

Summary
The increasing use of monoclonal antibodies (mAbs) in biology and medicine necessitates efficient methods for characterizing their binding epitopes. Here, we developed a high-throughput antibody footprinting method based on binding profiles. We used an antigen microarray to profile 23 human anti-influenza hemagglutinin (HA) mAbs using HA proteins of 43 human influenza strains isolated between 1918 and 2018. We showed that the mAb’s binding profile can be used to characterize its influenza subtype specificity, binding region, and binding site. We present mAb-Patch—an epitope prediction method that is based on a mAb’s binding profile and the 3D structure of its antigen. mAb-Patch was evaluated using four mAbs with known solved mAb-HA structures. mAb-Patch identifies over 67% of the true epitope when considering only 50–60 positions along the antigen. Our work provides proof of concept for utilizing antibody binding profiles to screen large panels of mAbs and to down-select antibodies for further functional studies.
Keywords: monoclonal antibody, antibody profiling, antibody characterization, antigenic cartography, antibody footprints, epitope prediction, mAb-Patch
Graphical abstract
Highlights
-
•
Antibody footprinting can be used to assess breadth and specificity of mAbs
-
•
We develop mAb-Patch, an algorithm for mAb epitope prediction using its binding profile
-
•
Influenza mAbs can be characterized using binding profiles to multiple influenza strains
Motivation
Monoclonal antibodies (mAbs) have gained significant importance in the field of biology and medicine, as they have been utilized in a wide range of applications such as diagnostic methods, therapeutic interventions, and vaccination strategies. Recent advances have enabled the rapid high-throughput isolation of antigen-specific mAbs. However, characterizing the binding and functional properties of mAbs, and in particular their binding epitope, is time consuming and cannot be readily scaled to study thousands or even hundreds of mAbs.
Azulay et al. describe a method for antibody footprinting based on antibody binding profiles measured using an antigen microarray. By comparing the amino acid distributions of strains that a mAb binds vs. those that it does not bind, the authors develop mAb-Patch—an algorithm for predicting the antibody’s epitope.
Introduction
Monoclonal antibodies (mAbs) play an increasingly key role in many fields of biology and medicine such as diagnosis, treatment, and vaccination. During the ongoing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, mAbs were authorized for clinical use both for passive immunization and for treatment following infection.1 While techniques for isolating mAbs were developed over nearly 50 years ago, recently, there have been a variety of novel single-cell technologies that allow rapid isolation of thousands of individual B cells in a single experiment.2,3,4 More recently, isolation of antigen-specific mAbs has been described.5,6,7
Common methods for mAb characterization include binding assays such as ELISA8 and functional assays such as neutralization assays9 and hemagglutinin inhibition assays (HAIs).10 These assays have been widely used to identify broadly neutralizing antibodies, some of which are now undergoing clinical testing.11,12,13 We have previously shown that an antigen microarray (AM)-based binding assay can be used for high-throughput profiling of antibodies and that the array-based binding profiles are comparable to those measured by ELISA.14
Determining the epitope targeted by a specific antibody, epitope mapping, has multiple applications including designing antibodies with improved affinity, identifying antibodies that target a specific antigenic site, and understanding mutations that lead to viral immune escape.15,16,17,18 Epitope mapping can be done by structure determination, including X-ray crystallography and cryoelectron microscopy.19 While both of these techniques provide high-resolution data on the antibody interaction with the antigen, they are technically challenging, time consuming, and expensive. Other experimental methods include peptide-based approaches,20 alanine scanning,21 epitope binning using competitive immunoassays such as ELISA, and bio-layer interferometry.22 In silico methods for epitope prediction include (1) methods that are not antibody specific, i.e., they predict which regions on the antigen can be part of any epitope,23,24,25,26,27 and (2) computational docking methods that require structures of the antigen and antibody separately. Overall, these methods have demonstrated limited success.28,29,30
Virus evolution is driven in part by immune pressure, and multiple studies have shown that viruses evade antibody responses by mutating key sites that are targeted by antibodies.31,32,33,34,35 In particular, the continuous evolution of influenza viruses, also known as antigenic drift, primarily leads to escape from antibodies to previous infections and vaccines and requires annual updates of the influenza vaccine. Considerable research has focused on the characterization of influenza-specific mAbs,36,37,38,39,40,41,42,43,44,45,46,47,48,49 allowing the definition of antigenic sites on the HA surface glycoprotein—which represent highly variable sites that are immunogenic—i.e., targeted by anti-influenza antibodies.
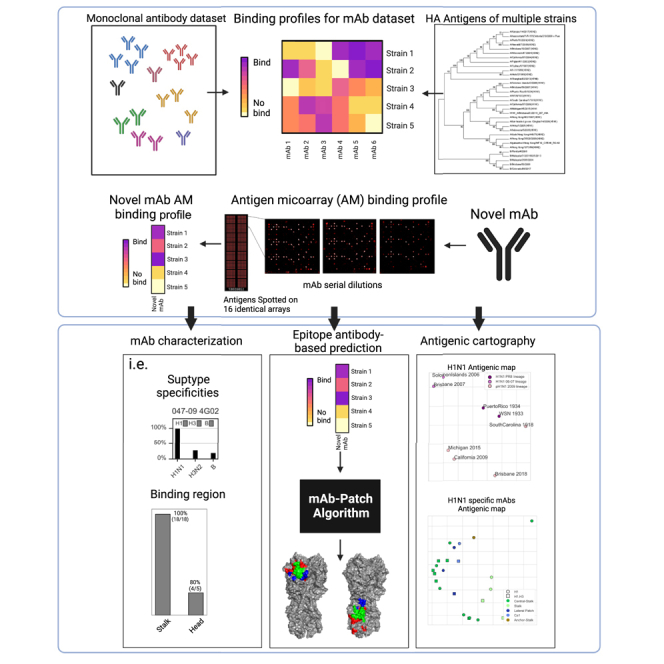
Here, we present a method for mapping the mAb footprints that is based on their binding profiles to a large number of related antigens. Specifically, we characterize a set of 23 influenza A/H1N1 mAbs using a set of 43 recombinant influenza HA proteins from multiple influenza subtypes using AMs, which allow measuring the binding propensity of a single antibody to all 43 HAs using a single assay. We show that the AM binding profile coupled with a database of previously characterized mAbs can be used to infer various properties of these mAbs including their subtype specificity, breadth, binding region, and binding site. We also show that the AM binding profiles can be used to infer antigenic cartography. Finally, we present mAb-Patch—an epitope prediction method that uses the antigen structure and an antibody’s binding profile to predict its contacts on the antigen. Using four HA-mAb complexes with solved structures, we show that mAb-Patch obtains reliable predictions with area under the curve (AUC) scores ranging from 0.7 to 0.82.
Results
Characterizing the binding profiles of human mAbs using an AM-based assay
We used a panel of 23 previously characterized anti-H1 mAbs (Figure S1A), which included both stalk- and head-based antibodies. The binding profiles of these mAbs to the A/H1, A/H3, and B influenza subtypes were previously characterized using ELISA,36,37,38,39,40,41,42,43,44,45,46,47,48,49 as well as their binding region (head or stalk) and binding site on the HA protein (see Table S1). We used AMs spotted with 43 recombinant HA proteins from multiple subtypes to generate binding profiles for each mAb. Each mAb was profiled using 3 serial dilutions: 6, 1.5, and 0.375 μg/mL. Binding profiles and subtype specificities for each strain were calculated based on the AUC statistic across the different dilutions. We used the complete-linkage clustering algorithm50 to cluster the mAbs based on their HA antigen binding profile and identified 4 major clusters of mAbs (Figure 1). The first cluster contains stalk-specific mAbs that target unknown epitopes. The second cluster contains mAbs that target the central stalk epitope and have a broad cross-reactive binding profile across all subtypes. The third cluster contains weakly binding central mAbs, an HA head-binding mAb (008–10 5G04), and an anchor epitope-binding mAb (SFV009-3D04). The fourth cluster includes HA head-binding mAbs.
Figure 1.

Binding profiles of mAbs generated using the influenza antigen microarray (AM)
Binding profiles of 23 influenza mAbs to a panel of 43 influenza recombinant HA proteins from influenza A/H1N1, A/H3N2, B, A/H5N1, A/H7N9, and A/H9N2 subtypes. Each mAb was profiled in multiple dilutions ranging from 6 to 0.375 μg/mL. For each mAb antigen pair, the area under the curve (AUC) is presented, with higher AUC values in darker colors. AUC values were calculated from the median fluorescence intensities (MFI) across all dilutions. AUC values that were smaller than 100 were considered background (light gray). The top bars represent the binding site and the binding profile of each mAb as previously characterized (see Table S1). mAbs are colored by epitope specificities as described in the legend. mAbs colored in dark green, light green, and brown bind the HA stalk, and those colored in dark blue and light blue bind the HA head. mAbs were clustered based on their binding profiles as represented by the dendrogram on the top of the figure. Four major clusters were identified that overall separate mAbs by their binding sites, other than the central stalk antibodies, which separate into two distinct clusters separated by binding breadth.
Antibody influenza subtype specificities can be inferred using binding profiles
To determine the subtype specificities of each mAb, we calculated the breadth summary statistic, which corresponds to the percentage of strains from a given subtype that an antibody binds. We compared these subtype profiles to previously characterized subtype specificities determined using ELISA with individual strains from the A/H1, A/H3, and B subtypes (Figures 2A, and S2). We found that 10/23 of the mAbs had binding profiles that were in perfect agreement with previously published binding data.36,37,38,39,40,41,42,43,44,45,46,47,48,49 However, 8/23 showed additional cross-reactivity to a single subtype that was not previously described, while 4/23 showed cross-reactivity to two additional subtypes. A single mAb that was previously shown to bind all three subtypes (047-09 4E01) only bound the H1 subtype (Figure 2B). To further examine the mAbs that exhibited broader binding profiles than previously reported, we selected 5 of the 12 mAbs with broad binding specificities using our AM assay and tested their binding to two H3 antigens and two B antigens using ELISA. We found that in all cases, mAbs that were found to bind these antigens using the AM also bound these antigens via ELISA (Figure 2C). We also found that H1-specific mAbs exhibited significantly lower breadth than mAbs that bound multiple subtypes (Figure 2D; p = 0.033, Wilcoxon rank-sum test). Head-specific mAbs had lower breadth as compared with stalk-specific mAbs, but this difference was not significant (Figure 2E; p = 0.086, Wilcoxon rank-sum test).
Figure 2.

mAb subtype specificities are broader than previously reported
The set of 23 mAbs studied here was previously profiled using ELISA against representative H1, H3, and B strains (see Table S1).
(A) Comparison of the subtype specificities of 8 mAbs as measured using our AM assay and ELISA (for all mAbs, see Figure S2). Black bars denote the overall binding breadth of each mAb to the A/H1N1, A/H3N2, and B subtypes as measured using our influenza AM. mAb specificities based on previous studies are presented by the gray boxes.
(B) Summary of comparisons of the AM binding profiles with previously known profiles. We found that for 12/23 mAbs, AM binding profiles were broader than those previously reported.
(C) Validating the mAb binding profiles of 5 of the 12 mAbs with broader AM subtype specificities using ELISA. We selected two H3N2 and two B strains from our antigen arrays and measured binding titers using ELISA (see STAR Methods). We found perfect concordance in the subtype specificities measured using the AM and ELISA.
(D and E) Antibody breadth is associated with broad subtype specificity and binding region. We computed the breadth summary statistic for each mAb, defined as the percentage of strains to which it binds (see STAR Methods), and tested for associations with various HA influenza antibody binding characteristics (see Table 2).
(D) Breadth of single subtype (H1N1 only)-binding vs. broad subtype-binding antibodies.
(E) Breadth of stalk- vs. head-binding antibodies.
Black lines represent the median, boxes denote the 25th and 75th percentiles, and error bars represent 1.5 times the interquartile range. Statistical significance was assessed using the Wilcoxon rank-sum test.
mAb binding profiles capture HA subtype similarities
We used the cosine similarity metric over the mAb binding profiles to define the pairwise distances between every pair of HA antigens from the H1N1, H3N2, B, H5N1, H7N9, and H9N2 subtypes (Figure 3A). Using complete-linkage clustering, we found 2 main clusters: the first contained only influenza A group 1 strains (H1N1 and H5N1), and the second contained one subcluster of all H3N2 strains and a second subcluster that contained all B strains, as well as two H9N2 and two H7N9 strains and a single H1N1 strain (H1N1 A/South Carolina/1/1918). We found that B strains from the same lineage (Yamagata and Victoria) had higher similarity as compared with B strains from the other lineage. We also generated a similarity matrix using the Euclidean similarity matrix and obtained similar clusters (Figure S3A).
Figure 3.

Inferring antigenic cartography based on mAb binding antibody profiles
(A and B) The mAb binding profiles across HA strains were used to compute pairwise similarities between influenza strains and monoclonal antibodies using cosine similarity. Dendrograms were computed using the complete-linkage algorithm.
(A) Pairwise cosine similarity matrix of 35 influenza HAs from different subtypes computed over the binding profile of all 23 mAbs to each strain. The top color bar represents the antigenic group of each strain (group 1: H1N1, H5N1, and H9N2; group 2: H3N2 and H7N9).
(B) Pairwise cosine similarity matrix of the 23 mAbs computed using the binding profiles to 35 HA strains (Table S1).
(C and D) Classification accuracy of KNN classification, using k = 3, was used to classify each mAb as follows: (C) binding region (head or stalk) and (D) binding site (central stalk, stalk, lateral patch, and Ca1). A single anchor-stalk-binding mAb was discarded from this analysis.
(E and F) Antigenic cartography maps based on cosine similarity were computed using the Ramacs antigenic cartography package.51
(E) Antigenic cartography of 8 H1N1 strains based on the binding profiles of 23 H1 mAbs. Influenza strains were divided into 3 known antigenic groups, annotated by color: PR8 lineage, SI06 and BRIS07 lineage, and pH1N1 lineage.
(F) Antigenic cartography of 23 influenza mAbs based on their binding profiles to 36 influenza A HA proteins from both human and avian subtypes (Tables 2 and S2). An embedding based on the Euclidean distance metric is provided in Figure S3, and a combined antigenic map of mAbs and antigens is provided in Figure S4. The shape of each mAb represents its known binding profile to H1 and H3 subtypes based on AM binding profiles. The color of each mAb represents its epitope specificity. PR8, Puerto Rico 1934; SI06, Solomon Islands 2006; BRIS07, Brisbane 2007; B Vic, B Victoria lineage; B Yam, B Yamagata lineage.
Binding profiles can be used to discriminate between head vs. stalk antibodies and predict their binding site
We used the binding profiles of each mAb to measure the similarity between every pair of mAbs. Specifically, we used the cosine similarity metric to generate a pairwise similarity matrix between all mAbs. We then applied the complete-linkage clustering algorithm to this matrix (Figure 3B). We identified three main clusters: one that includes all central stalk mAbs and a second that includes stalk mAbs with unknown epitope and a single anchor epitope mAb (SFV009 3D04), as well as a single cross-reactive head mAb (047-09 4G02) that also had high similarity to other head mAbs in cluster 3 (045-09 2B05, SFV15-2F04, and SFV-019 2A02). The third cluster includes all other head mAbs and three additional mAbs with unique binding profiles (008–10 5G04, 030-09 2B03, and 045-09 2B06).
Given the coherence of the clusters obtained using the binding profiles, we assessed whether the antibody binding profiles of the set of mAbs can be used to predict the mAb binding region (head/stalk) of a novel mAb. Specifically, we used a k-nearest neighbor (KNN) classifier, using k = 3, and performance was evaluated using leave-one-out cross-validation. Prediction accuracy was 96% (Figure 3C), with only one head mAb that was misclassified (008-10 5G04). We also conducted the same analysis using the Euclidean similarity metric and obtained a similar prediction accuracy (Figure S3B). We next asked whether the binding profiles could be used for predicting the binding site of an antibody within the head domain (Sa, Sb, Cb, Ca1, and Ca2, receptor-binding site [RBS] and the lateral patch44,52,53) or the stalk domain (central stalk and anchor45). We used the same KNN approach as above. We found that the overall prediction accuracy was 81%. Accuracy was high for all sites but Ca1 (Figure 3D).
Using mAb binding profiles for antigenic cartography
The binding matrix of mAbs to influenza strains can also be used to generate antigenic cartography of both strains and mAbs. Since all of the mAbs included in our dataset were H1N1 specific, we focus on H1N1 antigenic cartography. Specifically, each strain was represented using its binding profile to all mAbs, and we used multidimensional scaling (MDS)54 to embed strains into an antigenic map (Figure 3E). In line with previous findings, we found that the three post-2009 H1N1 pandemic strains clustered together (California 2009, Michigan 2015, and Brisbane 2018), the Puerto Rico 1934 and Wisconsin (WSN) 1933 strains formed another cluster, and the Solomon Islands 2006 and Brisbane 2007 strains also clustered together.55 The South Carolina 1918 pandemic strain was located between the pandemic cluster and the Puerto Rico 1934 and Wisconsin (WSN) 1933 cluster. We also generated an antigenic map of H1 strains using the Euclidean similarity matrix and obtained similar groups (Figure S3C).
We then used the binding profiles of each mAb across all strains spotted on our arrays to generate an antigenic map of the mAbs (Figure 3F). We found that mAbs clustered into three main groups: a group of central stalk mAbs (green mAbs), a group containing mostly head-binding mAbs (blue mAbs), and a group of stalk mAbs with unknown epitope (light green). Most H1-specific mAbs were located on the left side of the map, excluding the four mAbs that exhibited broad binding profiles across all subtypes (051-09 4B02, SFV005-2G02, SC1000-3D04, SC70-5B03). Similar groups were also identified using the Euclidean similarity matrix (Figure S3D). We also generated an antigenic map that included both H1 strains and mAbs based on the binding profiles of all mAbs across H1 strains only (Figure S4). We found that most central stalk mAbs were located in between the three H1 antigenic groups, while the head-specific mAbs were closer to the three pandemic lineage strains.
Epitope prediction using its binding profile across HA strains
Since antigenic escape from antibody binding can in many cases occur by a single mutation within the binding interface of the antibody, we sought to develop a prediction method of an antibody binding interface using its binding profile across HA strains. Specifically, we developed mAb-Patch, a binding patch prediction algorithm that uses the antibody binding profile of a mAb to predict its binding epitope (Figure 4; Box 1). Given a novel mAb with unknown epitope, mAb-Patch receives as input the following: (1) the novel mAb binding profile as measured on the AM, (2) the binding profile of a set of mAbs with known binding regions (e.g., head vs. stalk), and (3) a multiple sequence alignment (MSA) of the set of antigens included in the binding profiles. The method first uses the set of previously characterized mAbs to predict the binding region of the novel mAb (head or stalk) using a KNN classifier. The method then uses the set of isolates to which the novel mAb binds (denoted by Sbound) and the remaining set of antigens to which it does not bind (denoted by Sunbound) to define a position-specific score based on the sequence similarity between all pairs of strains within Sbound and between all pairs of strains between Sbound and Sunbound (Figure 4; STAR Methods). The method only ranks positions from the head or stalk, based on the novel mAb predicted binding site. Intuitively, antibody binding will be affected by mutations within its epitope. Therefore, positions with high sequence similarity within Sbound and low sequence similarity between Sbound and Sunbound may affect antibody binding and as such may also be part of the antibody’s epitope. Since some of the positions within the binding epitope may be highly conserved across all isolates, and since binding footprints are somewhat continuous in 3D space, we also considered the proximal neighborhood of each candidate site (patch) identified using our metric based on our finding that positions that are known to be part of the epitope have significantly smaller geometric mean distances from the patch generated by mAb-Patch (Figure S5).
Figure 4.

The mAb-Patch epitope prediction algorithm
The algorithm receives three inputs: (A) the binding profile of a novel mAb, generated using an AM; (B) AM binding profiles for a set of mAbs with known binding regions (head or stalk) across a set of HA strains, and (C) a multiple sequence alignment of the HA strains included in binding profile (A). Using these inputs, the algorithm includes the following steps: (1) classify binding region (head vs. stalk) of novel mAb using KNN classification using known mAb binding profiles (input B). (2) Binarize binding profile A using a predefined threshold. (3) Use binarized profile to define binding and non-binding HA strains. (4) Compute position-specific score, S(a), based on the ratio between the similarity between all pairs of amino acids from binding strains (Sw) and all pairs of amino acids from binding and non-binding strains (Sb). (5) Rank all positions in binding region by their score (6) Use the top ranked positions to define the seed epitope patch (SEP). (7) Rank all other positions withing the binding region by their geometric mean distance from the center of the SEP. Output-candidate epitope position list based on top ranked positions.
Box 1. mAb-Patch.
| Predict for . Ranked list of candidate epitope positions on the HA antigen. Input: • : mAb binding profile across a set of HA antigens from a given group. • : binding profiles for a set of mAbs with known binding region (head or stalk) across the set of HA strains. • : multiple sequence alignment of the set of HA antigen sequences. • : given parameters where is a given binding threshold, is given parameter to KNN classification, and is seed epitope patch length. (1) Classify binding region (head vs. stalk) of using KNN classification over . (2) Binarize using a given binding threshold : . (3) Define the set of binding HA strains and the set of non-binding strains . (4) Compute position -specific score, , over for all positions within the predicted binding region using the following formula: where , and : number of pairs of sequences of binding strains in . : the number of binding sequences in times the number of non-binding sequences in . : a modified BLOSUM62 distance measure (see STAR Methods) between amino acid in position from sequence and sequence in . (5) Rank all positions on the HA region and select the top positions as the seed epitope patch, . (6) Rank all non- remaining positions on the HA binding region using the geometric mean distance to the center to obtain final ranked list of all HA binding region positions. Output: ranked list of candidate epitope positions within the HA binding region. |
To evaluate mAb-Patch, we used four mAbs with solved 3D structures included in our dataset: two head mAbs (047-09 4G02, 045-09 2B05) and two stalk mAbs (CR-9114, FI6). For each mAb, we used the average binding profiles across two experiments, which included different mAb dilutions (see STAR Methods). Performance was assessed using receiver operating characteristic (ROC) curves with AUC scores ranging from 0.7 to 0.82 (Figure 5). Specifically, we found that within the top 25% of all ranked positions (n = 52 for stalk and n = 60 for head), mAb-Patch identified 67%–82% of the true epitope (Table 1). We also compared the performance of mAb-Patch to that of the SA/nMIproxsum/PS method—a previously published method for antibody epitope prediction that utilized a mutual-information-based score over antibody neutralization profiles.56 Specifically, we implemented the optimal method reported by Chuang et al. (SA/nMIproxsum/PS method)56 over our antibody binding profiles using the optimal parameters provided. To compare both methods, we computed their true positive (TP) rate at multiple false positive (FP) rates. We found that mAb-Patch outperformed this method across all of the 4 mAbs tested (Table 1).
Figure 5.

Inferring mAb epitope footprints using the mAb-Patch algorithm
We used 4 mAbs with known epitopes based on solved structures of the antibodies in complex with the influenza H1 protein (047-09 4G02, 045-09 2B05, CR-9114, and FI6). The binding profiles were used to define a position-specific score, which estimates the likelihood of the position to belong to the antibody binding epitope (see STAR Methods and Table S2 for strains used in MSA). The score was used to rank all sites on the HA stalk or head. Epitope patch predictions were based on the ranked positions and their proximal neighborhoods in 3D space (see also Figure S5). For each mAb, we calculated the receiver operating characteristic (ROC) curves for epitope position ranked by mAb-Patch. The dashed line represents 25% false positive (FP) rate. The predicted vs. experimentally measured binding footprints of each mAb using a 25% FP rate are visualized on the HA 3D structure of pH1N1 (PDB: 7MEM). Positions colored in green represent positions that were correctly identified (true positives [TPs]), positions in blue represent positions that are part of the solved footprint that were not predicted by our method (false negatives [FNs]), and positions in red represent positions that are not part of the known binding footprint but were identified by our score (FPs).
Table 1.
Prediction accuracy of mAb-Patch and the SA/nMIproxsum/PS method at different false positive rates
| mAb | Antigen region | No. antigen residue | No. epitope residue | Method | TP rate at FP rate of 0.05 | TP rate at FP rate of 0.1 | TP rate at FP rate of 0.15 | TP rate at FP rate of 0.2 | TP rate at FP rate of 0.25 | TP rate at FP rate of 0.3 |
|---|---|---|---|---|---|---|---|---|---|---|
| 045-09 2B05 | head | 238 | 18 | mAb-Patch | 0.06∗ | 0.17∗ | 0.56∗ | 0.61∗ | 0.72∗ | 0.72∗ |
| SA/nMIproxsum/PS | 0.00 | 0.00 | 0.06 | 0.11 | 0.22 | 0.22 | ||||
| 047-09 4G02 | head | 238 | 18 | mAb-Patch | 0.22∗ | 0.33∗ | 0.56∗ | 0.61∗ | 0.67∗ | 0.67∗ |
| SA/nMIproxsum/PS | 0.11 | 0.17 | 0.39 | 0.39 | 0.61 | 0.61 | ||||
| CR-9114 | stalk | 210 | 19 | mAb-Patch | 0.11 | 0.26 | 0.53∗ | 0.68∗ | 0.74∗ | 0.84∗ |
| SA/nMIproxsum/PS | 0.32∗ | 0.32∗ | 0.42 | 0.47 | 0.63 | 0.74 | ||||
| FI6 | stalk | 210 | 19 | mAb-Patch | 0.06∗ | 0.18∗ | 0.47∗ | 0.76∗ | 0.82∗ | 0.82∗ |
| SA/nMIproxsum/PS | 0.00 | 0.11 | 0.32 | 0.47 | 0.47 | 0.58 |
Asterisks (∗) indicate the better method performance.
We also compared mAb-Patch to the Z-dock docking prediction server.57 Specifically, we provided the HA monomer and a single mAb to Z-dock and obtained its top-ranked solution (Figure S6). We found that, overall, Z-dock failed to properly identify the mAb binding location for three out of four mAbs.
Discussion
In this study, we proposed a method for mapping mAb footprints that is based on antibody profiles generated using an AM. To showcase the feasibility of the method, we used a characterized dataset of 23 influenza-specific mAbs that have been previously characterized using ELISA assays and neutralization assays and, in four cases, have been solved.36,37,38,39,40,41,42,43,44,45,46,47,48,49 We found out that the binding profiles were mostly in agreement with the known binding patterns measured via ELISA. We also found that some mAbs that were previously reported to bind only influenza H1 exhibited broad binding patterns, some of which were validated using ELISA. We showed that mAbs targeting the same binding site clustered together based on their binding profiles and that the binding profiles could be used for accurate predictions of the binding region and binding site of each antibody. We then showed that the binding profiles can be used to infer similarities between both influenza strain and mAbs, allowing the generation of antigenic cartography that is in agreement with previously published antigenic maps of H1N1 strains. Finally, we presented mAb-Patch, an epitope prediction algorithm, that uses the antibody profiles to predict the binding patch of a mAb on the HA protein. We showed that mAb-Patch was able to correctly identify over 67% of the true epitope of 4 mAbs with solved structures when considering only 52–60 positions on the antigen.
Our work builds upon the elegant studies that previously characterized all of the 23 mAbs studied here, which utilized a broad set of antibody characterization tools,36,37,38,39,40,41,42,43,44,45,46,47,48,49 allowing us to evaluate the ability of our method to recapitulate some of their known properties. Interestingly, we found that some of the previously reported H1-specific mAbs also bound H3N2 and B strains, to which they were not previously tested. These data suggest that characterizing breadth of an anti-HA mAb using a panel consisting of single H1, H3, and B strains is limited and that the actual breadth should be determined using a much larger panel of antigens. In this respect, the parallel characterization of binding breadth using the approach presented here provides a better alternative than traditional ELISA. In our previous work, we demonstrated that our antigen arrays are indeed comparable to the traditional ELISA, providing a single-shot high-throughput alternative to ELISA.14 While, in principle, our method can also utilize ELISA binding profiles, characterization of the 23 mAbs analyzed here across 43 antigens would require 989 ELISA measurements.
Our work is also related to previous studies that investigated the use of AMs to characterize mAbs.40,41,42,58,59,60,61,62,63 In some of these studies, mAbs were spotted on arrays, and antigens were used as samples.40,41,42 Other studies used the serum polyclonal binding profiles to predict previous infection with pandemic H1N1 influenza.59 We have previously shown that baseline antibody binding profiles to SARS-CoV-2 are correlates of risk for symptomatic SARS-CoV-2 infection.64
Previous studies on mapping the antigenic landscape of influenza using antigenic cartography were based on HAIs.51,65,66 More recently, Einav et al.67 utilized neutralization profiles of mAbs to infer influenza antigenic cartography, and others have used such neutralization profiles for generating antigenic cartography of SARS-CoV-2.68,69 Here, we showed that antigenic cartography could be inferred using binding profiles, which are significantly easier to measure. We showed the similar antigenic maps could be obtained using both the cosine and Euclidean distance metrics. However, there were a few noticeable differences between the two metrics. In particular, we found that the cosine distance metric was more sensitive to changes in the binding pattern of each monoclonal (e.g., mAb 030-09 2B03) and that the Euclidean metric was more sensitive to changes in the overall magnitude of the binding patterns.
Previous work on epitope mapping focused on two major approaches: computational and experimental. Experimentally, studies utilized peptide-based approaches;20 structural biology approaches such as crystal structure and cryoelectron microscopy (cryo-EM); mutagenesis such as point mutations, alanine scanning,212 and deep mutational scanning70,71; and epitope binning-competitive immunoassays such as competitive ELISA, surface plasmon resonance, and bio-layer interferometry.22 On the computational front, multiple methods for in silico prediction of binding epitopes have been proposed,72 using the antigen structure, antibody sequence, antigen-antibody complexes, etc.23,24,72,73,74 We compared our method with a docking-based method and found that it failed to properly identify the binding epitopes of three out of the four mAbs analyzed here. While it is likely that newer approaches that utilize state-of-the-art computational protein-folding prediction methods may provide improved mAb docking, our method offers a clear alternative that is based on experimentally measured binding data.
Another commonly used strategy for epitope mapping of antibody responses is peptide microarrays, which typically include overlapping 15- to 20-mer peptides that span the immunogen of interest.75,76,77,78,79,80 One specific study showed that a peptide microarray signature could be used to predict survival in mice following influenza challenge.81 The major drawback of this approach is that it mainly identifies antibodies that target linear epitopes, and therefore it is not well suited to profile binding of conformational mAbs.
Our work is closely related to the work by Chuang et al.,56 who utilized antibody neutralization profiles of mAbs to predict their binding epitope. Their method also relies on the hypothesis that positions that affect neutralization potency are likely to be more affected by residue changes at epitope positions. While we found that our method outperformed the method proposed by Chuang et al.,56 we note that we were unable to optimize the parameters of this method due to the small set of mAbs with solved structures analyzed here. Importantly, these parameters were optimized over neutralization profiles and not binding profiles. While previous approaches utilized neutralization data for inferring antibody properties and their potential epitope,56,67 our method relies only on binding profiles. A clear advantage of our approach is that it can also be used to characterize non-neutralizing antibodies that may have potent antiviral activity via Fc-mediated functions.82,83,84 A key advantage of our method as compared to characterization using functional assays is the multiplex nature of our AM platform, which allows us to scale linearly with the number of mAbs that are characterized. Previous methods based on functional assays (e.g., neutralization assays) are quadratic in the number of mAbs and antigens and cannot be easily scaled for characterizing large panels of mAbs. More recently, deep mutational scanning was used for identification of escape mutations that affect SARS-CoV-2 therapeutic antibodies.85 A key advantage of our method is that it measures binding to multiple HA strains, many of which include multiple mutations that may be epistatically related.
The HA antigens used here were produced in mammalian cells and should therefore be properly glycosylated. Previous work in influenza and HIV has shown that viruses can shed or add glycans in order to escape from antibody recognition.53,86,87,88,89 Since it is possible to predict glycosylation sites from the antigen sequence, it would indeed be possible to extend our algorithm and also consider the glycosylation of each of our HA antigens and to utilize this information to improve our patch prediction. However, this would require designing a set of HA antigens that differ in glycosylation patterns with which this idea could be further developed.
Given the high accuracy of predicting the binding region of a monoclonal HA antibody (i.e., head vs. stalk), we used this predictor to focus the mAb-Patch search for the binding footprint. We also experimented with directly predicting the epitope from its binding profile and found that, especially for “sticky” mAbs that bound many antigens, this sometimes led to the identification of regions that could not all be bound by the same mAb (i.e., amino acids in both the stalk and head). Given the larger number of antibodies with known binding footprints, it is likely that we could improve this first step by predicting the actual epitope region of a mAb and only then trying to identify its unique binding footprint.
Our work provides proof of concept for the ability to rapidly characterize large panels of mAbs using their binding profiles as a first step in their characterization process. As we demonstrate, many antibody properties can be inferred from their binding profiles across multiple antigens. However, there are clear limitations to our approach, as it does not test the functionality of these mAbs. We propose that utilizing functional assays for mAb characterization should be performed at a later stage after identifying a small set of mAbs with broad binding profiles that may be potentially broadly cross-neutralizing or may have potent Fc-mediated functions.
In summary, here we presented an antibody footprinting method for rapid high-throughput antibody characterization. The mAb-Patch algorithm proposed here combines experimental and computational approaches to predict the antibody epitope. Our results demonstrate the feasibility of this approach. However, more work needs to be done in order to fully optimize our method and to further evaluate its performance. While it is clear that a binding footprint cannot fully recapitulate functional characterization of an antibody, it can be rapidly used to screen large panels of mAbs and to down-select antibodies for further functional studies. While we have demonstrated the ability to rapidly characterize mAbs using their binding profile for anti-influenza HA mAbs, our method can also be readily adapted for other pathogens such as SARS-CoV-2, HIV, and any other RNA viruses with sufficiently diverse viral strains.
Limitations of the study
The provided method is applicable to characterize influenza monoclonals, and additional work is required to extend it to profile monoclonals of other rapidly evolving pathogens such as SARS-CoV-2 and HIV. The data analyzed here are limited to a set of 23 mAbs that were previously extensively characterized and provide proof-of-concept data. Additional work on a larger set of previously uncharacterized mAbs will be required in the future. The mAb-Patch algorithm can only be used to characterize mAbs from rapidly evolving pathogens for which an extensive collection of strains is publicly available.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 008-10 5G04 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| SFV019-2A02 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 045-09 2B05 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | PDB:7MEM |
| 047-09 4G02 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | PDB:5W6G |
| SFV015-2F04 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| SFV009-3D04 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 029-09 4A01 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 030-09 2B03 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 045-09 2B06 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 047-09 4E01 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 051-09 4B02 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 051-09 5A02 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| CR-9114 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | PDB:5CJS |
| FI6 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | PDB:3ZTN |
| SC1000-3D04 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| SC1009-3B05 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| SC70-1F02 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| SC70-5B03 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| SFV005-2G02 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 030-09 1E05 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 047-09 1G05 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 051-09 4A03 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| 051-09 5A01 | contributed by Patrick Wilson and are described in many papers published elsewhere36,37,38,39,40,41,42,43,44,45,46,47,48,49 | N/A |
| Alexa Fluor® 647 affinipure Donkey Anti-Human IgG (H + L) | Jackson ImmunoResearch | Cat#:709-605-149 |
| Peroxidase-AffiniPure Goat Anti-Human IgG (H + L) | Jackson ImmunoResearch | Cat#:109–035-088 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| A/South Carolina/1/1918 H1N1 rHA | IRR | Cat#:FR-692; GenBank:AAD17229.1 |
| A/WSN/1933 H1N1 rHA | Sino | Cat#:11692-V08H; GenBank:ACF54598.1 |
| A/Puerto Rico/8/1934 H1N1 rHA | Sino | Cat#:11684-V08H; GenBank:ABD77675.1 |
| A/Solomon Islands/3/2006 H1N1 rHA | Sino | Cat#:11708-V08H; GenBank:ABU99109.1 |
| A/Brisbane/59/2007 H1N1 rHA | Sino | Cat#:11052-V08H; GenBank:ACA28844.1 |
| A/California/07/2009 H1N1 rHA | Sino | Cat#:11085-V08H; GenBank:ACP41953.1 |
| A/Michigan/45/2015 H1N1 rHA | Sino | Cat#:40567-V08H1; GenBank:APC60198.1 |
| A/Brisbane/02/2018 H1N1 rHA | Native Antigens | Cat#:custom_EPI1440504; GISAID:EPI1440504 |
| A/Aichi/2/1968 H3N2 rHA | Sino | Cat#:11707-V08H; GenBan::AAA43178.1 |
| A/X-31/1968 H3N2 rHA | Sino | Cat#:40059-V08H; GenBank:P03438 |
| A/Sydney/5/1997 H3N2 rHA | Sino | Cat#:40149-V08B; GenBank:ACO95259.1 |
| A/Fujian/411/2002 H3N2 rHA | Sino | Cat#:40120-V08B; GenBank:AFG72823.1 |
| A/California/7/2004 H3N2 rHA | Sino | Cat#:40118-V08B; GenBank:ABW80975.1 |
| A/Wisconsin/67/2005 H3N2 rHA | Sino | Cat#:11972-V08H GenBank:ACF54576.1 |
| A/Brisbane/10/2007 H3N2 rHA | Sino | Cat#:11056-V08H; GenBank:ABW23353.1 |
| A/Hawaii/07/2009 H3N2 rHA | IRR | Cat#:FR-401; GenBank:ACT67781.1 |
| A/Perth/16/2009 H3N2 rHA | Sino | Cat#:40043-V08H; GenBank:ACS71642.1 |
| A/Victoria/210/2009 H3N2 rHA | Sino | Cat#:40058-V08B; GenBank:ADI52838.1 |
| A/Switzerland/9715293/2013 H3N2 rHA | Sino | Cat#:40497-V08B; N/A |
| A/Maryland/26/2014 H3N2 rHA | N/A | N/A; N/A |
| A/Singapore/INFIMH-16-0019/2016 H3N2 rHA | Native Antigens | Cat#:custom_EPI1140322; GISAID::EPI1140322 |
| A/Kansas/14/2017 H3N2 rHA | Native Antigens | Cat#:custom_AVG71503.1; GenBank:AVG71503.1 |
| B/Malaysia/2506/2004 B rHA | Sino | Cat#:11716-V08H; GenBank:ACO05957.1 |
| B/Florida/4/2006 B rHA | Sino | Cat#:11053-V08H; GenBank:ACA33493.1 |
| B/Brisbane/60/2008 B rHA | Sino | Cat#:40016-V08H; GenBank:ACN29380.1 |
| B/Phuket/3073/2013 B rHA | Sino | Cat#:40498-V08B; GenBank:ANK57684.1 |
| B/Colorado/06/2017 B rHA | Native Antigens | Cat#:custom_ARQ85589; GenBank:ARQ85589 |
| A/Hong Kong/483/1997 H5N1 rHA | Sino | Cat#:11689-V08H; GenBank:AAC32099.1 |
| A/Anhui/1/2005 H5N1 rHA | Sino | Cat#:11048-V08H1; GenBank:ABD28180.1 |
| A/Indonesia/5/2005 H5N1 rHA | Sino | Cat#:11060-V08B; GenBank:ABW06108.1 |
| A/Bar-headed goose/Qingi/14/2008 H5N1 rHA | Sino | Cat#:11059-V08B1; GenBank:ACL28277.1 |
| A/Guineafowl/Hong Kong/WF10/1999 H9N2 rHA | Native Antigens | Cat#:FLU-H9N2-HA-100; GenBank:AOT22363.1 |
| A/Hong Kong/1073/1999 H9N2 rHA | Sino | Cat#:11229-V08H; GenBank:NP_859037.1 |
| A/Anhui/1/2013 H7N9 rHA | Sino | Cat#:40103-V08H; N/A |
| A/Shanghai/1/2013 H7N9 rHA | Sino | Cat#:40104-V08H; GenBank:YP_009118475 |
| A/Japan/305/1957 H2N2 rHA | Sino | Cat#:11088-V08H; GenBank:AAA43185.1 |
| A/mallard/Ohio/657/2002 H4N6 rHA | Sino | Cat#:11714-V08H; GenBank:ABI47995.1 |
| A/chicken/Hong Kong/17/1977 H6N4 rHA | Sino | Cat#:40027-V08H; GenBank:CAC84244.1 |
| A/New York/107/2003 H7N2 rHA | IRR | Cat#:FR-69; N\A |
| A/Netherlands/219/2003 H7N7 rHA | Sino | Cat#:11082-V08B; GenBank:AAR02640.1 |
| A/pintail duck/Alberta/114/1979 H8N4 rHA | Sino | Cat#:11722-V08H; GenBank:ABB87729.1 |
| A/mallard/Minnesota/Sg-00194/2007 H10N3 rHA | Sino | Cat#:40184-V08B; GenBank:ACT84107.1 |
| A/mallard/Alberta/294/1977 H11N9 rHA | Sino | Cat#:11704-V08H; GenBank:ABB87228.1 |
| Software and Algorithms | ||
| MUSCLE | EML-EBI | https://doi.org/10.1093/nar/gkh340 |
| In-house python script ncluding mAb-Patch | This paper | http://doi.org/10.5281/zenodo.7947980 |
| PyMOL2 | The PyMOL Molecular Graphics System, Version 2 Schrödinger, LLC | https://pymol.org/2 |
| ZDock Server | Pierce et al.57 | https://doi.org/10.1093/bioinformatics/btu097 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Tomer Hertz (thertz@post.bgu.ac.il).
Materials availability
This study did not generate new unique reagents. Unique code was generated, as detailed below.
Method details
Monoclonal antibody dataset
We used a dataset of twenty-three human anti-HA monoclonal antibodies contributed by Patrick Wilson (affiliation Table S1), which have been previously characterized36,37,38,39,40,41,42,43,44,45,46,47,48,49 in terms of the following properties: (1) subtype specificity; (2) binding region (e.g., head vs. stalk); and (3) binding site. Structures of antibody-HA complexes have been solved for three of these antibodies: 045-09 2B05 PDB:7MEM, 047-09 4G02 PDB:5W6G, CR-9114 PDB:5CJS and FI6 PDB:3ZTN.44,46,49 All the antibodies were anti-H1 antibodies, but some of them also bound antigens from other subtypes as summarized in Table S1and Figure S1.
Antigens
Our influenza AMs were spotted with 43 recombinant hemagglutinin (rHA) proteins from influenza A and B viruses, including 5 different influenza A subtypes and 7 avian subtypes (Tables 2 and S2).
Table 2.
Influenza strains spotted on our influenza antigen microarray
| Virus name | Subtype | Protein | Source | Cat no. | Accession no. |
|---|---|---|---|---|---|
| A/South Carolina/1/1918 | H1N1 | rHA | IRR | FR-692 | AAD17229.1 |
| A/WSN/1933 | H1N1 | rHA | Sino | 11692-V08H | ACF54598.1 |
| A/Puerto Rico/8/1934 | H1N1 | rHA | Sino | 11684-V08H | ABD77675.1 |
| A/Solomon Islands/3/2006 | H1N1 | rHA | Sino | 11708-V08H | ABU99109.1 |
| A/Brisbane/59/2007 | H1N1 | rHA | Sino | 11052-V08H | ACA28844.1 |
| A/California/07/2009 | H1N1 | rHA | Sino | 11085-V08H | ACP41953.1 |
| A/Michigan/45/2015 | H1N1 | rHA | Sino | 40567-V08H1 | APC60198.1 |
| A/Brisbane/02/2018 | H1N1 | rHA | Native Antigens | custom_EPI1440504 | EPI1440504 |
| A/Aichi/2/1968 | H3N2 | rHA | Sino | 11707-V08H | AAA43178.1 |
| A/X-31/1968 | H3N2 | rHA | Sino | 40059-V08H | P03438 |
| A/Sydney/5/1997 | H3N2 | rHA | Sino | 40149-V08B | ACO95259.1 |
| A/Fujian/411/2002 | H3N2 | rHA | Sino | 40120-V08B | AFG72823.1 |
| A/California/7/2004 | H3N2 | rHA | Sino | 40118-V08B | ABW80975.1 |
| A/Wisconsin/67/2005 | H3N2 | rHA | Sino | 11972-V08H | ACF54576.1 |
| A/Brisbane/10/2007 | H3N2 | rHA | Sino | 11056-V08H | ABW23353.1 |
| A/Hawaii/07/2009 | H3N2 | rHA | IRR | FR-401 | ACT67781.1 |
| A/Perth/16/2009 | H3N2 | rHA | Sino | 40043-V08H | ACS71642.1 |
| A/Victoria/210/2009 | H3N2 | rHA | Sino | 40058-V08B | ADI52838.1 |
| A/Switzerland/9715293/2013 | H3N2 | rHA | Sino | 40497-V08B | N/A |
| A/Maryland/26/2014 | H3N2 | rHA | N/A | N/A | N/A |
| A/Singapore/INFIMH-16-0019/2016 | H3N2 | rHA | Native Antigens | custom_EPI1140322 | EPI1140322 |
| A/Kansas/14/2017 | H3N2 | rHA | Native Antigens | custom_AVG71503.1 | AVG71503.1 |
| B/Malaysia/2506/2004 | B | rHA | Sino | 11716-V08H | ACO05957.1 |
| B/Florida/4/2006 | B | rHA | Sino | 11053-V08H | ACA33493.1 |
| B/Brisbane/60/2008 | B | rHA | Sino | 40016-V08H | ACN29380.1 |
| B/Phuket/3073/2013 | B | rHA | Sino | 40498-V08B | ANK57684.1 |
| B/Colorado/06/2017 | B | rHA | Native Antigens | custom_ARQ85589 | ARQ85589 |
| A/Hong Kong/483/1997 | H5N1 | rHA | Sino | 11689-V08H | AAC32099.1 |
| A/Anhui/1/2005 | H5N1 | rHA | Sino | 11048-V08H1 | ABD28180.1 |
| A/Indonesia/5/2005 | H5N1 | rHA | Sino | 11060-V08B | ABW06108.1 |
| A/Bar-headed goose/Qingi/14/2008 | H5N1 | rHA | Sino | 11059-V08B1 | ACL28277.1 |
| A/Guineafowl/Hong Kong/WF10/1999 | H9N2 | rHA | Native Antigens | FLU-H9N2-HA-100 | AOT22363.1 |
| A/Hong Kong/1073/1999 | H9N2 | rHA | Sino | 11229-V08H | NP_859037.1 |
| A/Anhui/1/2013 | H7N9 | rHA | Sino | 40103-V08H | N/A |
| A/Shanghai/1/2013 | H7N9 | rHA | Sino | 40104-V08H | YP_009118475 |
Microarray spotting
Influenza rHA proteins were spotted in a single dilution of 32.5ug/ml. Antigens were spotted in Scispot D1 spotting buffer (Scienion) i onto N-hydroxysuccinimide ester–derivatized Hydrogel slides (H slides) using a Scienion Sx non-contact array spotter. Spot volumes ranged between 300 and 360 pL. Each antigen was spotted in triplicate. Sixteen identical microarrays were spotted on each slide.
Antigen microarray assay
Array slides were blocked with 4 mL of chemical blocking solution (50 mM ethanolamine, 50 mM borate, pH 9.0) for 1 h at room temperature (RT) on a shaker. After blocking the liquid was vacuumed, the slide was washed 2 times for 3 min in a washing buffer (0.05% tween 20 in PBS), 2 times for 3 min in PBS and an additional 3 min wash in double deionized water (DDW). Every wash was performed with 3 mL of liquid per slide on a shaker at RT. mAb samples were diluted in 1% BSA and 0.025% tween 20 in PBS. Each mAb was run in a set of serial dilutions as follows: 6 μg/ml, 1.5ug/ml, 0.375 μg/ml. Slides were dried by centrifugation at RT for 5 min at speed 2000 rpm in a slide holder padded with kim wipes, loaded on divided incubation trays (PepperChips, PepperPrint, Germany), and then the samples were added and hybridized with the arrays for 2h at RT on shaker. After hybridization, the samples were discarded, and the slides were washed in washing buffer X 2 and PBS X 2 as described above. After washes, the slides were incubated for 45 min on shaker at RT with a fluorescently labeled polyclonal secondary anti-human IgG antibody (Alexa Fluor 647 affinipure Donkey Anti-Human IgG (H + L), cat# 709-605-149, Jackson) at 1:1000 dilution in hybridization buffer, depending on the antibody Fc source. To detect bound immunoglobulins, slides were scanned on a three-laser GenePix 4400 scanner. Images were analyzed using GenePix Pro version 7 to obtain the mean fluorescence intensity (MFI) of each spot after subtracting the mean local background fluorescence intensity (0 ≤MFI ≤65,000). Since each antigen was spotted in triplicate, the median intensity of the replicates was calculated.
ELISA
We used an ELISA assay optimized for 384 well plate format for validating some of the binding data obtained using the antigen microarray, focusing on mAbs that bound to antigens from subtypes they were not previously reported to bind. The 384-well white MaxiSorp-coated plates (120 μL wells; catalog number 460372; Thermo Fisher, USA) were coated with 17 μL of 4 μg mL−1 recombinant HA protein per well (diluted in PBS) and incubated overnight at 4°C. Plates were washed five times with PBS-T washing buffer (0.1% Tween 20 in PBS, 60 μL per well) using a plate washer (ELx405 Select Deep-Well Microplate Washer; BioTek, USA). Plates were then blocked with 100 μL of 10% skim milk powder (Sigma, Germany) in PBS-T and incubated for 1 h at 37°C. Following five PBS-T washes, mAbs were run using serial dilutions ranging from 3.75ul/ml - 0.005ul/ml in 2% skimmed milk in PBS-T, and added to the plates in triplicates (30 μL per well) for 1-h incubation at 37°C in the incubator, and then washed five times with PBS-T. The secondary antibody, Peroxidase-AffiniPure Goat Anti-Human IgG (H + L) (catalog number 109–035-088, Jackson ImmunoResearch, USA) was diluted 1:10,000 in 2% skimmed milk in PBS-T and added to the plates (30 μL per well). After incubation for 1 h at 37°C and five additional PBS-T washes, equal volumes of peroxide and luminol were mixed and added (30 μL stable peroxide +30 μL luminol/enhancer per well, SuperSignal West Pico Chemiluminescent Substrate, catalog number 34578, Thermo Fisher). Following 1-min incubation, the luminescence was measured by an ELISA reader (Infinite 200 PRO, TECAN, Switzerland) at 600-nm wavelength. Specifically, plates were coated with 2,H3 antigens: A/Switzerland/9715293/2013 and A/Brisbane/10/2007, and 2 B antigens B/Florida/4/2006, B/Phuket/3073/2013. We tested the following 5 mAbs: 051-09 5A01, 051-09 4B02, 051-09 5A02, SFV009-3D04 and 047-09 1G05. This protocol was tested and verified in previous paper by our group.14
Breadth and magnitude summary statistics
To summarize our array data, we utilized two commonly used summary statistics: breadth and magnitude. We denote the area under the curve (AUC) array measurements by where: i – mAb, = 1, ⋯, N;
a – antigen from influenza antigen microarray, a = 1, ⋯, Na; For a given mAb, i, we define the breadth and magnitude of AUC responses to each antigen a as follows:
-
•
Magnitude - denotes the sum of AUC responses to a given set of antigens :
-
•
Breadth – denotes the number of given antigens with AUC higher than a given threshold in respect to , the maximum AUC response antigen of mAb i.
Computational analyses
Antigen microarray analysis
The array results were analyzed using an in-house python analysis pipeline. All data visualizations were plotted using Matplotlib and Seaborn python packages.
Data normalization
During each experiment, a negative control array was hybridized with the hybridization buffer only. The background staining of the negative control array was subtracted from all other arrays.
Clustering and classification
Agglomerative Hierarchical clustering
The matrix of normalized reactivities of every mAb across the panel of influenza HA proteins spotted on the arrays was used to cluster by complete-linkage clustering algorithm both for mAbs and influenza strains (implemented by the Seaborn clustermap function in python). We used the cosine similarity metric to compute the pairwise similarity between mAbs, as well as the pairwise similarity between HA strains. We also experimented using the Euclidean distance metric as well. Clustering was performed using the complete-linkage clustering algorithm as implemented by the Seaborn clustermap function in python.
Classifying antibody binding sites
To classify antibody binding sites - i.e., head vs. stalk we used a k-nearest neighbor classifier with k = 3, using a leave-one-out cross validation framework. Similarity between each pair of antibodies was defined by the cosine similarity or the Euclidean similarity.50,90
Antigenic cartography and multidimensional-scaling analysis
Antigenic cartography is used to visualize relationships among related strains.51 We generated antigenic cartography of H1N1 influenza HAs and the set of mAbs used in this study, all of which bound to H1. Maps were generated by multidimensional scaling (MDS) on the matrix of antibody profiles of mAbs to the panel of influenza strains spotted on the array. Specifically, we utilized the Racmacs package - an antigenic cartography method originally implemented for HAI-based antigenic cartography.51 Briefly the binding matrix was calculated, and values were log-10 transformed. Antibody profiles were then normalized by the max value across all antigens, and the distance between strains and antibodies were measured using the pairwise cosine similarity or Euclidean distance. The resulting matrix was then embedded into a 2-dimensional space using MDS.
Position binding scores and epitope prediction
To utilize the binding profile of each mAb to predict its epitope, we developed a position specific binding score. We assume that a given antibody will only bind antigens in which its native epitope, or large parts of it, are present. We note however, that even single mutations within the epitope may disrupt antibody binding, as evidenced by multiple studies on immune escape.91,92 Furthermore, in some cases even mutations outside the epitope may lead to conformational changes that will reduce or abrogate binding.93
To identify potential positions that may be part of an antibody’s epitope, we defined a position specific binding score that ranks all positions within a protein domain by their likelihood to belong to the antibody’s epitope. Specifically, our method compares the similarity of amino acids at a given position in all influenza strains that bind a given mAb (Sw), to the similarity between these strains and strains that the mAb does not bind (Sb). The position specific score is defined by: Spos = 1 - Sb/Sw. For more details see Box 1 in results.
Multiple sequence alignment
To define a position-specific binding score, we first generate a multiple sequence alignment (MSA) of a set of 14 H1 proteins (Table S3) using MUSCLE.94
Defining the set of binding strains and non-binding strains for a given mAb
To define the set of HAs that a given mAb binds we used a binding percentile threshold. All strains with AUC above the given percentile were defined as binders. Specifically, a threshold of 10% was used for head binding mAbs and a threshold of 30% was used for stalk binding mAbs.
Defining the head and stalk domains
For head binding mAbs we only considered HA1 positions 60–290, and for stalk mAbs we considered HA1 positions 12–60, 290–332, and HA2 positions 1–175. Non-accessible surface positions were discarded as well in the final ranking.
Modified BLOSUM62 distance matrix
Site-specific binding score was calculated using BLOSUM62 substitution matrix that was modified to measure distance, similar to what was done by Dash et al.95 briefly the matrix was modified as the following.
-
•
The distance between identical amino acids was set to 0
-
•
Distance was normalized to the maximum value of BLOSUM62 substitution matrix (4) maximum, in order to get values from 0 to 1
-
•
Positions that were total conserved were discarded
Binding epitope prediction
To infer the binding epitope using the site-specific binding scores defined above, we sorted all amino acids on the antigen by their similarity scores and analyzed the top 15 ranked positions. For each position, we identified all of its neighbors on the solved structure of the H1N1 HA (PDB:7MEM) using a radius of 7.5A for the head domain and 9A for the stem domain. The final ordered set of candidate positions included the top 15 ranked positions, followed by their list of neighbors, which were further ranked by their geometric mean distance from these 15 ranked positions. Performance was assessed on 4 mAbs with solved structures: 045-09 2B05 (PDB:7MEM),44 047-09 4G02 (PDB:5W6G),44,96 CR-9114 (PDB:5CJS)49 and FI6 (PDB:3ZTN).46 False positive rate vs. True positive rate of mAb-Patch was assessed using Receiver operating characteristic (ROC) curves. The predicted epitope patches were plotted onto the H1 structure using PyMOL2 (Version 2.5.2).
Docking
In order to compare the performance of mAb-Patch docking based methods we used the Z-DOCK server, an interactive docking prediction of protein-protein complexes and symmetric multimers.57 Briefly we used 4 mAbs with solved structures: 045-09 2B05 (PDB:7MEM),44 047-09 4G02 (PDB:5W6G),44,96 CR-9114 (PDB:5CJS)49 and FI6 (PDB:3ZTN).46 PDB structures of H1 HA monomer and the mAb were uploaded separately to the Z-Dock server and for each mAb the top score prediction was used for plotting onto the H1 monomer using PyMOL2 (Version 2.5.2). See Figure S6.
Quantification and statistical analysis
Measurement quantification and statistical analysis methods are described in the star method Details and figure legends. Details such as the specific statistical test used, the sample size (n), and the measured variables are provided in the text or figure legend corresponding to each section.
Acknowledgments
This work was supported by Israel Science Foundation (ISF) grants no. 882/17 and 2683/21. Figures were created using BioRender.com.
Author contributions
Conceptualization, T.H.; methodology, T.H., L.M.F., and A.A.; software, T.H., L.C.-L., A.A., and L.M.F.; formal analysis, A.A., L.C.-L., and L.M.F.; investigation, A.A. and L.M.F.; writing – original draft, T.H. and A.A.; writing – review & editing, T.H., A.A., L.C.-L., L.M.F., and M.A.M.; visualization, A.A.; funding acquisition, T.H.; resources, M.A.M. and T.H.; supervision, T.H. and L.M.F.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 22, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100566.
Supplemental information
Data and code availability
-
•
This paper re-analyzes publicly available datasets that are listed in the key resources table. All other data generated within this study is reported in the paper.
-
•
All original code and files are available via Zenodo: http://doi.org/10.5281/zenodo.7947980. The DOI is also listed in the key resources table.
-
•
Any additional information needed to re-analyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Levin M.J., Ustianowski A., De Wit S., Launay O., Avila M., Seegobin S., Templeton A., Yuan Y., Ambery P., Arends R.H., et al. LB5. PROVENT: Phase 3 Study of Efficacy and Safety of AZD7442 (Tixagevimab/Cilgavimab) for Pre-exposure Prophylaxis of COVID-19 in Adults. Open Forum Infect. Dis. 2021;8 doi: 10.1093/ofid/ofab466.1646. [DOI] [Google Scholar]
- 2.Smith K., Garman L., Wrammert J., Zheng N.-Y., Capra J.D., Ahmed R., Wilson P.C. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat. Protoc. 2009;4:372–384. doi: 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wrammert J., Smith K., Miller J., Langley W.A., Kokko K., Larsen C., Zheng N.-Y., Mays I., Garman L., Helms C., et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guthmiller J.J., Dugan H.L., Neu K.E., Lan L.Y.-L., Wilson P.C. An Efficient Method to Generate Monoclonal Antibodies from Human B Cells. Methods Mol. Biol. 2019;1904:109–145. doi: 10.1007/978-1-4939-8958-4_5. [DOI] [PubMed] [Google Scholar]
- 5.Andreano E., Nicastri E., Paciello I., Pileri P., Manganaro N., Piccini G., Manenti A., Pantano E., Kabanova A., Troisi M., et al. Extremely potent human monoclonal antibodies from COVID-19 convalescent patients. Cell. 2021;184:1821–1835.e16. doi: 10.1016/j.cell.2021.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Setliff I., Shiakolas A.R., Pilewski K.A., Murji A.A., Mapengo R.E., Janowska K., Richardson S., Oosthuysen C., Raju N., Ronsard L., et al. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell. 2019;179:1636–1646.e15. doi: 10.1016/j.cell.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim W., Zhou J.Q., Horvath S.C., Schmitz A.J., Sturtz A.J., Lei T., Liu Z., Kalaidina E., Thapa M., Alsoussi W.B., et al. Germinal centre-driven maturation of B cell response to mRNA vaccination. Nature. 2022;604:141–145. doi: 10.1038/s41586-022-04527-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark M.F., Lister R.M., Bar-Joseph M. Plant Molecular Biology Methods in Enzymology. Elsevier; 1986. ELISA techniques; pp. 742–766. [DOI] [Google Scholar]
- 9.Truelove S., Zhu H., Lessler J., Riley S., Read J.M., Wang S., Kwok K.O., Guan Y., Jiang C.Q., Cummings D.A.T. A comparison of hemagglutination inhibition and neutralization assays for characterizing immunity to seasonal influenza A. Influenza Other Respi. Viruses. 2016;10:518–524. doi: 10.1111/irv.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spackman E., Sitaras I. Hemagglutination Inhibition Assay. Methods Mol. Biol. 2020;2123:11–28. doi: 10.1007/978-1-0716-0346-8_2. [DOI] [PubMed] [Google Scholar]
- 11.Crowell T.A., Colby D.J., Pinyakorn S., Sacdalan C., Pagliuzza A., Intasan J., Benjapornpong K., Tangnaree K., Chomchey N., Kroon E., et al. Safety and efficacy of VRC01 broadly neutralising antibodies in adults with acutely treated HIV (RV397): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. HIV. 2019;6 doi: 10.1016/S2352-3018(19)30053-0. e297–e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pizzuto M.S., Zatta F., Minola A., Peter A., Culap K., Bianchi S., Soriaga L., De Marco A., Guarino B., Passini N., et al. 1231. VIR-2482: A potent and broadly neutralizing antibody for the prophylaxis of influenza A illness. Open Forum Infect. Dis. 2020;7 doi: 10.1093/ofid/ofaa439.1416. S635–S636. [DOI] [Google Scholar]
- 13.Kumar S., Chandele A., Sharma A. Current status of therapeutic monoclonal antibodies against SARS-CoV-2. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy S., Abd Alhadi M., Azulay A., Kahana A., Bujanover N., Gazit R., McGargill M.A., Friedman L.M., Hertz T. FLU-LISA (fluorescence-linked immunosorbent assay): high-throughput antibody profiling using antigen microarrays. Immunol. Cell Biol. 2023;101:231–248. doi: 10.1111/imcb.12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams O., Bonzel L., Kovacevic A., Mayatepek E., Hoehn T., Vogel M. Palivizumab-resistant human respiratory syncytial virus infection in infancy. Clin. Infect. Dis. 2010;51:185–188. doi: 10.1086/653534. [DOI] [PubMed] [Google Scholar]
- 16.Diskin R., Scheid J.F., Marcovecchio P.M., West A.P., Klein F., Gao H., Gnanapragasam P.N.P., Abadir A., Seaman M.S., Nussenzweig M.C., Bjorkman P.J. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science. 2011;334:1289–1293. doi: 10.1126/science.1213782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu C.-M., Peng H.-P., Chen I.-C., Lee Y.-C., Chen J.-B., Tsai K.-C., Chen C.-T., Chang J.-Y., Yang E.-W., Hsu P.-C., et al. Rationalization and design of the complementarity determining region sequences in an antibody-antigen recognition interface. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ofek G., Guenaga F.J., Schief W.R., Skinner J., Baker D., Wyatt R., Kwong P.D. Elicitation of structure-specific antibodies by epitope scaffolds. Proc. Natl. Acad. Sci. USA. 2010;107:17880–17887. doi: 10.1073/pnas.1004728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y., Pan J., Jenni S., Raymond D.D., Caradonna T., Do K.T., Schmidt A.G., Harrison S.C., Grigorieff N. CryoEM Structure of an Influenza Virus Receptor-Binding Site Antibody-Antigen Interface. J. Mol. Biol. 2017;429:1829–1839. doi: 10.1016/j.jmb.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forsström B., Axnäs B.B., Stengele K.-P., Bühler J., Albert T.J., Richmond T.A., Hu F.J., Nilsson P., Hudson E.P., Rockberg J., Uhlen M. Proteome-wide epitope mapping of antibodies using ultra-dense peptide arrays. Mol. Cell. Proteomics. 2014;13:1585–1597. doi: 10.1074/mcp.M113.033308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilvebrant J., Rockberg J. An introduction to epitope mapping. Methods Mol. Biol. 2018;1785:1–10. doi: 10.1007/978-1-4939-7841-0_1. [DOI] [PubMed] [Google Scholar]
- 22.Brooks B.D., Miles A.R., Abdiche Y.N. High-throughput epitope binning of therapeutic monoclonal antibodies: why you need to bin the fridge. Drug Discov. Today. 2014;19:1040–1044. doi: 10.1016/j.drudis.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Haste Andersen P., Nielsen M., Lund O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006;15:2558–2567. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponomarenko J., Bui H.-H., Li W., Fusseder N., Bourne P.E., Sette A., Peters B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinf. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkarni-Kale U., Bhosle S., Kolaskar A.S. CEP: a conformational epitope prediction server. Nucleic Acids Res. 2005;33 doi: 10.1093/nar/gki460. W168-W171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker J.M., Guo D., Hodges R.S. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25:5425–5432. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- 27.Potocnakova L., Bhide M., Pulzova L.B. An Introduction to B-Cell Epitope Mapping and In Silico Epitope Prediction. J. Immunol. Res. 2016;2016 doi: 10.1155/2016/6760830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halperin I., Ma B., Wolfson H., Nussinov R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 29.Vajda S., Camacho C.J. Protein-protein docking: is the glass half-full or half-empty? Trends Biotechnol. 2004;22:110–116. doi: 10.1016/j.tibtech.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Wodak S.J., Méndez R. Prediction of protein-protein interactions: the CAPRI experiment, its evaluation and implications. Curr. Opin. Struct. Biol. 2004;14:242–249. doi: 10.1016/j.sbi.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Wei X., Decker J.M., Wang S., Hui H., Kappes J.C., Wu X., Salazar-Gonzalez J.F., Salazar M.G., Kilby J.M., Saag M.S., et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 32.Wibmer C.K., Bhiman J.N., Gray E.S., Tumba N., Abdool Karim S.S., Williamson C., Morris L., Moore P.L. Viral escape from HIV-1 neutralizing antibodies drives increased plasma neutralization breadth through sequential recognition of multiple epitopes and immunotypes. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doud M.B., Lee J.M., Bloom J.D. How single mutations affect viral escape from broad and narrow antibodies to H1 influenza hemagglutinin. Nat. Commun. 2018;9:1386. doi: 10.1038/s41467-018-03665-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hensley S.E., Das S.R., Bailey A.L., Schmidt L.M., Hickman H.D., Jayaraman A., Viswanathan K., Raman R., Sasisekharan R., Bennink J.R., Yewdell J.W. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science. 2009;326:734–736. doi: 10.1126/science.1178258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eguia R.T., Crawford K.H.D., Stevens-Ayers T., Kelnhofer-Millevolte L., Greninger A.L., Englund J.A., Boeckh M.J., Bloom J.D. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andrews S.F., Huang Y., Kaur K., Popova L.I., Ho I.Y., Pauli N.T., Henry Dunand C.J., Taylor W.M., Lim S., Huang M., et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl. Med. 2015;7 doi: 10.1126/scitranslmed.aad0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wrammert J., Koutsonanos D., Li G.-M., Edupuganti S., Sui J., Morrissey M., McCausland M., Skountzou I., Hornig M., Lipkin W.I., et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 2011;208:181–193. doi: 10.1084/jem.20101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henry Dunand C.J., Leon P.E., Huang M., Choi A., Chromikova V., Ho I.Y., Tan G.S., Cruz J., Hirsh A., Zheng N.-Y., et al. Both Neutralizing and Non-Neutralizing Human H7N9 Influenza Vaccine-Induced Monoclonal Antibodies Confer Protection. Cell Host Microbe. 2016;19:800–813. doi: 10.1016/j.chom.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nachbagauer R., Shore D., Yang H., Johnson S.K., Gabbard J.D., Tompkins S.M., Wrammert J., Wilson P.C., Stevens J., Ahmed R., et al. Broadly Reactive Human Monoclonal Antibodies Elicited following Pandemic H1N1 Influenza Virus Exposure Protect Mice against Highly Pathogenic H5N1 Challenge. J. Virol. 2018;92 doi: 10.1128/JVI.00949-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H. 2019. Development of a Label-free and Multiplex Antibody Microarray Biosensor on the Arrayed Imaging Reflectometry (AIR) Platform for Rapid Detection of Influenza Viruses. [Google Scholar]
- 41.Zhang H., Henry C., Anderson C.S., Nogales A., DeDiego M.L., Bucukovski J., Martinez-Sobrido L., Wilson P.C., Topham D.J., Miller B.L. Crowd on a Chip: Label-Free Human Monoclonal Antibody Arrays for Serotyping Influenza. Anal. Chem. 2018;90:9583–9590. doi: 10.1021/acs.analchem.8b02479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez-Sobrido L., Blanco-Lobo P., Rodriguez L., Fitzgerald T., Zhang H., Nguyen P., Anderson C.S., Holden-Wiltse J., Bandyopadhyay S., Nogales A., et al. Characterizing emerging canine H3 influenza viruses. PLoS Pathog. 2020;16 doi: 10.1371/journal.ppat.1008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guthmiller J.J., Utset H.A., Henry C., Li L., Zheng N.-Y., Sun W., Costa Vieira M., Zost S., Huang M., Hensley S.E., et al. An Egg-Derived Sulfated N-Acetyllactosamine Glycan Is an Antigenic Decoy of Influenza Virus Vaccines. mBio. 2021;12 doi: 10.1128/mBio.00838-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guthmiller J.J., Han J., Li L., Freyn A.W., Liu S.T.H., Stovicek O., Stamper C.T., Dugan H.L., Tepora M.E., Utset H.A., et al. First exposure to the pandemic H1N1 virus induced broadly neutralizing antibodies targeting hemagglutinin head epitopes. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abg4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guthmiller J.J., Han J., Utset H.A., Li L., Lan L.Y.-L., Henry C., Stamper C.T., McMahon M., O’Dell G., Fernández-Quintero M.L., et al. Broadly neutralizing antibodies target a haemagglutinin anchor epitope. Nature. 2022;602:314–320. doi: 10.1038/s41586-021-04356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corti D., Voss J., Gamblin S.J., Codoni G., Macagno A., Jarrossay D., Vachieri S.G., Pinna D., Minola A., Vanzetta F., et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 47.Chen Y.-Q., Lan L.Y.-L., Huang M., Henry C., Wilson P.C. Hemagglutinin Stalk-Reactive Antibodies Interfere with Influenza Virus Neuraminidase Activity by Steric Hindrance. J. Virol. 2019;93 doi: 10.1128/JVI.01526-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dreyfus C., Laursen N.S., Kwaks T., Zuijdgeest D., Khayat R., Ekiert D.C., Lee J.H., Metlagel Z., Bujny M.V., Jongeneelen M., et al. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Impagliazzo A., Milder F., Kuipers H., Wagner M.V., Zhu X., Hoffman R.M.B., van Meersbergen R., Huizingh J., Wanningen P., Verspuij J., et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science. 2015;349:1301–1306. doi: 10.1126/science.aac7263. [DOI] [PubMed] [Google Scholar]
- 50.Duda, Richard O., Hart, Peter E. A Wiley-Interscience Publication; 1973. Pattern Classification and Scene Analysis. [Google Scholar]
- 51.Smith D.J., Lapedes A.S., de Jong J.C., Bestebroer T.M., Rimmelzwaan G.F., Osterhaus A.D.M.E., Fouchier R.A.M. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- 52.Caton A.J., Brownlee G.G., Yewdell J.W., Gerhard W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype) Cell. 1982;31:417–427. doi: 10.1016/0092-8674(82)90135-0. [DOI] [PubMed] [Google Scholar]
- 53.Wu N.C., Wilson I.A. Influenza hemagglutinin structures and antibody recognition. Cold Spring Harb. Perspect. Med. 2020;10 doi: 10.1101/cshperspect.a038778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lapedes A., Farber R. The geometry of shape space: application to influenza. J. Theor. Biol. 2001;212:57–69. doi: 10.1006/jtbi.2001.2347. [DOI] [PubMed] [Google Scholar]
- 55.Anderson C.S., McCall P.R., Stern H.A., Yang H., Topham D.J. Antigenic cartography of H1N1 influenza viruses using sequence-based antigenic distance calculation. BMC Bioinf. 2018;19:51. doi: 10.1186/s12859-018-2042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chuang G.-Y., Acharya P., Schmidt S.D., Yang Y., Louder M.K., Zhou T., Kwon Y.D., Pancera M., Bailer R.T., Doria-Rose N.A., et al. Residue-level prediction of HIV-1 antibody epitopes based on neutralization of diverse viral strains. J. Virol. 2013;87:10047–10058. doi: 10.1128/JVI.00984-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pierce B.G., Wiehe K., Hwang H., Kim B.-H., Vreven T., Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakajima R., Supnet M., Jasinskas A., Jain A., Taghavian O., Obiero J., Milton D.K., Chen W.H., Grantham M., Webby R., et al. Protein Microarray Analysis of the Specificity and Cross-Reactivity of Influenza Virus Hemagglutinin-Specific Antibodies. mSphere. 2018;3 doi: 10.1128/mSphere.00592-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.te Beest D., de Bruin E., Imholz S., Wallinga J., Teunis P., Koopmans M., van Boven M. Discrimination of influenza infection (A/2009 H1N1) from prior exposure by antibody protein microarray analysis. PLoS One. 2014;9 doi: 10.1371/journal.pone.0113021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davies D.H., Liang X., Hernandez J.E., Randall A., Hirst S., Mu Y., Romero K.M., Nguyen T.T., Kalantari-Dehaghi M., Crotty S., et al. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc. Natl. Acad. Sci. USA. 2005;102:547–552. doi: 10.1073/pnas.0408782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meade P., Latorre-Margalef N., Stallknecht D.E., Krammer F. Development of an influenza virus protein microarray to measure the humoral response to influenza virus infection in mallards. Emerg. Microbes Infect. 2017;6 doi: 10.1038/emi.2017.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dotsey E.Y., Gorlani A., Ingale S., Achenbach C.J., Forthal D.N., Felgner P.L., Gach J.S. A high throughput protein microarray approach to classify HIV monoclonal antibodies and variant antigens. PLoS One. 2015;10 doi: 10.1371/journal.pone.0125581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Du P.-X., Chou Y.-Y., Santos H.M., Keskin B.B., Hsieh M.-H., Ho T.-S., Wang J.-Y., Lin Y.-L., Syu G.-D. Development and application of human coronavirus protein microarray for specificity analysis. Anal. Chem. 2021;93:7690–7698. doi: 10.1021/acs.analchem.1c00614. [DOI] [PubMed] [Google Scholar]
- 64.Hertz T., Levy S., Ostrovsky D., Oppenheimer H., Zismanov S., Kuzmina A., Friedman L., Trifkovic S., Brice D., Chun-Yang L., et al. Correlates of protection for booster doses of the BNT162b2 vaccine. medRxiv. 2022 doi: 10.1101/2022.07.16.22277626. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sandbulte M.R., Westgeest K.B., Gao J., Xu X., Klimov A.I., Russell C.A., Burke D.F., Smith D.J., Fouchier R.A.M., Eichelberger M.C. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc. Natl. Acad. Sci. USA. 2011;108:20748–20753. doi: 10.1073/pnas.1113801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao J., Couzens L., Burke D.F., Wan H., Wilson P., Memoli M.J., Xu X., Harvey R., Wrammert J., Ahmed R., et al. Antigenic Drift of the Influenza A(H1N1)pdm09 Virus Neuraminidase Results in Reduced Effectiveness of A/California/7/2009 (H1N1pdm09)-Specific Antibodies. mBio. 2019;10 doi: 10.1128/mBio.00307-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Einav T., Creanga A., Andrews S.F., McDermott A.B., Kanekiyo M. Harnessing low dimensionality to visualize the antibody–virus landscape for influenza. Nat. Comput. Sci. 2022;3:164–173. doi: 10.1038/s43588-022-00375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mykytyn A.Z., Rosu M.E., Kok A., Rissmann M., van Amerongen G., Geurtsvankessel C., de Vries R.D., Munnink B.B.O., Smith D.J., Koopmans M.P.G., et al. 2023. Antigenic Mapping of Emerging SARS-CoV-2 Omicron Variants BM.1.1.1, BQ.1.1, and XBB.1. Lancet Microbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mykytyn A.Z., Rissmann M., Kok A., Rosu M.E., Schipper D., Breugem T.I., van den Doel P.B., Chandler F., Bestebroer T., de Wit M., et al. Antigenic cartography of SARS-CoV-2 reveals that Omicron BA.1 and BA.2 are antigenically distinct. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abq4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hilton S.K., Huddleston J., Black A., North K., Dingens A.S., Bedford T., Bloom J.D. dms-view: Interactive visualization tool for deep mutational scanning data. J. Open Source Softw. 2020;5 doi: 10.21105/joss.02353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fowler D.M., Fields S. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sela-Culang I., Ofran Y., Peters B. Antibody specific epitope prediction-emergence of a new paradigm. Curr. Opin. Virol. 2015;11:98–102. doi: 10.1016/j.coviro.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rubinstein N.D., Mayrose I., Martz E., Pupko T. Epitopia: a web-server for predicting B-cell epitopes. BMC Bioinf. 2009;10:287. doi: 10.1186/1471-2105-10-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.El-Manzalawy Y., Honavar V. Recent advances in B-cell epitope prediction methods. Immunome Res. 2010;6(Suppl 2) doi: 10.1186/1745-7580-6-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gottardo R., Bailer R.T., Korber B.T., Gnanakaran S., Phillips J., Shen X., Tomaras G.D., Turk E., Imholte G., Eckler L., et al. Plasma IgG to linear epitopes in the V2 and V3 regions of HIV-1 gp120 correlate with a reduced risk of infection in the RV144 vaccine efficacy trial. PLoS One. 2013;8 doi: 10.1371/journal.pone.0075665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karasavvas N., Billings E., Rao M., Williams C., Zolla-Pazner S., Bailer R.T., Koup R.A., Madnote S., Arworn D., Shen X., et al. The Thai Phase III HIV Type 1 Vaccine trial (RV144) regimen induces antibodies that target conserved regions within the V2 loop of gp120. AIDS Res. Hum. Retrovir. 2012;28:1444–1457. doi: 10.1089/aid.2012.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nitahara Y., Nakagama Y., Kaku N., Candray K., Michimuko Y., Tshibangu-Kabamba E., Kaneko A., Yamamoto H., Mizobata Y., Kakeya H., et al. High-Resolution Linear Epitope Mapping of the Receptor Binding Domain of SARS-CoV-2 Spike Protein in COVID-19 mRNA Vaccine Recipients. Microbiol. Spectr. 2021;9 doi: 10.1128/Spectrum.00965-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haynes W.A., Kamath K., Bozekowski J., Baum-Jones E., Campbell M., Casanovas-Massana A., Daugherty P.S., Dela Cruz C.S., Dhal A., Farhadian S.F., et al. High-resolution epitope mapping and characterization of SARS-CoV-2 antibodies in large cohorts of subjects with COVID-19. Commun. Biol. 2021;4:1317. doi: 10.1038/s42003-021-02835-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karlsson E.A., Hertz T., Johnson C., Mehle A., Krammer F., Schultz-Cherry S. Obesity outweighs protection conferred by adjuvanted influenza vaccination. mBio. 2016;7 doi: 10.1128/mBio.01144-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alhadi M.A., Friedman L.M., Karlsson E.A., Cohen-Lavi L., Burkovitz A., Schultz-Cherry S., Noah T.L., Weir S.S., Shulman L.M., Beck M.A., et al. Obesity is associated with an altered baseline and post-vaccination influenza antibody repertoire. medRxiv. 2021 doi: 10.1101/2021.03.02.21252785. Preprint at. [DOI] [Google Scholar]
- 81.Legutki J.B., Johnston S.A. Immunosignatures can predict vaccine efficacy. Proc. Natl. Acad. Sci. USA. 2013;110:18614–18619. doi: 10.1073/pnas.1309390110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vanderven H.A., Kent S.J. The protective potential of Fc-mediated antibody functions against influenza virus and other viral pathogens. Immunol. Cell Biol. 2020;98:253–263. doi: 10.1111/imcb.12312. [DOI] [PubMed] [Google Scholar]
- 83.Parsons M.S., Chung A.W., Kent S.J. Importance of Fc-mediated functions of anti-HIV-1 broadly neutralizing antibodies. Retrovirology. 2018;15:58. doi: 10.1186/s12977-018-0438-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Erp E.A., Luytjes W., Ferwerda G., van Kasteren P.B. Fc-Mediated Antibody Effector Functions During Respiratory Syncytial Virus Infection and Disease. Front. Immunol. 2019;10:548. doi: 10.3389/fimmu.2019.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Starr T.N., Greaney A.J., Stewart C.M., Walls A.C., Hannon W.W., Veesler D., Bloom J.D. Deep mutational scans for ACE2 binding, RBD expression, and antibody escape in the SARS-CoV-2 Omicron BA.1 and BA.2 receptor-binding domains. PLoS Pathog. 2022;18 doi: 10.1371/journal.ppat.1010951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Altman M.O., Angel M., Košík I., Trovão N.S., Zost S.J., Gibbs J.S., Casalino L., Amaro R.E., Hensley S.E., Nelson M.I., Yewdell J.W. Human influenza A virus hemagglutinin glycan evolution follows a temporal pattern to a glycan limit. mBio. 2019;10 doi: 10.1128/mBio.00204-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pejchal R., Doores K.J., Walker L.M., Khayat R., Huang P.-S., Wang S.-K., Stanfield R.L., Julien J.-P., Ramos A., Crispin M., et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011;334:1097–1103. doi: 10.1126/science.1213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moore P.L., Gray E.S., Wibmer C.K., Bhiman J.N., Nonyane M., Sheward D.J., Hermanus T., Bajimaya S., Tumba N.L., Abrahams M.-R., et al. Evolution of an HIV glycan-dependent broadly neutralizing antibody epitope through immune escape. Nat. Med. 2012;18:1688–1692. doi: 10.1038/nm.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seabright G.E., Doores K.J., Burton D.R., Crispin M. Protein and glycan mimicry in HIV vaccine design. J. Mol. Biol. 2019;431:2223–2247. doi: 10.1016/j.jmb.2019.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sammut C., Webb G.I. Encyclopedia of machine learning; 2010. Leave-one-out Cross-Validation; pp. 600–601. [Google Scholar]
- 91.Lee M.-S., Chen M.-C., Liao Y.-C., Hsiung C.A. Identifying potential immunodominant positions and predicting antigenic variants of influenza A/H3N2 viruses. Vaccine. 2007;25:8133–8139. doi: 10.1016/j.vaccine.2007.09.039. [DOI] [PubMed] [Google Scholar]
- 92.Chakraborty C., Sharma A.R., Bhattacharya M., Lee S.-S. A Detailed Overview of Immune Escape, Antibody Escape, Partial Vaccine Escape of SARS-CoV-2 and Their Emerging Variants With Escape Mutations. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.801522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang J.-W., King C.-C., Yang J.-M. Co-evolution positions and rules for antigenic variants of human influenza A/H3N2 viruses. BMC Bioinf. 2009;10(Suppl 1) doi: 10.1186/1471-2105-10-S1-S41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dash P., Fiore-Gartland A.J., Hertz T., Wang G.C., Sharma S., Souquette A., Crawford J.C., Clemens E.B., Nguyen T.H.O., Kedzierska K., et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017;547:89–93. doi: 10.1038/nature22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Raymond D.D., Bajic G., Ferdman J., Suphaphiphat P., Settembre E.C., Moody M.A., Schmidt A.G., Harrison S.C. Conserved epitope on influenza-virus hemagglutinin head defined by a vaccine-induced antibody. Proc. Natl. Acad. Sci. USA. 2018;115:168–173. doi: 10.1073/pnas.1715471115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper re-analyzes publicly available datasets that are listed in the key resources table. All other data generated within this study is reported in the paper.
-
•
All original code and files are available via Zenodo: http://doi.org/10.5281/zenodo.7947980. The DOI is also listed in the key resources table.
-
•
Any additional information needed to re-analyze the data reported in this paper is available from the lead contact upon request.