

Abstract
Intracellular calcium (Ca2+) is an essential second messenger in eukaryotic cells regulating numerous cellular functions such as contraction, secretion, immunity, growth, and metabolism. Ca2+ signaling is also a key signal transducer in the intrinsic apoptosis pathway. The store-operated Ca2+ entry pathway (SOCE) is ubiquitously expressed in eukaryotic cells, and is the primary Ca2+ influx pathway in non-excitable cells. SOCE is mediated by the endoplasmic reticulum Ca2+ sensing STIM proteins, and the plasma membrane Ca2+-selective Orai channels. A growing number of studies have implicated SOCE in regulating cell death primarily via the intrinsic apoptotic pathway in a variety of tissues and in response to physiological stressors such as traumatic brain injury, ischemia reperfusion injury, sepsis, and alcohol toxicity. Notably, the literature points to excessive cytosolic Ca2+ influx through SOCE in vulnerable cells as a key factor tipping the balance towards cellular apoptosis. While the literature primarily addresses the functions of STIM1 and Orai1, STIM2, Orai2 and Orai3 are also emerging as potential regulators of cell death. Here, we review the functions of STIM and Orai proteins in regulating cell death and the implications of this regulation to human pathologies.
Graphical Abstract
Introduction
Intracellular calcium (Ca2+) is a ubiquitous and essential regulator of eukaryotic cell function. As a second messenger employed in a myriad of signaling pathways, intracellular Ca2+ regulates diverse functions such as metabolism, transcription, immune function, contraction, proliferation, and apoptosis[1-5]. In order to perform this broad array of functions, intracellular Ca2+ homeostasis is tightly controlled spatiotemporally by a variety of ion channels, exchangers, and pumps in the plasma membrane (PM) and subcellular organelles including mitochondria, endolysosomes and the endoplasmic reticulum (ER)[6, 7]. This intricate and tightly regulated network of Ca2+ transport ensures checks and balances necessary to maintain a low resting concentration of cytosolic Ca2+ (~100 nM) relative to the extracellular matrix (~1-2 mM). As such, this regulation enables Ca2+ ions to perform signal transduction while preventing toxic Ca2+ overload[1, 6, 8]. Typically, the ER comprises the largest store of intracellular Ca2+ in eukaryotic cells (100 μM − 1 mM)[9]. Store-operated calcium entry (SOCE), which is activated by ER Ca2+ depletion, is the primary mechanism of regulated Ca2+ influx in non-excitable cells[6, 10-12]. SOCE is driven by the intricate interactions of ubiquitously expressed proteins: the two ER membrane-localized Ca2+ sensing stromal interaction molecules (STIM1 and STIM2) and the three PM localized Ca2+-selective Orai channel proteins (Orai1, Orai2, and Orai3) [4, 5, 12-14].
In non-excitable cells, ER store depletion is typically initiated by the activation of PM receptors coupled to phosphoinositide-specific phospholipase C (PLC) isoforms which rapidly convert phosphatidylinositol-4,5bisphosphate (PIP2) into soluble inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG)[15, 16]. The newly mobilized IP3 subsequently triggers the release of ER Ca2+ via IP3 receptors (IP3RS), resulting in the depletion of the ER Ca2+ stores[17-19]. The falling Ca2+ levels in the ER lumen activate the Ca2+ sensing STIM proteins, causing them to form discrete multimers referred to as puncta at the ER-PM junction and resulting in a conformational change that exposes their C-terminal STIM Orai-activating region/CRAC-activating domain (SOAR/CAD), which physically traps and activates PM Orai channels[1, 5, 12, 13]. Of the SOCE-mediating components, STIM1 and Orai1 have garnered the most dedicated studies, due in large part to their prominent contribution to SOCE and the identification of patients with loss of function (LoF) mutations in the Orai1 and STIM1 genes. LoF mutations in Orai1 and STIM1 genes result in severe combined immunodeficiency, autoimmunity, ectodermal dysplasia, defective dental enamel development, and myopathies[20-25]. Furthermore, while both STIM1 and STIM2 are expressed ubiquitously, STIM1 expression is higher in most tissues compared to STIM2, making its influence more readily discernible[26, 27]. Despite the predominance of STIM1, STIM2 has been documented as a major contributor to SOCE in a variety of tissue types, including neurons, dendrites, NUT 3T3 fibroblasts, and colorectal carcinoma cells[28-31]. Notably, STIM1 requires significant depletion of the ER store to activate and is generally considered the major activator of SOCE during agonist stimulation. STIM2, which is activated with low to moderate levels of store depletion was initially thought to play a homeostatic cellular function by maintaining resting levels of ER Ca2+. However, emerging evidence supports a prominent function of STIM2 in physiological Ca2+ signaling to transcription (beyond store refilling) when low physiologically relevant concentrations of agonists cause modest ER Ca2+ store depletion [32, 33]. Further, STIM2 contributes to SOCE through enhanced recruitment of STIM1 to the ER-PM junction[32, 34-36]. Similar to the predominance of STIM1 in the literature, Orai1 is the best characterized of the three Orai homologs, and is noted to have a major contribution to SOCE in many tissues[24, 37-49]. By contrast, the functions of the remaining Orai homologs are less well characterized, due in large part to a lack of selective pharmacological agents and antibodies that target them[6]. Despite this, there are an increasing number of studies that have begun to shed light on the function of these homologs. Knockout of Orai2 and Orai3, both independently and in combination, reveal that SOCE is enhanced by their absence, suggesting that Orai2 and/or Orai3 form heteromultimeric channels with Orai1 and act as negative regulators under native conditions[50-53]. Orai3 has been shown to synergize with Orai1 in mediating SOCE, transcriptional activity and metabolic reprogramming in B cells during activation[54]. Additionally, Orai3 has been shown, in conjunction with Orai1, to form the store-independent arachidonic acid-regulated Ca2+ (ARC) influx pathway activated by either arachidonic acid or its metabolite, leukotriene C4 [55-62].
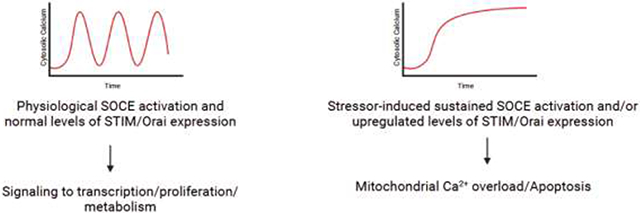
Cellular Ca2+ homeostasis must strike a delicate balance to enable a diverse array of signaling outcomes and influence cell fate decisions. In most instances of physiological stimulation, cells respond to low concentrations of receptor agonists originating from surrounding tissue or circulation[63]. These low levels of stimulation give rise to transient and regenerative spikes of cytosolic Ca2+ known as oscillations[15, 64-66]. Typically, Ca2+ oscillations in most cells are initially driven by the release of Ca2+ by IP3RS, but beyond the first few spikes, they are sustained by SOCE[67, 68]. The frequency of Ca2+ oscillations increase with the concentration of agonist until sufficiently high levels of stimulation induce a plateau of high intracellular Ca2+ levels[69-72]. Notably, the frequency of cytosolic Ca2+ oscillations has been tied to distinct physiological responses in cases such as regulating distinct gene expression in T-lymphocytes or the regulation of calmodulin kinase II activity[73-75]. However, while it is tempting to broadly tie the frequency of cytosolic Ca2+ oscillations with distinct physiological responses, it is important to note that oscillation frequency can vary stochastically in response to the same stimulus, and that in many cases the frequency of cytosolic Ca2+ oscillations is not directly proportional to the extent of the physiological response[16]. Despite this, it can be broadly stated that the most frequent outcomes induced by transient Ca2+ oscillations favor cell survival and proliferation[76-78]. Indeed, oscillatory rises in cytosolic Ca2+ levels sustained by SOCE have been known to promote survival by stimulating the calcineurin-dependent activation of nuclear factor of activated T-cells (NFAT) and NF-κB, transcription factors that regulates numerous cell survival genes[79-81] (Fig. 1A). Similarly, transient Ca2+ signals have long been known to activate the Akt pathway to promote survival and regulate metabolism[82-88]. By contrast, however, sustained increases of cytosolic Ca2+ over long periods of time initiate apoptosis by increasing Ca2+concentrations at the ER-mitochondrial junctions resulting in mitochondrial Ca2+ overload and opening the mitochondrial permeability transition pore (MPTP) [76, 89-91] (Fig. 1B).
Figure 1.

(A) In healthy tissues, the ER Ca2+-sensing STIM proteins activate the plasma membrane Orai channels to mediate cytosolic Ca2+ influx in a physiological moderate and regulated oscillatory pattern. The rise in cytosolic Ca2+ under physiological levels of stimulation enhances mitochondrial bioenergetics, and activates the phosphatase calcineurin, which induces the nuclear translocation of transcription factors such as NFAT and initiates the transcription of proliferative and metabolic genes. Under physiological conditions, this Ca2+ signal will cease when the agonist is absent. (B) In stressed cells and systems, upregulated expression/activation of STIM and Orai induces sustained Ca2+ influx over extended periods causing mitochondrial Ca2+ overload, increased mitochondrial ROS production, mPTP opening, the collapse of mitochondrial membrane potential, and the release of cytochrome c. The released cytochrome c leads to the activation of caspase 3, and ultimately results in cellular apoptosis.
Apoptosis is a form of programmed cell death that is necessary for normal tissue homeostasis and cell turnover and is employed to dispose of cells that are either no longer required or detrimental to the health of the surrounding tissue[91, 92]. Apoptosis plays essential roles in virtually all elements of an organism’s life, from embryonic development to termination of immune responses, and its dysregulation is intertwined with the pathophysiology of cancer[93-97]. Apoptosis is regulated primarily by two main pathways: the extrinsic pathway in which a death receptor is activated by an extracellular pro-apoptotic agonist resulting in a signaling cascade leading to caspase 3 activation; and the intrinsic pathway, which is activated by severe forms of mitochondrial dysregulation caused by stimuli such as accumulation of unfolded proteins, reactive oxygen species (ROS) or Ca2+ overload[91, 98]. Pertinent to this review, the mechanisms through which Ca2+ overload is initiated to trigger apoptosis are numerous and varied. One such mechanism is mediated by the excess release of ER Ca2+ stores by IP3R into the ER-mitochondria junctional space and the subsequent propagation of Ca2+ into the mitochondria via MCU[91]. The activation of IP3RS have been frequently observed in response to apoptotic stimuli, and it has long been noted that silencing the expression of IP3RS in lymphocytes imparts a resistance to apoptosis in response to various stimuli[99-102]. Voltage-gated Ca2+ channels have also been shown to induce Ca2+ overload and thereby mediate apoptosis[91]. In particular, L-type voltage-gated Ca2+ channels have been shown to convert depolarization-triggered Ca2+ entry into apoptotic signals in chromaffin cells, PC12 cells, and pancreatic beta cells[103-105]. Additionally, various members of transient receptor potential (TRP) channel superfamily were shown to play a significant role in Ca2+-mediated apoptosis in response to a variety of stimuli including oxidative stress[106], high glucose[107], natural compounds such as menthol[108-110], capsaicin[111, 112], and cannabidiol[113, 114] as well as apoptotic signaling mediated by the Fas receptor[115]. Below, we will focus on SOCE and its function in the induction of Ca2+ overload and apoptosis.
Of the two primary apoptotic pathways, it is the intrinsic pathway that is of note when examining the pathophysiology of SOCE[76]. Fundamentally, the intrinsic apoptotic pathway is driven by changes in mitochondrial membrane permeability by either the opening of the mitochondrial permeability transition pore (mPTP) or through pore-formation via the actions of pro-apoptotic BCL-2 protein family members[98, 116-119]. The permeabilization of mitochondria lead in turn to the collapse of the mitochondrial membrane potential, the arrest of mitochondrial ATP synthesis, and the release of pro-apoptotic factors such as cytochrome c, which in turn leads a signaling cascade that activates the apoptosis initiator caspase 3[98, 119, 120]. Thus, a pathophysiological connection can be drawn between the cytosolic influx of Ca2+ through SOCE and the initiation of the apoptotic response. Below, we will examine the function of each protein component of SOCE in initiating signaling cascades resulting in cell death, primarily through apoptosis.
STIM1 and cell death
As mentioned above, STIM1 is the more highly expressed STIM homolog and is generally regarded as the major regulator of SOCE in many tissues[26, 27]. This prevalence greatly facilitates its examination and as a result, most studies regarding SOCE-induced cell death have focused on STIM1, often in conjunction with Orai1, over other SOCE components. Reports of STIM1 regulation of SOCE-induced cell death cover a much wider variety of tissues than those for STIM2, Orai2, or Orai3.
Several studies have shown that STIM1 plays a significant role in inducing apoptosis in neurons[121-127]. This function of STIM1 appears to have been largely identified due to triggering events such as traumatic brain injury (TBI) and oxidative stress. TBI is known to cause secondary injury via the excessive release of the excitatory neurotransmitter glutamate and the influx of Ca2+ ions[121, 128-130]. The release of glutamate during TBI has been shown to activate STIM1-mediated SOCE in primary mouse cortical neurons, resulting in cytosolic Ca2+ overload and apoptosis[121]. This increased cytosolic Ca2+ load and resulting apoptosis are significantly decreased in neurons in which STIM1 was silenced with siRNA. Further, in vitro studies showed that mGluR1-dependent Ca2+ release from the ER of injured neurons was attenuated in the absence of STIM1, suggesting that STIM1 and SOCE contribute to ER refilling in neurons. It seems that an immediate early gene product Homer1a serves as neuroprotective in response to glutamate-induced oxidative stress injuries in HT-22 cells. Homer1a appears to cause dissociation of STIM1/Orai1 interactions to prevent STIM1-mediated Ca2+ overload[124]. A separate study found that knockdown of STIM1 in an ischemic rat model significantly decreased neuronal intracellular Ca2+ and reduced ischemic neuronal cell death[122]. Similarly, knockdown of STIM1 in rat primary neurons reduced cell death and the expression of cytochrome c in response to high glucose[131]. The function of STIM1 as pro-apoptotic in neurons was highlighted by the action of the sedative dexmedetomidine[123]. This study reported that PC12 cells pretreated with dexmedetomidine significantly downregulated STIM1 and Orai1 expression levels and were protected from cell death induced by oxygen-glucose deprivation. Taken together, these studies indicate a prominent role for STIM1 in the induction of cell death specifically in neurons.
STIM1 has also been shown to regulate apoptosis in hepatocytes[132-136]. Because the liver serves as the primary detoxification system, hepatocytes are regularly exposed to environmental stressors. Based on the observation that ethanol-induced hepatic cell death involved perturbations in Ca2+ homeostasis, Liu et al. found that the mRNA and protein expression of STIM1 and Orai1 were significantly upregulated in the presence of 200 mM ethanol[132]. To assess whether this upregulation of STIM1 and Orai1 were responsible for the disrupted Ca2+ homeostasis, they employed STIM1 and Orai1 siRNA knockdown, which abrogated ethanol-induced Ca2+ elevation and alleviated cell death. In a follow-up study, the same authors showed that the knockdown of STIM1/Orai1 protected hepatocytes from ethanol toxicity by preserving mitochondrial membrane potential and preventing the cells from undergoing the intrinsic apoptotic pathway[133]. Additional examples of STIM1/Orai1 regulation of hepatocyte apoptosis in response to hepatotoxic compounds were found in the cases of the weight loss agent usnic acid[134], and the industrial contaminant hexavalent chromium[135]. In both cases, inhibition of STIM1/Orai1 alleviated cytosolic Ca2+ overload and prevented apoptosis. In addition to regulating hepatocyte apoptosis in response to toxic compounds, STIM1 was also found to be upregulated and to induce apoptosis in hepatocytes under liver ischemia/reperfusion conditions[136]. In this study, liver tissue samples from STIM1−/− mice subjected to hepatic ischemia/reperfusion exhibited significantly less cell death, release of the inflammatory cytokines TNF-α, IL-6 and IL-1β, and cleaved caspase 3 in comparison to wildtype mice.
STIM1 was shown to induce apoptosis in the cardiovascular system[137, 138]. In a case similar to some of those conducted in neurons and hepatocytes, STIM1 and Orai1 regulated apoptosis in cardiomyocytes in response to ischemia/reperfusion injury[137]. As with previous cases in other tissues, STIM1/Orai1 were significantly upregulated in injured conditions, and knockdown of either protein alleviated cytosolic Ca2+ overload, decreased ROS production, and attenuated mPTP opening. Interestingly, a follow-up study found that when primary rat ventricular cardiomyocytes subjected in vitro to ischemia/reperfusion injury, were pre-treated with the antioxidant and anti-inflammatory compound resveratrol, STIM1 expression was downregulated and apoptosis was alleviated[138]. The ability resveratrol to inhibit STIM1-mediated apoptosis was reported by a separate group who found that pretreatment with resveratrol protected MS-1 microvascular endothelial cells from STIM1/Orai1-mediated apoptosis induced by high glucose levels[139]. A third group proposed a mechanism of how resveratrol inhibited STTM1 function[140]. Resveratrol inhibited ERK1/2 activation—normally triggered by ER Ca2+ store depletion—and subsequently prevented the phosphorylation of STIM1 on Ser575, Ser608, and Ser621 thereby preventing STIM1 dissociation from the microtubule-binding protein EB1, STIM1 multimerization and activation of Orai1. Additional studies that point to STIM1 function in regulating apoptosis in the vasculature include the discovery that STIM1/Orai1 proteins induce apoptosis is pulmonary microvascular endothelial cells in response to lipopolysaccharide (LPS) release during acute pancreatitis[141]. Once again, knockdown and pharmacological inhibition of STIM1/Orai1 alleviated SOCE-induced apoptosis initiated by LPS treatment. Interestingly, similar results were reported by two separate groups in human umbilical vein endothelial cells and primary mouse cardiac tissue in LPS models of sepsis[142, 143]. Together, these studies suggest that the inhibition of STIM1/Orai1 may be a tool in preventing cardiovascular cell death in cases of sepsis. Lastly, STIM1 was found to regulate apoptosis in rat endothelial progenitor cells in response to oxidative stress triggered by exposure to excess hydrogen peroxide (H2O2)[141]. Knockdown of STIM1 with shRNA decreased the expression of ER apoptosis signaling proteins caspase 9 and caspase 12, decreased ROS levels and rescued mitochondrial membrane potential[141]. It is worth noting that this protection is likely not limited to endothelial progenitor cells, as it has also been previously reported that STIM1 functions as a redox sensor and when under oxidative stress, S-glutathionylation of STIM1 will cause constitutive activation of Orai channels regardless of ER Ca2+ store levels [144].
STIM1 plays a key role in regulating immune cell function. Indeed, the activation of T lymphocytes is notably dependent upon Ca2+ signaling through SOCE, and existing reviews discuss the role of STIM/Orai-mediated SOCE in T lymphocyte proliferation and apoptosis[1, 3, 145-147]. Given the highly centralized role SOCE plays in healthy T lymphocyte function, it is perhaps fitting that it also represents one of the only documented instances in which the downregulation of SOCE proteins STIM1 and Orai1 were found to facilitate apoptosis via the extrinsic pathway initiated by Fas stimulation[148]. The Fas receptor is a key component of immune system regulation by inducing apoptosis in activated lymphocytes and thereby preventing prolonged immune responses that could cause autoimmunity[149]. In an examination of apoptosis in Jurkat T cells mediated by moderate Fas receptor ligation, STIM1 and Orai1 were downregulated and SOCE was reduced by the de-energization of mitochondria and the upregulation of caspase activity[148]. The authors of this study speculated that this decrease in SOCE might serve to protect T cells undergoing apoptosis from shifting into a necrotic cell death pathway by preventing cytosolic Ca2+ overload.
Another function for STIM1/Orai1 in regulating apoptosis in immune cells was reported in headkidney macrophages from the species of catfish Clarias gariepinus[150, 151]. Infection of these macrophages with Mycobacterium fortuitum leads to production of toxic levels of ROS by macrophages and a surge in cytosolic Ca2+ to induce apoptosis, thereby destroying the infecting bacteria[152]. Dahiya et al. found that toll-like receptor 2-dependent internalization of M. fortuitum by macrophages induced significant ER stress, resulting in the upregulation of STIM1 and Orai1, which in turn activate calpain, causing it to cleave nitric oxide synthase interacting protein and initiate the release of toxic levels of nitric oxide. Knockdown of STIM1 and Orai1 reduced calpain activation, and prevented the accumulation of ROS and macrophage apoptosis, facilitating bacterial survival. A follow-up study by the same group suggested that the accumulation of mitochondrial ROS was dependent upon calcium uptake by the mitochondrial Ca2+ uniporter, and that inhibiting mitochondrial ROS with the pharmacological compound YCG063 reciprocally resulted in the downregulation of STIM1 and Orai1[151]. This suggests that SOCE and mitochondrial ROS production create an amplifying positive feedback loop to induce apoptosis in infected macrophages. This is consistent with the previously mentioned reports proposing that STIM1 facilitates apoptosis in response to oxidative stress[126, 141, 144].
Scattered reports have also identified instances of STIM1 induction of apoptosis in a variety of other tissues including the pancreas[153], gastric cancer[154], and mammary tissue[155]. In an in vitro model of diabetic hyperlipidemia, a mouse pancreatic β-TC3 cell line significantly upregulated STIM1 and Orai1 expression in response to free fatty acids[153]. The resulting increase in SOCE enhanced the expression of the ER stress response protein CHOP and activated caspase 3, initiating apoptosis. In human gastric cancer cell lines BGC-823 and SGC-7901, treatment with 3,3’-diindolylmethane—a natural phytochemical known to induce apoptosis in gastric cancer—was proposed to act via the upregulation of STIM1 and subsequent cytosolic Ca2+ overload, which was rescued by STIM1 knockdown [154]. STIM1 and Orai1 alike were found to regulate apoptosis in the injured mammary tissue of dairy cows suffering from subacute ruminal acidosis resulting from a carbohydrate-rich diet[155]. Finally, STIM1 was found to regulate apoptosis in normal cervical epithelial cells but not cervical cancer cells cultured in soft collagen substrates designed to test cellular responses to mechanical stimuli[156]. Together, these studies indicate the widespread influence of STIM1 on SOCE and apoptosis across many tissues and disease states.
STIM2 and cell death
Despite the comparative lack of dedicated research focused on STIM2 relative to STIM1, there have been a small number of studies indicating that STIM2 can play a role in the induction of cell death. The earliest identified role for STIM2 in inducing cell death was described in neurons[28]. In this study, STIM2 was identified as an essential regulator of SOCE in neurons, and responsible for significant cytosolic Ca2+ accumulation and cell death under oxygen-glucose deprivation in isolated wildtype mouse neuronal cells. By contrast, neurons isolated from Stim2−/− mice were protected from Ca2+ accumulation and associated cell death. These results were replicated in vivo, where the Stim2−/− mice were protected from ischemic stroke. In agreement with these findings, an independent study found that the knockdown of STIM2 in neurons had neuroprotective effects in TBI/traumatic neuron injury models both in vivo and in vitro[157]. Furthermore, this work demonstrated that knockdown of STIM2 in mouse neurons prevented mitochondrial Ca2+ overload, thus drawing the connection between STIM2 activity and the intrinsic apoptosis pathway. More recently, the role of STIM2 in inducing ischemic cell death was also illustrated in the rat cardiomyoblast cell line H9c2[158]. In this study, the release of cytochrome c from the mitochondria was measured after subjecting the H9c2 cells to ischemia/reperfusion, and the authors reported an increase in cytochrome c under ischemic conditions in wildtype cells, while knockdown of STIM2 significantly reduced the release of cytochrome c under the same conditions. Taken together, these studies indicate that STIM2 plays an intriguing role in inducing apoptosis in ischemic conditions that warrants further investigation in other tissues.
STIM2 was proposed to play a role in regulating apoptosis in colorectal cancer[159, 160]. In a comparison of human colon carcinoma cell line HT29 versus the normal human mucosa cell line NCM460, the loss of STIM2 in the HT29 line resulted in partially depleted ER Ca2+ stores and resistance to apoptosis[159]. The knockdown of STIM2 in the NCM460 cells is similarly protective from apoptosis. Consistently, a separate study showed that increased expression of STIM2 in colorectal tumors resulted in the suppression of tumor growth[160]. In this study, colorectal tumor xenografts that were identified as exhibiting high STIM2 expression levels displayed significantly less vascular invasion and a heavily reduced proliferation rate compared to tumor xenografts exhibiting low STIM2 expression.
Orai1 and cell death
Orai1 is by far the best characterized of the Orai homologs, with many studies showing virtually equal contributions of STIM1 and Orai1 in mediating apoptosis. Several studies have identified instances in which Orai1 is a major contributor to the induction of apoptosis. The earliest example of Orai1-mediated cell death was in prostate cancer cells[161]. In this study, Orai1 was the primary mediator of apoptosis in response to chemotherapeutics such as cisplatin and oxaliplatin, and its expression was downregulated in steroid-deprived prostate cancer cells. Consistent with previously reported findings[162], the prostate cancer cells that survive steroid-deprivation via the downregulation of Orai1 simultaneously acquire a resistance to cisplatin-induced apoptosis as a result[161]. Orai1 was suggested to regulate apoptosis of pancreatic acinar cells of multiple mouse models of acute pancreatitis[163]. Pancreatic acinar cells were protected from pancreatitis-triggered apoptosis in the presence of various pharmacological inhibitors of SOCE such as GSK-7975A. However, it should be noted that current pharmacological agents, including GSK-7975A, are poorly selective for Orai1, and instead affect all Orai homologs[164]. Thus, it is possible that this observed apoptotic protection is mediated by other Orai homologs, rather than strictly by Orai1. Additional apoptotic roles induced by Orai1 in response to oxidative stress were described in the mouse hippocampal cell line HT22 in an in vivo mouse model[165, 166]. Of particular note, molecular docking, thermal shift and plasmon resonance assays suggested that the neuroprotective supplement icaritin prevented apoptosis in response to H2O2-induced oxidative stress through direct binding and inhibition of Orai1[166].
Orai1 has also been found to play diverse roles including the induction of apoptosis in immune cells[145]. In addition to the previously mentioned study that identified STIM1 and Orai1 as regulators of apoptosis in T lymphocytes[148], an additional study found that Orai1 alone was equally capable of mediating T lymphocyte death[167]. In this study, the CD4+ T cells of Orai1−/− mice were found to be resistant to T cell receptor stimulation-induced cell death as a result in changes to Fas receptor expression and reduced mitochondrial Ca2+ uptake. Additionally, upon challenge with an activating anti-CD3 antibody in vivo, the CD4+ T cells of Orai1−/− mice survived at significantly higher rates than the CD4+ T cells of Orai1+/+ mice, reinforcing the proapoptotic function of Orai1 in T lymphocytes. Lastly, Orai1 regulated apoptosis in foam cells in a mouse model of atherosclerosis[168]. In this system, Orai1-dependent Ca2+ influx occurred in response to acute administration of oxidized low-density lipoprotein, and resulted in the activation of apoptosis signal-regulating kinase 1. This effect was mitigated by siRNA knockdown of Orai1, and by inhibition of SOCE with another poorly selective pharmacological agent, SKF96365[168].
Orai2 and cell death
The potential function of Orai2 in cell death has gone nearly unexamined since its discovery, and largely remains a mystery. However, a pair of studies conducted in 2019 by separate groups have revealed that similar to STIM2, Orai2 appears to play a role in regulating apoptosis in the brain[169, 170]. Using a bioinformatics approach, and analyzing gathered RNA-seq data from the The Cancer Genome Atlas and the French, Sun and Gene Expression Omnibus, Yuan et al. identified significant upregulation of Orai2 in glioblastoma samples[169]. Furthermore, they found a strong positive correlation between expression of Orai2 in glioblastoma with the expression of cytochrome c, caspase 3, and caspase 9. While this study lacks in vitro and in vivo experiments to draw a direct mechanistic connection between Orai2 expression and apoptosis, the results suggest a relationship between the two. The second study, by contrast, examined the role of Orai2 in regulating neuronal cell death in response to ischemic stroke both in a mouse model and in vitro[170]. In this study, Orai2 was found to be a major contributor to SOCE and to strongly promote the accumulation of Ca2+ and expression of caspase-3 in the cortical neurons of wildtype mice in ischemic conditions. By comparison, the cortical neurons of Orai2−/− showed significantly less SOCE, Ca2+ accumulation, and expression of caspase 3. As a result, these findings indicate that Orai2 plays a similar role in the regulation of SOCE-mediated apoptosis in neurons as STIM2.
Orai3 and cell death
As with STIM2 and Orai2, there is currently a deficiency in the number of studies examining the function of Orai3 in cell death. Indeed, most studies of Orai3 and cell death suggest that Orai3 plays a vastly more prominent role in apoptosis resistance, growth and invasion of cancer cells than in inducing apoptosis[76, 171-176]. For instance, knockdown of Orai3 in pancreatic ductal adenocarcinoma (PDAC) cell lines caused enhanced SOCE [177], which is consistent with the function of Orai3 as a negative regulator of SOCE [14, 53, 177]. In this study, the authors showed that knockdown of Orai3 inhibited PDAC cells growth and promoted apoptosis, suggesting that in these cells Orai3 offers a survival advantage by negatively regulating SOCE activity and preventing Ca2+ overload-mediated apoptosis. Interestingly, Orai3 knockdown in normal pancreatic cells inhibited SOCE [177], cautioning against extrapolating findings in malignant cell lines to the physiology of primary cells and systems. Curiously, one of the few examples of Orai3 inducing apoptosis was identified in a human lens epithelial cell line (HLEpiC) resulting from an observation that Orai3 expression is dramatically increased in the cataracts of diabetic patients[178]. In this study, HLEpiCs cultured in high glucose (25.6 mM) exhibited significant upregulation of Orai3 and STIM1 expression compared to HLEpiCs cultured in normal glucose (5.5 mM). Interestingly, Orai1 protein expression was reduced in the high glucose condition. SOCE measurements in cells with either Orai3 or STIM1 knockdown seemed to indicate that SOCE was significantly reduced in HLEpiCs cultured in high glucose, although some of these recordings showed atypically transient Ca2+ entry signals. These results were compounded by the observation of increased apoptotic markers in the controls compared to cells with Orai3 knockdown. Primary cultured lens epithelial cells from Orai3−/− diabetic rats showed that SOCE was virtually abolished compared to cells from diabetic wildtype rats. Finally, the lens turbidity levels were significantly lower in Orai3−/− diabetic rats compared to their diabetic wildtype counterparts. These results suggest that Orai3 might be involved in apoptosis of lens epithelial cells, and contributes to the development of diabetic cataracts[178].
Another interesting instance in which Orai3 was found to induce apoptosis was identified in kidney proximal tubular cells[179]. In this study, the internalization of accumulated calcium oxalate monohydrate and calcium phosphate crystals by the proximal tubular cells altered their primary Ca2+ influx pathway from receptor-operated calcium influx to SOCE via the induction of ER stress and the upregulation of STIM1, STIM2, and Orai3. The upregulation of ER stress-related to Nucleus signaling 1 (ERN1) and claudin 1 (CLDN1) were observed in proximal tubular cells with internalized crystals, and were indicative of increased apoptotic signaling. As with previous examples, knockdown of either STIM protein, or Orai3 significantly reduced SOCE and alleviated the expression of ER stress signaling molecules. Notably, while either knockdown of STIM1 or STIM2 reduced the expression of ERN1, only the knockdown of Orai3 reduced the expression of ERN1 and CLDN1, suggesting that Orai3 plays a more prominent role than that of STIM1/2 in crystal-induced apoptosis.
Conclusions
The majority of studies examining SOCE clearly indicate that its prevailing role is to drive normal cell function and encourage proliferative gene expression (Fig. 1A)[77, 78, 180, 181]. Indeed, this has resulted in very few groups exploring the roles that the SOCE proteins play in inducing cell death. However, when the existing studies are examined as a whole, the current message communicated by the literature as it pertains to the role of SOCE in mediating cell death is that it is fundamentally a process driven by excess in circumstances where the cells are otherwise already vulnerable (Fig. 1B). Many of the studies reviewed herein demonstrate this concept in cases of Traumatic brain injury and/or in ischemia/reperfusion injury[28, 121, 122, 136, 157, 158, 170]. Others demonstrate this concept in the presence of other toxic excesses such as H2O2, ethanol, glutamate, glucose, and Ca2+crystals[124, 126, 131, 132, 141, 165, 178, 179]. Notably—albeit unsurprisingly—SOCE appears to almost exclusively drive apoptosis via the intrinsic pathway, rather than broadly contributing to the extrinsic pathway. Regardless, the majority of reported instances of SOCE-induced cell death demonstrate that it is seldom the key initiator of apoptosis, but that it instead tips the balance in favor of apoptosis in already stressed cells or systems.
SOCE has been found to induce apoptosis in a growing number of tissues, with STIM1 and Orai1 contributing the majority of cases in the widest variety of tissues. This is not surprising as STIM1 and Orai1 are robust mediators of SOCE with the notable exception of STIM2 and Orai2 in the brain, where they appear to be highly expressed and contribute a sizeable portion of SOCE. In particular, STIM1 has been noted to regulate apoptosis in the nervous system[121-127], the cardiovascular system[137, 138], the immune system[148], as well as in the liver[132-136] and pancreas[153] among other tissues. In many regards, the contributions of STIM1 and Orai1 to the induction of apoptosis can be seen as a reflection of their prevalence in most tissues relative to the lesser-studied homologs STIM2, Orai2, and Orai3. However, it should be noted that many of the studies that identified STIM1 and Orai1 as regulators of apoptosis relied upon knockdown of these homologs specifically, and they did not report on attempts to knockdown the remaining homologs.
Because SOCE primarily plays a role in inducing cell death in systems with pre-existing stressors an intriguing number of potential therapeutic interventions become worthy of consideration. Normally, the disruption of SOCE by pharmacological inhibitors would be associated with unfavorable outcomes in many tissues due to its wide-reaching regulation of healthy cell function and proliferation, especially in the treatment of chronic disease where the effects of SOCE inhibition would be amplified. Indeed, while a recent study has shown that chronic reduction of SOCE in a mouse model with a STIM1 loss of function (LoF) mutation could be therapeutically beneficial, these mice with STIM1 LoF mutation were hypertensive and exhibited tachycardia as a result of catecholamine accumulation in the circulatory system[182], underscoring the ubiquitous involvement of SOCE in human physiological systems. However, the use of pharmacological SOCE inhibitors administered over a confined period of time in cases of ischemia/reperfusion or traumatic brain injury, for example, would offer a potentially viable and effective treatment option. Ironically, this is particularly true of potential inhibitors for the lesser-studied components of SOCE: STIM2, Orai2, and Orai3. As discussed above, STIM1 and Orai1 represent the primary mediators of SOCE in most tissue types, and most identified instances of pathophysiology involving the dysregulation of SOCE are derived from STIM1 and Orai1 LoF mutations[20-27]. Accordingly, one would expect the highest probability of a negative outcome by the pharmacological inhibition of STIM1 and Orai1. By contrast, STIM2, Orai2, and Orai3 play more subtle regulatory roles in most tissues, while also exhibiting significant upregulation in expression and contribution to pathophysiological activity in instances such as ischemia/reperfusion or TBI. Thus, a window of opportunity is created to both efficiently target the pathophysiology while also minimizing potential off-target effects. This would work only if Orai2 and Orai3 are indeed forming channels on their own. One important caveat to consider within this context is that Orai2 and Orai3 might be forming heteromeric channels with Orai1, thus inhibiting Orai1 activity and SOCE. In this case, any therapeutic drug should be targeted to this heteromeric channel. Unfortunately, the currently available pharmacological tools do not possess sufficient selectivity to achieve this goal, as the oligomeric state and Orai isoform composition of SOCE channels in any given cell type remains unclear and most available agents target multiple Orai homologs[164]. Thus, future research efforts should be dedicated to the elucidation of the molecular make-up of native SOCE channels in different cell types and on the discovery of pharmacological agents that selectively target individual Orai homologs as well as various heteromeric Orai compositions that exist under native conditions.
In conclusion, the role of SOCE in inducing cell death remains a largely understudied area. Future research efforts will be necessary to understand the breadth and nuances of SOCE-mediated cell death more fully, but existing trends suggest that it plays a pronounced role in tissues already experiencing significant environmental stressors, with ischemia being the most prominent example. Furthermore, it must be emphasized that despite a general lack of studies focused on STIM2, Orai2, and Orai3, they appear to play vital roles in SOCE-mediated cell death that offer highly exploitable opportunities to develop effective treatments where agents targeting the more prominent STIM1 and Orai1 may fail. Regardless of therapeutic potential, the development of selective pharmacological compounds against each Orai homolog and potential native Orai heteromers will greatly facilitate the identification and discovery of novel functions that SOCE might have in cell death, cell function and human disease.
Highlights.
Store-Operated Ca2+ entry is mediated by two STIM and three Orai proteins
STIM1 and Orai1 are most studied and little is known about STIM2, Orai2 and Orai3
STIM/Orai mediate intrinsic cellular apoptosis through mitochondrial Ca2+ overload
Acknowledgements:
Work in the Trebak Laboratory is supported by National Institutes of Health (NIH) grant R35-HL150778 (to M.T.).
Abbreviations
- Ca2+
Calcium
- PM
plasma membrane
- ER
endoplasmic reticulum
- SOCE
store-operated calcium entry
- STIM
stromal interaction molecule
- PLC
Phospholipase C
- PIP2
phosphatidylinositol 4,5-bisphosphate
- IP3
inositol 1,4,5-triphosphate
- DAG
diacylglycerol
- IP3R
IP3 receptors
- SOAR/CAD
STIM Orai-activating region/CRAC-activating domain
- LoF
loss of function
- ARC
arachidonic acid/leukotriene C4-regulated channels
- NFAT
nuclear factor of activated T-cells
- MPTP
mitochondrial permeability transition pore
- ROS
reactive oxygen species
- TRP
transient receptor potential
- TBI
traumatic brain injury
- LPS
lipopolysaccharide
- H2O2
hydrogen peroxide
- ERN1
ER stress-related to nucleus signaling 1
- CLDN1
claudin 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Sample CRediT author statement
J. Cory Benson: Conceptualization, Writing- Original draft preparation. Mohamed Trebak: Conceptualization, Supervision, Writing- Reviewing and Editing
Conflict of Interest
Mohamed Trebak is a paid consultant for Seeker Biologicals Inc. J. Cory Benson has no potential conflict of interest to declare.
References
- [1].Trebak M, Kinet JP, Calcium signalling in T cells, Nat. Rev. Immunol, 19 (2019) 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Clapham DE, Calcium signaling, Cell, 131 (2007) 1047–1058. [DOI] [PubMed] [Google Scholar]
- [3].Feske S, Skolnik EY, Prakriya M, Ion channels and transporters in lymphocyte function and immunity, Nat. Rev. Immunol, 12 (2012) 532–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Prakriya M, Lewis RS, STORE-OPERATED CALCIUM CHANNELS, Physiol. Rev, 95 (2015) 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hogan PG, Rao A, Store-operated calcium entry: Mechanisms and modulation, Biochem. Biophys. Res. Commun, 460 (2015) 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Emrich SM, Yoast RE, Trebak M, Physiological Functions of CRAC Channels, Annu. Rev. Physiol, 84 (2022) 355–379. [DOI] [PubMed] [Google Scholar]
- [7].Sukumaran P, Da Conceicao VN, Sun YY, Ahamad N, Saraiva LR, Selvaraj S, Singh BB, Calcium Signaling Regulates Autophagy and Apoptosis, Cells, 10 (2021) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].La Rovere RML, Roest G, Bultynck G, Parys JB, Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy, Cell Calcium, 60 (2016) 74–87. [DOI] [PubMed] [Google Scholar]
- [9].Bygrave FL, Benedetti A, What is the concentration of calcium ions in the endoplasmic reticulum?, Cell Calcium, 19 (1996) 547–551. [DOI] [PubMed] [Google Scholar]
- [10].Ashby MC, Tepikin AV, ER calcium and the functions of intracellular organelles, Semin. Cell Dev. Biol, 12 (2001) 11–17. [DOI] [PubMed] [Google Scholar]
- [11].Putney JW, A MODEL FOR RECEPTOR-REGULATED CALCIUM ENTRY, Cell Calcium, 7 (1986) 1–12. [DOI] [PubMed] [Google Scholar]
- [12].Potier M, Trebak M, New developments in the signaling mechanisms of the store-operated calcium entry pathway, Pflugers Arch, 457 (2008) 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Trebak M, Putney JW, ORAI Calcium Channels, Physiology, 32 (2017) 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yoast RE, Emrich SM, Trebak M, The anatomy of native CRAC channel(s), Curr Opin Physiol, 17 (2020) 89–95. [PMC free article] [PubMed] [Google Scholar]
- [15].Parekh AB, Decoding cytosolic Ca2+ oscillations, Trends Biochem.Sci, 36 (2011) 78–87. [DOI] [PubMed] [Google Scholar]
- [16].Dupont G, Combettes L, Fine tuning of cytosolic Ca (2+) oscillations, F1000Research, 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Berridge MJ, Bootman MD, Lipp P, Calcium - a life and death signal, Nature, 395 (1998) 645–648. [DOI] [PubMed] [Google Scholar]
- [18].Berridge MJ, Inositol trisphosphate and calcium signalling mechanisms, Biochim. Biophys. Acta-Mol. Cell Res, 1793 (2009) 933–940. [DOI] [PubMed] [Google Scholar]
- [19].Berridge MJ, THE INOSITOL TRISPHOSPHATE/CALCIUM SIGNALING PATHWAY IN HEALTH AND DISEASE, Physiol. Rev, 96 (2016) 1261–1296. [DOI] [PubMed] [Google Scholar]
- [20].McCarl CA, Picard C, Khalil S, Kawasaki T, Rother J, Papolos A, Kutok J, Hivroz C, LeDeist F, Plogmann K, Ehl S, Notheis G, Albert MH, Belohradsky BH, Kirschner J, Rao A, Fischer A, Feske S, ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia, J. Allergy Clin. Immunol, 124 (2009) 1311–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lian J, Cuk M, Kahlfuss S, Kozhaya L, Vaeth M, Rieux-Laucat F, Picard C, Benson MJ, Jakovcevic A, Bilic K, Martinac I, Stathopulos P, Kacskovics I, Vraetz T, Speckmann C, Ehl S, Issekutz T, Unutmaz D, Feske S, ORAI1 mutations abolishing store-operated Ca2+ entry cause anhidrotic ectodermal dysplasia with immunodeficiency, J. Allergy Clin. Immunol, 142 (2018) 1297-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vaeth M, Feske S, Ion channelopathies of the immune system, Curr. Opin. Immunol, 52 (2018) 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, Fischer A, Feske S, Brief Report: STIM1 Mutation Associated with a Syndrome of Immunodeficiency and Autoimmunity, N. Engl. J. Med, 360 (2009) 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A, A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function, Nature, 441 (2006) 179–185. [DOI] [PubMed] [Google Scholar]
- [25].Silva-Rojas R, Laporte J, Bohm J, STIM1/ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases, Front. Physiol, 11 (2020) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Williams RT, Manji SSM, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ, Dziadek MA, Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins, Biochem. J, 357 (2001) 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A, Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance, Nat. Immunol, 9 (2008) 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Berna-Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G, Nieswandt B, STIM2 Regulates Capacitive Ca2+ Entry in Neurons and Plays a Key Role in Hypoxic Neuronal Cell Death, Sci. Signal, 2 (2009) 10. [DOI] [PubMed] [Google Scholar]
- [29].Bandyopadhyay BC, Pingle SC, Ahern GP, Store-operated Ca2+ signaling in dendritic cells occurs independently of STIM1, J. Leukoc. Biol, 89 (2011) 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nelson HA, Leech CA, Kopp RF, Roe MW, Interplay between ER Ca2+ Binding Proteins, STIM1 and STIM2, Is Required for Store-Operated Ca2+ Entry, Int. J. Mol. Sci, 19 (2018) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Emrich SM, Yoast RE, Xin P, Zhang XX, Pathak T, Nwokonko R, Gueguinou MF, Subedi KP, Zhou YD, Ambudkar IS, Hempel N, Machaca K, Gill DL, Trebak M, Cross-talk between N-terminal and C-terminal domains in stromal interaction molecule 2 (STIM2) determines enhanced STIM2 sensitivity, J. Biol. Chem, 294 (2019) 6318–6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Emrich SM, Yoast RE, Xin P, Arige V, Wagner LE, Hempel N, Gill DL, Sneyd J, Yule DI, Trebak M, Omnitemporal choreographies of all five STIM/Orai and IP(3)Rs underlie the complexity of mammalian Ca2+ signaling, Cell Reports, 34 (2021) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ahmad M, Ong HL, Saadi H, Son GY, Shokatian Z, Terry LE, Trebak M, Yule DI, Ambudkar I, Functional communication between IP(3)R and STIM2 at subthreshold stimuli is a critical checkpoint for initiation of SOCE, Proc Natl Acad Sci U S A, 119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brandman O, Liou J, Park WS, Meyer T, STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels, Cell, 131 (2007) 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Subedi KP, Ong HL, Son GY, Liu XB, Ambudkar IS, STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions, Cell Reports, 23 (2018) 522–534. [DOI] [PubMed] [Google Scholar]
- [36].Ong HL, de Souza LB, Zheng CY, Cheng KT, Liu XB, Goldsmith CM, Feske S, Ambudkar IS, STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions, Sci. Signal, 8 (2015) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Feske S, CRAC channels and disease - From human CRAC channelopathies and animal models to novel drugs, Cell Calcium, 80 (2019) 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tiffner A, Derler I, Isoform-Specific Properties of Orai Homologues in Activation, Downstream Signaling, Physiology and Pathophysiology, Int. J. Mol. Sci, 22 (2021) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Spinelli AM, Gonzalez-Cobos JC, Zhang XX, Motiani RK, Rowan S, Zhang W, Garrett J, Vincent PA, Matrougui K, Singer HA, Trebak M, Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration, Pflugers Arch, 464 (2012) 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M, Stim1 and Orai1 Mediate CRAC Currents and Store-Operated Calcium Entry Important for Endothelial Cell Proliferation, Circ.Res, 103 (2008) 1289–U1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, Trebak M, Evidence for STIM1-and Orai1-dependent store-operated calcium influx through I-CRAC in vascular smooth muscle cells: role in proliferation and migration, Faseb J, 23 (2009) 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Luo R, Gomez AM, Benitah JP, Sabourin J, Targeting Orai1-Mediated Store-Operated Ca(2+)Entry in Heart Failure, Front. Cell. Dev. Biol, 8 (2020) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang W, Trebak M, STIM1 and Orai1: novel targets for vascular diseases?, Sci. China-Life Sci, 54 (2011) 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Feske S, Picard C, Fischer A, Immunodeficiency due to mutations in ORAI1 and STIM1, Clin. Immunol, 135 (2010) 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shawer H, Norman K, Cheng CW, Foster R, Beech DJ, Bailey MA, ORAI1 Ca2+ Channel as a Therapeutic Target in Pathological Vascular Remodelling, Front. Cell. Dev. Biol, 9 (2021) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Conte E, Imbrici P, Mantuano P, Coppola MA, Camerino GM, De Luca A, Liantonio A, Alteration of STIM1/Orai1-Mediated SOCE in Skeletal Muscle: Impact in Genetic Muscle Diseases and Beyond, Cells, 10 (2021) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bisaillon JM, Motiani RK, Gonzalez-Cobos JC, Potier M, Halligan KE, Alzawahra WF, Barroso M, Singer HA, Jourd'heuil D, Trebak M, Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration, Am J Physiol Cell Physiol, 298 (2010) C993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Motiani RK, Hyzinski-Garcia MC, Zhang X, Henkel MM, Abdullaev IF, Kuo YH, Matrougui K, Mongin AA, Trebak M, STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion, Pflugers Arch, 465 (2013) 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shinde AV, Motiani RK, Zhang X, Abdullaev IF, Adam AP, Gonzalez-Cobos JC, Zhang W, Matrougui K, Vincent PA, Trebak M, STIM1 controls endothelial barrier function independently of Orai1 and Ca2+ entry, Sci Signal, 6 (2013) ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tsvilovskyy V, Solís-López A, Schumacher D, Medert R, Roers A, Kriebs U, Freichel M, Deletion of Orai2 augments endogenous CRAC currents and degranulation in mast cells leading to enhanced anaphylaxis, Cell Calcium, 71 (2018) 24–33. [DOI] [PubMed] [Google Scholar]
- [51].Vaeth M, Yang J, Yamashita M, Zee I, Eckstein M, Knosp C, Kaufmann U, Jani PK, Lacruz RS, Flockerzi V, Kacskovics I, Prakriya M, Feske S, ORAI2 modulates store-operated calcium entry and T cell-mediated immunity, Nat. Commun, 8 (2017) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Eckstein M, Vaeth M, Aulestia FJ, Costiniti V, Kassam SN, Bromage TG, Pedersen P, Issekutz T, Idaghdour Y, Moursi ARM, Feske S, Lacruz RS, Differential regulation of Ca2+ influx by ORAI channels mediates enamel mineralization, Sci. Signal, 12 (2019) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yoast RE, Emrich SM, Zhang X, Xin P, Johnson MT, Fike AJ, Walter V, Hempel N, Yule DI, Sneyd J, Gill DL, Trebak M, The native ORAI channel trio underlies the diversity of Ca2+ signaling events, Nat. Commun, 11 (2020) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Emrich SM, Yoast RE, Zhang X, Fike AJ, Wang YH, Bricker KN, Tao AY, Xin P, Walter V, Johnson MT, Pathak T, Straub AC, Feske S, Rahman ZSM, Trebak M, Orai3 and Orai1 mediate CRAC channel function and metabolic reprogramming in B cells, Elife, 12 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang XX, Gonzalez-Cobos JC, Schindl R, Muik M, Ruhle B, Motiani RK, Bisaillon JM, Zhang W, Fahrner M, Barroso M, Matrougui K, Romanin C, Trebak M, Mechanisms of STIM1 Activation of Store-Independent Leukotriene C-4-Regulated Ca2+ Channels, Mol. Cell. Biol, 33 (2013) 3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang W, Zhang XX, Gonzalez-Cobos JC, Stolwijk JA, Matrougui K, Trebak M, Leukotriene-C-4 Synthase, a Critical Enzyme in the Activation of Store-independent Orai1/Orai3 Channels, Is Required for Neointimal Hyperplasia, J. Biol. Chem, 290 (2015) 5015–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mignen O, Thompson JL, Shuttleworth TJ, The molecular architecture of the arachidonate-regulated Ca2+-selective ARC channel is a pentameric assembly of Orai1 and Orai3 subunits, J. Physiol.-London, 587 (2009) 4181–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cantonero C, Sanchez-Collado J, Gonzalez-Nunez MA, Salido GM, Lopez JJ, Jardin I, Rosado JA, Store-independent Orai1-mediated Ca2+ entry and cancer, Cell Calcium, 80 (2019) 1–7. [DOI] [PubMed] [Google Scholar]
- [59].Mignen O, Thompson JL, Shuttleworth TJ, Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels, J. Physiol.-London, 586 (2008) 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Motiani RK, Stolwijk JA, Newton RL, Zhang XX, Trebak M, Emerging roles of Orai3 in pathophysiology, Channels, 7 (2013) 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Thompson MA, Prakash YS, Pabelick CM, Arachidonate-Regulated Ca2+ Influx in Human Airway Smooth Muscle, Am. J. Respir. Cell Mol. Biol, 51 (2014) 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang X, Zhang W, Gonzalez-Cobos JC, Jardin I, Romanin C, Matrougui K, Trebak M, Complex role of STIM1 in the activation of store-independent Orai1/3 channels, J Gen Physiol, 143 (2014) 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].McCarron JG, Wilson C, Heathcote HR, Zhang X, Buckley C, Lee MD, Heterogeneity and emergent behaviour in the vascular endothelium, Curr. Opin. Pharmacol, 45 (2019) 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bird GS, Hwang SY, Smyth JT, Fukushima M, Boyles RR, Putney JW, STIM1 Is a Calcium Sensor Specialized for Digital Signaling, Curr. Biol, 19 (2009) 1724–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Christo SN, Diener KR, Nordon RE, Brown MP, Griesser HJ, Vasilev K, Christo FC, Hayball JD, Scrutinizing calcium flux oscillations in T lymphocytes to deduce the strength of stimulus, Sci Rep, 5 (2015) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sneyd J, Han JM, Wang LW, Chen J, Yang XS, Tanimura A, Sanderson MJ, Kirk V, Yule DI, On the dynamical structure of calcium oscillations, Proc. Natl. Acad. Sci. U. S. A, 114 (2017) 1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Putney JW, Calcium Signaling: Deciphering the Calcium-NFAT Pathway, Curr. Biol, 22 (2012) R87–R89. [DOI] [PubMed] [Google Scholar]
- [68].Dupont G, Combettes L, Bird GS, Putney JW, Calcium Oscillations, Cold Spring Harbor Perspect. Biol, 3 (2011) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Shuttleworth TJ, What drives calcium entry during Ca2+ (i) oscillations? challenging the capacitative model, Cell Calcium, 25 (1999) 237–246. [DOI] [PubMed] [Google Scholar]
- [70].Berridge MJ, Galione A, CYTOSOLIC CALCIUM OSCILLATORS, Faseb J, 2 (1988) 3074–3082. [DOI] [PubMed] [Google Scholar]
- [71].Thomas AP, Bird GSJ, Hajnoczky G, RobbGaspers LD, Putney JW, Spatial and temporal aspects of cellular calcium signaling, Faseb J, 10 (1996) 1505–1517. [PubMed] [Google Scholar]
- [72].Putney JW, Bird GS, Cytoplasmic calcium oscillations and store-operated calcium influx, J. Physiol.-London, 586 (2008) 3055–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Dolmetsch RE, Xu KL, Lewis RS, Calcium oscillations increase the efficiency and specificity of gene expression, Nature, 392 (1998) 933–936. [DOI] [PubMed] [Google Scholar]
- [74].Dupont G, Houart G, De Koninck P, Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations: a simple model, Cell Calcium, 34 (2003) 485–497. [DOI] [PubMed] [Google Scholar]
- [75].De Koninck P, Schulman H, Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations, Science, 279 (1998) 227–230. [DOI] [PubMed] [Google Scholar]
- [76].Tanwar J, Motiani RK, Role of SOCE architects STIM and Orai proteins in Cell Death, Cell Calcium, 69 (2018) 19–27. [DOI] [PubMed] [Google Scholar]
- [77].Pinto MCX, Kihara AH, Goulart VAM, Tonelli FMP, Gomes KN, Ulrich H, Resende RR, Calcium signaling and cell proliferation, Cell. Signal, 27 (2015) 2139–2149. [DOI] [PubMed] [Google Scholar]
- [78].Capiod T, Cell proliferation, calcium influx and calcium channels, Biochimie, 93 (2011) 2075–2079. [DOI] [PubMed] [Google Scholar]
- [79].Srikanth S, Gwack Y, Orai1-NFAT signalling pathway triggered by T cell receptor stimulation, Mol. Cells, 35 (2013) 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Park YJ, Yoo SA, Kim M, Kim WU, The Role of Calcium-Calcineurin-NFAT Signaling Pathway in Health and Autoimmune Diseases, Front. Immunol, 11 (2020) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Berry CT, May MJ, Freedman BD, STIM- and Orai-mediated calcium entry controls NF-kappa B activity and function in lymphocytes, Cell Calcium, 74 (2018) 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yano S, Tokumitsu H, Sodeling TR, Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway, Nature, 396 (1998) 584–587. [DOI] [PubMed] [Google Scholar]
- [83].Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, Cameron S, Turvey SE, Serfling E, Berberich-Siebelt F, Possemato R, Feske S, Store-Operated Ca2+ Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming, Immunity, 47 (2017) 664-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zou WY, Meng XJ, Cai CQ, Zou MC, Tang SH, Chu XW, Wang XB, Zou F, Store-operated Ca2+ Entry (SOCE) Plays a Role in the Polarization of Neutrophil-like HL-60 Cells by Regulating the Activation of Akt, Src, and Rho Family GTPases, Cell. Physiol. Biochem, 30 (2012) 221–237. [DOI] [PubMed] [Google Scholar]
- [85].Zhou YB, Gu P, Li J, Li F, Zhu J, Gao P, Zang YC, Wang YC, Shan YX, Yang DR, Suppression of STIM1 inhibits the migration and invasion of human prostate cancer cells and is associated with PI3K/Akt signaling inactivation, Oncol. Rep, 38 (2017) 2629–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Radoslavova S, Folcher A, Lefebvre T, Kondratska K, Guenin S, Dhennin-Duthille I, Gautier M, Prevarskaya N, Ouadid-Ahidouch H, Orai1 Channel Regulates Human-Activated Pancreatic Stellate Cell Proliferation and TGF(beta 1) Secretion through the AKT Signaling Pathway, Cancers, 13 (2021) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Sandoval AJ, Riquelme JP, Carretta MD, Hancke JL, Hidalgo MA, Burgos RA, Store-operated calcium entry mediates intracellular alkalinization, ERK1/2, and Akt/PKB phosphorylation in bovine neutrophils, J. Leukoc. Biol, 82 (2007) 1266–1277. [DOI] [PubMed] [Google Scholar]
- [88].Huang YJ, Li Q, Feng ZY, Zheng LR, STIM1 controls calcineurin/Akt/mTOR/NFATC2-mediated osteoclastogenesis induced by RANKL/M-CSF, Exp. Ther. Med, 20 (2020) 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Naon D, Scorrano L, At the right distance: ER-mitochondria juxtaposition in cell life and death, Biochim. Biophys. Acta-Mol. Cell Res, 1843 (2014) 2184–2194. [DOI] [PubMed] [Google Scholar]
- [90].Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R, Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis, Oncogene, 27 (2008) 6407–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kondratskyi A, Kondratska K, Skryma R, Prevarskaya N, Ion channels in the regulation of apoptosis, Biochim. Biophys. Acta-Biomembr, 1848 (2015) 2532–2546. [DOI] [PubMed] [Google Scholar]
- [92].Kerr JFR, Wyllie AH, Currie AR, APOPTOSIS - BASIC BIOLOGICAL PHENOMENON WITH WIDE-RANGING IMPLICATIONS IN TISSUE KINETICS, Br. J. Cancer, 26 (1972) 239-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Suzanne M, Steller H, Shaping organisms with apoptosis, Cell Death Differ, 20 (2013) 669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A, BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes, Nature, 415 (2002) 922–926. [DOI] [PubMed] [Google Scholar]
- [95].Pellegrini M, Belz G, Bouillet P, Strasser A, Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim, Proc. Natl. Acad. Sci. U. S. A, 100 (2003) 14175–14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Cotter TG, Apoptosis and cancer: the genesis of a research field, Nat. Rev. Cancer, 9 (2009) 501–507. [DOI] [PubMed] [Google Scholar]
- [97].Hanahan D, Weinberg RA, Hallmarks of Cancer: The Next Generation, Cell, 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [98].Elmore S, Apoptosis: A review of programmed cell death, Toxicol. Pathol, 35 (2007) 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Joseph SK, Hajnoczky G, IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond, Apoptosis, 12 (2007) 951–968. [DOI] [PubMed] [Google Scholar]
- [100].Blackshaw S, Sawa A, Sharp AH, Ross CA, Snyder SH, Khan AA, Type 3 inositol 1,4,5-trisphosphate receptor modulates cell death, Faseb Journal, 14 (2000) 1375–1379. [DOI] [PubMed] [Google Scholar]
- [101].Jayaraman T, Marks AR, T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis, Molecular and Cellular Biology, 17 (1997) 3005–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Khan AA, Soloski MJ, Sharp AH, Schilling G, Sabatini DM, Li SH, Ross CA, Snyder SH, Lymphocyte apoptosis: Mediation by increased type 3 inositol 1,4,5-trisphosphate receptor, Science, 273 (1996) 503–507. [DOI] [PubMed] [Google Scholar]
- [103].Cano-Abad MF, Villarroya M, Garcia AG, Gabilan NH, Lopez MG, Calcium entry through L-type calcium channels causes mitochondrial disruption and chromaffin cell death, Journal of Biological Chemistry, 276 (2001) 39695–39704. [DOI] [PubMed] [Google Scholar]
- [104].Diaz-Prieto N, Herrera-Peco I, de Diego AMG, Ruiz-Nuno A, Gallego-Sandin S, Lopez MG, Garcia AG, Cano-Abad MF, Bcl2 mitigates Ca2+ entry and mitochondrial Ca2+ overload through downregulation of L-type Ca2+ channels in PC12 cells, Cell Calcium, 44 (2008) 339–352. [DOI] [PubMed] [Google Scholar]
- [105].Junttiberggren L, Larsson O, Rorsman P, Ammala C, Bokvist K, Wahlander K, Nicotera P, Dypbukt J, Orrenius S, Hallberg A, Berggren PO, INCREASED ACTIVITY OF L-TYPE CA2+ CHANNELS EXPOSED TO SERUM FROM PATIENTS WITH TYPE-I DIABETES, Science, 261 (1993) 86–90. [DOI] [PubMed] [Google Scholar]
- [106].Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y, LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death, Molecular Cell, 9 (2002) 163–173. [DOI] [PubMed] [Google Scholar]
- [107].Yang H, Zhao B, Liao C, Zhang R, Meng KX, Xu J, Jiao JD, High glucose-induced apoptosis in cultured podocytes involves TRPC6-dependent calcium entry via the RhoA/ROCK pathway, Biochemical and Biophysical Research Communications, 434 (2013) 394–400. [DOI] [PubMed] [Google Scholar]
- [108].Yamamura H, Ugawa S, Ueda T, Morita A, Shimada S, TRPM8 activation suppresses cellular viability in human melanoma, American Journal of Physiology-Cell Physiology, 295 (2008) C296–C301. [DOI] [PubMed] [Google Scholar]
- [109].Li Q, Wang XH, Yang ZH, Wang B, Li SL, Menthol Induces Cell Death via the TRPM8 Channel in the Human Bladder Cancer Cell Line T24, Oncology, 77 (2009) 335–341. [DOI] [PubMed] [Google Scholar]
- [110].Zhu SY, Wang YX, Pan LT, Yang S, Sun YL, Wang XY, Hu F, Involvement of transient receptor potential melastatin-8 (TRPM8) in menthol-induced calcium entry, reactive oxygen species production and cell death in rheumatoid arthritis rat synovial fibroblasts, European Journal of Pharmacology, 725 (2014) 1–9. [DOI] [PubMed] [Google Scholar]
- [111].Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella A, Giangaspero F, Santoni G, Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation, Journal of Neurochemistry, 102 (2007) 977–990. [DOI] [PubMed] [Google Scholar]
- [112].Amantini C, Ballarini P, Caprodossi S, Nabissi M, Morelli MB, Lucciarini R, Cardarelli MA, Mammana G, Santoni G, Triggering of transient receptor potential vanilloid type 1 (TRPV1) by capsaicin induces Fas/CD95-mediated apoptosis of urothelial cancer cells in an ATM-dependent manner, Carcinogenesis, 30 (2009) 1320–1329. [DOI] [PubMed] [Google Scholar]
- [113].Nabissi M, Morelli MB, Santoni M, Santoni G, Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents, Carcinogenesis, 34 (2013) 48–57. [DOI] [PubMed] [Google Scholar]
- [114].Yamada T, Ueda T, Shibata Y, Ikegami Y, Saito M, Ishida Y, Ugawa S, Kohri K, Shimada S, TRPV2 Activation Induces Apoptotic Cell Death in Human T24 Bladder Cancer Cells: A Potential Therapeutic Target for Bladder Cancer, Urology, 76 (2010) 7. [DOI] [PubMed] [Google Scholar]
- [115].Xu M, Li XX, Walsh SW, Zhang Y, Abais JM, Boini KM, Li PL, Intracellular two-phase Ca2+ release and apoptosis controlled by TRP-ML1 channel activity in coronary arterial myocytes, American Journal of Physiology-Cell Physiology, 304 (2013) C458–C466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Szabo I, Zoratti M, MITOCHONDRIAL CHANNELS: ION FLUXES AND MORE, Physiol. Rev, 94 (2014) 519–608. [DOI] [PubMed] [Google Scholar]
- [117].Tait SWG, Green DR, Mitochondria and cell death: outer membrane permeabilization and beyond, Nat. Rev. Mol. Cell Biol, 11 (2010) 621–632. [DOI] [PubMed] [Google Scholar]
- [118].Brenner C, Grimm S, The permeability transition pore complex in cancer cell death, Oncogene, 25 (2006) 4744–4756. [DOI] [PubMed] [Google Scholar]
- [119].Ichim G, Tait SWG, A fate worse than death: apoptosis as an oncogenic process, Nat. Rev. Cancer, 16 (2016) 539–548. [DOI] [PubMed] [Google Scholar]
- [120].Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang XD, Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade, Cell, 91 (1997) 479–489. [DOI] [PubMed] [Google Scholar]
- [121].Hou PF, Liu ZH, Li N, Cheng WJ, Guo SW, Knockdown of STIM1 Improves Neuronal Survival After Traumatic Neuronal Injury Through Regulating mGluR1-Dependent Ca2+ Signaling in Mouse Cortical Neurons, Cell. Mol. Neurobiol, 35 (2015) 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zhang M, Song JN, Wu Y, Zhao YL, Pang HG, Fu ZF, Zhang BF, Ma XD, Suppression of STIM1 in the early stage after global ischemia attenuates the injury of delayed neuronal death by inhibiting store-operated calcium entry-induced apoptosis in rats, Neuroreport, 25 (2014) 507–513. [DOI] [PubMed] [Google Scholar]
- [123].Hu YD, Tang CL, Jiang JZ, Lv HY, Wu YB, Qin XD, Shi S, Zhao B, Zhu XN, Xia ZY, Neuroprotective Effects of Dexmedetomidine Preconditioning on Oxygen-glucose Deprivation-reoxygenation Injury in PC12 Cells via Regulation of Ca2+-STIM1/Orai1 Signaling, Curr. Med. Sci, 40 (2020) 699–707. [DOI] [PubMed] [Google Scholar]
- [124].Rao W, Peng C, Zhang L, Su N, Wang K, Hui H, Dai SH, Yang YF, Luo P, Fei Z, Homer1a attenuates glutamate-induced oxidative injury in HT-22 cells through regulation of store-operated calcium entry, Sci Rep, 6 (2016) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Song Q, Gou WL, Zou YL, FAM3A Protects Against Glutamate-Induced Toxicity by Preserving Calcium Homeostasis in Differentiated PC12 Cells, Cell. Physiol. Biochem, 44 (2017) 2029–2041. [DOI] [PubMed] [Google Scholar]
- [126].Rao W, Zhang L, Su N, Wang K, Hui H, Wang L, Chen T, Luo P, Yang YF, Liu ZB, Fei Z, Blockade of SOCE protects HT22 cells from hydrogen peroxide-induced apoptosis, Biochem. Biophys. Res. Commun, 441 (2013) 351–356. [DOI] [PubMed] [Google Scholar]
- [127].Li B, Xiao L, Wang ZY, Zheng PS, Knockdown of STIM1 inhibits 6-hydroxydopamine-induced oxidative stress through attenuating calcium-dependent ER stress and mitochondrial dysfunction in undifferentiated PC12 cells, Free Radic. Res, 48 (2014) 758–768. [DOI] [PubMed] [Google Scholar]
- [128].Chen T, Cao L, Dong WP, Luo P, Liu WB, Qu Y, Fei Z, Protective Effects of mGluR5 Positive Modulators Against Traumatic Neuronal Injury Through PKC-Dependent Activation of MEK/ERK Pathway, Neurochem. Res, 37 (2012) 983–990. [DOI] [PubMed] [Google Scholar]
- [129].Stoica BA, Faden AI, Cell Death Mechanisms and Modulation in Traumatic Brain Injury, Neurotherapeutics, 7 (2010) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Raghupathi R, Cell death mechanisms following traumatic brain, Brain Pathol, 14 (2004) 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Xu ZK, Xu WZ, Song Y, Zhang B, Li F, Liu YG, Blockade of store-operated calcium entry alleviates high glucose-induced neurotoxicity via inhibiting apoptosis in rat neurons, Chem.-Biol. Interact, 254 (2016) 63–72. [DOI] [PubMed] [Google Scholar]
- [132].Liu HM, Jia XQ, Luo Z, Guan H, Jiang H, Li XH, Yan M, Inhibition of store-operated Ca2+ channels prevent ethanol-induced intracellular Ca2+ increase and cell injury in a human hepatoma cell line, Toxicol. Lett, 208 (2012) 254–261. [DOI] [PubMed] [Google Scholar]
- [133].Cui RB, Yan LH, Luo Z, Guo XL, Yan M, Blockade of store-operated calcium entry alleviates ethanol-induced hepatotoxicity via inhibiting apoptosis, Toxicol. Appl. Pharmacol, 287 (2015) 52–66. [DOI] [PubMed] [Google Scholar]
- [134].Chen S, Zhang ZH, Wu YF, Shi Q, Yan H, Mei N, Tolleson WH, Guo L, Endoplasmic Reticulum Stress and Store-Operated Calcium Entry Contribute to Usnic Acid-Induced Toxicity in Hepatic Cells, Toxicol. Sci, 146 (2015) 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Liang Q, Zhang YJ, Zeng M, Guan L, Xiao YY, Xiao F, The role of IP3R-SOCCs in Cr(VI)-induced cytosolic Ca(2+)overload and apoptosis in L-02 hepatocytes, Toxicol. Res, 7 (2018) 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Li YY, Lou CY, Wang WY, STIM1 deficiency protects the liver from ischemia/reperfusion injury in mice, Biochem. Biophys. Res. Commun, 496 (2018) 422–428. [DOI] [PubMed] [Google Scholar]
- [137].He F, Wu QF, Xu BL, Wang XC, Wu JX, Huang L, Cheng J, Suppression of Stim1 reduced intracellular calcium concentration and attenuated hypoxia/reoxygenation induced apoptosis in H9C2 cells, Biosci. Rep, 37 (2017) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Xu H, Cheng J, Wang XB, Liu HQ, Wang SY, Wu JX, Xu BL, Chen AH, He F, Resveratrol pretreatment alleviates myocardial ischemia/reperfusion injury by inhibiting STIM1-mediated intracellular calcium accumulation, J. Physiol. Biochem, 75 (2019) 607–618. [DOI] [PubMed] [Google Scholar]
- [139].Lu T, Zhou DY, Gao P, Si LY, Xu Q, Resveratrol attenuates high glucose-induced endothelial cell apoptosis via mediation of store-operated calcium entry, Mol. Cell. Biochem, 442 (2018) 73–80. [DOI] [PubMed] [Google Scholar]
- [140].Casas-Rua V, Alvarez IS, Pozo-Guisado E, Martin-Romero FJ, Inhibition of STIM1 phosphorylation underlies resveratrol-induced inhibition of store-operated calcium entry, Biochem. Pharmacol, 86 (2013) 1555–1563. [DOI] [PubMed] [Google Scholar]
- [141].Wang YW, Zhang JH, Yu Y, Yu J, Huang L, Inhibition of Store-Operated Calcium Entry Protects Endothelial Progenitor Cells from H2O2-Induced Apoptosis, Biomol. Ther, 24 (2016) 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Qiu XC, Dong KS, Sun RJ, STIM1 Regulates Endothelial Calcium Overload and Cytokine Upregulation During Sepsis, J. Surg. Res, 263 (2021) 236–244. [DOI] [PubMed] [Google Scholar]
- [143].Ye JJ, Li MF, Li Q, Jia ZJ, Hu XY, Zhao GJ, Zhi SC, Hong GL, Lu ZQ, Activation of STIM1/Orai1-mediated SOCE in sepsis-induced myocardial depression, Mol. Med. Rep, 26 (2022) 11. [DOI] [PubMed] [Google Scholar]
- [144].Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang YJ, Bhanumathy CD, Subbiah R, Ritchie MF, Soboloff J, Baba Y, Kurosaki T, Joseph SK, Gill DL, Madesh M, S-glutathionylation activates STIM1 and alters mitochondrial homeostasis, J. Cell Biol, 190 (2010) 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Qui B, Al-Ansary D, Kummerow C, Hoth M, Schwarz EC, ORAI-mediated calcium influx in T cell proliferation, apoptosis and tolerance, Cell Calcium, 50 (2011) 261–269. [DOI] [PubMed] [Google Scholar]
- [146].Schwarz EC, Qu B, Hoth M, Calcium, cancer and killing: The role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells, Biochim. Biophys. Acta-Mol. Cell Res, 1833 (2013) 1603–1611. [DOI] [PubMed] [Google Scholar]
- [147].Feske S, Wulff H, Skolnik EY, Ion channels in innate and adaptive immunity, Annu Rev Immunol, 33 (2015) 291–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Onopiuk M, Wierzbicka K, Brutkowski W, Szczepanowska J, Zablocki K, Caspase-dependent inhibition of store-operated Ca2+ entry into apoptosis-committed Jurkat cells, Biochem. Biophys. Res. Commun, 399 (2010) 198–202. [DOI] [PubMed] [Google Scholar]
- [149].Russell JH, ACTIVATION-INDUCED DEATH OF MATURE T-CELLS IN THE REGULATION OF IMMUNE-RESPONSES, Curr. Opin. Immunol, 7 (1995) 382–388. [DOI] [PubMed] [Google Scholar]
- [150].Dahiya P, Datta D, Hussain MA, Verma G, Shelly A, Mehta P, Mazumder S, The coordinated outcome of STIM1-Orai1 and superoxide signalling is crucial for headkidney macrophage apoptosis and clearance of Mycobacterium fortuitum, Dev. Comp. Immunol, 114 (2021) 13. [DOI] [PubMed] [Google Scholar]
- [151].Dahiya P, Hussain MA, Mazumder S, mtROS Induced via TLR-2-SOCE Signaling Plays Proapoptotic and Bactericidal Role in Mycobacterium fortuitum-Infected Head Kidney Macrophages of Clarias gariepinus, Front. Immunol, 12 (2021) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Oh SM, Lim YJ, Choi JA, Lee J, Cho SN, Go D, Kim SH, Song CH, TNF--mediated ER stress causes elimination of Mycobacterium fortuitum reservoirs by macrophage apoptosis, Faseb J, 32 (2018) 3993–4003. [DOI] [PubMed] [Google Scholar]
- [153].Cui W, Ma J, Wang XQ, Yang WJ, Zhang J, Ji QH, Free Fatty Acid Induces Endoplasmic Reticulum Stress and Apoptosis of beta-cells by Ca2+/Calpain-2 Pathways, PLoS One, 8 (2013) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ye Y, Li X, Wang ZH, Ye F, Xu WR, Lu RZ, Shen HJ, Miao SH, 3,3'-Diindolylmethane induces gastric cancer cells death via STIM1 mediated store-operated calcium entry, Int. J. Biol. Sci, 17 (2021) 1217–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Meng MJ, Wang LR, Wang Y, Ma NN, Xie W, Chang GJ, Shen XZ, A high-concentrate diet provokes inflammation, endoplasmic reticulum stress, and apoptosis in mammary tissue of dairy cows through the upregulation of STIM1/ORAI1, J. Dairy Sci, 105 (2022) 3416–3429. [DOI] [PubMed] [Google Scholar]
- [156].Chiu WT, Tang MJ, Jao HC, Shen MR, Soft substrate upregulates the interaction of STIM1 with store-operated Ca2+ channels that lead to normal epithelial cell apoptosis, Mol. Biol. Cell, 19 (2008) 2220–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Rao W, Zhang L, Peng C, Hui H, Wang K, Su N, Wang L, Dai SH, Yang YF, Chen T, Luo P, Fei Z, Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction, Biochim. Biophys. Acta-Mol. Basis Dis, 1852 (2015) 2402–2413. [DOI] [PubMed] [Google Scholar]
- [158].Tu CC, Wan BY, Zeng Y, STIM2 knockdown protects against ischemia/reperfusion injury through reducing mitochondrial calcium overload and preserving mitochondrial function, Life Sci, 247 (2020) 7. [DOI] [PubMed] [Google Scholar]
- [159].Sobradillo D, Hernandez-Morales M, Ubierna D, Moyer MP, Nunez L, Villalobos C, A Reciprocal Shift in Transient Receptor Potential Channel 1 (TRPC1) and Stromal Interaction Molecule 2 (STIM2) Contributes to Ca2+ Remodeling and Cancer Hallmarks in Colorectal Carcinoma Cells, J. Biol. Chem, 289 (2014) 28765–28782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Aytes A, Mollevi DG, Martinez-Iniesta M, Nadal M, Vidal A, Morales A, Salazar R, Capella G, Villanueva A, Stromal interaction molecule 2 (STIM2) is frequently overexpressed in colorectal tumors and confers a tumor cell growth suppressor phenotype, Mol. Carcinog, 51 (2012) 746–753. [DOI] [PubMed] [Google Scholar]
- [161].Flourakis M, Lehen'kyi V, Beck B, Raphael M, Vandenberghe M, Vanden Abeele F, Roudbaraki M, Lepage G, Mauroy B, Romanin C, Shuba Y, Skryma R, Prevarskaya N, Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells, Cell Death Dis, 1 (2010) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Denmeade SR, Lin XS, Isaacs JT, Role of programmed (apoptotic) cell death during the progression and therapy for prostate cancer, Prostate, 28 (1996) 251–265. [DOI] [PubMed] [Google Scholar]
- [163].Wen L, Voronina S, Javed MA, Awais M, Szatmary P, Latawiec D, Chvanov M, Collier D, Huang W, Barrett J, Begg M, Stauderman K, Roos J, Grigoryev S, Ramos S, Rogers E, Whitten J, Velicelebi G, Dunn M, Tepikin AV, Criddle DN, Sutton R, Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models, Gastroenterology, 149 (2015) 481-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Zhang XX, Xin P, Yoast RE, Emrich SM, Johnson MT, Pathak T, Benson JC, Azimi I, Gill DL, Monteith GR, Trebak M, Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels, Cell Calcium, 91 (2020) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Henke N, Albrecht P, Bouchachia I, Ryazantseva M, Knoll K, Lewerenz J, Kaznacheyeva E, Maher P, Methner A, The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress, Cell Death Dis, 4 (2013) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Fan JW, Miao Y, Zhao Y, Guan YX, Zhang L, Pan LH, Feng Q, Yao JC, Sun CH, Icaritin inhibits oxidative stress in murine astrocytes by binding to Orai1 to block store-operated calcium channel, Chem. Biol. Drug Des, 10. [DOI] [PubMed] [Google Scholar]
- [167].Kim KD, Srikanth S, Yee MKW, Mock DC, Lawson GW, Gwack Y, ORAI1 Deficiency Impairs Activated T Cell Death and Enhances T Cell Survival, J. Immunol, 187 (2011) 3620–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Liang SJ, Zeng DY, Mai XY, Shang JY, Wu QQ, Yuan JN, Yu BX, Zhou P, Zhang FR, Liu YY, Lv XF, Liu J, Ou JS, Qian JS, Zhou JG, Inhibition of Orai1 Store-Operated Calcium Channel Prevents Foam Cell Formation and Atherosclerosis, Arterioscler. Thromb. Vasc. Biol, 36 (2016) 618–628. [DOI] [PubMed] [Google Scholar]
- [169].Yuan F, Yi L, Hai L, Wang YS, Yang YH, Li T, Tong LQ, Ma HW, Liu PD, Ming HL, Ren BC, Yu SP, Lin Y, Yang XJ, Identification of Key Pathways and Genes in the Orai2 Mediated Classical and Mesenchymal Subtype of Glioblastoma by Bioinformatic Analyses, Dis. Markers, 2019 (2019) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Stegner D, Hofmann S, Schuhmann MK, Kraft P, Herrmann AM, Popp S, Hohn M, Popp M, Klaus V, Post A, Kleinschnitz C, Braun A, Meuth SG, Lesch KP, Stoll G, Kraft R, Nieswandt B, Loss of Orai2-Mediated Capacitative Ca2+ Entry Is Neuroprotective in Acute Ischemic Stroke, Stroke, 50 (2019) 3238–3245. [DOI] [PubMed] [Google Scholar]
- [171].Dubois C, Vanden Abeele F, Lehen'kyi V, Gkika D, Guarmit B, Lepage G, Slomianny C, Borowiec AS, Bidaux G, Benahmed M, Shuba Y, Prevarskaya N, Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer, Cancer Cell, 26 (2014) 19–32. [DOI] [PubMed] [Google Scholar]
- [172].Monteith GR, Prostate Cancer Cells Alter the Nature of Their Calcium Influx to Promote Growth and Acquire Apoptotic Resistance, Cancer Cell, 26 (2014) 1–2. [DOI] [PubMed] [Google Scholar]
- [173].Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H, Down-Regulation of Orai3 Arrests Cell-Cycle Progression and Induces Apoptosis in Breast Cancer Cells But Not in Normal Breast Epithelial Cells, J. Cell. Physiol, 226 (2011) 542–551. [DOI] [PubMed] [Google Scholar]
- [174].Faouzi M, Kischel P, Hague F, Ahidouch A, Benzerdjeb N, Sevestre H, Penner R, Ouadid-Ahidouch H, ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells, Biochim. Biophys. Acta-Mol. Cell Res, 1833 (2013) 752–760. [DOI] [PubMed] [Google Scholar]
- [175].Motiani RK, Abdullaev IF, Trebak M, A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells, J Biol Chem, 285 (2010) 19173–19183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, Trebak M, Orai3 is an estrogen receptor alpha-regulated Ca(2)(+) channel that promotes tumorigenesis, FASEB J, 27 (2013) 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Dubois C, Kondratska K, Kondratskyi A, Morabito A, Mesilmany L, Farfariello V, Toillon RA, Ziental Gelus N, Laurenge E, Vanden Abeele F, Lemonnier L, Prevarskaya N, ORAI3 silencing alters cell proliferation and promotes mitotic catastrophe and apoptosis in pancreatic adenocarcinoma, Biochim Biophys Acta Mol Cell Res, 1868 (2021) 119023. [DOI] [PubMed] [Google Scholar]
- [178].Wang Y, Bai SW, Zhang R, Xia L, Chen LH, Guo JZ, Dai F, Du J, Shen B, Orai3 exacerbates apoptosis of lens epithelial cells by disrupting Ca2+ homeostasis in diabetic cataract, Clin. Transl. Med, 11 (2021) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Gombedza FC, Shin S, Kanaras YL, Bandyopadhyay BC, Abrogation of store-operated Ca2+ entry protects against crystal-induced ER stress in human proximal tubular cells, Cell Death Discov, 5 (2019) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Xie JS, Pan HM, Yao JL, Zhou YB, Han WD, SOCE and cancer: Recent progress and new perspectives, Int. J. Cancer, 138 (2016) 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Shapovalov G, Gordienko D, Prevarskaya N, Store operated calcium channels in cancer progression, in: Marchi S, Galluzzi L (Eds.) Inter-Organellar Ca2+ Signaling in Health and Disease - Pt B, Elsevier Academic Press Inc, San Diego, 2021, pp. 123–168. [DOI] [PubMed] [Google Scholar]
- [182].Yu F, Courjaret R, Elmi A, Adap EA, Orie NN, Zghyer F, Hubrack S, Hayat S, Asaad N, Worgall S, Suthanthiran M, Ali VM, Machaca K, Chronic reduction of store operated Ca2+ entry is viable therapeutically but is associated with cardiovascular complications, Journal of Physiology-London, 600 (2022) 4827–4848. [DOI] [PubMed] [Google Scholar]