

Abstract
Protein higher order structure (HOS), encoded in a three-dimensional structure, is critical for the functioning of proteins. Mass spectrometry (MS) has become an important tool for determining protein HOS owing to its high throughput, mid-to-high spatial resolution, low sample amount requirement, and broad compatibility with various protein systems. Modern MS-based protein HOS analysis relies, in part, on footprinting, where a reagent reacts “to mark” the solvent-accessible surface of the protein, and MS-enabled proteomic analysis locates the modifications to afford a footprint. Fast photochemical oxidation of proteins (FPOP), first introduced in 2005, has become a powerful approach for protein footprinting, and it has been employed in solving several important problems including mapping epitopes, following aggregating proteins, locating small molecule binding, measuring ligand binding affinity, monitoring protein folding/unfolding, and determining hidden conformational changes invisible to other methods. Broader adoption will be promoted by dissemination of the technical details for assembling the FPOP platform and for dealing with the complexities of analyzing FPOP data. In this protocol, we will describe the FPOP platform, the conditions for successful footprinting, the post labeling sample handling and digestion, and the data analysis. This protocol is intended not only as a guide for investigators trying to establish a FPOP platform in their own lab but also for those planning to use the approach in collaboration.
Introduction
Proteins are biomacromolecules that are constructed of 20 amino acids as building-block sequence to enable programmed functions. The biological function is determined by higher order structure (HOS), which is encoded in its primary sequence.1 Thus, understanding structure-function relationships of proteins is critical and often requires integrated approaches. To date, X-ray crystallography2 and nuclear magnetic resonance (NMR)3 have been the “gold standards” in protein HOS determination because they offer the highest structural resolution. On the other hand, X-ray crystallography requires protein crystals that are always hard to obtain.4 NMR requires milligrams of sample, and the data acquisition requires extensive signal averaging and processing. Recent advances in cryogenic electron microscopy (Cryo-EM) seems to resolve these drawbacks by delivering high-resolution protein structure with low sample amounts.5, 6 There are, however, limited demonstrations for cryo-EM to obtain high quality images for small proteins,7 and resources for this approach are not widely available at this writing. Other than these high-resolution approaches, many optical methods including circular dichroism,8 ultra-violet spectroscopy,9 infrared spectroscopy,10 and dynamic light scattering,11 also play important roles in protein HOS analysis. These approaches, however, seldom afford spatial resolution of their structural information.
Mass spectrometry (MS) is now positioned to play a key role in protein HOS analysis owing to its mid-to-high structural resolution, low sample amount requirement, high throughput, and proteomics capabilities.12–18 Footprinting is becoming a commonly used MS-based approach for elucidating protein HOS, during which different covalent labeling approaches “mark” the protein’s solvent accessible surface area (SASA). In the commonly used “bottom-up” approach, a footprinted protein molecule is digested by a protease, and the resulting peptides are separated, detected, and sequenced by integrated liquid chromatography (LC), MS, and tandem MS (MS/MS), respectively.19 The readout is an identification of labeling with respect to the peptide sequence and often a quantification of the modification fraction, providing a protein footprint that reflects protein HOS. Protein footprinting can be executed not only for pure protein system but also for complex mixtures.20–22
Among protein footprinting approaches, hydrogen deuterium exchange (HDX), is the most common example.13, 18, 23, 24 HDX is a powerful approach not only for footprinting but also for assessing protein dynamics in solution, as demonstrated by numerous NMR25, 26 and MS-based27, 28 studies. In a typical “exchange-in” scenario, deuterium from solvent D2O replaces hydrogen of the backbone amides to mark the solvent-accessible surfaces inter alia and the weakly hydrogen bonded peptide bonds of the protein. Although HDX probes more than solvent accessible backbone amides, it provides a “footprint” when executed in a differential manner where the protein deuterium uptakes are compared for different states (e.g., ligand bound versus unbound). In the comparison, contributions of intrinsic protein dynamics cancel, and differences in deuterium updates between the two states represent the changes in solvent accessibilities and binding-induced dynamics changes. HDX is the least disruptive of protein structure as compared with other footprinting approaches, and the coverage is nearly complete (i.e., all amino acids except proline are covered). The effective concentration of HDX labeling reagent, the solvent “D2O”, is over 50 M and changes negligibly over the time of footprinting. Therefore, the exchange is not limited by reagent amount and follows pseudo-first-order kinetics over the time course of the exchange.
On the other hand, HDX is reversible, imposing constraints on protein and peptide analysis to retain the deuterium labeling during post-exchange sample handling. Such restrictions greatly limit the broad adoption of HDX in complex protein systems; for example, membrane proteins, which require post-exchange lipid/detergent removal,29, 30 and glycosylated proteins, which necessitate complex deglycosylation after the exchange.31 Nevertheless, MS has become the most used measurement approach in HDX, replacing NMR for many applications.
For irreversible footprinting, targeted reagents label selected amino-acid side chains with high specificity. The labeling reaction is easily implemented, and the resulting mass tag is usually bio-orthogonal. Moreover, as the reaction is highly specific, data analysis is usually straightforward. All these advantages also make targeted-reagent-based protein footprinting a high throughput method, and it is widely adopted, although less than HDX, in protein HOS studies.12, 18 On the other hand, the labeling is usually slow (minutes or more), and there is always a concern that the target protein will undergo a conformational change in the early stages of labeling and then continue to be modified (become over-labeled) to yield a composite and misleading footprint.12 Moreover, specific amino acid labeling by its nature provides low coverage, modifying often one or a few amino acid side chains. In practice, protein footprinting by targeted labeling reagents requires post labeling approaches (e.g., circular dichroism, NMR) to ensure that the structural integrity of the protein has not been affected by the labeling. Multiple labeling reagents can be used to increase the amino acid coverage. The limitations of HDX and specific amino acid labeling motivate another approach that overcomes their drawbacks.
Protein HOS analysis by fast radical footprinting enjoys features of HDX and specific amino acid labeling but has important advantages over both.14, 16, 18, 32 The modifying reagents are reactive species, usually radicals, that react with solvent-accessible amino acid residues to report on protein SASA. Protein footprinting by radical species was first demonstrated with hydroxyl radical (•OH) generated in synchrotron water radiolysis.33 The advantages of fast reaction rates and broad amino acid coverage recommend such approach, but the limited availability of synchrotron sources is a drawback. Fast photochemical oxidation of proteins (FPOP), originally demonstrated by Hambly and Gross34 in 2005, makes such powerful footprinting approach more available in general research labs. In a FPOP setup, the •OH is generated from laser photolysis of hydrogen peroxide (H2O2) and further oxidatively footprints proteins by irreversibly substituting an H for OH onto reactive amino acid side chains. In practice, •OH reacts with 14 out of 20 amino acid residues, a significant advance over specific amino acid labeling. Moreover, as the primary radical species are highly reactive, the labeling reaction timescale is shortened to the submillisecond range,16, 34 a time that is faster than most protein conformational changes. Fast reaction rates ensure that the FPOP design eliminates the concern of over labeling because FPOP takes a fast “snapshot” of the protein system at residue-level resolution.
These features allow FPOP footprinting to address a number of significant biological questions32 that are shown in Figure 1. FPOP can map epitopes,39, 40 track protein folding/unfolding kinetics,37, 41, 42 and assay protein aggregation with some residue-level spatial resolution36 usually by executing differential experiments. Moreover, by combining FPOP with ligand titration and computer modeling, one can also locate ligand binding sites, measure site-specific binding affinities, and follow binding-induced protein conformational changes.35, 43, 44 In addition to studying proteins in vitro, one can also label live C. elegans by FPOP to facilitate a protein HOS analysis in vivo.20 In addition to affording biophysical results, incorporation of FPOP footprinting data into computer-based protein structure prediction greatly elevates the confidence in a model.38, 45 The FPOP platform also accommodates the development of novel labeling reagents,46–48 as well as facilitates the understanding of radical reaction mechanisms in solution49 and the transport pathways of naturally occurring radical species in live cells,50 forecasting a useful and broad-based future.
Fig. 1 |. Major applications of FPOP.
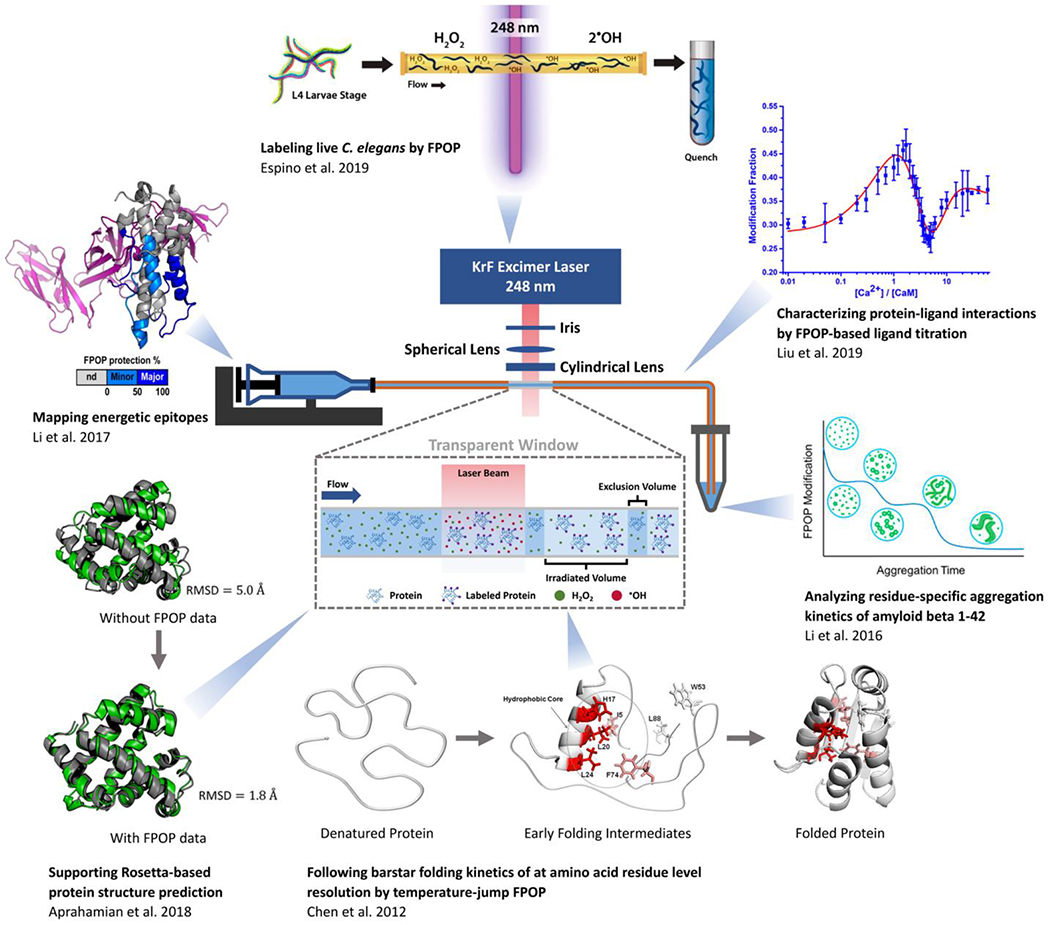
Clockwise from top: Espino, et al.20 labeled live Caenorhabditis elegans (C. elegans) with FPOP and identified oxidative labeling on several hundred proteins. Liu, et al.35 combined ligand titration with FPOP and characterized binding sites, site-specific affinities, and binding orders of the calcium - calmodulin (Ca2+ – CaM) system. Li, et al.36 used FPOP to follow the amyloid beta 1-42 aggregation kinetics and revealed the critical role of its middle domain in the aggregation process. Chen, et al.37 established a temperature-jump platform and demonstrated its utility for the folding kinetics of barstar with residue-level FPOP. Aprahamian, et al.38 developed a Rosetta scoring algorithm that utilizes FPOP data, after which the root mean square deviation (RMSD, as compared with crystal structure) of the best scoring model improved significantly. Li, et al.39 demonstrated the applicability of FPOP in mapping epitopes by successfully identified the binding regions between an antibody and interleukin-23.
On the other hand, FPOP is demanding, requiring careful control of the laser energy, proper alignment of sample capillary, thoughtful determination of sample flow rate during laser irradiation, establishment of standardized post-labeling sample handling, optimized MS. Serving as the “birthplace” of the technique, we are in position to present in this protocol the technical details needed to setup an FPOP platform, configure the LC separation and the mass spectrometer, and analyze the FPOP data.
Development of the protocol
Protein footprinting by •OH was first demonstrated in 1994 with Fenton chemistry,51 and later with synchrotron-mediated water hydrolysis in 199933. Both these methods suffer limitations. The Fenton reaction takes minutes to complete, during which time the protein may become over-labeled because it altered its conformation during the footprinting owing to labeling-induced changes in protein hydrophilicity. Furthermore, the number of synchrotrons is limited, making it sometimes difficult and time-consuming for investigators to take advantage of such a powerful approach.
Protein footprinting by H2O2 photolysis dates back to 2004, when a UV lamp was used to facilitate the photolysis, but those experiments required large concentrations of H2O2 (15%).52 The amount was lowered (to 0.3%) when a Nd:YaG laser (at fourth harmonic, 266 nm) was introduced to avoid direct oxidation by H2O2,53 but even then, the sample (contained in a tube) may not get even exposure to the laser.
The FPOP platform presented in this protocol significantly advances •OH protein footprinting by introducing a flow system into the design and including a radical scavenger, usually an amino acid that has high reactivity towards •OH, to control the time (dosage) of exposure. The flow system also allows an exclusion volume between adjacent laser shots to reduce the chance of double-exposure of the protein to the laser. By using a scavenger, the primary radical (•OH) lifetime can be reduced to microseconds, a time that is faster than any oxidation-triggered protein conformational changes. Taken together, the chances of over labeling are minimized on the FPOP platform, increasing significantly the reproducibility and reliability of protein footprinting.
Since introduction, the FPOP platform has been optimized for improved radical scavengers (histidine),54 more effective post-labeling clean up (e.g., acetone precipitation),35 and precise MS quantification through global, peptide, and residue-level measurements.55 Deeper understanding of the radical reaction pathways has been achieved through selective isotopic labeling,49 and better data processing is available via several software platforms. These improvements mean better controls of the footprinting reaction and more reproducible and reliable experimental output, increasing its applicability for addressing biological questions. These advances are summarized in this Protocol.
Levels of expertise needed to implement the protocol
The protocol presented here can be followed by most biochemists with hands-on experience with protein handling and digestion. For working with the laser, we recommend involving a safety specialist so that implementation does not violate safety protocols. The preparation of the materials, handling of the proteins, and digestion of the protein are standard biochemical practices and do not require expertise beyond that found in a biochemistry laboratory. For the MS analysis, we recommend that an experienced mass spectrometrist be engaged in the setup of the micro-HPLC, the nano-HPLC, and the mass spectrometer. MS expertise is helpful in dealing with complex protein samples where it is necessary to fine-tune the instrument parameters for optimal detection. We present in the “Equipment Setup” section some suggestions for optimizing the nano-HPLC-MS system for better signal acquisitions.
Limitations
Because LC-MS instruments are now widely available for proteomics measurements, the adoption of FPOP is mainly limited by availability of the laser, its setup, operation, and maintenance. We designed this protocol to help overcome a lack of expertise not only in the laser setup but also in the sample handing, LC-MS measurement, and data processing.
As compared with widely used HDX, the irreversible labeling by FPOP sometimes allows characterization of protein HOS at the amino acid residue level without the need of special fragmentation techniques such as electron capture/transfer dissociation. Although the information obtained from any MS-based protein HOS analysis is not sufficient to construct a high-resolution structural model, these approaches are usually executed in differential manner to report changes in SASA for different protein states (e.g., bound vs. unbound, wild-type vs. mutant). Incorporating the covalent modeling data into protein structure predictions may overcome the need for differential measurements.38, 45
The precursor of the most common reactive species in FPOP is H2O2, which itself is an oxidant. Proteins that are prone to oxidation may not be compatible with typical FPOP because the incubation of H2O2 with the protein before laser irradiation will already introduce -OH groups and lead to biased footprinting. This can be minimized by placing a “mixing T” immediately before the laser window to introduce H2O2 just before the footprinting and minimize the protein exposure to H2O2.56 Moreover, •OH reacts with 14 out of 20 amino acids with a preference toward aromatic, heterocyclic, and sulfur-containing sidechains.16, 57 For proteins that are not rich in these residues, •OH may not be the optimal reporter of their HOS. Because protein oxidation often occurs not only in protein isolation, purification, and storage but also during post labeling sample handling and even during LC separations, it is sometimes ambiguous to distinguish the oxidations from footprinting and those from other processes. These limitations can be overcome by introducing other bio-orthogonal labeling reagents into the FPOP platform, as demonstrated recently.46, 48, 58, 59
Experimental design
The experimental design involves proper setup of the laser, preparation of the working solutions, determination of the H2O2 concentration for optimized FPOP modification, handling of the post-labeling sample including enzymatic digestion, and finally measurement of the FPOP-labeled protein samples at both global and peptide/residue levels. Specifically, H2O2 concentrations and the enzymatic digestion conditions are system-specific and require optimization upon working with a new protein system. The LC gradient for protein/peptide analysis and the instrument acquisition parameters should also be optimized to meet the requirements of analyzing complex peptide samples. Once established, only minimal tuning of the laser platform including checking energy and alignment are required. Data analysis is becoming more routine with commercial software and can be generalized to all proteins of interest.
Addressing biological questions by FPOP usually requires adjustment of the basic FPOP labeling to operate in a differential manner. Investigators can conduct parallel sets of FPOP experiments under different conditions (e.g., with and without a binding partner) to elucidate protein-ligand binding sites39, 40 by a titration format to afford ligand/protein affinity measurements,35, 43, 44 and to follow changes in oxidation fractions for protein aggregation kinetics in a time-dependent manner (measured by changes in modification fractions at peptide and, if possible, residue levels).36 One can also design experiments with living cells for in vivo labeling20, 60 and incorporate FPOP modification data into protein structural predictions.38, 45 Other applications may require modifying the FPOP platform presented here; such measurements can interrogate changes in protein conformations upon denaturation for unfolding analysis, where a T-mixer is necessary before the transparent window on the capillary.41 These applications take as a starting point the fundamental implementation of FPOP as described in this protocol. Technical details of these applications, however, are beyond the scope of this article but can be found in the research works referenced above.
Materials
Biological materials
A protein of interest (exemplified by calcium-free bovine calmodulin in this protocol), proteases for protein digestion, and catalase for H2O2 decomposition are needed. ▲CRITICAL STEP Check the protein concentration by Nano Drop or other UV-Vis spectroscopy before conducting any experiments. !CAUTION Structural integrity of the protein of interest should be checked by other low-resolution, high-throughput methods including but not limited to circular dichroism, dynamic light scattering, and native MS prior to footprinting. Because proteins may bind to plastic surfaces of storage vessels, we recommend protein LoBind tubes for protein handling to achieve acceptable recovery rates and sensitivities.
Reagents
General laboratory safety precautions need to be followed when handling flammable, corrosive, and toxic chemicals. !CAUTION Always work in a fume hood with proper personal protection equipment.
0.1% Formic Acid (v/v) in acetonitrile (LC-MS grade, Thermo Scientific, Cat. No. 85174) !CAUTION Formic acid is flammable, corrosive, and toxic. Acetonitrile is flammable and toxic.
0.1% Formic Acid (v/v) in water (LC-MS grade, Thermo Scientific, Cat. No. 85170) !CAUTION Formic acid is flammable, corrosive, and toxic.
Acetone (HPLC Plus, for HPLC, GC, and residue analysis, ≥ 99.9%, Sigma-Aldrich, Cat. No. 650501) !CAUTION Acetone is flammable and toxic.
Acetonitrile (for HPLC, gradient grade, ≥ 99.9%, Sigma-Aldrich, Cat. No. 439134) !CAUTION Acetonitrile is flammable and toxic.
Calmodulin (bovine, Ocean Biologics, Seattle, WA) as a test protein
Catalase from bovine liver (lyophilized powder, 2,000 – 5,000 units/mg protein, Sigma-Aldrich, Cat. No. C9322)
Compressed Gas, N.O.S., UN1956, Fluorine/Neon (F2 0.18%, Kr 3.75%, Xe 15 ppm, Ne Bal, Messer Electronics Holdings, Cat. No. 24107902) !CAUTION The gas mixture is toxic. Store and dispense the gas from the cylinder situated in a ventilated cabinet.
DL-Dithiothreitol (DTT, BioUltra, for molecular biology, ≥ 99.5% (RT), Sigma-Aldrich, Cat. No. 43815) !CAUTION DTT is toxic.
Formic Acid (LCMS grade, CovaChem, Cat. No. 11202) !CAUTION Formic acid is flammable, corrosive, and toxic.
Hydrogen Peroxide Solution (contains inhibitor, 30 wt. % in H2O, ACS reagent, Sigma-Aldrich, Cat. No. 216763) !CAUTION Hydrogen peroxide is corrosive.
Iodoacetamide (IAM, BioUltra, Sigma-Aldrich, Cat. No. I1149) !CAUTION IAM is toxic and an aspiration hazard.
L-Histidine (ReagentPlus, ≥ 99% (TLC), Sigma-Aldrich, Cat. No. H8000)
L-Methionine (BioUltra, ≥ 99.5% (NT), Sigma-Aldrich, Cat. No. 64319)
Phosphate-buffered saline (PBS, tablet, Sigma-Aldrich, Cat. No. P4417)
Trifluoroacetic Acid (LCMS grade, CovaChem, Cat. No. 11204) !CAUTION Trifluoroacetic acid is corrosive and toxic.
Trypsin/Lys-C Mix (20 μg, Mass Spec grade, Promega, Cat. No. V5071)
Urea (8 M after reconstitution with 16 mL high purity water, Sigma-Aldrich, Cat. No. U4883)
Water (for HPLC, Sigma-Aldrich, Cat. No. 270733)
Equipment
General equipment
Benchtop centrifuge (SPROUT, Heathrow Scientific)
Benchtop vortex mixer (Digital Vortex Mixer, 120V US/JP plug, Thermo Scientific, Cat. No. 88882009)
High-speed Centrifuge (Centrifuge 5418, Eppendorf)
Incubator (Q. Instruments BioShake iQ, Quantifoil Instruments GmbH)
Low-speed Centrifuge (Centrifuge 5430, Eppendorf)
Nanodrop spectrophotometer (NanoDrop OneC Microvolume UV-Vis Spectrophotometer, Thermo Scientific)
Benchtop pH meter (Orion Star A211, Thermo Scientific)
Protein LoBind Tubes (0.5 mL and 1.5 mL, Eppendorf, Cat. Nos. 022431064 and 022431081)
Secondary liquid containers (20 mL glass vials)
Speed Vac (SC110, Savant Instruments)
Vernier caliper (No. 721, Starrett)
Two-valve LC equipment
C8 column (Eclipse XDB-C8, 3.5 μm, Agilent, Cat. No. 931975-936)
UHPLC injector (Cheminert microbore, Valco Instruments Co. Inc., Cat. No. C82X-1576)
PEEK tubing 1/16 × 0.004 (black, P.J. Cobert Associates, Inc., Cat. No. 1561XL)
High pressure 1/16” nuts (Standard internal nuts, Valco Instruments Co. Inc., Cat. No. IZN1-10)
High pressure ferrules (Stainless steel, 1/16”, type 316, Valco Instruments Co. Inc., Cat. No. ZF1S6-10)
Injection port (PEEK needle port, Rheodyne 9013, P.J. Cobert Associates, Inc., Cat. No. 8605632)
Union (Union PEEK 0.020” 10-32 threads hi press, P.J. Cobert Associates, Inc., Cat. No. P-704)
Low pressure nuts and ferrules (F-330X, Fingertight PEEK nuts, long, with F-142 PEEK ferruels, 10-32, P.J. Cobert Associates, Inc., Cat. No. 998784X)
Cartridge column hardware kit (Rapid resolution and rapid resolution HT cartridge (400 bar), RR system, Agilent, Cat. No. 820555-901)
Loading pump (LS class, 5 mL/min, 6000 PSI, Teledyne SSI, Cat. No. LS005SFT1A)
Binary gradient pump (FLX. 10371, Leap Technologies)
50 μL PEEK sample loop (P.J. Cobert Associates, Inc., Cat. No. 9055-023)
FPOP platform equipment
⊘1/2” Optical post (SS, 8-32 setscrew, ¼”−20 tap, L = 3”, ThorLabs, Cat. No. TR3)
⊘1/2” Universal post holder (spring-loaded locking thumbscrew, L = 4”, ThorLabs, Cat. No. UPH4)
10 MHz Function Generator (with 4-Digit LED display, Model 4030, B&K Precision)
Aluminum breadboard (12” × 48” × ½”, ¼”−20 taps, ThorLabs, Cat. No. MB1248)
Beam Block (400 – 700 nm, 10 W Max Avg. Power, CW Only, ThorLabs, Cat. No. LB1)
Capillary (Polymicro Flexible Fused Silica Capillary Tubing, 150 μm I.D., 360 μm O.D., Polymicro Products and Solutions - Molex, Cat. No. 1068150024)
Capillary holder (Custom-made, dimensions provided in Supporting Information as Supplementary Fig. 1)
Capillary sleeve (Tubing sleeve FEP green, 380 μm (0.015”) I.D. × 1/16” O.D. × 1.55”, P.J. Cobert Associates, Inc., Cat. No. F-242)
Cylindrical Lens: UVFS Plano-Convex Cylindrical Lens (L = 30 mm, H = 20 mm, Uncoated, f = 75 mm, ThorLabs, Cat. No. LJ4878)
Dovetail Linear Stage (1.0 in travel, fast – drive 20 TPI screw, ¼ - 20 threads, Newport Corporation, Cat. No. TSX-1D)
Fixed ⊘1” mirror mount (8-32 tap, ThorLabs, Cat. No. FMP1)
Injection port (PEEK needle port, Rheodyne 9013, P.J. Cobert Associates, Inc., Cat. No. 8605632)
Kinematic Mount for up to 1.3” (33 mm) Tall Rectangular Optics (Right-handed, ThorLabs, Cat. No. KM100C)
Krypton fluoride excimer laser (KrF laser, EX50/250, GAM Laser) !CAUTION Proper shielding of the laser is necessary. Always wear laser goggles when working with a laser.
Laser Curtain (LAZ-R-Barrier LP200, Wilson Industries, Inc.)
Laser Goggles (ThorLabs, Cat. No. LG10)
Mounting base (1” × 2.3” × 3/8”, ThorLabs, Cat. No. BA1S)
Mounted standard iris (25.0 mm max aperture, TR3 post, ThorLabs, Cat. No. ID25)
One-piece finger tight nuts (Fittings, PEEK, black, 10 – 32 threads, for 1/16” O.D. tubing, P.J. Cobert Associates, Inc., Cat. No. 997734)
Optical Post (ThorLabs, Cat. No. TR3)
Pyroelectric energy sensor (PE25-C, Ophir Optronics Solutions)
Self-adhesive Arrow Flag (0.47 in × 1.7 in, red, 3M Post-it)
Spherical focusing lens: UV Plano-Convex Lens (25 mm Dia. × 250 mm FL Uncoated, Edmund Optics, Cat. No. 48-281)
Stainless steel cap screw (¼”−20, ThorLabs, Cat. No. SH25S100 and SH25S075)
Stainless steel cap screw (8-32, ½” in length, ThorLabs, Cat. No. SH8S050)
Stainless steel single- and double-ended setscrew (¼”−20, ThorLabs, Cat. No. SS25S10)
Syringe (100 μL, Gastight #1710, Hamilton Co.)
Syringe Pump (Pump 11 Elite, Harvard Apparatus)
Thread adapter – internal to external (thread A 8-32 (internal) to thread B ¼”−20 (external), ThorLabs, Cat. No. AE8E25ES)
Union (Union assy PEEK 0.010” thru 10-32 threads, P.J. Cobert Associates, Inc., Cat. No. 998499)
Mass spectrometer and related equipment
11 mm Snap caps (Thermo Scientific SUN-SRi, Fisher Scientific, Cat. No. 14-823-480)
Crimp/snap cap vials (0.25 mL, Thermo Scientific SUN-SRi Crimp/Snap Cap Vials, Fisher Scientific, Cat. No. 05-704-225)
Bruker MaXis 4G Quadrupole Time-of-Flight (Q-ToF) mass spectrometer with electrospray source (Bruker Cooperation)
Nano-flow HPLC system (Dionex UltiMate 3000 RSLCnano UPLC, Thermo Scientific)
Thermo Scientific Q-Exactive Plus hybrid quadrupole-orbitrap mass spectrometer with nano-electrospray source (Thermo Scientific)
Trapping column for nano-flow HPLC (Acclain PreMap 100 C18, 5 μm particle size, 100 μm × 2 cm with nanoViper fittings, Thermo Scientific, Cat. No. 164564)
Reagent setup
Sample-loading solution for global level measurement of the extent of labeling
This solution is 0.1% (vol/vol) LC-MS-grade trifluoroacetic acid in LC-MS-grade water. Add 4 mL LC-MS-grade trifluoroacetic acid into 4 L of LC-MS-grade water. This solution can be prepared in advance and stored at room temperature for 2 months.
LC mobile phase A for global level measurement
This solution is 0.1% (vol/vol) LC-MS-grade formic acid in LC-MS-grade water. Add 4 mL LC-MS-grade formic acid into 4 L of LC-MS-grade water. This solution can be prepared in advance and stored at room temperature for 2 months.
LC mobile phase B for global level measurement
This solution is 0.1% (vol/vol) LC-MS-grade formic acid in LC-MS-grade water-acetonitrile mixture (20% water, 80% acetonitrile, by volume). Mix 1 mL LC-MS-grade formic acid with 200 mL of LC-MS-grade water and 800 mL of LC-MS-grade acetonitrile in a clean glass bottle. Volumes of water and acetonitrile may be measured by using a clean graduated cylinder. This solution can be prepared in advance and stored at room temperature for 2 months.
PBS buffer
Obtain a 250 mL, triple-rinsed (with HPLC water) bottle, add to the bottle 200 mL HPLC water and a PBS tablet to prepare a PBS buffer solution that contains 137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM phosphate buffer (10 mM di-sodium hydrogen phosphate and 2 mM potassium di-hydrogen phosphate) at pH of 7.4. This solution can be prepared in advance and stored at 4 °C for 3 months.
L-Histidine, 50 mM, in PBS buffer
Measure 0.0310 g L-histidine in a 1.5 mL protein LoBind tube; dissolve by adding 1.0 mL PBS buffer to give 200 mM L-histidine stock solution. Obtain another 1.5 mL protein LoBind tube, add 250 μL, 200 mM L-histidine stock solution and 750 μL PBS buffer. This solution can be prepared in advance and stored at 4 °C for 2 weeks.
L-Methionine, 70 mM, in PBS buffer
Measure 0.0104 g L-methionine in a 1.5 mL protein LoBind tube, dissolve by adding 1.0 mL PBS buffer. This solution can be prepared in advance and stored at 4 °C for 2 weeks.
H2O2, 200 mM, in PBS buffer
Obtain a 1.5 mL protein LoBind tube and add 980 μL of PBS buffer. Add to the solution 20 μL of 30 wt. % hydrogen peroxide solution and mix well. This solution must be prepared immediately before use and can be stored on ice for up to 8 h.
DTT, 100 mM, in PBS buffer
To a 1.5 mL protein LoBind tube, add 0.0154 g DTT powder and 1.0 mL PBS buffer. Vortex until completely dissolved. This solution must be prepared immediately before use.
IAM, 300 mM, in PBS buffer
To a 1.5 mL protein LoBind tube, add 0.0555 g IAM powder and 1.0 mL PBS buffer. Vortex until completely dissolved. This solution must be prepared immediately before use.
Catalase, 0.25 – 0.63 units/μL, in PBS buffer
To a 1.5 mL protein LoBind tube, add 0.010 g lyophilized catalase and 1.0 mL of PBS buffer. Vortex and spin-down to give a catalase stock solution of concentration 2.0 – 5.0 units/μL (8× of the target). To another 1.5 mL protein LoBind tube, add 875 μL of PBS buffer and 125 μL of aforementioned catalase stock solution. Upon mixing, aliquot the solution into 0.5 mL protein LoBind tubes for storage. This solution can be prepared in advance and stored at −80 °C for up to 12 months.
Trypsin/Lys-C mix solution, 0.5 μg/μL, in HPLC water
Add 40 μL of HPLC water into the enzyme container; vortex thoroughly. To several 0.5 mL protein LoBind tubes, aliquot enzyme solution. The solution should be prepared immediately before use, but the aliquoted solution can be stored at −80 °C for up to 30 days.
Equipment setup
FPOP platform setup
Laser setup
The setup of the laser platform includes a function generator, a krypton fluoride (KrF) excimer laser, a Fluorine/Krypton/Xenon/Neon premix gas cylinder with regulator, a pyroelectric energy sensor, a mounted standard iris, a spherical focusing lens, a cylindrical lens, a linear stage, a beam stop, a capillary holder, a solid aluminum optical breadboard. Establish personnel protection by surrounding the laser system and associated major components with a laser curtain. A schematic illustration of the setup with detailed laser pathways is shown in Figure 2 below.
Fig. 2 |. Schematic illustration of the laser optics setup.
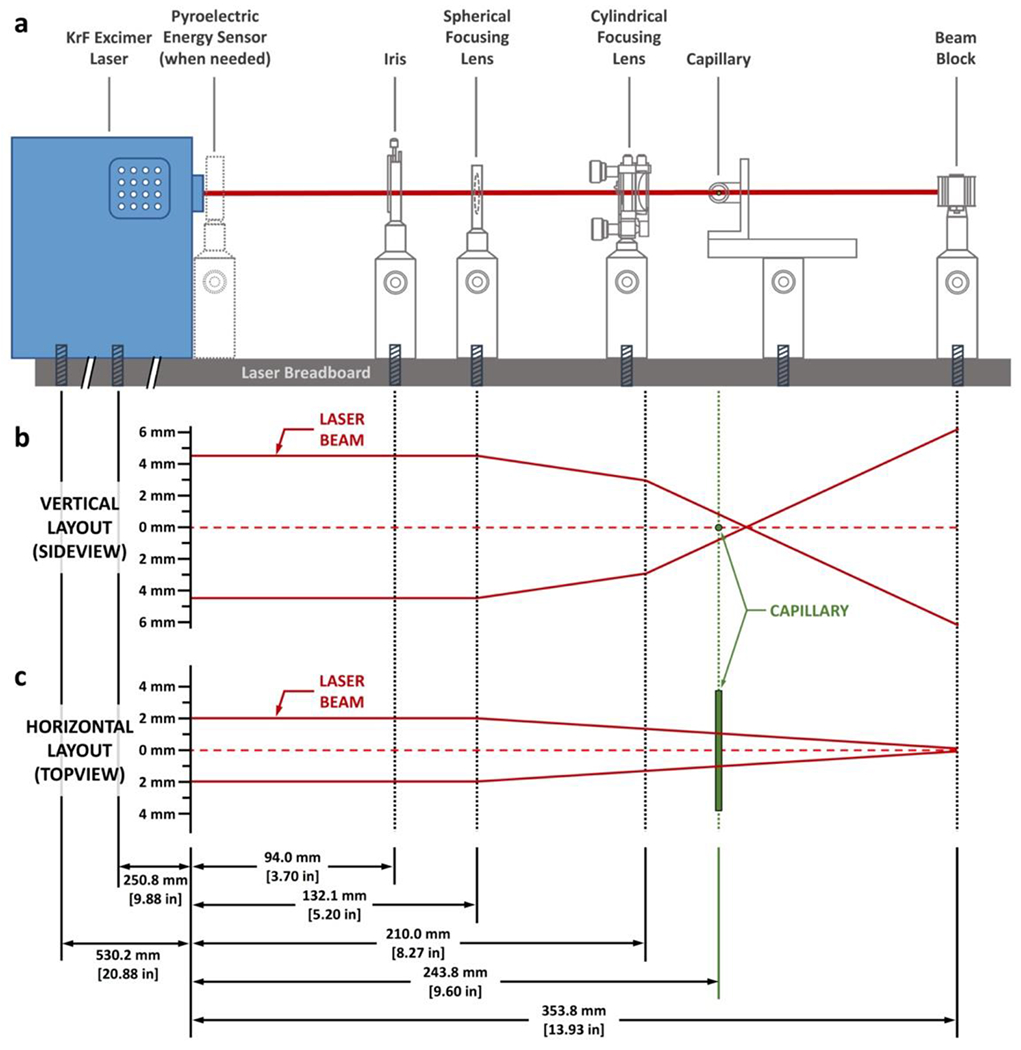
a, A sideview of the laser configuration. All laser optics are aligned in a linear fashion along the laser axis and are attached to the laser breadboard with screws. The positions of the laser optics, not their physical sizes, are drawn to scale. Detailed drawing of the optics setup and a blueprint of their alignment are available as Supplementary Fig. 2. b, Vertical layout of the laser beam and the effect of the laser optics on its pathway. The drawing is to scale, as indicated by the axis on the left. c, Horizontal layout of the laser beam and the effect of the laser optics on its pathway. Note that the cylindrical focusing lens has no effect on the laser beam on the horizontal direction as illustrated in this figure. The plot is drawn to scale as indicated by the axis on the left.
Connect the premix gas cylinder, stored in a ventilated cabinet, to the gas inlet of the laser with regulator and copper tubing, which was first evacuated following the laser manufacturer’s instructions. The next step is to connect the function generator to the KrF excimer laser so that the laser can receive a trigger signal from the function generator. The correct functioning of the assembly can be tested by using the pyroelectric energy sensor.
Design of the laser optics follows Figure 2a. The laser beam from the KrF excimer laser is 4 mm × 9 mm (width × height, based on our current laser as described in the “Equipment” section). Further focus the laser to achieve effective FPOP labeling, where laser optics play key roles. In brief, arrange all laser optics in a linear configuration. The iris is 94 mm from the laser outlet, the spherical focusing lens and the cylindrical focusing lens are 132 mm and 210 mm from the outlet of the laser, respectively. The capillary center is 244 mm from the laser outlet whereas the beam stop is 354 mm from the laser outlet. All the laser optics are held by optical posts and mounted onto a solid aluminum optical breadboard. Note that the linear stage that hosts the capillary holder requires two optical posts for better stability. The laser is secured to the breadboard by screws, and the assembly is secured on the bench. There is no need to use an optical table; a standard laboratory table is adequate. A custom-built capillary holder (made of aluminum, stainless steel, or engineering plastic; dimensions provided in supporting information as Supplementary Fig. 1) is secured onto the linear stage to hold the capillary in position.
The spherical lens focuses the laser beam from all perpendicular-to-beam directions, and the cylindrical lens further compresses the laser beam vertically to achieve a final laser spot of ca. 2.0 mm × 1.8 mm (width × height) at the capillary (Figure 2b and c, Figure 3b and c). The spot size can be further adjusted by using the iris. The location of the beam stop can be varied as long as the actual laser height is smaller than the size of the beam stop and gives an effective stop. The gap between the iris and the laser outlet allows the pyroelectric energy sensor to be placed immediately after the laser outlet, when necessary (Figure 3a). The gap also allows convenient service of the laser. Detailed setup of the laser and its optics is available in supporting information as Supplementary Fig. 2.
Fig. 3. Red arrow flag for checking the laser alignment.
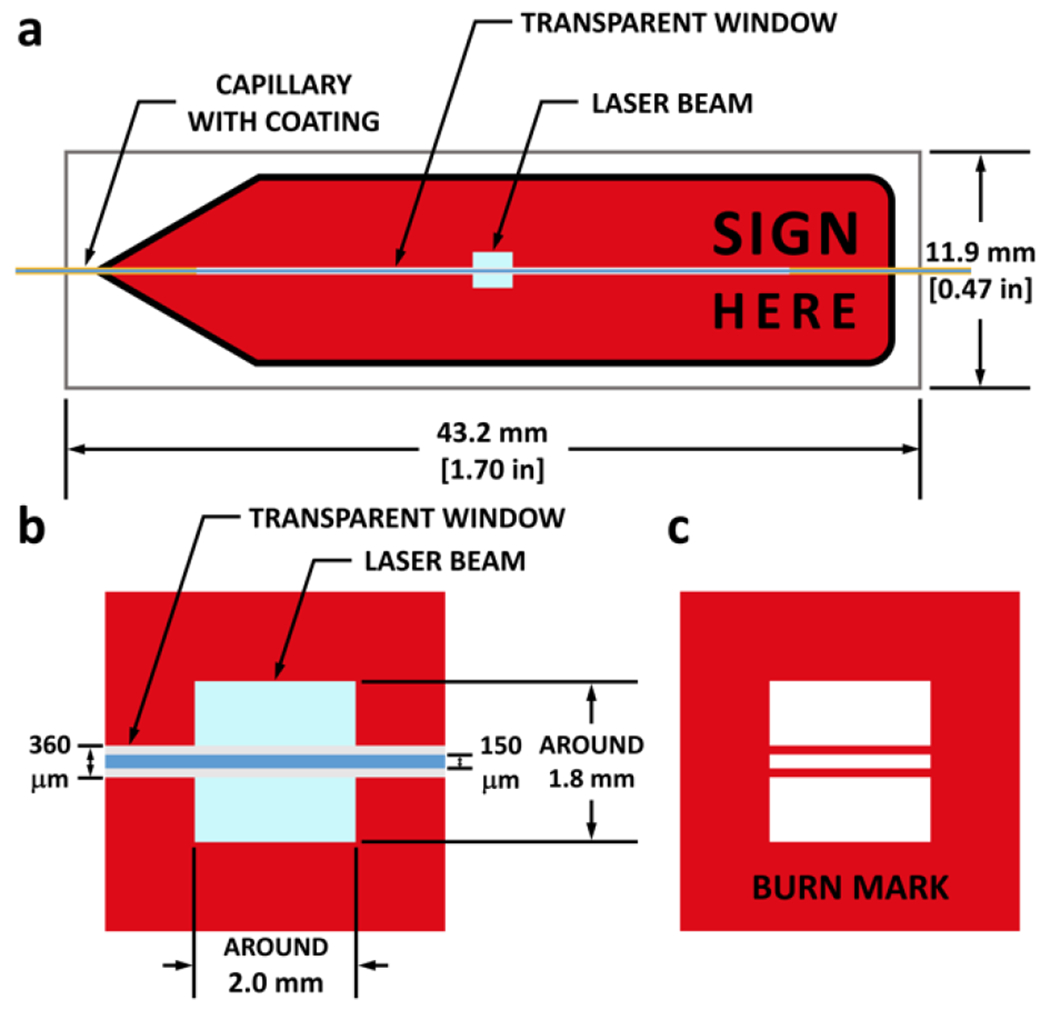
a, Front view of the red arrow flag when aligned with the laser beam. Region colored in light blue represents the laser beam. The regions colored in orange, light gray, and blue are the polyimide-coated wall, naked wall (transparent window), and the liquid inside the capillary, respectively. b, Zoom-in of the region illuminated by the laser beam, with detailed dimensions of the laser spot and the capillary diameters. c, Schematic of the laser burn mark after a 50-shot laser sequence. Dimensions are in line with those in b.
Connect the glass capillary to the injection port via nuts, capillary sleeves, and a union. To hold the capillary in position, pass it through a nut with a capillary sleeve, and secure the nut in the circular opening of the capillary holder. The glass capillary comes with polyimide coating on its outer surface to provide flexibility. Because the coating is not suitable for laser transmission, burn it off (over a width of 30 mm) with an ethanol burner before fitting the capillary onto the system to create a transparent window for better laser transmission. When the transparent glass capillary gets darkened by the laser, move the linear stage so that the laser beam intersects the capillary at other sites of the transparent window. Replace the whole capillary when necessary. !CAUTION Ethanol is flammable.
Check the final alignment of the laser and the laser optics by putting a red-arrow flag immediately after the capillary (Figure 3a) and then triggering a 50-shot laser sequence, after which the laser will leave a “burn mark” on the arrow flag (Figure 3b and c for zoom-in view). This protocol allows visualization of the laser spot size and the location of the capillary with respect to the laser. Note that the laser will travel through the capillary wall of the transparent window but not on the top and bottom as they are parallel to the incident beam; thus, two horizontal red lines are produced in the burn mark, allowing the condition of the transparent window to be checked in terms of the area between two red lines in the burn mark. This is an important indicator of capillary condition as excessive laser shots will darken the capillary wall, after which the two red lines are no longer seen. Finally, place laser curtains around the FPOP platform to protect coworkers in the lab.
!CAUTION Always wear laser goggles when working with lasers. The fill Fluorine/Neon gas is toxic and must be handled with care. Refill the laser regularly (the half-life of the gas in the absence of using the laser is approximately 120 days). Ensure safety by requiring two people to be present when refilling the gas cylinder. A Nd:YAG solid state laser at its fourth harmonic (266 nm) may be used in lieu of the KrF laser (248 nm), and this choice avoids the need for the gas refilling, but its wavelength is in a region showing a small decrease in the absorbance of H2O2 and the yield of radicals. Another possibility is a UV discharge lamp, which would require a special design. ▲CRITICAL STEP The laser alignment needs to be checked before every FPOP experiment (by checking the burn mark on the arrow flag).
Determine the sample flow rate
Upon setting up the laser, measure the laser spot width (in mm) with the Vernier calipers. By considering the inner diameter (I.D., in μm) of the capillary, calculate the illuminated volume of a single laser shot as:
| (1) |
A unique feature of the FPOP flow system is it accommodates an exclusion volume, whereby over-labeling can be minimized (Figure 1, center). The exclusion volume is the fraction of non-illuminated length compared to illuminated + non-illuminated (i.e., exclusion fraction) length, and it is typically set to 15 - 25%. With the exclusion volume, the total volume within a single laser shot is:
| (2) |
Together with the laser frequency (in Hz), calculate the flow rate (in μL/min) as:
| (3) |
Adjust the syringe pump flow rate with the calculated flow and then proceed to finalize the FPOP preparation.
LC-MS setup
Manual two-valve LC system setup for loading intact proteins to the mass spectrometer
To set up a simple two-valve LC system, obtain parts mentioned above and follow the diagram presented in Figure 4. Note that one should use high pressure (stainless steel) ferrules and nuts when connecting to ports 1, 2, 3, 4 of valve 2, the outlet of the gradient pump, and to the column cartridge, as the pressure of the gradient pump changes based upon the flow rate and can be higher than the limit of the PEEK nuts. During LC-MS analysis, keep the desalting pump running (at both injection and loading modes) at a flow rate of 200 μL/min (Sample Loading Solution, 0.1% (vol/vol) LC-MS-grade trifluoroacetic acid in LC-MS-grade water) whereas switch the gradient pump on only during LC separation (injection mode). For the analysis of intact protein, the C-8 column listed above can function as both the trapping column (when flushing with aqueous phase during desalting, loading mode) and the analytical column (when flushing with LC gradient, injection mode).
Fig. 4 |.
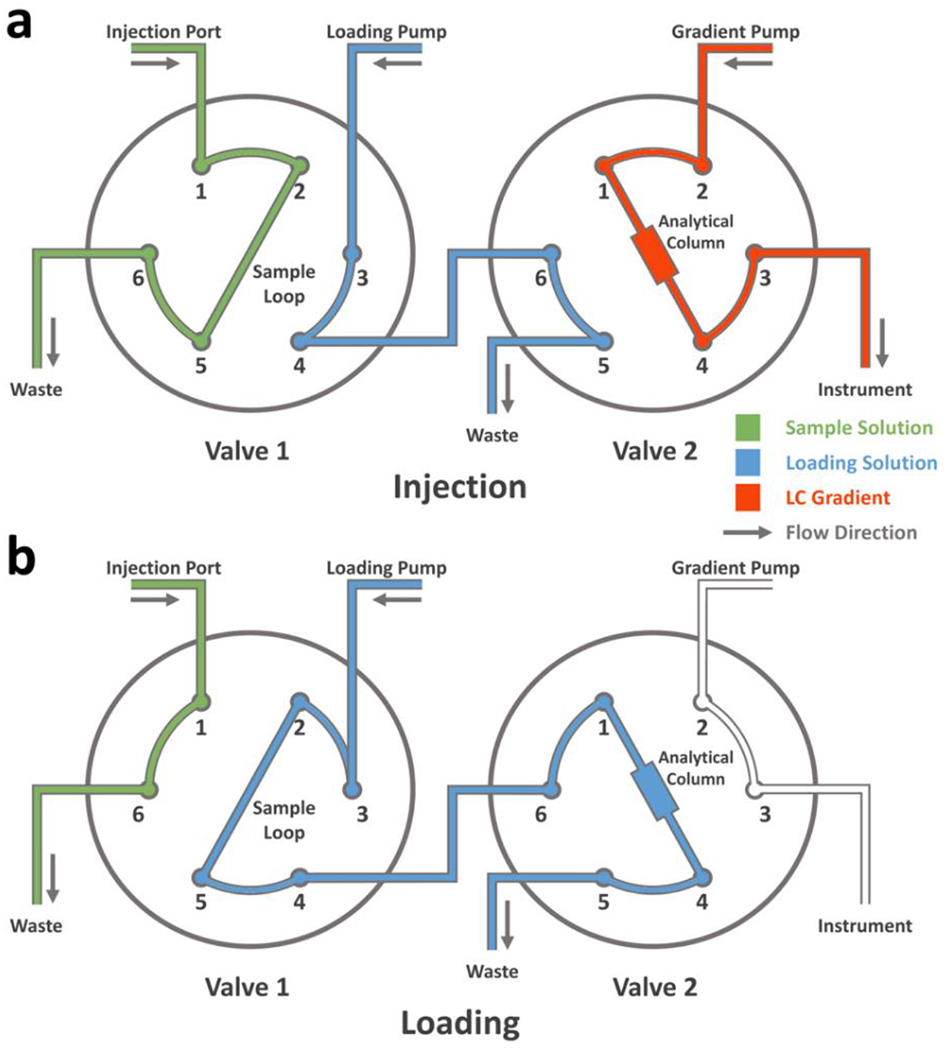
Schematic illustration of a two-valve LC configuration in the a, “Injection” and b, “Loading” modes.
While loading the sample into the LC system, switch the system to “injection” mode, after which the sample solution is injected into the sample loop (Figure 4a). Upon sample injection, switch both valves to the “loading” mode for a 3 min desalting (Figure 4b), during which the sample solution in the sample loop is pushed into Valve 2 by the loading pump and trapped on in the analytical column. Salts and small molecules cannot be effectively trapped by the C-8 column and are washed away with the sample loading solution. After the desalting, switch the valves back to “injection” mode (Figure 4a) and start the MS acquisition at the same time. The sample loop can be cleaned with the washing solution to minimize carry over for subsequent injections. Below (Table 1) is a sample LC gradient used for intact protein analysis, where phase A is 0.1% (vol/vol) LC-MS-grade formic acid in LC-MS-grade water and phase B is 0.1% (vol/vol) LC-MS-grade formic acid in LC-MS-grade water-acetonitrile mixture (20% water, 80% acetonitrile, by volume).
Table 1 |.
A sample micro-flow LC gradient for analyzing FPOP samples on a Bruker MaXis 4G mass spectrometer
| Retention Time (min) | Flow Rate (μl·min−1) | % Phase A | % Phase B | |
|---|---|---|---|---|
| 1 | 0.0 | 200.0 | 95.0 | 5.0 |
| 2 | 1.0 | 200.0 | 95.0 | 5.0 |
| 3 | 6.0 | 200.0 | 0.0 | 100.0 |
| 4 | 7.0 | 200.0 | 0.0 | 100.0 |
| 5 | 8.0 | 200.0 | 95.0 | 5.0 |
| 6 | 10.0 | 200.0 | 95.0 | 5.0 |
Bruker MaXis acquisition parameters for intact proteins
To meet the need of a successful global-level protein measurement at denaturing environment (0.1 % vol/vol formic acid in the LC gradient), all key parameters in the MS acquisition are presented below. The parameters are suitable for a Bruker MaXis 4G, a widely used quadrupole time-of-flight mass spectrometer for protein analysis. Note that the measurements are for soluble proteins. Complex and heterogeneous membrane and glycosylated proteins require adjustments.
▲CRITICAL STEP Note that any mass spectrometer that covers the mass range of denatured proteins can execute such measurements.
Nano LC setup for digested proteins
Separate the digested mixture via a nano-LC system prior to MS analysis. Provided here is a typical gradient that is readily applicable to most FPOP samples (Table 3). Note that the gradient is binary, with phase A being 0.1% formic acid (v/v) in water whereas phase B is 0.1% formic acid (v/v) in acetonitrile. ▲CRITICAL STEP The gradient and its time should be adjusted based on the complexity of the digest.
Table 3 |.
A sample nano-flow LC gradient for analyzing FPOP samples on a Thermo Scientific Q Exactive mass spectrometer
| Retention Time (min) | Flow Rate (μl·min−1) | % Phase A | % Phase B | |
|---|---|---|---|---|
| 1 | 0 | 0.40 | 97.5 | 2.5 |
| 2 | 10 | 0.40 | 97.5 | 2.5 |
| 3 | 40 | 0.40 | 82.5 | 17.5 |
| 4 | 62 | 0.40 | 50.0 | 50.0 |
| 5 | 67 | 0.40 | 20.0 | 80.0 |
| 6 | 75 | 0.40 | 20.0 | 80.0 |
| 7 | 80 | 0.40 | 97.5 | 2.5 |
| 8 | 90 | 0.40 | 97.5 | 2.5 |
Within the gradient illustrated above, the first and last 10 min are for the column equilibration. The effective separation of the digested peptides lies between 10 and 62 min, where % phase B increases gradually from 2.5 to 17.5% and finally to 50%. For the complex digest of a large protein, this part of the gradient can be extended so that its overall time is 120 min. From 62 to 75 min, the percent phase B is sharply increased to 80% and maintained for 8 min, during which time the column is cleaned with a flow of high organic content. This section of the LC gradient should remain unchanged even if the gradient is modified to fit the need of complex samples.
Meanwhile, the loading pump in the nano-LC system should have a constant flow of phase A. As a rough guide, the flow rate in the 0 – 10 min should be 4 μL/min for online desalting, 0.5 μL/min between 10 – 80 min (during the gradient run) and back to 4 μL/min in 80 – 90 min to equilibrate for the next injection.
Q Exactive acquisition parameters for digested proteins
Upon establishing a robust LC platform for peptide separations, next adjust the MS acquisition parameters. For a Thermo Scientific Q Exactive quadrupole orbitrap mass spectrometer (used in our lab) and the common mass spectrometer in proteomics laboratories, the key acquisition parameters are listed (Table 4) for a successful peptide and residue-level analysis. The parameters including “Method Duration” and “Run Time” are based on the LC gradient. The values for these two parameters in Table 4 are for a 90 min LC gradient and should be adjusted for other LC gradients. Moreover, some of the parameters should be optimized to meet better the specific need of the sample.
Table 4 |.
Key parameters of peptide and residue-level FPOP analysis based on a Thermo Scientific Q Exactive mass spectrometer
| No. | Parameter | Value | No. | Parameter | Value |
|---|---|---|---|---|---|
| Global Settings | 11 | AGC Target | 5×104 | ||
| 1 | Chromatographic Peak Width | 30 s | 12 | Maximum IT | 100 ms |
| 2 | Method Duration | 80.0 min | 14 | Loop Count | 10 |
| General | 15 | TopNc | 10 | ||
| 3 | Run Time | 0 – 80.0 min | 12 | Isolation Window | 3.0 m/z |
| 4 | Polarity | Positive | 13 | Isolation Offset | 1.0 m/z |
| 5 | Default Charge State | 2 | 14 | NCEd | 27 |
| Full MS | 15 | Spectrum Data Type | Centroid | ||
| 6 | Resolution | 70,000 | dde Settings | ||
| 7 | AGCa Target | 2×105 | 16 | Minimum AGC Target | 1×103 |
| 8 | Maximum ITb | 200 ms | 17 | Charge Exclusion | Unassigned, 6 – 8, >8 |
| 9 | Scan Range | 400 – 1600 m/z | |||
| dd-MS2 | 18 | Exclude Isotopes | On | ||
| 10 | Resolution | 17,500 | 19 | Dynamic Exclusion | 5.0 s |
Automatic Gain Control
Ion Injection Time
Top N Precursors for MS/MS
Normalized Collision Energy
Data-dependent Analysis
Parameters mentioned in the table above are chosen to handle a relatively simple protein like calmodulin. For proteins that have limited sample amounts or peptides that are difficult to identify in database searching, choose a Maximum ion injection time (IT) of dd-MS2 (data dependent-MS2) to 200 ms for the accumulation of sufficient ions for detection.
For proteins that are large and give a large number of peptides upon enzymatic digestion, increase the LC gradient length (note that the method duration in the global settings and the run time in general need to be updated consistent with the LC gradient length). Increasing TopN in dd-MS2 to 15 and increasing the dynamic exclusion in dd settings to up to 30.0 s are recommended for better sequence coverage. For critical peptide whose product-ion (MS/MS) spectral quality is insufficient for a positive identification, scanning the Normalized Collision Energy (NCE) in the dd-MS2 from 25 to 35 for an optimized fragmentation is recommended. For proteins that are insufficiently digested, whose resulting peptides are usually multiply charged, changing the charge exclusion in dd settings to “unassigned, >8” is recommended.
In principle, many mass spectrometers (not only quadrupole-orbitraps) that are currently used for peptide analysis (with MS/MS capability, high sensitivity, and compatibility with HPLC separation) are suitable. Compatibility with HPLC here indicates that the MS/MS of the mass spectrometer needs to be sufficiently sensitive and fast to capture, fragment, and measure the fragment ions. In other words, the time it takes for a MS/MS measurement should be shorter than the peak width of a single species in the HPLC separation so that all existing species, especially isomeric peptides identical in sequence but bearing +16 or other labeling on different residues can be fragmented by the mass spectrometer. A good rule of thumb is that the instrument is sufficient for fragmenting 10 precursor ions per chromatography peak. Under these requirements, some Fourier-transfer ion-cyclotron resonance instruments are not adequate for FPOP analysis owing to their relatively long signal acquisition time to achieve high mass resolving power.
Software
Required software includes that for both instrument control and data processing. Instrument control software is usually specific. FPOP data processing includes searching the database, identifying MS/MS spectra, generating extracted ion chromatograms (EICs), assigning chromatographic peaks, and integrating LC chromatographic peaks for quantification. Commercially available software can facilitate these tasks, and they include but are not limited to PEAKS studio (Bioinformatics Solutions), Proteome Discoverer (Thermo Scientific), XCalibur (Thermo Scientific), Mass Spec Studio (University of Calgary), Mascot (Matrix Science), Protein Metrics suite (Protein Metrics), and Protein Prospector (University of California, San Francisco). We recommend using the Protein Metrics suite because it is industry-leading for processing protein covalent labeling data and provides user-friendly data visualization.
Bruker Data Analysis (Version 4.2, Build 383.1)
Bruker Otof control (Version 3.4, Build 14)
Flux Instruments Janeiro II (Version 2.6, Build 2.60.0007)
GAM Laser, Inc. Excimer Laser Control Software (Version 3.70)
Ophir Optronics USBI (Version 1.11, Build 5)
Protein Metrics Product Suite, Byonic and Byologic (Version v3.7-5-g0ce0c0fde2 x64)
Thermo Scientific Xcalibur (Version 4.0.27.19)
Software setup
The software setup mainly focuses on the proper setup of the database searching for data processing at peptide and residue level (as stated above, the settings presented here are for the Protein Metrics suite). The underlying principles for the settings, however, are applicable to other data processing software listed in the previous section.
Byonic by Protein Metrics is for database searching. Start a database search by preparing a .fasta file that includes not only the protein of interest, but also other related proteins or proteins that may exist in the sample. Together with the datafile (.raw by Thermo Scientific Q Exactive), carry out a database search, assign the product-ion (MS/MS) spectra, and identify the location of the FPOP modifications (key search parameters are presented in Table 5). Note that the sample digestion is exemplified by the commonly used trypsin. Change the protease to meet the specific need and change the search parameters accordingly.
Table 5 |.
Key parameters in database searching by Byonic
| No. | Parameter | Value | No. | Parameter | Value |
|---|---|---|---|---|---|
| Sample Digestion | Instrument Parameters | ||||
| 1 | Cleavage Site (s) | KR | 5 | Precursor Mass Tolerance | 10 ppm |
| 2 | Cleavage Side | C-terminal | 6 | Fragmentation Type | Both CID & HCD |
| 3 | Digestion Specificity | Sully Specific (Fastest) | 7 | Fragment Mass Tolerance (CID) | 60 ppm |
| 4 | Missed Cleavages | 2 | 8 | Fragment Mass Tolerance (HCD) | 60 ppm |
Another key consideration for database searching is the modifications (a complementary summary of •OH-based FPOP modifications can be found in review articles by Xu and Chance16 in 2007 and by Liu, Zhang, and Gross18 in 2020). The most commonly seen FPOP modifications and their corresponding software “settings” are in Table 6 (note that the modifications in the table are FPOP-induced). All known post translational modifications (PTMs) should be included, if needed, as fixed modifications together with the FPOP-induced modifications.
Table 6 |.
Common FPOP-induced modifications and their settings in Byonic
| No. | Modifications | Targets | Fine Control |
|---|---|---|---|
| 1 | FPOP Oxidation Usual Suspects/+15.994915 | F, H, I, L, M, V, W, Y | Variable – common 1 |
| 2 | FPOP Dioxidation/+31.989829 | F, M, W | Variable – common 2 |
| 3 | FPOP Oxidation Less Common/+15.994915 | D, E, K, N, P, Q, R | Variable – rare 1 |
| 4 | FPOP Oxidation Acid Loss CO2/−43.989829 | D, E | Variable – rare 2 |
| 5 | Carbamidomethyl/+57.021464 | C | Fixed |
Total Common Max: 2, Total Rare Max: 2
As disulfides commonly occur, include carbamidomethylation by IAM as a fixed modification when digesting proteins with disulfide linkages followed by alkylation of resulting free cysteines by IAM.
▲CRITICAL STEP We recommend executing an wildcard search prior to the modification search to identify all possible PTMs and include all of them in the final modification search to increase the sequence coverage, identification confidence, and quantification precision.
Procedure
Protein footprinting by FPOP • TIMING 2 - 4 h
Step 1 - 12 describe the general workflow for a successful FPOP protein footprinting. For an unknown protein system, titrate with H2O2 to establish the optimal conditions as described in step 13. For all protein systems, include three control experiments, including a protein negative control, a no-laser control, and a no-H2O2 control, as described in steps 14, 15, and 16, respectively.
-
1
Switch on the laser, pulse generator, controlling computer, and syringe pump. Allow laser to warm up (for the KrF laser, ~ 8 min).
-
2
Prepare an ice chest for storing pre-made L-histidine, L-methionine solutions. Obtain the protein stock solution (50 μM, in PBS buffer) and the catalase stock solution (0.25 – 0.63 units/μL, in PBS buffer) from the −80 °C freezer and allow them to defrost slowly in the ice chest.
-
3
Prepare the 200 mM H2O2 working solution as induced above. Mix 20 μL H2O2 stock solution with 980 μL PBS buffer in a 1.5 mL protein LoBind tube, vortex for 10 s for a thorough mix. Label the tubes and situate them in the ice chest.
-
4
Obtain three secondary solution containers (20 mL vials), fill two with PBS buffer and label as “PBS buffer” and “aqueous wash”. Fill third container with a mixture of PBS buffer/acetonitrile (50/50, by volume), label as “organic wash”.
-
5
Use a 100 μL Hamilton glass syringe to wash the capillary by flushing twice with the “aqueous wash”, followed by flushing twice with the “organic wash”, and complete by flushing twice again with the “aqueous wash”. Each flush should contain 70 – 80 μL of liquid. ▲CRITICAL STEP The flow pathway needs to be thoroughly rinsed to minimize carryover.
? TROUBLESHOOTING
-
6
Measure the laser energy by placing the pyroelectric energy sensor immediately at the laser outlet (before passing any laser optics, Figure 2a). Initiate a laser sequence with 50 laser shots at 7.2 Hz. Tune the laser energy by changing the excitation voltage of the laser to achieve an average energy of 25 mJ. Record the average and standard deviations of the energies obtained from the 50 shots (standard deviations are usually ~ 200 μJ). ▲CRITICAL STEP The laser energies need to be comparable between runs to allow accurate comparisons. They should be measured and fixed at the beginning of the day of experiments.
? TROUBLESHOOTING
-
7
Stick a self-adhesive arrow flag onto a supporting brick and take a “burn mark” on the red arrow flag (Figure 3c) as described above. Move the linear stage to allow the capillary to be located at the center of the laser path. When the laser is aligned, use the Vernier calipers to measure the width of the laser spot (the burn mark on the red arrow flag). The width of the spot can be adjusted with the iris. ▲CRITICAL STEP The laser needs to be well-aligned for an optimal yield of radicals. Burn multiple arrow flags if necessary. A typical laser-spot-width ranges from 1.5 to 2.5 mm.
-
8Determine the liquid flow rate by Eq. 1–3 and adjust syringe pump accordingly. Calculate the required laser shots by Eq. 4 shown below:
(4) Input the calculated number of laser shots into the laser control software to complete the setup.
-
9
Obtain two 0.5 mL Protein LoBind tubes, fill tube 1 with 10 μL 70 mM L-methionine and 1.0 μL 0.25-0.63 units/μL catalase. Add to tube 2 36.5 μL PBS buffer (from secondary container “PBS buffer”), 10 μL, 50 μM calmodulin stock solution (final concentration of 10 μM), 1.0 μL, 50 mM L-histidine solution (final concentration of 1 mM). ▲CRITICAL STEP Add the PBS buffer first, then pipet the protein and the scavenger, L-histidine, solutions into the existing buffer to achieve precise liquid transfer. If many experiments are planned in series, the aliquots can be prepared together.
-
10
Add 2.5 μL, 200 mM H2O2 solution into tube 2 (final concentration of 10 mM) to complete the recipe. Vortex both tubes (10 s) and spin down the aliquots (10 s). Use the Hamilton syringe to draw all 50 μL liquid from tube 2 and load in the syringe pump (discard tube 2). Place tube 1 at the end of the capillary as a collection tube. Trigger both laser and syringe pump to initiate FPOP. ▲CRITICAL STEP If there are many parallel experiments, do not add H2O2 until immediately prior to laser irradiation. Ensure that the outlet of the capillary is submerged in the liquid in the collection tube to achieve an efficient quench of the remaining radicals and H2O2. For each condition, execute two or more replicates to assess precision.
? TROUBLESHOOTING
-
11
After laser irradiation, the sample in tube 1 is a solution of the footprinted protein. Allow the sample to sit at room temperature for 1-3 min. You will usually observe gas bubbles at the inner wall of the tube. Vortex for 10 s and spin-down for 10 s to remove the gas bubbles. ▲CRITICAL STEP Gas bubbles are composed of oxygen from the catalase-mediated H2O2 decomposition. Owing to the limited yield of photodissociation, there will always be residual H2O2 even after laser irradiation, and the gas bubbles on the inner wall of the tube are a good measure of the effectiveness of catalase. H2O2 is an oxidant and excess amount of H2O2 after laser irradiation needs to be removed promptly, even before low-temperature storage,61 for a precise FPOP footprint.
? TROUBLESHOOTING
■PAUSE POINT The labeled protein sample can be stored at 4 °C for 1 week or at −80 °C for up to three months.
-
12
While waiting for catalase mediated H2O2 decomposition (described in step 11), repeat step 5 to clean the aliquot flow path and prepare it for a subsequent sample.
-
13
In practice, fix the protein concentration and scavenger concentration (10 μM and 1 mM in this protocol, respectively). For an unknown protein, start the titration of H2O2 from zero and continue to 10,000 to 20,000 times the protein concentration by changing the amount of added H2O2. Maintain the total volume of the aliquot at 50 μL. ▲CRITICAL STEP The optimal H2O2 concentration is determined after the measurement of protein oxidation extents as a function of H2O2 concentration at the global (whole protein) level. The optimal condition should afford enough labeling but not yet reach the plateau in the titration curve (modification fraction vs. H2O2 concentration). The titration curve is protein-dependent as it is closely related to the number of •OH-modifiable sites on its solvent accessible surface. The titration curve also depends on the protein concentration and can level-off at different [H2O2]:[protein] when the protein concentrations are different.
-
14
For the protein negative control sample, mix together 39.0 μL PBS buffer, 10 μL of 50 μM calmodulin solution, 1.0 μL of 50 mM L-histidine solution, 10 μL of 70 mM L-methionine, and 1.0 μL 0.25-0.63 units/μL catalase in a 0.5 mL Protein LoBind tube.
-
15
For the no-laser control, prepare two 0.5 mL Protein LoBind tubes and follow the recipe described in step 9. Upon completion, follow the guidelines in step 10 to add H2O2, then vortex and spin-down both aliquots, load the syringe, and attach the collection tube. Trigger only the syringe pump and allow the aliquot to run through the capillary and finally into the collection tube. Do not trigger the laser for these experiments.
-
16
In the no-H2O2 control experiment, obtain two 0.5 mL Protein LoBind tubes, fill tube 1 with 10 μL 70 mM L-methionine and 0.5 μL 0.25 – 0.63 units/μL catalase. Add into tube 2 containing 39.0 μL PBS buffer, 10 μL, 50 μM calmodulin stock solution (final concentration of 10 μM), 1.0 μL, 50 mM L-histidine solution (final concentration of 1 mM). Do not add H2O2 for this control. Vortex both tubes (10 s) and spin down the aliquots (10 s). Draw all 50 μL liquid from tube 2 into the Hamilton syringe and load onto the syringe pump (discard tube 2). Place tube 1 (collection tube) at the end of the capillary. Trigger both laser and syringe pump flow.
Global level characterization of FPOP-labeled protein and condition determination • TIMING 1 - 4 h
-
17
Prepare the LC mobile phase and configure the two-valve LC system as described above. Attach the C-8 column and equilibrate it by flushing the column with the intended LC gradient for at least three consecutive cycles. Meanwhile, keep the loading pump running at 200 μL/min (sample loading solution as instructed above). ▲CRITICAL STEP Both valves should be in the “injection” position (Figure 4).
-
18
Obtain three secondary solution containers (20 mL vials), fill two with 0.1% (vol/vol) LC-MS-grade formic acid in HPLC water and label them with “water/formic acid-sample” and “water/formic acid-wash”. Fill the third container with acetonitrile, label the container as “ACN-wash”.
-
19
Obtain a 100 μL Hamilton glass syringe, wash the sample loop by flushing twice with the “water/formic acid-wash”, followed by flushing twice with the “ACN-wash”, and complete by flushing twice more with the “water/formic acid-wash”. Each flush should contain 70 – 80 μL of liquid. ▲CRITICAL STEP The sample loop needs to be thoroughly rinsed to minimize carryover. Both valves should be in the “injection” position (Figure 4).
-
20
Obtain a 0.5 mL Protein LoBind tube, add to it 48.2 μL of “water/formic acid-sample” and 1.8 μL of FPOP solution (~15 pmole), vortex and spin-down. Use the syringe to draw up all 50 μL solution and inject it into the LC sample loop.
-
21
Switch both valves to the “loading” position and start a 3 min timer immediately after the switch. The loading pump now pushes the sample loading solution through the sample loop to the C-8 column. As the protein in the mix is trapped by the C-8 column under aqueous-phase conditions, this loading process desalts the mixture. Setup the mass spectrometer (Bruker MaXis) by loading the proper acquisition parameters and naming the data file.
-
22
After 3 min of desalting, switch both valves back to the “injection” position. Start the LC gradient and the instrument acquisition simultaneously (refer above for setting up instrument parameters). Monitor the LC gradient pump pressure to assure that the LC system is working properly. Repeat step 19 to clean- the sample loop and prepare it for the next injection. ▲CRITICAL STEP Monitoring the LC pressure to check on gradient so that the protein elutes at 3.5 – 5.0 min. Note that the signal intensities for labeled samples are usually lower those of control samples because the FPOP oxidation disperses the protein into multiple oxidation states, leading to a decrease in the signal intensity of the unmodified protein when comparing the most intense peaks at two different conditions.
? TROUBLESHOOTING
Post-labeling sample handling and protease digestion • TIMING 24 h
Post-labeling protein handling includes acetone precipitation to clean-up the protein mixture, urea denaturation of the protein, and trypsin digestion. These steps are described in steps 23-27 and 30-33. For proteins with disulfide bonds, reduce and alkylate prior to adding the protease. These steps are described as 28 and 29.
-
23
Fill a secondary solvent container with acetone, store at −20 °C for at least 6 h for temperature equilibration.
-
24
Obtain a 0.5 mL protein LoBind tube, add 12.2 μL of FPOP-labeled protein solution (~ 100 pmole), followed by 100 μL of cold acetone, vortex and spin-down (10 s each). Store the mixture at −20 °C for 10 hours. ▲CRITICAL STEP One should see cloudy protein precipitate immediately after adding the cold acetone.
-
25
Spin-down the mixture with the high-speed centrifuge at 4 °C, 15000 relative centrifugal force (rcf) for 10 min. Remove the supernatant by pipet, leaving 5-10 μL of liquid together with the protein pellet at the bottom of the tube. !CAUTION For some proteins, the pellet may not be seen. If not visible, remove by pipet 100 μL of liquid from the upper part of the liquid mixture.
-
26
Situate the tube with the protein pellet and remaining liquid in a SpeedVac, set the temperature at 25 °C, and evaporate the remaining liquid. This usually takes less than 20 minutes.
-
27
Remove the tubes containing the dried protein pellet, add 10 μL of 8 M urea solution to cover the protein pellet, vortex and spin-down (10 s each). Leave the mixture in the incubator for 30 min. Set the incubator at 25 °C and 500 rpm for a complete protein denaturation.
-
28
Add 2 μL of 100 mM DTT solution into the tube, vortex and spin-down (10 s). Leave the mixture in the incubator for 30 min. Set the incubator at 37 °C and 500 rpm for a complete disulfide reduction.
-
29
Add 2 μL of 300 mM IAM solution to the tube, vortex and spin-down (10 s each). Leave the mixture in the incubator and wrap the incubator with aluminum foil to minimize light exposure. Allow 30 min for a complete alkylation of free cysteine. Set the incubator at 25 °C and 500 rpm.
-
30
Prepare a 0.5 μg/μL enzyme stock solution by reconstituting the lyophilized enzyme powder (20 μg) with 40 μL HPLC water.
-
31
Add 89.7 μL of PBS buffer to the mixture (85.7 μL if there are disulfide bonds) so that the final urea concentration is less than 1 M. Add 0.35 μL of 0.5 μg/μL enzyme stock solution to the protein mixture so that the final weight ratio between the protein and the enzyme is 10:1 (total volume at this point is 100 μL). ▲CRITICAL STEP The amount of enzyme is appropriate for calmodulin and should be adjusted based on the molecular weight of the protein. The protein:enzyme weight ratio should be 10 : 1.
-
32
Situate the mixture in the incubator for 12 h, set at 37 °C and 500 rpm for a complete protein digestion.
-
33
Upon completing the digestion, add 1 μL of formic acid to quench the enzymatic digestion. The solution is now ready for further peptide and residue-level MS analysis (peptide concentration is approximately 1 μM/μL).
■PAUSE POINT The digested protein sample can be stored at 4 °C for 2 days or at −80 °C for up to one month.
Peptide and residue-level characterization of FPOP-labeled protein • TIMING 2 h
-
34
Obtain an autosampler vial, add 25 μL of digested sample, place in the low speed centrifuge at 8000 revolutions per minute (rpm) for 3 min to spin down any large particles that may clog the injection needle of the autosampler.
-
35
Obtain a second autosampler vial, add 100 μL of 0.1% formic acid (v/v) in water. Mark the vial as “Blank”.
-
36
Load samples to the autosampler rack, setup the sample sequence, and start the LC elution and MS acquisition. The injection volume is 5 μL so that each injection contains ~ 5 pmol of protein. The LC gradient length ranges from 60 to 120 mins depending on the complexity of the analyte. For the LC gradient and instrument parameters, refer to “LC-MS Setup” section.
-
37
After each sample injection, conduct a “Blank” run to minimize sample carryover.
Data analysis • TIMING 2 d
-
38
Open the global-level data file (recorded by Bruker MaXis) by Bruker Data Analysis software, extract the EIC for m/z = 1050.3750 ± 0.0050, which represents the average m/z of the +16 state of unmodified calmodulin. ▲CRITICAL STEP One should extract a different m/z ion for a different protein of interest.
-
39
Integrate the chromatographic peaks of the EIC, locate the ion cluster for further processing. Zoom in the specific region of an ion cluster and identify the average mass for the unmodified protein.
-
40Based on the average mass of the unmodified peptide, compute the masses of the +16, +32, +48, and +64 modified peaks, read out their intensities from the software. The global level modification fraction can be calculated as:
(5) ▲CRITICAL STEP Account for all resolvable oxidations. As the number of oxidations increase, the intensities become smaller.
? TROUBLESHOOTING
-
41
Set up the Byonic software as instructed above, load the datafile from the Q Exactive, execute the database search. ▲CRITICAL STEP Include multiple proteins in the .fasta database to increase the searching confidence.
? TROUBLESHOOTING
-
42
Load the search output together with the datafile (.raw) from the Q Exactive into Biologic software, select the proper protein entry in the .fasta database, and create a Biologic file for further processing.
-
43
Open the newly created Biologic file, examine all entries (MS/MS identifications) by manually validating the product-ion (MS/MS) spectra. Mark and differentiate the entries as “true positive” and “false positive”.
▲CRITICAL STEP The “true positive” and “false positive” assignments should be justified based on precursor m/z error (with respect to theoretical value) and the quality of the product-ion spectrum. Specifically, a “true positive” entry should have a matching precursor ion mass and confident MS/MS supporting assignment of its modified residue. Confident MS/MS that locates +16 modification to a specific residue in a peptide should have either a series of b- or y-ions or both to support assignment (e.g., an assignment of +16 on methionine 3 on peptide 1-10 should at least have both b2 and b3 ions or both y7 and y8 ions). Moreover, all major peaks should be assignable. Examples of confident product-ion (MS/MS) spectra are provided in Figure 6b.
? TROUBLESHOOTING
-
44
After verifying all the entries, filter the results to show only the “true positive” entries.
-
45Rank the entries by the start and ending numbers of the amino acid in the sequence. Identify a peptide group that includes the unmodified, the +16 modified species, and the +32 modified species (if available) at the same charge state, and work with this group in the analysis. Peptide-level oxidation extents can be calculated by obtaining the integration areas of the EICs for the unmodified, +16 modified, and +32 modified species and calculated by using the following equation:
(6) ▲CRITICAL STEP A peptide with a mass shift of +48 or more is rare. If there are more than two oxidations (e.g., +48 mass shift or more), account for all of them. In the calculation, include in-source oxidation as part of the signal for an unmodified species. In-source oxidation is usually identified as identical chromatographic peak shape and elution time as those of the unmodified peptide.
-
46For the group of peptides identified in step 45, sort all chromatographic signals in the EIC of the +16 and +32 species. Assign each available peptide signal by using the MS/MS spectra that were taken at the specific LC elution time of that peptide (an example is provided in Figure 6). In other words, relate a “true positive” MS/MS spectrum to the LC elution profile. Once assigned, start a worksheet by grouping the resolvable chromatographic peaks. Integrate each and record the integration areas on the worksheet for calculating residue-level modification by using equation 7.
(7) Selected residues (including but not limited to phenylalanine, tryptophan, tyrosine, leucine, isoleucine) usually appear as multiple chromatographic signals of +16 modification because there are multiple +16 modifiable sites for these residues, resulting in isomers that may elute differently in the LC separation. In such cases, all available chromatographic signals should be accounted for.
▲CRITICAL STEP In general, a high-confidence chromatographic signal assignment usually includes two or more confident MS/MS spectra at the elution time of a specific peptide. It is also commonly seen that the MS/MS spectra for a specific modified species appear at several LC elution times. In such cases, this specific modification is not resolvable at the residue level. An alternative quantification approach is to fragment the peptide through electron transfer dissociation (ETD) and quantify the labeled residues through peak intensities of corresponding ETD fragments.62, 63
Fig. 6 |. Anticipated FPOP results.
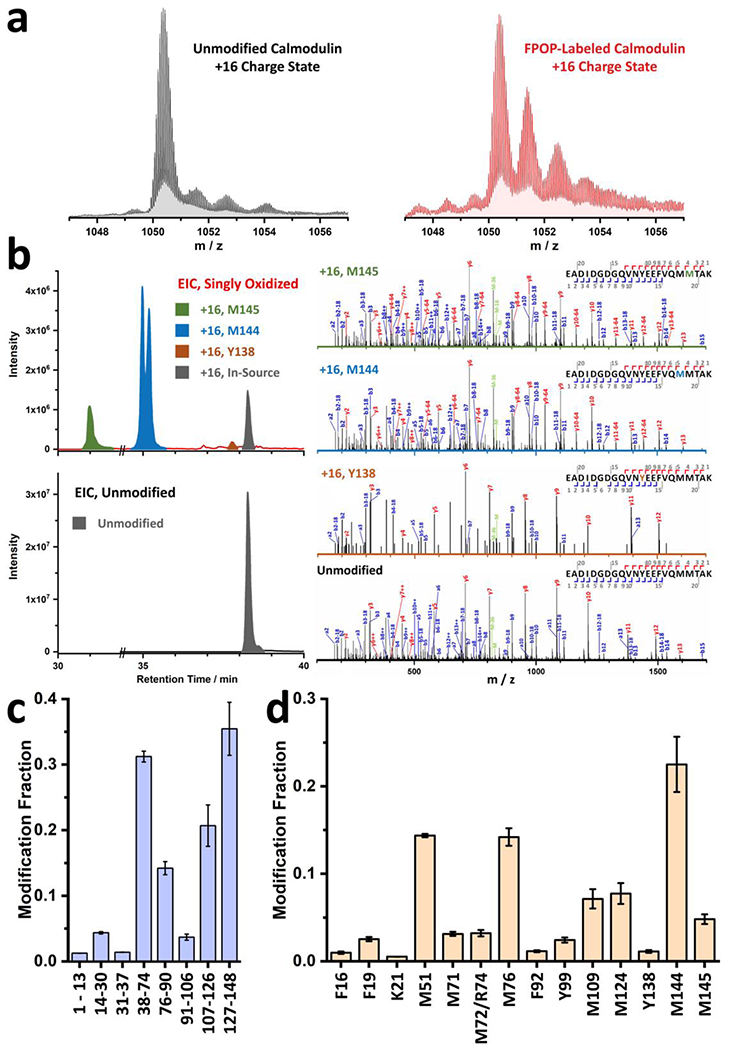
a, Global level spectra for unmodified (bottom, black) and FPOP-labeled (top, maroon) calmodulin in + 16 charge state. b, Sample EICs for unmodified (bottom, black) and +16 modified (top) peptide 129-148 from FPOP-labeled calmodulin. EIC for modified species are colored in olive, blue and orange to represent the +16 modifications on residue M145, M144, and Y138, respectively. Representative product-ion (MS/MS) spectra supporting these assignments together with spectrum for the unmodified peptide are provided on the right. c, Peptide-level FPOP results of calcium-free calmodulin with 99% sequence coverage. d, Residue-level FPOP results of calcium-free calmodulin with 14 resolvable residues. Error bars in c and d are standard deviations from two replicates.
Timing
Steps 1-12, footprint the protein by FPOP: 1-1.5 h for reagent and equipment setup, 10 min per sample
Steps 13-16, optimize FPOP labeling conditions: 2 h (depending on number of titration points and number of replicates)
Steps 17-22, measure the FPOP-labeled proteins at the protein level: 40 min for LC equilibration and instrument setup, 20 min per sample
Steps 23-33, conduct post-labeling acetone precipitation and protease digestion: 24 h
Steps 34-37, measure labeled proteins at peptide/residue level: 30 min for instrument setup, 90-120 min per sample
Steps 38-46, analyze data: 0.2 – 2 days per sample (depending on complexity of the sample and depth of analysis)
Troubleshooting
Troubleshooting advice can be found in Table 7.
Table 7 |.
Troubleshooting Table
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 5 | Capillary is clogged | Salt in wash buffer crystallizes after previous use owing to solvent change | Replace glass capillary |
| 6 | Laser energy is lower than needed, even at the maximum voltage of laser | The fluorine/neon gas is low | Refill the fluorine/neon gas cylinder |
| The pyroelectric energy sensor is mis-aligned or needs cleaning | Check to ensure the pyroelectric sensor is positioned properly and is clean | ||
| 10 | The transparent window on the capillary darkens quickly, or the capillary breaks after a few runs | The laser is focused on the capillary instead of the beam stop | Check the laser optics to ensure they are well aligned and secured in proper positions |
| 11 | There are few gas bubbles after the solution containing the footprinted protein gets mixed with the quenching solution in Tube 2 | The catalase is no longer active | Obtain a 0.5 mL protein LoBind tube and add 8.0 μL 0.25 – 0.63 units/μL catalase and 20 μL 200 mM H2O2 solution to check the catalase activity. Prepare new catalase solution if necessary |
| The H2O2 stock solution is less concentrated than expected | Check storage conditions and expiration date of the H2O2 stock solution. Obtain new batch if necessary | ||
| The amount of H2O2 in the current FPOP recipe is not enough | Run a parallel experiment by doubling the amount of H2O2 and check the global-level oxidation. If the oxidation fraction increases, execute a H2O2 titration as described in step 13 to optimize the H2O2 concentration | ||
| 22 | The protein signal is low as determined by global level measurement | The LC system is clogging | Disassemble the LC system and check for points of clogging. Replace tubing, ferrules and nuts if necessary |
| The protein is heavily oxidized, changing its hydrophobicity and causing poor trapping in desalting step (a heavily oxidized protein is seen with modifications of +16, +32, +48,…; excess oxidation disperses further the signal, leading to a decrease in signal intensities | Optimize the H2O2 concentration by executing a H2O2 titration as described in step 13 | ||
| 40 | The protein is barely oxidized in the global-level measurement | The FPOP recipe is not optimized; H2O2 concentration is insufficient | Run an H2O2 titration as described in step 13 to optimize the H2O2 concentration |
| 41 | The sequence coverage is low | Execute an in silico digestion with an online server (e.g. https://web.expasy.org/prptide_mass). Open the data file with XCalibur and manually search for missing peptides by using m/z’s from the in silico digestion list | |
| If peptides can be located by their precursor m/z, the digestion is likely adequate, but the instrument failed to trigger MS/MS on that peptide | Optimize instrument parameters by increasing maximum IT or by changing NCE for better MS/MS. It is also possible that the peptide of interest is coeluting with higher abundance species. If so, create a precursor-ion inclusion list of the m/z values from in silico digestion | ||
| If the peptides cannot be located by their precursor m/z, the digestion is inadequate | Check the digestion conditions, switch to a different protease if necessary. From calculation of the hydrophobicity of missing peptides, optimize the LC gradient to better capture them | ||
| The mass measurements errors are greater than the search criteria | Calibrate the mass spectrometer by following the manufacturer’s protocol | ||
| There are unidentified post translational modifications on the missing peptide | Execute a wildcard search to identify possible PTMs and add newly identified PTMs to the modification list as fixed modifications | ||
| 43 | The product-ion (MS/MS) spectra are of poor quality, insufficient for residue-level identification | The precursor peptides are not sufficiently fragmented | Execute a series of injections with different NCEs to optimize the value. Increase the maximum IT during MS/MS acquisition. As a last resort, create a precursor-ion inclusion list that includes expected peptides and switch off the dynamic exclusion during acquisition so that the precursors in the inclusion list are fragmented multiple times during acquisition. Alternatively, switch to a different digestion enzyme to obtain a new set of peptides that may give improved fragmentation |
Anticipated Results
Figure 6 above summarizes the anticipated FPOP results, consisting of global level spectra of unmodified and FPOP-labeled calmodulin (Figure 6a), an example of peptide-level EIC and its assignment for residue-level quantifications (Figure 6b, left), representative spectra to support peak assignments of the peptide-level EICs (Figure 6b, right), and a summary of the labeling fraction at both peptide and residue levels (Figure 6c and d). Modification fractions are correlated with both solvent accessibilities and intrinsic reactivities. When executing FPOP labeling, differences in modification for the same peptide report the changes in solvent accessibilities under different conditions. When possible, the absolute modification fractions/differences under different conditions can be mapped onto available protein structural models for better visualization.
In the supplementary information, we provide a full dataset of FPOP-labeled calmodulin, containing global-level measurements of unmodified and FPOP-labeled calmodulin, and a peptide/residue level measurement of the FPOP-labeled calmodulin (digested by trypsin, in duplicate). This dataset and the results presented in Figure 6 can be taken as references to establish a data-processing workflow when using other software.
Supplementary Material
Supplementary Figure 1. Dimensions of the custom-made capillary holder. The figure is drawn to scale.
Supplementary Figure 2. Detailed drawing of the optics setup and their alignments. The figure is drawn to scale.
Fig. 5 |. Schematic illustration of the FPOP workflow.
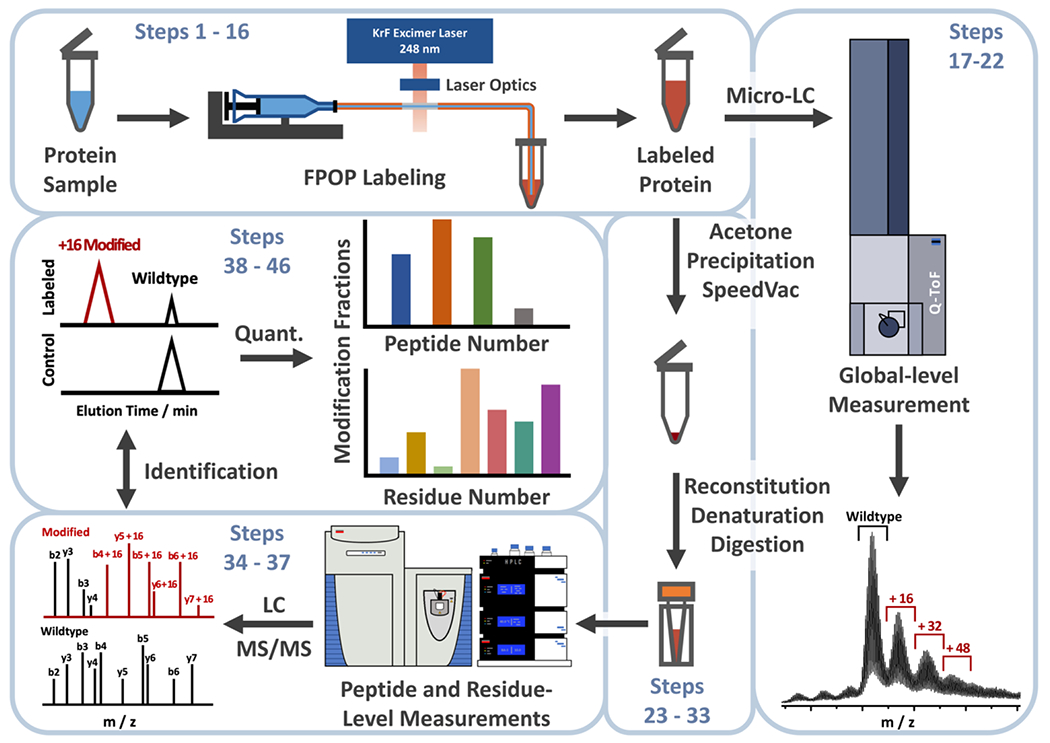
Steps 1-16 describe the protein footprinting by FPOP, 17-22 cover the global level analysis of FPOP-labeled protein sample, 23-33 show the post-labeling sample handling and protease digestion, 34-37 represent the LC-MS/MS analysis of the FPOP-labeled sample at the peptide and residue levels, and finally steps 38-46 denote the data processing and final presentation.
Table 2 |.
Key parameters for global-level FPOP analysis by a Bruker MaXis 4G mass spectrometer
| No. | Parameter | Value | No. | Parameter | Value |
|---|---|---|---|---|---|
| 1 | Ion Polarity | Positive | 8 | Dry Gas | 8.0 L/min |
| 2 | Scan Mode | MS | 9 | Dry Temp. | 200 °C |
| 3 | Mass Range | 300 – 3000 m/z | 10 | Transfer Funnel 1 RF | 400.0 Vpp |
| 4 | Spectral Rate | 1.00 Hz | 11 | Transfer isCID Energy | 0.0 eV |
| 5 | End Plate Offset | 500 V | 12 | Transfer Multiple RF | 400.0 Vpp |
| 6 | Capillary | 3800 V | 13 | Quadrupole Ion Energy | 3.0 eV |
| 7 | Nebulizer | 1.2 Bar | 14 | Quadrupole Low Mass | 100.0 m/z |
Acknowledgements
This work was supported by the National Institutes of Health NIGMS Grant 5P41GM103422 and R24GM135766. Instrumentation was supplied by 1S10OD016298-01A1 (to M.L.G.). We are grateful to Dr. Henry Rohrs for the help in identifying parts for the HPLC and to Protein Metrics for software support.
Footnotes
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Software Availability
The Protein Metrics Suite is commercially available at https://www.proteinmetrics.com.
Competing Interests
The authors declare an ongoing collaboration with Protein Metrics in establishing an HDX data processing platform, a topic that is not related to this protocol. The recommendation of using Protein Metrics as preferred data processing software for FPOP data predates the ongoing collaboration.
Supplementary information is available for this paper at XXX.
Data Availability
A calmodulin dataset that contains a global level measurement by Bruker MaXis, a peptide and residue level measurement by Thermo Scientific Q Exactive is freely available online in the Mendeley Data (https://data.mendeley.com/) with DOI 10.17632/xfd5sh76pm.1.
References
- 1.Anfinsen CB, Haber E, Sela M & White FH THE KINETICS OF FORMATION OF NATIVE RIBONUCLEASE DURING OXIDATION OF THE REDUCED POLYPEPTIDE CHAIN. Proceedings of the National Academy of Sciences 47, 1309–1314 (1961). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drenth J Principles of protein X-ray crystallography. (Springer Science & Business Media, 2007). [Google Scholar]
- 3.Wüthrich K The way to NMR structures of proteins. Nature Structural Biology 8, 923–925 (2001). [DOI] [PubMed] [Google Scholar]
- 4.McPherson A Introduction to protein crystallization. Methods 34, 254–265 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Cheng Y Single-Particle Cryo-EM at Crystallographic Resolution. Cell 161, 450–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai X. c., McMullan G & Scheres SHW How cryo-EM is revolutionizing structural biology. Trends in Biochemical Sciences 40, 49–57 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Merk A et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell 165, 1698–1707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenfield NJ Using circular dichroism spectra to estimate protein secondary structure. Nature Protocols 1, 2876–2890 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noble JE & Bailey MJA Chapter 8 Quantitation of Protein, in Methods in Enzymology, Vol. 463. (eds. Burgess RR & Deutscher MP) 73–95 (Academic Press, 2009). [DOI] [PubMed] [Google Scholar]
- 10.KONG J & YU S Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures. Acta Biochimica et Biophysica Sinica 39, 549–559 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Stetefeld J, McKenna SA & Patel TR Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophysical Reviews 8, 409–427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendoza VL & Vachet RW Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrometry Reviews 28, 785–815 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konermann L, Pan J & Liu Y-H Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chemical Society Reviews 40, 1224–1234 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Maleknia SD & Downard KM Advances in radical probe mass spectrometry for protein footprinting in chemical biology applications. Chemical Society Reviews 43, 3244–3258 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Bolla JR, Agasid MT, Mehmood S & Robinson CV Membrane Protein–Lipid Interactions Probed Using Mass Spectrometry. Annual Review of Biochemistry 88, 85–111 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Xu G & Chance MR Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev 107, 3514–3543 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Kaur U et al. Evolution of Structural Biology through the Lens of Mass Spectrometry. Analytical Chemistry 91, 142–155 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu XR, Zhang MM & Gross ML Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chemical Reviews (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelleher NL et al. Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry. Journal of the American Chemical Society 121, 806–812 (1999). [Google Scholar]
- 20.Espino JA & Jones LM Illuminating Biological Interactions with in Vivo Protein Footprinting. Analytical Chemistry 91, 6577–6584 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West GM et al. Quantitative proteomics approach for identifying protein–drug interactions in complex mixtures using protein stability measurements. Proceedings of the National Academy of Sciences 107, 9078–9082 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin L, Wang D, Gooden DM, Ball CH & Fitzgerald MC Targeted Mass Spectrometry-Based Approach for Protein–Ligand Binding Analyses in Complex Biological Mixtures Using a Phenacyl Bromide Modification Strategy. Analytical Chemistry 88, 10987–10993 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Katta V, Chait BT & Carr S Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Communications in Mass Spectrometry 5, 214–217 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z & Smith DL Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Science 2, 522–531 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Englander SW & Kallenbach NR Hydrogen exchange and structural dynamics of proteins and nucleic acids. Quarterly Reviews of Biophysics 16, 521–655 (1983). [DOI] [PubMed] [Google Scholar]
- 26.Schanda P & Brutscher B Very Fast Two-Dimensional NMR Spectroscopy for Real-Time Investigation of Dynamic Events in Proteins on the Time Scale of Seconds. Journal of the American Chemical Society 127, 8014–8015 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Wales TE & Engen JR Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrometry Reviews 25, 158–170 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Engen JR Analysis of Protein Conformation and Dynamics by Hydrogen/Deuterium Exchange MS. Analytical Chemistry 81, 7870–7875 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson KW, Gallagher ES & Hudgens JW Automated Removal of Phospholipids from Membrane Proteins for H/D Exchange Mass Spectrometry Workflows. Analytical Chemistry 90, 6409–6412 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Möller IR et al. Improving the Sequence Coverage of Integral Membrane Proteins during Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. Analytical Chemistry 91, 10970–10978 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Jensen PF et al. Removal of N-Linked Glycosylations at Acidic pH by PNGase A Facilitates Hydrogen/Deuterium Exchange Mass Spectrometry Analysis of N-Linked Glycoproteins. Analytical Chemistry 88, 12479–12488 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Li KS, Shi L & Gross ML Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization. Accounts of Chemical Research 51, 736–744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maleknia SD, Brenowitz M & Chance MR Millisecond Radiolytic Modification of Peptides by Synchrotron X-rays Identified by Mass Spectrometry. Analytical Chemistry 71, 3965–3973 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Hambly DM & Gross ML Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom 16, 2057–2063 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Liu XR, Zhang MM, Rempel DL & Gross ML A Single Approach Reveals the Composite Conformational Changes, Order of Binding, and Affinities for Calcium Binding to Calmodulin. Analytical Chemistry 91, 5508–5512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li KS, Rempel DL & Gross ML Conformational-sensitive fast photochemical oxidation of proteins and mass spectrometry characterize amyloid beta 1–42 aggregation. Journal of the American Chemical Society 138, 12090–12098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Rempel DL, Gau BC & Gross ML Fast Photochemical Oxidation of Proteins and Mass Spectrometry Follow Submillisecond Protein Folding at the Amino-Acid Level. Journal of the American Chemical Society 134, 18724–18731 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aprahamian ML, Chea EE, Jones LM & Lindert S Rosetta Protein Structure Prediction from Hydroxyl Radical Protein Footprinting Mass Spectrometry Data. Analytical Chemistry 90, 7721–7729 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J et al. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Analytical Chemistry 89, 2250–2258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones LM, B. Sperry J, A. Carroll J & Gross ML Fast Photochemical Oxidation of Proteins for Epitope Mapping. Analytical Chemistry 83, 7657–7661 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stocks BB & Konermann L Structural Characterization of Short-Lived Protein Unfolding Intermediates by Laser-Induced Oxidative Labeling and Mass Spectrometry. Analytical Chemistry 81, 20–27 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Rempel DL & Gross ML Temperature jump and fast photochemical oxidation probe submillisecond protein folding. Journal of the American Chemical Society 132, 15502–15504 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu XR, Zhang MM, Rempel DL & Gross ML Protein-Ligand Interaction by Ligand Titration, Fast Photochemical Oxidation of Proteins and Mass Spectrometry: LITPOMS. Journal of The American Society for Mass Spectrometry 30, 213–217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu XR, Rempel DL & Gross ML Composite Conformational Changes of Signaling Proteins upon Ligand Binding Revealed by a Single Approach: Calcium-Calmodulin Study. Analytical Chemistry 91, 12560–12567 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aprahamian ML & Lindert S Utility of Covalent Labeling Mass Spectrometry Data in Protein Structure Prediction with Rosetta. Journal of Chemical Theory and Computation 15, 3410–3424 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng M, Zhang B, Cui W & Gross ML Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting. Angewandte Chemie International Edition 56, 14007–14010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang MM, Rempel DL & Gross ML A Fast Photochemical Oxidation of Proteins (FPOP) platform for free-radical reactions: the carbonate radical anion with peptides and proteins. Free Radical Biology and Medicine 131, 126–132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang B, Rempel DL & Gross ML Protein Footprinting by Carbenes on a Fast Photochemical Oxidation of Proteins (FPOP) Platform. Journal of The American Society for Mass Spectrometry 27, 552–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu XR, Zhang MM, Zhang B, Rempel DL & Gross ML Hydroxyl-Radical Reaction Pathways for the Fast Photochemical Oxidation of Proteins Platform As Revealed by 18O Isotopic Labeling. Analytical Chemistry 91, 9238–9245 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weisz DA, Gross ML & Pakrasi HB Reactive oxygen species leave a damage trail that reveals water channels in Photosystem II. Science Advances 3, eaao3013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heyduk E & Heyduk T Mapping Protein Domains Involved in Macromolecular Interactions: A Novel Protein Footprinting Approach. Biochemistry 33, 9643–9650 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Sharp JS, Becker JM & Hettich RL Analysis of Protein Solvent Accessible Surfaces by Photochemical Oxidation and Mass Spectrometry. Analytical Chemistry 76, 672–683 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Aye TT, Low TY & Sze SK Nanosecond Laser-Induced Photochemical Oxidation Method for Protein Surface Mapping with Mass Spectrometry. Analytical Chemistry 77, 5814–5822 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Yan Y et al. Fast Photochemical Oxidation of Proteins (FPOP) Maps the Epitope of EGFR Binding to Adnectin. Journal of the American Society for Mass Spectrometry 25, 2084–2092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H, Gau BC, Jones LM, Vidavsky I & Gross ML Fast Photochemical Oxidation of Proteins for Comparing Structures of Protein-Ligand Complexes: The Calmodulin-Peptide Model System. Anal. Chem 83, 311–318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Rempel DL, Zhang H & Gross ML An Improved Fast Photochemical Oxidation of Proteins (FPOP) Platform for Protein Therapeutics. Journal of The American Society for Mass Spectrometry 26, 526–529 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garrison WM Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chemical Reviews 87, 381–398 (1987). [Google Scholar]
- 58.Chen J, Cui W, Giblin D & Gross ML New protein footprinting: fast photochemical iodination combined with top-down and bottom-up mass spectrometry. Journal of the American Society for Mass Spectrometry 23, 1306–1318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manzi L et al. Carbene footprinting accurately maps binding sites in protein–ligand and protein–protein interactions. Nature communications 7, 13288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Espino JA, Mali VS & Jones LM In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Analytical Chemistry 87, 7971–7978 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Hambly DM & Gross ML Cold Chemical Oxidation of Proteins. Analytical Chemistry 81, 7235–7242 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Srikanth R, Wilson J & Vachet RW Correct identification of oxidized histidine residues using electron-transfer dissociation. Journal of Mass Spectrometry 44, 755–762 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X, Li Z, Xie B & Sharp JS Improved Identification and Relative Quantification of Sites of Peptide and Protein Oxidation for Hydroxyl Radical Footprinting. Journal of The American Society for Mass Spectrometry 24, 1767–1776 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Dimensions of the custom-made capillary holder. The figure is drawn to scale.
Supplementary Figure 2. Detailed drawing of the optics setup and their alignments. The figure is drawn to scale.
Data Availability Statement
A calmodulin dataset that contains a global level measurement by Bruker MaXis, a peptide and residue level measurement by Thermo Scientific Q Exactive is freely available online in the Mendeley Data (https://data.mendeley.com/) with DOI 10.17632/xfd5sh76pm.1.