

Abstract
Petroleum substances, as archetypical UVCBs (substances of unknown or variable composition, complex reaction products, or biological substances), pose a challenge for chemical risk assessment as they contain hundreds to thousands of individual constituents. It is particularly challenging to determine the biodegradability of petroleum substances since each constituent behaves differently. Testing the whole substance provides an average biodegradation, but it would be effectively impossible to obtain all constituents and test them individually. To overcome this challenge, comprehensive two-dimensional gas chromatography (GC × GC) in combination with advanced data-handling algorithms was applied to track and calculate degradation half-times (DT50s) of individual constituents in two dispersed middle distillate gas oils in seawater. By tracking >1000 peaks (representing ∼53–54% of the total mass across the entire chromatographic area), known biodegradation patterns of oil constituents were confirmed and extended to include many hundreds not currently investigated by traditional one-dimensional GC methods. Approximately 95% of the total tracked peak mass biodegraded after 64 days. By tracking the microbial community evolution, a correlation between the presence of functional microbial communities and the observed progression of DT50s between chemical classes was demonstrated. This approach could be used to screen the persistence of GC × GC-amenable constituents of petroleum substance UVCBs.
Keywords: comprehensive two-dimensional gas chromatography (GC × GC); oil constituent DT50s; biodegradation experiment; unknown or variable composition; complex reaction products, or biological materials (UVCBs); peak tracking; hydrocarbon degrader bacterial succession; complex mixtures; persistence screening
Short abstract
This study reports a new method for conducting a more comprehensive biodegradation assessment of petroleum substance constituents.
Introduction
Biodegradation affects the fate of petroleum substances in the environment and is a fundamental parameter for environmental risk assessment under chemical registration regulations (e.g., OSPAR, REACH, TSCA, and CEPA).1−4 Under typical contamination of aquatic environments by dissolved or dispersed petroleum substances, the combination of relatively low environmental exposure concentrations of the chemical constituents and low concentrations of competent bacteria will usually cause biodegradation to follow first-order or logistic rate models.5 Primary biodegradation half-lives are usually derived directly from the first-order rate coefficients in those models, while degradation models with non-responsive periods also include a lag period for half-life determination (degradation half-time; DT50).6 This is a simple metric that is used to describe a complex biochemical process.
Evaluation of substances of “unknown or variable composition, complex reaction products, or biological materials” (UVCBs), like petroleum products, remains challenging.7 This is primarily due to the high chemical complexities of these substances,8,9 which are by definition not fully characterizable on an individual constituent basis. While there have been many studies examining the biodegradation of petroleum products, it is challenging to attribute biodegradation potentials and rates to individual constituents of the whole substance.10,11 Furthermore, the testing of all individual constituents of a whole petroleum substance is practically infeasible due to the sheer number of constituents for sourcing and testing; individual constituent testing also excludes important microbial processes occurring during UVCB biodegradation, like co-metabolism or competitive inhibition.12−15
Traditionally, the biodegradation of petroleum substances has been tracked by one-dimensional gas chromatography (GC) coupled with flame ionization detection (GC–FID) or mass spectrometry (GC–MS), quantifying either the total extractable material (TEM) or selected constituents restricted to relatively few targets.10,16−21 However, modern high-resolution chromatographic methods expand the analytical window relative to one-dimensional approaches, allowing a much wider range of target and non-target UVCB constituents to be determined in complex substances.22,23 Comprehensive two-dimensional GC (GC × GC) can separate multiple constituents coeluting in the first chromatographic dimension (typically volatility-based) along the second chromatographic dimension (often polarity-based), thus vastly increasing the total number of resolved constituents that can be accurately monitored. As an example, GC × GC–FID has been used to show that oil constituents that are unresolved by one-dimensional GC analyses are depleted by biodegradation in seawater (SW) at 8 °C after 60 days of incubation.19 Similarly, the biodegradation of saturated hydrocarbons in weathered oil samples from the Deepwater Horizon (DWH) oil spill was assessed by GC × GC–FID, allowing for the assessment of the persistence of hydrophobic saturated constituents that comprise the unresolved complex mixture (UCM) observed in conventional one-dimensional analyses.22 Advances in peak tracking23 provide a basis for quantitatively using the 10-fold more data provided by GC × GC relative to one-dimensional GC–MS methods.24−26
High-resolution analyses can be combined with microbial analyses to investigate relations between microbial community successions and biodegradation patterns on a finer scale than with one-dimensional analytical tools. Although multiple bacterial classes have the ability to degrade hydrocarbons, often a specific class of bacteria may only be able to utilize certain chemical classes. Field and laboratory studies in natural SW with targeted hydrocarbon constituents have shown early abundances of hydrocarbonoclastic bacteria associated with alkane degradation, followed by later community changes toward typical aromatic hydrocarbon constituent degraders and those able to utilize the degradation products of hydrocarbon biotransformation.27−29 However, the direct associations between microbial taxa and their specific degradation capacities are often unknown, although several studies have revealed hydrocarbonoclastic bacteria associated with the degradation of either alkanes or aromatic hydrocarbons.29−31 Therefore, degradation profiles are often empirically correlated with the dynamic changes in the composition of the microbial population.
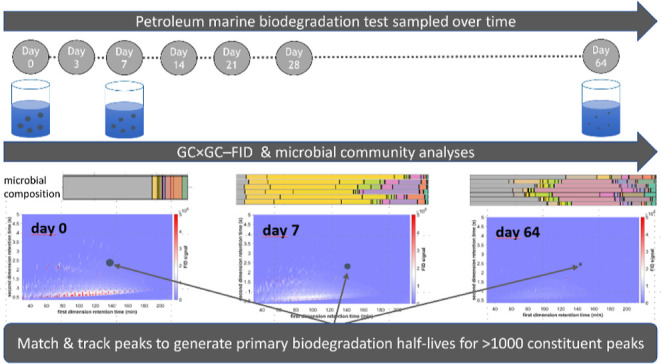
The objectives of this study are to establish a data analysis pipeline which uses a high-resolution GC × GC–FID method to characterize the DT50 profiles of two gas oils using peak tracking. Corresponding shifts in microbial ecology that follow the biodegradation process are also determined and related to the biodegradation profiles. This methodology permits simultaneous calculation of primary biodegradation half-lives for multiple UVCB constituents, for which biodegradation data generation may otherwise be difficult or nigh impossible.
Materials and Methods
Petroleum Substances Tested and SW Inoculum
The two petroleum substances tested in the study were petroleum distillates: VHGO (vacuum gas oils, hydrocracked gas oils, and distillate fuels) and SRGO (straight run gas oil). The VHGO is a distillate fuel (CAS no. 68334-30-5) with a density of 0.842 g/cm3, hydrocarbon numbers predominantly in the range of C9–C20 and boiling points in the range of approximately 160 to 360 °C. The SRGO is a full-range straight-run gas oil middle distillate (CAS no. 68814-87-9) with a density of 0.845 g/cm3, hydrocarbon numbers predominantly in the range of C9–C25 and boiling points in the range of approximately 150 to 400 °C (EPA Substance Registry Services). The C10+ mass represented 96.09 and 93.61% of the total substance mass for VHGO and SRGO, respectively, based on simulated distillation data.32 Conventional GC–FID chromatograms for both gas oils are shown in the Supporting Information (Figure S1). Prior to use, the gas oils were spiked with known amounts of hexachlorobenzene (CAS no. 118-74-1, Supelco) and o-terphenyl (CAS no. 84-15-1, Chiron AS, Trondheim, Norway) as non-degradable controls (described in Table S1, Supporting Information). During data processing, o-terphenyl was found to have undergone a small degree of biodegradation, which is possible given the timeframe of the experiment.33,34 Therefore, only the hexachlorobenzene was used in the calculation of constituent DT50s. Natural, non-amended SW was used as the inoculum in the biodegradation experiment. The SW was collected in April 2020 at a depth of 80 m in Trondheimsfjord, Norway (63°26′N and 10°23′E), outside the harbor area of Trondheim, as previously described.10
Biodegradation Experiment
The biodegradation of the test substances at 13 °C was monitored for 64 days using the previously published methodology.10,35,36 Briefly, stock dispersions of VHGO and SRGO (200 ppmV oil) with a median droplet size of ∼10 μm were prepared in SW, filtered through a 5 μm cellulose filter (Aqua-Pure model AP110), and then pre-acclimated (overnight) in SW at 13 °C using a bespoke oil droplet generator as described by Nordtug et al.37 Oil droplet concentrations and size distributions were analyzed by a Multisizer Coulter Counter (100 μm aperture). The stock dispersions were mixed with acclimated non-filtered SW (13 °C) to achieve an oil droplet concentration of 2–3 mg/L. The dispersions were transferred to 2.3 L capped Pyrex bottles that were completely filled (no headspace). The bottles were mounted on slowly continuously rotating (0.75 rpm) carousels (Figure S2) at 13 °C, as previously described.10 Sterile controls were prepared by passing the SW through 0.22 μm Sterivex filters, and 100 mg/L of HgCl2 biocide was added before adding the dispersed oil. Finally, non-sterile samples of SW without oil were subjected to the same exposure and sample treatment regime to serve as controls for the bacterial population evolution.
During the incubation period, triplicate test bottles for each oil were removed for analysis after 0, 3, 7, 14, 21, 28, and 64 days.10,36,38 Duplicate (oil-containing) sterile controls for each oil were removed for analysis after 7, 14, 28, and 64 days, while single blank control samples (non-oil-containing SW) were taken at each time point. At each time point, dissolved oxygen (DO; model 59 Dissolved Oxygen Meter, YSI Inc., Yellow Springs, OH, USA), particle concentration, and particle size (Coulter Counter) were measured in each sample bottle. The DO concentration over time is presented in Figure S3. Samples were split for DNA isolation (1 L at 0–7 days and 0.5 L at 14–64 days) and chemical analysis (1 L at 0–7 days and 1.5 L at 14–64 days). Samples for DNA extraction were filtered through 0.22 μm filters (Durapore(R) PVDF Membrane), and the filters were frozen at −20 °C until extraction. Samples for chemical characterization were immediately acidified (HCl, pH 2) upon sampling to quench degradation and preserve the samples, which were stored in a refrigerator (4–5 °C) until further processing (maximum 14 days). Emptied glass bottles were rinsed with 30 mL of dichloromethane (DCM) to remove potential chemicals sorbed to the bottle walls and pooled with the water samples for chemical analysis.
DNA Extraction and Microbial Community Analysis
DNA was extracted by the ZymoBIOMICS 96 MagBead DNA kit (ZYMO Research), mainly following the instructions for the manual extraction procedure provided by the manufacturer. Briefly, frozen filters were aseptically cut into small pieces, and 1 mL 1× DNA/RNA Shield (ZYMO Research) was added in a ZR BashingBead Lysis Tube. Samples in BashingBead Lysis Tubes were homogenized in a bead beater (FastPrep) at maximum speed (6.0 m/s) for 5 min. Extracted DNA was subjected to quantification and quality control using Nanodrop 1000 and Qubit 3.0 instruments (Thermo Fisher Scientific). Deviation from the manufacturer’s protocol included the use of stand-alone 1.5 mL vials (Eppendorf) instead of a 96-Deep well block (ZYMO Research) for extraction and purification purposes after the homogenization step.
Total DNA extracts were sent to the Beijing Genomics Institute (BGI, Hong Kong) for 16S rDNA microbiome sequencing. In brief, a 30 ng DNA template was used for microbiome analyses, and the 16S rRNA fusion primers, which included primers 341F (ACTCCTACGGGAGGCAGCAG) and 806R (GGACTACHVGGGTWTCTAAT), were added for the polymerase chain reaction (PCR). All PCR products were purified by Agencourt AMPure XP beads, dissolved in Elution buffer, and eventually labeled to finish library construction. Library size and concentration were detected by the Agilent 2100 Bioanalyzer. Qualified libraries were sequenced on a HiSeq platform according to their insert size, with the primers 341F and 806R. The sequenced raw data were subjected to bioinformatic and downstream statistical analysis using Quantitative Insights Into Microbial Ecology 2 (QIIME2), as previously described,39 with a cut-off of 5% relative abundance. Taxa below this threshold were assigned to the group “Other”. To analyze potential differences in the dynamics of microbial communities between individual samples and sample groups at separate time points, multivariate statistics in the form of principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarity was carried out.40
Analysis of Water Samples Using One-Dimensional and Comprehensive Two-Dimensional GC–FID
To account for analyte losses during sample extraction and processing steps, a range of deuterated polycyclic aromatic hydrocarbons (PAHs) and phenols (25.334 μg of phenol-d6, 1.042 μg of p-cresol-d8, 1.374 μg of 4-n-propylphenol-d12, 2.522 μg of naphthalene-d8, 1.000 μg of acenaphthene-d10, 1.000 μg of fluorene-d10, 0.480 μg of phenanthrene-d10, 0.500 μg of chrysene-d12, and 0.508 μg of perylene-d12) were spiked into each water sample immediately prior to extraction. The water samples for chemical analysis were then serially extracted into DCM (60–30–30 mL, 3–2–2 min shaking in separatory funnels). Solvent extracts were dried over anhydrous sodium sulfate and concentrated by solvent evaporation (TurboVap 500) to 1 mL. Prior to analysis, 5α-androstane (10 μg) was spiked in each extract to calculate the recovery of the deuterated standards. Extracts were stored in the dark and frozen (−20 °C) until analysis. Analysis was performed in a single sequence, and samples were run in a randomized order.
One-dimensional GC–FID analysis was performed using an Agilent 7890A GC, and the method details are presented in the Supporting Information (Method S1). GC × GC–FID analysis was conducted using an Agilent 7890B GC fitted with a Zoex ZX2 cryogenic modulator and a FID. The first-dimension separation was achieved using a Crossbond dimethyl polysiloxane Rx1-ms column (60 m, 0.25 mm id, and 0.25 μm film thickness) coupled through an inert fused silica (1 m and 0.25 mm) loop to a phenyl polysilphenylene-siloxane BPX-50 column (0.9 m, 0.1 mm, and 0.1 μm) for the second-dimension separation. The DCM extract (1 μL) was introduced via an autosampler using splitless injection at 310 °C. Helium (6.0 purity) was used as a carrier gas at constant flow (1.1 mL/min). The oven temperature was held initially at 60 °C (10 min) and raised by 1.2 °C/min to 330 °C. The modulation period was set to 7 s with a 350 ms hot jet pulse. The hot jet and second-dimension oven were held offset by +75 °C (the maximum temperature of the second-dimension oven is 335 °C and the hot jet is 380 °C). The FID was operated at 300 °C with the following flows: H2: 30 mL/min, air: 400 mL/min, and N2: (make-up) 25 mL/min. The signal was recorded at 100 Hz using Agilent MassHunter software and further processed using a combination of GC Image and custom software for peak and blob detection and integration.
GC × GC–FID Data Processing Pipeline
A peak-tracking algorithm was used to automatically track peaks across the set of 21 chromatograms generated for each petroleum substance over the course of the experiment for the C10+ constituents. Several data-handling steps were performed in the following order, with more detailed methodological descriptions of each step presented in the Supporting Information.
Baseline Correction
Baseline is an operationally defined term whose specific definition depends on the aim of the analysis being conducted.41 The baseline needs to be removed before quantification and often corresponds to either the instrument background signal or the sum of the instrument background signal and any unresolved signal. To support the quantification of individual peaks, the baseline-correction algorithm of Eilers22,41−43 was selected using λ = 104.5 and p = 0.001, with the aim to remove the sum of instrument background signal and unresolved signal.41,44 For quantification of total GC × GC–FID-amenable mass, the baseline-correction algorithm of Reichenbach et al.45 was used, which is designed to remove instrument background signal (see Method S2 in the Supporting Information for more details).41,44,45
Automated Peak Delineation and Peak Integration
To delineate and integrate (quantify) thousands of peaks, an automatic algorithm is necessary. The inverted watershed algorithm included in the GC Image software46 was applied to chromatograms already baseline-corrected with the Eilers algorithm, an approach that enables automated signal integration of resolved peaks.41 It should be noted that these quantifications do not have accuracy comparable to more conventional approaches to signal integration in GC, such as expert manual integration or user-guided semi-automated methods. A user-supervised semi-automatic algorithm was applied to quantify selected peaks (<10 per chromatogram) that were used for key quantitative steps (normalization and correction for evaporative losses) to guarantee the highest precision in peak quantification. The semi-automatic 2-D Gaussian curve-fit algorithm developed by Arey et al.47 was used as it has been shown to enable accurate determination of individual peak volumes (see Method S3 in the Supporting Information for more details).22,23,41,46,47
Correction of Chromatograms and Peak Tables for Analyte Losses Incurred by Sample Processing
Quantification of the deuterated PAH standards spiked into the water samples immediately prior to extraction confirmed evaporative losses of the semi-volatile oil constituents during sample processing. The exact loss varied between samples, resulting from small variations in the extraction and concentration steps. This loss reflects the decreased concentrations of semi-volatile oil constituents relative to the less volatile constituents, thereby affecting the constituent signal intensities that were recorded by the GC × GC–FID. To compensate for these evaporative losses, the GC × GC–FID data of each sample was corrected as described in Method S4 in the Supporting Information.
Chromatogram and Peak Table Normalization
To enable comparisons of peak volume and total mass across chromatograms representing the full duration of the experiment, it was necessary to normalize the chromatograms because peak volumes depend on the mass of extract injected.44,48 Chromatograms were normalized to the hexachlorobenzene internal standard peak (see Method S5 in the Supporting Information for more details). The C10+ mass fraction was studied based on the chromatographic data, whereas the initial mass fraction of smaller constituents was quantified with simulated distillation data.
Chromatogram and Peak Table Alignment
Very high retention time reproducibility within GC × GC–FID chromatograms is crucial for automated peak tracking between chromatograms, where retention times play a prominent role for peak matching.23,49 However, the retention times of analytes within GC × GC–FID chromatograms experience small run-to-run variations resulting from uncontrollable instrument fluctuations.49−51 Consequently, retention time alignment is typically a necessary step in the processing of GC × GC–FID data. In the current study, the alignment algorithm developed by Gros et al.49,52 was applied and demonstrated to improve retention time precision by a factor of >2 (see Method S5 and Table S2 in the Supporting Information for more details).49−52
Peak Tracking
Automatic tools were needed to efficiently track hundreds to thousands of peaks across the 21 chromatograms spanning the experiment (days 0 to 64) for each of the two petroleum substances. An updated version of the algorithm developed by Wardlaw et al.23 was used in the current study. The algorithm relies on the matching of peaks based on retention time similarity, with several criteria aiming to minimize the risk of false positives. The detailed algorithm is described in Method S6 of the Supporting Information.
Fitting of First-Order DT50s
Exponential biodegradation curves without a lag time were assumed (eq 3 in Method S7 of the Supporting Information), where rate constants were fitted to the average peak volumes of peaks from the triplicate samples at each of the seven time points (see Method S7 in the Supporting Information for more details). Given that any lag phase is included in this calculation, the calculated “half-lives” are therefore DT50s according to the definitions given in Brown et al.53
Results and Discussion
General Observations of Biodegradation Based on Droplet Size, Droplet Concentration, and One-Dimensional GC–FID Analysis
The oil droplet generation produced dispersions with median oil droplet sizes of 10.0 ± 0.1 μm (VHGO) and 9.0 ± 0.1 μm (SRGO), respectively (Figure S4, Supporting Information). This corresponded to droplet sizes with near-to-neutral buoyancies (vertical velocities of 3.6 μm s–1 within stagnant SW, calculated with TAMOC54,55). The median droplet sizes as measured by the Coulter counter decreased in the dispersions of both oils over the 64 day incubation period from median sizes of 10 ± 0.1 and 9.0 ± 0.1 to 3.01 ± 0.37 and 2.84 ± 0.23 μm for VHGO and SRGO, respectively (Figure S4, Supporting Information). Similarly, the oil droplet concentrations were also reduced over the course of the experiment from 2.68 ± 0.08 mg/L (VHGO) and 2.79 ± 0.03 mg/L (SRGO) to 0.14 ± 0.05 mg/L (VHGO) and 0.24 ± 0.09 mg/L (SRGO).
Quantification of the TEM by one-dimensional GC–FID analyses revealed depletions of 82.8 ± 6.9 and 85.3 ± 5.5% of the C10+ mass after 64 days, while sterile controls for VHGO and SRGO showed negligible depletion (Figure S5, Supporting Information). The negligible depletion observed in the sterile controls confirms that the TEM depletion in the biotic samples was the result of biodegradation and not experimental artifacts. The observed reductions in oil droplet size and concentration, as well as TEM concentration reduction, were greater after 2 months of incubation than in previous experiments with crude oil dispersions produced using the same experimental system.10,36,38 This greater reduction was expected as the gas oils in the current experiments are distillation cuts in which recalcitrant constituents (e.g., asphaltenes, resins, etc.) with high boiling points are significantly removed during production, thus making the starting mass more biodegradable. It should be noted that a small degree of constituent loss due to sorption onto the exposure vessel walls is possible, but losses through volatilization are considered negligible due to there being no headspace in the exposure vessels.56
Chemical Analysis
Total (Resolved + Unresolved) Chromatographic Mass
The C10+ mass, corresponding to the overall mass eluting within the oil-containing region of the GC × GC–FID chromatograms (Figure S6), decreased with time throughout the experiment (Figure 1). The C10+ mass was assumed to be proportional to the FID signal after the Reichenbach et al.45 baseline correction, alignment, normalization, and evaporation correction steps. After 7 days, 20–29 and 34–51% of the initial C10+ mass were lost for VHGO and SRGO, respectively, while 73–81 and 84–88% of the initial C10+ mass had been lost at the end of the incubation period (day 64), respectively. These values match well with those determined by one-dimensional GC–FID analysis of 82.8 ± 6.9 and 85.3 ± 5.5% for VHGO and SRGO, respectively. This mass decrease follows an approximately logarithmic decay curve (Figure 1), which is consistent with the overlap of a large number of exponential decay curves (for individual constituents).
Figure 1.
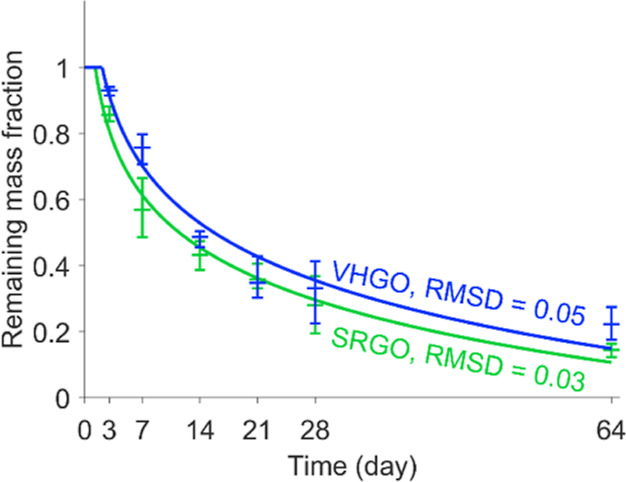
Remaining total C10+ mass fraction throughout the experiment for VHGO (blue) and SRGO (green) determined by GC × GC–FID. The symbols are the averages of three replicates at each time point (middle horizontal bar) and the range of the three replicates (outer horizontal bars), and the lines are fitted logarithmic decay curves with lag time. The root-mean-squared deviations between the data points and the fitted curves are provided in the figure.
The percentage of mass change for each chromatogram pixel was calculated across the experiment (Figure 2). The analysis indicates that initial losses at day 3 included the n-alkanes, which are known to biodegrade rapidly,57,58 such that the n-alkanes containing 9 to 26 carbon atoms were largely degraded by day 7 (Figures 2 and S7). Peak chemical identity attributions and the reason for using a FID are discussed in Method S8 of the Supporting Information, and it should be noted that other constituents may also occur within the defined constituent groups. Degradation of the n-alkanes is followed by degradation of the small aromatics (one and two rings) and some of the other saturated hydrocarbon constituents, especially methyl-branched alkanes, alkylcyclopentanes, and alkylcyclohexanes (Figure 2). This is consistent with existing knowledge regarding the sequential biodegradation of oil constituents, including observations made for saturated hydrocarbons in weathered oil samples by GC × GC–FID.22,57,58 Additionally, the mass loss patterns for the VHGO and SRGO oils are similar, which confirms the repeatability of the selected procedure.
Figure 2.

Percent mass change across the GC × GC–FID chromatogram throughout the experiment, calculated based on the average chromatogram at each time point in the experiment relative to the average chromatogram at day 0 (the average chromatogram is the average of the three replicate chromatograms). To limit noise, only the pixels representing the largest 10% in the day 0 chromatogram are displayed (the remaining 90% of pixels were shaded black), and smoothing was added to the data using a smoothing window of 2 and 10 pixels in the first and second dimensions. The general locations of selected constituents or constituent groups are indicated based on previous literature22,59 and on standard elution times, although it should be noted that other constituents may also occur within the defined constituent groups (see Methods S8 and Table S3, Supporting Information). Note: due to the smoothing applied, naphthalene and naphthalene-d8 overlapped, such that their mass losses are not accurate (naphthalene-d8 is not an oil constituent).
Tracking of Individual Peaks (Resolved Analytes)
Due to the stringency of the selection criteria for peaks that can be tracked, not all of the peaks present in the day 0 chromatogram for either VHGO or SRGO could be followed over the course of the biodegradation process. A total of 1114 and 1157 peaks were successfully tracked across the 21 chromatograms of VHGO and SRGO oils, respectively. Individually delineated peaks may represent either a single constituent or groups of multiple constituents due to the limits of the sample separation and the automated peak delineation algorithm. The tracked peaks were distributed across the entire chromatographic area, including the main constituent families (Figure 3). The peak volumes (relative to day 0) of each tracked peak for each of the sampling time points (3, 7, 14, 21, 28, and 64) are summarized in the Supporting Information (excel document). The tracked peaks corresponded to approximately 20–28% of the total number of peaks in the chromatograms (4168–5587). The mass of the tracked peaks corresponded to 53–54 and 53–54% of the total mass at day 0 for VHGO and SRGO oils, respectively. In other words, more than half of the petroleum substance mass could be tracked on an individual-peak basis. The peaks excluded by the peak-tracking algorithm were, on average, smaller at day 0 than the tracked peaks. In particular, most peaks in chromatographic regions known not to contain oil-constituent peaks could be excluded with this procedure (small, “artifactual” or SW-related peaks). However, a small number of excluded peaks were also scattered throughout the oil-constituent-containing region of the chromatograms. Overall, 184–188 and 126–135 peaks having mass fractions in the petroleum substances >0.1% (by mass) represented 43–44 and 41–42% of the mass of the VHGO and SRGO oils (at day 0), respectively. Among those peaks, 122–130 (33–34% of the oil mass) and 78–85 (36–37% of the oil mass) were tracked for the two oils. The non-tracked peaks were those that did not pass the selected criteria and were subsequently excluded, chiefly due to the risk of confusion with other peaks at ≥3 days.
Figure 3.
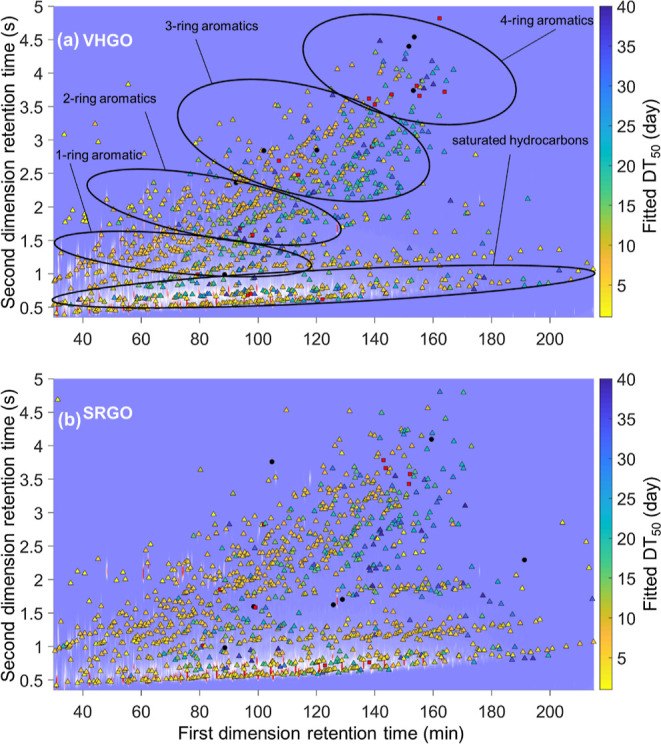
DT50s for the tracked peaks overlaid on the chromatogram of the fresh, non-biodegraded oil for both VHGO (top) and SRGO (bottom). Primary constituent classes are labeled on the top panel (see Methods S8). It should be noted that other constituents may also occur within the defined constituent groups (see Methods S8 and Table S3, Supporting Information). DT50s > 64 days (conserved constituents or constituents gaining mass) are displayed as black circles, and peaks having DT50s ≥ 40 days are displayed as red squares.
Only 4.0–12 and 1.6–3.0% of the mass of tracked peaks at day 0 remained at day 64 for the VHGO and SRGO oils, respectively. This corresponds to an 88–98.4% reduction in the mass of tracked peaks. By day 64, 52–65 and 69–76% of the non-tracked C10+ mass had been removed through biodegradation for VHGO and SRGO oils, respectively (Figure S8). Consequently, the mass of the tracked peaks decreased more than the overall petroleum mass (Figures 1 and 3). This is consistent with previous findings, where chromatographically resolved analytes are preferentially lost during the biodegradation of petroleum substances, often resulting in the emergence of prominent unresolved complex mixtures.24 For more detail and clarity, the tracked peaks in the VHGO experiment that have DT50s in the ranges <1–10, 10–20, 20–30, 30–40, 40–60, and >60 days are overlaid on individual day 0 chromatograms in Figures S9–S14 (Supporting Information). Overall, the calculated DT50s of the tracked peaks were consistent with pre-existing knowledge,10,20,58 with n-alkanes and small aromatics having the shortest DT50s and higher molecular weight and more complex constituents having longer DT50s. In agreement with previous experiments, the n-alkanes and small, non-substituted PAHs present in the VHGO and SRGO oils had DT50s < 16.4 days (Table S4, Supporting Information). The DT50s of individual constituent types confirm the overall knowledge of the relative biodegradation rates of hydrocarbon constituents. For example, the n-alkanes biodegraded more rapidly than the methyl-branched alkanes, which themselves degraded faster than isoprenoid molecules, consistent with previous work.22 However, the number of tracked peaks extends far beyond the reach of most existing studies and enables confirmation of a broad metabolic specificity of aerobic biodegradation, including for constituents not studied with most traditional approaches. Under the laboratory conditions employed in the current study, the vast majority of the >1000 peaks and masses had observed DT50s of <40 days.
Comparison of DT50 between VHGO and SRGO Oils
To validate the proposed approach for determining individual peak DT50s in a substance, the DT50s found for the experiments performed with VHGO and SRGO oils were compared for the 739 tracked peaks that are assumed to be the same constituent based on their ability to be matched between the two substances. These 739 peaks covered the whole chromatographic space, including the main constituent families (saturated hydrocarbons and 1–4-ring PAHs). The DT50s determined for 84% of these tracked peaks were within a factor of 2 of each other for VHGO and SRGO oils (Figure 4). This finding highlights the robustness and repeatability of the experimental, laboratory, and data treatment procedures.
Figure 4.
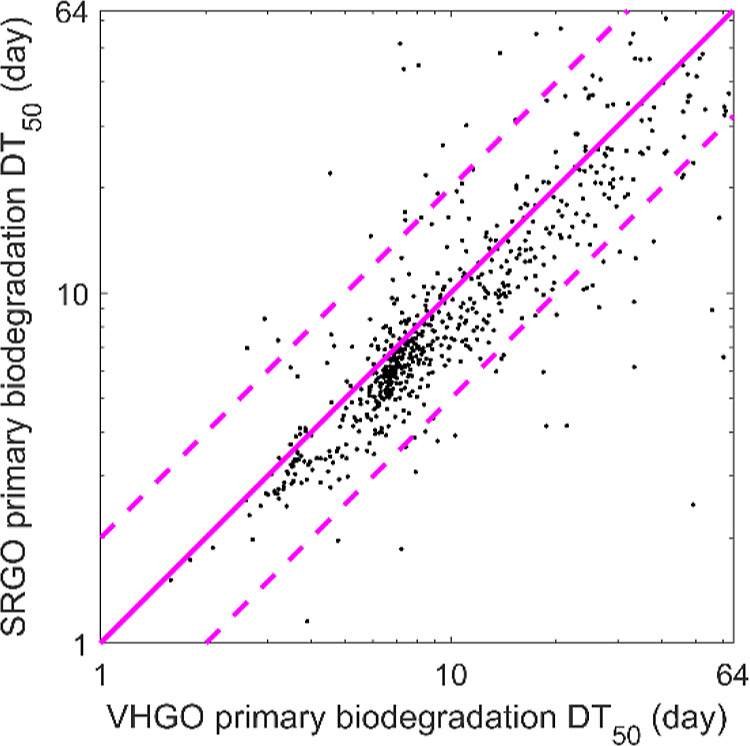
Comparison of peak DT50s calculated from peaks across the VHGO and SRGO experiments (solid dots). The pink solid line indicates the equality line, and the two pink dashed lines delineate the region where the VHGO and SRGO DT50s agree within a factor of 2 of each other. (Note the logarithmic axes.)
The common constituent peaks in SRGO and VHGO that exhibit DT50 discrepancies larger than a factor of 2 are typically low abundance peaks, which are more subject to artifacts such as baseline correction or low availability to microbes due to their preference for higher abundance constituents. For constituents with DT50s ≤ 64 d, the average peak volume of peaks having DT50s within a factor of 2 of each other was 9 times larger than the average peak volume of those peaks showing a larger discrepancy in the DT50. These peaks, retained by the criteria defined within the peak-tracking algorithm, are mostly located in regions of the chromatogram where small peaks occurred on top of a locally raised baseline, a situation that is known to pose a challenge for quantification. In contrast, the saturated hydrocarbons, small one-ring aromatic hydrocarbons, and constituents up to C4-substituted PAHs show, on average, a larger fraction of peaks agreeing within a factor of 2 of each other between the two oils. Overall, the method provides similar DT50s for the vast majority of common constituents present in different substances, which is consistent with the literature data. This confirms the observation that substance composition appears to be a minor factor influencing biodegradation and that the method appears to provide high-quality information on biodegradation. It is worth noting that DT50 determination is a relatively crude representation of the complex biodegradation process and that there will always be some degree of variability in biodegradation testing due to variations in the microbial community and test conditions.
Changes in Microbial Community Composition
A significant reduction in the diversity of the microbial communities (p < 0.01) was observed in oil-containing samples relative to that in non-oil-containing controls over the 64 day incubation period (Figure S15, Supporting Information). This reduction reflects the rapid emergence and abundance of a few groups of oil-degrading bacteria, mainly predominated by hydrocarbonoclastic bacteria specialized in hydrocarbon biodegradation. Furthermore, there were small differences in diversity between the communities present in SRGO and VHGO exposures, which were already observed by day 3. A comparison of the diversity between samples using two-dimensional PCoA plots showed non-oil-containing control samples clustering separately from the oil-containing VHGO and SRGO samples (Figure 5). The data for the oil-containing samples showed clustering related to incubation time, with incubations during the first 7 days distant from the 14–64 day incubations. The two-dimensional PCoA plots also showed that there were no clear separations in microbial community diversity related to oil type.
Figure 5.

PCoA plots (beta-diversity) of microbial communities in non-oil-containing controls (CTRL) and in dispersions with SRGO and VHGO at the start (0) and after 3–64 days of incubation. N.B. there is no day 0 sample for the SRGO and VHGO treatments, only a day 0 SW control (dark purple circle).
The relative abundances (Figure S15, Supporting Information) showed a predominance of the genus Colwellia in the control samples, with a spike of the Flavobacteriaceae genus Aurantivirga after 21 days of incubation. The oil-containing samples showed an early predominance of Saccharospirillaceae (>90% of the sequences after 3 days of incubation), represented by Oleispira and Thalassolituus. Both genera harbor obligate hydrocarbonoclastic members associated with alkane biodegradation.60,61 Although Saccharospirillaceae were still abundant in oil-containing samples after 7 days (50% of the sequences), the C1–B045 genus of Porticoccaceae became abundant, particularly in the SRGO incubations (27–43% of the sequences), after 14 days and remained abundant until day 64. Higher abundances of Cycloclasticus were found in VHGO (up to 30% after 14 days) than in SRGO. Both Porticoccaceae and Cycloclasticus include members of hydrocarbonoclastic bacteria associated with the biodegradation of aromatic hydrocarbons.31,62,63 Overall, the microbial community changes observed in the VHGO and SRGO samples over 64 days reflected the occurrence of biodegradation of the oil components and followed the expected evolution consistent with mineralization of alkane constituents followed by degradation of aromatic constituents.
Constituent Biodegradation and Microbial Community Changes
When comparing the DT50s of the tracked peaks (Figures 3 and S9–S14, Supporting Information) with changes in the microbial community (Figure S15, Supporting Information) over time, there are clear links. For example, the microbial community in both SRGO and VHGO rapidly shifts from day 0 to day 7 to being dominated by microbes from the Saccharospirillaceae family that are associated with alkane degradation (Figure S15, Supporting Information).31 Over the same time period, a wide range of n-alkanes and simply branched alkanes were determined to have DT50s in the range <1–10 days (Figure S9). The continued presence of Saccharospirillaceae up to day 28, although much less abundant compared to other genera of microbes, is also consistent with the complete mineralization of the majority of alkanes and consistent with the presence of more highly branched and cyclic alkane constituents with DT50s ranging from 10 to 40 days (Figures S10–S12, Supporting Information).30 Between day 7 and day 14, members of the Flavobacteriaceae genus emerge (Figure S15, Supporting Information), which are known to degrade PAHs.30,64 This coincides with the determination of many of the smaller, low-substituted PAHs having DT50s in the range of <1–20 days (Figures S9 and S10, Supporting Information). The Flavobacteriaceae remain present beyond 14 days, but the emergence of Porticoccaceae and Cycloclasticus from day 14 onward (Figure S15, Supporting Information) corresponds to the degradation of aromatic hydrocarbons with DT50s in the range of 10–40 days (Figures S10–S12, Supporting Information).38
Relative to previous studies on petroleum substance biodegradation, comparison of the microbial succession data with the higher-resolution analytical chemical techniques and peak tracking approach offers an advanced insight into the links between the biodegradation of constituent classes and the role particular types of microorganisms play in that process. However, it is important to note that constituent DT50s are somewhat arbitrary and may not be a robust indicator or a chemical’s environmental persistence due to the dynamic nature of the microbial population that eventually produces a community that is capable of degrading the complex substance.
Significance of This Approach for UVCB Biodegradation Assessment
The analysis approach demonstrated in the current study shows that combining advanced, high-resolution analytical chemical techniques (such as GC × GC–FID) with state-of-the-art data processing and peak tracking algorithms can improve the biodegradation characterization of petroleum substances. In the application of the approach here, it allows the DT50 determination of hundreds to thousands of constituents across a wide range of physicochemical properties. This represents a significant improvement upon the standardized approaches currently accepted for assessing the biodegradation of complex substances. The standard approaches are typically limited to either gross biodegradation of the whole substance or individually assessing the biodegradability of a small number of constituents present in a UVCB. However, for highly complex substances, the true chemical composition is largely unknown. The main limitation with pre-existing approaches is that the persistence assessment may become biased toward these known constituents, and the persistence of the full substance is defined by only a small proportion of the chemical mass or profile. The approach described herein showcases a new tool that can generate biodegradation information on individual peaks and blocks of peaks representing constituents belonging to the same class of hydrocarbons, as well as an improved comparison to available literature data.
The approach presented in the current study extends the persistence screening of complex substances so that it includes a much larger range of chemical classes, as well as the characterized mass than is found in single-dimension GC techniques. For example, typical GC–MS techniques characterize select constituents, including n-alkanes, iso-alkanes, and aromatic constituents (BTEX and PAH). The present study used GC × GC to characterize the biodegradation behavior for several different chemical classes, including naphthenic and mixed functionality naphthenic-aromatic constituents. Furthermore, the method characterized the biodegradation behavior of 53–58% of the C10+ mass. In contrast, single GC–MS techniques typically only characterize <5% of the total mass. Therefore, the present work represents a significant advancement over previous biodegradation studies and can enable more detailed characterization by comparing the chemical and microbial biodegradation profiles.
While it is also of interest to follow the evolution of hydrocarbon degradation products of the constituents in the test substances, this is beyond the scope of the present work. Biodegradation of hydrocarbons typically leads to the addition of functional groups (hydroxy-, ketone-, quinone-, and carboxyl-), producing polar compounds that are discriminated against during the sample preparation (solvent extraction of water) and which are also generally less amenable to gas chromatographic analysis, causing non-quantitative recoveries. For the most biodegraded samples (e.g., days 28 and 64), we do indeed observe “new” peaks in an area of the GC × GC-chromatogram that could correspond to such metabolites. The increased polarity relative to hydrocarbons causes these degradation product analytes to become positively offset (higher retention time) in the second dimension compared to the parent hydrocarbons (see Figure S7, Supporting Information). Therefore, we interpreted that these small peaks did not interfere with the quantification of tracked hydrocarbons in most cases, but this possibility was not rigorously investigated.
Although it was not possible to determine the degradation of the individual peaks representing 42–47% of the non-tracked C10+ mass, it was possible to determine the figure representing the total of this mass by subtracting the mass of C10+ tracked peaks from the total C10+ mass at each time point. For example, 52–65% (VHGO) and 69–76% (SRGO) of the non-tracked C10+ peak mass had been biodegraded by day 64 (Figure S8). The average DT50s for the tracked C10+ mass are 13 and 6 days for VHGO and SRGO, respectively, while the average DT50s for the non-tracked C10+ mass are 46 and 25 days. Chemical structure assignment for individual peaks may be performed with some combination of QSAR analysis and high-resolution mass spectrometry, but this is out of scope for the present study. This means that the method has the potential to generate much more data to support biodegradation assessments for UVCBs. While the method has been demonstrated to work very well for the VHGO and SRGO in the current study, FID limits the ability to unambiguously follow the degradation of specific UVCB constituents. Further development of the approach using GC × GC coupled to high-resolution mass spectrometry would represent a step forward by allowing more accurate tracking and assessment of specific constituents while also providing an opportunity to identify those constituents that either biodegrade rapidly or exhibit a high level of resistance to microbial degradation. To maximize the utility of the outcomes of this testing and analytical protocol, future work will include identification of the tracked peaks by either assignment to a hydrocarbon class/carbon number or using MS. The DT50 information can then more confidently be used for screening the persistence of constituents of petroleum substances.
Acknowledgments
Funding was provided by Concawe. The authors are grateful to Roman Netzer, Marianne Aas, Kjersti Almås, Marianne Rønsberg, Marianne Molid, Lisbet Støen, and Kristin Bonaunet (SINTEF Ocean) for assistance with experiments and analyses and to Bob Nelson (WHOI), Shahin Tavakoli (University of Geneva), Jacco Woldendorp, and Jan Blomberg (Shell Global Solutions, Amsterdam) for discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.3c01624.
Additional experimental details, methods, and underlying equations; summarizing material properties, DT50 values, sample numbers and information, and retention times before and after alignment; and experimental setup, oil droplet size and concentration data, microbial community succession, GC × GC–FID chromatograms, and evaporative mass losses (PDF)
Peak volumes (relative to day 0) of each tracked peak for each of the sampling time points (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- OSPAR Commission . Dynamic Selection and Prioritisation Mechanism for Hazardous Substances (New DYNAMEC Manual); OSPAR Commission, 2006. [Google Scholar]
- ECHA . Chapter R.11: PBT/vPvB assessment. Version 3.0. In Guidance on information requirements and chemical safety assessment; Europeqan Chemical Agency: Helsinki, Finland, 2017. [Google Scholar]
- EPA, U. S. Reviewing new chemicals under the Toxic Substances Control Act (TSCA). Actions under TSCA Section 5; U.S. Environmental Protection Agency.
- EPCRA . United States Toxics Release Inventory (TRI) Reporting (Section 313 of the Emergency Planning and Community Right to Know Act (EPCRA)), 1986.
- Alexander M. Biodegradation of organic chemicals. Environ. Sci. Technol. 1985, 19, 106–111. 10.1021/es00132a602. [DOI] [PubMed] [Google Scholar]
- OECD . Revised Introduction to the OECD Guidelines for Testing of Chemicals, Section 3; 2006. [Google Scholar]
- Salvito D.; Fernandez M.; Jenner K.; Lyon D. Y.; Knecht J.; Mayer P.; MacLeod M.; Eisenreich K.; Leonards P.; Cesnaitis R.; et al. Improving the environmental risk assessment of substances of unknown or variable composition, complex reaction products, or biological materials. Environ. Toxicol. Chem. 2020, 39, 2097–2108. 10.1002/etc.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A. M.; Nelson R. K.; Reddy C. M.; Savory J. J.; Kaiser N. K.; Fitzsimmons J. E.; Marshall A. G.; Rodgers R. P. Expansion of the analytical window for oil spill characterization by ultrahigh resolution mass spectrometry: Beyond gas chromatography. Environ. Sci. Technol. 2013, 47, 7530–7539. 10.1021/es305284t. [DOI] [PubMed] [Google Scholar]
- Salvito D.; Fernandez M.; Arey J. S.; Lyon D. Y.; Lawson N.; Deglin S.; MacLeod M. The path to UVCB ecological risk assessment: Grappling with substance characterization. Environ. Toxicol. Chem. 2022, 41, 2649–2657. 10.1002/etc.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakstad O. G.; Nordtug T.; Throne-Holst M. Biodegradation of dispersed Macondo oil in seawater at low temperature and different oil droplet sizes. Mar. Pollut. Bull. 2015, 93, 144–152. 10.1016/j.marpolbul.2015.02.006. [DOI] [PubMed] [Google Scholar]
- Prince R. C.; Butler J. D.; Redman A. D. The rate of crude oil biodegradation in the sea. Environ. Sci. Technol. 2017, 51, 1278–1284. 10.1021/acs.est.6b03207. [DOI] [PubMed] [Google Scholar]
- Dalton H.; Stirling D. Co-metabolism. Philos. Trans. R. Soc. London, Ser. B 1982, 297, 481–496. 10.1098/rstb.1982.0056. [DOI] [PubMed] [Google Scholar]
- Møller M. T.; Birch H.; Sjøholm K. K.; Hammershøj R.; Jenner K.; Mayer P. Biodegradation of an essential oil UVCB-Whole substance testing and constituent specific analytics yield biodegradation kinetics of mixture constituents. Chemosphere 2021, 278, 130409. 10.1016/j.chemosphere.2021.130409. [DOI] [PubMed] [Google Scholar]
- Chang M. K.; Voice T. C.; Criddle C. S. Kinetics of competitive inhibition and cometabolism in the biodegradation of benzene, toluene, and p-xylene by two Pseudomonas isolates. Biotechnol. Bioeng. 1993, 41, 1057–1065. 10.1002/bit.260411108. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Wang W.; Li Y.; Yang Q. Co-metabolic degradation of naphthalene and pyrene by acclimated strain and competitive inhibition kinetics. J. Environ. Sci. Health, Part B 2019, 54, 505–513. 10.1080/03601234.2019.1586033. [DOI] [PubMed] [Google Scholar]
- Boehm P. D.; Murray K. J.; Cook L. L. Distribution and attenuation of polycyclic aromatic hydrocarbons in Gulf of Mexico seawater from the Deepwater Horizon oil accident. Environ. Sci. Technol. 2016, 50, 584–592. 10.1021/acs.est.5b03616. [DOI] [PubMed] [Google Scholar]
- Douglas G. S.; Bence A. E.; Prince R. C.; McMillen S. J.; Butler E. L. Environmental stability of selected petroleum hydrocarbon source and weathering ratios. Environ. Sci. Technol. 1996, 30, 2332–2339. 10.1021/es950751e. [DOI] [Google Scholar]
- Mills M. A.; McDonald T. J.; Bonner J. S.; Simon M. A.; Autenrieth R. L. Method for quantifying the fate of petroleum in the environment. Chemosphere 1999, 39, 2563–2582. 10.1016/s0045-6535(99)00163-0. [DOI] [Google Scholar]
- Prince R. C.; McFarlin K. M.; Butler J. D.; Febbo E. J.; Wang F. C.; Nedwed T. J. The primary biodegradation of dispersed crude oil in the sea. Chemosphere 2013, 90, 521–526. 10.1016/j.chemosphere.2012.08.020. [DOI] [PubMed] [Google Scholar]
- Venosa A.; Holder E. Biodegradability of dispersed crude oil at two different temperatures. Mar. Pollut. Bull. 2007, 54, 545–553. 10.1016/j.marpolbul.2006.12.013. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Fingas M. F. Development of oil hydrocarbon fingerprinting and identification techniques. Mar. Pollut. Bull. 2003, 47, 423–452. 10.1016/s0025-326x(03)00215-7. [DOI] [PubMed] [Google Scholar]
- Gros J.; Reddy C. M.; Aeppli C.; Nelson R. K.; Carmichael C. A.; Arey J. S. Resolving biodegradation patterns of persistent saturated hydrocarbons in weathered oil samples from the Deepwater Horizon disaster. Environ. Sci. Technol. 2014, 48, 1628–1637. 10.1021/es4042836. [DOI] [PubMed] [Google Scholar]
- Wardlaw G. D.; Arey J. S.; Reddy C. M.; Nelson R. K.; Ventura G. T.; Valentine D. L. Disentangling oil weathering at a marine seep using GC×GC: Broad metabolic specificity accompanies subsurface petroleum biodegradation. Environ. Sci. Technol. 2008, 42, 7166–7173. 10.1021/es8013908. [DOI] [PubMed] [Google Scholar]
- Reddy C. M.; Eglinton T. I.; Hounshell A.; White H. K.; Xu L.; Gaines R. B.; Frysinger G. S. The West Falmouth Oil Spill after Thirty Years: The Persistence of Petroleum Hydrocarbons in Marsh Sediments. Environ. Sci. Technol. 2002, 36, 4754–4760. 10.1021/es020656n. [DOI] [PubMed] [Google Scholar]
- Frysinger G. S.; Gaines R. B.; Xu L.; Reddy C. M. Resolving the Unresolved Complex Mixture in Petroleum-Contaminated Sediments. Environ. Sci. Technol. 2003, 37, 1653–1662. 10.1021/es020742n. [DOI] [PubMed] [Google Scholar]
- Nelson R. K.; Kile B. M.; Plata D. L.; Sylva S. P.; Xu L.; Reddy C. M.; Gaines R. B.; Frysinger G. S.; Reichenbach S. E. Tracking the weathering of an oil spill with comprehensive two-dimensional gas chromatography. Environ. Forensics 2006, 7, 33–44. 10.1080/15275920500506758. [DOI] [Google Scholar]
- Brakstad O. G.; Throne-Holst M.; Netzer R.; Stoeckel D. M.; Atlas R. M. Microbial communities related to biodegradation of dispersed Macondo oil at low seawater temperature with norwegian coastal seawater. Microb. Biotechnol. 2015, 8, 989–998. 10.1111/1751-7915.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bælum J.; Borglin S.; Chakraborty R.; Fortney J. L.; Lamendella R.; Mason O. U.; Auer M.; Zemla M.; Bill M.; Conrad M. E.; et al. Deep-sea bacteria enriched by oil and dispersant from the Deepwater Horizon spill. Environ. Microbiol. 2012, 14, 2405–2416. 10.1111/j.1462-2920.2012.02780.x. [DOI] [PubMed] [Google Scholar]
- Dubinsky E. A.; Conrad M. E.; Chakraborty R.; Bill M.; Borglin S. E.; Hollibaugh J. T.; Mason O. U.; M Piceno Y.; Reid F. C.; Stringfellow W. T.; Tom L. M.; et al. Succession of hydrocarbon-degrading bacteria in the aftermath of the Deepwater Horizon oil spill in the Gulf of Mexico. Environ. Sci. Technol. 2013, 47, 10860–10867. 10.1021/es401676y. [DOI] [PubMed] [Google Scholar]
- Mason O. U.; Hazen T. C.; Borglin S.; Chain P. S. G.; Dubinsky E. A.; Fortney J. L.; Han J.; Holman H.-Y. N.; Hultman J.; Lamendella R.; et al. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 2012, 6, 1715–1727. 10.1038/ismej.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakimov M. M.; Timmis K. N.; Golyshin P. N. Obligate oil-degrading marine bacteria. Curr. Opin. Biotechnol. 2007, 18, 257–266. 10.1016/j.copbio.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Concawe . Concawe Substance Identification Group Analytical Program Report (Abridged Version); Concawe: Brussels, 2019, https://www.concawe.eu/wp-content/uploads/Concawe-Substance-Identification-Group-Analytical-Program-Report-Abridged-Version.pdf.
- Comber M. I. H.; den Haan K. H.; Djemel N.; Eadsforth C. V.; King D.; Paumen M. L.; Parkerton T.; Dmytrasz B.. In Primary Biodegradation of Petroleum Hydrocarbons in Seawater; CONCAWE: Brussels, Belgium, 2012. https://www.concawe.eu/wp-content/uploads/2017/01/rpt_12-10-2012-05442-01-e.pdf.
- Comber M.; Holt M.. Developing a Set of Reference Chemicals for Use in Biodegradability Tests for Assessing the Persistency of Chemicals MCC, 2010. http://cefic-lri.org/wp-content/uploads/uploads/Projectpublications/MCC_007_Eco12_Final_Report.pdf.
- Brakstad O. G.; Farooq U.; Ribicic D.; Netzer R. Dispersibility and biotransformation of oils with different properties in seawater. Chemosphere 2018, 191, 44–53. 10.1016/j.chemosphere.2017.10.012. [DOI] [PubMed] [Google Scholar]
- Brakstad O. G.; Ribicic D.; Winkler A.; Netzer R. Biodegradation of dispersed oil in seawater is not inhibited by a commercial oil spill dispersant. Mar. Pollut. Bull. 2018, 129, 555–561. 10.1016/j.marpolbul.2017.10.030. [DOI] [PubMed] [Google Scholar]
- Nordtug T.; Olsen A. J.; Altin D.; Meier S.; Overrein I.; Hansen B. H.; Johansen Ø. Method for generating parameterized ecotoxicity data of dispersed oil for use in environmental modelling. Mar. Pollut. Bull. 2011, 62, 2106–2113. 10.1016/j.marpolbul.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Ribicic D.; Netzer R.; Hazen T. C.; Techtmann S. M.; Drabløs F.; Brakstad O. G. Microbial community and metagenome dynamics during biodegradation of dispersed oil reveals potential key-players in cold Norwegian seawater. Mar. Pollut. Bull. 2018, 129, 370–378. 10.1016/j.marpolbul.2018.02.034. [DOI] [PubMed] [Google Scholar]
- Henry I. A.; Netzer R.; Davies E. J.; Brakstad O. G. Formation and fate of oil-related aggregates (ORAs) in seawater at different temperatures. Mar. Pollut. Bull. 2020, 159, 111483. 10.1016/j.marpolbul.2020.111483. [DOI] [PubMed] [Google Scholar]
- Schroeder P. J.; Jenkins D. G. How robust are popular beta diversity indices to sampling error?. Ecosphere 2018, 9, e02100 10.1002/ecs2.2100. [DOI] [Google Scholar]
- Samanipour S.; Dimitriou-Christidis P.; Gros J.; Grange A.; Samuel Arey J. Analyte quantification with comprehensive two-dimensional gas chromatography: Assessment of methods for baseline correction, peak delineation, and matrix effect elimination for real samples. J. Chromatogr. A 2015, 1375, 123–139. 10.1016/j.chroma.2014.11.049. [DOI] [PubMed] [Google Scholar]
- Gros-Eilers-Arey Code to Perform Baseline Correction of GC × GC Chromatograms; GitHub, 2015https://github.com/jsarey/GCxGC-baseline-correction.
- Eilers P. H. C. Parametric time warping. Anal. Chem. 2004, 76, 404–411. 10.1021/ac034800e. [DOI] [PubMed] [Google Scholar]
- Swarthout R. F.; Gros J.; Arey J. S.; Nelson R. K.; Valentine D. L.; Reddy C. M.. Comprehensive two-dimensional gas chromatography to assess petroleum product weathering. In Hydrocarbon and Lipid Microbiology Protocols: Petroleum, Hydrocarbon and Lipid Analysis; McGenity T. J., Timmis K. N., Nogales B., Eds.; Springer Berlin Heidelberg, 2017, pp 129–149. [Google Scholar]
- Reichenbach S. E.; Ni M.; Zhang D.; Ledford E. B. Image background removal in comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2003, 985, 47–56. 10.1016/S0021-9673(02)01498-X. [DOI] [PubMed] [Google Scholar]
- Reichenbach S. E.; Ni M.; Kottapalli V.; Visvanathan A. Information technologies for comprehensive two-dimensional gas chromatography. Chemom. Intell. Lab. Syst. 2004, 71, 107–120. 10.1016/j.chemolab.2003.12.009. [DOI] [Google Scholar]
- Arey J. S.; Nelson R. K.; Reddy C. M. Disentangling oil weathering using GC×GC. 1. Chromatogram analysis. Environ. Sci. Technol. 2007, 41, 5738–5746. 10.1021/es070005x. [DOI] [PubMed] [Google Scholar]
- Prince R. C.; Elmendorf D. L.; Lute J. R.; Hsu C. S.; Haith C. E.; Senius J. D.; Dechert G. J.; Douglas G. S.; Butler E. L. 17.alpha.(H)-21.beta.(H)-hopane as a conserved internal marker for estimating the biodegradation of crude oil. Environ. Sci. Technol. 1994, 28, 142–145. 10.1021/es00050a019. [DOI] [PubMed] [Google Scholar]
- Gros J.; Nabi D.; Dimitriou-Christidis P.; Rutler R.; Arey J. S. Robust algorithm for aligning two-dimensional chromatograms. Anal. Chem. 2012, 84, 9033–9040. 10.1021/ac301367s. [DOI] [PubMed] [Google Scholar]
- Pierce K. M.; Wood L. F.; Wright B. W.; Synovec R. E. A Comprehensive Two-Dimensional Retention Time Alignment Algorithm To Enhance Chemometric Analysis of Comprehensive Two-Dimensional Separation Data. Anal. Chem. 2005, 77, 7735–7743. 10.1021/ac0511142. [DOI] [PubMed] [Google Scholar]
- Reichenbach S. E.; Rempe D. W.; Tao Q.; Bressanello D.; Liberto E.; Bicchi C.; Balducci S.; Cordero C. Alignment for comprehensive two-dimensional gas chromatography with dual secondary columns and detectors. Anal. Chem. 2015, 87, 10056–10063. 10.1021/acs.analchem.5b02718. [DOI] [PubMed] [Google Scholar]
- Gros-Arey Code to Align GC × GC Chromatograms; GitHub, 2015https://github.com/jsarey/GCxGC-alignment.
- Brown D. M.; Camenzuli L.; Redman A. D.; Hughes C.; Wang N.; Vaiopoulou E.; Saunders D.; Villalobos A.; Linington S. Is the Arrhenius-correction of biodegradation rates, as recommended through REACH guidance, fit for environmentally relevant conditions? An example from petroleum biodegradation in environmental systems. Sci. Total Environ. 2020, 732, 139293. 10.1016/j.scitotenv.2020.139293. [DOI] [PubMed] [Google Scholar]
- Dissanayake A. L.; Gros J.; Socolofsky S. A. Integral models for bubble, droplet, and multiphase plume dynamics in stratification and crossflow. Environ. Fluid Mech. 2018, 18, 1167–1202. 10.1007/s10652-018-9591-y. [DOI] [Google Scholar]
- Gros J.; Socolofsky S. A.; Dissanayake A. L.; Jun I.; Zhao L.; Boufadel M. C.; Reddy C. M.; Arey J. S. Petroleum dynamics in the sea and influence of subsea dispersant injection during Deepwater Horizon. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 10065–10070. 10.1073/pnas.1612518114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD . Test No. 301: Ready Biodegradability, 1992.
- Prosser C. M.; Redman A. D.; Prince R. C.; Paumen M. L.; Letinski D. J.; Butler J. D. Evaluating persistence of petroleum hydrocarbons in aerobic aqueous media. Chemosphere 2016, 155, 542–549. 10.1016/j.chemosphere.2016.04.089. [DOI] [PubMed] [Google Scholar]
- Perry J. J.Microbial metabolism of cyclic alkanes. In Petroleum Microbiology; Atlas R. M., Ed.; Macmillan, 1984, pp 61–98. [Google Scholar]
- Nelson R. K.; Aeppli C.; Arey J. S.; Chen H.; de Oliveira A. H. B.; Eiserbeck C.; Frysinger G. S.; Gaines R. B.; Grice K.; Gros J.; et al. 8—Applications of comprehensive two-dimensional gas chromatography (GC × GC) in studying the source, transport, and fate of petroleum hydrocarbons in the environment. In Standard Handbook Oil Spill Environmental Forensics, 2nd ed.; Stout S. A., Wang Z., Eds; Academic Press, 2016; pp 399–448. [Google Scholar]
- Golyshin P.; Ferrer M.; Chernikova T.; Golyshina O.; Yakimov M.. Oleispira. Oleispira. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis K. N., Ed.; Springer: Berlin, Heidelberg, 2010, pp 1755–1763. [Google Scholar]
- Yakimov M.; Genovese M.; Denaro R.. Thalassolituus. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis K. N., Ed.; Springer: Berlin, Heidelberg, 2010; pp 1765–1772. [Google Scholar]
- Gutierrez T.Aerobic hydrocarbon-degrading Gammaproteobacteria: Porticoccus. In Taxonomy, Genomics and Ecophysiology of Hydrocarbon-Degrading Microbes; McGenity T. J., Ed.; Springer Cham: Berlin, 2019; pp 181–189. [Google Scholar]
- Gutierrez T.; Nichols P. D.; Whitman W. B.; Aitken M. D. Porticoccus hydrocarbonoclasticus sp. nov., an aromatic hydrocarbon-degrading bacterium identified in laboratory cultures of marine phytoplankton. Appl. Environ. Microbiol. 2012, 78, 628–637. 10.1128/aem.06398-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzesicka-Mlynarz D.; Ward O. P. Degradation of polycyclic aromatic hydrocarbons (PAHs) by a mixed culture and its component pure cultures, obtained from PAH-contaminated soil. Can. J. Microbiol. 1995, 41, 470–476. 10.1139/m95-063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.