

ABSTRACT
Lysosomal dysfunction is a pathogenic link that may explain the causal relationship between diabetes and Alzheimer disease; however, there is no information about the regulation of hyperglycemia in neuronal lysophagy modulating lysosomal function. We examined the effect and related mechanisms of action of high glucose on lysophagy impairment and subsequent Aβ accumulation in human induced pluripotent stem cell (hiPSC)-derived neurons, mouse hippocampal neurons, and streptozotocin (STZ)-induced diabetic mice. High-glucose induced neuronal lysosomal dysfunction through reactive oxygen species-mediated lysosomal membrane permeabilization and lysophagy impairment. Among lysophagy-related factors, the expression of TRIM16 (tripartite motif containing 16) was reduced in high-glucose-treated neuronal cells and the diabetic hippocampus through MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1)-mediated inhibition of TFEB (transcription factor EB) activity. TRIM16 overexpression recovered lysophagy through the recruitment of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3), SQSTM1/p62, and ubiquitin to damaged lysosomes, which inhibited the high-glucose-induced accumulation of Aβ and p-MAPT/tau. In the diabetic mice model, TFEB enhancer recovered lysophagy in the hippocampus, resulting in the amelioration of cognitive impairment. In conclusion, TRIM16-mediated lysophagy is a promising candidate for the inhibition of diabetes-associated Alzheimer disease pathogenesis.
Abbreviations
Aβ: amyloid β; AD: Alzheimer disease; AMPK: 5’ adenosine monophosphate-activated protein kinase; CTSB: cathepsin B; CTSD: cathepsin D; DM: diabetes mellitus; ESCRT: endosomal sorting complex required for transport; FBXO27: F-box protein 27; iPSC-NDs: induced pluripotent stem cell-derived neuronal differentiated cells; LAMP1: lysosomal-associated membrane protein 1; LMP: lysosomal membrane permeabilization; LRSAM1: leucine rich repeat and sterile alpha motif containing 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTORC1: mechanistic target of rapamycin kinase complex 1; p-MAPT/tau: phosphorylated microtubule associated protein tau; ROS: reactive oxygen species; STZ: streptozotocin; TFE3: transcription factor E3; TFEB: transcription factor EB; TRIM16: tripartite motif containing 16; UBE2QL1: ubiquitin conjugating enzyme E2 Q family like 1; VCP: valosin containing protein.
KEYWORDS: Amyloid β, autophagy, diabetes, hippocampal neuron, TFEB, TRIM16
Introduction
Although diabetes mellitus (DM) is considered as a risk factor for dementia, the relationship between diabetes and the development of Alzheimer disease (AD) has been controversial due to inconsistencies among human cohort studies, biochemical analyses, and animal studies [1,2]. Nevertheless, as the recent trend of diabetes is decreasing the age of onset [3], which significantly increases the incidence of dementia, more sophisticated studies focusing on the early pathogenesis of the disease would help in identifying the possibility of diabetes-associated AD. In fact, lysosomal disorders inducing amyloid plaques and neurofibrillary tangles are observed before the onset of AD [4,5]. In addition, previous studies have demonstrated that hyperglycemia upregulates the expression of neuronal amyloid β (Aβ) and phosphorylated-MAPT/tau (p-MAPT/tau) through endolysosomal dysfunction [6,7], an early pathological feature of AD [8]. Therefore, targeting lysosomes may help in elucidating the potential disease mechanism. To manage lysosomal dysfunction, lysosomal quality control is primarily performed through lysophagy [9]. Lysophagy, a selective macroautophagy/autophagy that detects and removes damaged lysosomes, is activated through ubiquitination-inducing proteins such as TRIM16 (tripartite motif containing 16), FBXO27 (F-box protein 27), LRSAM1 (leucine-rich repeat and sterile alpha motif containing 1), UBE2QL1 (ubiquitin conjugating enzyme E2Q family like 1), and VCP (valosin containing protein) [10]. Although genetic or chemical inhibition of lysophagy factors increases proteopathic seeding in neurons [11], the direct causal relationship between lysophagy and neurodegenerative diseases has not yet been elucidated. However, accumulating data suggest that high-efficiency lysophagy is essential for maintaining lysosomal function and protects against neurodegeneration [10].
In chronic diabetic state, hyperglycemia induces lysosomal membrane permeabilization (LMP) and inhibits lysosomal biogenesis, resulting in lysosomal dysfunction [12]. It has been shown that db/db diabetic mice exhibit hippocampal lysosomal dysfunction characterized by inhibition of the expression and activity of lysosomal hydrolases such as CTSD (cathepsin D) and HEXA (hexosaminidase subunit alpha) [13]. Lysosomal abnormalities are also induced through HMGB1 (high mobility group box 1)-mediated LMP in diabetic retinal pigment epithelial cells [14]. In diabetic renal tubular epithelial cells, SMAD3-mediated suppression of TFEB (transcription factor EB) activity was found to induce lysosomal depletion [12]. Although lysosomal dysfunction has been implicated in the pathogenesis of diabetes, no studies have examined how diabetes affects lysophagy and the subsequent effects on neuronal Aβ and p-MAPT degradation. Considering that suppression of VCP function through autosomal-dominant genetic mutation increases MAPT aggregation in the human AD brain [15], investigating impaired lysophagy in diabetes can provide mechanistic insights into AD pathology. In the present study, we explored the effects and molecular mechanisms of action of high glucose on neuronal lysophagy and subsequent accumulation of Aβ and p-MAPT and cognitive impairment using human induced pluripotent stem cell-derived neuronal differentiated cells (iPSC-NDs), mouse hippocampal neurons, and streptozotocin (STZ)-induced diabetic mice. Our study elucidates the lysophagy-related mechanisms and potential regulatory candidates of DM-associated AD pathology.
Results
High glucose induces neuronal lysosomal dysfunction, Aβ accumulation, and cell death
To elucidate the effect of high glucose on neuronal lysosomal function using LysoTracker that has a strong affinity for acidic organelles, we first examined acidic lysosomes in iPSC-NDs. High glucose decreased the degree of colocalization between the LysoTracker and LAMP1 (lysosomal-associated membrane protein 1), a lysosomal marker protein and the mean LysoTracker intensities (Figure 1A,B). The results of LysoSensor and BCECF staining showing that lysosomal pH increased and cytosolic pH decreased by high glucose treatment indirectly implied lysosomal damage (Fig. S1A and B). Moreover, to explore the relationship between lysosomal dysfunction and Aβ and p-MAPT degradation under high glucose conditions, we evaluated the subcellular localization of CTSB and CTSD, which respectively degrade Aβ and p-MAPT [16,17]. We observed that the degree of colocalization between CTSB or CTSD and LAMP1 was decreased in both iPSC-NDs and mouse hippocampal neurons under high glucose conditions (Figure 1C–F). Similar to a previous report [5], we found that high-glucose-induced Aβ accumulation in lysosomes (Figure 1G,H). Given that ROS overproduction, which is considered as the primary pathogenic mechanism of hyperglycemia-induced disorders, induces LMP [7,18] and TFEB activation compensates lysosomal stress [19], we investigated whether high-glucose-mediated lysosomal dysfunction induces neuronal cell death. Our data showed that high-glucose-induced neuronal cell death was inhibited by pretreatment with NAC, an ROS scavenger, and curcumin C1, an MTOR (mechanistic target of rapamycin kinase)-independent TFEB activator [19], respectively (Figure 1I,J). These results demonstrate that high glucose induces lysosomal dysfunction, which may be involved in intraneuronal Aβ accumulation and cell death.
Figure 1.
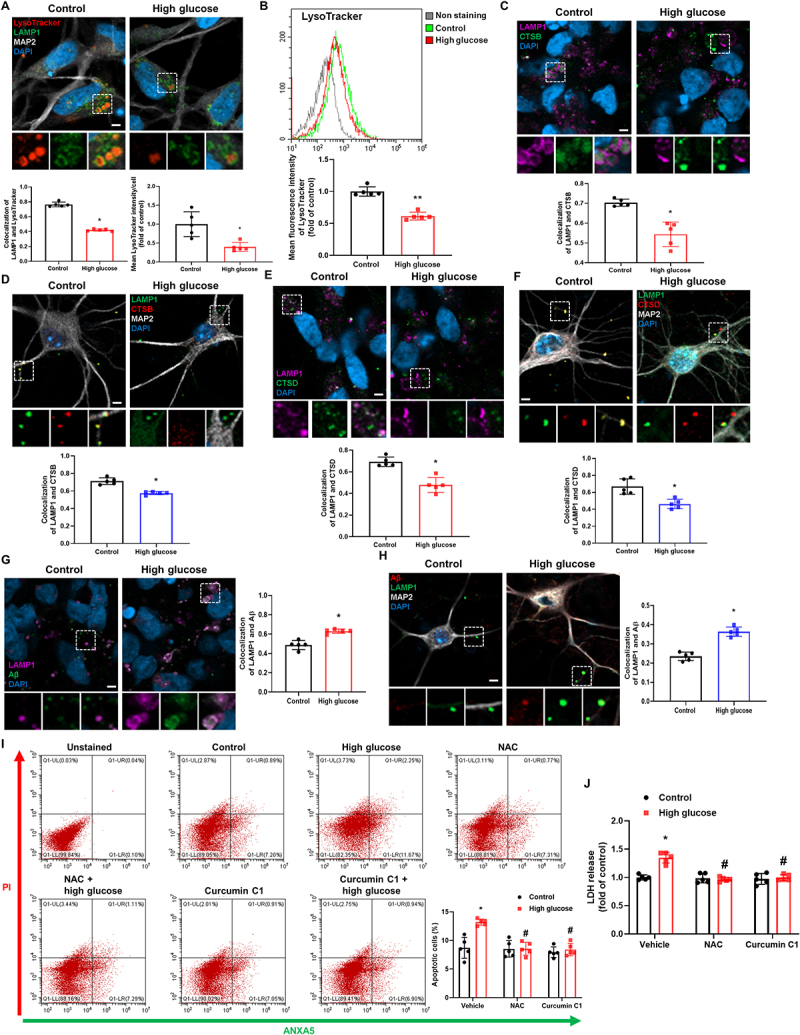
High glucose induces lysosomal dysfunction resulting in Aβ accumulation and neuronal cell death. (A-C, and E) iPSC-NDs were exposed to high glucose (HG; 25 mM) for 24 h. (D and F) Hippocampal neurons were exposed to HG for 24 h at DIV 21. (A) Immunofluorescence staining of LAMP1, MAP2, and LysoTracker were visualized. DAPI was used to stain nuclei. (B) The mean fluorescence intensities of LysoTracker were measured by flow cytometric analysis. (C and D) Double immunofluorescence staining of LAMP1 and CTSB were analyzed in iPSC-NDs and hippocampal neurons. (E and F) iPSC-NDs and hippocampal neurons were immunostained with LAMP1 and CTSD. (G and H) iPSC-NDs and hippocampal neurons at DIV 21 were exposed to HG for 48 h. Immunofluorescence staining of LAMP1 and Aβ were visualized. (I and J) iPSC-NDs were pretreated with NAC (2 mM) or curcumin C1 (1 μM) for 30 min prior to high glucose (25 mM) exposure for 72 h. (I) The percentage of apoptotic cells (ANXA5 and PI positive) were measured by flow cytometric analysis. (J) LDH from cell supernatant were measured with LDH assay kit. n = 5. Scale bars: 8 μm. All data are representative. Quantitative data are represented as mean ± SD. *P < 0.05; **P < 0.01 versus control; #P <0.05 versus HG.
High glucose suppresses neuronal lysophagy
To elucidate how high glucose induces lysosomal dysfunction, we investigated whether ROS scavenging alleviates lysosomal dysfunction. Pretreatment with NAC reversed the high-glucose-induced decrease in the signal intensities of the LysoTracker, indicating that lysosomal dysfunction was triggered by ROS-mediated LMP under high glucose conditions (Figure 2A). We next focused on the lysophagy machinery to explore how neurons cope with high-glucose-mediated lysosomal damage. In hippocampal neurons, we transfected LGALS3, a marker of lysosomal damage [20]. High glucose treatment decreased the degree of colocalization between GFP-LGALS3 and MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) (Figure 2B). Moreover, high glucose inhibited the recruitments of SQSTM1/p62 and ubiquitin to GFP-LGALS3 (Figure 2C). In iPSC-NDs, high glucose also inhibited the recruitment of lysophagy machinery to endogenous LGALS3 (Fig. S1C and D). In lysosome elimination assay using LLOMe [21], a lysosomotropic reagent, after 3 h of LLOMe washout, the signal intensities of LGALS3 were extensively increased in both control and high-glucose-treated cells. However, 12 h after LLOMe washout, there were significant differences in LGALS3 signal intensities between the control and high-glucose-treated groups, where LGALS3 signal intensities in the control groups were similar to those of 0 h groups (Figure 2D). We conducted RFP and GFP tandem fluorescent-tagged LGALS3 (tfGal3) plasmid transfection [21] and LysoTracker staining [22] to further investigate lysophagy and confirmed that lysophagy was working under normal conditions (Fig. S1E). Compared the data with high-glucose-treated groups, which showed that after 12 h of LLOMe washout, there were less RFP-and GFP-positive puncta in the control groups, which were canceled by pretreatment with bafilomycin A1, a vacuolar-type ATPase inhibitor (Figure 2E). Transmission electron analysis revealed that lysosomes swollen by LLOMe in control cells were sequestered by double membranes, a typical autophagosome structure. However, swollen lysosomes relatively were not engulfed by autophagosomes under high glucose conditions. (Figure 2F). The signal intensities of the LysoTracker were decreased 12 h after LLOMe washout under high glucose conditions, and pretreatment with pepstatin A+leupeptin, inhibitors of lysosomal hydrolases, also decreased LysoTracker signal intensities in the control groups (Fig. S1F). These findings suggest that high glucose treatment not only induces neuronal lysosomal membrane damage but also suppresses lysophagy activity.
Figure 2.
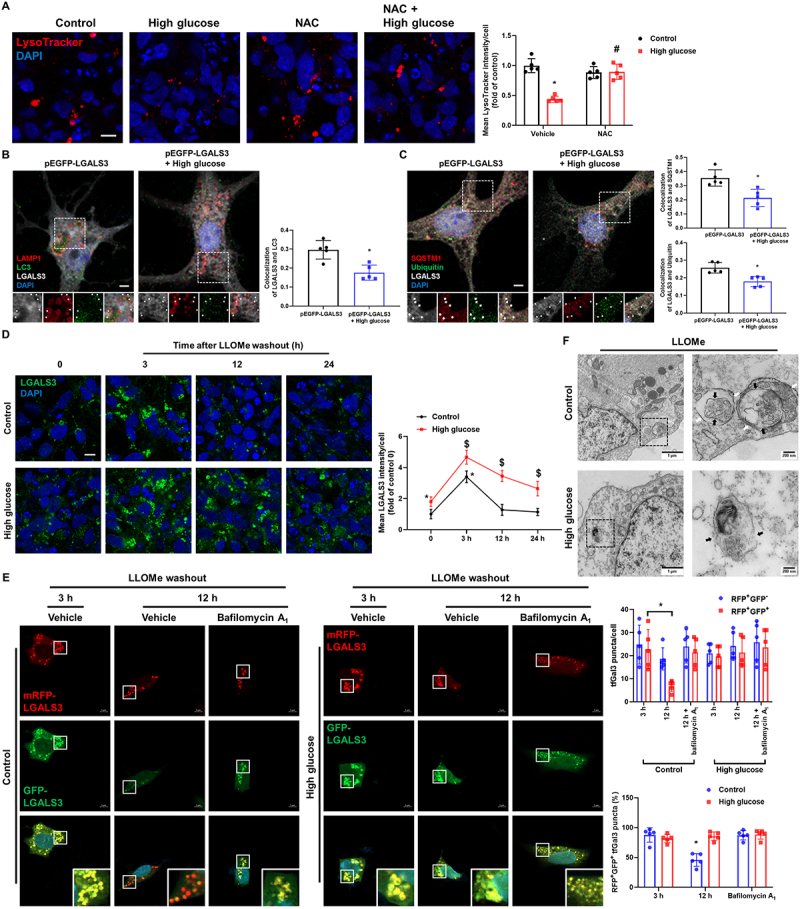
High glucose suppresses lysophagy. (A) iPSC-NDs were pretreated with NAC (2 mM) for 30 min before HG exposure for 24 h. LysoTracker was visualized by confocal microscope. (B and C) Hippocampal neurons were transfected with pEGFP-LGALS3 at DIV 4 and exposed to HG for 24 h at DIV 21. (B) Representative immunofluorescence images showing LC3 and GFP-LGALS3. (C) The cells were immunostained with GFP-LGALS3, SQSTM1, and ubiquitin. Arrow indicates GFP-LGALS3 puncta with autophagic machinery. (D) iPSC-NDs were treated with LLOMe (1 mM) for 1 h after HG exposure for 24 h and then washed away. The cells were further incubated for 3 or 12 or 24 h in the incubator. Immunofluorescence staining of LGALS3 was visualized. *P < 0.05 versus control at 0; $P <0.05 versus HG at 0. (E) SH-SY5Ys were transfected with tfGal3 prior to HG exposure for 24 h followed by LLOMe (1 mM) washout assays for 3 or 12 h. Bafilomycin A1 (10 nM) was pretreated for 30 min before incubation for 12 h. mRFP-LGALS3 and GFP-LGALS3 were visualized. *P < 0.05 versus control at 3 h. (F) SH-SY5Ys were exposed to HG for 24 h prior to treatment of LLOMe (1 mM) for 1 h. The cells were then fixed and observed by transmission electron microscopy. black arrow: lysosomal membrane; white arrow: autophagosome. n = 5. Scale bars: 8 μm. All data are representative. Quantitative data are represented as mean ± SD. *P < 0.05 versus control; #P <0.05 versus HG.
High-glucose-stimulated MTORC1 inhibits TFEB activity, resulting in TRIM16 downregulation
Upon lysosomal damage, several ubiquitination-related enzymes such as TRIM16, FBXO27, UBE2QL1, LRSAM1, and VCP detect damaged lysosomal membranes, triggering lysophagy [10]. To examine how high glucose treatment dysregulates neuronal lysophagy, we measured the mRNA expression levels of lysophagy-related genes in iPSC-NDs, which showed that the mRNA expression of TRIM16 was significantly downregulated under high glucose conditions (Figure 3A). Furthermore, the protein level of TRIM16 was reduced in iPSC-NDs, mouse hippocampal neurons, STZ-induced diabetic hippocampus, and SH-SY5Ys (Figure 3B–D and Fig. S2A). Because MTORC1 activation inhibits autophagy and lysosomal biogenesis, we hypothesized that MTORC1 downregulates TRIM16 as an upstream signaling molecule under high glucose conditions. Considering that recruitment to the lysosomal membrane is required for MTORC1 activation in response to glucose [23], we explored whether MTOR is transported to lysosomes in iPSC-NDs treated with high glucose. As shown in Figure 3E, the degree of colocalization between LAMP1 and MTOR was increased under high glucose conditions. Simultaneously, high-glucose treatment decreased PRKAA (protein kinase AMP-activated catalytic subunit alpha) phosphorylation at Thr172 and increased MTOR phosphorylation at Ser2448 (Figure 3F). Activated MTORC1 at the lysosomal membrane inhibits lysosomal biogenesis through phosphorylation-mediated inactivation of TFEB and TFE3 (transcription factor E3), master regulators of lysosomes [24]. Other transcription factors that regulate lysosomal genes, such as MITC and TFEC, were excluded because they are rarely expressed in the brain [25]. However, as high glucose can stimulate calcium signaling [18], which activates calcineurin, a calcium-dependent phosphatase, and inhibits GSK3B/GSK3β (glycogen synthase kinase 3 beta), resulting in activation of TFEB and TFE3 [26], we also examined whether high glucose treatment increases intracellular calcium levels. Considering that intracellular calcium levels were increased at 15 and 30 min after high-glucose treatment (Fig. S2B), we elucidated the effects of high glucose treatment on the nuclear translocation of TFEB and TFE3, for which we performed pretreatment with rapamycin, an MTOR inhibitor, and BAPTA-AM, a calcium chelator, before high glucose or vehicle treatment. Interestingly, high glucose treatment inhibited TFEB nuclear translocation, which was recovered by rapamycin pretreatment, and calcium chelation decreased TFEB nuclear translocation in the control group, but not in the high glucose conditions. (Figure 3G). This indicates that under high glucose conditions, MTORC1-mediated signaling is superior to calcium-mediated signaling for TFEB activation, suggesting that there is no effect of calcium influx on TFEB activation. However, the nuclear translocation of TFE3 remained unaffected by high glucose treatment and calcium chelation (Fig. S2C). As TFEB and TFE3 can regulate the expression of targeted genes bearing the coordinated lysosomal expression and regulation motif (GTCACGTGAC) [24], we speculated that there are several motifs in the TRIM16 promoter region, 500 bp upstream of transcription starting site, to which TFEB and TFE3 can bind (Fig. S2D). High glucose treatment suppressed the binding of TFEB to the TRIM16 promoter (Figure 3H,I), but there were no significant changes in the binding of TFE3 to the TRIM16 promoter (Fig. S2E). We performed pretreatments with rapamycin and curcumin C1 to determine whether there are other MTOR-mediated mechanisms for the regulation of TRIM16, except for TFEB inactivation. Results showed that rapamycin and curcumin C1 recovered the mRNA expression and protein levels of TRIM16 reduced by high glucose treatment (Figure 3J,K). These results indicate that MTOR-mediated TFEB inactivation directly downregulates TRIM16 expression under high glucose conditions.
Figure 3.
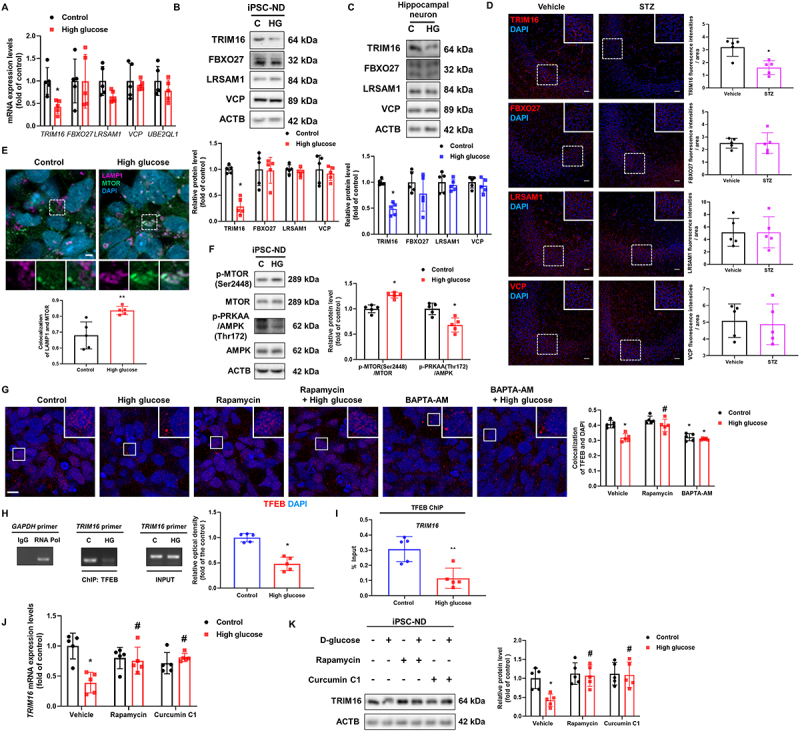
MTORC1-mediated TFEB inhibition downregulates TRIM16 under high glucose conditions. (A) iPSC-NDs were exposed to HG for 24 h. The mRNA expression levels of TRIM16, FBXO27, LRSAM1, VCP, and UBE2QL1 were investigated. (B and C) iPSC-NDs and hippocampal neurons at DIV 21 were exposed to HG for 24 h. The protein levels of TRIM16, FBXO27, LRSAM1, and VCP were detected by western blotting. (D) Representative immunohistochemistry images of the hippocampus of vehicle-or STZ-injected mice showing TRIM16, FBXO27, LRSAM1, and VCP. Scale bars: 20 μm. (E and F) iPSC-NDs were exposed to HG for 12 h. (E) LAMP1, MTOR, and DAPI were subjected to immunofluorescence analysis. (F) The protein levels of p-MTOR (Ser2448), MTOR, p-PRKAA (Thr172), AMPK, and ACTB were detected by western blotting. (G) iPSC-NDs were pretreated with rapamycin (200 nM) or BAPTA-AM (5 μM) for 30 min before HG exposure for 12 h. TFEB was immunostained. (H and I) SH-SY5Ys were exposed to HG for 12 h. DNA was immunoprecipitated with IgG, POLR/RNA polymerase, and TFEB antibody. The samples of immunoprecipitation and input were amplified with primers of GAPDH and TRIM16. Data were analyzed by conventional PCR and qPCR, respectively. (J and K) iPSC-NDs were pretreated with rapamycin (200 nM) or curcumin C1 (1 μM) for 30 min before HG treatment for 24 h. The TRIM16 mRNA expression and protein levels of TRIM16 were analyzed by qPCR and western blotting, respectively. n = 5. Scale bars: 8 μm. All data are representative. Quantitative data are represented as mean ± SD. *P < 0.05; **P < 0.01 versus control; #P <0.05 versus HG.
Upregulation of TRIM16 recovers lysophagy, which induces Aβ and p-MAPT degradation
Through pcDNA3.1-Trim16-GFP and pcDNA3.1-TRIM16-GFP transfection in hippocampal neurons and SH-SY5Ys, we observed that the inhibited neuronal lysophagy process was normalized by TRIM16 overexpression. The decreased colocalization between LC3 and LGALS3 induced by high glucose treatment were reversed by TRIM16 overexpression (Figure 4A and Fig. S3A). Moreover, the degree of colocalization between SQSTM1 or ubiquitin and LGALS3 in neurons treated with high glucose and transfected with pcDNA3.1-GFP was lower than that in cells treated with high glucose and transfected with pcDNA3.1-Trim16-GFP or pcDNA3.1-TRIM16-GFP (Figure 4B and Fig. S3B). After 12 h of LLOMe washout, the high-glucose-inhibited reduction of LGALS3 signal intensities was recovered by TRIM16 overexpression (Figure 4C). However, there is no significant change in the protein level of LGALS3 under high glucose conditions in iPSC-NDs, hippocampal neurons, and SH-SY5Ys (Fig. S3C-E). Subsequently, the LysoTracker signal intensities and the mean fluorescence intensities of LysoTracker decreased with high glucose treatment were reversed by TRIM16 overexpression (Figure 4D,E and Fig. S3F). These findings suggest that high-glucose-mediated TRIM16 downregulation impedes neuronal lysophagy.
Figure 4.
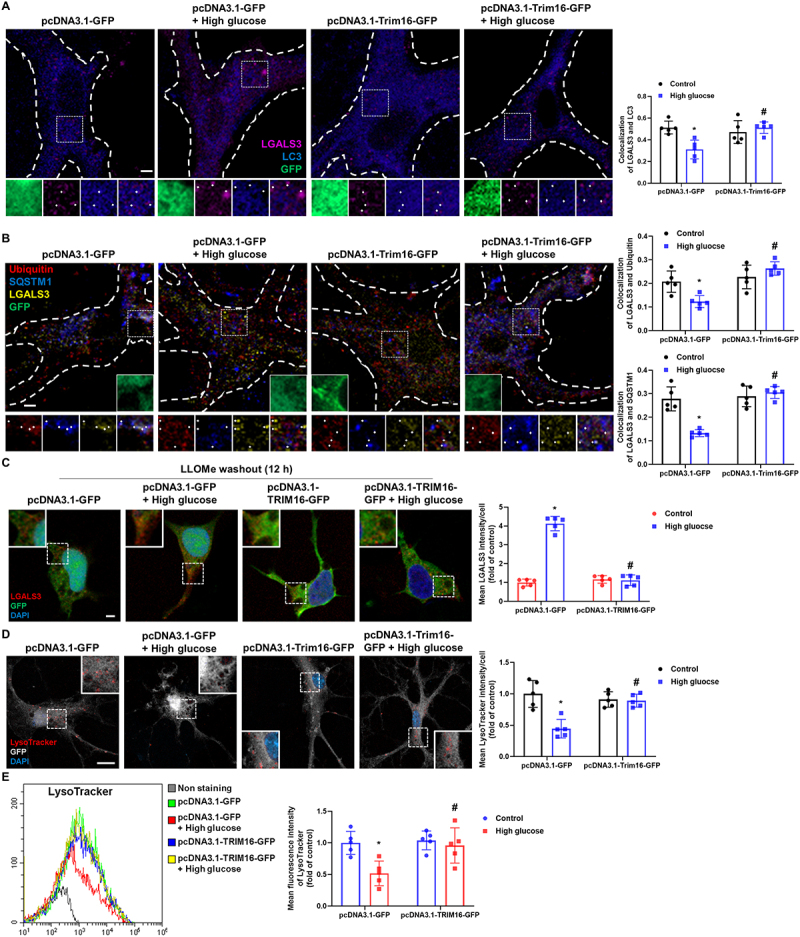
Upregulation of TRIM16 recovers high-glucose-inhibited lysophagy. (A, B, and D) Hippocampal neurons were transfected with pcDNA3.1-GFP or pcDNA3.1-Trim16-GFP at DIV 4 and exposed to HG for 24 h at DIV 21. (A) Representative immunofluorescence images showing LC3 and LGALS3. (B) The cells were immunostained with ubiquitin, SQSTM1, and LGALS3. (C and E) SH-SY5Ys were transfected with pcDNA3.1-GFP or pcDNA3.1-TRIM16-GFP before HG exposure for 24 h. (C) LLOMe (1 mM) washout assay was performed for 12 h followed by immunostaining with LGALS3. (D) LysoTracker was visualized and DAPI was used to stain nuclei. (E) The mean fluorescence intensities of LysoTracker were analyzed by flow cytometric analysis. n = 5. Scale bars: 8 μm. All data are representative. Quantitative data are represented as mean ± SD. *P < 0.05 versus control; #P <0.05 versus HG.
Deficiencies in lysosomal proteolysis and autophagy are potential mechanisms of Aβ and p-MAPT accumulation caused by lysosomal dysfunction [16,17,27], and defective lysophagy inhibits lysosomal biogenesis, exacerbating lysosomal dysfunction [9,21]. Under high glucose conditions, we showed that the degree of colocalization between RAB7, a marker of late endosomes, and LAMP1 [12] was significantly increased, indicating an increase in secondary lysosomes and a decrease in lysosomal biogenesis, and the activities of CTSD and CTSB and the formation of autolysosomes were inhibited, all of which were reversed by TRIM16 overexpression (Fig. S4A-F). These data mean that the mechanistic link between lysophagy and Aβ and p-MAPT accumulation may be lysosomal biogenesis, proteolysis, and autophagy. Thus, we investigated whether TRIM16-mediated lysophagy inhibits high-glucose-accumulated Aβ and p-MAPT in neuronal cells. The upregulated expressions of C99, p-MAPT Thr181, and p-MAPT Thr212 were inhibited by TRIM16 overexpression (Figure 5A). Hippocampal neurons exposed to high glucose with control plasmid transfection showed less degree of colocalization between LysoTracker and cellular Aβ than neurons exposed to high glucose with Trim16 plasmid transfection (Figure 5B). Cellular and secreted Aβ increased by high glucose were reduced by TRIM16 overexpression (Figure 5C,D). The high-glucose-induced neuronal cell death was also protected by pcDNA3.1-TRIM16-GFP transfection (Figure 5E). And then, we investigated whether lysophagy-related proteins other than TRIM16 did not actually affect lyosphagy to confirm their regulatory potential for Aβ and p-MAPT degradation, since they were located to some extent with the lysosome in our experimental conditions. Among these proteins, the degree of colocalization between FBXO27 or LRSAM1 or VCP and LAMP1 was not significantly altered under high glucose conditions, unlike TRIM16 (Fig. S5A – D). After 12 h of LLOMe washout, FBXO27 silencing led to increased and reduced signal intensities of LGALS3 and LysoTracker, respectively, whereas LRSAM1 and VCP silencing exerted no effects (Fig. S5E and F). Considering that the endosomal sorting complex required for transport (ESCRT) machinery promotes lysosomal repair [28], we investigated whether ESCRT complexes were associated with high-glucose-mediated lysosomal quality control. We found that the degree of colocalization of CHMP4B (charged multivesicular body protein 4B) or CHMP4 interacting protein PDCD6IP/ALIX and LAMP1 was increased in high-glucose-treated iPSC-NDs (Fig. S5G and H). These observations suggest that although the FBXO27 and ESCRT machinery exerts effects on neuronal lysosomal quality control, TRIM16 downregulation primarily impede lysophagy, which inhibits Aβ and p-MAPT degradation and induces neuronal cell death under high glucose conditions.
Figure 5.
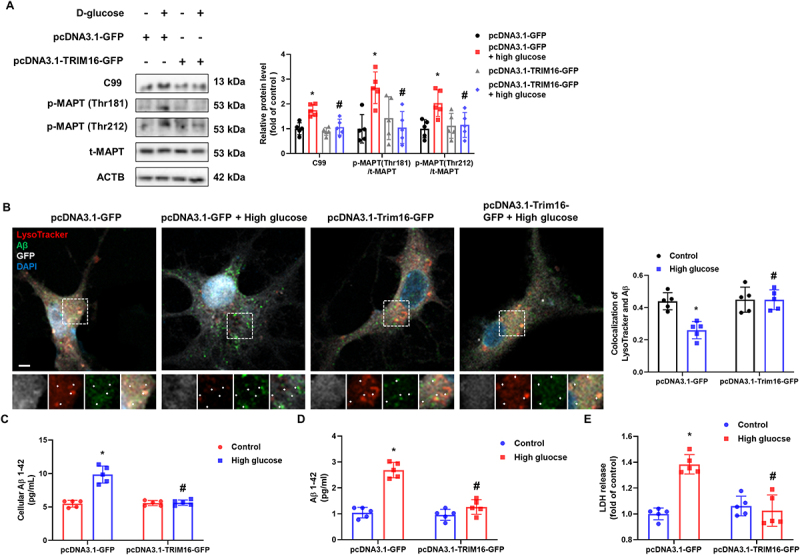
Upregulation of TRIM16 facilitates Aβ and p-MAPT degradation. (A) SH-SY5Ys were transfected with pcDNA3.1-GFP or pcDNA3.1-TRIM16-GFP prior to HG exposure. The protein levels of C99, p-MAPT (Thr181), p-MAPT (Thr212), t-MAPT, and ACTB were detected by western blotting. (B) Hippocampal neurons were transfected with pcDNA3.1-GFP or pcDNA3.1-Trim16-GFP at DIV 4 and exposed to HG for 48 h at DIV 21. Representative immunofluorescence images showing LysoTracker and Aβ. (C and D) SH-SY5Ys were transfected with pcDNA3.1-GFP or pcDNA3.1-TRIM16-GFP prior to HG exposure for 48 h. Cellular or secreted Aβ 1–42 were measured, respectively. (E) LDH from cell supernatant were measured after exposure to HG for 72 h. n = 5. All data are representative. Quantitative data are represented as mean ± SD. *P < 0.05 versus control; #P <0.05 versus HG.
TFEB enhancement recovers TRIM16-mediated lysophagy, ameliorating cognitive impairment in STZ-induced diabetic mice
To explore whether TRIM16-mediated lysophagy is involved in the pathogenesis of diabetes-associated AD-like phenotype, we treated STZ-induced diabetic mice with the TFEB activator curcumin C1 [19] orally for 8 weeks (Fig. S6A). We measured the body weight and blood glucose levels of mice at 8 and 18 weeks of age (Fig. S6B and C). Results showed that TFEB nuclear translocation was inhibited in the hippocampus of STZ-induced diabetic mice, which was reversed by curcumin C1 (Fig. S6D). Curcumin C1 increased the diabetes-reduced TRIM16 protein levels in the hippocampus (Figure 6A). Since LGALS3 colocalized with LAMP1 to some extent (Fig. S6E), we used LGALS3 staining to indicate damaged lysosomes in the hippocampus. The degree of colocalization between LGALS3 with ubiquitin and LC3 in mice treated with STZ + curcumin C1 was higher than that in STZ-treated mice (Figure 6B and Fig. S6F). Curcumin C1 treatment also alleviated the cytosolic release of CTSB and CTSD in the hippocampus of STZ-treated mice (Figure 6C,D). Subsequently, curcumin C1 inhibited the upregulation of hippocampal C99, Aβ, and p-MAPT expression in STZ-treated mice (Figure 6E,F). Results of the open field test showed that diabetic mice spent less time in the center compared with other mice groups (Figure 6G). Furthermore, results of the novel object recognition (NOR) test showed that STZ-treated mice had decreased discrimination index and novelty preference, which were alleviated by curcumin C1 treatment (Figure 6H). The Y-maze test results revealed that curcumin C1 recovered the diabetes-reduced spontaneous alternation (Figure 6I). These results suggest that Aβ and p-MAPT accumulation, anxiety, and cognitive impairment in diabetic mice were caused by the impairment of TRIM16-mediated lysophagy.
Figure 6.
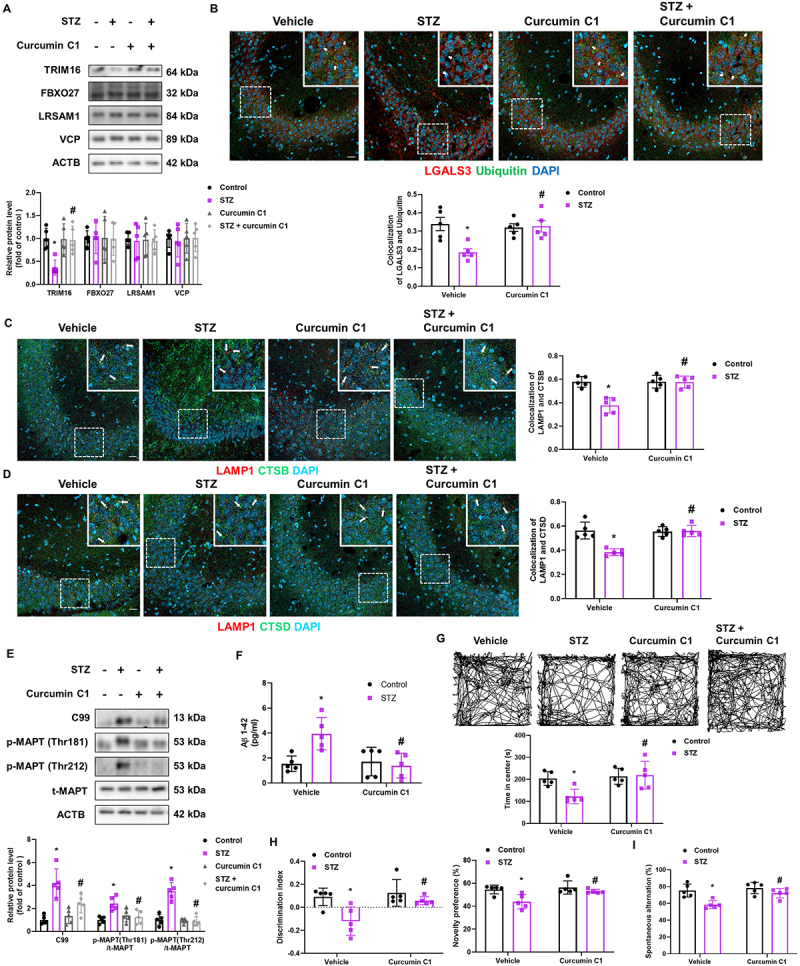
Pharmacological enhancement of TFEB activation inhibited Aβ and p-MAPT accumulation and cognitive impairment in diabetic mice. (A-G) Mice injected with vehicle or STZ were orally administrated vehicle or curcumin C1 (10 mg/kg) once daily for 8 weeks after diabetes induction (A) Protein levels of the hippocampal TRIM16, FBXO27, LRSAM1, VCP, and ACTB were determined by western blotting. (B) Representative immunohistochemistry images showing LGALS3 and ubiquitin. (C) The hippocampal slides were immunostained with LAMP1 and CTSB. (D) Immunofluorescence staining of LAMP1 and CTSD in the hippocampus. (E) Hippocampal C99, p-MAPT (Thr181), p-MAPT (Thr212), t-MAPT, and ACTB were subjected to western blotting. (F) Hippocampal Aβ 1–42 were measured. (G-I) The mice were subjected to open field test, NOR test, and Y-maze test, respectively. n = 5. Scale bars: 20 μm. All data are representative. Quantitative data are represented as mean ± SD. *P < 0.05 versus vehicle-injected mice; #P <0.05 versus STZ-injected mice.
Discussion
This study demonstrated that lysophagy dysfunction is a potential pathophysiological mechanism of DM-associated AD. In fact, in vitro findings showed that high glucose impaired TRIM16-mediated lysophagy, which impaired the degradation of Aβ and p-MAPT. In vivo findings also showed that the accumulation of Aβ and p-MAPT, resulting in cognitive impairment, was alleviated by TRIM16 recovery in diabetic mice. As mature neurons depend exceptionally on lysosomal systems to remove waste products such as protein aggregates and damaged organelles, lysosomal dysfunction is closely related to the pathogenesis of neurodegenerative diseases [29]. Patients with AD who have PSEN1 mutations show lysosomal dysfunction, and accumulation of Aβ/C99 in the enlarged lysosomes causes LMP, cathepsin release, and cell death [5,30]. Moreover, glucolipotoxicity in human primary β-cells induces lysosomal dysfunction, resulting in islet failure [31]. We observed that high glucose treatment reduced LysoTracker signal intensities and cytosolic release of CTSD and CTSB in both iPSC-NDs and mouse hippocampal neurons. Considered that CTSD in the neocortical region of the human AD brain is colocalized with p-MAPT in neurons [32] and lentivirus-mediated CTSB overexpression reduces amyloid deposits in hAPP mice [16], our data indicated that high glucose treatment impaired Aβ and p-MAPT degradation through lysosomal dysfunction. Regarding the mechanism by which high-glucose-induced lysosomal dysfunction, a previous study showed that high-glucose-evoked ROS accumulation triggers mechanistic damage to lysosomes, resulting in LMP in neuroblastoma cells [7]. Similarly, we observed that high glucose treatment resulted in ROS-mediated neuronal LMP. It has been reported that upon LMP, maintenance of lysosomal quality control through genetic and pharmacological restoration of lysosomes can recover damaged target cells and delay disease progression in both AD and DM [33]. Considering that Atg5 deficiency in disease involving lysosomal damage inhibits lysophagy, which suppresses lysosomal restoration and exacerbates disease progression [21], lysophagy is essential for lysosomal restoration upon lysosomal damage. However, to the best of our knowledge, no study has investigated whether lysophagy occurs in neurons exposed to high glucose. Thus, we demonstrated for the first time that high glucose impaired neuronal lysophagy. These findings suggest that high glucose causes lysosomal dysfunction through LMP induction and lysophagy suppression, which inhibit the degradation of Aβ and p-MAPT.
Lysosomes adapt their status in response to environmental cues to maintain cellular homeostasis through the regulation of lysosomal gene expression [34]. Under high-glucose conditions, 5’ adenosine monophosphate-activated protein kinase (AMPK) is dephosphorylated and MTORC1 is recruited to the lysosomal membrane by anchoring protein and activated, which inhibits catabolic processes [23]. A previous study showed that the AMPK-MTORC1 signaling pathway was stimulated in high-glucose-treated neuronal cells and in the hippocampus of diabetic mice [7]. Our data also demonstrated that MTORC1 was recruited to the lysosomes and activated in iPSC-NDs exposed to high glucose conditions. Although high-glucose-induced ROS and lysosomal injury can inhibit AMPK-MTORC1 signaling [35], lysosomal adaptation preceded and prevailed for modulation of MTORC1 activity in our experimental conditions. TFEB and TFE3 are members of the MiTF/TFE family that are considered as master regulators of lysosomal biogenesis and are implicated in the pathogenesis of both AD and DM [12,36,37]. Regarding the regulation of TFEB/TFE3 activity, the MTORC1 and calcium mediated protein kinase C and calcineurin signaling pathways regulate their activity independently and inversely [25,38]. Whether high glucose activates or inactivates TFEB/TFE3 remains controversial because high glucose can stimulate both signaling pathways under various conditions [18]. We observed that high glucose treatment inhibited the nuclear translocation of TFEB in an MTORC1-dependent manner, and calcium chelation also suppressed its translocation. This might be because high-glucose-mediated calcium signaling activated TFEB, but lysosomal adaptation-mediated MTORC1 signaling was predominant. Meanwhile, our data demonstrated that the nuclear translocation of TFE3 was not altered in response to high glucose treatment, MTOR inhibition, and calcium chelation. In TFEB-depleted cells, TFE3 increases the number of lysosomes [39]. Furthermore, considering that nuclear TFE3 was present in all our experimental groups, TFE3 might be constitutively activated. However, although the altered activities of TFEB may regulate lysosomal biogenesis, whether it modulates lysophagy has not been elucidated, especially under high glucose conditions. We confirmed that TFEB was directly bound to the TRIM16 promoter, and TFEB activation recovered the high-glucose-reduced mRNA expression and protein levels of TRIM16. These data indicate that lysophagy promotion is a novel mechanistic target of TFEB-mediated therapeutic strategy for neurogenerative disorders.
Lysophagy-related factors such as TRIM16, FBXO27, UBE2QL1, LRSAM1, and VCP detect galectins, which attach to the glycans exposed upon vesicular damage in the intraluminal regions of lysosomes and recruit ubiquitin and autophagic receptors to degrade damaged lysosomes and maintain lysosomal function. Nevertheless, the role of lysophagy factors in the regulation of lysosome quality in the pathogenesis of neurodegenerative diseases has been investigated only recently [11], and the alterations and regulatory mechanisms of neuronal lysophagy in diabetic conditions have never been clarified. Among lysophagy-related factors, we observed that only TRIM16 expression was decreased in iPSC-NDs and mouse hippocampal neurons exposed to high glucose and in the hippocampus of diabetic mice, followed by decreased recruitment to lysosomes. Other lysophagy-related factors that do not undergo significant changes in expression levels and recruitment to lysosomes may also affect the maintenance of basal lysosomal function [15,40,41]. However, our data revealed that only FBXO27 silencing led to impaired clearance of LGALS3 upon lysosomal damage and reduction of LysoTracker signal intensity, whereas knockdown of other lysophagy-related factors had no effects in neuronal cells. The variability in the knockdown effect may be attributed to differences in the degree of response of each factor to basal lysophagy. Furthermore, considering that LRSAM1 protects lymphoblasts against intracellular bacterial infection through lysosomal ubiquitination [42], cell-type-specific differences may also contribute to it. In addition to lysophagy, the ESCRT machinery is recruited into the lysosome for repair upon acute lysosomal injury [28]; however, whether this affects the maintenance of lysosomal function in disease situations remains to be investigated. We first demonstrated that PDCD6IP and CHMP4B were recruited to damaged lysosomes under high glucose conditions, but it was difficult to overcome LMP and lysophagy dysfunction. A recent study showed that TRIM16-mediated sensing of damaged lysosomes induces the unconventional seeding of intracellular wastes in human midbrain dopaminergic neurons [43]; however, gain-of-function studies focusing on lysophagy may provide an insight into the removal of wastes. Our data revealed that TRIM16 overexpression increased LysoTracker signal intensities by promoting LC3, SQSTM1, and ubiquitin recruitment to damaged lysosomes. We also observed that activities of CTSD and CTSB, and Aβ and p-MAPT accumulation were reversed by TRIM16-mediated lysophagy activation. In addition to lysophagy, further studies are required because TRIM16-mediated protein ubiquitination is a crucial disease mechanism [44,45]. However, considering that impairment of lysosomal maturation is a major mechanism of AD pathogenesis, maintenance of lysophagy through TRIM16 regulation could provide synergistic effects in disease prevention.
A gain-of-function study demonstrated that TFEB delivery into the cerebral hemispheres of 5×FAD and rTg4510 AD transgenic mice ameliorated the AD phenotype [37]. Moreover, pharmacological enhancement of TFEB activity through curcumin C1 treatment for 3 months in AD mice restored lysosomal activity and reduced APP, C99, Aβ, and p-MAPT levels followed by improvement of cognitive function [46]. In addition to the inhibition of Aβ and p-MAPT levels and improvement of cognitive function, our results demonstrated that curcumin C1 restored hippocampal TRIM16 protein levels, which elevated lysophagy and normalized lysosomal function. These findings indicate that TRIM16-mediated lysophagy suppresses diabetes-associated AD-like phenotype. To summarize, our study has demonstrated that impaired lysophagy due to MTORC1-TFEB-mediated TRIM16 downregulation inhibited Aβ and p-MAPT degradation in neuronal cells exposed to high glucose conditions, resulting in cognitive impairment (Figure 7). Our findings also suggest that lysophagy promotion through TRIM16 targeting is a promising strategy for the modulation of DM-associated AD.
Figure 7.
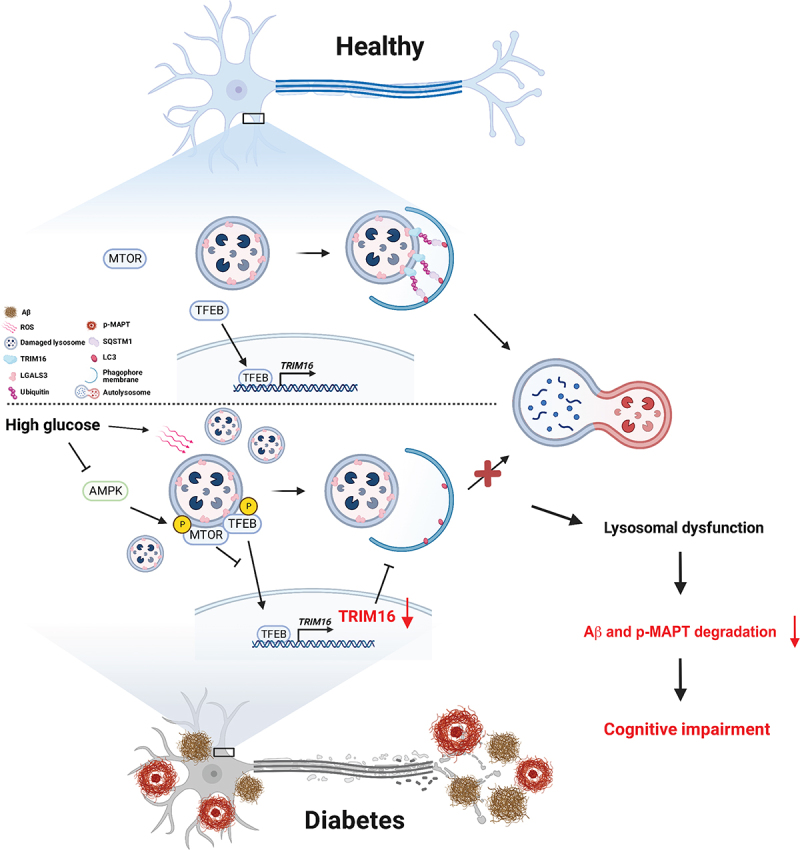
The schematic model for effects and molecular mechanism of action of high glucose on neuronal lysopahgy and subsequent accumulation of Aβ and p-MAPT. High glucose induces dysfunction of neuronal lysosomes through ROS-mediated LMP and lysophagy impairment. The expression of TRIM16 is downregulated by MTORC1-inhibited TFEB activity under high glucose conditions, but its overexpression recovered lysophagy, which in turn degrades Aβ and p-MAPT and ameliorates cognitive impairment. Conclusively, lysophagy promotion through TRIM16 targeting is a promising strategy for the modulation of DM-associated AD.
Materials and Methods
Materials
The iPSCs and SH-SY5Ys were acquired from the National Stem Cell Bank of Korea and Korean Cell Line Bank (KSCBi005-A and KCLB-22266), respectively. Antibiotics (15240096) and FBS (SV30207.02) were purchased from Gibco and Hyclone, respectively. The antibodies of p-PRKAA/AMPK (Thr172) (sc-33524), ACTB/β-actin (sc-47778), LAMP1 (sc-20011), CTSB (sc-365558), LGALS3/galectin 3 (sc-23938), ubiquitin (sc-8017), VCP (sc-57492), TRIM16 (sc-398851), and CTSD (sc-13148) were obtained from Santa Cruz Biotechnology. MAPT/Tau 5 (AHB-0042) antibody was acquired from Thermo Fisher Scientific. The antibodies of RAB7 (NB120–13253), SQSTM1/p62 (NBP1–48320), Aβ (NBP2–13075), LAMP1 (AF4800), and LC3 (NB100–2220) were purchased from Novus Biologicals. p-MAPT/Tau (Thr212) antibody was obtained from Signalway Antibody (11257). The antibodies of LAMP1 (9091), p-MTOR (Ser 2448) (2971S), MTOR (2983S), AMPK (2532S), and p-MAPT/Tau (Thr181) (12885) were acquired from Cell Signaling Technology. The antibodies of APP (ab32136) and MAP2 (ab32454) were obtained from Abcam. FBXO27 (14570–1-AP), LRSAM1 (24666–1-AP), TFEB (13372–1-AP), PDCD6IP/ALIX (12422–1-AP), CHMP4B (13683–1-AP) and TFE3 (14480–1-AP) antibodies were purchased from Proteintech. Secondary antibodies for anti-rat Alexa Fluor488 (A11006), anti-rat Alexa647 (A78947), anti-rabbit Alexa Fluor 647 (A21244), anti-mouse Alexa Fluor 488 (A32723), anti-Sheep Alexa Fluor488 (A11015), anti-rabbit Alexa Fluor 555 (A32732), anti-mouse Alex Fluor 555 (A32727), and LysoTracker Red DND-99 (L7528) were obtained from Thermo Fisher Scientific. H-Leu-Leu-OMe · HBr (LLOMe; G2550) was purchased from Bachem. Leupeptin (L2884), N-acetyl cysteine (NAC; A7250), bafilomycin A1 (B1793), D-glucose (G8769), rapamycin (37094), normal goat serum (NGS; 566380), pepstatin A (P5318), DAPI (D9542), and STZ (S0130) were acquired from Sigma Aldrich. curcumin C1 (M9451) was obtained from Abmole Bioscience. ptf-LGALS3/Galectin3 was a gift from Tamotsu Yoshimori [21]. pcDNA3.1(+)-TRIM16-eGFP, pcDNA3.1(+)-Trim16-eGFP and pcDNA3.1(+)-eGFP were purchased from Koma Biotech (OHu05385D and OMu09283C). mRNA primers for TRIM16, FBXO27, LRSAM1, VCP, UBE2QL1, and ChIP primers for the TRIM16 promoter region were obtained from Cosmo Genetech. siRNAs for FBXO27, LRSAM1, and VCP were purchased from Bioneer. Non-targeting siRNA (NT siRNA) was provided from Dharmacon.
Cell culture
iPSCs were grown on recombinant human vitronectin (Thermo Fisher Scientific, A14700)-covered plates with neural induction media (Thermo Fisher Scientific, A1647801) to induce neuronal stem cells (NSCs). The cells were re-plated on dishes coated with laminin (Thermo Fisher Scientific, 23017) and poly-L-ornithine (Sigma Aldrich, P3655) and grown in neural differentiation media (Neurobasal medium [Thermo Fisher Scientific, 21103] supplemented with 2% B27 [Thermo Fisher Scientific, 17504] and 1% GlutaMax-1 [Thermo Fisher Scientific, 35050]) for more than 10 days. 5 mM dibutyryl-cAMP (Sigma Aldrich, D0627) was used daily to facilitate neural differentiation between 7 and 10 days after initial differentiation. Mouse hippocampal neurons from E18 embryos were cultured as described following a modified protocol [47] and performed in compliance with the approval of the Institutional Animal Care and Use Committee of Seoul National University (SNU-201013-7-2). In brief, hippocampal neurons were cultured at low density on poly-D-lysine-coated coverslips with cortical rings of neurons and glias or high density on six-well plates coated with poly-D-lysine in neurobasal medium (Thermo Fisher Scientific, 21103) supplemented with 2% B27 supplement (Thermo Fisher Scientific, 17504) and 0.25% GlutaMax-1 (Thermo Fisher Scientific, 35050). SH-SY5Ys were cultured in minimum essential medium (MEM; Hyclone, SH30024) supplemented with 10% FBS and 1% antibiotic-antimycotics (Gibco, 15240062) at 37°C with 5% CO2. After cells reached 70% confluence, the media was replaced for 24 h with MEM containing 1% antibiotic-antimycotics prior to drug treatment. Given that glucose concentration above 20 mM were used to investigate hyperglycemia-induced cellular pathogenesis [6,14,48], and high glucose induced intracellular Aβ accumulation and increased cell death at concentration of 25 mM (Fig. S7A and B), the cells were exposed to 25 mM D-glucose to mimic diabetic conditions.
Plasmid DNA transfection
The tfGal3 plasmid, pcDNA3.1-Trim16-GFP, or pcDNA3.1-TRIM16-GFP were transfected into hippocampal neuron and SH-SY5Ys using lipofectamine 2000 (Thermo Fisher Scientific, 11668019). Neurons were transfected at DIV 4 for 2 h and replaced with fresh media [47]. SH-SY5Ys were transfected at 60% confluence for 12 h and replaced with fresh media. After transfection, cells were exposed to high glucose.
siRNA transfection
SH-SY5Ys were transfected with 25 nM of FBXO27, LRSAM1, or VCP siRNAs by using TurboFect™ (Thermo Fisher Scientific, R0531) for 12 h. The cells were transfected before being exposed to high glucose.
Immunofluorescence assay
The cells were fixed with 4% paraformaldehyde (PFA) for 10 min and permeabilized with 0.2% Tween-20 (Sigma Aldrich, P1379) or 0.1% Triton X-100 (Sigma Aldrich, T8787) for 10 min. They were incubated with primary antibodies (1:300 dilution) overnight at 4°C following blocking with 5% NGS for 1 h. After three washes with phosphate-buffered saline (PBS; Hyclone, SH30256), cells were incubated at room temperature (RT) for 2 h with fluorescent secondary antibodies (1:200 dilution). A confocal microscope system (Carl Zeiss, LSM 710) and a super-resolution radial fluctuations imaging system (SRRF) (Andor Technology) [49] were used to visualized immune-stained cells. Quantification of fluorescence intensities and measurements of LysoTracker- and LGALS3-positive signals were carried out with Fiji software [50]. JACop plugin with the Pearson correlation coefficient or the Mander’s overlap coefficient was used to measure and analyze colocalization [51,52].
Transmission electron microscopy
Cell pellets were resuspended in Karnovsky’s fixative (2% glutardialdehyde and 2% paraformaldehyde in 0.05 M sodium cacodylate buffer) overnight at 4°C. The pellet was washed in 0.05 M cacodylate buffer and subsequently post-fixed for 2 h with 1% osmium tetroxide in 0.1 M sodium cacodylate buffer. After washing in distilled water, en bloc staining was performed with 0.5% uranyl acetate overnight at 4°C. The pellet was washed with distilled water and then dehydrated with increasing ethanol concentrations from 30% to 100%. Transition was performed using 100% ethanol and Spurr’s resin (Electron Microscopy Sciences, 14300; 1:1 and 1:2), and 100% Spurr’s resin followed by embedding in 100% Spurr’s resin at 70°C for 24 h. Ultrathin sections were acquired (60 nm) using an ultramicrotome (Leica, EM UC7) and transferred to carbon grids followed by staining with uranyl acetate and lead citrate. Sections were observed using a transmission electron microscope (JEOL, JEM1010).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using an EZ-ChIP Kit (Sigma Aldrich, 17–371) according to the manufacturer’s instructions. Briefly, chromatin-protein complexes were incubated with the POLR/RNA polymerase (positive control) antibody, the normal IgG (negative control; Sigma Aldrich, 12–371), and primary antibodies for overnight at 4°C. Immunoprecipitated-DNA was eluted and then amplified using PCR. The sequences of the TRIM16 primer are as follows: forward primer, 5′- GAGACACGTGGTCTTCAGCA-3′ and reverse primer, 5′- GCTCGGCCACTCATTACTGT −3′.
cDNA synthesis and quantitative PCR (qPCR) analysis
RNA was extracted in compliance with the manufacturer’s instructions (TaKaRa, 9767). Then, cDNA was generated at 45°C for 1 h and then at 95°C for 5 min using a PCR premix solution (iNtRON, 25081). Using a Rotor-Gene 6000 real-time system (Corbett Research), TB™ Green Premix Ex Taq™ (TaKaRa, RR420A), and mRNA primers (Table S1), cDNAs were amplified. The qPCR was performed as follows: 10 min at 95°C, followed by 55 cycles of 10 s at 95°C, 15 s at 58°C, and 15 s at 72°C. ΔCt analysis was used to quantify mRNA expression levels and the data were normalized to the ACTB gene.
Western blot analysis
Sample dissociation was performed using RIPA lysis buffer (RIPA; ATTO, WSE-7420) and a cocktail of phosphatase and protease inhibitors (100X; Thermo Fisher Scientific, 78440). Protein quantification was performed using bicichoninic acid (BCA) assay (Thermo Fisher Scientific, 23227) with technical duplicates. Samples (5–10 μg) were loaded onto SDS-polyacrylamide gel for electrophoresis, and transferred to a polyvinylidene fluoride membrane. After the membrane was blocked with 5% skim milk (Gibco, 232100), it was incubated with the primary antibodies overnight at 4°C. The membrane was washed three times with Tris-buffered saline (TBST; 10 mM Tris-HCl, pH 7.6, 0.2% Tween-20, 150 mM NaCl) and incubated with a horseradish peroxidase (HRP)-coupled secondary antibody (Thermo Fisher Scientific, 31460) for 2 h at RT. The ChemiDoc imaging system and enhanced chemiluminescence reagent (Bio-Rad Laboratories, 1705061) were used to detect bands. Band quantification was performed using the Image J program. Protein levels were normalized against that of ACTB. The bands are in the linear range of detection.
Lysosomes and Lysosomal pH measurements
Lysosomal staining was performed using LAMP1 primary antibody. LAMP1-positive lysosomes were visualized with confocal microscopy equipment (Carl Zeiss, LSM 710). Acidic lysosomes and lysosomal pH were measured with LysoTracker Deep Red (Thermo Fisher Scientific, L12492) [30] and LysoSensor Green DND-189 (Thermo Fisher Scientific, L7535), respectively. Cells were incubated with 100 nM LysoTracker or 500 nM LysoSensor at 37°C for 30 min. LysoTracker and LysoSensor signals were analyzed by flow cytometry or confocal microscopy equipment.
Lysosome rupture and elimination assay
Lysosomal rupture and elimination assay were performed using LLOMe treatment and tfGal3 plasmid transfection [20]. Briefly, after drug treatment, iPSC-NDs were treated with LLOMe (1 mM) for 1 h to disrupt the lysosomal membrane, rinsed the reagent, and incubated the cells for additional hours in the absence of LLOMe. The lysosomal clearance was evaluated by immunostaining with LGALS3 antibody. The SH-SY5Ys were transfected with tfGal3 plasmids followed by drug and LLOMe treatment and then observed by confocal microscope. When LMP takes place, tfGal3 enters the ruptured lysosomal lumen and quickly attaches to the inner membrane, displaying both GFP and RFP fluorescence and producing yellow signals. Because RFP is relatively stable in acidic environment, GFP fluorescence is quenched in the lumen of intact lysosome during lysophagy.
Measurements of intracellular calcium
The pH-sensitive fluorescent probe Fluo-4 AM (Thermo Fisher Scientific, F14201) exhibiting fluorescence upon binding Ca2+ were used for measuring intracellular calcium. Measurements were performed according to the manufacturer’s instructions. After incubation with 4 mM Fluo-4 at 37°C for 1 h, cells were stabilized and exposed to drugs. Fluo-4-stained cells were measured via flow cytometry.
Lactate dehydrogenase (LDH) release assay
The optimization of cell concentration was determined according to the protocol provided in the LDH release assay kit (DoGenBio, DG-LDH500). After drug treatment, cells were centrifugated at 600 × g and then supernatants were incubated with the LDH assay mixture at RT for 30 min. The level of released LDH was determined by measuring the optical density at 450 nm with an Epoch 2 spectrophotometer (BioTek).
ANXA5/annexin V-PI staining
ANXA5-PI staining was performed using FITC Annexin V apoptosis detection kit according to the manufacturer’s instruction (BD Biosciences, 556547). After drug treatment, equal amounts of cells were resuspended in binding buffer with ANXA5-FITC and PI at RT for 30 min. Stained cells were detected with flow cytometry and analyzed with CytExpert 2.3 software. ANXA5-positive cells represent early apoptosis and PI-positive cells represent necrosis. Cells positive for both represent late apoptosis.
CTSB and CTSD activities assay
CTSB and CTSD activities were measured using CTSB (Biovision, K140) and CTSD (Biovision, K143) activity fluorometric assay kit. Assays were performed according to manufacturer’s protocol. Cells were lysed on ice for 10 min and centrifuged at 16,000 × g for 5 min. After protein quantification with BCA assay, lysates were incubated with substrate and reaction buffer at 37°C for 2 h. Fluorescent intensities of CTSB and CTSD were measured by fluorometer equipped with a 400-nm excitation/505-nm emission and 328-nm excitation/460-nm emission filter, respectively.
Measurements of Aβ level
Aβ level was determined using a human Aβ (1–42) ELISA assay kit (Thermo Fisher Scientific, KHB3544) and a mouse & rat Aβ (1–42) ELISA assay kit (Thermo Fisher Scientific, KMB3441) in compliance with manufacturer’s instructions. For secreted Aβ (1–42), cell media were centrifuged at 1,000 × g for 20 min and supernatants were collected with a mixture of phosphatase inhibitors (100X). For cellular Aβ (1–42), cell lysates were collected with a mixture of phosphatase inhibitors (100X). Hippocampal samples (100 μg) were prepared using lysis buffer and homogenizer. Samples were incubated with Aβ primary antibody at RT for 1 h and then incubated with secondary antibody conjugated with HRP for 30 min followed by adding substrate. The absorbance at 450-nm was measured, and the standard curve was used to convert optical density to concentration.
Animal study
Animal handling and experimentation were undertaken according to the ethical approval of the Institutional Animal Care and Use Committee of Seoul National University (IACUC, Approval No.: SNU-220307-1). Group size estimation was performed in compliance with the instructions given by IACUC (effect size: 15%, standard deviation [SD]: 6%, number of groups: 4; alpha: 0.05, power [1-β]: 0.8). Male ICR mice were used to investigate the effects of diabetes on hippocampal lysophagy impairment and subsequent Aβ and p-MAPT accumulations, and cognitive impairment. Mice (9 weeks of age) were housed under standard conditions of temperature (20–25°C), humidity (less than 60%), and a 12 h dark/12 h light cycle and free to access autoclaved chow and tap water. STZ was injected into mice to induce diabetes, as previously described with slight modifications [53]. Mice were randomized to receive STZ (75 mg/kg in 0.1 M sodium citrate buffer, pH 4.5) or vehicle intraperitoneally (i.p.) once daily for three days after 6 h of fasting. 1 week after STZ injection, blood glucose level was measured using a glucose monitor (Roche) and when the blood glucose level was 300 mg/dl or higher, severe diabetes was determined. Once there were enough diabetic mice, they were separated into 4 groups at random (n = 5 per group): vehicle, STZ, curcumin C1, and curcumin C1 + STZ. Curcumin C1 (10 mg/kg in 1% sodium carbonyl methylcellulose [CMCNa;Sigma Aldrich, C5678]) was orally administrated by gavage once daily for 8 weeks following diabetes induction, as previously reported with slight modifications [19]. At 18 weeks of age, body weight and blood glucose levels were measured followed by behavioral tests. For additional biochemical analysis, mice were anaesthetized by i.p. injection of avertin solution (250 mg/kg, mixture containing 2-methyl-2-butanol [Sigma Aldrich, 152463] and 2,2,2,-tribromoethanol[Sigma Aldrich, T48402]) and euthanized by exsanguination.
Y-maze test
Assessment of hippocampal-dependent spatial learning and memory is done using the Y maze behavioral test, since mice naturally explore the new arms of the Y-maze. The mice were kept in the testing room for 2 h before test to reduce stress. Each mouse was placed on a different arm of the Y maze equipment at random (GaonBio). During the test, the mouse had 10 min to explore the maze at will. Four limbs have to be in the arm for entry to be considered complete. We divide the number of triads by the maximum alternation (total entries-2) of 100 to obtain the percentage of spontaneous alternation. An animal’s ability to learn and remember spatial information is impaired when its alternation % is low.
Open field test
The open field test is used to assess anxiety in rodents because they navigate around the open field when they feel anxious. To reduce stress before the test, animals were acclimatized to the testing chamber for 2 h. Mice were put into the rectangular plastic boxes (H30 × L30 × W30 cm) and their activities were monitored for 10 min. The amount of time spent in the open field’s center and periphery was examined using Smart 3.0 video tracking system.
NOR test
The novel objectrecognition test relies on mice’s intrinsic desire for novelty and is afrequently used mouse behavioral test for evaluating object working memory. For habituation, the mice were housed in an open-field box in the testing chamber for 4 h. On the next day, the mice were given the same open area with two similar objects from the first session and were free to move around for 10 min. One of the two objects was changed after 4 h, and the mouse was then given a second chance to explore freely for 10 min in the open field. The time spent exploring a new object minus the time spent examining a familiar object was divided from the total exploration time to generate the discrimination index, which is used to evaluate cognition. The novelty preference was determined as the proportion of total exploration time spent on novel objects. A decrease in the discrimination index indicates impairment of object working memory.
Immunohistochemistry (IHC) analysis
Mice were anaesthetized before being transcardially perfused with PBS and then being fixed in 4% PFA. After being carefully removed, the brains were post-fixed with 4% PFA for 2 h followed by dehydration for 1–2 days in 30% sucrose solution. Continuous cutting with a cryostat (Leica Biosystems) was used to obtain coronal sections of the hippocampus that were 40 μm thick. Free-floating method was used to stain the hippocampus. Section was incubated with 5% NGS dissolved in 1% TritonX-100 at RT for 1 h, and then for 2 days at 4°C with the primary antibody (1:1,000 dilution). Samples were incubated with the secondary antibody (1:300 dilution) for 2 h at RT following three PBS washes. A confocal microscope system (Carl Zeiss, LSM710) and a super-resolution radial fluctuations imagingsystem (SRRF) (Andor Technology) were used to visualize the sections. Image analysis was performed with the Fiji software [50]. Application of the same threshold was used to measure signal intensities.
Statistical analysis
The sample size n refers to the number of biologically independent replicates and is used for statistical analysis. All data were checked for normality using the Shapiro-Wilk test before being subjected to parametric statistical analysis. An unpaired Student’s t-test with two-tailed was performed to compare the means of two groups. One-way or two-way ANOVA were used to compare the differences between various groups. Only when an ANOVA resulted in a significant F and no significant variance inhomogeneity were post hoc tests conducted. Using the Prism 8 analysis program (Graphpad), quantitative data are presented as a meanand SD. A level of probability of P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
This research was supported by National R&D Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (RS-2023-00208475) and BK21 FOUR Future Program for Creative Veterinary Science Research.
Funding Statement
The author(s) reported this wok was funded by National R&D Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (RS-2023-00208475)
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2229659
References
- [1].Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168–181. doi: 10.1038/nrneurol.2017.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lacy ME, Gilsanz P, Karter AJ, et al. Long-term glycemic control and dementia risk in Type 1 diabetes. Diabetes Care. 2018;41(11):2339–2345. doi: 10.2337/dc18-0073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Barbiellini Amidei C, Fayosse A, Dumurgier J, et al. Association between age at diabetes onset and subsequent risk of dementia. JAMA. 2021;325:1640–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lambeth TR, Riggs DL, Talbert LE, et al. Spontaneous isomerization of long-lived proteins provides a molecular mechanism for the lysosomal failure observed in Alzheimer’s Disease. ACS Cent Sci. 2019;5(8):1387–1395. doi: 10.1021/acscentsci.9b00369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lee JH, Yang DS, Goulbourne CN, et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci. 2022;25(6):688–701. doi: 10.1038/s41593-022-01084-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chae CW, Choi GE, Jung YH, et al. High glucose-mediated VPS26a down-regulation dysregulates neuronal amyloid precursor protein processing and tau phosphorylation. Br J Pharmacol. 2022;179(15):3934–3950. doi: 10.1111/bph.15836 [DOI] [PubMed] [Google Scholar]
- [7].Chae CW, Lee HJ, Choi GE, et al. High glucose-mediated PICALM and mTORC1 modulate processing of amyloid precursor protein via endosomal abnormalities. Br J Pharmacol. 2020;177(16):3828–3847. doi: 10.1111/bph.15131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kwart D, Gregg A, Scheckel C, et al. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, Not a Aβ. Neuron. 2019;104:256–70 e5. doi: 10.1016/j.neuron.2019.07.010 [DOI] [PubMed] [Google Scholar]
- [9].Yim WW, Mizushima N.. Lysosome biology in autophagy. Cell Discov. 2020;6(1):6. doi: 10.1038/s41421-020-0141-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Papadopoulos C, Meyer H. Detection and clearance of damaged lysosomes by the endo-lysosomal damage response and lysophagy. Curr Biol. 2017;27(24):R1330–R41. doi: 10.1016/j.cub.2017.11.012 [DOI] [PubMed] [Google Scholar]
- [11].Zhu J, Pittman S, Dhavale D, et al. VCP suppresses proteopathic seeding in neurons. Mol Neurodegener. 2022;17(1):30. doi: 10.1186/s13024-022-00532-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang C, Chen XC, Li ZH, et al. SMAD3 promotes autophagy dysregulation by triggering lysosome depletion in tubular epithelial cells in diabetic nephropathy. Autophagy. 2021;17(9):2325–2344. doi: 10.1080/15548627.2020.1824694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sims-Robinson C, Bakeman A, Rosko A, et al. The Role of oxidized cholesterol in diabetes-induced lysosomal dysfunction in the brain. Mol Neurobiol. 2016;53(4):2287–2296. doi: 10.1007/s12035-015-9207-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Feng L, Liang L, Zhang S, et al. HMGB1 downregulation in retinal pigment epithelial cells protects against diabetic retinopathy through the autophagy-lysosome pathway. Autophagy. 2022;18(2):320–339. doi: 10.1080/15548627.2021.1926655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Darwich NF, Phan JM, Kim B, et al. Autosomal dominant VCP hypomorph mutation impairs disaggregation of PHF-tau. Science. 2020;370(6519):370. doi: 10.1126/science.aay8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mueller-Steiner S, Zhou Y, Arai H, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron. 2006;51:703–714. doi: 10.1016/j.neuron.2006.07.027 [DOI] [PubMed] [Google Scholar]
- [17].Vagnozzi AN, Li JG, Chiu J, et al. VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol Psychiatry. 2019;26(11):6992–7005. doi: 10.1038/s41380-019-0453-x [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [18].Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res. 2016;118(11):1808–1829. doi: 10.1161/CIRCRESAHA.116.306923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song JX, Sun YR, Peluso I, et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of mTOR inhibition. Autophagy. 2016;12(8):1372–1389. doi: 10.1080/15548627.2016.1179404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Miranda AM, Lasiecka ZM, Xu Y, et al. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat Commun. 2018;9(1):291. doi: 10.1038/s41467-017-02533-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. Embo J. 2013;32(17):2336–2347. doi: 10.1038/emboj.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jia J, Claude-Taupin A, Gu Y, et al. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev Cell. 2020;52:69–87 e8. doi: 10.1016/j.devcel.2019.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu GY, DM S. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203 doi: 10.1038/s41580-020-0219-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. 2016;32(1):255–278. doi: 10.1146/annurev-cellbio-111315-125407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- [26].Saftig P, Haas A. Turn up the lysosome. Nat Cell Biol. 2016;18(10):1025–1027. doi: 10.1038/ncb3409 [DOI] [PubMed] [Google Scholar]
- [27].Fleming A, Bourdenx M, Fujimaki M, et al. The different autophagy degradation pathways and neurodegeneration. Neuron. 2022;110(6):935–966. doi: 10.1016/j.neuron.2022.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Skowyra ML, Schlesinger PH, Naismith TV, et al. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360(6384). doi: 10.1126/science.aar5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019;21(2):133–142. doi: 10.1038/s41556-018-0244-7 [DOI] [PubMed] [Google Scholar]
- [30].Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zummo FP, Cullen KS, Honkanen-Scott M, et al. Glucagon-like peptide 1 protects pancreatic β-cells from death by increasing autophagic flux and restoring lysosomal function. Diabetes. 2017;66(5):1272–1285. doi: 10.2337/db16-1009 [DOI] [PubMed] [Google Scholar]
- [32].Chai YL, Chong JR, Weng J, et al. Lysosomal cathepsin D is upregulated in Alzheimer’s disease neocortex and may be a marker for neurofibrillary degeneration. Brain Pathol. 2019;29(1):63–74. doi: 10.1111/bpa.12631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cao M, Luo X, Wu K, et al. Targeting lysosomes in human disease: from basic research to clinical applications. Sig Transduct Target Ther. 2021;6(1). doi: 10.1038/s41392-021-00778-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020;21(2):101–118. doi: 10.1038/s41580-019-0185-4 [DOI] [PubMed] [Google Scholar]
- [35].Jia J, Bissa B, Brecht L, et al. AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol Cell. 2020;77:951–69 e9. doi: 10.1016/j.molcel.2019.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bordi M, Berg MJ, Mohan PS, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12(12):2467–2483. doi: 10.1080/15548627.2016.1239003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Polito VA, Li H, Martini-Stoica H, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 2014;6(9):1142–1160. doi: 10.15252/emmm.201303671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Park K, Lee M-S, Kim J. Lysosomal Ca2+-mediated TFEB activation modulates mitophagy and functional adaptation of pancreatic β-cells to metabolic stress. Autophagy. 2022;13(1):1–3. doi: 10.1038/s41467-022-28874-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Martina JA, Diab HI, Lishu L, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014;7(309):ra9. doi: 10.1126/scisignal.2004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu EA, Schultz ML, Mochida C, et al. Fbxo2 mediates clearance of damaged lysosomes and modifies neurodegeneration in the Niemann-Pick C brain. JCI Insight. 2020;5(20). doi: 10.1172/jci.insight.136676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mishra R, Upadhyay A, Prajapati VK, et al. LRSAM1 E3 ubiquitin ligase: molecular neurobiological perspectives linked with brain diseases. Cell Mol Life Sci. 2019;76(11):2093–2110. doi: 10.1007/s00018-019-03055-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Huett A, Heath RJ, Begun J, et al. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe. 2012;12(6):778–790. doi: 10.1016/j.chom.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Burbidge K, Rademacher DJ, Mattick J, et al. LGALS3 (galectin 3) mediates an unconventional secretion of SNCA/α-synuclein in response to lysosomal membrane damage by the autophagic-lysosomal pathway in human midbrain dopamine neurons. Autophagy. 2022;18(5):1020–1048. doi: 10.1080/15548627.2021.1967615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang L, Zhang X, Lin ZB, et al. Tripartite motif 16 ameliorates nonalcoholic steatohepatitis by promoting the degradation of phospho-TAK1. Cell Metab. 2021;33(7):1372–88 e7. doi: 10.1016/j.cmet.2021.05.019 [DOI] [PubMed] [Google Scholar]
- [45].Liu J, Li W, Deng KQ, et al. The E3 Ligase TRIM16 is a key suppressor of pathological cardiac hypertrophy. Circ Res. 2022;130(10):1586–1600. doi: 10.1161/CIRCRESAHA.121.318866 [DOI] [PubMed] [Google Scholar]
- [46].Song JX, Malampati S, Zeng Y, et al. A small molecule transcription factor EB activator ameliorates β-amyloid precursor protein and tau pathology in Alzheimer’s disease models. Aging Cell. 2020;19(2):e13069. doi: 10.1111/acel.13069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fath T, Ke YD, Gunning P, et al. Primary support cultures of hippocampal and substantia nigra neurons. Nat Protoc. 2009;4(1):78–85. doi: 10.1038/nprot.2008.199 [DOI] [PubMed] [Google Scholar]
- [48].Griess K, Rieck M, Müller N, et al. Sphingolipid subtypes differentially control proinsulin processing and systemic glucose homeostasis. Nat Cell Biol. 2023;25(1):20–29. doi: 10.1038/s41556-022-01027-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gustafsson N, Culley S, Ashdown G, et al. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun. 2016;7(1):1–9. doi: 10.1038/ncomms12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224(3):213–232. doi: 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- [52].Sharma S, Quintana A, Findlay GM, et al. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature. 2013;499(7457):238–242. doi: 10.1038/nature12229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Furman BL. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc. 2021;1:e78. doi: 10.1002/cpz1.78 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.