukemia transcription factor‐interacting protein 1 | 1.71 | 0.0152 |
| Q9BPW8 | NIPS1_HUMAN | NIPSNAP1 | Protein NipSnap homolog 1 | 1.70 | 0.0383 |
| P06865 | HEXA_HUMAN | HEXA | Beta‐hexosaminidase subunit alpha | 1.69 | 0.0181 |
| Q7L9L4 | MOB1B_HUMAN | MOB1B | MOB kinase activator 1B | 1.69 | 0.0190 |
| P84085 | ARF5_HUMAN | ARF5 | ADP‐ribosylation factor 5 | 1.67 | 0.0304 |
| Q9BY32 | ITPA_HUMAN | ITPA | Inosine triphosphate pyrophosphatase | 1.67 | 0.0433 |
| Q9H8H3 | MET7A_HUMAN | METTL7A | Methyltransferase‐like protein 7A | 1.66 | 0.0059 |
| P29401 | TKT_HUMAN | TKT | Transketolase | 1.66 | 0.0030 |
| O43399 | TPD54_HUMAN | TPD52L2 | Tumor protein D54 | 1.66 | 0.0496 |
| P11766 | ADHX_HUMAN | ADH5 | Alcohol dehydrogenase class‐3 | 1.65 | 0.0246 |
| O95336 | 6PGL_HUMAN | PGLS | 6‐phosphogluconolactonase | 1.65 | 0.0153 |
| Q96Q06 | PLIN4_HUMAN | PLIN4 | Perilipin‐4 | 1.64 | 0.0455 |
| Q9UL46 | PSME2_HUMAN | PSME2 | Proteasome activator complex subunit 2 | 1.64 | 0.0368 |
| P51178 | PLCD1_HUMAN | PLCD1 | 1‐phosphatidylinositol 4,5‐bisphosphate phosphodiesterase delta‐1 | 1.63 | 0.0057 |
| P49721 | PSB2_HUMAN | PSMB2 | Proteasome subunit beta type‐2 | 1.63 | 0.0303 |
| P55008 | AIF1_HUMAN | AIF1 | Allograft inflammatory factor 1 | 1.62 | 0.0234 |
| P10768 | ESTD_HUMAN | ESD | S‐formylglutathione hydrolase | 1.62 | 0.0016 |
| P20073 | ANXA7_HUMAN | ANXA7 | Annexin A7 | 1.62 | 0.0309 |
| O75223 | GGCT_HUMAN | GGCT | Gamma‐glutamylcyclotransferase | 1.62 | 0.0402 |
| Q00796 | DHSO_HUMAN | SORD | Sorbitol dehydrogenase | 1.62 | 0.0153 |
| P49189 | AL9A1_HUMAN | ALDH9A1 | 4‐trimethylaminobutyraldehyde dehydrogenase | 1.62 | 0.0056 |
| O14807 | RASM_HUMAN | MRAS | Ras‐related protein M‐Ras | 1.61 | 0.0455 |
| P30626 | SORCN_HUMAN | SRI | Sorcin | 1.61 | 0.0493 |
| Q9BQA1 | MEP50_HUMAN | WDR77 | Methylosome protein 50 | 1.61 | 0.0064 |
| P63027 | VAMP2_HUMAN | VAMP2 | Vesicle‐associated membrane protein 2 | 1.60 | 0.0287 |
| Q04760 | LGUL_HUMAN | GLO1 | Lactoylglutathione lyase | 1.60 | 0.0040 |
| Q96DB5 | RMD1_HUMAN | RMDN1 | Regulator of microtubule dynamics protein 1 | 1.60 | 0.0404 |
| Q14118 | DAG1_HUMAN | DAG1 | Dystroglycan | 1.59 | 0.0195 |
| P61204 | ARF3_HUMAN | ARF3 | ADP‐ribosylation factor 3 | 1.58 | 0.0460 |
| P43490 | NAMPT_HUMAN | NAMPT | Nicotinamide phosphoribosyltransferase | 1.58 | 0.0313 |
| Q8N4P3 | MESH1_HUMAN | HDDC3 | Guanosine‐3′,5′‐bis(diphosphate) 3′‐pyrophosphohydrolase MESH1 | 1.57 | 0.0486 |
| Q13683 | ITA7_HUMAN | ITGA7 | Integrin alpha‐7 | 1.57 | 0.0051 |
| Q6IQ22 | RAB12_HUMAN | RAB12 | Ras‐related protein Rab‐12 | 1.55 | 0.0338 |
| Q15599 | NHRF2_HUMAN | SLC9A3R2 | Na(+)/H(+) exchange regulatory cofactor NHE‐RF2 | 1.52 | 0.0300 |
| P09211 | GSTP1_HUMAN | GSTP1 | Glutathione S‐transferase P | 1.50 | 0.0136 |
| P25786 | PSA1_HUMAN | PSMA1 | Proteasome subunit alpha type‐1 | 1.50 | 0.0402 |
| P27816 | MAP4_HUMAN | MAP4 | Microtubule‐associated protein 4 | 1.50 | 0.0219 |
| Downregulated proteins | |||||
|---|---|---|---|---|---|
| P23468 | PTPRD_HUMAN | PTPRD | Receptor‐type tyrosine‐protein phosphatase delta | 0.67 | 0.0323 |
| Q7KZF4 | SND1_HUMAN | SND1 | Staphylococcal nuclease domain‐containing protein 1 | 0.67 | 0.0434 |
| O75122 | CLAP2_HUMAN | CLASP2 | CLIP‐associating protein 2 | 0.67 | 0.0368 |
| Q9H0E2 | TOLIP_HUMAN | TOLLIP | Toll‐interacting protein | 0.66 | 0.0124 |
| Q15111 | PLCL1_HUMAN | PLCL1 | Inactive phospholipase C‐like protein 1 | 0.66 | 0.0275 |
| P36551 | HEM6_HUMAN | CPOX | Oxygen‐dependent coproporphyrinogen‐III oxidase, mitochondrial | 0.66 | 0.0115 |
| Q08380 | LG3BP_HUMAN | LGALS3BP | Galectin‐3‐binding protein | 0.65 | 0.0277 |
| P50453 | SPB9_HUMAN | SERPINB9 | Serpin B9 | 0.65 | 0.0340 |
| O95670 | VATG2_HUMAN | ATP6V1G2 | V‐type proton ATPase subunit G 2 | 0.65 | 0.0475 |
| O43615 | TIM44_HUMAN | TIMM44 | Mitochondrial import inner membrane translocase subunit TIM44 | 0.65 | 0.0490 |
| Q96RU3 | FNBP1_HUMAN | FNBP1 | Formin‐binding protein 1 | 0.65 | 0.0382 |
| P14866 | HNRPL_HUMAN | HNRNPL | Heterogeneous nuclear ribonucleoprotein L | 0.64 | 0.0460 |
| Q86VS8 | HOOK3_HUMAN | HOOK3 | Protein Hook homolog 3 | 0.63 | 0.0045 |
| Q9NP81 | SYSM_HUMAN | SARS2 | Serine—tRNA ligase, mitochondrial | 0.63 | 0.0464 |
| Q01433 | AMPD2_HUMAN | AMPD2 | AMP deaminase 2 | 0.62 | 0.0035 |
| O95757 | HS74L_HUMAN | HSPA4L | Heat shock 70 kDa protein 4 L | 0.62 | 0.0322 |
| Q9GZM8 | NDEL1_HUMAN | NDEL1 | Nuclear distribution protein nudE‐like 1 | 0.62 | 0.0150 |
| Q96B97 | SH3K1_HUMAN | SH3KBP1 | SH3 domain‐containing kinase‐binding protein 1 | 0.62 | 0.0109 |
| Q04323 | UBXN1_HUMAN | UBXN1 | UBX domain‐containing protein 1 | 0.62 | 0.0083 |
| Q9H9P8 | L2HDH_HUMAN | L2HGDH | L‐2‐hydroxyglutarate dehydrogenase, mitochondrial | 0.61 | 0.0360 |
| Q5T4S7 | UBR4_HUMAN | UBR4 | E3 ubiquitin‐protein ligase UBR4 | 0.60 | 0.0310 |
| Q92609 | TBCD5_HUMAN | TBC1D5 | TBC1 domain family member 5 | 0.60 | 0.0143 |
| P46379 | BAG6_HUMAN | BAG6 | Large proline‐rich protein BAG6 | 0.59 | 0.0202 |
| Q04609 | FOLH1_HUMAN | FOLH1 | Glutamate carboxypeptidase 2 | 0.59 | 0.0275 |
| P48147 | PPCE_HUMAN | PREP | Prolyl endopeptidase | 0.59 | 0.0143 |
| P02787 | TRFE_HUMAN | TF | Serotransferrin | 0.59 | 0.0027 |
| Q8NBJ7 | SUMF2_HUMAN | SUMF2 | Inactive C‐alpha‐formylglycine‐generating enzyme 2 | 0.58 | 0.0450 |
| Q9BXJ9 | NAA15_HUMAN | NAA15 | N‐alpha‐acetyltransferase 15, NatA auxiliary subunit | 0.58 | 0.0233 |
| Q13617 | CUL2_HUMAN | CUL2 | Cullin‐2 | 0.58 | 0.0488 |
| Q8N7J2 | AMER2_HUMAN | AMER2 | APC membrane recruitment protein 2 | 0.57 | 0.0148 |
| Q96FC7 | PHIPL_HUMAN | PHYHIPL | Phytanoyl‐CoA hydroxylase‐interacting protein‐like | 0.56 | 0.0067 |
| Q15438 | CYH1_HUMAN | CYTH1 | Cytohesin‐1 | 0.56 | 0.0019 |
| Q8IXJ6 | SIR2_HUMAN | SIRT2 | NAD‐dependent protein deacetylase sirtuin‐2 | 0.56 | 0.0062 |
| Q9C0D3 | ZY11B_HUMAN | ZYG11B | Protein zyg‐11 homolog B | 0.56 | 0.0181 |
| P48426 | PI42A_HUMAN | PIP4K2A | Phosphatidylinositol 5‐phosphate 4‐kinase type‐2 alpha | 0.55 | 0.0008 |
| Q6L8Q7 | PDE12_HUMAN | PDE12 | 2′,5′‐phosphodiesterase 12 | 0.55 | 0.0242 |
| Q9C0E8 | LNP_HUMAN | LNPK | Endoplasmic reticulum junction formation protein lunapark | 0.54 | 0.0241 |
| Q13619 | CUL4A_HUMAN | CUL4A | Cullin‐4A | 0.53 | 0.0284 |
| Q3ZCW2 | LEGL_HUMAN | LGALSL | Galectin‐related protein | 0.53 | 0.0445 |
| P60228 | EIF3E_HUMAN | EIF3E | Eukaryotic translation initiation factor 3 subunit E | 0.53 | 0.0091 |
| Q9Y276 | BCS1_HUMAN | BCS1L | Mitochondrial chaperone BCS1 | 0.53 | 0.0427 |
| Q8N111 | CEND_HUMAN | CEND1 | Cell cycle exit and neuronal differentiation protein 1 | 0.52 | 0.0320 |
| O00505 | IMA4_HUMAN | KPNA3 | Importin subunit alpha‐4 | 0.52 | 0.0167 |
| Q9NTM9 | CUTC_HUMAN | CUTC | Copper homeostasis protein cutC homolog | 0.52 | 0.0427 |
| Q16773 | KAT1_HUMAN | KYAT1 | Kynurenine‐oxoglutarate transaminase 1 | 0.52 | 0.0048 |
| O76094 | SRP72_HUMAN | SRP72 | Signal recognition particle subunit SRP72 | 0.52 | 0.0355 |
| Q9H9Q2 | CSN7B_HUMAN | COPS7B | COP9 signalosome complex subunit 7b | 0.52 | 0.0287 |
| O95292 | VAPB_HUMAN | VAPB | Vesicle‐associated membrane protein‐associated protein B/C | 0.52 | 0.0074 |
| Q5TCQ9 | MAGI3_HUMAN | MAGI3 | Membrane‐associated guanylate kinase, WW and PDZ domain‐containing protein 3 | 0.51 | 0.0224 |
| Q9NR45 | SIAS_HUMAN | NANS | Sialic acid synthase | 0.51 | 0.0040 |
| Q9Y2J0 | RP3A_HUMAN | RPH3A | Rabphilin‐3A | 0.51 | 0.0192 |
| Q7Z4S6 | KI21A_HUMAN | KIF21A | Kinesin‐like protein KIF21A | 0.51 | 0.0033 |
| P19823 | ITIH2_HUMAN | ITIH2 | Inter‐alpha‐trypsin inhibitor heavy chain H2 | 0.50 | 0.0448 |
| P28676 | GRAN_HUMAN | GCA | Grancalcin | 0.50 | 0.0177 |
| Q9UIA9 | XPO7_HUMAN | XPO7 | Exportin‐7 | 0.50 | 0.0093 |
| Q9UPV7 | PHF24_HUMAN | PHF24 | PHD finger protein 24 | 0.49 | 0.0332 |
| Q9NQW6 | ANLN_HUMAN | ANLN | Anillin | 0.48 | 0.0098 |
| O60262 | GBG7_HUMAN | GNG7 | Guanine nucleotide‐binding protein G(I)/G(S)/G(O) subunit gamma‐7 | 0.48 | 0.0169 |
| O75689 | ADAP1_HUMAN | ADAP1 | Arf‐GAP with dual PH domain‐containing protein 1 | 0.48 | 0.0085 |
| O75208 | COQ9_HUMAN | COQ9 | Ubiquinone biosynthesis protein COQ9, mitochondrial | 0.45 | 0.0050 |
| A5YM72 | CRNS1_HUMAN | CARNS1 | Carnosine synthase 1 | 0.44 | 0.0063 |
| Q9UDY2 | ZO2_HUMAN | TJP2 | Tight junction protein ZO‐2 | 0.43 | 0.0382 |
| Q96GW9 | SYMM_HUMAN | MARS2 | Methionine—tRNA ligase, mitochondrial | 0.42 | 0.0091 |
| P20916 | MAG_HUMAN | MAG | Myelin‐associated glycoprotein | 0.42 | 0.0025 |
| Q96FJ2 | DYL2_HUMAN | DYNLL2 | Dynein light chain 2, cytoplasmic | 0.41 | 0.0078 |
| O94967 | WDR47_HUMAN | WDR47 | WD repeat‐containing protein 47 | 0.40 | 0.0130 |
| Q8TAM6 | ERMIN_HUMAN | ERMN | Ermin | 0.39 | 0.0217 |
| P02689 | MYP2_HUMAN | PMP2 | Myelin P2 protein | 0.28 | 0.0170 |
| Q96HU8 | DIRA2_HUMAN | DIRAS2 | GTP‐binding protein Di‐Ras2 | 0.26 | 0.0396 |
| P53597 | SUCA_HUMAN | SUCLG1 | Succinate—CoA ligase [ADP/GDP‐forming] subunit alpha, mitochondrial | 0.23 | 0.0307 |
Note: FC < 1.5, p value <0.05, total identified proteins available via ProteomeXchange with identifier PXD038322.
In order to relate the DEPs to the specific neuronal, microglial and/or astroglial cell populations, we crossed them with lists of proteins preferentially expressed in each cell type, as well as with lists of proteins which interacts with Aβ (APP interactome) and Tau (MAPT interactome) to see their involvement in the pathology (for details, see Online Resource 11). Thus, cell cycle exit and neuronal differentiation protein 1 (CEND1), WDR47, and DIRAS2 were identified as DEPs and preferentially expressed in neurons. Nineteen proteins were recognized as DEPs and preferentially expressed in microglia. Specifically, Annexin A5 (ANXA5) was associated with both pathological markers and microglia, and proteasome activator complex subunit 2 (PSME2) and galectin‐3‐binding protein (LGALS3BP) were associated with Aβ and microglia. Eighteen proteins were linked to astrocytes. The marker clusterin (CLU) was related to both pathological markers, Flotillin‐2 (FLOT2) to Tau interactions and astrocytes, and peroxiredoxin‐6 (PRDX6) to Aβ and astrocytes (Table 3; Online Resource 12).
TABLE 3.
Identified proteins from DEPs that interact with pathological proteins and expressed in neurons, microglia, and astrocytes.
| DEPs‐neurons | DEPs‐neurons‐Aβ | DEPs‐neurons‐tau | DEPs‐neurons‐Aβ‐tau |
|---|---|---|---|
| CEND1, WDR47, DIRAS2 | ‐ | ‐ | ‐ |
| DEPs‐microglia | DEPs‐Microglia‐Aβ | DEPs‐Microglia‐Tau | DEPs‐Microglia‐Aβ‐Tau |
|---|---|---|---|
| PLD3, ANXA1, COTL1, S100A11, CLIC1, SERPINA3, MSN, ANXA11, CAPG, LCP1, GALM, PPT1, ISYNA1, CSTB, HEXA, AIF1, SORD, SH3KBP1 | PSME2, LGALS3BP | ‐ | ANXA5 |
| DEPs‐astrocytes | DEPs‐Astrocytes‐Aβ | DEPs‐Astrocytes‐Tau | DEPs‐Astrocytes‐Aβ‐Tau |
|---|---|---|---|
| F3, NEBL, CBR3, AHCYL2, NES, CNN3, AGLN, CMBL, PBXIP1, MRAS, DAG1, ITGA7, MAP4, FOLH1, AMER2, PHYHIPL, LGALSL, TJP2 | FLOT2 | PRDX6 | CLU |
Note: Four main groups are presented: proteins preferentially expressed in cell type, proteins preferentially expressed in cell type that interact with Aβ, proteins preferentially expressed in cell type that interact with tau, and proteins preferentially expressed that interact with both markers are shown.
SynGo analysis revealed certain synaptic alterations in AD (29 proteins of 178 DEPs) with a clear effect on the synaptic vesicle system (Table 4; for detailed analysis, see Online Resource 10). On the other hand, Metascape analysis revealed affected processes such as cellular responses to stress, regulation of proteolysis, regulation of vesicle‐mediated transport, apoptotic signaling pathway or response to wounding, among others (Table 5; for detailed analysis, see Online Resources 11 and 12).
TABLE 4.
SynGo analysis revealed synaptic affectation in AD.
| GO term ID | GO domain | GO term name | FDR corrected p value | Genes |
|---|---|---|---|---|
| GO:0045202 | CC | Synapse | 0.000103167 | FLOT2; PRKAR1A; MAGI3; RPLP2; RPS13; RPL18; RPS23; CLU; RPL13A; HNRNPL; PHB; ANXA1; CADPS; VDAC1; ANXA5; RPH3A; ATP6V1G2; VAMP2; TMEM163; ATP6V0D1; CNTN1; CYTH1; PTPRD; DYNLL2; CNN3; DAG1; EIF3E |
| GO:0098793 | CC | Presynapse | 0.000139198 | CADPS; VDAC1; FLOT2; PHB; ANXA5; RPH3A; ATP6V1G2; VAMP2; TMEM163; ATP6V0D1; CNTN1; CYTH1; PTPRD; RPL13A; RPL18; RPLP2; RPS13 |
| GO:0048787 | CC | Presynaptic active zone membrane | 0.027329597 | VDAC1; FLOT2; PHB |
| GO:0030672 | CC | Synaptic vesicle membrane | 0.002092777 | ANXA5; RPH3A; ATP6V1G2; VAMP2; TMEM163; ATP6V0D1 |
| GO:0030285 | CC | Integral component of synaptic vesicle membrane | 0.027329597 | VAMP2; TMEM163; ATP6V0D1 |
| GO:0098794 | CC | Postsynapse | 0.027329597 | DYNLL2; CNN3; DAG1; PHB; RPS13; EIF3E; VDAC1; CNTN1; RPL13A; RPL18; RPLP2; RPS23 |
| GO:0099504 | BP | Synaptic vesicle cycle | 0.027829211 | VAMP2; CADPS; RPH3A; TMEM163; ATP6V0D1; ATP6V1G2 |
| GO:0140236 | BP | Translation at presynapse | 0.01123458 | RPL13A; RPL18; RPLP2; RPS13 |
| GO:0140242 | BP | Translation at postsynapse | 0.01123458 | RPL13A; RPL18; RPLP2; RPS13 |
Note: Outstanding information about SynGo analysis including the GO information, false discovery rate and genes involved.
Abbreviations: BP, biological process; CC, cellular component; FDR, false discovery rate; GO, GeneOntology.
TABLE 5.
Functional analysis by Metascape
| Term | Description | Log (P) | Log(q‐value) | Proteins |
|---|---|---|---|---|
| R‐HSA‐2262752 | Cellular responses to stress | −9.55086 | −5.355 | ATP6V1G2, COX4I1, COX5B, GSK3A, GSTP1, COX2, PSMA1, PSMA3, PSMB2, PSME2, RBBP4, RPL18, RPLP2, RPS13, RPS23, SOD2, TKT, CUL2, ATP6V0D1, PRDX6, HSPA4L, RPL13A, DYNLL2 |
| GO:0030162 | Regulation of proteolysis | −5.65205 | −2.406 | SERPINA3, ANXA2, C4B, CD44, CLU, CSTB, F3, GSK3A, ITIH2, SERPINB9, PSMA3, PSME2, BAG6, ANP32B, SIRT2, UBXN1, ZYG11B |
| GO:0032386 | Regulation of intracellular transport | −5.31044 | −2.263 | ANXA2, STOM, GSK3A, LCP1, MSN, VAMP2, EZR, ANP32B, ARFIP1, RAB23, NDEL1, DAG1, SRI |
| GO:0048260 | Positive regulation of receptor‐mediatedendocytosis | −4.81393 | −1.928 | ANXA2, CLU, PPT1, TF, TBC1D5, GLO1 |
| GO:0060627 | Regulation of vesicle‐mediated transport | −4.72155 | −1.926 | ANXA1, ANXA2, C4B, CLU, MSN, PPT1, VAMP2, TF, EZR, TBC1D5, CLASP2, ARFIP1, RAB12, BCS1L, FLOT2, SRP72, BAG6, SLC9A3R2, CHMP4B |
| GO:0030036 | Actin cytoskeleton organization | −4.69625 | −1.926 | AIF1, ANXA1, CAPG, CNN3, LCP1, PRKAR1A, TF, EZR, NEBL, MRAS, SH3KBP1, ANLN, ERMN, CLASP2, HDGFL3, NDEL1, HOOK3 |
| GO:0097190 | Apoptotic signaling pathway | −4.46144 | −1.794 | ANXA6, CLU, GSK3A, SOD2, VDAC2, BAG6, CUL4A, CUL2, GGCT, AIF1, CD44, GSTP1, ICAM1, VDAC1, EZR, UBXN1, NDEL1, MAGI3, BCS1L, PHB1, TIMM44, DAG1 |
| GO:0006914 | Autophagy | −4.31508 | −1.718 | ANXA7, CLU, PIP4K2A, TBC1D5, SIRT2, RAB23, TOLLIP, CHMP4B, RAB12, ATP6V0D1, HOOK3 |
| GO:0009611 | Response to wounding | −4.2287 | −1.678 | AIF1, ANXA5, ANXA6, CD44, CLIC1, DAG1, F3, MAG, SOD2, LNPK, CHMP4B |
Note: Proteins related to main affected pathways and biological processes.
The selection of proteins for validation was based on available literature and FC threshold. Proteins with no evidence or relation with the disease were excluded. Since the aim of the study was to provide new insights about AD in AC, well‐known proteins associated with the pathology were also excluded. Furthermore, potential relation or expression in the studied cell types (neurons, microglia, and astrocytes) was also considered for protein selection (Figure 4). Considering these criteria, Arf‐GAP with dual PH domain‐containing protein 1 (ADAP1), CEND1, and ANXA2 were selected for neuronal; chloride intracellular channel protein 1 (CLIC1) and ANXA5 for microglial; and Annexin A1 (ANXA1) and PRDX6 for astroglial evaluation by confocal analysis.
FIGURE 4.
Procedure for proteomic data analysis and criteria for protein selection validation. In a first step, dia‐PASEF analysis of human AC samples revealed 2153 proteins. After applying restricted condition of FC > 1.5 and p value <0.05, 178 proteins were identified as DEPs and cell type expression, SynGo and Metascape analyses were performed (data shown in Tables 3, 4, 5). Then, literature review of DEPs was carried out in order to select proteins for validation. Proteins were chosen based on three main criteria: previous evidence linking protein and AD must be reported; proteins widely described in the disease were excluded; and potential relation or expression in the studied cell types (neurons, microglia, and astrocytes) was also considered.
3.5. Neuronal and glial responses to pathology in the AC
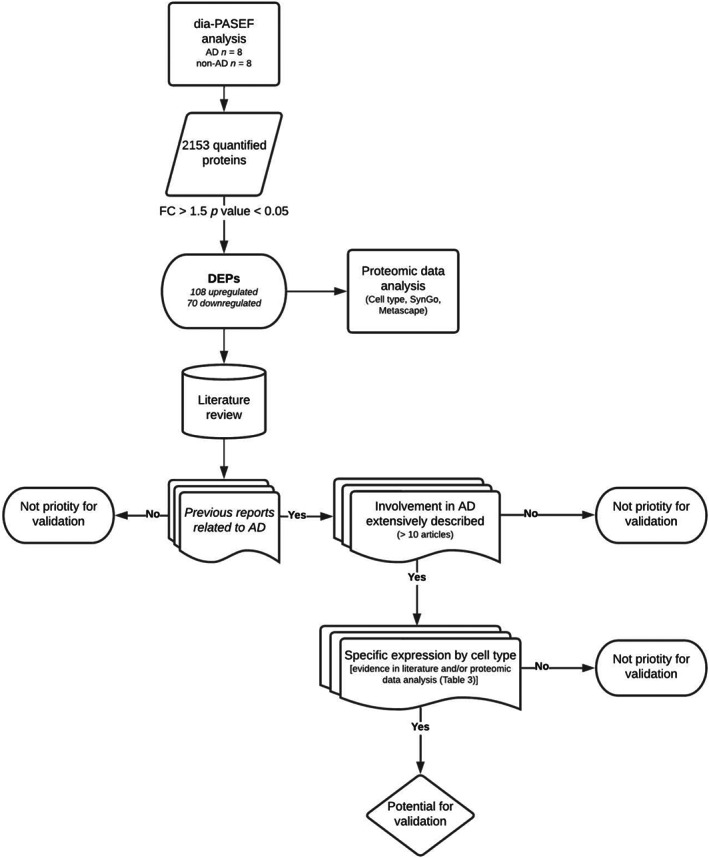
According to the proteomic analysis, ADAP1 and CEND1 were identified as downregulated, while ANXA2 was identified as upregulated by dia‐PASEF analysis. ADAP1 expression was identified not only in the soma but also associated with dendrites and axons in non‐AD samples (Figure 5A). However, ADAP1 labeling was dramatically reduced in AD samples (Figure 5B,C). Its expression was observed to be associated with Tau (Figure 5B) and soma (Figure 5C, dashed line). Likewise, CEND1 was widely expressed in neurons in non‐AD samples (Figure 5D). Nevertheless, few neurons were labeled with CEND1 in AD samples (Figure 5E,F). Interestingly, when labeling was identified in neurons in the vicinity of Aβ, CEND1 expression was reduced (Figure 5F, dashed line) compared to that surrounding Tau deposits (Figure 5E). ANXA2 was expressed by neurons in non‐AD samples (Figure 5G), and qualitatively, the ANXA2 intensity of labeling was higher in AD samples (Figure 5H,I). ANXA2 was closely distributed with Aβ plaques (Figure 5H,I), being more intense in the periphery of the plaques (Figure 5H) than inside (Figure 5I).
FIGURE 5.
Neuronal involvement in the amygdaloid complex nuclei in AD: ADAP1, CEND1, and ANXA2. Triple immunofluorescence against ADAP1 (A–C), CEND1 (D,E), ANXA2 (G–I), and pathological markers. In non‐AD, ADAP1 (A, green) was mainly associated with vesicles in axons and dendrites, although it was also observed in soma. CEND1 (D, green) revealed neuronal expression in non‐AD samples. ADAP1 expression was drastically reduced in AD (B,C), with spatial coexpression with Tau (red) and MAP2 (purple) in the soma (B). Neurons close to Aβ (C, dashed line) presented a reduced number of ADAP1 vesicles in the soma and axon. A reduced number of CEND1‐stained neurons was observed in AD (E,F). CEND1 staining was remarkably associated with Tau deposits (E) compared with neurons near Aβ plaques (F, dashed line). ANXA2 (G, green) expression in neurons was identified in non‐AD samples. In AD, ANXA2 expression was increased close to Aβ (red) deposits (H,I). ANXA2 staining was higher on the outside of the plaques (H) than on the inside (I). Scale bar = 10 μm.
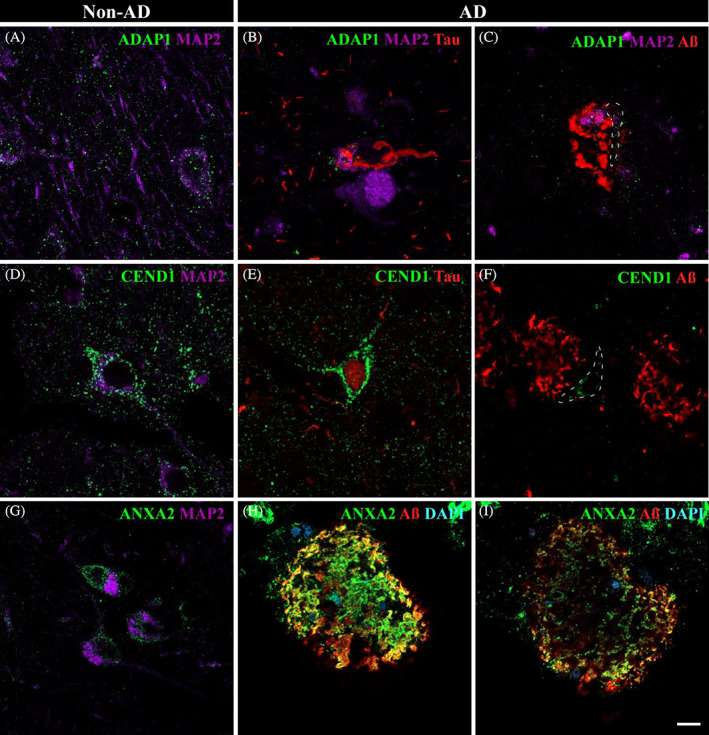
CLIC1 and ANXA5 were assessed as upregulated by proteomic analysis. In non‐AD samples, CLIC1 labeling suggested possible expression in neurons (Figure 6A, dashed line). In AD, we observed two different situations: first, CLIC1 colocalized with Tau deposits, with microglia frequently present close to those affected neurons (Figure 6B, dashed line), and second, microglia expressed CLIC1 in the vicinity of Aβ (Figure 6C, arrow). On the other hand, ANXA5 was expressed in microglia in non‐AD samples (Figure 6D, arrow) and more intensely expressed in AD samples (Figure 6E,F, arrow). ANXA5 was frequently observed with Tau deposits (Figure 6E, arrowhead), whereas ANXA5‐microglia coexpression was closely associated with Aβ in AD samples (Figure 6F, arrow).
FIGURE 6.
Microglial involvement in amygdaloid pathology in AD. Immunofluorescences against CLIC1 (A–C) and ANXA5 (D–F) and pathological markers are shown. In non‐AD samples, CLIC1 (A, green) labeling suggested possible expression in neurons (dashed line). In AD, CLIC1 colocalized intimately with Tau pathology (B, red) and microglia (purple, dashed line). Additionally, CLIC1 expression was observed in the microglial cells nearest to Aβ plaques (red) (C, arrow). ANXA5 (D, green) was related to microglia (purple, arrow) in non‐AD tissue. Microglial ANXA5 expression was increased in AD (E,F, arrow) with a closed spatial expression with Aβ plaques (E, reed) and Tau deposits (F, arrowhead). Scale bar = 10 μm.
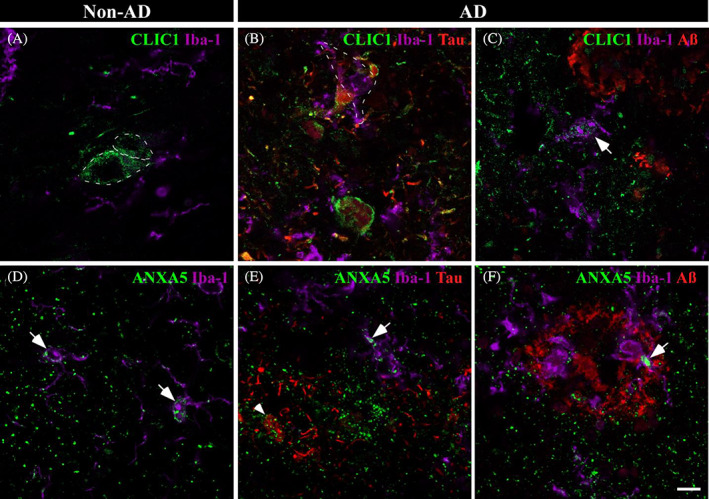
Concerning ANXA1 and PRDX6, dia‐PASEF analysis revealed upregulated expression in AD samples. In non‐AD samples, ANXA1 was expressed in neurons (Figure 7A, dashed line) and, to a lesser extent, in astrocytes (Figure 7A, arrow). Increased ANXA1 was observed in astrocytes in AD samples (Figure 7B, arrow) with tight spatial coexpression with Tau (Figure 7B, arrowhead). Frequently, neurons with Tau deposits were marked with ANXA1 (Figure 7C, dashed line). On the other hand, PRDX6 was associated with astrocytes in non‐AD and AD samples (Figure 7D–F, respectively). Colocalization with pathological markers was observed (Figure 7E,F), with remarkable coexpression with small accumulations of Aβ (Figure 7F, arrow).
FIGURE 7.
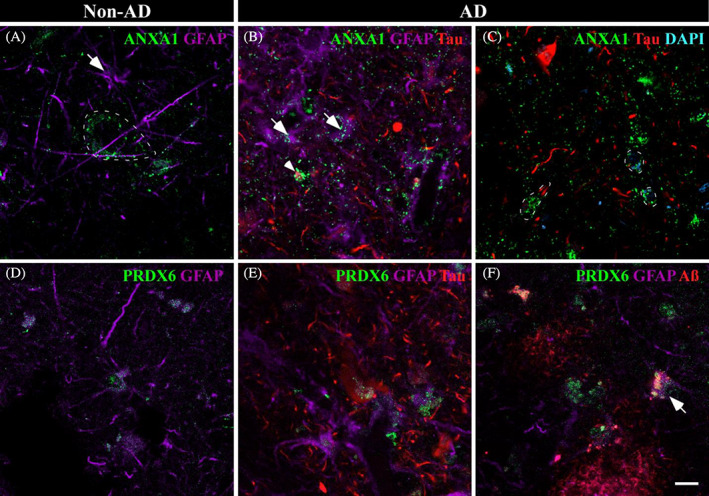
Astroglial participation in AD. Immunofluorescences against ANXA1 and PRDX6 are shown in non‐AD (A and D, respectively) and AD (B,C and E,F, respectively) samples. In non‐AD samples, ANXA1 (A, green) was expressed in neurons (dashed line) and in astrocytes to a lesser extent (purple, arrow). In AD, ANXA1 expression in astrocytes was increased (B, arrow), and ANXA1 was coexpressed with Tau deposits (B, arrowhead). Frequently, neurons with slight Tau staining expressed increased levels of ANXA1 (C, dashed line). PRDX6 (green) expression by astrocytes (purple) was observed in non‐AD (D) and AD (E,F) samples. PRDX6 was related to Tau (red) (E) and Aβ (red) (F) pathology. Scale bar = 10 μm.
4. DISCUSSION
The present work includes a dual approach using stereological and proteomic techniques with the aim of assessing neuronal and glial involvement in the AC in AD. Synaptic alterations as well as the potential participation of glial cells in response to pathology have been identified as particularly relevant in AC pathology in AD.
Amygdala volume reduction has been postulated as a diagnostic criterion in AD [19], since amygdala atrophy has been described as comparable to that in the hippocampus [13]. Specifically, histological analysis and diffeomorphometry highlight the BL and BM as the most affected nuclei in AD [20, 37], and it is also linked to neuronal loss in the different nuclei analyzed [21, 22, 38]. In the present study, amygdala atrophy was confirmed, and the Co and La were identified as the most affected nuclei (Figure 1C). However, the volume reduction was not associated with differences in neuronal populations (Figure 2C) but with neuropil, which could be related to synaptic alterations, as highlighted by proteomic data analysis (Table 4). This is in consonance with the reduction in intrinsic connections in the BLA described in the literature [39]. The discrepancy with previous studies could be explained since no specific cell type markers have been employed to identify neurons, establishing a possible bias in the analysis.
In addition, the glial population has been described to be affected in AD. A reduction in glial cells has been identified in the BL and Co [21], and morphological changes have been described in the latest stages of AD [40]. However, the analysis was conducted with cresyl violet, and glia were differentiated from neurons by morphology, without distinguishing between astrocytes and microglia. Here, we conducted separate analyses of microglia and astrocytes with specific markers, resulting in an increase in astrocytes (Figure 2I) and no variation in the microglial population (Figure 2F). The increase in the number of astrocytes, as well as the microgliosis observed in all analyzed nuclei, might be generated as a response to pathology. Pathological markers have been described to affect different nuclei, since plaques are predominantly present in the BLA, whereas tangles are mainly present in the corticomedial complex [41, 42, 43]. However, we observed a similar distribution pattern of pathology in the AC concerning Tau and Aβ, which appeared as a gradient from the cortical to lateral areas, with the Co, BM, and BL being more affected than the La (Figure 3C,D). Furthermore, TDP‐43‐P pathology observed in the amygdala nuclei resembles Tau and Aβ distribution (Online Resource 10). The involvement of these nuclei could be related to the spread of the disease via connections with the hippocampus and/or olfactory areas [44]. Pathology might propagate from the olfactory and hippocampal areas (early affected in AD) to the Co and BL, respectively. The projections from the Co to CA1 and layer I‐II of the entorhinal cortex (EC), together with the loops established between CA1‐BA‐CA1 and layer V‐BL‐layer III to V of the EC (diffuse projections) [16, 44, 45], might indicate that the AC is a regulator of pathology distribution in these areas [46] (Figure 8).
FIGURE 8.
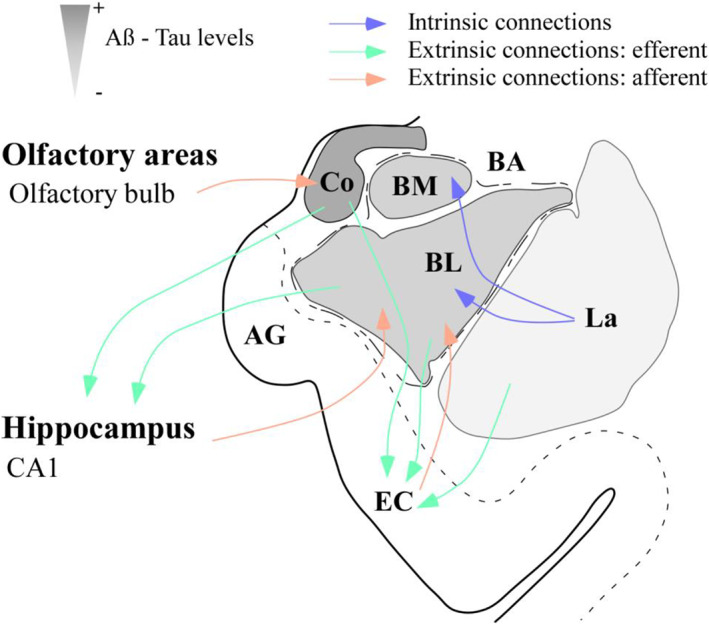
Amygdaloid complex as a “switch” in AD. Scheme of the amygdaloid complex (AC) and its main connections with olfactory areas, the hippocampus, and the entorhinal cortex (EC). Different amygdaloid nuclei are represented in grayscale from more (darker) to less (weaker) affected by pathology. Efferences and afferences regarding olfactory areas, CA1 and the EC might act as vehicles for pathology from and to the AC. AG, ambiens gyrus; BL, basolateral nucleus; BM, basomedial nucleus; Co, cortical nucleus; La, lateral nucleus.
Considering proteomic data analysis, neuronal and glial implications in amygdala pathology were evaluated by confocal microscopy. In this sense, ADAP1, CEND1, and ANXA2 revealed a close linkage with neurons, whereas ANXA1, ANXA5, CLIC1, and PRDX6 may have a potential role in the pathology response through glia.
ADAP1 is a brain‐specific GTPase‐activating protein and a member of the ADP ribosylation factor family; ADAP1 is localized in axonal processes and is frequently associated with presynaptic vesicles. ADAP1 participates in dendritic differentiation since its downregulation inhibits dendritic branching and reduces the length of dendrites, with no effect on axon morphology [47]. Recently, a pathological role of ADAP1 has been described because the increase in its expression has been identified as a response to Aβ, resulting in synaptic dysfunction and negative regulation of memory formation in mouse models [48, 49]. However, to the best of our knowledge, only one previous report has confirmed the increased expression of ADAP1 in human tissue by immunostaining [50]. In contrast, our results revealed a reduction in ADAP1 expression (FC = 0.47574, p value = 0,009) in human amygdala AD samples identified by dia‐PASEF. Furthermore, immunofluorescence revealed reduced labeling in AD samples (Figure 5B,C), possibly because of synaptic dysfunction. These results highlight the need for further studies to elucidate the involvement of ADAP1 in human AD.
CEND1 is a brain‐specific protein that plays an important role in neuronal differentiation [51]. Previous data have reported that CEND1 expression is decreased in the brains of AD mice, resulting in synaptic dysfunction [52]. Here, we found that CEND1 is decreased in AD human samples by dia‐PASEF analysis, confirming previous results in animal models. Although neurons labeled by CEND1 were scarce in AD samples, reduced expression was notable in neurons near Aβ plaques (Figure 5F) compared to Tau deposits (Figure 5E), suggesting a potential involvement of Aβ in CEND1 expression. The reduction in CEND1 in AD may potentiate synaptic dysfunction in human amygdala pathology.
ANXA2 has been described to participate in the redistribution of Tau under pathological conditions [53] and to facilitate autophagosome‐lysosome fusion to reduce Aβ accumulation [54]. Here, we observed increased expression of ANXA2 in AD human amygdala samples according to proteomic data, which is consistent with previous results from our laboratory [28]. ANXA2 was associated with neurons in non‐AD samples (Figure 5G) and particularly with Aβ in AD samples (Figure 6H,I), suggesting the possible engulfment of this marker in the autophagosome‐lysosome system.
CLIC1 is an intracellular chloride channel proposed as a potential marker of neurodegenerative processes [55]. It has been described to participate in the microglial activation induced by Aβ, causing a harmful phenotype that produces reactive oxygen species and, consequently, neuronal death [56]. The blockage of CLIC1 promotes Aβ phagocytosis, inhibiting the neurotoxic phenotype of microglia [57]. In this sense, the increase in CLIC1 observed by proteomic analysis might be linked to inflammatory and neurotoxic processes. CLIC1 expression observed in microglia in AD samples (Figure 6C, arrow) and microglia disposed in close contact with tangles (Figure 6B, dashed line) might induce apoptosis.
A protective role of ANXA5 against Ca2+‐induced damage and reducing Aβ toxicity has been highlighted [58]. Furthermore, ANXA5 has been evaluated as a potential biomarker for AD since its plasma levels are increased in AD [59] and as a potential candidate for monitoring the progression of the disease [60]. A previous report in our laboratory revealed enriched ANXA5 in AD extracts, which was especially noticeable surrounding Aβ plaques [28]. Our results confirmed the elevated expression of ANXA5 in AD samples and identified ANXA5 expression in microglia (Figure 6D, arrow). This increased expression and the ANXA5 interaction with pathological markers (Figure 6E,F) observed by immunofluorescence suggest an attempt to reduce Tau and Aβ toxicity by microglia.
ANXA1 is a proresolving protein that modulates microglial activation and stimulates the phagocytosis of apoptotic neurons by microglia by acting as an “eat me” signal [61]. Consistent with our proteomic results, increased levels of ANXA1 have been previously noted in AD [62]. Preceding reports have assessed expression by microglia, astrocytes, and neurons [63, 64], but we have identified expression exclusively in astrocytes and neurons (Figure 6A–C). In AD, an accumulation of ANXA1 was predominantly observed in neurons with slight Tau deposits (Figure 6C, dashed line). In pathological conditions, astrocytes might express ANXA1 in an attempt to tag neurons “to be degraded” by microglia.
PRDX6 is an antioxidant enzyme, and its increased expression has been associated with astrocytes in AD [65]. Recently, a protective role of astrocytes via PRDX6 in Aβ proteostasis has been highlighted, since increased PRDX6 expression might mediate phagocytic activation of periplaque microglia [66]. Previous proteomic analysis in our laboratory revealed increased PRDX6 in the EC in AD, which was linked to microglia and astrocytes [29]. Here, we also identified an increase in PRDX6 levels in the AD amygdala. PRDX6 was associated with astrocytes in non‐AD and AD samples (Figure 7D–F, respectively), with remarkable colocalization with pathological markers (Figure 7E,F). In Aβ pathology, PRDX6 accumulation was specifically related to astrocytes in close contact with small plaques (Figure 7F, arrow), suggesting its involvement in Aβ proteostasis.
Considering these results, AD pathology in the AC could cause synaptic dysfunction (ADAP1 and CEND1 reduction) accompanied by a glial response to damage. ANXA2 might mediate autophagosome‐lysosome fusion to contain the pathology. Astrocytes, via upregulated PRDX6 expression, might be mediating phagocytic microglia activation, as well as labeling neurons with ANXA1 for microglial degradation. However, microglia might have a dual role involving a protective function of ANXA5 in reducing pathology toxicity and a neurotoxic phenotype related to the increased CLIC1 expression that may promote neuronal damage (Figure 9).
FIGURE 9.
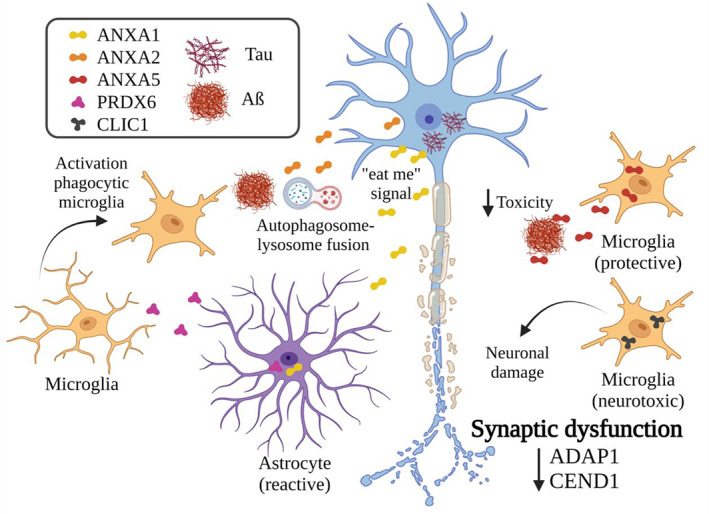
Synaptic and glial responses against injury. Representative scheme of neuronal and glial responses against pathology in the amygdala according to proteomic data analysis and the literature. Reductions in ADAP1 and CEND1 suggest synaptic dysfunction. To control the disease, ANXA2 might mediate autophagosome‐lysosome fusion. Astrocytes might promote the activation of phagocytic microglia (PRDX6) and mark neurons for their clearance by microglia (ANXA1). Microglia might have a dual role since protective (ANXA5) and neurotoxic (CLIC1) roles have been linked. Created with BioRender.com
To the best of our knowledge, this work comprises the first stereological analysis that includes volume and cell population estimations (employing specific cell markers), as well as pathology evaluation considering the same amygdaloid nuclei, facilitating the comprehension of the results. Furthermore, this study constitutes the first proteomic analysis of the human amygdala in AD. The combination of methodologies allowed us to elucidate the possible synaptic alterations as well as the potential participation of glial cells in response to pathology. Astrocytes might facilitate the protective actions of microglia, whereas microglia might play neuroprotective and neurotoxic roles. Moreover, the gradient observed in pathology distribution points out the relevance of the connections with olfactory areas and the hippocampal formation, suggesting a particular participation of the AC in AD.
AUTHOR CONTRIBUTIONS
Conceptualization: Melania Gonzalez‐Rodriguez, Daniel Saiz‐Sanchez, Alino Martinez‐Marcos; Methodology: Melania Gonzalez‐Rodriguez, Sandra Villar‐Conde, Patricia Villanueva‐Anguita; Formal analysis and investigation: Melania Gonzalez‐Rodriguez; Writing—original draft preparation: Melania Gonzalez‐Rodriguez; Writing—review and editing: Sandra Villar‐Conde, Veronica Astillero‐Lopez, Isabel Ubeda‐Banon, Alicia Flores‐Cuadrado, Daniel Saiz‐Sanchez, Alino Martinez‐Marcos; Funding acquisition: Alino Martinez‐Marcos, Daniel Saiz‐Sanchez, Isabel Ubeda‐Banon; Supervision: Alino Martinez‐Marcos, Daniel Saiz‐Sanchez.
FUNDING INFORMATION
The study was sponsored by the University of Castilla‐La Mancha/European Regional Development Fund (2021‐GRIN‐31233 to Alino Martinez–Marcos), Spanish Ministries of Economy and Competitiveness/European Regional Development Fund (grant no. SAF2016‐75768‐R to Alino Martinez‐Marcos) and Science and Innovation (grant no. PID2019‐108659RB‐I00 to Alino Martinez‐Marcos) and Autonomous Government of Castilla‐La Mancha/European Regional Development Fund (grant no. SBPLY/17/180501/000430 to Alino Martinez‐Marcos and Daniel Saiz‐Sanchez and SBPLY/21/180501/000093 to Alino Martinez‐Marcos and Isabel Ubeda‐Banon). Melania Gonzalez‐Rodriguez and Sandra Villar‐Conde held predoctoral fellowships granted by the University of Castilla‐La Mancha/European Social Fund.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interests.
Supporting information
Online Resource 1. Antibodies detail.
Online Resource 2. LC‐MS/MS.
Online Resource 3. Volume data.
Online Resource 4. Increased astroglia density in the amygdaloid nuclei in AD. The MAP2‐positive cells/mm3 (a), Iba‐1‐positive cells/mm3 (b) and GFAP‐positive cells/mm3 (c) in the global AC and the different nuclei are shown (the graphs show the mean ± SEM, *p value <0.05, **p value <0.01, ***p value <0.001). AC, amygdaloid complex (Co, BLA); Co, cortical nucleus; BLA, basolateral complex (BM, BL, La); BM, basomedial nucleus; BL, basolateral nucleus; La, lateral nucleus.
Online Resource 5. MAP2 stereological quantification data.
Online Resource 6. Iba‐1 stereological quantification data.
Online Resource 7. GFAP stereological quantification data.
Online Resource 8. Aβ stereological quantification data.
Online Resource 9. Tau stereological quantification data.
Online Resource 10. Phosphorylated TDP‐43 (TDP‐43‐P) deposits in amygdala in AD. (a) Immunohistochemistry against TDP‐43‐P in AC in AD (case 2). Note different deposition pattern between nuclei. (b) Abundant cellular accumulations of TDP‐43‐P can be observed in Co, while in (c) BM and (d) BLTDP‐43‐P appears in clusters. In contrast, (e) scarce deposits are in La. Co: Cortical nucleus, BM: Basomedial nucleus, BL: Basolateral nucleus, La: Lateral nucleus, EC: Entorhinal cortex. Scale bar = 1000 μm in (a); and 50 μm in (b,c,d,e).
Online Resource 11. (a) Protein lists that interact with APP or MAPT, and preferentially expressed in neurons, microglia and astrocytes. (b) SynGo analysis from 178 DEP (dataset version: 20210225). (c) Metascape analysis of 178 DEP.
Online Resource 12. Proteomic analysis. Volcano plot (a) showed 108 up‐(green) and 70 downregulated (red) DEPs (FC > 1.5, p value <0.05). Venn diagrams identified that are present in the different cellular types, neurons (b), microglia (c) and astrocytes (d), and interact with pathological proteinsin AD. Cellular responses to stress, regulation of proteolysis and response to wounding were exposed as impaired processes by functional analysis with Metascape (f). DEPs: differentially expressed proteins.
ACKNOWLEDGMENTS
The authors are indebted to our donors and to the Spanish National Biobanks Network for providing the samples used in this study. Artwork was created with Canvas and BioRender.com. This study is part of the doctoral thesis of Melania Gonzalez‐Rodriguez.
Gonzalez‐Rodriguez M, Villar‐Conde S, Astillero‐Lopez V, Villanueva‐Anguita P, Ubeda‐Banon I, Flores‐Cuadrado A, et al. Human amygdala involvement in Alzheimer's disease revealed by stereological and dia‐PASEF analysis. Brain Pathology. 2023;33(5):e13180. 10.1111/bpa.13180
Contributor Information
Alino Martinez‐Marcos, Email: alino.martinez@uclm.es.
Daniel Saiz‐Sanchez, Email: daniel.saiz@uclm.es.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article (and its additional files). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [39] partner repository with the dataset identifier PXD038322.
REFERENCES
- 1. Tromp D, Dufour A, Lithfous S, Pebayle T, Despres O. Episodic memory in normal aging and Alzheimer disease: insights from imaging and behavioral studies. Ageing Res Rev. 2015;24:232–62. 10.1016/j.arr.2015.08.006 [DOI] [PubMed] [Google Scholar]
- 2. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. 10.1212/wnl.58.12.1791 [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. 10.1007/s00401-006-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239–59. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 5. Peng C, Trojanowski JQ, Lee VM. Protein transmission in neurodegenerative disease. Nat Rev Neurol. 2020;16:199–212. 10.1038/s41582-020-0333-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walker LC. Prion‐like mechanisms in Alzheimer disease. Handb Clin Neurol. 2018;153:303–19. 10.1016/B978-0-444-63945-5.00016-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain. 2015;138:2814–33. 10.1093/brain/awv236 [DOI] [PubMed] [Google Scholar]
- 8. Gal J, Chen J, Katsumata Y, Fardo DW, Wang WX, Artiushin S, et al. Detergent insoluble proteins and inclusion body‐like structures immunoreactive for PRKDC/DNA‐PK/DNA‐PKcs, FTL, NNT, and AIFM1 in the amygdala of cognitively impaired elderly persons. J Neuropathol Exp Neurol. 2018;77:21–39. 10.1093/jnen/nlx097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gal J, Katsumata Y, Zhu H, Srinivasan S, Chen J, Johnson LA, et al. Apolipoprotein E proteinopathy is a major dementia‐associated pathologic biomarker in individuals with or without the APOE epsilon 4 allele. Am J Pathol. 2022;192:564–78. 10.1016/j.ajpath.2021.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meneses A, Koga S, O'Leary J, Dickson DW, Bu G, Zhao N. TDP‐43 pathology in Alzheimer's disease. Mol Neurodegener. 2021;16:84. 10.1186/s13024-021-00503-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503–27. 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mears D, Pollard HB. Network science and the human brain: using graph theory to understand the brain and one of its hubs, the amygdala, in health and disease. J Neurosci Res. 2016;94:590–605. 10.1002/jnr.23705 [DOI] [PubMed] [Google Scholar]
- 13. Poulin SP, Dautoff R, Morris JC, Barrett LF, Dickerson BC, Alzheimer's Disease Neuroimaging Initiative . Amygdala atrophy is prominent in early Alzheimer's disease and relates to symptom severity. Psychiatry Res. 2011;194:7–13. 10.1016/j.pscychresns.2011.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Son G, Jahanshahi A, Yoo SJ, Boonstra JT, Hopkins DA, Steinbusch HWM, et al. Olfactory neuropathology in Alzheimer's disease: a sign of ongoing neurodegeneration. BMB Rep. 2021;54:295–304. 10.5483/BMBRep.2021.54.6.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ubeda‐Banon I, Saiz‐Sanchez D, Flores‐Cuadrado A, Rioja‐Corroto E, Gonzalez‐Rodriguez M, Villar‐Conde S, et al. The human olfactory system in two proteinopathies: Alzheimer's and Parkinson's diseases. Transl Neurodegener. 2020;9:22. 10.1186/s40035-020-00200-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McDonald AJ, Mott DD. Functional neuroanatomy of amygdalohippocampal interconnections and their role in learning and memory. J Neurosci Res. 2017;95:797–820. 10.1002/jnr.23709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mori E, Ikeda M, Hirono N, Kitagaki H, Imamura T, Shimomura T. Amygdalar volume and emotional memory in Alzheimer's disease. Am J Psychiatry. 1999;156:216–22. 10.1176/ajp.156.2.216 [DOI] [PubMed] [Google Scholar]
- 18. Simic G, Tkalcic M, Vukic V, Mulc D, Spanic E, Sagud M, et al. Understanding emotions: origins and roles of the amygdala. Biomolecules. 2021;11:823. 10.3390/biom11060823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang DW, Ding SL, Bian XL, Zhou SY, Yang H, Wang P. Diagnostic value of amygdala volume on structural magnetic resonance imaging in Alzheimer's disease. World J Clin Cases. 2021;9:4627–36. 10.12998/wjcc.v9.i18.4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scott SA, DeKosky ST, Scheff SW. Volumetric atrophy of the amygdala in Alzheimer's disease: quantitative serial reconstruction. Neurology. 1991;41:351–6. 10.1212/wnl.41.3.351 [DOI] [PubMed] [Google Scholar]
- 21. Scott SA, DeKosky ST, Sparks DL, Knox CA, Scheff SW. Amygdala cell loss and atrophy in Alzheimer's disease. Ann Neurol. 1992;32:555–63. 10.1002/ana.410320412 [DOI] [PubMed] [Google Scholar]
- 22. Vereecken TH, Vogels OJ, Nieuwenhuys R. Neuron loss and shrinkage in the amygdala in Alzheimer's disease. Neurobiol Aging. 1994;15:45–54. 10.1016/0197-4580(94)90143-0 [DOI] [PubMed] [Google Scholar]
- 23. Li Q, Haney MS. The role of glia in protein aggregation. Neurobiol Dis. 2020;143:105015. 10.1016/j.nbd.2020.105015 [DOI] [PubMed] [Google Scholar]
- 24. Wei Y, Liu M, Wang D. The propagation mechanisms of extracellular tau in Alzheimer's disease. J Neurol. 2022;269:1164–81. 10.1007/s00415-021-10573-y [DOI] [PubMed] [Google Scholar]
- 25. Fleeman RM, Proctor EA. Astrocytic propagation of tau in the context of Alzheimer's disease. Front Cell Neurosci. 2021;15:645233. 10.3389/fncel.2021.645233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fakhoury M. Microglia and astrocytes in Alzheimer's disease: implications for therapy. Curr Neuropharmacol. 2018;16:508–18. 10.2174/1570159X15666170720095240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fernandez‐Irigoyen J, Zelaya MV, Santamaria E. Applying mass spectrometry‐based qualitative proteomics to human amygdaloid complex. Front Cell Neurosci. 2014;8:80. 10.3389/fncel.2014.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pedrero‐Prieto CM, Flores‐Cuadrado A, Saiz‐Sanchez D, Ubeda‐Banon I, Frontinan‐Rubio J, Alcain FJ, et al. Human amyloid‐beta enriched extracts: evaluation of in vitro and in vivo internalization and molecular characterization. Alzheimers Res Ther. 2019;11:56. 10.1186/s13195-019-0513-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Astillero‐Lopez V, Gonzalez‐Rodriguez M, Villar‐Conde S, Flores‐Cuadrado A, Martinez‐Marcos A, Ubeda‐Banon I, et al. Neurodegeneration and astrogliosis in the entorhinal cortex in Alzheimer's disease: stereological layer‐specific assessment and proteomic analysis. Alzheimers Dement. 2022;18:2468–80. 10.1002/alz.12580 [DOI] [PubMed] [Google Scholar]
- 30. Gonzalez‐Rodriguez M, Villar‐Conde S, Astillero‐Lopez V, Villanueva‐Anguita P, Ubeda‐Banon I, Flores‐Cuadrado A, et al. Neurodegeneration and astrogliosis in the human CA1 hippocampal subfield are related to hsp90ab1 and bag3 in Alzheimer's disease. Int J Mol Sci. 2021;23:165. 10.3390/ijms23010165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Villar‐Conde S, Astillero‐Lopez V, Gonzalez‐Rodriguez M, Villanueva‐Anguita P, Saiz‐Sanchez D, Martinez‐Marcos A, et al. The human hippocampus in Parkinson's disease: an integrative stereological and proteomic study. J Parkinsons Dis. 2021;11:1345–65. 10.3233/JPD-202465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perez‐Riverol Y, Bai J, Bandla C, Garcia‐Seisdedos D, Hewapathirana S, Kamatchinathan S, et al. The PRIDE database resources in 2022: a hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Res. 2022;50:D543–52. 10.1093/nar/gkab1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun. 2019;10:1523. 10.1038/s41467-019-09234-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–9. 10.1093/nar/gkj109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, et al. A multi‐network approach identifies protein‐specific co‐expression in asymptomatic and symptomatic Alzheimer's disease. Cell Syst. 2017;4:60–72.e4. 10.1016/j.cels.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mai JK, Paxinos G, Voss T. Atlas of the human brain. San Diego, CA: Academic Press; 2007. [Google Scholar]
- 37. Miller MI, Younes L, Ratnanather JT, Brown T, Trinh H, Lee DS, et al. Amygdalar atrophy in symptomatic Alzheimer's disease based on diffeomorphometry: the BIOCARD cohort. Neurobiol Aging. 2015;36(Suppl 1):S3–S10. 10.1016/j.neurobiolaging.2014.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coleman PD, Flood DG. Neuron numbers and dendritic extent in normal aging and Alzheimer's disease. Neurobiol Aging. 1987;8:521–45. 10.1016/0197-4580(87)90127-8 [DOI] [PubMed] [Google Scholar]
- 39. Ortner M, Pasquini L, Barat M, Alexopoulos P, Grimmer T, Forster S, et al. Progressively disrupted intrinsic functional connectivity of basolateral amygdala in very early Alzheimer's disease. Front Neurol. 2016;7:132. 10.3389/fneur.2016.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murphy GM Jr, Ellis WG, Lee YL, Stultz KE, Shrivastava R, Tinklenberg JR, et al. Astrocytic gliosis in the amygdala in Down's syndrome and Alzheimer's disease. Prog Brain Res. 1992;94:475–83. 10.1016/s0079-6123(08)61774-4 [DOI] [PubMed] [Google Scholar]
- 41. Kromer Vogt LJ, Hyman BT, Van Hoesen GW, Damasio AR. Pathological alterations in the amygdala in Alzheimer's disease. Neuroscience. 1990;37:377–85. 10.1016/0306-4522(90)90408-v [DOI] [PubMed] [Google Scholar]
- 42. Sahin HA, Emre M, Ziabreva I, Perry E, Celasun B, Perry R. The distribution pattern of pathology and cholinergic deficits in amygdaloid complex in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2006;111:115–25. 10.1007/s00401-005-0003-2 [DOI] [PubMed] [Google Scholar]
- 43. Tsuchiya K, Kosaka K. Neuropathological study of the amygdala in presenile Alzheimer's disease. J Neurol Sci. 1990;100:165–73. 10.1016/0022-510x(90)90029-m [DOI] [PubMed] [Google Scholar]
- 44. Benarroch EE. The amygdala: functional organization and involvement in neurologic disorders. Neurology. 2015;84:313–24. 10.1212/WNL.0000000000001171 [DOI] [PubMed] [Google Scholar]
- 45. Amaral DG. Amygdalohippocampal and amygdalocortical projections in the primate brain. Adv Exp Med Biol. 1986;203:3–17. 10.1007/978-1-4684-7971-3_1 [DOI] [PubMed] [Google Scholar]
- 46. Nelson PT, Abner EL, Patel E, Anderson S, Wilcock DM, Kryscio RJ, et al. The amygdala as a locus of pathologic misfolding in neurodegenerative diseases. J Neuropathol Exp Neurol. 2018;77:2–20. 10.1093/jnen/nlx099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stricker R, Reiser G. Functions of the neuron‐specific protein ADAP1 (centaurin‐alpha1) in neuronal differentiation and neurodegenerative diseases, with an overview of structural and biochemical properties of ADAP1. Biol Chem. 2014;395:1321–40. 10.1515/hsz-2014-0107 [DOI] [PubMed] [Google Scholar]
- 48. Szatmari EM, Moran C, Cohen S, Jacob A, Parra‐Bueno P, Kamasawa N, et al. ADAP1/centaurin‐alpha1 negatively regulates dendritic spine function and memory formation in the hippocampus. eNeuro. 2021;8:ENEURO.0111–20.2020. 10.1523/ENEURO.0111-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Szatmari EM, Oliveira AF, Sumner EJ, Yasuda R. Centaurin‐alpha1‐Ras‐Elk‐1 signaling at mitochondria mediates beta‐amyloid‐induced synaptic dysfunction. J Neurosci. 2013;33:5367–74. 10.1523/JNEUROSCI.2641-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reiser G, Bernstein HG. Neurons and plaques of Alzheimer's disease patients highly express the neuronal membrane docking protein p42IP4/centaurin alpha. Neuroreport. 2002;13:2417–9. 10.1097/00001756-200212200-00008 [DOI] [PubMed] [Google Scholar]
- 51. Politis PK, Makri G, Thomaidou D, Geissen M, Rohrer H, Matsas R. BM88/CEND1 coordinates cell cycle exit and differentiation of neuronal precursors. Proc Natl Acad Sci U S A. 2007;104:17861–6. 10.1073/pnas.0610973104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xie W, Guo D, Li J, Yue L, Kang Q, Chen G, et al. CEND1 deficiency induces mitochondrial dysfunction and cognitive impairment in Alzheimer's disease. Cell Death Differ. 2022;29:2417–28. 10.1038/s41418-022-01027-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gauthier‐Kemper A, Suarez Alonso M, Sundermann F, Niewidok B, Fernandez MP, Bakota L, et al. Annexins A2 and A6 interact with the extreme N terminus of tau and thereby contribute to tau's axonal localization. J Biol Chem. 2018;293:8065–76. 10.1074/jbc.RA117.000490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bustos V, Pulina MV, Bispo A, Lam A, Flajolet M, Gorelick FS, et al. Phosphorylated Presenilin 1 decreases beta‐amyloid by facilitating autophagosome‐lysosome fusion. Proc Natl Acad Sci U S A. 2017;114:7148–53. 10.1073/pnas.1705240114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carlini V, Verduci I, Cianci F, Cannavale G, Fenoglio C, Galimberti D, et al. CLIC1 protein accumulates in circulating monocyte membrane during neurodegeneration. Int J Mol Sci. 2020;21:1484. 10.3390/ijms21041484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Milton RH, Abeti R, Averaimo S, DeBiasi S, Vitellaro L, Jiang L, et al. CLIC1 function is required for beta‐amyloid‐induced generation of reactive oxygen species by microglia. J Neurosci. 2008;28:11488–99. 10.1523/JNEUROSCI.2431-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Paradisi S, Matteucci A, Fabrizi C, Denti MA, Abeti R, Breit SN, et al. Blockade of chloride intracellular ion channel 1 stimulates Abeta phagocytosis. J Neurosci Res. 2008;86:2488–98. 10.1002/jnr.21693 [DOI] [PubMed] [Google Scholar]
- 58. Bedrood S, Jayasinghe S, Sieburth D, Chen M, Erbel S, Butler PC, et al. Annexin A5 directly interacts with amyloidogenic proteins and reduces their toxicity. Biochemistry. 2009;48:10568–76. 10.1021/bi900608m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sohma H, Imai S, Takei N, Honda H, Matsumoto K, Utsumi K, et al. Evaluation of Annexin A5 as a biomarker for Alzheimer's disease and dementia with lewy bodies. Front Aging Neurosci. 2013;5:15. 10.3389/fnagi.2013.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Muraoka S, DeLeo AM, Sethi MK, Yukawa‐Takamatsu K, Yang Z, Ko J, et al. Proteomic and biological profiling of extracellular vesicles from Alzheimer's disease human brain tissues. Alzheimers Dement. 2020;16:896–907. 10.1002/alz.12089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McArthur S, Cristante E, Paterno M, Christian H, Roncaroli F, Gillies GE, et al. Annexin A1: a central player in the anti‐inflammatory and neuroprotective role of microglia. J Immunol. 2010;185:6317–28. 10.4049/jimmunol.1001095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chua XY, Chong JR, Cheng AL, Lee JH, Ballard C, Aarsland D, et al. Elevation of inactive cleaved Annexin A1 in the neocortex is associated with amyloid, inflammatory and apoptotic markers in neurodegenerative dementias. Neurochem Int. 2022;152:105251. 10.1016/j.neuint.2021.105251 [DOI] [PubMed] [Google Scholar]
- 63. Eberhard DA, Brown MD, VandenBerg SR. Alterations of annexin expression in pathological neuronal and glial reactions. Immunohistochemical localization of Annexins I, II (p36 and p11 subunits), IV, and VI in the human hippocampus. Am J Pathol. 1994;145:640–9. [PMC free article] [PubMed] [Google Scholar]
- 64. Ries M, Loiola R, Shah UN, Gentleman SM, Solito E, Sastre M. The anti‐inflammatory Annexin A1 induces the clearance and degradation of the amyloid‐beta peptide. J Neuroinflammation. 2016;13:234. 10.1186/s12974-016-0692-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Power JH, Asad S, Chataway TK, Chegini F, Manavis J, Temlett JA, et al. Peroxiredoxin 6 in human brain: molecular forms, cellular distribution and association with Alzheimer's disease pathology. Acta Neuropathol. 2008;115:611–22. 10.1007/s00401-008-0373-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pankiewicz JE, Diaz JR, Marta‐Ariza M, Lizinczyk AM, Franco LA, Sadowski MJ. Peroxiredoxin 6 mediates protective function of astrocytes in Abeta proteostasis. Mol Neurodegener. 2020;15:50. 10.1186/s13024-020-00401-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Resource 1. Antibodies detail.
Online Resource 2. LC‐MS/MS.
Online Resource 3. Volume data.
Online Resource 4. Increased astroglia density in the amygdaloid nuclei in AD. The MAP2‐positive cells/mm3 (a), Iba‐1‐positive cells/mm3 (b) and GFAP‐positive cells/mm3 (c) in the global AC and the different nuclei are shown (the graphs show the mean ± SEM, *p value <0.05, **p value <0.01, ***p value <0.001). AC, amygdaloid complex (Co, BLA); Co, cortical nucleus; BLA, basolateral complex (BM, BL, La); BM, basomedial nucleus; BL, basolateral nucleus; La, lateral nucleus.
Online Resource 5. MAP2 stereological quantification data.
Online Resource 6. Iba‐1 stereological quantification data.
Online Resource 7. GFAP stereological quantification data.
Online Resource 8. Aβ stereological quantification data.
Online Resource 9. Tau stereological quantification data.
Online Resource 10. Phosphorylated TDP‐43 (TDP‐43‐P) deposits in amygdala in AD. (a) Immunohistochemistry against TDP‐43‐P in AC in AD (case 2). Note different deposition pattern between nuclei. (b) Abundant cellular accumulations of TDP‐43‐P can be observed in Co, while in (c) BM and (d) BLTDP‐43‐P appears in clusters. In contrast, (e) scarce deposits are in La. Co: Cortical nucleus, BM: Basomedial nucleus, BL: Basolateral nucleus, La: Lateral nucleus, EC: Entorhinal cortex. Scale bar = 1000 μm in (a); and 50 μm in (b,c,d,e).
Online Resource 11. (a) Protein lists that interact with APP or MAPT, and preferentially expressed in neurons, microglia and astrocytes. (b) SynGo analysis from 178 DEP (dataset version: 20210225). (c) Metascape analysis of 178 DEP.
Online Resource 12. Proteomic analysis. Volcano plot (a) showed 108 up‐(green) and 70 downregulated (red) DEPs (FC > 1.5, p value <0.05). Venn diagrams identified that are present in the different cellular types, neurons (b), microglia (c) and astrocytes (d), and interact with pathological proteinsin AD. Cellular responses to stress, regulation of proteolysis and response to wounding were exposed as impaired processes by functional analysis with Metascape (f). DEPs: differentially expressed proteins.
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its additional files). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [39] partner repository with the dataset identifier PXD038322.