

Abstract
DNA methylation of cytosine-guanine sites (CpGs) is associated with type 1 diabetes (T1D). The sequence of methylated and non-methylated sites in a specific genetic region constitutes its methyl-haplotype. The aim of the present study was to identify insulin gene promoter (IGP) methyl-haplotypes among children and adolescents with T1D and suggest a predictive model for the discrimination of cases and controls according to methyl-haplotypes. A total of 40 individuals (20 T1D) participated. The IGP region from peripheral whole blood DNA of 40 participants (20 T1D) was sequenced using next-generation sequencing, sequences were read using FASTQ files and methylation status was calculated by python-based pipeline for targeted deep bisulfite sequenced amplicons (ampliMethProfiler). Methylation profile at 10 CpG sites proximal to transcription start site of the IGP was recorded and coded as 0 for unmethylation or 1 for methylation. A single read could result in ‘1111111111’ methyl-haplotype (all methylated), ‘000000000’ methyl-haplotype (all unmethylated) or any other combination. Principal component analysis was applied to the generated methyl-haplotypes for dimensionality reduction, and the first three principal components were employed as features with five different classifiers (random forest, decision tree, logistic regression, Naive Bayes, support vector machine). Naive Bayes was the best-performing classifier, with 0.9 accuracy. Predictive models were evaluated using receiver operating characteristics (AUC 0.96). Methyl-haplotypes ‘1111111111’, ‘1111111011’, ‘1110111111’, ‘1111101111’ and ‘1110101111’ were revealed to be the most significantly associated with T1D according to the dimensionality reduction method. Methylation-based biomarkers such as IGP methyl-haplotypes could serve to identify individuals at high risk for T1D.
Keywords: children, adolescents, type 1 diabetes, insulin gene, epigenetics, CpG, methylation
Introduction
Type 1 diabetes (T1D) is a multifactorial autoimmune disease caused by the complex interaction between genes and environment. Although the major role of genetic alterations in T1D pathogenesis has been extensively supported, the influence of environmental factors remains less clear. As a result, the research interest has shifted to the elucidation of the effect of environmental triggering gene expression (1,2).
Epigenetics, the study of stable and mitotic inherited changes in the expression of genes that do not directly alter the original DNA sequence, is one of the proposed mechanisms (3-5). The main and, at the same time, the most studied epigenetic mechanism is DNA methylation, which involves the addition of a methyl group at specific sites in the molecular sequence (3,4). As a result, cytosine-guanine sites (CpGs) in the genome may be identified as methylated or unmethylated (3,4). Methylation is associated with the development of T1D by altering the expression of genes associated with the immune response implicating pancreatic β-cells (1). The combination of the sequence of methylated and non-methylated sites in a specific genetic region constitutes its methylation haplotype (methyl-haplotype), following the pattern of the DNA base sequence (6).
The insulin gene (INS) is the second most important gene after the human leucocyte antigen complex in the development of T1D and is responsible for ~10% of the genetic risk of the disease (7). It is involved in the pathogenesis of T1D by acting as an immunoregulatory agent that inhibits cellular stress or even the death of pancreatic β-cells (7). INS promoter (IGP) is a molecular site of great importance, including the information that regulates the loci expression. Epigenetic changes in this site are of exceptional clinical significance.
The aim of the present study was to create a predictive model that classified individuals with T1D and healthy individuals using methylation haplotypes. There is great interest in the area, with studies using different observational techniques (8,9). Towards this direction, there are reports that explore the use of machine learning methods in T1D (10) and identify epigenetic differentiations with prognostic value (11). Specifically, studies are using machine learning algorithms, fed with a gene signature deriving from gene expression (12) or daily life data (13) to diagnose diabetes, while others use deep learning algorithms to classify diabetic and healthy cohorts (14). Regarding the prognostic value of methylation haplotypes, research has focused on the early detection of carcinogenesis (15) and only very recently as an autoimmunity biomarker (16).
In the present study, methylation haplotypes were used as features to feed the classification algorithms. After the initial data preprocessing, an unsupervised clustering method was implemented to explore the differentiation between patients and healthy individuals. For the feature extraction, a classic method was not used for the selection of the significant features, as implemented in a previous study (6), but a dimensionality reduction algorithm was performed that used the new fewer variables as features.
Materials and methods
Data
The present study (ClinicalTrials.gov identifier, ΝCT04139369) is an original observational clinical study with cross-sectional protocol design, which took place at the Second Department of Pediatrics, Faculty of Health Sciences, School of Medicine, Aristotle University of Thessaloniki (Thessaloniki, Greece) in collaboration with the Laboratory of Computing, Medical Informatics and Biomedical Imaging Technologies, Faculty of Health Sciences, School of Medicine, Aristotle University of Thessaloniki (Thessaloniki, Greece).
All participants and their guardians were informed in detail about the aims and content of the study and gave written consent for their participation. The study was carried out in accordance with the rules of the Declaration of Helsinki of 1975, revised in 2013, after the approval from the Bioethics Committee of the School of Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki (approval no. 185/30.12.2015).
The study population consisted of 20 children and adolescents with T1D and 20 healthy children and adolescents matched for age and sex (Table I). Recruitment was performed between January 2016 and February 2019. According to the inclusion criteria of the present study, all participants were non-consanguineous and at least three generations of Greek origin. An additional inclusion criterion of the healthy group was a negative family history of T1D or any other autoimmune disease. Exclusion criteria were the presence of any chronic disease for the healthy group or the presence of any chronic disease apart from T1D for the T1D group. Patients were followed up at the Unit of Pediatric Endocrinology and Metabolism and the Unit of Diabetes Mellitus of children and adolescents of the Second Department of Pediatrics, Faculty of Health Sciences, School of Medicine, Aristotle University of Thessaloniki, AHEPA University Hospital.
Table I.
Demographic characteristics of the study population, presented per group.
| Characteristic | Group Α (healthy controls) | Group Β (T1D) | P-value |
|---|---|---|---|
| Number | 20.00 | 20.00 | |
| Sex (female/male) | 12.00/8.00 | 8.00/12.00 | 0.206 |
| Age, years | 13.93±6.20 | 13.18±3.79 | 0.559 |
| Age at T1D diagnosis, years | - | 7.03±4.00 | - |
| Duration of T1D, years | - | 6.15±4.12 | - |
T1D, type 1 diabetes.
Sample representativeness
The calculation of sample size for continuous variables with respect to the INS gene was based on data on the standard deviation of the INS gene methylation variation in healthy participants (5%) (17). To detect a real potential difference of 5% in the variation of the methylation levels of the INS gene between the compared groups (statistical power, 80%; false negative rate β=0.2; probability of error type Ia=0.05), a total of 16 cases of each study group was necessary.
Definitions
The diagnosis of T1D was made based on the current diagnostic criteria of the International Society for Pediatric and Adolescent Diabetes and the American Diabetes Association (18,19).
Analysis
DNA of all participants was extracted from a whole peripheral blood sample which was immediately stored in a deep freezer (-80˚C) until processed. Total DNA was isolated using a special isolation kit (QIAamp® DNA Blood Mini kit; Qiagen, Inc.) according to the manufacturer’s instructions. The isolated DNA samples were quantified spectrophotometrically using the ratio OD 260/280 (1OD=50 µg/ml) (BioPhotometer 6131; Eppendorf) and the amount of isolated material was checked by 1.5% agarose gel electrophoresis. DNA modification was followed in a quantity of 300 ng from each sample, with the use of the EZ DNA Methylation-Gold™ Kit (Methylation Gold kit; Zymo Research Corp.).
The studied molecular locus (IGP) was then amplified by a standard PCR protocol (PCR conditions: Initial denaturation at 95˚C for 3 min; 40 cycles of 95˚C denaturation for 30 sec, 55˚C annealing for 30 sec and 72˚C extension for 2.5 min; 1 min final extension step at 72˚C) using the following primers: INS forward, 5'-TATTTTGGAATTTTGAGTTTATT-3' and INS reverse, 5'-AACAAAAATCTAAAAACAACAA-3'. In addition, an overhang adapter sequence was added to the gene-specific primers for the regions to be targeted (Nextera Transposase Adaptors; Illumina, Inc.), for prompt construction of the next-generation sequencing (NGS) libraries. The promoter-specific primers targeted the NGS libraries using the Transposase adapter (read_1 forward 5'-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3', read_2 reverse 5'-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3'). PCR products were amplified using a low temperature ramping instrument (9700 Thermal Cycler; Eppendorf AG; cat. no. 5341) using the preset 9600 emulation mode. The reaction solution consisted of AmpliTaq Gold DNA Polymerase with Buffer II and MgCl2 (Applied Biosystems; Thermo Fisher Scientific, Inc.). The total reaction volume was 25 µl, which consisted of 1.3 µl bisulfite-treated DNA, 2.5 µl 10X Buffer (100 mM Tris-HCl, pH 8.3, 500 mM KCl), 0.2 µM of each primer, 200 µM dNTPs mix, 2 mM MgCl2 and 1.25 units AmpliGold Taq Polymerase.
After purification of the PCR products using NucleoMag NGS Clean-up and Size Select (cat. no. 744970.5; Macherey-Nagel GmbH), they were pooled at similar molar quantities and submitted for library construction according to the manufacturer's protocol (Nextera XT DNA Library Preparation kit; cat. no. FC-131-1096; Illumina, Inc.). For NGS, the readings (pair-end reads) were chosen to have a read length format of 2x250 bp on a MiSeq® System Platform (cat. no. SY-410-1003; Illumina, Inc.). FASTQ files were used to read the sequence. The state of methylation was calculated with the ampliMethProfiler tool (20), which is a conductor based on the Python programming language (Python-based pipeline) and aims to extract and analyze the synthesis of epitype sequences resulting from treatment with sulfite. Methylation status was analyzed at 10 distinct CpG sites of the IGP surrounding the transcription start site (TSS) at the 5'-end of the sequence of the INS gene (Supplementary data).
The methylation status at each of the 10 distinct and default CpGs sites of IGP was encoded with the character 0 (zero) when it was found non-methylated or encoded with the character 1 (one) when it was found methylated. The combination of the codes for the 10 default CpGs sites was the methylation pattern of each individual for the IGP promoter, forming its methyl-haplotype. Thus, each reading of the 10 CpGs of each participant could yield methyl-haplotype ‘1111111111’ (complete methylation in all sites), methyl-haplotype ‘000000000’ (complete non-methylation in all sites) or any other methyl-haplotype combination. Data derived from the total reads of the samples are the methyl-haplotypes that were thereafter analyzed as distinct features in bioinformatics analyses.
Descriptional statistics
Demographic characteristics of the study population were analyzed. Normality of distribution for continuous data was examined using the Shapiro-Wilk test. Continuous variables were expressed as mean ± standard deviation and comparisons between groups were performed applying the independent-samples Student's t-test or its non-parametric equivalent Mann-Whitney-U test. Distribution of categorical variables among groups was compared using the χ2 test. Data analysis was performed using SPSS 19.0 (IBM Corp.). P#x003C;0.05 was considered to indicate a statistically significant difference.
Data preprocessing
The first step in data preprocessing was to remove the low variance features. This is achieved by applying a low-variance filter. This filter removes the features that have constant or close to constant values among samples, based on a threshold. The threshold selected was ‘0.3’, which means that features (methyl-haplotypes) that have a constant value in 70% of the samples were detected and removed.
Unsupervised clustering as an exploratory step
The second step was to perform spectral clustering (SP) (21). SC is a clustering method in which the algorithm detects clusters with similar characteristics in a dataset. This method was applied as an exploratory unsupervised clustering method, showing how the data were clustered without imposing the knowledge of the two groups. The purpose of this implementation was to explore the dataset that is derived from an innovative method and has specific properties, and to identify if the two groups were divided properly. The parameters used for the spectral clustering analysis implementation were: i) The nearest neighbors' method for the construction of the affinity matrix; and ii) the k-means method for the assignment of labels in the embedding space.
Transformation and feature extraction
As a third step, a dimensionality reduction algorithm, principal component analysis (PCA) (22), was applied to the dataset. With this method, the features were reduced to new, fewer variables [principal components (PC)] and data are expressed in terms of these variables. Considered together, the new variables represent the same amount of information as the original variables, by keeping the same summed value of variance with the initial dataset.
Classification
Finally, PCs were used as the features that fed the classification algorithms for the implementation of the predictive models. Five classification algorithms were tested: i) Random forest; ii) decision tree; iii) linear regression; iv) Naive Bayes; and v) support vector machine (linear). The evaluation of the models was performed using the k-fold cross-validation method and the evaluation metrics were: i) Accuracy; ii) precision; iii) recall; iv) F1-score; and v) area under curve. All methods were implemented in python 3 and scikit-learn library (23) for machine learning in python.
Results
Data
The study population was composed of two groups of 20 participants each (class A, healthy individuals; class B, individuals with T1D), aged 2-17 years. The demographic characteristics of the sample per patient group are presented in detail in Table I.
Based on the design of the study, there was no difference between the two groups in the distribution of both gender (P=0.206) and age (P=0.559). The body mass index also showed no difference between the two groups (P=0.119, data not shown). In group B, the diagnosis of T1D had been made in all patients before the age of 15 years (mean age of diagnosis, 7.03 years), with the manifestation of diabetic ketoacidosis in most cases (16/20). Glycemic regulation of T1D patients was optimum (HbA1c%, 7.76±0.94). None of the participants had T1D complications.
The initial frequency range of methyl-haplotypes detected by reading the 10 IGP methylation sites in the general population was [0, 1478]. After normalization of all methyl-haplotype data, the value range was changed from [0, 1478] to [0, 99.27].
Analysis. Preprocessing
The initial shape of the dataset was [40, 469]. After the low variance columns removal, 133 columns were removed, and the final shape of the dataset was [40, 336].
Unsupervised clustering
To explore the differentiation between the two groups, a spectral clustering algorithm was applied as an unsupervised clustering method. In Fig. 1, two scatter plots are presented. In these plots and for visualization purposes, a dimensionality reduction algorithm was implemented to present the results, creating two PC. These plots show the distribution of the subjects divided into the two groups using: i) The ground truth for the characterization of each subject; and ii) the prediction of spectral clustering. Table II demonstrates the validation metrics of this method, based on the confusion matrix between actual groups and separation in two clusters. The two groups seem to be clustered with high accuracy.
Figure 1.

Spectral clustering. Each point is one subject, and the two groups have a different sign (dot, type 1 diabetes; cross, control). X- and y-axis correspond to the value of the two PC. PC, principal component.
Table II.
Evaluation of spectral clustering.
| Method | Accuracy | Precision | Recall | F1 score | Area under curve |
|---|---|---|---|---|---|
| Spectral clustering | 0.90 | 0.94 | 0.85 | 0.89 | 0.912 |
Transformation and feature extraction
Since the dataset contains numerous features with a number of zero values, PCA was applied as a dimensionality reduction algorithm. The PCA algorithm, before applying the dimensionality reduction methodology, performs whitening, which is a transformation method. In this method, the vectors are multiplied by the square root of samples and then divided by the singular values to ensure uncorrelated outputs with unit component-wise variances. For the implementation of the PCA method, the sklearn library of python was used. An initial implementation was performed without specifying the number of components. In this configuration, the default number of components is calculated as the minimum value between features and samples, which in this case is 40. This way all the components are kept. Each component/new variable created by this algorithm explains variance in a proportion. With this initial implementation, it was noticed that the majority of the components had low explained variance. As a next step, five components were selected. Each component/new variable created by this algorithm explains variance in a proportion depicted in Table III.
Table III.
Loadings of most significant feature in each PC.
| Component | Explained variance (%) | Significant feature | Loading value |
|---|---|---|---|
| PC1 | 63.42 | 1111111111 | 0.769 |
| PC2 | 11.52 | 1111111011 | 0.498 |
| PC3 | 9.02 | 1110111111 | 0.873 |
| PC4 | 4.80 | 1111101111 | 0.706 |
| PC5 | 1.56 | 1110101111 | 0.446 |
PC, principal component.
For each PC the most significant feature is the one with the maximum value of loading. Loading is the correlation coefficient between original variables and the component and is calculated by the algorithm during the dimensionality reduction procedure, as the loading value (explained variance) of each component for each feature (24). Table III describes the features with the highest loadings for the respective PC, while Fig. 2 presents the loading values of the five most significant features for each PC, among all PCs.
Figure 2.

Loadings correlation matrix plot for the five most significant features of each principal component.
The summary of the explained variance of the five PC is ~90%, which means that they represent a large number of the initial features and they are concentrating a high percentage of the information included in the initial dataset. However, PC4 and PC5 represent only 6% of the information; therefore, only the first three PC were kept for the next steps of the analysis. PC1, PC2 and PC3 represent ~85% of the initial features. Fig. 3A shows the result of the analysis in 3D space for the first three PC against the classes of patients (control and diabetes), while Fig. 3B-D presents the relations between the three PC in pairs in 2D space.
Figure 3.

A total of three PCs against the two patient classes (A) in 3 dimensions and (B-D) in pairs of components in 2D space: (B) PC2 vs. PC1, (C) PC3 vs. PC 1 and (D) PC3 vs. PC2. PC, principal component.
Classification
The three variables (PC) identified by the previous step were used as the features that fed the classification algorithms. Thus, five different classifiers were trained and evaluated using the 5-fold cross-validation method. This method splits the dataset into five groups and uses the fourth for training and the fifth for testing and repeats the procedure until all groups are used as the testing dataset. The evaluation metrics of the five classifiers that were trained are presented in Table IV. For every classifier, the training was repeated 100 times and the results recorded are based on the mean value of every metric.
Table IV.
Evaluation metrics.
| Classifier | Accuracy | Precision | Recall | F1 score | Area under curve |
|---|---|---|---|---|---|
| Random forest | 0.87 | 0.86 | 0.89 | 0.87 | 0.93 |
| Decision tree | 0.88 | 0.86 | 0.82 | 0.88 | 0.84 |
| Logistic regression | 0.65 | 0.61 | 0.80 | 0.70 | 0.75 |
| Naive Bayes | 0.90 | 0.94 | 0.85 | 0.90 | 0.96 |
| Support vector machine (linear) | 0.77 | 0.72 | 0.90 | 0.80 | 0.85 |
The best-performing classifier, when considering accuracy, was Naive Bayes (25). Fig. 4A shows the model performance on the train and validation dataset over times of evaluation. They are converging after a number of iterations, following the same course and resulting in the final score. Fig. 4B presents the receiver operating characteristic curve. The curve is close to the upper left quadrant of the plot which means that the model trained is efficient.
Figure 4.
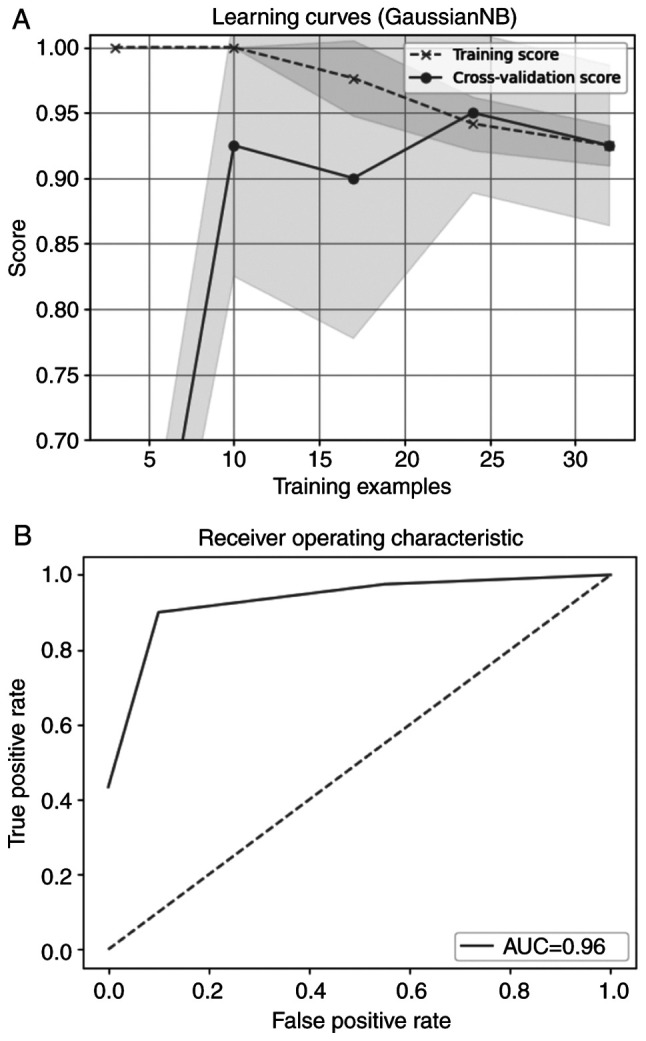
(A) Learning curve and (B) AUC for Naive Bayes classifier. AUC, area under curve.
In one of our previous works, the most significant methyl-haplotypes were detected with the Mann-Whitney-Wilcoxon method and used as features (6). The representative features detected with this methodology, as having statistically significant differences between the two groups (P#x003C;0.001), were the specific methyl-haplotypes (‘1110101110’, ‘1110111110’ and ‘1111111100’). The accuracy of the best-performing classifier was 82%. Table V describes the loadings for each one of the eight features, 3 from previous work and five from the current work, and for every PC, while Fig. 5 depicts their loading values for each PC.
Table V.
Loadings of the features selected by the two methods in each PC.
| A, Present method features | |||||
|---|---|---|---|---|---|
| Feature | PC1 | PC2 | PC3 | PC4 | PC5 |
| 1111111111 | 0.769500 | -0.362961 | -0.304812 | -0.339602 | -0.071543 |
| 1111111011 | 0.202861 | 0.498655 | 0.009574 | -0.238478 | -0.329020 |
| 1110111111 | 0.370387 | -0.080940 | 0.873374 | 0.165998 | -0.044353 |
| 1111101111 | 0.361261 | 0.149943 | -0.259968 | 0.706187 | 0.181835 |
| 1110101111 | 0.138275 | 0.012254 | 0.049107 | -0.162598 | 0.446792 |
| B, Previous method features | |||||
| Feature | PC1 | PC2 | PC3 | PC4 | PC5 |
| 1110101110 | 0.008783 | -0.008252 | 0.027514 | 0.026659 | 0.110130 |
| 1110111110 | 0.001483 | -0.002835 | 0.001250 | -0.000759 | 0.001347 |
| 1111111100 | 0.013053 | -0.020119 | 0.054517 | 0.008947 | -0.025733 |
PC, principal component.
Figure 5.

Loadings correlation matrix plot for the methyl-haplotypes highlighted by the two methods in each PC.
Discussion
To the best of our knowledge, the present study investigated for the first time methyl-haplotypes in the IGP region in children and adolescents with T1D and identified specific methylation patterns significantly associated with T1D. Furthermore, based on machine-learning methods, the present study attempted to develop a predictive model for the discrimination of patients with T1D and healthy individuals using methyl-haplotypes as classification parameters. Naive Bayes turned out to be the best-performing classifier in the context of accuracy, precision, recall, F1 score and area under curve.
T1D is the result of a chronic, progressive, T-cell-mediated selective destruction of pancreatic islet β-cells, leading to loss of insulin secretion and lifelong need for exogenous administration (1,7). It is characterized by a strong genetic background responsible for the increased rate of recurrence within families (1,26). Although the exact genetic etiology remains unknown, >60 gene sites have been found to be implicated through complex and synergistic interactions (27,28). Among them, the INS gene (11p15.5) is one of the most consistently replicating regions associated with T1D (7) that is involved in both induction and early phases of pancreatic islet β-cellular immunity (27). Furthermore, the increase in the incidence of T1D in a genetically stable population for a short period of time highlights the importance of epigenetics beyond that of genomics (29). At present, there is an increasing number of studies investigating DNA methylation of several gene loci in patients with T1D with a wide range of results (30).
INS gene methylation has been extensively studied in adults with T1D (17,31-36). Specific CpGs of this gene have been recorded in either a state of hypermethylation or hypomethylation compared with a healthy population (17,31-36). In children with T1D, even at the time of diagnosis, different levels of methylated and non-methylated DNA in the INS gene have also been detected compared with controls (37). A recent study by our team estimated the rate of methylation of the INS gene at IGP-CpGs sites, in children and adolescents of Greek origin with T1D and mean disease duration of 6 years and found hypermethylated sites compared with healthy individuals (38).
The state of methylation of the IGP region is emerging as a suitable biomarker for the detection of individuals with pancreatic β-cell autoimmunity. Given the large number of CpGs that are potential substrates for methylation, methyl-haplotype processing offers the possibility of combinational searching and comparison of patterns instead of single sites (39). As a result, the conclusions drawn are of higher clinical significance and more compatible with the complexity of the molecular and biological systems to be elucidated (39). The present study studied the overall methylation pattern at ten IGP-CpGs sites and detected five distinct methyl-haplotypes highly associated with T1D.
Methyl-haplotype based association studies have been proposed to have implications in the genetic investigation of complex diseases (40). With the development of biotechnology, even Epigenome-Wide Association Study software has been developed in order to systematically approach and reveal common disease/phenotype-related methyl-haplotypes (41). Methyl-haplotypes are used in cancer diagnostics at an early disease stage (39,42-44). Methods such as MHap and MHap_DMR have been developed for the construction of methylation haplotypes in CpG dense regions of homologous chromosomes, permitting the elucidation of the two-way direction, cell differentiation or cancerization (45). In this context, a cell-free DNA methylation panel, ThyMet classifier, has been established to differentiate papillary thyroid carcinoma from benign thyroid nodule (46). Furthermore, a follicular thyroid carcinoma (FTC) predicting model based on DNA methylation markers is used in cases of thyroid tumors with uncertain malignant potential in order to distinguish between FTC and benign follicular adenoma (FA) (47). Plasma methylation haplotyping has been suggested as a promising method for the early detection of tumor and its tissue of origin, as well as for the continuous monitoring of tumor progression and metastasis to multiple organs (42). More specifically, methylation haplotyping is studied as a diagnostic tool in cervical precancer (39,43) and hepatocellular carcinoma (HCC) (44), pancreatic ductal adenocarcinoma (48) and colorectal cancer (49). In human papillomavirus (HPV)-positive women, a methylation assay including the basic carcinogenic HPV types can be applied as a screening test (39), while in high-risk HPV-positive women, certain methyl-haplotypes are found to consistently serve as a potential biomarker for the stratification of risk (39). In HCC and pancreatic ductal adenocarcinoma (48), methyl-haplotypes can early, accurately and non-invasively detect microvascular invasion and predict prognosis of cancer (44,50). Recently, full methylation haplotypes levels in the protein region of homeodomain-interacting protein kinase 3 are supported as a diagnostic biomarker and CRP level indicator for rheumatoid arthritis, opening the way for studies in other autoimmune diseases, such as T1D (16). More recommended is the use of methyl-haplotypes as a screening tool in clinical decision making (40).
In the present study, the evaluation metrics of the predictive model show that this method had a greater performance compared with our initial approach (6). The accuracy of the best-performing classifier in the present case was 90%. In the previous work, the accuracy of the best-performing classifier was 82% (6). By this, we hypothesize that the transformation of the whole dataset with a dimensionality reduction method included more useful information compared with the selection of some representative methyl-haplotypes. This may suggest that the phenomenon is expressed by several correlated features rather than a few specific methyl-haplotypes.
The representative features detected in our previous work, methyl-haplotypes (‘1110101110’, ‘1110111110’ and ‘1111111100’) (6) do not coincide with the features marked as the most significant ones for each PC that was created by the dimensionality reduction method (‘1111111111’, ‘1111111011’, ‘1110111111’, ‘1111101111’, ‘1110101111’). The present study observed that none of the features selected by the previous analysis has a significant participation in the PCs. However, it has to be highlighted that the currently proposed classifier used as features the transformed data, PCs, instead of the original methyl-haplotypes, and in this sense it combines information by more methyl-haplotypes in each PC.
With the implementation of the spectral clustering method on the dataset, it is apparent that the two groups, T1D and control, were classified with high accuracy, underlying that they had significantly different characteristics in the structure of the INS gene. Moreover, the high accuracy of the trained predictive model indicated that methyl-haplotypes from INS gene may constitute a reliable marker that can be used to identify the existence of T1D.
The methodology implemented is a first approach to the investigation of statistically significant IGP-CpGs methyl-haplotypes as predictor parameters in a classification system. After the preprocessing of our dataset and the removal of the low variance features, the shape of the dataset was (40, 336). PCA can be applied in all cases where n#x003C;p (51). A similar attempt of applying PCA in relevant dataset, on the grounds of the above-mentioned rationale, has already been published, providing valuable results (52). The only restriction that could be applied to the present bioinformatic approach was that the resulting components with non-zero variance should be at least n-1. In the present case, only the first five components (#x003C;#x003C;n-1) represent 90% of the initial dataset and the rest of them have near-to-zero variance. Only the three first components were hereby used, in order to avoid fitting noise. For all these reasons, the results of the hereby presented analysis are solid and could be repeatable in other non-relating datasets.
Studies on the widespread use of machine-learning methods in diabetes have already been published (10,12,14). The large data available after the implementation of internationally accepted recommendations in diabetes, allow the use of artificial intelligence machine-learning methods to extract new knowledge and develop predictive tools (10). Among the epigenetic variations, changes in DNA methylation can feed future prediction tools to be applied in primary disease prevention (11). The results of the present clustering tool succeeded in highlighting a specific method with extremely high percentages of metrics (accuracy, sensitivity and specificity), despite the fact that the implementation was performed using a small amount of data. Data of the present study could serve as a base of evidence to implement distinct statistical approaches for reconstructing methyl-haplotypes frequency in population data (53). Utilization of specific software with Bayesian methodology, allows the use of priori expectations in order to inform haplotype reconstruction (53,54). Through this approach, the hereby described methylation status in the IGP locus could serve as the basis to extrapolate and calculate the expected frequency of methylation haplotype in large datasets of T1D population, in order to optimize the use of our experimental resources.
This protocol presents some limitations that are recognized and include the homogeneity of the national origin of the sample, the small number of participants, the recruitment by a single center, as well as the homogeneity of patients in terms of glycemic control and body mass index. Factors that reduced the statistical significance of the results by increasing the accuracy, but on the other hand limited the investigation of the influence of environmental factors involved in the dynamic process of methylation.
To summarize the results of these studies, we can assume that the specific dataset and methyl-haplotypes of the present study could be used as features for distinguishing T1D diabetes. The first part of the analysis, SP, proved that these features could differentiate the two classes of interest, control and diabetes. The second part of the analysis, machine learning based on PCA, showed evidence that effective predictive models could be built based on the methyl-haplotypes. This methodology is a promising step towards early T1D diagnosis.
Epigenetic changes can serve as biomarkers of early diagnosis of T1D and as potential targets for therapeutic intervention. Methyl-haplotyped studies such as the present are expected to provide the evidence to put them in the service of daily clinical practice as a tool of diagnosis and treatment with the ultimate goal of improving the level of health services in individuals with T1D or in those that they are prone to develop T1D. Verification of the results of this protocol in large datasets of patients with T1D could be extended accordingly.
Supplementary Material
Acknowledgements
Not applicable.
Funding Statement
Funding: This research was funded by the Hellenic Association for the Study and Education of Diabetes Mellitus (grant no. 2015).
Availability of data and materials
The data generated in the present study may be found in Zenodo under accession no. DOI:10.5281/zenodo.8001833 or the following URL: https://zenodo.org/record/8001833. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
AGT and IC conceptualized the study. AK, EPK and KM designed the methodology. AK and IC performed the bioinformatic analysis. EPK, KM, SG, VRT and AS performed investigation. EPK, AK, KM, SG, VRT and AS curated the data. EPK, AK, KM, SG, VRT and AS wrote the original draft preparation. AGT and IC reviewed and edited the manuscript. AK and IC prepared the figures. AGT was the supervisor and project administrator to the study and acquired the funding. ΚΜ, EPK and AK confirm the authenticity of all the raw data. All authors have read and approved the final manuscript and agree to be personally accountable for their own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved and documented in the literature.
Ethics approval and consent to participate
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the guidelines of the Declaration of Helsinki, and the protocol was approved by the Bioethics Committee of the School of Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki (approval no. 185/30.12.2015).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Jerram ST, Dang MN, Leslie RD. The role of epigenetics in type 1 diabetes. Curr Diab Rep. 2017;17(89) doi: 10.1007/s11892-017-0916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie Z, Chang C, Huang G, Zhou Z. The role of epigenetics in type 1 diabetes. Adv Exp Med Biol. 2020;1253:223–257. doi: 10.1007/978-981-15-3449-2_9. [DOI] [PubMed] [Google Scholar]
- 3.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 4.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 5.D'Angeli MA, Merzon E, Valbuena LF, Tirschwell D, Paris CA, Mueller BA. Environmental factors associated with childhood-onset type 1 diabetes mellitus: An exploration of the hygiene and overload hypotheses. Arch Pediatr Adolesc Med. 2010;164:732–738. doi: 10.1001/archpediatrics.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotanidou EP, Mouzaki K, Chouvarda I, Koutsiana E, Kosvyra A, Tsinopoulou VR, Giza S, Serbis A, Galli-Tsinopoulou A. CpG methylation haplotypes of the insulin gene promoter as predictive biomarker in a cohort of children and adolescents with type 1 diabetes. Paediatriki. 2021;83:150–162. [Google Scholar]
- 7.Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clin Chem. 2011;57:176–185. doi: 10.1373/clinchem.2010.148221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moulder R, Bhosale SD, Erkkilä T, Laajala E, Salmi J, Nguyen EV, Kallionpää H, Mykkänen J, Vähä-Mäkilä M, Hyöty H, et al. Serum proteomes distinguish children developing type 1 diabetes in a cohort with HLA-conferred susceptibility. Diabetes. 2015;64:2265–2278. doi: 10.2337/db14-0983. [DOI] [PubMed] [Google Scholar]
- 9.Rodríguez-Ventura AL, Yamamoto-Furusho JK, Coyote N, Dorantes LM, Ruiz-Morales JA, Vargas-Alarcón G, Granados J. HLA-DRB1*08 allele may help to distinguish between type 1 diabetes mellitus and type 2 diabetes mellitus in Mexican children. Pediatr Diabetes. 2007;8:5–10. doi: 10.1111/j.1399-5448.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- 10.Kavakiotis I, Tsave O, Salifoglou A, Maglaveras N, Vlahavas I, Chouvarda I. Machine learning and data mining methods in diabetes research. Comput Struct Biotechnol J. 2017;15:104–116. doi: 10.1016/j.csbj.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Pollin TI. Epigenetics variation and pathogenesis in diabetes. Curr Diab Rep. 2018;18(121) doi: 10.1007/s11892-018-1091-4. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Ding J, Zhi DU, Gu K, Wang H. Identification of type 2 diabetes based on a ten-gene biomarker prediction model constructed using a support vector machine algorithm. Biomed Res Int. 2022;2022(1230761) doi: 10.1155/2022/1230761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lethebe BC, Williamson T, Garies S, McBrien K, Leduc C, Butalia S, Soos B, Shaw M, Drummond N. Developing a case definition for type 1 diabetes mellitus in a primary care electronic medical record database: An exploratory study. CMAJ Open. 2019;7:E246–E251. doi: 10.9778/cmajo.20180142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fufurin I, Berezhanskiy P, Golyak I, Anfimov D, Kareva E, Scherbakova A, Demkin P, Nebritova O, Morozov A. Deep learning for type 1 diabetes mellitus diagnosis using infrared quantum cascade laser spectroscopy. Materials (Basel) 2022;15(2984) doi: 10.3390/ma15092984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seoighe C, Tosh NJ, Greally JM. DNA methylation haplotypes as cancer markers. Nat Genet. 2018;50:1062–1063. doi: 10.1038/s41588-018-0185-x. [DOI] [PubMed] [Google Scholar]
- 16.Jiang P, Wei K, Xu L, Chang C, Zhang R, Zhao J, Jin Y, Xu L, Shi Y, Qian Y, et al. DNA methylation change of HIPK3 in Chinese rheumatoid arthritis and its effect on inflammation. Front Immunol. 2023;13(1087279) doi: 10.3389/fimmu.2022.1087279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fradin D, Le Fur S, Mille C, Naoui N, Groves C, Zelenika D, McCarthy MI, Lathrop M, Bougnères P. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS One. 2012;7(e36278) doi: 10.1371/journal.pone.0036278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, Collins BS, Hilliard ME, Isaacs D, Johnson EL, et al. Classification and diagnosis of diabetes: Standards of care in diabetes-2023. Diabetes Care. 2023;46:19–40. doi: 10.2337/dc23-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Libman I, Haynes A, Lyons S, Pradeep P, Rwagasor E, Tung JY, Jefferies CA, Oram RA, Dabelea D, Craig ME. ISPAD clinical practice consensus guidelines 2022: Definition, epidemiology, and classification of diabetes in children and adolescents. Pediatr Diabetes. 2022;23:1160–1174. doi: 10.1111/pedi.13454. [DOI] [PubMed] [Google Scholar]
- 20.Scala G, Affinito O, Palumbo D, Florio E, Monticelli A, Miele G, Chiariotti L, Cocozza S. AmpliMethProfiler: A pipeline for the analysis of CpG methylation profiles of targeted deep bisulfite sequenced amplicons. BMC Bioinformatics. 2016;17(484) doi: 10.1186/s12859-016-1380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Luxburg U. A tutorial on spectral clustering. Stat Comput. 2007;17:395–416. [Google Scholar]
- 22.Jollife IT, Cadima J. Principal component analysis: A review and recent developments. Philos Trans A Math Physc Eng Sci. 2016;374(20150202) doi: 10.1098/rsta.2015.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. Scikit-learn: Machine learning in python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 24.Frost HR. Eigenvectors from eigenvalues Sparse principal component analysis (EESPCA) J Comput Graph Stat. 2022;31:486–501. doi: 10.1080/10618600.2021.1987254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webb GI. Naïve Bayes. In: Encyclopedia of Machine Learning. Springer, Boston, MA, pp713-714, 2010. [Google Scholar]
- 26.Jerram ST, Leslie RD. The genetic architecture of type 1 diabetes. Genes (Basel) 2017;8(209) doi: 10.3390/genes8080209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nokoff N, Rewers M. Pathogenesis of type 1 diabetes: Lessons from natural history studies of high-risk individuals. Ann N Y Acad Sci. 2013;1281:1–15. doi: 10.1111/nyas.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morahan G. Insights into type 1 diabetes provided by genetic analyses. Curr Opin Endocrinol Diabetes Obes. 2012;19:263–270. doi: 10.1097/MED.0b013e328355b7fe. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Xie Z, Lu Q, Chang C, Zhou Z. Beyond genetics: What causes type 1 diabetes. Clin Rev Allergy Immunol. 2017;52:273–286. doi: 10.1007/s12016-016-8592-1. [DOI] [PubMed] [Google Scholar]
- 30.Cerna M. Epigenetic regulation in etiology of type 1 diabetes mellitus. Int J Mol Sci. 2019;21(36) doi: 10.3390/ijms21010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stefan M, Zhang W, Concepcion E, Yi Z, Tomer Y. DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J Autoimmun. 2014;50:33–37. doi: 10.1016/j.jaut.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Husseiny MΙ, Kaye A, Zebadua E, Kandeel F, Ferreri K. Tissue-specific methylation of human insulin gene and PCR assay for monitoring beta cell death. PLoS One. 2014;9(e94591) doi: 10.1371/journal.pone.0094591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neiman D, Moss J, Hecht M, Magenheim J, Piyanzin S, Shapiro AMJ, de Koning EJP, Razin A, Cedar H, Shemer R, Dor Y. Islet cells share promoter hypomethylation independently of expression, but exhibit cell-type-specific methylation in enhancers. Proc Natl Acad Sci USA. 2017;114:13525–13530. doi: 10.1073/pnas.1713736114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgård B, Blennow K, Zetterberg H, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci USA. 2016;113:E1826–E1834. doi: 10.1073/pnas.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuroda A, Rauch TA, Todorov I, Ku HT, Al-Abdullah IH, Kandeel F, Mullen Y, Pfeifer GP, Ferreri K. Insulin gene expression is regulated by DNA methylation. PLoS One. 2009;4(e6953) doi: 10.1371/journal.pone.0006953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herold KC, Usmani-Brown S, Ghazi T, Lebastchi J, Beam CA, Bellin MD, Ledizet M, Sosenko JM, Krischer JP, Palmer JP. β cell death and dysfunction during type 1 diabetes development in at-risk individuals. J Clin Invest. 2015;125:1163–1173. doi: 10.1172/JCI78142. Type 1 Diabetes TrialNet Study Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher MM, Watkins RA, Blum J, Evans-Molina C, Chalasani N, DiMeglio LA, Mather KJ, Tersey SA, Mirmira RG. Elevations in circulating methylated and unmethylated preproinsulin DNA in new-onset type 1 diabetes. Diabetes. 2015;64:3867–3872. doi: 10.2337/db15-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mouzaki K, Kotanidou EP, Fragou A, Kyrgios I, Giza S, Kleisarchaki A, Tsinopoulou VR, Serbis A, Tzimagiorgis G, Galli-Tsinopoulou A. Insulin gene promoter methylation status in Greek children and adolescents with type 1 diabetes. Biomed Rep. 2020;13(31) doi: 10.3892/br.2020.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mirabello L, Frimer M, Harari A, McAndrew T, Smith B, Chen Z, Wentzensen N, Wacholder S, Castle PE, Raine-Bennett T, et al. HPV16 methyl-haplotypes determined by a novel next-generation sequencing method are associated with cervical precancer. Int J Cancer. 2015;136:E146–E153. doi: 10.1002/ijc.29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao L, Liu D, Xu J, Wang Z, Chen Y, Lei C, Li Y, Liu G, Jiang Y. The framework for population epigenetic study. Brief Bioinform. 2018;19:89–100. doi: 10.1093/bib/bbw098. [DOI] [PubMed] [Google Scholar]
- 41.Xu J, Zhao L, Liu D, Hu S, Song X, Li J, Lv H, Duan L, Zhang M, Jiang Q, et al. EWAS: Epigenome-wide association study software 2.0. Bioinformatics. 2018;34:2657–2658. doi: 10.1093/bioinformatics/bty163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet. 2017;49:635–642. doi: 10.1038/ng.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clarke MA, Gradissimo A, Schiffman M, Lam J, Sollecito CC, Fetterman B, Lorey T, Poitras N, Raine-Bennett TR, Castle PE, et al. Human papillomavirus DNA methylation as a biomarker for cervical precancer: Consistency across 12 genotypes and potential impact on management of HPV-positive women. Clin Cancer Res. 2018;24:2194–2202. doi: 10.1158/1078-0432.CCR-17-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu L, Pen S, He Q, Chen Z, Kuang M. Identification of DNA methylation signatures for microvascular invasion in hepatocellular carcinoma. Gut. 2019;68:A1–A166. [Google Scholar]
- 45.Peng X, Li Y, Kong X, Zhu X, Ding X. Investigating different DNA methylation patterns at the resolution of methylation haplotypes. Front Genet. 2021;12(697279) doi: 10.3389/fgene.2021.697279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hong S, Lin B, Xu M, Zhang Q, Huo Z, Su M, Ma C, Liang J, Yu S, He Q, et al. Cell-free DNA methylation biomarker for the diagnosis of papillary thyroid carcinoma. EBioMedicine. 2023;90(104497) doi: 10.1016/j.ebiom.2023.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Zhang Z, Liu X, Duan H, Xiang T, He Q, Su Z, Wu H, Liang Z. DNA methylation haplotype block markers efficiently discriminate follicular thyroid carcinoma from follicular adenoma. J Clin Endocrinol Metab. 2021;106:1011–1021. doi: 10.1210/clinem/dgaa950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu H, Guo S, Liu X, Li Y, Su Z, He Q, Liu X, Zhang Z, Yu L, Shi X, et al. Noninvasive detection of pancreatic ductal adenocarcinoma using the methylation signature of circulating tumour DNA. BMC Med. 2022;20(458) doi: 10.1186/s12916-022-02647-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mo S, Dai W, Wang H, Lan X, Ma C, Su Z, Xiang W, Han L, Luo W, Zhang L, et al. Early detection and prognosis prediction for colorectal cancer by circulating tumour DNA methylation haplotypes: A multicentre cohort study. EClinicalMedicine. 2022;55(101717) doi: 10.1016/j.eclinm.2022.101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hao Y, Yang Q, He Q, Hu H, Weng Z, Su Z, Chen S, Peng S, Kuang M, Chen Z, Xu L. Identification of DNA methylation signatures for hepatocellular carcinoma detection and microvascular invasion prediction. Eur J Med Res. 2022;27(276) doi: 10.1186/s40001-022-00910-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Winter JCF, Dodou D, Wieringa PA. Exploratory factor analysis with small sample sizes. Multivariate Behav Res. 2009;44:147–181. doi: 10.1080/00273170902794206. [DOI] [PubMed] [Google Scholar]
- 52.Preacher KJ, MacCallum RC. Exploratory factor analysis in behavior genetics research: Factor recovery with small sample sizes. Behav Genet. 2002;32:153–161. doi: 10.1023/a:1015210025234. [DOI] [PubMed] [Google Scholar]
- 53.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in the present study may be found in Zenodo under accession no. DOI:10.5281/zenodo.8001833 or the following URL: https://zenodo.org/record/8001833. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.