

Summary
We present TopicFlow, a computational framework for flow cytometry data analysis of patient blood samples for the identification of functional and dynamic topics in circulating T cell population. This framework applies a Latent Dirichlet Allocation (LDA) model, adapting the concept of topic modeling in text mining to flow cytometry. To demonstrate the utility of our method, we conducted an analysis of ∼17 million T cells collected from 138 peripheral blood samples in 51 patients with melanoma undergoing treatment with immune checkpoint inhibitors (ICIs). Our study highlights three latent dynamic topics identified by LDA: a T cell exhaustion topic that independently recapitulates the previously identified LAG-3+ immunotype associated with ICI resistance, a naive topic and its association with immune-related toxicity, and a T cell activation topic that emerges upon ICI treatment. Our approach can be broadly applied to mine high-parameter flow cytometry data for insights into mechanisms of treatment response and toxicity.
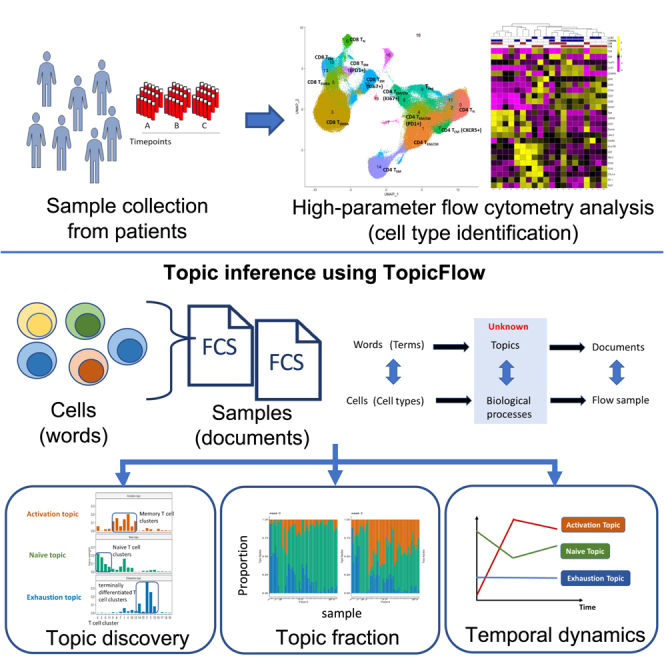
Graphical abstract
Highlights
-
•
TopicFlow adapts the concept of topic modeling to flow cytometry data
-
•
We use TopicFlow to study dynamic changes of T cell states during cancer immunotherapy
-
•
Dynamic T cell topics (exhaustion, naive, activation) are revealed in patient blood
-
•
TopicFlow identifies T cell topics related to treatment resistance and toxicity
Motivation
Immune checkpoint inhibitors (ICIs), now mainstays of cancer treatment, show great potential but only benefit a subset of patients. A more complete understanding of the immunological mechanisms and pharmacodynamics of ICI in patients with cancer may help identify the patients most likely to benefit and generate knowledge for the development of next-generation ICI regimens. We set out to interrogate the temporal dynamics of T cell populations in patients treated with ICI from flow cytometry data. Advanced statistical and computational approaches are needed for mining high-parameter flow cytometry data to uncover immunological insights with clinical relevance.
Peng et al. introduce a computational framework, TopicFlow, to explore T cell population dynamics in peripheral blood samples obtained from patients with cancer receiving immune checkpoint inhibitor treatment. Their findings reveal distinct T cell topics associated with ICI resistance, immune-related toxicity, and treatment-induced immune activation.
Introduction
Cancer immunotherapies with immune checkpoint inhibitors (ICIs) are revolutionizing cancer treatment.1 ICIs, given as monotherapy or in combination, have proven efficacious in multiple types of cancer, and it is estimated that approximately 44% of patients with cancer in the United States are eligible to receive ICIs.2 However, patient tumor response and toxicity under different treatment regimens are highly heterogeneous. For example, patients with melanoma who receive α-CTLA-4 and α-PD-1 combination blockade have a higher response rate but are more likely to experience immune-related adverse events (irAEs) compared with those who receive α-PD-1 monotherapy.3,4,5 The identification of biomarkers that link immune characteristics of individual patients to clinical outcomes during ICI treatment may help guide treatment selection for individual patients to improve therapeutic benefit while minimizing toxicity for patients.6
Flow cytometry analysis has become an important tool to study the tumor microenvironment as well as patients’ peripheral blood samples in the context of immunotherapy. Several biomarkers examining functional cell types have been identified to predict treatment response or define resistance mechanisms to ICIs.7,8,9 These analyses commonly focus on a limited number of pre-specified cell types determined from prior domain knowledge, potentially overlooking important unmined subpopulations. Furthermore, recent advances in flow and mass cytometry have significantly improved the throughput, allowing 30–50 markers to be measured simultaneously at single-cell resolution,10 which allows for the exploration of a much larger number of possible cell subsets. Such high-parameter flow cytometry data, when performed on longitudinally collected samples, are exceedingly complex and pose a great analytical challenge to (1) delineate cell-type composition from millions of single cells and (2) map the temporal evolution of cell types over time. Sophisticated statistical and computational tools are needed to fully leverage the complexity and richness of high-parameter single-cell data in order to expedite biomarker discovery in cancer immunotherapy.
In recent years, there have been concerted efforts to advance the development of cutting-edge computational methods for flow cytometry including visualization, clustering, and lineage tracing of cell populations, as reviewed in Aghaeepour et al.11 The current state-of-the-art approach allows refined cell-type classification and visualization. Although identifying cell types is an important task, a more difficult challenge lies in modeling the relationship between them and how they collectively characterize the immune compartment at baseline and any subsequent shifts pharmacodynamically upon specific ICI regimens. Furthermore, it remains a challenge to quantitatively link complex immune cell composition and the temporal dynamics with clinical outcomes at the individual sample level.
To fill these gaps, we present an innovative statistical and computational framework that is inspired by works developed in monitoring temporal dynamics of bacterial strains.12,13 We adapt the Latent Dirichlet Allocation (LDA) model14 to investigate the pharmacodynamics of T cell compositions in peripheral blood of ICI-treated patients with cancer early after treatment initiation. LDA is a generative statistical model for the identification of hidden structures in large data and is widely applied for topic discovery in text-mining analysis. Here, we present a unique application of LDA to understand the temporal evolution of T cells in flow cytometry data to track early pharmacodynamic changes after exposure to ICIs (Figure 1A). In an unsupervised fashion, LDA explores the hidden structure and identifies latent topics with interpretable features relating to biologically relevant function states (Figure 1B), allowing for the discovery of potential biomarkers of clinical relevance.
Figure 1.

Latent Dirichlet Allocation reveals hidden structures in flow cytometry data
(A) Data overview.
(B) Deconvolution of flow cytometry data with Latent Dirichlet Allocation (LDA) model after pooled clustering analysis.
(C) The analogy between text analysis and flow cytometry analysis.
(D) Fractional membership of topics within each sample and its evolution over time.
(E) Graphic representation of LDA model.
See also Table S4.
Contrasting the conventional flow cytometry analysis of individual cell population one at a time, the LDA framework allows joint modeling of the relationship between different cell types and the evaluation of dynamic changes of cell populations in consort. Our proposed work provides a data-mining tool for flow cytometry analysis of tens of millions of single cells across a large collection of patient samples. It allows the investigator to discover functional themes that connect different cell types that characterize the immune composition in patient samples, to evaluate pharmacodynamic changes upon treatment, and to interrogate the association between specific patterns and clinical outcomes.
Results
Method overview
We present TopicFlow, a topic model approach for mining large-scale high-dimensional flow cytometry data from longitudinally collected patient samples. Motivated by the similarities between text data mining and flow cytometry analysis, LDA considers cells as words, cell types as terms, patient samples as documents, and biological processes as topics (Figure 1C). It assumes that each cell in a patient sample arises from a mixture of topics, each of which is a probability distribution over cell types. LDA takes as input a cell-type-by-sample count matrix, which is similar to the term-by-document matrix in text analysis. The cell types can be obtained through a graph-based clustering of single cells from pooled samples (Figure 1B). Then, the cell-type-by-sample count matrix is decomposed by LDA into three matrices:
-
(1)
cell-type-by-topic matrix, B, for topic content
-
(2)
topic-by-sample matrix, , for topic prevalence
-
(3)
vector of cell counts, N
The cell-type-by-topic matrix represents topics as distinct discrete distributions across cell types, thus facilitating the linkage between topics and cell types. Each topic is a weighted combination of a specific set of cell types that may be functionally related. Within each topic, cell types exhibiting similar abundance patterns across patient samples are likely to be involved in the same biological process. In contrast to the conventional approach of assessing one cell type at a time, utilization of LDA offers a comprehensive approach to systematically evaluate all cell types simultaneously. This enables researchers to gain insights into the underlying biological processes by examining the co-occurring patterns of cell types within topics.
The topic-by-sample matrix displays topic proportions estimated within each individual sample. This matrix enables us to characterize and quantify topic composition at the sample level and track the topic evolution over time (Figure 1D). By using this topic prevalence matrix, we can directly correlate topics (composed of T cell subtypes with shared functional states) with clinical outcomes. Patients with similar topic composition and temporal dynamics may also share similar clinical outcomes and pharmacodynamic profiles, as we will describe in detail in the STAR Methods. In the following section, we provide an illustration of how LDA deconvolutes the longitudinal flow cytometry data to characterize topics that generate biological insights. To demonstrate this, we present a specific data example.
Data
The large-scale flow cytometry dataset we analyzed contains ∼17 million T cells from a cohort of 51 patients with melanoma (138 samples) treated with a combination of anti-CTLA-4 and anti-PD-1 ICI as part of a phase II clinical trial (ClinicalTrials.gov: NCT03122522).15 The clinical outcome data (response, overall survival [OS], progression-free survival [PFS], toxicity) of the cohort have been previously reported.15 Nearly half of patients (45%) experienced severe (≥ grade 3) irAEs, and 61% of patients responded (complete response [CR] or partial response [PR]) to the ICI treatment (Figure 1A). Based on pre-treatment peripheral blood samples, our prior work on a large cohort has classified patients into three “immunotypes” (LAG+/LAG−/PRO) that are correlated to survival and response,16 which we also include in the analysis. Flow cytometry was performed using an X50 panel that measures 29 markers for each single cell (a complete list of markers described in STAR Methods), including checkpoint blockade biomarkers (e.g., PD-1, CTLA-4, LAG-3) and T cell lineage markers (e.g., CD45RA, CCR7). Staining was performed on cryo-banked peripheral blood mononuclear cells (PBMCs) collected at three time points for each patient: weeks 0 (pre-treatment) and 3 and 6 (on treatment).
Identification of T cell types and composition across patient samples
Before applying the LDA model, we first identified T cell types via the Louvain algorithm, a popular data-driven graph-based clustering method,17 after pooling viable CD3+ cells from all patient samples at all time points together to allow the comparison of consistent T cell clusters across multiple samples. The 20 main T cell clusters with relative abundance >0.1% are displayed in the uniform manifold approximation and projection (UMAP) (Figure 2A), where CD4 and CD8 T cells are separated into two main distinct parts (Figure 2B). The marker expression profile in the T cell clusters is shown in Figure 2C. Based on the lineage markers CD45RA and CCR7 (Figure 2D), we are able to further identify T cell clusters with different differentiation states, including naive T cell (Tn) clusters (CCR7+CD45RA+), central and effector memory T cell (Tcm and Tem) clusters (CCR7+CD45RA− and CCR7−CD45RA−, respectively), and effector memory T cell re-expressing CD45RA (Temra) clusters (CCR7−CD45RA+). Among identified cell clusters, we note one CD4 Tcm cluster (cluster 8) and one CD8 Tem cluster (cluster 12) that highly express Ki67, a proliferation marker recognized in previous studies9 (Figure 2D). We also identified T cell subsets with less well-recognized roles in tumor immunity, including a cytotoxic CD4 Tem cluster (cluster 14) and a double-positive (CD4+CD8+) T cell cluster (cluster 19). These cell types are currently attracting attention to understand their role in tumor immunity.18,19
Figure 2.

Identification of T cell clusters in the X50 flow cytometry data
(A) UMAP plot of T cell clusters.
(B) UMAP plot of T cells overlaid with the expression of CD4 and CD8.
(C) Heatmap displaying average marker expression (scaled) of markers in each cluster.
(D) UMAP plot of T cells overlaid with the expression of CD45RA, CCR7, Ki67, and FoxP3.
See also Table S4.
LDA reveals hidden structures in flow cytometry data
The T cell clusters we identified are inter-correlated as governed by the underlying functional and differentiation states. To discover the connections, we applied LDA and uncovered K = 3 latent topics, which capture the major patterns embedded in the data. The determination of the number of topics K is described in the STAR Methods.
To evaluate each topic, we visualize the topic weights for every single topic, where a topic is represented as a distinct probability distribution over the T cell clusters (Figure 3A). We note that a large cell type may have high positive weights in multiple topics simply because of its high abundance (e.g., cluster 1). Therefore, we introduced the lift20 metric, the log ratio of the topic weight of a T cell cluster v in topic k over its empirical frequency, to rank the importance of individual T cell clusters in each topic (Figures 3B and S1). The top-ranked clusters, characterized by their high lift value, were selected as representatives of the corresponding topic. This approach avoids selecting high-abundance clusters with little variation that could also have high topic weight. We define the three topics as activation topic, naive topic, and exhaustion topic based upon the most representative cell clusters ranked by lift (Figure 3B). The activation topic is highly represented by memory T cell clusters (Tcm/Tem), and later, we will show that these clusters capture the major pattern of T cell expansion after ICI. The naive topic has high probability weights over the Tn clusters, while the exhaustion topic consists of exclusively terminally differentiated T cell clusters (Temra). The biological significance of each topic will be discussed in the next section.
Figure 3.

LDA identifies three topics in flow cytometry data
(A) Estimated weights (compositions) of clusters in single topics.
(B) Clusters with the top 10 highest lift for each topic. Clusters with top lift are identified as representative clusters for each topic.
(C) Sample topic proportions () within each patient sample collected at weeks 0, 3, and 6.
Each sample can be represented as a mixture of the three topics. The topic-by-sample matrix provides the estimated topic proportions within each sample. Figure 3C shows that at week 0 (pre-treatment), most patient samples are characterized by a strong presence of the naive topic (green). Upon treatment exposure, the activation topic (orange) clearly emerges as seen by the striking increase of this topic proportion at weeks 3 and also after a second dose at week 6 (though this was weaker in some samples). The naive topic proportion correspondingly decreases upon ICI treatment as cells transition into more “activated” states. This topic diagram is useful in mapping the immune cell composition and the pharmacodynamic changes upon ICI exposure at the patient level.
In order to classify patients based on the identified immune topics, we performed a hierarchical clustering of the topic-by-sample matrix, which led to four subgroups as shown in Figure 4. Figure 4A (and Figure S2A) highlights the pattern of topic fraction changes pre- and on treatment arranged by each topic individually. As described earlier, the activation topic mainly captures the expansion of Tcm/Tem upon treatment. For most patients, the proportion of the activation topic is near zero (dark blue) in pre-treatment samples (week 0). This topic emerges on treatment as seen by the increase of topic proportions in week 3 and 6 samples. At baseline (week 0), most of the patient samples are characterized by a high presence of the naive topic, which subsequently decreases after ICI treatment as cells transition into more “activated” states. In contrast, a small subgroup of patient samples has a low proportion of the naive topic but a high fraction of the exhaustion topic presented at week 0. There is no visible reduction in the exhausted T cell population after ICI treatment.
Figure 4.

LDA reveals patient subgroups with distinct pharmacodynamics
(A) Heatmap showing the sample topic proportions () for each single topic (patients, n = 50). Patient 17-162-08 has only one sample at week 0, and thus it is not included. Missing samples are colored gray in the heatmap.
(B) Dynamics of sample topic proportions () of the three topics in the four patient subgroups across time.
We identified four patient subgroups by hierarchical clustering on patient topic proportions, while each subgroup exhibits distinct dynamic patterns within the three interpretable topics (Figure 4B). Patients in groups 1 and 2 both have inferior increases in activation topic. Group 1 has the highest proportion of the exhaustion topic and group 2 has the highest naive topic across time. Patients in group 3 have the highest increase in the activation topic compared with other groups and are accompanied by the highest decrease in the naive topic fraction. Group 4 has a high proportion of the naive topic at week 0 and a moderate increase in the activation topic. Patients in group 4 are more likely to experience severe ICI-related toxicity compared with the other groups: 73.1% (19/26) vs. 37.5% (9/24) (p = 0.025, chi-squared test). There is a trend that patients in group 4 have higher response rates, 69.2% (18/26) vs. 54.2% (13/24), and better survival outcomes (Figure S2B), although these do not reach statistical significance.
Activation topic reveals T cell expansion after ICI treatment
The activation topic captures the pattern of T cell expansion in peripheral blood after ICI treatment, as seen by the increase of cells in the representative clusters highlighted in Figure 5A. The five representative cell clusters we identified include two CD4 T cell clusters (clusters 8 and 4), one CD8 T cell cluster (cluster 12), one regulatory T cell (Treg) cluster (cluster 9), and one CD4−CD8− T cell cluster (cluster 16) (Figure 5B). Upon treatment at week 3, the five representative clusters dramatically increased for the entire patient cohort (Figure 5C), which was captured by the increase in topic proportions (p = 1.3e−33) (Figure 5D). It might be of clinical interest that most immunological changes happen just after the first dose (from baseline to week 3). The comprehensive pharmacodynamics of all 20 clusters are provided in Figures S3–S5.
Figure 5.

The activation topic
(A) UMAP plots of T cells at three time points of patient 17-162-05 (PR, severe irAE), with five representative clusters of the activation topic highlighted. Each UMAP plot contains 20,000 random-sampled cells from each sample.
(B) Heatmap showing average marker expression (scaled) of the five representative clusters.
(C) Relative abundances (percentages of cells in each cluster out of total T cells) of the five representative clusters of the activation topic change over time. The clusters are ordered by lift. p values were provided to test the time effect. One outlier (30.2% at week 3) for cluster 12 was removed for better visualization.
(D) Activation topic proportions of each individual patient, paired with gray lines.
(E) Ridge plots of Ki67 marker expression over the five representative clusters.
Cell clusters 8 (CD4+), 12 (CD8+), and 16 (CD4−CD8−) highly express Ki67+, which has been established as a T cell proliferation marker.9,21,22 These cell clusters also express CD38 and HLA-DR, which are markers of activation (Figure 5B). In particular, the Ki67+ (CD38+HLA-DR+) CD8+ population (cluster 12) is Eomes high, Tbet high, and CD27 low (Figure 5B), consistent with an effector phenotype. It also shows expression of PD-1, TIM-3, and LAG-3, consistent with previous findings that the increase in Ki67 expression was most prominent in the PD-1+CD8 T cells.9 Cluster 12 showed an average 8-fold increase at week 3 post-treatment from the level in pre-treatment samples (Figure 5C). In addition to cluster 12, three other clusters with high Ki67 expression, cluster 8 (CD4+), cluster 16 (CD4−CD8−), cluster 9 (Treg), and a Tem/Tcm CD4 (cluster 4), co-expanded upon combination of anti-PD-1 and anti-CTLA-4 treatment, with an observed 3.1-, 2.6-, 1.7-, and 1.8-fold increase in cell cluster size at week 3, respectively. The activation topic presents a unique combination of all these T cell subsets, which can be used as a complex pharmacodynamic index to monitor patients’ immune responses during treatment.
Naive topic is associated with ICI-related toxicity
The second topic we labeled the naive topic, with CD4+ and CD8+ naive T cell clusters identified as the most representative clusters highlighted in Figure 6A. The four representative clusters include two naive CD4 clusters (clusters 0 and 2), one naive CD8 cluster (cluster 6), and one Treg cluster (cluster 11) (Figure 6B). The abundances of the four representative clusters, as well as the proportions of the naive topic, decrease slightly after treatment (p = 5.1e−17 for the difference in proportions across time) (Figures 6C and 6D), indicating the differentiation of naive T cells during the immune response. The four representative clusters shared a high level of marker expression in CCR7, CD45RA, and CD127, which are key markers of naive T cell lineage (Figure 6B). Interestingly, individuals that experience severe ICI-related toxicity (grade 3–4) have a higher proportion of the naive topic at baseline week 0 (p = 0.029) (Figure 6E), while there is no significant difference in changes over time between patients with/without severe toxicity (p = 0.095 for the interaction effect). In contrast, we failed to identify the association between each individual naive cell cluster and toxicity (Table S1), likely due to lack of power by univariate analysis of each individual cell cluster in isolation. There is no association found between toxicity and the other two topics in this cohort and no association observed between the three topics and response (Figure S6).
Figure 6.

The naive topic
(A) UMAP plots of T cells at three time points of patient 17-162-EXT09 (PR, severe irAE), with four representative clusters of the naive topic highlighted. Each UMAP plot contains 5,000 random-sampled cells from each sample.
(B) The heatmap shows the average marker expression (scaled) of the four representative clusters.
(C) Relative abundances (percentages of cells in each cluster out of total T cells) of the four representative clusters of the naive topic change over time. The clusters are ordered by lift. p values were provided to test the time effect.
(D) Naive topic proportions of each individual patient, paired with gray lines.
(E) Sample proportions of the naive topic between patients experiencing severe/no severe irAE (Y/N). p values were provided by Wilcoxon rank-sum test for each time point.
Exhaustion topic is related to LAG+ immunotype
The exhaustion topic includes four representative clusters (Figure 7A): two CD8 Temra clusters (clusters 3 and 5), one CD4 Tem cluster (cluster 14), and one CD4−CD8− cluster (cluster 13). The representative clusters in this topic highly express LAG-3, a T cell exhaustion marker. Besides LAG-3, the four representative clusters also highly express Tbet, GZM-B, and Eomes, markers for functional cytotoxic T cells (Figure 7B). Cluster 14 is a cytotoxic CD4 T cell cluster previously reported to be correlated with clinical outcomes in bladder cancer.18,23 Compared with the other two topics, the topic proportions of the exhaustion topic, as well as the abundances of its four representative clusters, are not significantly changing over time (p = 0.14 for the difference in proportions across time) (Figures 7C and 7D), but there is a subgroup of patients with strikingly high exhaustion topic proportions, contrasting the rest of the patients (Figures 3C and 4A). For better illustration, we compared pre-treatment samples from two patients (LAG+ vs. LAG− immunotypes) with four representative clusters highlighted (Figure 7A). The LAG+ patient sample is dominated by the exhaustion topic (), while the LAG− patient sample is not (). We observed substantial differences in abundances of clusters 3, 5, and 14 when comparing the two patients.
Figure 7.

The exhaustion topic
(A) UMAP plots of T cells of patients 17-162-05 (PR, severe irAE, LAG+ immunotype) and 17-162-27 (stable disease [SD], severe irAE, LAG− immunotype) at time point A, each with 20,000 random-sampled cells. The four representative clusters are highlighted.
(B) Heatmap of the average marker expression (scaled) of the four representative clusters of the exhaustion topic.
(C) Relative abundances (percentages of cells in each cluster out of total T cells) of the four representative clusters of the naive topic change over time. The clusters are ordered by lift. p values were provided to test the time effect.
(D) Exhaustion topic proportions of each individual patient, paired with gray lines.
(E) The abundance ratio of CD8 Temra (clusters 3 and 5) to CD8 Tn (cluster 6) across different immunotypes (p = 0.006 for immunotype main effect and p < 0.001 for the interaction effect between time and immunotype). The sample ratios of patient 17-162-EXT05 are extremely high (around ten times the second highest) and thus are not shown in the boxplot.
The exhaustion topic is highly related to the LAG+ immunotype, which has been associated with worse clinical outcomes in the previous study.16 In that study, three immunotypes (LAG−, LAG+, and PRO) were identified in peripheral blood samples using a four-marker classifier (%LAG-3+CD8+, %Ki67+CD8+, %TIM-3+CD8+, %ICOS+CD8+). Shen et al. reported that LAG+ patients with high levels of LAG-3+CD8+ cells prior to treatment are more likely to have a poor response, particularly when treated with anti-PD-1 regimens.16 The exhaustion topic provides advanced insights into the underlying T cell composition of LAG+/LAG− immunotypes. Moreover, we show the ratio of CD8 Temra/Tn (abundance of clusters 3 and 5/abundance of cluster 6) may be a superior biomarker (stable across time and not limited to pre-treatment samples) for distinguishing between LAG− and LAG+ immunotypes (Figure 7E), with p = 0.006 for the immunotype main effect and p = 2e−5 for the interaction effect between time and immunotype. This can be attributed to the fact that the majority of LAG-3+CD8+ cells in the pre-treatment samples belong to the Temra cell subset (in clusters 3 and 5).
Discussion
Immune cells are highly heterogeneous, containing a mixture of signals from all unknown ongoing biological processes. Here, we addressed the problem of deciphering hidden structures from longitudinal flow cytometry data in patients treated with ICI. We adopted the LDA model from text analysis and presented a unique computational framework for investigating potentially clinically relevant pharmacodynamical characteristics underlying the data. We demonstrated that LDA is effective in deconvoluting noisy flow cytometry data and can characterize topics that provide biological insights. With LDA, T cell subsets can be distilled into topics, which reveal patient subgroups with distinct dynamics.
Our method was inspired by the application of LDA in longitudinal microbiome analysis,12,13 where it was able to decipher the temporal changes in microbe composition. Alternative models to monitor dynamics of T cell compositions include the fitness model24 from population genetics and the Lotka-Volterra model (known as the predator-prey model).25 However, these models require more time points for model fitting and/or assume no differentiation between cell types. The LDA model, on the other hand, allows analysis of data from patients with limited time points and was demonstrated to work well on the longitudinal flow cytometry data.
LDA performs an unsupervised decomposition of T cell compositions to identify “topics” made up of T cell subtypes with shared functional and differentiation states. This allows us to mine tens of millions of single cells from a large collection of patient samples to discover the underlying functional themes that characterize the immune composition and pharmacodynamics changes in patients with cancer. Unlike the conventional flow cytometry analysis looking at one cell type at a time, our proposed LDA approach allows a joint modeling of the relationship between different cell types and for the evaluation of dynamic changes of cell populations in consort. The approach is data driven and does not require any domain knowledge, thus avoiding the possible “user” bias in the analysis step introduced by the preferential selection of specific markers.
In the case of our application of LDA to flow cytometry, topics are identified as combinations of distinct cell types collectively defined by markers relating to cell lineage and function, and the relationship between these cell types offers some insight into potential connections among them and unique pharmacodynamic patterns in pre- and on-treatment samples from patients treated with specific immunotherapy regimens. The key to this approach lies in the data-driven discovery of the combination of cell clusters that define a topic. The topics defined by unbiased interrogation of all data offer at least two strong advantages and opportunities for new biological discovery.
In the naive topic example, we demonstrate that the LDA method increases the power for discover in a large, complex dataset. The topic identified by LDA that we labeled naive includes both CD4 and CD8 T cell clusters with high expression of CCR7, CD45RA, and CD127, key markers known to be expressed on naive T cells, confirming a biologic coherence identified by the model and, hence, the eponymous topic. Through a data-driven aggregation of these naive cell types, we found a statistically significant association between the naive topic and treatment-related toxicity. This association was not found when we analyzed the individual naive cell clusters in isolation (see Table S1 for results for each individual cluster). It is only in the context of using the LDA model that this clinical association emerges.
A second opportunity for discovery that the LDA method presents is the potential to find connections or relationships that were previously unrecognized. For example, the co-expansion of CD38+ HLA-DR+ CD8+ and CD4+ T cells (clusters 12 and 8) and the Treg population (FoxP3+CD4+) in PBMCs captured by the activation topic upon the combination of anti-PD-1 and anti-CTLA-4 treatment is uniquely identified through the LDA framework. Furthermore, the co-existence of circulating exhaustion-like CD8 cells (clusters 3 and 5) and a cytotoxic CD4 cell type (cluster 14) in the exhaustion topic is new. Cytotoxic CD4 cells and their role in tumor immunity have currently been receiving much attention,18 but their potential relationship to an exhausted T cell population identified in our data has not been well explored. We note that this study is a proof of principle for the methodology, and validation of these findings requires additional analysis of independent cohorts of data, which is outside the scope of this paper.
LDA allows samples to have fractional membership across topics and is sometimes referred to as a soft clustering algorithm. Unlike clustering, which simply forces cell types into one of the cellular modules (clusters), LDA provides a biologically meaningful decomposition, quantifying not only the contribution of each cell type to an immune topic but also the fraction of topics within each sample. To better illustrate this, we performed a hierarchical clustering based on Pearson correlation of T cell subtypes (Figure S7A). The clustering approach may identify similar cellular modules (clusters of high-correlated cell types) but does not provide a clear way to correlate the cellular modules back to patient-level metadata (such as clinical outcomes) to make inferences. Figure S7B shows a traditional clustering analysis that fails to capture any underlying functional or pharmacodynamic themes that are immunologically meaningful or clinically relevant.
LDA can be further extended and embedded in more complex models for inference. Firstly, incorporating covariates in the topic model could further extend the model application on flow cytometry data, especially under complex experimental design. The structural topic model (STM), for example, allows us to incorporate patient/sample metadata into the model. The metadata can be added as covariates associated with topic prevalence (parameter ) or topic content (parameter B) with a log link,26 and a variational expectation maximization algorithm can be implemented for model inference.27 Secondly, in a setting where long-term monitoring of treatment effects is of interest with a large number of samples collected over time, a dynamic topic model28 can be more powerful with a more complex modeling of the temporal relationship across samples. Finally, incorporating additional constraints, e.g., sparsity constraint on cell-type-by-topic matrix , may further improve the efficiency of the model.29
The application of LDA is not limited to flow cytometry analysis, and it can be applied to any single-cell data (another example of the application of LDA on a single-cell RNA sequencing [scRNA-seq] dataset30 is provided in the tutorial). For future work, we can further extend LDA to explore the tumor microenvironment in multiplexed imaging data.31 Spatial information can be incorporated into the model to investigate the tumor and immune cell interactions. Moreover, LDA can also be applied for multi-omics data analysis,32,33 integrating data from multiple assays to better understand cancer heterogeneity and predict patient clinical outcomes.
Limitations of the study
As higher-parameter flow cytometry analysis is being adopted at a fast pace, allowing 40+ markers to be simultaneously measured with modern technologies including CyTOF and spectral flow, our study is a timely contribution to the field and provides a powerful method along with software tools for translating flow cytometry data into meaningful biological and clinical insights. A limitation of the current study is that the application of the TopicFlow method is illustrated using a flow cytometry dataset generated from a single center. Although the method we developed and presented in this paper is broadly applicable to high-parameter flow cytometry data, its utility remains to be seen with additional data applications. To facilitate future applications, we have developed an extensive tutorial on using the TopicFlow method for flow cytometry data analysis. It is a step-by-step demonstration of the workflow including pre-gating FCS files to the T cell population, quality control steps, cell-type classification using graph-based clustering, and finally, the key piece of topic modeling of cell types with an LDA model. The cell-type data matrix along with the code scripts that reproduce the main figures in the article are also now made publicly available. The tutorial page can be found here: https://xiyupeng.github.io/LDA_examples/melanoma.html.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| De-identified clinical data and summarized flow cytometry data on cell cluster counts and percentages | This paper | Table S4 |
| Analysis codes and flow cytometry data for the topic model | This paper |
https://github.com/xiyupeng/topic_modeling https://doi.org/10.5281/zenodo.7942456 |
| Single-cell RNA-seq data | Xue et al.30 | http://meta-cancer.cn:3838/scPLC |
| Software and algorithms | ||
| TopicFlow (tutorial) | This paper |
https://xiyupeng.github.io/LDA_examples/ https://doi.org/10.5281/zenodo.7942425 |
| FlowJo v10.8.0 | Becton Dickinson & Company (BD) | https://www.flowjo.com/ |
| FlowAI v1.22.0 | Monaco et al.34 | https://bioconductor.org/packages/flowAI/ |
| openCyto v2.4.0 | Finak et al.35 | http://opencyto.org/ |
| Seurat v4.0 | Hao et al.36 | https://satijalab.org/seurat/ |
| nparLD v2.1 | Noguchi et al.37 | https://cran.r-project.org/web/packages/nparLD/ |
| Topicmodels v0.2-12 | Grün and Hornik38 | https://cran.r-project.org/web/packages/topicmodels/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ronglai Shen (shenr@mskcc.org).
Materials availability
Any sharing of materials or data may be subject to material transfer agreements and data sharing agreements per requirement of Memorial Sloan Kettering Cancer Center.
Experimental model and subject details
The study includes melanoma patients (n = 51) in a cohort receiving combined immune checkpoint blockade (Anti PD-1/CTLA-4) therapy from 2017 to 2019 at the Memorial Sloan Kettering Cancer Center in a phase II clinical trial study (NCT03122522).15 For each patient, blood samples were collected at three different time points at week 0 (pre-treatment), and at weeks 3 and 6 (on-treatment) after the first dose. Best Overall Response (BOR) [partial response (PR), complete response (CR), stable disease (SD), and progression of disease (PD)], survival, PFS, and toxicity grade [grade 1–2 (N), grade 3–4 (Y)] were determined and reported for each patient. The clinical data for this cohort, including 20 females (19–80 years old) and 31 males (35–84 years old), has been previously reported15 and are shown in Table S4. We also included patient immunotype defined based on the 11-color panel flow cytometry data of pre-treatment samples in our previous study.16
Method details
Flow cytometry data
The goal of the study is to identify the characteristics of peripheral blood T cells that are related to clinical outcomes (response, toxicity). Flow cytometry with an X50 panel was performed on the collected peripheral blood mononuclear cells (PBMCs) as previously described.39,40 Our own X50 panel uses a cocktail of antibodies for the following markers: CD45RA-BUV395, CD4-BUV496, ICOS-BUV563, CD25-BUV615, TIM3-BUV661, CD27-BUV737, CD8-BUV805, CD57-BV421, CXCR5-BV480, Live/Dead-FVS510, CD14-BV570, CD19-BV570, CCR4-BV605, CCR7-BV650, HLA-DR-BV711, CD3-BV750, CD28-BV786, PD1-BB515, LAG3-BB660, CD127-BB700, CD38-BB790, TIGIT-PE, EOMES-PE-CF594, CTLA4-PE-Cy5, FOXP3-PE-Cy5.5, GITR-PE-Cy7, TBET-APC, KI67-AF700, GZMB-APC-Fire750. A list of the markers in the designed panel is provided in Table S2. Samples with very poor quality were pre-identified by the flow specialist (M.A.) and were not included in the analysis.
Pre-gating analysis and quality control
Each Flow Cytometry Standard (FCS) file acquired from the flow cytometry experiments was independently preprocessed using our in-house automated gating pipeline (built with R 4.1.3). The main preprocessing steps include (Figure S8): (1) compensation with matrices exported from FlowJo v10.8.0 software (BD Biosciences), (2) biexponential transformation on all marker channels with parameters extra negative decades = 0.5, width basis = −30, positive decades = 4.5, (3) quality control via the R package flowAI (v1.22.0),34 and (4) pre-gating up to CD3+ T cells via the R package openCyto (v2.4.0).35 The pre-gating strategy is detailed in Table S3: a modified version of the T cell gating template originally provided in the openCyto R package.
For each marker, we carefully checked the consistency of transformed intensity values across all patient samples, for evaluating the possible batch effects. We downsampled 10k cells from each sample and performed UMAP visualization and clustering analysis on the downsampled data, the same procedure as described in the following clustering analysis section. We visually assessed the UMAP plots and observed no significant batch effect in this cohort. Three samples were excluded in the following analysis due to a lack of cells (<10k cells) for accurate clustering and frequency calculations.
Clustering analysis
UMAP visualization (min.dist = 0.1) and clustering analysis were performed via Seurat R package (v4.0)36 on pre-gated T cells (CD14−CD19−, CD3+) pooled from all samples. Note that the Seurat R package is specifically designed for analyzing scRNA-seq data. However, we utilized its embedded functions, including PCA, UMAP, and the Louvain method, for our data analysis. The unique preprocessing steps for flow cytometry data have been described in the previous section prior to visualization and clustering analysis. The expression of each marker was further scaled to mean 0 and variance 1. Both UMAP and clustering analysis were conducted based on the 26 principal components (PCs), using the scaled and transformed intensity values of all 27 markers as input. The first 26 PCs contribute 99.43% variation of the data. We used the Louvain algorithm, a graph-based clustering method that identifies cell clusters or modules from a Shared-Nearest Neighbor (SNN) graph, a variant of the K-Nearest Neighbor (KNN) graph. We set K = 5 for constructing the SNN graph since it is computationally feasible for over 10 million cells. Clustering on a KNN graph with a larger K is computationally intensive and results in many tiny clusters. We ran the clustering algorithms with different resolutions (resolution = 0.5, 0.8, 1.0, 1.2, 1.5, 2, 2.5, 3) and obtained the best clustering result from 10 random starts under each resolution. The range of clustering resolution was set from 0.5 to 3, slightly higher than the typical range (0.4–1.4) recommended by the Seurat package, since the optimum resolution usually increases for larger datasets.36
We chose the clustering solution under resolution 1.5 with the highest average Silhouette scores.41 Heatmap was used to show the average (scaled) marker expression of each individual cluster. Clusters of less than 0.1% abundance were not displayed in both UMAP and heatmap to increase the clarity of the figures. We did not include clusters with very low abundance since there is not enough evidence to support that they are real and not generated by technical noises. Moreover, there is no evidence that the low-frequency T cell subpopulations show clinical or biological interests in our analysis. We manually annotated the 20 major T cell clusters (abundance >0.1%) out of 35 clusters in total. We use the main cell lineage markers, CD4, CD8, FoxP3, CCR7, and CD45RA to annotate main T cell clusters: T regulatory (CD4+FoxP3+), CD4/CD8 T naive (CCR7+CD45RA+), CD4/CD8 T effector memory (CCR7-CD45RA-), CD4/CD8 T central memory (CCR7+CD45RA-), and CD4/CD8 effector memory T cells re-expressing CD45RA (CCR7-CD45RA+) clusters. These markers are commonly used to manually gate functional T cell subsets in flow cytometry analysis. For better visualization, UMAP was rerun for each individual patient with different parameter settings (min.dist = 0.3).
Latent Dirichlet Allocation
LDA is a generative model that helps to identify hidden structures that explain why some parts of the data are similar. We briefly describe the model and its application to the flow cytometry data below and refer readers to the original paper for more details.14
The LDA models the clustered flow cytometry data by considering cells as words, flow samples as documents, and topics as biological profiles or processes. Suppose there are V T cell types (clusters) identified across M samples from S patients. Let represent the nth cell in the dth sample classified to the vth cell types (clusters). The LDA model assumes each sample has fractional membership across K underlying topics and word in samples is generated from th topic, where are latent variables. In LDA, each sample can be explained by the following generative process (Figure 1E).
For each sample d,
-
a)
Choose sample proportion α.
-
b)For each cell in sample d:
-
i)Choose a topic d,
-
ii)Choose a cell conditional on the topic ,
-
i)
are mixing proportions of sample d over K underlying topics and each topic is characterized as a distribution over V T cell types (clusters), where denote the weights in the kth topic over V T cell types (clusters).
In practice, we use the formulation that marginalizes over the . Setting , the cell count of the vth cell type in the dth sample, the marginal distribution for each sample d is
where denote weights of all topics.
Model fitting
Gibbs sampling implemented in R package topicmodels (v0.2-12)38 was used for inferring the two sets of parameters for the LDA model: , a matrix, and , a matrix. We used the following setting for Gibbs sampling: iter = 1000, burnin = 1000, thin = 100 (1000 Gibbs sampling draws are made with the first 1000 iterations discarded and then every 100th iteration kept). To evaluate the model reproducibility, we repeated the algorithm ten times and the results of multiple runs are consistent (Figure S9).
The number of topics K needs to be selected before running the algorithm and it is a model selection problem. There is no ``right’’ answer to the number of topics that are the most appropriate for data.42 We failed to select the number of topics with a 10-fold cross-validation, likely a reflection of the size of the dataset (only 138 samples). Thus, we guided the choice of the number of topics based on what is most useful for scientific interpretation. Moreover, during the cross-validation, we observed the biggest drop in perplexity38 (a measure of how successfully a trained topic model predicts new data) is between K = 2 and K = 3. Thus we set K = 3 in this study.
Lift statistic
We are interested in representatives, clusters that are primarily associated with a single topic. We use metric lift,20 a popular metric for ranking words within single topics in text analysis, to select representative clusters with the following formula
where is the empirical frequency of the vth T cell type in data, with being the size of the vth T cell type in the dth sample. The lift metric gives higher weights to cell types that appear less frequently in other topics.
Survival and response analysis
For each cluster, we also tested its association to time, clinical outcomes (response, toxicity), and immunotypes via the nonparametric test in nparLD R package (v2.1),37 which is designed for longitudinal data in factorial experiments. The same method was used to test the association of the ratio (CD8 Temra/Tn), topic proportions to patient clinical outcomes or immunotypes. Only patients with all three time points (n= 37) were included since the package does not support missing data. We included p-values from ANOVA-type tests provided by the nparLD R package. For main effects (e.g. immunotypes, response, toxicity) involving only the whole-plot factors, p-values were provided with modified ANOVA-type tests with an adjusted degree of freedom. The Kaplan-Meier method was used for survival estimation and the log-rank test was used for comparisons with the help of survminer R package (v0.4.9).43 Wilcoxon rank-sum test was performed when comparing topic proportions or cluster abundances at single time point. All p-values from multiple comparisons were adjusted by the Benjamini-Hochberg method with a false discovery rate controlled at 5%.
Identification of patient subgroups
Patients were grouped by hierarchical clustering (hclust () function with default parameters in R) on their estimated sample topic proportions . Heatmap was drawn to display the sample topic proportions for each patient, as well as clinical outcomes (response, toxicity) and immunotypes, using the ComplexHeatmap R package (v2.10.0).44 Boxplot was used to show the dynamics of sample proportions of the three topics within each patient group. One patient (17-162-08) with only one sample at time point A was excluded from the heatmap and the boxplot. Chi-squared tests were performed to test the association between patient subgroups and clinical outcomes (response, toxicity).
Additional resources
The samples were obtained from a clinical trial study registered under the number NCT03122522, with results published in Postow et al.15
Acknowledgments
We are thankful for computational support from MSK-MIND. We thank Jedd. D. Wolchok for help and support on this project. We also thank Nicole Rusk for reviewing and editing the manuscript. This work is supported in part by the MSKCC Society, the V Foundation, the Parker Institute for Cancer Immunotherapy, NIH P30 CA008748, NIH R01 CA276286, and the MSK-MIND consortium.
Author contributions
X.P. contributed to the original draft and performed bioinformatics data analysis. X.P., R.S., and K.S.P. conceived and designed the algorithm. J.L. contributed to the pre-gating analysis of flow cytometry data. R.S., K.S.P., and M.K.C. developed the initial study concept and oversaw all data generation and analysis. C.M. extracted and analyzed clinical data. M.A. generated and analyzed flow cytometry data. M.A.P. contributed to the initial concept and to the acquisition and analysis of clinical data. X.P., J.L., R.S., K.S.P., and M.K.C. reviewed and edited the manuscript. All authors reviewed and approved the final manuscript.
Declaration of interests
M.K.C. reports research support (for MSK) from Bristol Myers Squibb (BMS) and consulting fees from Merck, InCyte, Moderna, ImmunoCore, BMS, and AstraZeneca. M.A.P. is a consultant, advisor, and speaker for BMS, Chugai, Eisai, Merck, Nektar, Novartis, Pfizer, and Replimune. M.A.P. receives research funding from BMS, Infinity, Merck, Novartis, and RGenix.
Published: August 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100546.
Contributor Information
Margaret K. Callahan, Email: callaham@mskcc.org.
Katherine S. Panageas, Email: panageak@mskcc.org.
Ronglai Shen, Email: shenr@mskcc.org.
Supplemental information
Data and code availability
-
•
Table S4 contains the de-identified clinical and correlative data (flow cytometry clusters) analyzed in this manuscript. Additional data for reproducing figures are available in the repository: https://github.com/xiyupeng/topic_modeling (https://doi.org/10.5281/zenodo.7942456).
-
•
Analysis codes to reproduce this work are available in the repository: https://github.com/xiyupeng/topic_modeling (https://doi.org/10.5281/zenodo.7942456). A detailed tutorial of the method is available on the GitHub page: https://xiyupeng.github.io/LDA_examples/ (https://doi.org/10.5281/zenodo.7942425).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haslam A., Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open. 2019;2:e192535. doi: 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammers H.J., Plimack E.R., Infante J.R., Rini B.I., McDermott D.F., Lewis L.D., Voss M.H., Sharma P., Pal S.K., Razak A.R.A., et al. Safety and Efficacy of Nivolumab in Combination With Ipilimumab in Metastatic Renal Cell Carcinoma: The CheckMate 016 Study. J. Clin. Oncol. 2017;35:3851–3858. doi: 10.1200/JCO.2016.72.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sznol M., Ferrucci P.F., Hogg D., Atkins M.B., Wolter P., Guidoboni M., Lebbé C., Kirkwood J.M., Schachter J., Daniels G.A., et al. Pooled Analysis Safety Profile of Nivolumab and Ipilimumab Combination Therapy in Patients With Advanced Melanoma. J. Clin. Oncol. 2017;35:3815–3822. doi: 10.1200/JCO.2016.72.1167. [DOI] [PubMed] [Google Scholar]
- 5.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.J., Cowey C.L., Lao C.D., Schadendorf D., Dummer R., Smylie M., Rutkowski P., et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Signorelli D., Giannatempo P., Grazia G., Aiello M.M., Bertolini F., Mirabile A., Buti S., Vasile E., Scotti V., Pisapia P., et al. Patients Selection for Immunotherapy in Solid Tumors: Overcome the Naïve Vision of a Single Biomarker. BioMed Res. Int. 2019;2019:e9056417. doi: 10.1155/2019/9056417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Henau O., Rausch M., Winkler D., Campesato L.F., Liu C., Cymerman D.H., Budhu S., Ghosh A., Pink M., Tchaicha J., et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443–447. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitano S., Postow M.A., Ziegler C.G.K., Kuk D., Panageas K.S., Cortez C., Rasalan T., Adamow M., Yuan J., Wong P., et al. Computational algorithm-driven evaluation of monocytic myeloid-derived suppressor cell frequency for prediction of clinical outcomes. Cancer Immunol. Res. 2014;2:812–821. doi: 10.1158/2326-6066.CIR-14-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang A.C., Postow M.A., Orlowski R.J., Mick R., Bengsch B., Manne S., Xu W., Harmon S., Giles J.R., Wenz B., et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spitzer M.H., Nolan G.P. Mass Cytometry: Single Cells, Many Features. Cell. 2016;165:780–791. doi: 10.1016/j.cell.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aghaeepour N., Finak G., FlowCAP Consortium. DREAM Consortium. Hoos H., Mosmann T.R., Brinkman R., Gottardo R., Scheuermann R.H. Critical assessment of automated flow cytometry data analysis techniques. Nat. Methods. 2013;10:228–238. doi: 10.1038/nmeth.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sankaran K., Holmes S.P. Latent variable modeling for the microbiome. Biostatistics. 2019;20:599–614. doi: 10.1093/biostatistics/kxy018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woloszynek S., Mell J.C., Zhao Z., Simpson G., O’Connor M.P., Rosen G.L. Exploring thematic structure and predicted functionality of 16S rRNA amplicon data. PLoS One. 2019;14:e0219235. doi: 10.1371/journal.pone.0219235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blei D.M., Ng A.Y., Jordan M.I. Latent Dirichlet Allocation. J. Mach. Learn. Res. 2003;3:993–1022. [Google Scholar]
- 15.Postow M.A., Goldman D.A., Shoushtari A.N., Betof Warner A., Callahan M.K., Momtaz P., Smithy J.W., Naito E., Cugliari M.K., Raber V., et al. Adaptive Dosing of Nivolumab + Ipilimumab Immunotherapy Based Upon Early, Interim Radiographic Assessment in Advanced Melanoma (The ADAPT-IT Study) J. Clin. Oncol. 2022;40:1059–1067. doi: 10.1200/JCO.21.01570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen R., Postow M.A., Adamow M., Arora A., Hannum M., Maher C., Wong P., Curran M.A., Hollmann T.J., Jia L., et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci. Transl. Med. 2021;13:eabf5107. doi: 10.1126/scitranslmed.abf5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waltman L., van Eck N.J. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B. 2013;86:471. doi: 10.1140/epjb/e2013-40829-0. [DOI] [Google Scholar]
- 18.Oh D.Y., Fong L. Cytotoxic CD4+ T cells in cancer: Expanding the immune effector toolbox. Immunity. 2021;54:2701–2711. doi: 10.1016/j.immuni.2021.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schad S.E., Chow A., Mangarin L., Pan H., Zhang J., Ceglia N., Caushi J.X., Malandro N., Zappasodi R., Gigoux M., et al. Tumor-induced double positive T cells display distinct lineage commitment mechanisms and functions. J. Exp. Med. 2022;219:e20212169. doi: 10.1084/jem.20212169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matt T. In: Proceedings of the Fifteenth International Conference on Artificial Intelligence and Statistics. Lawrence N.D., Girolami M., editors. PMLR; 2012. On Estimation and Selection for Topic Models; pp. 1184–1193. [Google Scholar]
- 21.Blackburn S.D., Shin H., Haining W.N., Zou T., Workman C.J., Polley A., Betts M.R., Freeman G.J., Vignali D.A.A., Wherry E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Twyman-Saint Victor C., Rech A.J., Maity A., Rengan R., Pauken K.E., Stelekati E., Benci J.L., Xu B., Dada H., Odorizzi P.M., et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh D.Y., Kwek S.S., Raju S.S., Li T., McCarthy E., Chow E., Aran D., Ilano A., Pai C.-C.S., Rancan C., et al. Intratumoral CD4+ T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell. 2020;181:1612–1625.e13. doi: 10.1016/j.cell.2020.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salehi S., Kabeer F., Ceglia N., Andronescu M., Williams M.J., Campbell K.R., Masud T., Wang B., Biele J., Brimhall J., et al. Clonal fitness inferred from time-series modelling of single-cell cancer genomes. Nature. 2021;595:585–590. doi: 10.1038/s41586-021-03648-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joseph T.A., Shenhav L., Xavier J.B., Halperin E., Pe’er I. Compositional Lotka-Volterra describes microbial dynamics in the simplex. PLoS Comput. Biol. 2020;16:e1007917. doi: 10.1371/journal.pcbi.1007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts M.E., Stewart B.M., Airoldi E.M. A Model of Text for Experimentation in the Social Sciences. J. Am. Stat. Assoc. 2016;111:988–1003. doi: 10.1080/01621459.2016.1141684. [DOI] [Google Scholar]
- 27.Roberts M.E., Stewart B.M., Tingley D. stm: An R Package for Structural Topic Models. J. Stat. Softw. 2019;91:1–40. doi: 10.18637/jss.v091.i02. [DOI] [Google Scholar]
- 28.Blei D.M., Lafferty J.D. Proceedings of the 23rd international conference on Machine learning ICML ’06. Association for Computing Machinery; 2006. Dynamic topic models; pp. 113–120. [DOI] [Google Scholar]
- 29.Wu X., Wu H., Wu Z. Penalized Latent Dirichlet Allocation Model in Single-Cell RNA Sequencing. Stat. Biosci. 2021;13:543–562. [Google Scholar]
- 30.Xue R., Zhang Q., Cao Q., Kong R., Xiang X., Liu H., Feng M., Wang F., Cheng J., Li Z., et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022;612:141–147. doi: 10.1038/s41586-022-05400-x. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z., Soifer I., Hilton H., Keren L., Jojic V. Modeling Multiplexed Images with Spatial-LDA Reveals Novel Tissue Microenvironments. J. Comput. Biol. 2020;27:1204–1218. doi: 10.1089/cmb.2019.0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spakowicz D., Lou S., Barron B., Gomez J.L., Li T., Liu Q., Grant N., Yan X., Hoyd R., Weinstock G., et al. Approaches for integrating heterogeneous RNA-seq data reveal cross-talk between microbes and genes in asthmatic patients. Genome Biol. 2020;21:150. doi: 10.1186/s13059-020-02033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Funnell T., Zhang A.W., Grewal D., McKinney S., Bashashati A., Wang Y.K., Shah S.P. Integrated structural variation and point mutation signatures in cancer genomes using correlated topic models. PLoS Comput. Biol. 2019;15:e1006799. doi: 10.1371/journal.pcbi.1006799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monaco G., Chen H., Poidinger M., Chen J., de Magalhães J.P., Larbi A. flowAI: automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics. 2016;32:2473–2480. doi: 10.1093/bioinformatics/btw191. [DOI] [PubMed] [Google Scholar]
- 35.Finak G., Frelinger J., Jiang W., Newell E.W., Ramey J., Davis M.M., Kalams S.A., De Rosa S.C., Gottardo R. OpenCyto: An Open Source Infrastructure for Scalable, Robust, Reproducible, and Automated, End-to-End Flow Cytometry Data Analysis. PLoS Comput. Biol. 2014;10:e1003806. doi: 10.1371/journal.pcbi.1003806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noguchi K., Gel Y.R., Brunner E., Konietschke F. nparLD: An R Software Package for the Nonparametric Analysis of Longitudinal Data in Factorial Experiments. J. Stat. Softw. 2012;50:1–23. doi: 10.18637/jss.v050.i12. [DOI] [Google Scholar]
- 38.Grün B., Hornik K. topicmodels: An R Package for Fitting Topic Models. J. Stat. Softw. 2011;40:1–30. doi: 10.18637/jss.v040.i13. [DOI] [Google Scholar]
- 39.Segal N.H., Cercek A., Ku G., Wu A.J., Rimner A., Khalil D.N., Reidy-Lagunes D., Cuaron J., Yang T.J., Weiser M.R., et al. Phase II Single-arm Study of Durvalumab and Tremelimumab with Concurrent Radiotherapy in Patients with Mismatch Repair–proficient Metastatic Colorectal Cancer. Clin. Cancer Res. 2021;27:2200–2208. doi: 10.1158/1078-0432.CCR-20-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andrews M.C., Duong C.P.M., Gopalakrishnan V., Iebba V., Chen W.-S., Derosa L., Khan M.A.W., Cogdill A.P., White M.G., Wong M.C., et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 2021;27:1432–1441. doi: 10.1038/s41591-021-01406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rousseeuw P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987;20:53–65. doi: 10.1016/0377-0427(87)90125-7. [DOI] [Google Scholar]
- 42.Grimmer J., Stewart B.M. Text as Data: The Promise and Pitfalls of Automatic Content Analysis Methods for Political Texts. Polit. Anal. 2013;21:267–297. doi: 10.1093/pan/mps028. [DOI] [Google Scholar]
- 43.Kassambara A., Kosinski M., Biecek P. 2021. Survminer: Drawing Survival Curves Using “Ggplot2”. [Google Scholar]
- 44.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Table S4 contains the de-identified clinical and correlative data (flow cytometry clusters) analyzed in this manuscript. Additional data for reproducing figures are available in the repository: https://github.com/xiyupeng/topic_modeling (https://doi.org/10.5281/zenodo.7942456).
-
•
Analysis codes to reproduce this work are available in the repository: https://github.com/xiyupeng/topic_modeling (https://doi.org/10.5281/zenodo.7942456). A detailed tutorial of the method is available on the GitHub page: https://xiyupeng.github.io/LDA_examples/ (https://doi.org/10.5281/zenodo.7942425).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.