

Abstract
The hemostatic and inflammatory systems work hand in hand to maintain homeostasis at mucosal barrier sites. Among the factors of the hemostatic system, fibrin is well recognized for its role in mucosal homeostasis, wound healing, and inflammation. Here, we present a basic overview of the fibrinolytic system, discuss fibrin as an innate immune regulator, and provide recent work uncovering the role of fibrin–neutrophil activation as a regulator of mucosal/periodontal homeostasis. We reason that the role of fibrin in periodontitis becomes most evident in individuals with the Mendelian genetic defect, congenital plasminogen (PLG) deficiency, who are predisposed to severe periodontitis in childhood due to a defect in fibrinolysis. Consistent with plasminogen deficiency being a risk factor for periodontitis, recent genomics studies uncover genetic polymorphisms in PLG, encoding plasminogen, being significantly associated with periodontal disease, and suggesting PLG variants as candidate risk indicators for common forms of periodontitis.
Keywords: plasminogen, fibrin, extracellular matrix (ECM), immunity, proteases/proteinases, inflammation
Introduction
Periodontitis is an inflammatory disease characterized by progressive destruction of the structures that support teeth. Several behavioral, environmental, microbial, systemic, and genetic factors have been reported to influence its development. Host susceptibility also plays a significant role in periodontitis, with up to a third of variance explained by genetic factors and heritability increasing with disease severity (Nibali et al. 2019). The strongest evidence for genetically conferred risk and increased innate susceptibility to periodontitis comes from studies in patients with single gene defects who are predisposed to periodontitis at an early age (Silva, Brenchley, Moutsopoulos, 2019). Periodontitis is a frequent clinical manifestation in several monogenic syndromes that are transmitted as Mendelian traits. Such human single-gene defects not only demonstrate the existence of genetic susceptibility to periodontitis but also reveal insights into the roles of specific genes and related pathways in periodontal homeostasis and immunopathology. To date, Mendelian diseases associated with periodontitis include syndromes with neutropenia, abnormal neutrophil function, and metabolic, structural, or immune protein defects. Among all the Mendelian diseases discovered in the genes coding for the components of the hemostatic system, type 1 congenital plasminogen (PLG) deficiency (CPD) has been definitively identified to predispose to early-onset, severe periodontitis, which is associated with defective fibrinolysis. In this review, we discuss plasminogen deficiency and associated defective fibrinolysis as a mechanism underlying periodontitis pathogenesis in context of Mendelian-type gene defects (i.e., mutations). We further overview data related to common genetic variation (i.e., single-nucleotide polymorphisms [SNPs]) in and around the PLG gene, which is associated with non-Mendelian forms of periodontitis, and propose that relevant pathways may be involved in subsets of patients with common forms of periodontitis.
Congenital Plasminogen Deficiency, a Cause for Periodontitis
CPD in humans is of 2 types: type I or hypoplasminogenemia (severe PLG deficiency) and type II or dysplasminogenemia, which is rarely, if ever, symptomatic (Schuster and Seregard 2003). Type I CPD is a rare autosomal recessive disease caused by mutations in PLG, the gene coding for the zymogen plasminogen, with resultant decrease in antigenic and functional levels (Schuster et al. 1997; Schuster and Seregard 2003; Tefs et al. 2006). The prevalence of heterozygous mutations for type I CPD is ~0.3% to 0.4% of the general population (Okamoto et al. 2003; Tefs et al. 2004). The prevalence of severe type I CPD (i.e., with either homozygous or compound heterozygous mutations) has been estimated to 1.6 per one million people in Europe (Tefs et al. 2004).
CPD is associated with abnormal accumulation of an amorphous, fibrin-rich, amyloid-like substance in the lamina propria of mucous membranes, including the oral mucosa, the eyes, respiratory system, urinary and genital systems, and the gastrointestinal tract (Tefs et al. 2006) (Fig. 1A). The term ligneous was first used in 1933 to describe the wood-like (ligneous) characteristic of the pseudomembranous lesions (Borel 1933) first described by Bouisson (1847). The link between type I CPD and ligneous lesions on mucosal sites was first reported in 1995 (Mingers et al. 1995). A large study conducted in 2006 (Tefs et al. 2006) showed that the most common clinical manifestation of type I CPD is ligneous conjunctivitis (80%), followed by ligneous gingivitis (34%). (Tefs et al. 2006). Herein we provide a summary of published reports of ligneous gingivitis cases between 1973 and 2022 (Appendix Table 1).
Figure 1.
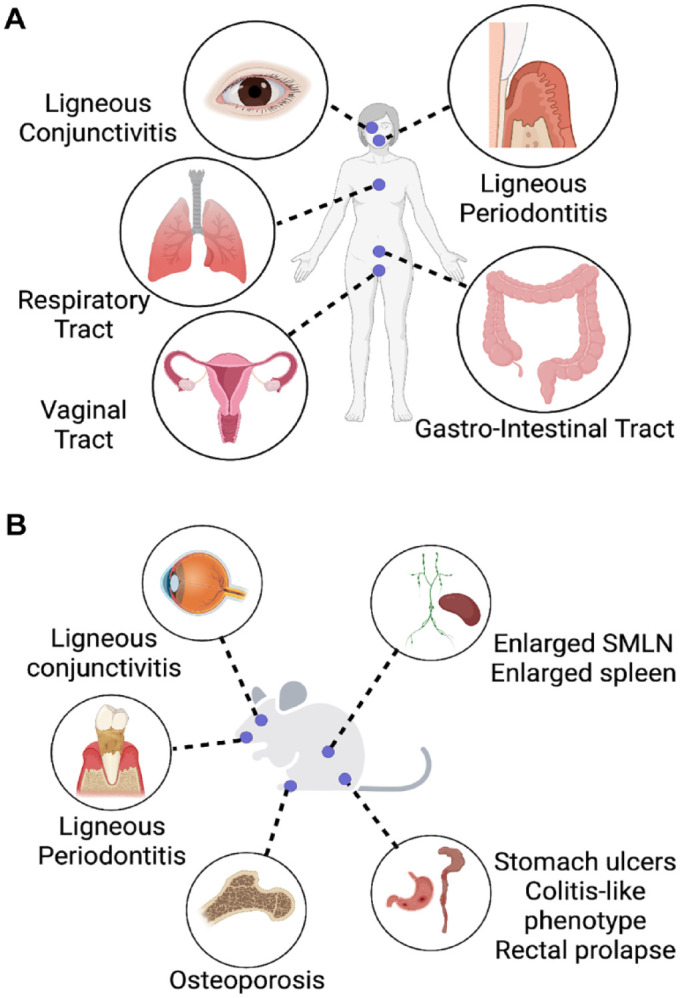
Patients with congenital plasminogen deficiency have immunopathologies at many mucosal barrier sites. (A) Congenital plasminogen deficiency (CPD) is associated with abnormal accumulation of an amorphous, fibrin- and leukocyte-rich, amyloid-like substance in the lamina propria of mucous membranes, including the oral mucosa, the eyes, respiratory system, urinary and genital systems, and the gastrointestinal system. (B) Plasminogen (PLG)–deficient mice display an array of mucosal immunopathologies phenocopying the human disease, including ligneous conjunctivitis; periodontitis; osteoporosis; systemic inflammation, as denoted by enlarged submandibular lymph nodes (SMLN) and splenomegaly; and inflammation of the gastrointestinal tract—stomach ulcers, colitis-like phenotype, and rectal prolapse.
Initially, these ligneous deposits were considered compatible with primary localized amyloidosis. However, the material deposited did not share the histochemical features of amyloid. Some years after the discovery of a link between PLG deficiency and ligneous conjunctivitis (Mingers et al. 1997), Scully et al. (2001) described the etiology of the disease. The homogeneous, amyloid-like, eosinophilic material was identified to be fibrin (Pierro et al. 2006; Karaer et al. 2007; Kurtulus et al. 2007; Fine et al. 2009). Type I CPD causes the inability to remove fibrin deposits mainly at mucosal barrier sites secondary to injury or inflammation, as well as delayed tissue repair. Due to limited fibrin degradation, the wound-healing process has been shown to cease at the granulation tissue formation stage (Pantanowitz et al. 2004; Mehta and Shapiro 2008).
Ligneous periodontitis is characterized by membranous, nodular gingival lesions and progressive destructive periodontal disease, periodontitis, leading to tooth loss at a young age despite a diverse array of attempted treatments (Appendix Table 1). These patients developed ligneous periodontitis at a very young age with severe bone loss evident in teenage years (Appendix Table 1). Surgical and periodontal treatment efforts were shown to be unsuccessful for the management of the reported oral lesions. In some cases, gingival lesions showed regression or disappeared following tooth loss or full-mouth extractions (Gunhan et al. 1999). For instance, in one case (Baykul and Bozkurt 2004), mandibular lesions regressed slowly 4 wk after the last tooth extraction and disappeared, whereas maxillary lesions remained unchanged for 12 wk.
The Fibrinolytic System
Plasmin plays an important role in hemostasis via controlled dissolution of fibrin blood clots into fibrin degradation products (Collen and Lijnen 1994). PLG is a proenzyme that is synthesized primarily by the liver. PLG circulates in the blood and is converted into the active enzyme, plasmin, on proteolytic cleavage by 2 PLG activators: tissue (tPA) and urokinase (uPA) plasminogen activator (Collen and Lijnen 1994; Scully et al. 2007). Fibrinogen is a provisional extracellular matrix protein that is produced in the liver and is present in the circulation and in interstitial fluids. During tissue or vascular injury, fibrinogen is converted to fibrin by the enzymatic reaction of thrombin and then to a fibrin-based blood clot (Fig. 2). Despite fibrin being the main physiological substrate of plasmin, it also initiates the fibrinolytic process (Cesarman-Maus and Hajjar 2005; Green et al. 2008) (Fig. 2). This is not at all surprising as the deposits of fibrin are meant to be a temporary matrix for stemming blood loss as in the form of a blood clot or a scaffold for wound repair that ultimately needs to be removed in a timely manner.
Figure 2.

Fibrinolysis and regulatory mechanisms. (A) While fibrin is the main physiological target of plasmin (Pln), fibrin also facilitates the fibrinolytic process. This self-directed cleavage process involves facilitating of tissue plasminogen activator (tPA)–mediated activation of plasminogen (PLG), enabling pro–urokinase plasminogen activator (uPA) activation by plasmin to allow for a PLG and pro-uPA feedback activation cycle and, finally, the protection of plasmin from α2-antiplasmin inhibition. (B) The fibrinolysis process is tightly regulated by (i) α2-antiplasmin-mediated inactivation of Pln, (ii) plasminogen activator inhibitor 1 (PAI-1)–mediated inhibition of tPA and uPA, and (iii) thrombin activatable fibrinolysis inhibitor (TAFI)–mediated blockage of tPA and PLG scaffolding to fibrin.
The self-directed fibrin dissolution process is orchestrated by the presence of C-terminal lysine residues on fibrin, which are recognized by lysine binding sites located in the Kringle domains of PLG (Law et al. 2012) (Fig. 2). This process not only brings fibrin and PLG together but also enhances the proteolytic conversion of PLG to plasmin by tPA and uPA by several means. These include unfolding of the PLG Kringle domains to expose the activation cleavage site on PLG, scaffolding tPA-dependent PLG activation through mutual binding to fibrin, and enabling sustained pro-uPA activation by fibrin-bound plasmin to allow for a PLG/pro-uPA feedback activation cycle and, finally, by the protection of plasmin from α2-antiplasmin inhibition (Collen and Lijnen 1992; Cesarman-Maus and Hajjar 2005; Green et al. 2008; Law et al. 2012). α2-Antiplasmin targets plasmin directly and thus the most critical fibrinolytic inhibitor. Together with a very high plasma level (~1 mmol/L) and half-life (~2.6 d) (Collen and Wiman 1979), antiplasmin ensures that the half-life of plasmin activity in solution is less than 10 ms, which significantly differs from the half-life of the inactive proform, plasminogen (~2.2 d). Moreover, the association between plasmin and α2-antiplasmin is the fastest described among the serine proteases and their inhibitors. However, within the fibrin matrix, plasmin is protected from α2-antiplasmin inhibition and gets ample time to perform its proteolytic task. The fibrinolytic system is also under the regulation of other mechanisms that restrict plasmin formation. The first level of regulation comes from PLG activator inhibitor (PAI) 1 (Coleman et al. 1982; Loskutoff et al. 1983) that targets both tPA and uPA. The second level of regulation is exerted by a carboxypeptidase, called thrombin activatable fibrinolysis inhibitor (TAFI) (Nesheim and Bajzar 2005). TAFI cleaves C-terminal lysine residues from fibrin, thus reducing the capability of plasminogen and tPA to bind to the fibrin surface, thereby inhibiting fibrinolysis.
Fibrin(ogen) is a Component of the Innate Immune System
Extravascular fibrin deposits in response to tissue damage are not exclusively formed to control hemorrhage but also act as a critical functional component in regulating the resulting inflammatory response (Esmon 2005, 2008). While almost all the components of the coagulation system can clearly influence the inflammatory events, fibrinogen is one of the most potent contributors to injury-induced inflammation (Fig. 3). Fibrinogen and fibrin contribute to inflammation by stimulating leukocyte extravasation and directly modulating leukocyte effector functions by acting as a local, spatially defined cue within damaged tissue. Fibrin(ogen) can exert a wide range of effects by functioning as a ligand for a myriad of cell surface receptors, including VE-cadherin, ICAM-1, and the integrins αIIbβ3, α5β1, αvβ3, αMβ2, and αXβ2 (Luyendyk et al. 2019). These cell surface receptors are expressed by cell types, including leukocytes, endothelial cells, fibroblasts, platelets, and smooth muscle cells (Luyendyk et al. 2019), thus increasing the complexity of the inflammatory responses mediated by fibrin.
Figure 3.

Fibrin is a critical regulator of mucosal immunity. Mucosal barriers are frequently damaged due to microbial, environmental, or mechanical stress, leading to fibrin deposition to stem blood loss, facilitate pathogen clearance, and repair tissue. Fibrin is proinflammatory and involved in immune regulatory processes, such as leukocyte migration and neutrophil effector function activation via the αMβ2 binding site on fibrin gamma chain. NETosis, neutrophil extracellular trap formation; ROS, reactive oxygen species.
Several mechanisms of fibrinogen-mediated leukocyte migration have been proposed, and many studies highlight a critical role for fibrin in mediating inflammation potentially through recruitment of leukocytes (Luyendyk et al. 2019), but additional experimental validations are warranted. In addition to fibrin’s role in leukocyte extravasation, our team recently discovered that efficient migration of macrophages to sites of inflammation requires plasmin-mediated fibrinolysis (Silva, Lum, et al. 2019). Using a murine thioglycollate-induced peritonitis and an in vitro macrophage migration model, we showed that the macrophage requirement for plasmin-mediated fibrinolysis, both in vivo and in vitro, was negated by deletion of the major myeloid integrin αMβ2-binding motif on the gamma chain of fibrin(ogen) (Silva, Lum, et al. 2019).
The deposition of extravascular fibrin(ogen) in and around inflammatory foci suggests a potential direct involvement of fibrin in mediating inflammatory cell effector functions. In vitro studies have demonstrated that fibrin induces expression of proinflammatory cytokines/chemokines by endothelial and peripheral blood mononuclear cells, nuclear factor κB–mediated transcription, mitogen-activated protein kinase, and protein kinase C signaling during inflammation (Luyendyk et al. 2019). As mentioned above, fibrin is a ligand for several leukocyte surface receptors. However, the role of fibrin(ogen) and β2 integrin engagement in modulating leukocyte proinflammatory response has been widely supported by a growing body of data. Among the β2 integrin subfamily, αXβ2 (p150,95 and CD11c/CD18) and αMβ2 (Mac-1, CD11b/CD18, or CR3) have been identified as fibrin(ogen) receptors (Ugarova and Yakubenko 2001; Ugarova et al. 2003). This integrin family plays a central role in leukocyte function. Indeed, a congenital disorder affecting the expression of the common β2 subunit results in leukocyte adhesion deficiency type 1, causing profound defects in leukocyte recruitment, thus leading to early-onset periodontitis (Hajishengallis and Moutsopoulos 2014; Moutsopoulos et al. 2014). However, the role of fibrin(ogen) in periodontitis pathogenesis, if any, is yet to be clarified.
In 1988, two research groups independently identified αMβ2 as the receptor responsible for interaction between fibrin(ogen) and myeloid cells (Altieri et al. 1988; Wright et al. 1988). The αMβ2 integrin “I-domain” within the αM subunit (Diamond et al. 1993; Lishko et al. 2001) engages with fibrin(ogen) residues 377 to 395 within the γ chain (“P2” site) of the D nodule (Ugarova et al. 1998). Further biochemical validations recognized that P2 core recognition motif P2-C:γ383–395 was adequate for high-affinity binding (Ugarova et al. 2003).
The engagement between fibrin(ogen) and αMβ2 integrin is mechanistically regulated, as the fibrinogen–αMβ2 binding motif remains cryptic unless fibrinogen becomes immobilized on a surface or is converted to a fibrin polymer, so that it is not available when fibrinogen is in the form of a soluble monomer as in circulation (Lishko et al. 2002). This mechanism ensures that leukocyte activation by fibrinogen occurs only after fibrinogen is released from circulation and deposited at sites of tissue damage. A mouse model expressing a version of fibrin that cannot bind to αMβ2 (Fibγ390–396A) (Flick, Du, Witte, et al. 2004) has been instrumental in discovering the role of fibrin in antimicrobial host defense, inflammatory arthritis, colitis, neuroinflammatory disease, and musculoskeletal disease (Adams et al. 2007; Flick et al. 2007; Vidal et al. 2008; Steinbrecher et al. 2010; Cole et al. 2014). In addition, there is evidence that fibrin(ogen) plays a role in chronic low-grade inflammation, such as diet-induced obesity. Fibγ390–396A mice gained significantly less weight, developed comparatively less adipose tissue inflammation, and were protected from obesity compared with similarly challenged wild-type mice (Kopec et al. 2017). Moreover, fibrin deposits play a role in bacterial clearance within infected organ systems. The clearance of purulent Yersinia pestis foci in the liver of infected hosts by neutrophils and macrophages was shown to be driven by fibrin deposits in and around the lesions (Degen et al. 2007). Likewise, Staphylococcus aureus clearance from the peritoneal cavity in Fibγ390–396A mice was reduced, suggesting a failure of fibrin(ogen)-driven αMβ2 innate immune cell function (Flick, Du, Degen, 2004).
In our recent work, we discovered another mechanism by which fibrin potentiates neutrophil effector functions (Silva et al. 2021). Fibrin engages with neutrophils via the αMβ2 binding site on the fibrin gamma chain and enhances neutrophil effector functions associated with antimicrobial defense, such as reactive oxygen species production, myeloperoxidase production, and neutrophil extracellular trap formation (NETosis) (Fig. 3). NETs consist of modified chromatin “decorated” with bactericidal proteins from granules and cytoplasm and are released into the extracellular environment upon various triggers, mainly to act as a defensive mechanism against bacteria that are too large to be engulfed (Silva, Brenchley, Moutsopoulos, 2019). Mucosal barrier sites are frequent encounters of bacteria and thus armed with a strong defense system to maintain mucosal homeostasis. These mechanisms, enhanced by fibrin–neutrophil engagement, may be beneficial in maintaining homeostasis at the mucosal barrier sites.
Given the proinflammatory properties of extravascular fibrin, it is plausible to expect exacerbated immunopathologies in the presence of persistent fibrin deposits. Accordingly, PLG-deficient humans suffer from many mucosal pathologies as discussed above. Interestingly, PLG-deficient mice phenocopy the human disease and have become instrumental in understanding the mechanisms underlying mucosal immunopathologies (Fig. 1B). All these mucosal immunopathologies are corrected by imposing fibrinogen deficiency on PLG-deficient mice (Bugge et al. 1996; Silva et al. 2021). In addition to mucosal defects, PLG-deficient mice uniformly develop severe osteoporosis at a young age (Cole et al. 2014). Removal of fibrinogen from or expressing Fibγ390–396A in PLG-deficient mice reduced the inflammatory bone loss phenotype (Cole et al. 2014). In addition, in vitro analyses indicated that immobilized fibrinogen plays a role in differentiating monocytes into osteoclasts, thereby driving bone resorption (Cole et al. 2014). We recently showed that these fibrin-mediated homeostatic immune responses can become pathogenic when exaggerated (Silva et al. 2021).
Fibrin-Mediated Neutrophil Activation Drives the Oral Mucosal Disease, Periodontitis
Sulniute et al. (2011) first showed that PLG deficiency in mice causes periodontitis and suggested supplementation of plasminogen as a potential therapy for treating periodontitis in PLG deficiency. Recently, we discovered that this PLG deficiency-associated periodontal immunopathology is driven by persistent fibrin(ogen) accumulation in the oral mucosal tissues (Silva et al. 2021) (Fig. 4). In the absence of plasmin-mediated fibrinolysis, fibrin deposits persist in the extravascular microenvironment, and neutrophils are recruited and retained in increasing numbers compared to other immune cell types. Interestingly, fibrin-mediated activation of neutrophil effector functions that are associated with antimicrobial defense becomes exaggerated and drives oral mucosal immunopathology. Indeed, we established that NETosis drives periodontal immunopathology in the absence of PLG with the aid of several genetic and pharmacological models. In addition, the commensal oral microbiota acted as a trigger for extravascular fibrin deposition caused by the absence of PLG. However, the mechanism underlying this microbiota-driven fibrin deposition and the role of fibrin in bone disruption in the oral cavity is yet to be understood. Importantly, this fibrin–neutrophil engagement via the αMβ2 binding site on the fibrin gamma chain contributes to immunopathology in wild-type mice, suggesting a potential role for this pathway in common forms of periodontitis.
Figure 4.
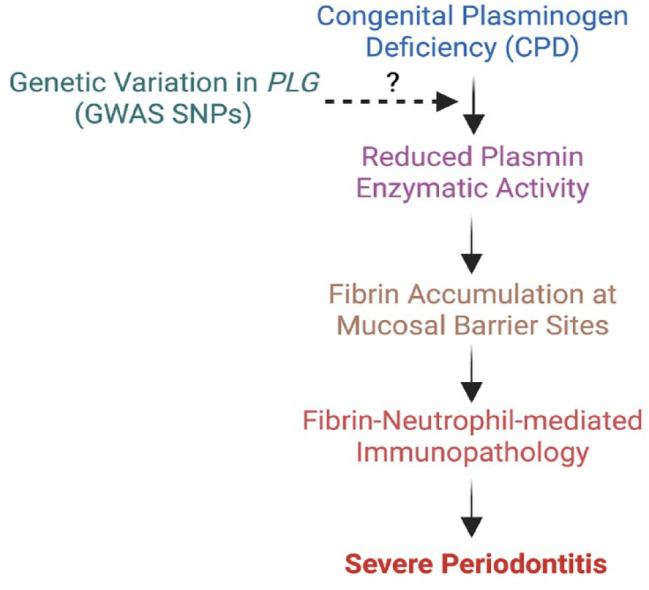
Proposed connection between (PLG) deficiency with periodontitis in rare and common forms of periodontitis. In the absence of PLG and/or in the common forms of periodontitis, fibrin deposits in the mucosa and enhances immunopathology via the engagement with neutrophils.
PLG Polymorphisms in Common Forms of Periodontitis
Besides several reports of monogenic disease-type cases (e.g., ligneous periodontitis) and associated rare loss-of-function variants involving the human PLG (Appendix Table 1), there is now evidence linking common genetic variation (i.e., SNPs) in the PLG locus to periodontitis. Our recent gene-centric interrogation of PLG associations (i.e., 153 SNPs in the gene locus) with subtypes of periodontal disease (Silva et al. 2021), combined with an earlier report from a genome-wide association study (GWAS) investigating loci for aggressive and chronic periodontitis (Munz et al. 2019), provides evidence in support of a role for PLG variation in common forms of periodontitis. This body of literature has implicated several SNPs in the PLG locus as statistically associated with periodontal end points (e.g., rs1247559, rs2465836, rs4252120, rs4252200), including severe periodontal disease and high colonization with Aggregatibacter actinomycetemcomitans.
The functional or regulatory role of these polymorphisms remains to be empirically studied, as SNP associations reported by GWASs are conservatively interpreted as locus-tagging. Future fine-mapping, targeted sequencing, and mechanistic studies may reveal truly functional or regulatory roles for these SNPs or other neighboring or correlated variants. In publicly available multitissue expression data (Consortium 2020), rs1247559 has been reported as a quantitative trait locus (eQTL) associated (P = 10–7) with the expression of RP1-81D8.4 (a pseudogene similar to PLG) in the liver. A correlated variant (rs1740430, R2 = 0.96) has been reported as an eQTL for lipoprotein a (LPA) expression in esophageal mucosa. Recently, Müller et al. (2023) reported evidence of interactions between variants rs1247559 in PLG and rs4284742 in SIGLEC5 (the only genome-wide significant locus of periodontitis in consortium meta-analysis reported by Shungin et al. 2019) with the functional variant rs1122900 (a predicted transcription factor PRDM14 binding site) of the long noncoding RNA (lncRNA) CTD-2353F22.1, also known as SLC1A3 antisense RNA 1 (SLC1A3-AS1). This case-only investigation among ~900 European adults found gene–gene interactions (odds ratio due to interaction = 1.6) of PLG and SIGLEC5 with SLC1A3-AS1 that are linked to the expression of SLC1A3-AS1 (mostly expressed in mast cells, B cells, and dendritic cells) and with downstream regulatory effects on the coagulation cascade and on pathways involved in response to wounding and revascularization. Taken together, this accumulating evidence implicates common variants in the PLG locus in common yet severe forms of periodontitis and sheds light onto plausible mechanisms at play.
Concluding Remarks
Elucidation of the genetic basis of the Mendelian forms of periodontitis provides a critical understanding of the mechanisms underlying these disease states. This knowledge may ultimately lead to the development of treatments to compensate for the underlying biological defects. This would be a gamechanger in our ability to provide care, as many affected individuals do not respond well to conventional periodontal therapies. While these conditions are rare, each of these gene defects provides a starting point to elucidate how the abnormal gene product disrupts normal homeostasis in the periodontium. This will allow the identification of specific biological pathways that are critical for the normal homeostasis, such as the fibrin-mediated neutrophil effector function activation, and can also accelerate the discovery of potential therapeutic targets. Ultimately, these findings may provide a foundation for gene-associated studies, as discussed above, for more common, genetically complex forms of periodontitis.
Author Contributions
L.M. Silva, K. Divaris, contributed to conception and data design, drafted and critically revised the manuscript; T.H. Bugge, N.M. Moutsopoulos, contributed to conception and design, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, sj-docx-1-jdr-10.1177_00220345231171837 for Plasmin-Mediated Fibrinolysis in Periodontitis Pathogenesis by L.M. Silva, K. Divaris, T.H. Bugge and N.M. Moutsopoulos in Journal of Dental Research
Acknowledgments
We thank T. Greenwell-Wild for critically reviewing this manuscript.
Footnotes
A supplemental appendix to this article is available online.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the intramural program of NIDCR/NIH (T.H. Bugge and N.M. Moutsopoulos) and NIDCR 1K99DE030124-01A1 (L.M. Silva). Figures have been created using BioRender.
ORCID iDs: L.M. Silva  https://orcid.org/0000-0001-6282-2657
https://orcid.org/0000-0001-6282-2657
K. Divaris
https://orcid.org/0000-0003-1290-7251
Data Availability Statement: Data sharing not applicable—no new data generated.
References
- Adams RA, Schachtrup C, Davalos D, Tsigelny I, Akassoglou K. 2007. Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: lessons from multiple sclerosis. Curr Med Chem. 14(27): 2925–2936. [DOI] [PubMed] [Google Scholar]
- Altieri DC, Bader R, Mannucci PM, Edgington TS. 1988. Oligospecificity of the cellular adhesion receptor Mac-1 encompasses an inducible recognition specificity for fibrinogen. J Cell Biol. 107(5):1893–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baykul T, Bozkurt Y. 2004. Destructive membranous periodontal disease (ligneous periodontitis): a case report and 3 years follow-up. Br Dent J. 197(8):467–468. [DOI] [PubMed] [Google Scholar]
- Borel MG. 1933. A new palpebral syndrome (in french). Bull Soc Fr Ophthalmol 46:168–180. [Google Scholar]
- Bouisson M. 1847. Ophthalmie sur-aigue avec formation de pseudomembranes àla surface de la conjonctive. Ann Oculist. 17(1847):100–104. [Google Scholar]
- Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL. 1996. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 87(4):709–719. [DOI] [PubMed] [Google Scholar]
- Cesarman-Maus G, Hajjar KA. 2005. Molecular mechanisms of fibrinolysis. Br J Haematol. 129(3):307–321. [DOI] [PubMed] [Google Scholar]
- Cole HA, Ohba T, Nyman JS, Hirotaka H, Cates JM, Flick MJ, Degen JL, Schoenecker JG. 2014. Fibrin accumulation secondary to loss of plasmin-mediated fibrinolysis drives inflammatory osteoporosis in mice. Arthritis Rheumatol. 66(8):2222–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman PL, Barouski PA, Gelehrter TD. 1982. The dexamethasone-induced inhibitor of fibrinolytic activity in hepatoma cells. A cellular product which specifically inhibits plasminogen activation. J Biol Chem. 257(8):4260–4264. [PubMed] [Google Scholar]
- Collen D, Lijnen HR. 1992. Fibrin-specific fibrinolysis. Ann N Y Acad Sci. 667:259–271. [DOI] [PubMed] [Google Scholar]
- Collen D, Lijnen HR. 1994. Fibrinolysis and the control of hemostasis. In: Stamatoyannopoulos G, Nienhuis AW, Majerus PW, Varmus HE, editors. The molecular basis of blood diseases. Philadelphia (PA): WB Saunders Co. p. 725–752. [Google Scholar]
- Collen D, Wiman B. 1979. Turnover of antiplasmin, the fast-acting plasmin inhibitor of plasma. Blood. 53(2):313–324. [PubMed] [Google Scholar]
- Consortium GT. 2020. The GTEx consortium atlas of genetic regulatory effects across human tissues. Science. 369(6509):1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degen JL, Bugge TH, Goguen JD. 2007. Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost. 5(Suppl 1):24–31. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. 1993. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J Cell Biol. 120(4):1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmon CT. 2005. The interactions between inflammation and coagulation. Br J Haematol. 131(4):417–430. [DOI] [PubMed] [Google Scholar]
- Esmon CT. 2008. Crosstalk between inflammation and thrombosis. Maturitas. 61(1–2):122–131. [DOI] [PubMed] [Google Scholar]
- Fine G, Bauer K, Al-Mohaya M, Woo SB. 2009. Successful treatment of ligneous gingivitis with warfarin. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 107(1):77–80. [DOI] [PubMed] [Google Scholar]
- Flick MJ, Du X, Degen JL. 2004. Fibrin(ogen)-αMβ2 interactions regulate leukocyte function and innate immunity in vivo. Exp Biol Med (Maywood). 229(11):1105–1110. [DOI] [PubMed] [Google Scholar]
- Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. 2004. Leukocyte engagement of fibrin(ogen) via the integrin receptor αMβ2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 113(11):1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, Thornton S, Degen JL. 2007. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin αMβ2 binding motif. J Clin Invest. 117(11):3224–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KA, Almholt K, Ploug M, Rono B, Castellino FJ, Johnsen M, Bugge TH, Romer J, Lund LR. 2008. Profibrinolytic effects of metalloproteinases during skin wound healing in the absence of plasminogen. J Invest Dermatol. 128(8):2092–2101. [DOI] [PubMed] [Google Scholar]
- Gunhan O, Gunhan M, Berker E, Gurgan CA, Yildirim H. 1999. Destructive membranous periodontal disease (ligneous periodontitis). J Periodontol. 70(8):919–925. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Moutsopoulos NM. 2014. Etiology of leukocyte adhesion deficiency-associated periodontitis revisited: not a raging infection but a raging inflammatory response. Expert Rev Clin Immunol. 10(8):973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaer A, Mert I, Akinsu F, Tug M, Tefs K, Schuster V, Mollamahmutoglu L. 2007. Ligneous inflammation involving the female genital tract. J Obstet Gynaecol Res. 33(4):581–584. [DOI] [PubMed] [Google Scholar]
- Kopec AK, Abrahams SR, Thornton S, Palumbo JS, Mullins ES, Divanovic S, Weiler H, Owens AP, 3rd, Mackman N, Goss A, et al. 2017. Thrombin promotes diet-induced obesity through fibrin-driven inflammation. J Clin Invest. 127(8):3152–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtulus I, Gokbuget A, Efeoglu A, Cintan S, Tefs K, Schuster V, Scully C. 2007. Hypoplasminogenemia with ligneous periodontitis: a failed local therapeutic approach. J Periodontol. 78(6):1164–1175. [DOI] [PubMed] [Google Scholar]
- Law RH, Caradoc-Davies T, Cowieson N, Horvath AJ, Quek AJ, Encarnacao JA, Steer D, Cowan A, Zhang Q, Lu BG, et al. 2012. The x-ray crystal structure of full-length human plasminogen. Cell Rep. 1(3):185–190. [DOI] [PubMed] [Google Scholar]
- Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. 2002. Regulated unmasking of the cryptic binding site for integrin αMβ2 in the gamma c-domain of fibrinogen. Biochemistry. 41(43):12942–12951. [DOI] [PubMed] [Google Scholar]
- Lishko VK, Yakubenko VP, Hertzberg KM, Grieninger G, Ugarova TP. 2001. The alternatively spliced αEC domain of human fibrinogen-420 is a novel ligand for leukocyte integrins αMβ2 and αXβ2. Blood. 98(8):2448–2455. [DOI] [PubMed] [Google Scholar]
- Loskutoff DJ, van Mourik JA, Erickson LA, Lawrence D. 1983. Detection of an unusually stable fibrinolytic inhibitor produced by bovine endothelial cells. Proc Natl Acad Sci U S A. 80(10):2956–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyendyk JP, Schoenecker JG, Flick MJ. 2019. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood. 133(6):511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R, Shapiro AD. 2008. Plasminogen deficiency. Haemophilia. 14(6):1261–1268. [DOI] [PubMed] [Google Scholar]
- Mingers AM, Heimburger N, Lutz E. 1995. Familial homozygous and heterozygous type I plasminogen deficiency [in German]. In: Scharrer I, Schramm W, editors. Haemophilie Symposium, Hamburg, 1994. Berlin: Springer-Verlag. p. 96–104. [Google Scholar]
- Mingers AM, Heimburger N, Zeitler P, Kreth HW, Schuster V. 1997. Homozygous type I plasminogen deficiency. Semin Thromb Hemost. 23(3):259–269. [DOI] [PubMed] [Google Scholar]
- Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, Abusleme L, Zenobia C, Hosur KB, Abe T, et al. 2014. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. 6(229):229ra240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller R, Freitag-Wolf S, Weiner J, 3rd, Chopra A, Top T, Dommisch H, Schaefer AS. 2023. Case-only design identifies interactions of genetic risk variants at SIGLEC5 and PLG with the lncRNA CTD-2353f22.1 implying the importance of periodontal wound healing for disease aetiology. J Clin Periodontol. 50(1):90–101. [DOI] [PubMed] [Google Scholar]
- Munz M, Richter GM, Loos BG, Jepsen S, Divaris K, Offenbacher S, Teumer A, Holtfreter B, Kocher T, Bruckmann C, et al. 2019. Meta-analysis of genome-wide association studies of aggressive and chronic periodontitis identifies two novel risk loci. Eur J Hum Genet. 27(1):102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesheim M, Bajzar L. 2005. The discovery of TAFI. J Thromb Haemost. 3(10):2139–2146. [DOI] [PubMed] [Google Scholar]
- Nibali L, Bayliss-Chapman J, Almofareh SA, Zhou Y, Divaris K, Vieira AR. 2019. What is the heritability of periodontitis? A systematic review. J Dent Res. 98(6):632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto A, Sakata T, Mannami T, Baba S, Katayama Y, Matsuo H, Yasaka M, Minematsu K, Tomoike H, Miyata T. 2003. Population-based distribution of plasminogen activity and estimated prevalence and relevance to thrombotic diseases of plasminogen deficiency in the Japanese: the Suita Study. J Thromb Haemost. 1(11):2397–2403. [DOI] [PubMed] [Google Scholar]
- Pantanowitz L, Bauer K, Tefs K, Schuster V, Balogh K, Pilch BZ, Adcock D, Cirovic C, Kocher O. 2004. Ligneous (pseudomembranous) inflammation involving the female genital tract associated with type-1 plasminogen deficiency. Int J Gynecol Pathol. 23(3):292–295. [DOI] [PubMed] [Google Scholar]
- Pierro VS, Vazquez-Sullca R, Vieira AS, Takiya CM, Carakushansky G, Feres-Filho EJ. 2006. Ligneous periodontitis and Ehlers-Danlos syndrome. J Periodontol. 77(1):123–128. [DOI] [PubMed] [Google Scholar]
- Schuster V, Mingers AM, Seidenspinner S, Nussgens Z, Pukrop T, Kreth HW. 1997. Homozygous mutations in the plasminogen gene of two unrelated girls with ligneous conjunctivitis. Blood. 90(3):958–966. [PubMed] [Google Scholar]
- Schuster V, Seregard S. 2003. Ligneous conjunctivitis. Surv Ophthalmol. 48(4):369–388. [DOI] [PubMed] [Google Scholar]
- Scully C, Gokbuget A, Kurtulus I. 2007. Hypoplasminogenaemia, gingival swelling and ulceration. Oral Dis. 13(6):515–518. [DOI] [PubMed] [Google Scholar]
- Scully C, Gokbuget AY, Allen C, Bagan JV, Efeoglu A, Erseven G, Flaitz C, Cintan S, Hodgson T, Porter SR, et al. 2001. Oral lesions indicative of plasminogen deficiency (hypoplasminogenemia). Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 91(3):334–337. [DOI] [PubMed] [Google Scholar]
- Shungin D, Haworth S, Divaris K, Agler CS, Kamatani Y, Keun Lee M, Grinde K, Hindy G, Alaraudanjoki V, Pesonen P, et al. 2019. Genome-wide analysis of dental caries and periodontitis combining clinical and self-reported data. Nat Commun. 10(1):2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LM, Brenchley L, Moutsopoulos NM. 2019. Primary immunodeficiencies reveal the essential role of tissue neutrophils in periodontitis. Immunol Rev. 287(1):226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LM, Doyle AD, Greenwell-Wild T, Dutzan N, Tran CL, Abusleme L, Juang LJ, Leung J, Chun EM, Lum AG, et al. 2021. Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science. 374(6575):eabl5450. [DOI] [PubMed] [Google Scholar]
- Silva LM, Lum AG, Tran C, Shaw MW, Gao Z, Flick MJ, Moutsopoulos NM, Bugge TH, Mullins ES. 2019. Plasmin-mediated fibrinolysis enables macrophage migration in a murine model of inflammation. Blood. 134(3):291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbrecher KA, Horowitz NA, Blevins EA, Barney KA, Shaw MA, Harmel-Laws E, Finkelman FD, Flick MJ, Pinkerton MD, Talmage KE, et al. 2010. Colitis-associated cancer is dependent on the interplay between the hemostatic and inflammatory systems and supported by integrin αMβ2 engagement of fibrinogen. Cancer Res. 70(7):2634–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulniute R, Lindh T, Wilczynska M, Li J, Ny T. 2011. Plasmin is essential in preventing periodontitis in mice. Am J Pathol. 179(2):819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefs K, Gueorguieva M, Klammt J, Allen CM, Aktas D, Anlar FY, Aydogdu SD, Brown D, Ciftci E, Contarini P, et al. 2006. Molecular and clinical spectrum of type I plasminogen deficiency: a series of 50 patients. Blood. 108(9):3021–3026. [DOI] [PubMed] [Google Scholar]
- Tefs K, Georgieva M, Seregard S, Tait CR, Luchtman-Jones L, Ziegler M, Hugle B, Schuster V. 2004. Characterization of plasminogen variants in healthy subjects and plasminogen mutants in patients with inherited plasminogen deficiency by isoelectric focusing gel electrophoresis. Thromb Haemost. 92(2):352–357. [DOI] [PubMed] [Google Scholar]
- Ugarova TP, Lishko VK, Podolnikova NP, Okumura N, Merkulov SM, Yakubenko VP, Yee VC, Lord ST, Haas TA. 2003. Sequence γ377-395(P2), but not γ190-202(P1), is the binding site for the αMI-domain of integrin αMβ2 in the γC-domain of fibrinogen. Biochemistry. 42(31):9365–9373. [DOI] [PubMed] [Google Scholar]
- Ugarova TP, Solovjov DA, Zhang L, Loukinov DI, Yee VC, Medved LV, Plow EF. 1998. Identification of a novel recognition sequence for integrin αMβ2 within the γ-chain of fibrinogen. J Biol Chem. 273(35):22519–22527. [DOI] [PubMed] [Google Scholar]
- Ugarova TP, Yakubenko VP. 2001. Recognition of fibrinogen by leukocyte integrins. Ann N Y Acad Sci. 936:368–385. [DOI] [PubMed] [Google Scholar]
- Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, Baeza-Raja B, Martinez de Lagran M, Lafuste P, Ruiz-Bonilla V, et al. 2008. Fibrinogen drives dystrophic muscle fibrosis via a TGFβ/alternative macrophage activation pathway. Genes Dev. 22(13):1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SD, Weitz JI, Huang AJ, Levin SM, Silverstein SC, Loike JD. 1988. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc Natl Acad Sci U S A. 85(20):7734–7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-jdr-10.1177_00220345231171837 for Plasmin-Mediated Fibrinolysis in Periodontitis Pathogenesis by L.M. Silva, K. Divaris, T.H. Bugge and N.M. Moutsopoulos in Journal of Dental Research