

Abstract
The fields of Neurobiology and Neuromodulation have never been closer. Consequently, the phrase “synaptic plasticity” has become very familiar to non-basic scientists, without actually being very familiar. We present the “Story of the AMPA receptor,” an easy-to-understand “10,000 ft” narrative overview of synaptic plasticity, oriented toward the brain stimulation clinician or scientist without basic science training. Neuromodulation is unparalleled in its capacity to both modulate and probe plasticity, yet many are not comfortable with their grasp of the topic. Here, we describe the seminal discoveries that defined the canonical mechanisms of long-term potentiation (LTP), long-term depression (LTD), and homeostatic plasticity. We then provide a conceptual framework for how plasticity at the synapse is accomplished, describing the functional roles of N-methyl-D-aspartate (NMDA) receptors and calcium, their effect on calmodulin, phosphatases (ie, calcineurin), kinases (ie, calcium/calmodulin-dependent protein kinase [CaMKII]), and structural “scaffolding” proteins (ie, post-synaptic density protein [PSD-95]). Ultimately, we describe how these affect the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor. More specifically, AMPA receptor delivery to (LTP induction), removal from (LTD), or recycling within (LTP maintenance) the synapse is determined by the status of phosphorylation and protein binding at specific sites on the tails of AMPA receptor subunits: GluA1 and GluA2. Finally, we relate these to transcranial magnetic stimulation (TMS) treatment, highlighting evidences for LTP as the basis of high-frequency TMS therapy, and briefly touch on the role of plasticity for other brain stimulation modalities. In summary, we present Synaptic Plasticity 101 as a singular introductory reference for those less familiar with the mechanisms of synaptic plasticity.
Keywords: homeostatic plasticity, long-term depression, long-term potentiation, synaptic plasticity, neuromodulation
INTRODUCTION
It may be that the phrase “synaptic plasticity” has never been uttered so frequently by clinicians and neuroscientists as it is today. Thanks to the emergence of brain stimulation, a new subfield of brain medicine, the disparate fields of cellular neurobiology and systems neuroscience are now relevant to each other as never before. We recognize that understanding mechanisms can inform experimental approaches and ultimately improve treatment effectiveness. Despite the frequency of its use, “synaptic plasticity” still remains conceptually unclear for many clinicians and scientists who were trained from the “top-down.” Our objective is to demystify synaptic plasticity by providing a conceptual framework and then getting into the “nuts and bolts” of its mechanisms.
Disclaimers
We make no effort at a comprehensive or systematic review, which would fill at least one large textbook and require tens of thousands of references. Although we have intentionally referenced the seminal findings throughout, we refer the interested reader to these recent in-depth reviews covering the topics to follow.1–18 We also acknowledge here that canonical synaptic plasticity was primarily studied in relation to learning and memory, rather than depression; and from the hippocampal CA1 region, rather than the cortex.
PART 1: PLASTICITY
A patient remains depressed despite medications. The patient is then successfully treated with transcranial magnetic stimulation (TMS), returns to full functional and normal life, and remains well. What happened? What did TMS do to the brain to cause this transformation?
Plasticity. We could also say “ability to change”, occurs on many interdependent levels (Fig. 1). In the field of brain stimulation, we assess plasticity at differing levels, creating some confusion with the terminology. We therefore propose standardization using the following terms: (1) neuroplasticity (aka neural plasticity)—broadly encompassing plasticity across all cell types and levels of the nervous system (ie, neuronal and non-neuronal, genetic to behavioral, neurogenesis); (2) neuronal plasticity—referring to changes within the neuron (ie, mRNA and protein production, dendritic branching, spine formation, axonal sprouting); (3) synaptic plasticity—referring to changes at the synapse (ie, presynaptic neurotransmitter release, postsynaptic receptor number or conductance). In addition to these general terms, level-specific terms can also be applied to plasticity (ie, transcriptional, network, behavioral, etc.); as well as type-specific terms (ie, glial plasticity, myelin plasticity, structural plasticity [ie, new or enlarged dendritic spines, branches, axon terminals], functional plasticity [ie, neurotransmitter levels]). Among these levels, synaptic plasticity is the “hub,” as it directs subcellular plasticity with regional specificity, and underlies much of circuit-level plasticity.
Figure 1.
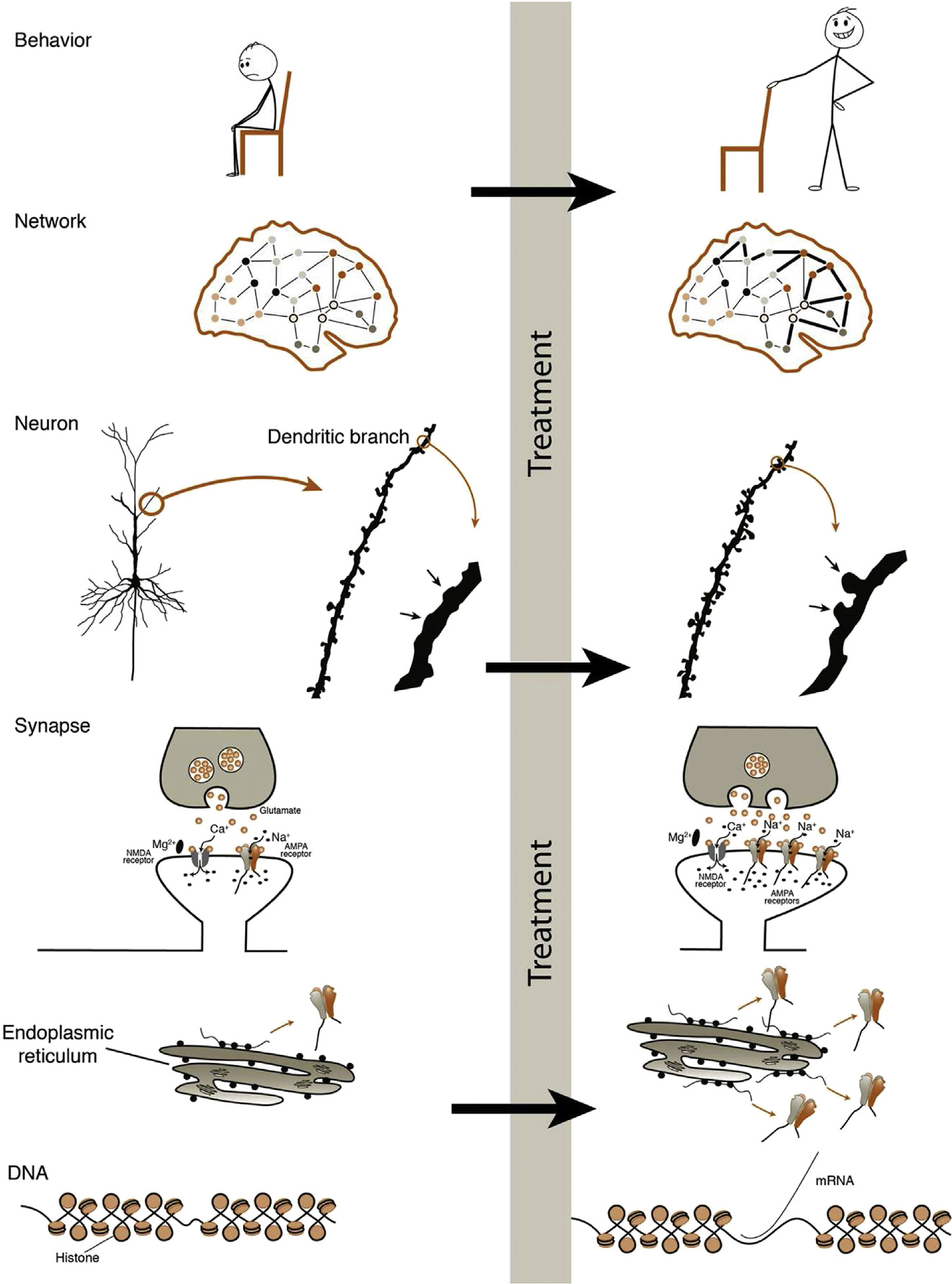
Plasticity. Behavioral plasticity is the product of network plasticity; networks are comprised of circuits. Plasticity of circuits is caused by neuronal plasticity including neurogenesis, dendritic branching, and dendritic spine growth. At the center of it all is synaptic plasticity, or the change in strength of connection between axon terminal and dendritic spine. Synaptic plasticity directly underlies macroscopic changes and triggers signaling molecules to alter mRNA and protein production.
PART 2. WHAT IS SYNAPTIC PLASTICITY?
Synaptic plasticity is the ability to change the strength of connection between neurons. How does the synapse change in strength? It is primarily through the change in conductance and expression (the synaptic presence or absence) of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (thankfully, just “AMPA”) receptors that determines how readily a post-synaptic electrical response is elicited by presynaptic neurotransmitter release. This is the meaning of synaptic strength. When the connections between neurons (synapses) change their strength, we find corresponding downstream changes in DNA transcription, protein production and trafficking; changes in the physical structure of neurons (dendritic branches and spines), circuits and networks; and ultimately, changes in behavior.
The Background
The story of the AMPA receptor began 33 years before its discovery. In 1949, built on the discovery by Santiago Ramón y Cajal that the brain was composed of individual nerve cells, Donald Hebb the First theorized what Carla Shatz famously coined: “neurons that fire together, wire together.”19 In other words, the synapse is strengthened by repeated activity (such as magnetic or electrical stimulation, experience, or learning). This activity may be adaptive (ie, improved function: the goal of rTMS) or maladaptive (ie, impaired function: as with post-traumatic associations).
The Hebbian theory of synaptic plasticity was quickly and widely accepted, despite having no direct evidence until 1973, when Terje Lømo and Tim Bliss published the first report of “long-lasting potentiation” in the rabbit hippocampus. They administered repetitive short-lasting, high-frequency electrical stimulation at 10 to 20 pulses per second (Hz) for 10 to 15 seconds, as well as 100 Hz for 3 to 4 seconds, to produce what came to be known as long-term potentiation, or LTP20,21 (Fig. 2). A core property of LTP is persistence—the enduring nature of neural changes. In fact, although LTP may only last minutes to hours in vitro, it can persist beyond 1 year in live animals.22
Figure 2.
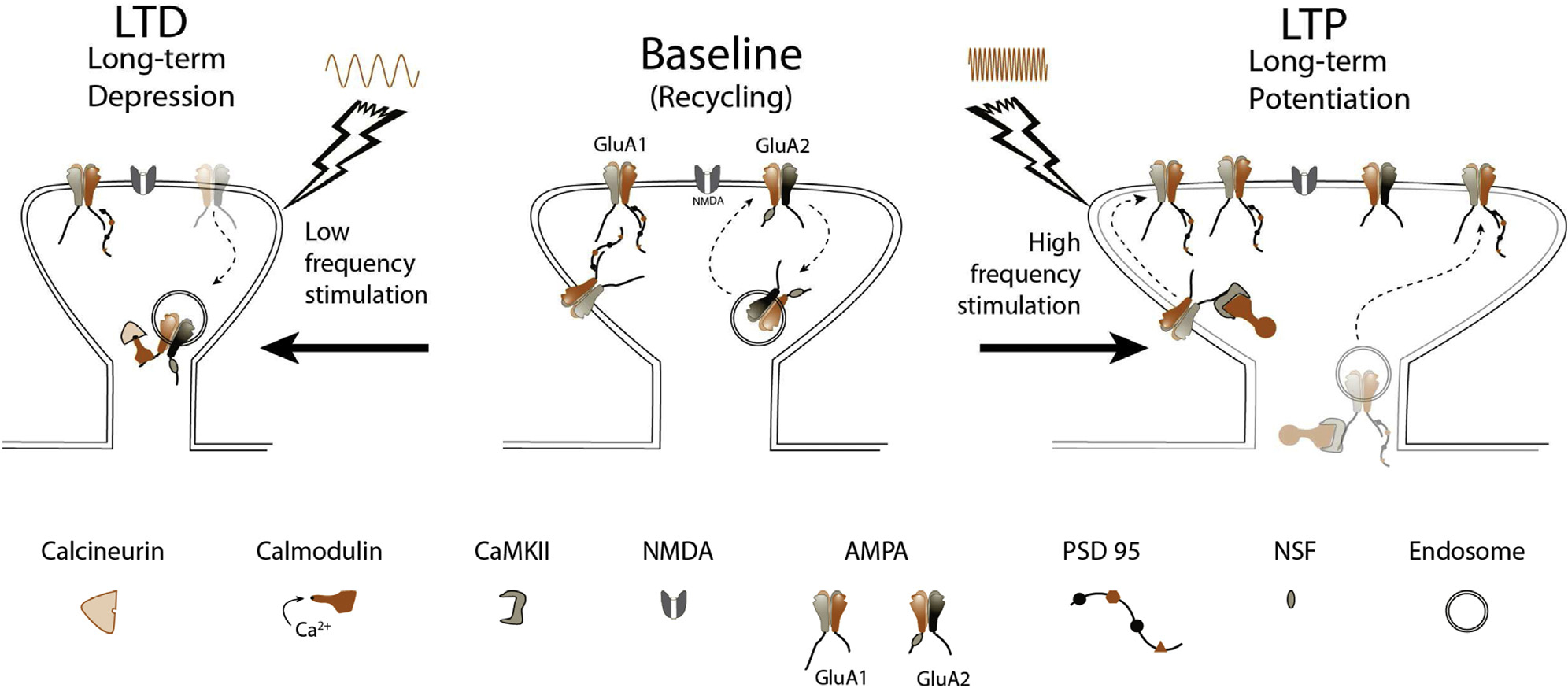
Synaptic plasticity. Center: At baseline, synaptic NMDA and AMPA receptor numbers are stable, with GluA2-subtype AMPA receptors recycling constitutively, dependent on NSF. Left: Following low-frequency stimulation, chronic low levels of calcium enter through the NMDA receptor and bind calmodulin. Calcineurin has the highest affinity for Ca2+/CaM, beating out CaMKII, and causing dephosphorylation of the AMPA receptor, and subsequent removal from the synapse. The result is a smaller spine and decreased synaptic strength. Right: High-frequency stimulation causes AMPA receptor depolarization, consequent ejection of the Mg2+ plug, and opening of the Ca2+-permeable NMDA receptor channel. A flood of Ca2+ leads to acutely elevated concentrations of Ca2+/CaM complexes, allowing interactions with CaMKII to dominate, ultimately causing phosphorylation and delivery of GluA1 to the synapse through translocation along the membrane (initial 20 minutes), or direct exocytosis and insertion in the synapse. Scaffolding proteins like PSD-95 accompany this insertion. Spine size grows, and synaptic strength is increased.
Soon after this discovery, while inducing LTP in synapses at one end of the neuron, scientists observed that distant synapses not being stimulated had decreased responses. This effect was also persistent and represents a second type of synaptic plasticity called long-term depression, or LTD23 (Fig. 2). This type of LTD was termed heterosynaptic LTD because it resulted from stimulation of a different area. It was not until 15 years later that scientists discovered how to induce LTD directly, called homosynaptic LTD, or just LTD (unless otherwise specified, this is generally the type of LTD being referred to). LTD is produced by repeated low-frequency (ie, 1 Hz) and chronic (ie, 900 pulses) stimulation.24 LTP and LTD can be thought of as artificial mechanisms that simulate what the brain does when learning, remembering, and forgetting. For example, learning can cause measurable LTP and LTD.25,26 Naturally, these changes don’t occur “willy-nilly,” but as Hebb predicted, are precisely placed and timed. For example, repeated pre-synaptic action potentials (“spikes”) just preceding (ie, by several milliseconds) a post-synaptic action potential may produce LTP, whereas the reverse sequence may produce LTD. We call this “spike-timing dependent plasticity” (STPD). Speaking more generally, this is the principle of “activity-dependent synaptic plasticity.” It’s the rationale behind pairing a cognitive therapy task with brain stimulation. Association between events (ie, ate unknown berries, vomited profusely) appears to be nature’s way of ensuring fidelity and specificity in memory formation.
What happens after a synapse has undergone LTP? Does it retain the ability to undergo LTP again? These questions introduce a third type of synaptic plasticity, called homeostatic plasticity. Conceptually least understood, homeostatic plasticity simply refers to the capacity of the neuron to maintain homeostasis at the synapse—avoiding “ceiling” or “floor” effects. For example, researchers found that after rats had learned a particular task, LTP of the same synaptic circuit could not be induced thereafter due to a saturation effect called “occlusion.”27 Fortunately, for our ability to remember important above non-important things, a homeostatic plasticity mechanism called synaptic scaling allows the neuron to “scale down” synaptic strength across the entire neuron, making repeat potentiation possible, and very importantly, preserving memory by keeping the relative differences in strength among synapses intact.28,29 The neuron can also “scale up” after decreased neuronal activity.30,31 Synaptic scaling takes a while (4 to 24 hours), which may explain in part why long-term memory is best facilitated by revisiting the information periodically, rather than all at once. Homeostatic plasticity falls under the umbrella of another term we hear a lot: “metaplasticity.” It is described as a “plasticity of plasticity” (compare with meta-analysis, which is an analysis of analyses). More concretely, metaplasticity controls how a synapse will respond (ie, direction, magnitude, duration) to a given stimulus.32 Ultimately, Hebbian plasticity (LTP and LTD) and homeostatic plasticity are inextricably interconnected.
PART 3: MECHANISMS OF SYNAPTIC PLASTICITY: THE STORY OF THE AMPA RECEPTOR
The synapse is the command center of plasticity. However, before Hebb’s theory, the glutamatergic synapse was an unknown landscape. Ten years later, the first constituent was finally identified: an excitatory neurotransmitter known as L-glutamate.33,34 This finding led to the search for a corresponding receptor. Three were found: NMDA, Kainate, and AMPA. We mention Kainate receptors only briefly here to say that initially their role in synaptic plasticity was controversial,35,36 but with the recent emergence of appropriate pharmacologic agents, their pre- and post-synaptic role in LTP, LTD, and homeostatic plasticity has been better established, which can be discovered in the following reviews.37–39 Additionally, recent years have brought a growing recognition of non-ionotropic receptors (those which do not admit ions), but instead are metabotropic (mGluR), and thus, initiate intracellular signaling cascades.15 These too have an important role in LTP, LTD, homeostatic plasticity, and clinically.40,41
The NMDA receptor was the first to receive attention for its role in synaptic plasticity after the observation that an NMDA receptor antagonist successfully blocked LTP.42 Evidence for LTP as a cellular correlate of learning and memory took hold when this NMDA receptor antagonist also prevented disgruntled wet rats from learning and remembering their way to a rescue platform in the Morris water maze.43
LTD was likewise found to depend on NMDA receptors.44 Although NMDA receptor-independent forms (such as mGluR-dependent) are also well characterized,45 some evidence suggests they may ultimately have a common convergence point.46 The central role of the NMDA receptor in synaptic plasticity is evident from the mechanistic studies in human noninvasive brain stimulation (NIBS) focused on these receptors (Part 5).
Magnesium was serendipitously found to antagonize NMDA receptor-mediated transmission.47 This blocking effect is voltage-dependent (Fig. 2).48,49 Adjacent AMPA receptors can depolarize the membrane (through sodium admittance) sufficient to eject the NMDA receptor’s magnesium “plug,” allowing entry of sodium, and most notably, calcium. This magnesium-gating mechanism explains some of the other core properties of LTP including (1) input specificity (LTP occurs only at the synapses being activated); (2) cooperativity (summation of simultaneous nearby stimulated pathways—a property which may explain how relatively low TMS frequencies like 10 Hz can have excitatory effects); and (3) associativity (a weak stimulus pattern [ie, 3 Hz] can induce LTP if paired with global depolarization [ie, 0 mV], as used in many LTP protocols). TMS protocols like paired associative stimulation (PAS) capitalize on this property of LTP.
These early studies noticed that NMDA receptors were required for changes in synaptic strength (ie, synaptic plasticity), but curiously, they did not mediate baseline synaptic activity, also known as synaptic transmission. If NMDA receptors do not mediate synaptic transmission, what does?
Doing the Work of the Synapse
The answer came in 1988, when Muller and colleagues found that 85% of the synaptic transmission in the hippocampus was abolished with antagonism of the AMPA receptor.50 Across brain regions, AMPA receptors mediate about 70% of the excitatory transmission. Therefore, AMPA receptors do most of the synaptic “work” (ie, ion admittance and current conduction), but NMDA receptors direct that work. How, then, does the NMDA receptor direct the AMPA receptor? This sophisticated communication is the mechanism of synaptic plasticity. Two side notes: First, why are so many synaptic plasticity studies in the hippocampus? Aside from its role in learning and memory; the hippocampus has a unique ‘trisynaptic’ structure that is ideal for stimulating presynaptic axons (CA3 Schaffer collateral pathway) and recording from innervated postsynaptic neurons (CA1 region). The prefrontal cortex (PFC), by contrast, is messy. It is innervated by axons from all over the brain, and consequently, it’s much more difficult to stimulate axons connected to the neuron being recorded. The CA1 neurons are biochemically very similar to the PFC. Second, Mediators vs Modulators: In clinical brain medicine, we often focus on modulators, such as the monoamines. Modulators alter the efficiency of the mediator. The actual glutamatergic signal is mediated by the AMPA receptor. But modulators can tune up, or tune down, the signal strength, such as through norepinephrine or GABA. To put it another way, modulators are like medical students. They can make things more or less efficient for the physician, but they can’t actually sign any notes, or put in any orders. Modulators could alter the clinical response to TMS as suggested by naturalistic reports on benzos and stimulant effects. We also see their influence on excitability of the motor cortex.51
Synaptic Communication
If synaptic plasticity is a language, Calcium concentration is the binary code. Ca2+ itself is necessary and sufficient for LTP52,53 and LTD.44 How can the same signal be used for opposing processes? It is the concentration of Ca2+, in the right place, over the right time period, that determines which biochemical cascade is initiated, LTP or LTD.54,55 Acutely high Ca2+ concentrations cause LTP, whereas chronically low Ca2+ concentrations cause LTD. Notice how TMS protocols mirror Ca2+ concentration patterns. For example, “excitatory” protocols are higher frequency (10 Hz), given in short bursts (4 sec “on”, 26 sec “off”) or in an intermittent theta burst pattern, while “inhibitory” protocols chronically (with no “off” periods) stimulate with lower-frequencies (1 Hz) or in a continuous theta burst pattern.56
Before the signaling sequence is initiated, Ca2+ must first become recognizable to other molecules. This happens when Ca2+ binds with calmodulin (Ca2+/CaM).57,58 For this reason, calmodulin must be precisely localized to come into contact with Ca2+. This localization is the singular job of neurogranin,59 a protein that can enhance LTP and learning if increased, or it could cause working memory problems, Alzheimer’s, and schizophrenia if dysfunctional.60,61 Acutely high Ca2+/CaM concentrations activate kinases. Kinases phosphorylate the tail of the AMPA receptor shown in Figure 3, leading to both increased AMPA receptor conductance, and insertion into the synapse and LTP. Chronically low Ca2+/CaM concentrations activate phosphatases and resultant dephosphorylation, leading to LTD.
Figure 3.
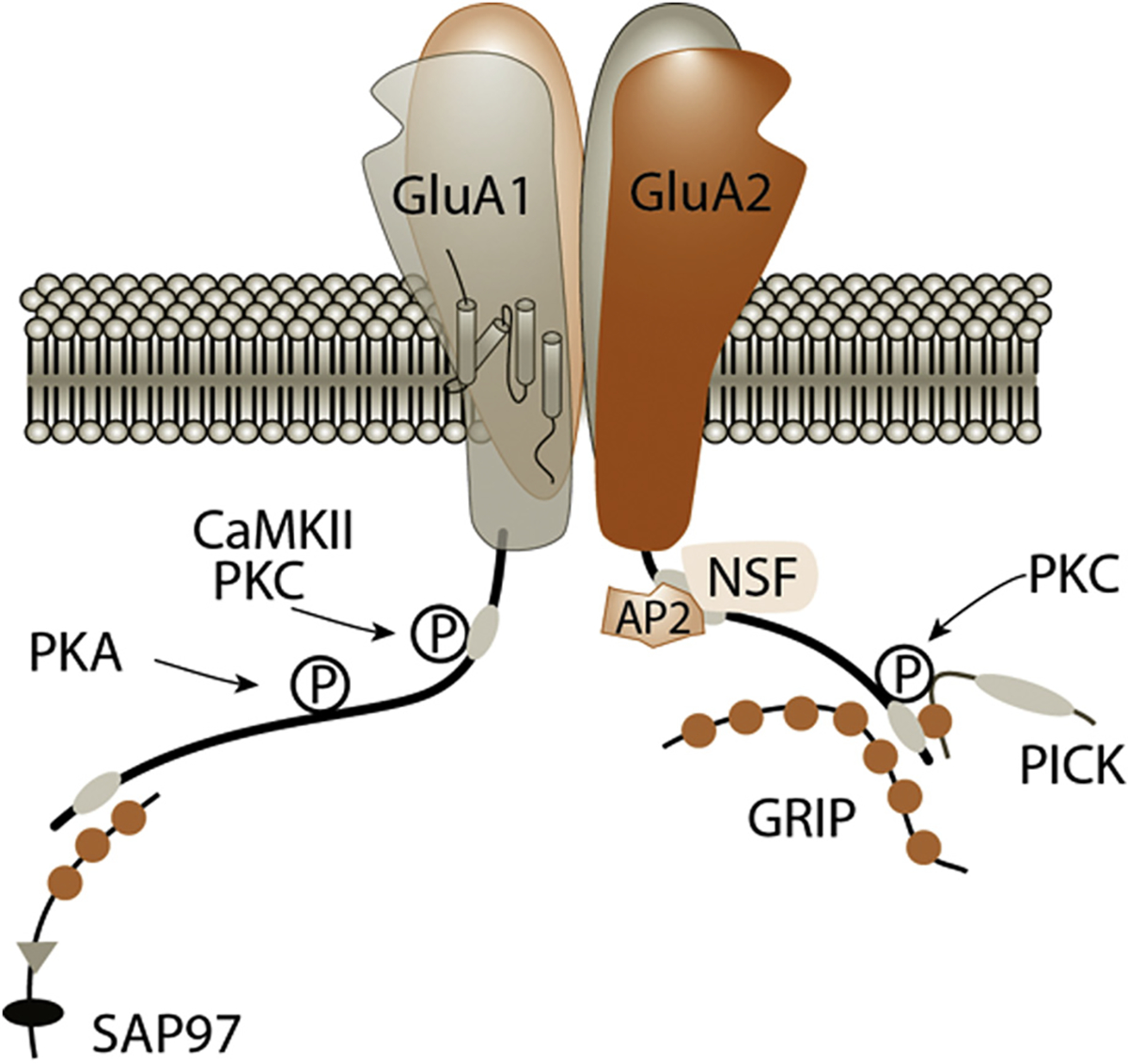
AMPA receptor binding sites. AMPA receptors are composed of 4 subunits, forming a central pore region where sodium enters. Glutamate binding induces a conformational change that opens the pore, allowing sodium to enter according to an electrical gradient. Each subunit contains 4 domains that span the membrane, an extracellular side where glutamate binds, and a tail inside the neuron with key binding and phosphorylation sites that determine the fate of the receptor. Binding Proteins: GluA1: *SAP-97 (synapse-associated Protein-97 [kDa]), *Protein 4.1 N (not shown). GluA2: *NSF (N-ethylmaleimide sensitive factor), AP-2 (adaptor protein-2), **GRIP (glutamate receptor interacting protein), **PICK (protein interacting with C-kinase), BRAG-2 (not shown). Phosphorylation sites (‘P’): GluA1: *S831: CaMKII (Ca2+/CaM–dependent protein kinase II), PKC (protein kinase-C); T840: p70S6 (not shown), PKC; S845: **PKA (cAMP-dependent protein kinase/protein kinase A). GluA2: *S863: PKC; Y876: *Src family kinase (not shown); S880: PKC. *Promotes AMPAR insertion. **Able to facilitate both insertion and removal.
AMPA Receptor Anatomy: the Key to Understanding the Mechanism
Like any story, synaptic plasticity involves the complex interactions of the main character (the AMPA receptor) with others (enzymatic and structural proteins). To understand these interactions, we must understand the anatomy of the AMPA receptor (Fig. 3). See also62,63 if interested in more details. The authors advise that despite our best efforts to simplify a complex topic, the following inevitably involves a level of detail that may discourage the casual reader, who may skip to Part 4.
Inducing LTP
AMPA receptors come in several varieties based on the subunits they contain. Each subunit has distinct binding and phosphorylation sites that determine what they can do. The subunits are called “GluA” types 1 to 4 to denote a glutamate receptor of AMPA-type (formerly known as “GluR”, which did not distinguish from NMDA and Kainate receptors) with 4 possible subtypes. When it comes to LTP, there is only one subtype worth talking about: the GluA1/GluA2 heterodimer (meaning 2 types of each subunit), or simply GluA1 (Fig. 3). The GluA1 tail has specific amino acids (S831 and S845 if you’re a “gunner” [ie, overambitious learner]) that can be phosphorylated by kinases, and no kinase says “LTP” more than calcium–calmodulin-dependent protein kinase II, also known as CaMKII (pronounced “Cam-kinase-2”). [Other prominent kinases involved in LTP include protein kinase A (PKA), and protein kinase C (PKC); and to a lesser extent, phosphatidylinositiol-3-kinase (PI3K), extracellular signal–regulated kinase (ERK), and protein-tyrosine kinases (PTKs).] As a reminder, CaMKII is activated by acutely high levels of Ca2+/CaM complexes resulting from NMDA receptor activation and opening. Phosphorylation by CaMKII causes a conformational change in the AMPA receptor. The new conformation opens the pore to let more sodium in. Even more to the point, it triggers a series of complex reactions leading to delivery and insertion of AMPA receptors into the synapse.64 Some of these details are still being worked out, but by way of an overview, there are several steps to get a receptor from the endoplasmic reticulum (ER) to the synapse. First, AMPA receptor-containing vesicles are formed and transported along microtubule tracks from the ER (in the nucleus) to the distant dendritic spines (small protrusions budding off the dendritic branch). The vesicles must eventually fuse with the cell membrane, exposing their contents (receptors) via exocytosis. Some receptors are inserted directly into the synapse during LTP. Others are inserted into the membrane near the synapse, waiting “on-deck” for the signal to migrate along the membrane into the synapse. These peri-synaptic “on-deck” receptors are thought to account for the first 20 minutes of LTP.65 Transportation and synaptic delivery of these receptors depend on signaling proteins directing motor proteins allowing the membrane fusion proteins to finish the job.66–69
Maintaining LTP
The receptor has been delivered to the synapse. LTP has occurred…or has it? By definition, LTP must be “long-term.” AMPA receptors are highly mobile, and they turn over frequently to avoid breakdown. So once LTP is induced, that is, the AMPA receptor makes it to the synapse, how is it kept there? What good is the formation of a memory, or circuit, if it is lost just as quickly?
If GluA1 is the LTP “inducer,” then GluA2 is the “maintainer.” This AMPA receptor subtype replaces GluA1 after ~18 hours.70,71 Once there, protein Kinase M-ζ facilitates GluA2 recycling in and out of the synapse constitutively, in order to maintain molecular memory.72,73 Unlike GluA1-mediated LTP, which depends on neuronal and NMDA receptor activity, GluA2 recycling occurs regardless of neuronal or NMDA receptor activity. They are only removed “intentionally” through regulated mechanisms, like LTD. Although this subunit exchange is critical to retain important information (like the location of the cleanest restroom in the building), we do not yet know exactly how it happens. One protein, called BRAG1, has been proposed to fulfill this role. BRAG1 both removes GluA1 through LTD, and increases GluA2, independent of activity.74,75 [If you are curious about what BRAG1 stands for, don’t be: Brefeldin A-resistant ADP-ribosylation factor (Arf)-guanine nucleotide exchange factor (GEF)-type 1.] It would be a brilliant design of nature to have a single protein both remove and insert the desired receptors to ensure an equal ratio. To this end, mutations in BRAG1 cause autism and X-linked intellectual disability.76,77
For some people, maintaining LTP is not enough; they want to specify for how long. “Early” and “late” LTP are differentiated most notably by the transcription of DNA and the production of new protein. If you block DNA transcription during stimulation, you still see potentiation initially, but it does not endure. After 100 minutes, responses return to baseline.78 By comparison, if you stimulate first, then block 2 hours after, the result is normal, durable LTP.78 The same thing happens with protein production: Translation blockers don’t impair the induction of LTP, but the effects are short-lived.79 Thus, durable, “late” LTP depends on new proteins being produced and transported to the appropriate synapse.
We confess that in our excitement to talk about LTP maintenance, we glossed over an important “character” involved in LTP induction: scaffolding proteins. Like AMPA receptors, these proteins are recruited to the synapse during LTP induction. Without the structure they provide, AMPA and NMDA receptors would float around the synapse willy-nilly. They also support trafficking of receptors and associated proteins to their destination like a signaling beacon. In aggregate, these proteins can be easily seen on electron microscopy, and even have a name: the post-synaptic density, or PSD. The PSD is the city center of the neuron, where the action happens. Scaffolding proteins are large and highly connected with other proteins to provide the structure of the synapse. The best-known scaffolding protein, named after the PSD, weighs 95 kDa. PSD-95 is intimately involved with LTP induction and eventual maintenance. Without it, LTP doesn’t happen.80 This is at least in part because of the structural stability it provides for the synaptic AMPA receptors.81 PSD-95 is one of a family of proteins (called membrane-associated guanylate kinases, or MAGUKs) that have redundant functions but all have an essential role in AMPA receptor trafficking and stability.82 Like PSD-95, most proteins don’t directly bind the limited real estate of the AMPA receptor directly. Additional “hooks” to bind are created by the “transmembrane AMPA receptor regulatory protein,” or TARP. These create an entire complex around the receptor for signaling and binding. However, 2 proteins operate independently of TARPs, and bind GluA1 directly: SAP-97, a member of the MAGUK family, moves AMPA receptors into proper position in the synapse.69 The other, Protein 4.1 N, helps build the actin cytoskeleton of the dendritic spine.83 After LTP is induced, this network of actin enlarges the dendritic spine (a hallmark of LTP, as shown in Fig. 2) and provides a framework for the new GluA1 receptors and binding proteins.84
LTD
So far, we have discussed the “scaffolding” and signaling role of these binding proteins. Now, we briefly delve into how these proteins walk the fine line between “will I stay or will I go?” (“I” being an AMPA receptor, of course). We described how they stay, through recycling GluA2 receptors, but how do they go? How does LTD work? The surface-level answer is that phosphorylation status and binding of key proteins to the GluA2 tail control these processes.85,86 [By the way, because both GluA1 and GluA2 receptors contain the actual GluA2 subunit protein (Fig. 3), both can undergo LTD, though GluA2 seems to have a more prominent role, perhaps because they are longer term.] We hope you’ll agree that one mechanistic example of how this can happen will be both enlightening and sufficient. As shown in Figure 3, on the GluA2 subunit tail, amino acid S880 is subject to phosphorylation. When this happens, a change in conformation pushes off GRIP1, making way for PICK1 to move in. GRIP1 promotes receptor insertion;87 whereas its opponent, PICK1, causes removal through LTD,88,89 and even problems with learning90 and sociability in autism.91 You see, PICK1 has a fetish for curved membranes (ie, vesicles),92 which tends to lead to endocytosis while bringing GluA2 along for the ride.93 How does endocytosis actually occur - you ask? Well, clathrin-coated pits (bristly proteins that embed and surround the vesicle) act sort of like a detergent to break the vesicle away from the rest of the membrane, making internalization possible.94 How do the clathrin proteins even know where to congregate, and which receptor to internalize? The GluA2 tail is tagged by yetanother AMPA receptor binding protein (adaptor Protein-2 [AP2]), while a special lipophilic messenger (phosphatidylinositol 4,5-bisphosphate [PIP2]) marks the membrane, all orchestrated by a covert puppet-master enzyme called ADP-ribosylating factor-6 (Arf6).95–97 And that is how the receptor is internalized, resulting in LTD.
PART 4: THE AMPA ALLEGORY
We have provided an overview, with a few deep dives, on the mechanisms of synaptic plasticity. We will summarize and review with an allegorical anthropomorphization of the AMPA receptor. We compare the AMPA receptor to a medical resident (a post-doc can be interchanged with a little imagination). We hope it facilitates understanding. The mechanism or molecule being represented is in parentheses.
At the hospital (the synapse), patients (ions) typically present for admission (entry into cell) at a fairly stable rate. Patients enter through the Emergency Room where the ER doc (glutamate) will call a consult (activates the receptor). However, patients (ions) only gain admission (cell entry) through the house staff, also known as the resident physician (AMPA receptor). Some hospitals also have mid-level providers (Kainate receptors) who can admit patients (ions), but their roles were only recently understood. If the residents (AMPA receptors) are antagonized, will hospital productivity decrease by 85% (as with AMPA receptor antagonists)? The attending (NMDA receptor), on the other hand, does very little of the actual work of admitting patients (ions). This may be in part because of their significant other (Mg2+), who is often around the attending and “in the way” (in the pore, plugging it) of their productivity (ion admittance). It usually takes a resident (AMPA receptor) to activate the attending (depolarizes, ejecting Mg2+ plug). However, the attending does oversee and direct (plasticity) the work. The attending works through written communication (calcium concentrations). However, attendings notes and orders are notoriously microscopic and illegible (Ca2+ alone) unless a transcriptionist (calmodulin) makes them readable. If the transcriptionist is not at the right place at the right time, the message is not received (no synaptic plasticity cascade). Thus, a dedicated supervisor makes sure the transcriptionist is where they need to be (neurogranin).
Although resident shifts have mercifully decreased from 36 to 18 hours, residents still fatigue (protein degradation) and must be switched out (GluA1 to GluA2 exchange occurs around 18 hours). Importantly, physicians (receptors) do not work alone. A team of nurses, CNAs, secretaries, and others (enzymatic and signaling proteins) carry out physician (receptor)-initiated orders. These dedicated folks keep the hospital humming (carry out plasticity cascades). Finally, work rooms and call rooms (structural/scaffolding proteins) provide a place to do the work.
There may be times of increased demand, such as patients being diverted to a hospital (high-frequency stimulation), resulting in increased hospital admissions (ion flow), which requires “surging” in the back-up residents (GluA1) to handle the work (LTP). Other hospital workers (enzymatic and signaling proteins) must also be surged in to handle the load, and increased work space becomes necessary (structural/scaffolding proteins). However, even hospitals (synapses) have their limits and can only hold so many patients and residents (occlusion). To maintain the capacity to bring in more if needed, they will try to prevent saturation by sending some residents home (homeostatic plasticity).
The neighboring hospitals, the ones not being diverted to, will see fewer admissions, and will therefore send their residents home (heterosynaptic LTD). There may also be cases where a hospital might also divert away admissions (low-frequency stimulation), also leading to decreased work, and decreased need for staff (enzymatic and signaling proteins) and residents (AMPA receptors), and because the administration (nucleus—control center for protein trafficking) are shrewd business people, residents and staff are sent home to not waste (cellular) resources (LTD).
PART 5: SYNAPTIC PLASTICITY IN BRAIN STIMULATION
Now for practical application; how is knowledge of synaptic plasticity relevant in the real world? We return to our hypothetical depressed patient from the beginning who remitted after TMS treatment. What did TMS do to the brain to cause a lasting and therapeutic change? The prevailing view is that synaptic plasticity is the basis of this change. What is the evidence for this? Perhaps the most direct evidence comes from mouse hippocampal slices. Vlachos and colleagues found that 10 Hz rMS (no cranium to “trans” here!) increased AMPA receptor insertion in the synapse, increased GluA1 levels, and enlarged dendritic spines.98 Moreover, potentiation was blocked by NMDA receptor antagonists and sodium and calcium channel blockers.98,99
Human studies are less substantiative. As a proxy to opening the brain and laying out hippocampal slices to record from individual neurons, the majority of human studies have measured rTMS-induced changes in brain excitability indirectly with thumb or finger twitches called motor-evoked potentials (MEPs). Various experimental “plasticity-promoting” rTMS protocols (ie, continuous theta burst stimulation (c)TBS, PAS, laser PAS, and ischemic nerve block with 0.1 Hz rTMS) and a recently FDA-approved protocol (intermittent (i)TBS) have been used in the presence of NMDA receptor blockers. Like the mouse experiments, these studies have unanimously shown that NMDA receptors are necessary for TMS-induced potentiation and inhibition.100–104 More specifically, is NMDA receptor activity sufficient to enhance rTMS facilitation? Two groups used iTBS with NMDA receptor partial agonist, d-cycloserine (DCS), but instead of facilitation, they unexpectedly observed inhibition.105,106 By contrast, the only study to assess traditional 10 Hz rTMS found subjects had greater facilitation with DCS than with placebo in a crossover design, indicating NMDA receptor agonism was sufficient.107 Interestingly, a follow-up analysis of short-term plasticity paired-pulse protocols from the same cohort found effects mimicking occlusion of facilitation and a potential homeostatic-based enhanced inhibition in the DCS condition.108 Taken together with the iTBS studies, NMDA receptor activation appears sufficient to enhance 10 Hz and demonstrates previously described LTP-like properties of occlusion27 and homeostatic depression23 with more robust plasticity protocols. Bearing in mind that these small studies would benefit from replication with larger “n,” they are consistent with the LTP hypothesis for TMS.
MEPs provide an easily quantifiable measure of brain excitability, but do they translate to clinical outcomes? After all, MEPs are in the motor, not the prefrontal cortex, and parameters differ from those used clinically. Evidence translating MEPs to clinical outcomes is scarce, but one group found that depression symptom improvement correlated with increased MEP plasticity.109 TMS-evoked potentials (TEPs) use electroencephalography (EEG) to quantify excitability anywhere in the brain. TEPs appear to correspond with MEPs when done over the motor cortex, and have a similar response to drugs.110–113 TEPs have tremendous potential to reveal brain plasticity outside the motor cortex, but great care must be taken to avoid interpretation of auditory and somatosensory artifacts as brain signal.114
Why does it seem like TMS is suspiciously applicable to so many neuropsychiatric conditions?115 Unlike drugs which alter the “firing” propensity of neurons all over the brain, TMS “speaks the language of the brain”, sufficient to “fire” neurons at specified brain regions. Able to both promote and probe plasticity, TMS is the “poster child” of synaptic plasticity.
What about other forms of brain stimulation? What role does synaptic plasticity have in their mechanisms of action? It is beyond the scope here to provide a comprehensive review, but a few points bear mentioning. Transcranial direct current stimulation (tDCS) is not yet clinically indicated, but like TMS, can be spatially targeted to specific brain regions and circuits. Unlike TMS, tolerable intensities of tDCS are not strong enough to produce action potentials, but are thought to “prime” the neuron. Interestingly, tDCS still displays many of the properties of synaptic plasticity, reviewed here.116 For example, like the human TMS studies described above, NMDA receptor activity was required for both LTP- and LTD-like effects117,118 and NMDA receptor agonists prolonged the effect of excitatory tDCS.119 Other clinical brain stimulation therapies like electroconvulsive therapy (ECT) and vagus nerve stimulation (VNS) produce numerous changes throughout the brain and across systems (ie, neuroplasticity), but the role of synaptic plasticity is not well defined.120–123 Interestingly, even the effects of deep brain stimulation (DBS), traditionally thought limited to the duration of stimulation, now have evidence suggesting a role for synaptic plasticity, even giving rise to a proposed rebranding: “deep brain neuromodulation.”124
CONCLUSIONS
How can we use our knowledge of synaptic plasticity to help ourselves and our patients? Those activities forever known to promote brain health and optimize learning, such as sleep and exercise, have also been found to enhance plasticity in TMS/neurophysiology measures.125,126 Some might even take it a step further by enhancing their performance with stimulants, which increase MEPs,51 or with electrical stimulation, such as with gamers and tDCS.127 Anecdotally, gamers have been an interesting living experiment; they have learned that stimulating for too long can actually have deleterious effects on performance. Finally, perhaps the holy grail of brain stimulation is knowing how to select the best parameters for a desired effect. Because a systematic approach to parameter selection with clinical trials is simply not feasible, could we use plasticity outcomes first, with translation to the clinic afterward? We have already seen principles of synaptic plasticity leading to the advent of iTBS128,129 and even the rationale for 1-hour spacing between multiple daily iTBS sessions.130 What will be next?
Acknowledgements
The authors thank Gregory Sahlem, MD, Lisa McTeague, PhD, Noah Philip, MD, Shiwen Yuan, MD, Morgan Healey, Andy Fukuda, MD, PhD, Rich Weiner, MD, PhD, and Linda Carpenter, MD, for helpful suggestions and feedback.
Source(s) of financial support:
This work was supported by the National Institute of General Medical Sciences (P20GM130452) and by the DART Training Grant at MUSC: National Institute on Drug Abuse (R25DA020537).
Footnotes
Conflict of Interest: The authors reported no conflict of interest.
REFERENCES
- 1.Alkadhi KA. NMDA receptor-independent LTP in mammalian nervous system. Prog Neurobiol. 2021;200:101986. [DOI] [PubMed] [Google Scholar]
- 2.Baez MV, Cercato MC, Jerusalinsky DA. NMDA receptor subunits change after synaptic plasticity induction and learning and memory acquisition. Neural Plast. 2018;2018:5093048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baltaci SB, Mogulkoc R, Baltaci AK. Molecular mechanisms of early and late LTP. Neurochem Res. 2019;44:281–296. [DOI] [PubMed] [Google Scholar]
- 4.Bayer KU, Schulman H. CaM kinase: still inspiring at 40. Neuron. 2019;103:380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diering GH, Huganir RL. The AMPA receptor code of synaptic plasticity. Neuron. 2018;100:314–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franchini L, Carrano N, Di Luca M, Gardoni F. Synaptic GluN2A-containing NMDA receptors: from physiology to pathological synaptic plasticity. Int J Mol Sci. 2020;21:1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greger IH, Watson JF, Cull-Candy SG. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron. 2017;94:713–730. [DOI] [PubMed] [Google Scholar]
- 8.Hell JW. How Ca2+-permeable AMPA receptors, the kinase PKA, and the phosphatase PP2B are intertwined in synaptic LTP and LTD. Sci Signal. 2016;9:e2. [DOI] [PubMed] [Google Scholar]
- 9.Kavalali ET, Monteggia LM. Targeting homeostatic synaptic plasticity for treatment of mood disorders. Neuron. 2020;106:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei W, Omotade OF, Myers KR, Zheng JQ. Actin cytoskeleton in dendritic spine development and plasticity. Curr Opin Neurobiol. 2016;39:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mateos-Aparicio P, Rodríguez-Moreno A. Calcium dynamics and synaptic plasticity. Adv Exp Med Biol. 2020;1131:965–984. [DOI] [PubMed] [Google Scholar]
- 12.Nicoll RA. A brief history of long-term potentiation. Neuron. 2017;93:281–290. [DOI] [PubMed] [Google Scholar]
- 13.Penny CJ, Gold MG. Mechanisms for localising calcineurin and CaMKII in dendritic spines. Cell Signal. 2018;49:46–58. [DOI] [PubMed] [Google Scholar]
- 14.Piochon C, Kano M, Hansel C. LTD-like molecular pathways in developmental synaptic pruning. Nat Neurosci. 2016;19:1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiner A, Levitz J. Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron. 2018;98:1080–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vallejo D, Codocedo JF, Inestrosa NC. Posttranslational modifications regulate the postsynaptic localization of PSD-95. Mol Neurobiol. 2017;54:1759–1776. [DOI] [PubMed] [Google Scholar]
- 17.Won S, Levy JM, Nicoll RA, Roche KW. MAGUKs: multifaceted synaptic organizers. Curr Opin Neurobiol. 2017;43:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y, Cheng X, Zhang L, et al. The functional and molecular properties, physiological functions, and pathophysiological roles of GluN2A in the central nervous system. Mol Neurobiol. 2017;54:1008–1021. [DOI] [PubMed] [Google Scholar]
- 19.Hebb DO. The Organization of Behavior; A Neuropsychological Theory. New York: Wiley; 1949. [Google Scholar]
- 20.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bliss TV, Gardner-Medwin AR. Long-lasting potentiation of synaptic transmission in the dentate area of the unanaestetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:357–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abraham WC. How long will long-term potentiation last? Philos Trans R Soc Lond B Biol Sci. 2003;358:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch GS, Dunwiddie T, Gribkoff V. Heterosynaptic depression: a postsynaptic correlate of long-term potentiation. Nature. 1977;266:737–739. [DOI] [PubMed] [Google Scholar]
- 24.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci U S A. 1992;89:4363–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong Z, Gong B, Li H, et al. Mechanisms of hippocampal long-term depression are required for memory enhancement by novelty exploration. J Neurosci. 2012;32:11980–11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. [DOI] [PubMed] [Google Scholar]
- 27.Rioult-Pedotti MS, Friedman D, Donoghue JP. Learning-induced LTP in neocortex. Science. 2000;290:533–536. [DOI] [PubMed] [Google Scholar]
- 28.Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turrigiano G Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4:a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. [DOI] [PubMed] [Google Scholar]
- 31.Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. [DOI] [PubMed] [Google Scholar]
- 32.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. [DOI] [PubMed] [Google Scholar]
- 33.Curtis DR, Phillis JW, Watkins JC. Chemical excitation of spinal neurones. Nature. 1959;183:611–612. [DOI] [PubMed] [Google Scholar]
- 34.Watkins JC. l-glutamate as a central neurotransmitter: looking back. Biochem Soc Trans. 2000;28:297–309. [PubMed] [Google Scholar]
- 35.Bortolotto ZA, Clarke VR, Delany CM, et al. Kainate receptors are involved in synaptic plasticity. Nature. 1999;402:297–301. [DOI] [PubMed] [Google Scholar]
- 36.Nicoll RA, Mellor J, Frerking M, Schmitz D. Kainate receptors and synaptic plasticity. Nature. 2000;406:957. [DOI] [PubMed] [Google Scholar]
- 37.Nair JD, Wilkinson KA, Henley JM, Mellor JR. Kainate receptors and synaptic plasticity. Neuropharmacology. 2021;196:108540. [DOI] [PubMed] [Google Scholar]
- 38.Sihra TS, Flores G, Rodríguez-Moreno A. Kainate receptors: multiple roles in neuronal plasticity. Neuroscientist. 2014;20:29–43. [DOI] [PubMed] [Google Scholar]
- 39.Valbuena S, Lerma J. Kainate receptors, homeostatic gatekeepers of synaptic plasticity. Neuroscience. 2021;456:17–26. [DOI] [PubMed] [Google Scholar]
- 40.Vose LR, Stanton PK. Synaptic plasticity, metaplasticity and depression. Curr Neuropharmacol. 2017;15:71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee S, Manahan-Vaughan D. Role of metabotropic glutamate receptors in persistent forms of hippocampal plasticity and learning. Neuropharmacology. 2013;66:65–81. [DOI] [PubMed] [Google Scholar]
- 42.Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris RG. Synaptic plasticity and learning: selective impairment of learning rats and blockade of long-term potentiation in vivo by the N-methyl-D-aspartate receptor antagonist AP5. J Neurosci. 1989;9:3040–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. [DOI] [PubMed] [Google Scholar]
- 45.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. [DOI] [PubMed] [Google Scholar]
- 46.Scholz R, Berberich S, Rathgeber L, Kolleker A, Köhr G, Kornau HC. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron. 2010;66:768–780. [DOI] [PubMed] [Google Scholar]
- 47.Ault B, Evans RH, Francis AA, Oakes DJ, Watkins JC. Selective depression of excitatory amino acid induced depolarizations by magnesium ions in isolated spinal cord preparations. J Physiol. 1980;307:413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. [DOI] [PubMed] [Google Scholar]
- 49.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. [DOI] [PubMed] [Google Scholar]
- 50.Muller D, Joly M, Lynch G. Contributions of quisqualate and NMDA receptors to the induction and expression of LTP. Science. 1988;242:1694–1697. [DOI] [PubMed] [Google Scholar]
- 51.Ziemann U, Reis J, Schwenkreis P, et al. TMS and drugs revisited 2014. Clin Neurophysiol. 2015;126:1847–1868. [DOI] [PubMed] [Google Scholar]
- 52.Dunwiddie TV, Lynch G. The relationship between extracellular calcium concentrations and the induction of hippocampal long-term potentiation. Brain Res. 1979;169:103–110. [DOI] [PubMed] [Google Scholar]
- 53.Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. [DOI] [PubMed] [Google Scholar]
- 54.Lisman J A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci U S A. 1989;86:9574–9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans RC, Blackwell KT. Calcium: amplitude, duration, or location? Biol Bull. 2015;228:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di Lazzaro V, Pilato F, Dileone M, et al. Low-frequency repetitive transcranial magnetic stimulation suppresses specific excitatory circuits in the human motor cortex. J Physiol. 2008;586:4481–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malenka RC, Kauer JA, Perkel DJ, et al. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. [DOI] [PubMed] [Google Scholar]
- 58.Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009;28:3027–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Casaletto KB, Elahi FM, Bettcher BM, et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology. 2017;89:1782–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Gong X, Yin Z, et al. Association between NRGN gene polymorphism and resting-state hippocampal functional connectivity in schizophrenia. BMC Psychiatry. 2019;19:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. [DOI] [PubMed] [Google Scholar]
- 64.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. [DOI] [PubMed] [Google Scholar]
- 65.Shi SH, Hayashi Y, Petralia RS, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. [DOI] [PubMed] [Google Scholar]
- 66.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. [DOI] [PubMed] [Google Scholar]
- 67.Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Z, Edwards JG, Riley N, et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu H, Nash JE, Zamorano P, Garner CC. Interaction of SAP97 with minus-end-directed actin motor myosin VI. Implications for AMPA receptor trafficking. J Biol Chem. 2002;277:30928–30934. [DOI] [PubMed] [Google Scholar]
- 70.McCormack SG, Stornetta RL, Zhu JJ. Synaptic AMPA receptor exchange maintains bidirectional plasticity. Neuron. 2006;50:75–88. [DOI] [PubMed] [Google Scholar]
- 71.Takahashi T, Svoboda K, Malinow R. Experience strengthening transmission by driving AMPA receptors into synapses. Science. 2003;299:1585–1588. [DOI] [PubMed] [Google Scholar]
- 72.Yao Y, Kelly MT, Sajikumar S, et al. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of post-synaptic AMPA receptors. J Neurosci. 2008;28:7820–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. [DOI] [PubMed] [Google Scholar]
- 74.Brown JC, Petersen A, Zhong L, et al. Bidirectional regulation of synaptic transmission by BRAG1/IQSEC2 and its requirement in long-term depression. Nat Commun. 2016;7:11080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Myers KR, Wang G, Sheng Y, Conger KK, Casanova JE, Zhu JJ. Arf6-GEF BRAG1 regulates JNK-mediated synaptic removal of GluA1-containing AMPA receptors: a new mechanism for nonsyndromic X-linked mental disorder. J Neurosci. 2012;32:11716–11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shoubridge C, Tarpey PS, Abidi F, et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet. 2010;42:486–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rogers EJ, Jada R, Schragenheim-Rozales K, et al. An IQSEC2 mutation associated with intellectual disability and autism results in decreased surface AMPA receptors. Front Mol Neurosci. 2019;12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. [DOI] [PubMed] [Google Scholar]
- 79.Krug M, Lössner B, Ott T. Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Res Bull. 1984;13:39–42. [DOI] [PubMed] [Google Scholar]
- 80.Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen L, Chetkovich DM, Petralia RS, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. [DOI] [PubMed] [Google Scholar]
- 82.Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352. [DOI] [PubMed] [Google Scholar]
- 83.Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci. 2000;20:7932–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang Y, Wang XB, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. J Neurosci. 2008;28:5740–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lüscher C, Xia H, Beattie EC, et al. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. [DOI] [PubMed] [Google Scholar]
- 86.Nishimune A, Isaac JT, Molnar E, et al. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. [DOI] [PubMed] [Google Scholar]
- 87.Tan HL, Chiu SL, Zhu Q, Huganir RL. GRIP1 regulates synaptic plasticity and learning and memory. Proc Natl Acad Sci U S A. 2020;117:25085–25091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matsuda S, Mikawa S, Hirai H. Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem. 1999;73:1765–1768. [DOI] [PubMed] [Google Scholar]
- 89.Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23:9220–9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang J, Wang Y, Chi Z, et al. The AAA+ ATPase Thorase regulates AMPA receptor-dependent synaptic plasticity and behavior. Cell. 2011;145:284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mejias R, Adamczyk A, Anggono V, et al. Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proc Natl Acad Sci U S A. 2011;108:4920–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Peter BJ, Kent HM, Mills IG, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. [DOI] [PubMed] [Google Scholar]
- 93.Iwakura Y, Nagano T, Kawamura M, et al. N-methyl-D-aspartate-induced alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptor down-regulation involves interaction of the carboxyl terminus of GluR2/3 with Pick1. Ligand-binding studies using Sindbis vectors carrying AMPA receptor decoys. J Biol Chem. 2001;276:40025–40032. [DOI] [PubMed] [Google Scholar]
- 94.Cocucci E, Aguet F, Boulant S, Kirchhausen T. The first five seconds in the life of a clathrin-coated pit. Cell. 2012;150:495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krauss M, Kinuta M, Wenk MR, De Camilli P, Takei K, Haucke V. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Igamma. J Cell Biol. 2003;162:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. [DOI] [PubMed] [Google Scholar]
- 97.Kastning K, Kukhtina V, Kittler JT, et al. Molecular determinants for the interaction between AMPA receptors and the clathrin adaptor complex AP-2. Proc Natl Acad Sci U S A. 2007;104:2991–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vlachos A, Müller-Dahlhaus F, Rosskopp J, Lenz M, Ziemann U, Deller T. Repetitive magnetic stimulation induces functional and structural plasticity of excitatory postsynapses in mouse organotypic hippocampal slice cultures. J Neurosci. 2012;32:17514–17523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lenz M, Platschek S, Priesemann V, et al. Repetitive magnetic stimulation induces plasticity of excitatory postsynapses on proximal dendrites of cultured mouse CA1 pyramidal neurons. Brain Struct Funct. 2015;220:3323–3337. [DOI] [PubMed] [Google Scholar]
- 100.Huang YZ, Chen RS, Rothwell JC, Wen HY. The after-effect of human theta burst stimulation is NMDA receptor dependent. Clin Neurophysiol. 2007;118:1028–1032. [DOI] [PubMed] [Google Scholar]
- 101.Ziemann U, Hallett M, Cohen LG. Mechanisms of deafferentation-induced plasticity in human motor cortex. J Neurosci. 1998;18:7000–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Salavati B, Daskalakis ZJ, Zomorrodi R, et al. Pharmacological modulation of long-term potentiation-like activity in the dorsolateral prefrontal cortex. Front Hum Neurosci. 2018;12:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stefan K, Kunesch E, Benecke R, Cohen LG, Classen J. Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol. 2002;543:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Suppa A, Biasiotta A, Belvisi D, et al. Heat-evoked experimental pain induces long-term potentiation-like plasticity in human primary motor cortex. Cereb Cortex. 2013;23:1942–1951. [DOI] [PubMed] [Google Scholar]
- 105.Selby B, MacMaster FP, Kirton A, McGirr A. d-cycloserine blunts motor cortex facilitation after intermittent theta burst transcranial magnetic stimulation: a double-blind randomized placebo-controlled crossover study. Brain Stimul. 2019;12:1063–1065. [DOI] [PubMed] [Google Scholar]
- 106.Teo JT, Swayne OB, Rothwell JC. Further evidence for NMDA-dependence of the after-effects of human theta burst stimulation. Clin Neurophysiol. 2007;118:1649–1651. [DOI] [PubMed] [Google Scholar]
- 107.Brown JC, DeVries WH, Korte JE, et al. NMDA receptor partial agonist, d-cycloserine, enhances 10 Hz rTMS-induced motor plasticity, suggesting long-term potentiation (LTP) as underlying mechanism. Brain Stimul. 2020;13:530–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brown JC, Yuan S, DeVries WH, et al. NMDA-receptor agonist reveals LTP-like properties of 10-Hz rTMS in the human motor cortex. Brain Stimul. 2021;14:619–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oliveira-Maia AJ, Press D, Pascual-Leone A. Modulation of motor cortex excitability predicts antidepressant response to prefrontal cortex repetitive transcranial magnetic stimulation. Brain Stimul. 2017;10:787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rogasch NC, Zipser C, Darmani G, et al. The effects of NMDA receptor blockade on TMS-evoked EEG potentials from prefrontal and parietal cortex. Sci Rep. 2020;10:3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Darmani G, Bergmann TO, Zipser C, Baur D, Müller-Dahlhaus F, Ziemann U. Effects of antiepileptic drugs on cortical excitability in humans: a TMS-EMG and TMS-EEG study. Hum Brain Mapp. 2019;40:1276–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Darmani G, Ziemann U. Pharmacophysiology of TMS-evoked EEG potentials: a mini-review. Brain Stimul. 2019;12:829–831. [DOI] [PubMed] [Google Scholar]
- 113.Biabani M, Fornito A, Coxon JP, Fulcher BD, Rogasch NC. The correspondence between EMG and EEG measures of changes in cortical excitability following transcranial magnetic stimulation. J Physiol. 2021;599:2907–2932. [DOI] [PubMed] [Google Scholar]
- 114.Conde V, Tomasevic L, Akopian I, et al. The non-transcranial TMS-evoked potential is an inherent source of ambiguity in TMS-EEG studies. Neuroimage. 2019;185:300–312. [DOI] [PubMed] [Google Scholar]
- 115.Lefaucheur JP, Aleman A, Baeken C, et al. Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS): an update (2014–2018). Clin Neurophysiol. 2020;131:474–528. [DOI] [PubMed] [Google Scholar]
- 116.Stagg CJ, Antal A, Nitsche MA. Physiology of transcranial direct current stimulation. J ECT. 2018;34:144–152. [DOI] [PubMed] [Google Scholar]
- 117.Liebetanz D, Nitsche MA, Tergau F, Paulus W. Pharmacological approach to the mechanisms of transcranial DC-stimulation-induced after-effects of human motor cortex excitability. Brain. 2002;125:2238–2247. [DOI] [PubMed] [Google Scholar]
- 118.Nitsche MA, Fricke K, Henschke U, et al. Pharmacological modulation of cortical excitability shifts induced by transcranial direct current stimulation in humans. J Physiol. 2003;553:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nitsche MA, Jaussi W, Liebetanz D, Lang N, Tergau F, Paulus W. Consolidation of human motor cortical neuroplasticity by D-cycloserine. Neuropsychopharmacology. 2004;29:1573–1578. [DOI] [PubMed] [Google Scholar]
- 120.Jiang J, Wang J, Li C. Potential mechanisms underlying the therapeutic effects of electroconvulsive therapy. Neurosci Bull. 2017;33:339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang J, Tang Y, Curtin A, et al. ECT-induced brain plasticity correlates with positive symptom improvement in schizophrenia by voxel-based morphometry analysis of grey matter. Brain Stimul. 2019;12:319–328. [DOI] [PubMed] [Google Scholar]
- 122.Grimonprez A, Raedt R, Baeken C, Boon P, Vonck K. The antidepressant mechanism of action of vagus nerve stimulation: evidence from preclinical studies. Neurosci Biobehav Rev. 2015;56:26–34. [DOI] [PubMed] [Google Scholar]
- 123.Leaver AM, Vasavada M, Joshi SH, et al. Mechanisms of antidepressant response to electroconvulsive therapy studied with perfusion magnetic resonance imaging. Biol Psychiatry. 2019;85:466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ashkan K, Rogers P, Bergman H, Ughratdar I. Insights into the mechanisms of deep brain stimulation. Nat Rev Neurol. 2017;13:548–554. [DOI] [PubMed] [Google Scholar]
- 125.Lulic T, El-Sayes J, Fassett HJ, Nelson AJ. Physical activity levels determine exercise-induced changes in brain excitability. PLOS ONE. 2017;12, e0173672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuhn M, Wolf E, Maier JG, et al. Sleep recalibrates homeostatic and associative synaptic plasticity in the human cortex. Nat Commun. 2016;7:12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Friehs MA, Dechant M, Vedress S, Frings C, Mandryk RL. Shocking advantage! Improving digital game performance using non-invasive brain stimulation. Int J Hum Comput Stud. 2021;148:102582. [Google Scholar]
- 128.Huang YZ, Edwards MJ, Rounis E, Bhatia KP, Rothwell JC. Theta burst stimulation of the human motor cortex. Neuron. 2005;45:201–206. [DOI] [PubMed] [Google Scholar]
- 129.Blumberger DM, Vila-Rodriguez F, Thorpe KE, et al. Effectiveness of theta burst versus high-frequency repetitive transcranial magnetic stimulation in patients with depression (THREE-D): a randomised non-inferiority trial. Lancet. 2018;391:1683–1692. [DOI] [PubMed] [Google Scholar]
- 130.Cole EJ, Stimpson KH, Bentzley BS, et al. Stanford accelerated intelligent neuromodulation therapy for treatment-resistant depression. Am J Psychiatry. 2020;177:716–726. [DOI] [PubMed] [Google Scholar]