

Abstract
Fibrillarin (FBL) is a highly conserved nucleolar methyltransferase responsible for methylation of ribosomal RNA and proteins. Here, we reveal a role for FBL in DNA damage response and its impact on cancer proliferation and sensitivity to DNA‐damaging agents. FBL is highly expressed in various cancers and correlates with poor survival outcomes in cancer patients. Knockdown of FBL sensitizes tumor cells and xenografts to DNA crosslinking agents, and leads to homologous recombination‐mediated DNA repair defects. We identify Y‐box‐binding protein‐1 (YBX1) as a key interacting partner of FBL, and FBL increases the nuclear accumulation of YBX1 in response to DNA damage. We show that FBL promotes the expression of BRCA1 by increasing the binding of YBX1 to the BRCA1 promoter. Our study sheds light on the regulatory mechanism of FBL in tumorigenesis and DNA damage response, providing potential therapeutic targets to overcome chemoresistance in cancer.
Keywords: FBL, DNA damage repair, YBX1, BRCA1, colon cancer
Subject Categories: Cancer; DNA Replication, Recombination & Repair
Fibrillarin (FBL) is a nucleolar methyltransferase involved in rRNA and protein methylation. This study reveals FBL's role in promoting cancer cell resistance to DNA damage and facilitating BRCA1 transcription via its interaction with YBX1.
Introduction
Fibrillarin (FBL) is a crucial and highly conserved component of small nucleolar ribonucleoproteins (snoRNPs) involved in the methylation of pre‐rRNA and proteins. FBL functions in ribosome biogenesis and other cellular processes, such as viral progression, pathological processes, and aging dynamics (Watanabe‐Susaki et al, 2014; Rodriguez‐Corona et al, 2015). FBL is located predominantly in the fibrillary region of the nucleolus (Kiss‐László et al, 1996; Yanagida et al, 2004; Feric et al, 2016) and also present in Cajal bodies (Yanagida et al, 2004). The human FBL crystal structure was obtained by X‐ray diffraction with a resolution of 1.82 Å in 2006 (PDB code: 2IPX). Highly conserved in eukaryotes with respect to sequences, structures, and functions (Schimmang et al, 1989; David et al, 1997), human FBL comprises 321 amino acids and is 66% identical to yeast homolog Nop1p (Aris & Blobel, 1991; Wang et al, 2014). FBL contains two main domains: the N‐terminal glycine and arginine‐rich (GAR) domain and the C‐terminal region containing a methyltransferase (MTase) domain (Yanagida et al, 2004; Rodriguez‐Corona et al, 2015; Shubina et al, 2016, 2018). The GAR domain is essential for nuclear localization and interaction with the SMN protein and the DEAD‐box helicase p68 (Yanagida et al, 2004; Rodriguez‐Corona et al, 2015).
The link between FBL and cancer has been previously reported. For instance, overexpression of FBL has been shown to promote cellular proliferation and resistance to chemotherapy in MCF‐7 breast cancer cells (Marcel et al, 2013; Su et al, 2014). FBL is also overexpressed in mouse and human prostatic intraepithelial neoplasia, which can lead to prostate cancer (Koh et al, 2011). Mechanistically, the elevated expression of FBL leads to a distinct methylation pattern in cancer cells, resulting in the production of more heterogeneous ribosomes and enhanced translation of key cancer‐related genes, such as IGF‐1R, c‐Myc, VEGF‐A, and FGF1 (Marcel et al, 2013), ultimately leading to tumor initiation and progression (Marcel et al, 2013; Shubina et al, 2018). FBL is a highly dynamic protein involved in various nuclear processes. However, its role in cell stress mechanisms, particularly DNA damage, has not been extensively investigated. For instance, changes in the dotted pattern of FBL staining were observed in U2OS cells following ionizing radiation‐induced damage (Foltánková et al, 2013). Checkpoint kinase 1 also migrates to the nucleolus and colocalizes with FBL upon DNA damage (Peddibhotla et al, 2014), suggesting additional roles of FBL in DNA repair. Dysregulation of DNA damage repair can contribute to cancer cell hypersensitivity or resistance to genotoxic agents, and targeting DNA repair pathway can increase tumor sensitivity to cancer therapies. Therefore, targeting DNA repair pathways may be a potential therapeutic approach for cancer treatment. However, the biological roles of FBL in response to DNA damage and its involvement in DNA repair pathways in chemoresistance remain largely undefined in cancer.
YBX1 is a multifunctional protein consisting of three domains: an N‐terminal alanine/proline‐rich domain (A/P domain) involved in protein/protein interactions and RNA‐binding stabilization, a central evolutionarily conserved cold‐shock domain (CSD), and a large C‐terminal domain (B/A repeat) involved in nucleic acid binding and protein/protein interactions (Kuwano et al, 2019). The flexible structure of YBX1 allows it to interact with a multitude of molecular partners, including proteins, DNA, and RNA (Lyabin et al, 2014). YBX1 has broad functions in post‐transcriptional regulation, mRNA splicing, mRNA stabilization, stress response signaling, inflammation, and DNA damage (Sangermano et al, 2020). It also functions as a transcription factor controlling the expression of genes involved in cell cycle progression and stress response (Eliseeva et al, 2012) and is recognized as an oncogenic factor in several tumors (Sangermano et al, 2020). YBX1 is an important sensor of the cellular DNA damage response (Koike et al, 1997; Kuwano et al, 2019). Accumulating evidence shows that YBX1 acts as a non‐canonical DNA repair factor participating in different DNA repair mechanisms, including homologous recombination repair (HRR), base‐excision repair (BER), mismatch repair (MMR), and nucleotide excision repair (NER), and is involved in chemoresistance (Lasham et al, 2003; Gaudreault et al, 2004; En‐Nia et al, 2005; Eliseeva et al, 2012).
Our study revealed that FBL is frequently overexpressed in a wide variety of cancers by analyzing the TCGA database. Depletion of FBL significantly sensitized both in vitro and in vivo cancer cells to DNA‐damaging agents. We uncovered that FBL plays a crucial role in HR‐mediated DNA damage repair, and interacts with the nuclear YBX1. FBL promotes YBX1 nuclear accumulation in response to DNA damage by augmenting the FBL‐YBX1 interaction. Functional experiments further demonstrated that FBL exerts a regulatory role on BRCA1 expression through YBX1, affecting the DNA damage repair process in cancer cells. Collectively, our findings suggest that the FBL‐YBX1‐BRCA1 axis is essential for the cellular response to DNA damage and provide further insights into the novel regulatory mechanism of FBL‐mediated tumorigenesis and DNA damage repair.
Results
FBL overexpression in human tumors and its functional role in colon and lung cancers
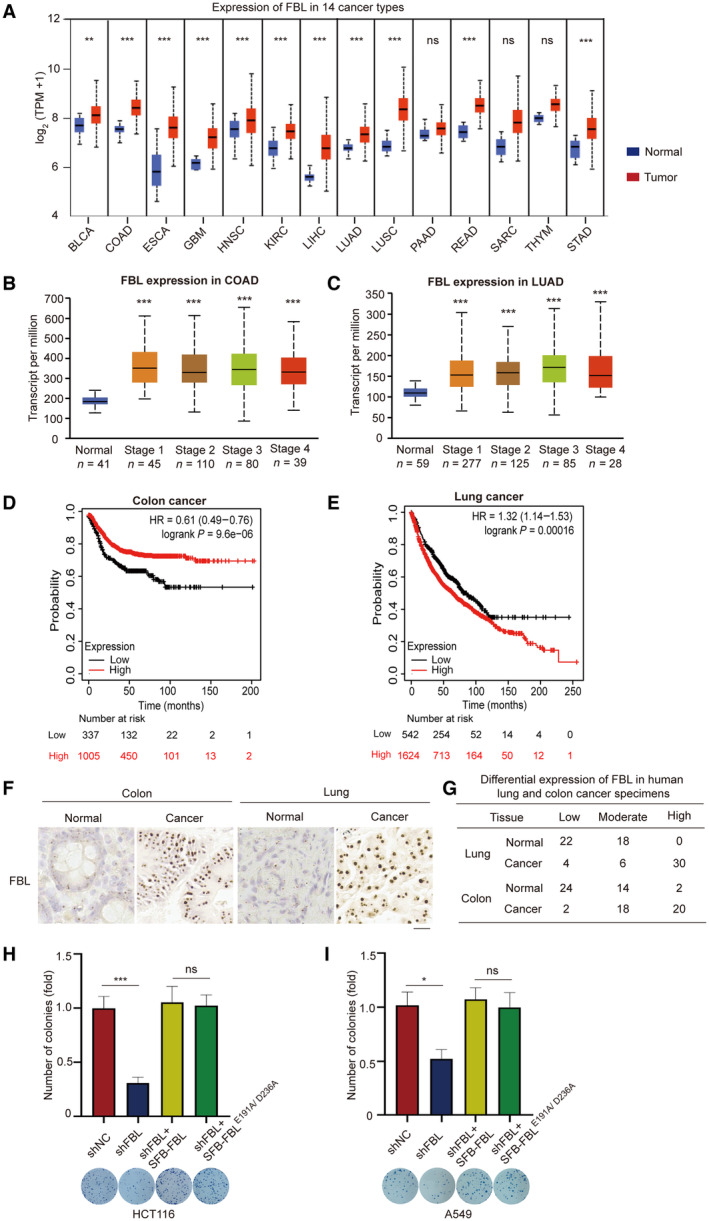
Previous reports have indicated that aberrant FBL expression contributes to tumorigenesis and is associated with poor survival in breast cancer patients (Marcel et al, 2013). To further investigate the role of FBL in human cancers, we examined FBL expression across 14 types of human tumors using The Cancer Genome Atlas (TCGA) database. Our analysis revealed that FBL mRNA was significantly upregulated in 14 cancer types compared to their corresponding normal tissues (Fig 1A and Table EV1). Furthermore, clinical data analysis showed that FBL expression was elevated in all stages of lung and colon cancer patients (Fig 1B and C). Notably, higher FBL expression was associated with poorer survival probability for colon and lung cancer patients (Fig 1D and E), suggesting that FBL may serve as a prognostic biomarker for patients with colon or lung adenocarcinoma.
Figure 1. FBL is overexpressed in human tumors and promotes cancer cell proliferation.
-
ABox and whisker plots showing statistical comparisons of the FBL expression from the human tumors and their respective normal tissues in TCGA database. Horizontal lines within the box indicate median values. Boxes encapsulate the central tendency (first and third quartile), and whiskers indicate values that lie within 1.5 times the interquartile ranges. The statistical significance was determined using Student's t‐test. ns indicates no statistical significance, **P < 0.01, and ***P < 0.001. BLCA (normal n = 19, tumor n = 408), COAD (normal n = 41, tumor n = 286), ESCA (normal n = 11, tumor n = 184), GBM (normal n = 5, tumor n = 156), HNSC (normal n = 44, tumor n = 520), KIRC (normal n = 72, tumor n = 533), LIHC (normal n = 50, tumor n = 371), LUAD (normal n = 59, tumor n = 515), LUSC (normal n = 52, tumor n = 503), PAAD (normal n = 4, tumor n = 178), READ (normal n = 10, tumor n = 166), SARC (normal n = 2, tumor n = 260), THYM (normal n = 2, tumor n = 120), and STAD (normal n = 34, tumor n = 415). The abbreviations and full names of cancer types used are listed in Table EV1.
-
B, CThe relative expression levels of FBL in healthy subjects and patients with stage I–IV colon adenocarcinoma (COAD) or lung adenocarcinoma (LUAD). Data were obtained from the TCGA database. Horizontal lines within the box indicate median values. Boxes encapsulate the central tendency (first and third quartile), and whiskers indicate values that lie within 1.5 times the interquartile ranges. The statistical significance was determined using Student's t‐test. ***P < 0.001.
-
D, EKaplan–Meier curve analysis was conducted to evaluate the relapse‐free survival or overall survival probabilities of colon cancer (n = 1,342) or lung cancer (n = 2,166) patients based on low or high levels of FBL expression. Data were sourced from an online tool website (https://kmplot.com/analysis/). The lower quartile‐based partitioning method was used to select upper and lower thresholds to subset tumor samples into low and high groups. A cut‐off value of the 25th percentile was defined as the low level of FBL in colon or lung cancer patients.
-
FRepresentative images of immunohistochemical analysis of FBL expression levels in human lung or colon tumors and their respective normal tissues (n = 40). Scale bar, 25 μm.
-
GThe FBL expression in human tumors and their corresponding normal tissues were categorized as low, moderate, or high based on the staining intensity, which was scored on a scale of 0–3. The staining intensity was evaluated as 0 for negative, 1 for weak, 2 for moderate, and 3 for strong. Staining intensities of 0 and 1 were classified as low.
-
H, IEnzymatic activity of FBL is not required for cell proliferation. Colony formation assays were conducted to investigate the role of enzymatic activity in cell proliferation. FBL knockdown HCT116 or A549 cells with ectopic expression of FBL or the catalytically inactive mutant (FBL‐E191A/D236A) were used, with a bar graph showing the relative number of colonies per well (mean ± SD). All cells were allowed to form colonies for 10 days and representative images of colonies in plates stained with Giemsa were displayed. The bar graphs present data as mean ± SD from three independent experiments. The statistical significance was determined using one‐way ANOVA. ns denotes no statistical significance, *P < 0.05 and ***P < 0.001.
Source data are available online for this figure.
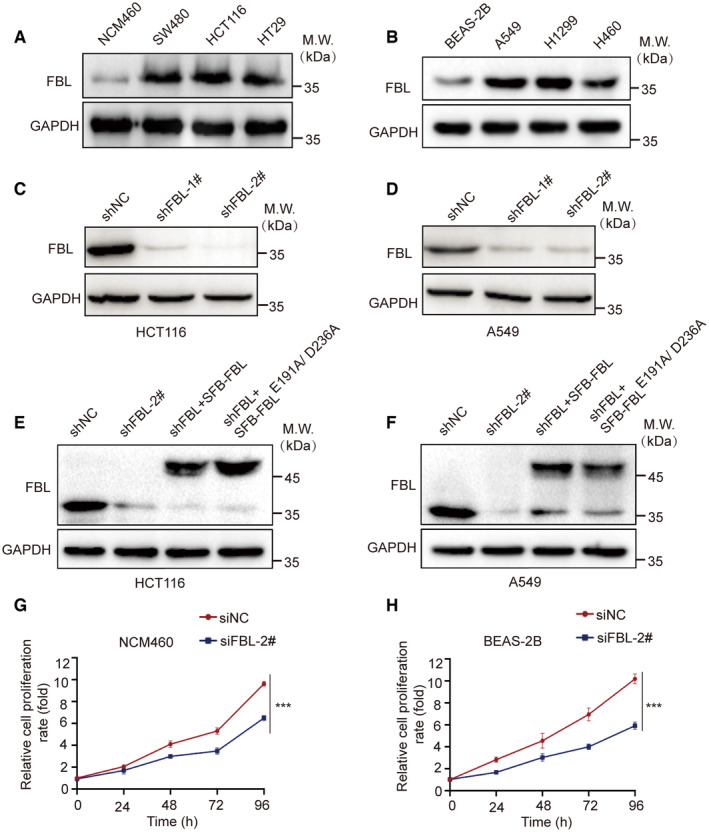
To validate our results, we conducted immunohistochemistry (IHC) on 40 individual colon and lung cancer samples as well as their matched adjacent normal tissue controls. Consistent with the TCGA database analysis, FBL exhibited significant overexpression in both lung and colon cancer samples (Fig 1F). The IHC staining was quantified using computerized scanning analysis and manual scoring by expert pathologists. The summarized IHC data indicated that FBL was more frequently overexpressed at moderate‐to‐high levels in colon and lung cancers compared to their non‐malignant counterparts (Fig 1G). Furthermore, we observed remarkably higher expressions of FBL in several lung cancer or colon cancer cell lines compared to wild type (Fig EV1A and B). Taken together, these results indicate that FBL is highly expressed in various types of human tumors.
Figure EV1. FBL is overexpressed in cancer cell lines and promotes cell proliferation.
-
A, BWestern blotting analyses of FBL expression in colon/lung cancer and normal cell lines.
-
C, DCell lysates of monoclonal cell lines were immunoblotted with FBL antibodies to confirm the knockdown efficiency in HCT116/A549 cells.
-
E, FCells were reconstituted with full‐length FBL and FBLE191A/D236A in HCT116/A549 cells. Protein expressions were examined by western blot with indicated antibodies.
-
G, HCell viability assays were performed on control (siNC) and FBL knockdown (siFBL) NCM460/BEAS‐2B cells using CCK‐8 assays. The data are presented as mean ± SD from three independent experiments. The statistical significance was determined using Student's t‐test. ***P < 0.001.
To further investigate the role of FBL in colon and lung cancers, we generated stable FBL knockdown (KD) cell lines by lentiviral‐mediated expression of FBL‐targeting shRNAs in HCT116 (shFBL‐HCT116) and A549 (shFBL‐A549) cells. Two independent specific shRNAs were used to silence FBL in HCT116 or A549 cells, and western blotting was performed to test the knockdown efficiency. The more efficient shFBL‐HCT116#2 and shFBL‐A549#2 were used for further experiments (Fig EV1C and D). The colony formation assays demonstrated a significant reduction in cell growth in shFBL‐HCT116 cells and shFBL‐A549 cells (Fig 1H and I). Furthermore, cell proliferation of FBL KD cells was restored to control levels by reconstituting with wild‐type FBL. Interestingly, the cell proliferation mediated by FBL did not depend on its methyltransferase activity, as the FBL catalytically inactive mutant (E191A/D236A) also successfully rescued the ability of cell proliferation (Yao et al, 2019) (Figs 1H and I, and EV1E and F). Additionally, knocking down FBL in normal (colon/lung) cells suppressed cell proliferation (Fig EV1G and H). These findings suggest that FBL plays a role in promoting cell proliferation that is independent of its function as a methyltransferase and may have an oncogenic role in colon and lung cancers.
FBL enhances cellular resistance to DNA damage and promotes HR‐mediated DNA repair
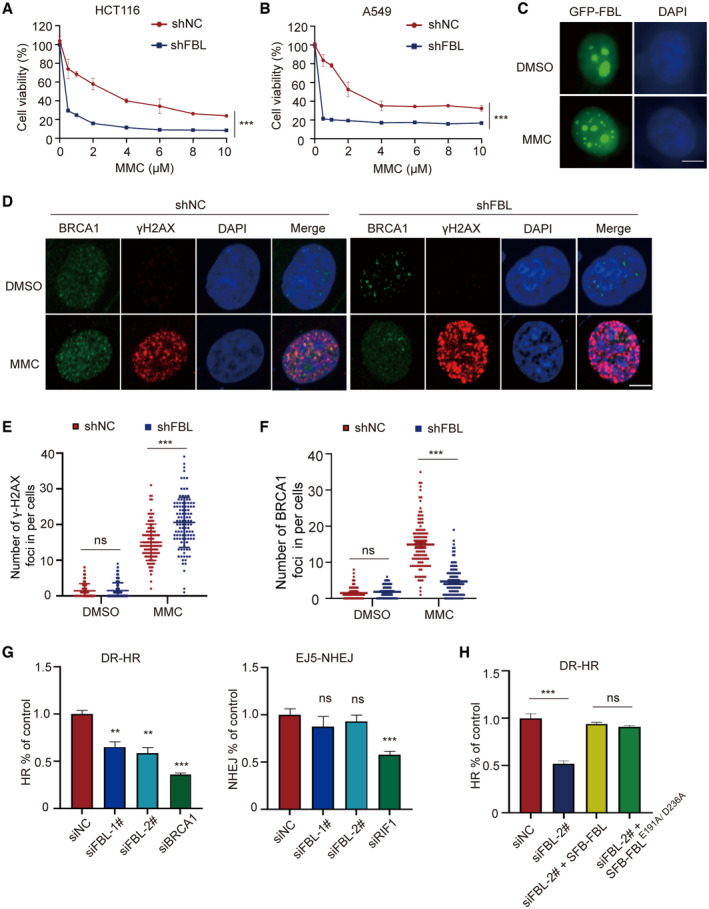
The expression of FBL is significantly upregulated in a wide range of cancers compared to normal tissues, indicating its significant involvement in oncogenesis. To investigate its potential role in cell viability, we depleted FBL expression in cells and examined their response to antitumor chemotherapy drugs. We tested the viability of FBL knockdown (KD) cells versus control cells in response to DNA‐damaging drugs, including MMC, cisplatin, camptothecin (CPT), etoposide, and H2O2. Notably, FBL depletion sensitized cancer cells to DNA‐damaging drugs, particularly to the interstrand crosslinker MMC or cisplatin treatment, with a significant dose‐dependent decrease in survival of FBL KD cells compared to control cells (Figs 2A and B, and EV2A and B). These results suggest that depletion of FBL sensitizes cancer cells to DNA‐damaging drugs, especially MMC or cisplatin. Additionally, we confirmed that overexpression of FBL conferred resistance to MMC (Fig EV2C–F). Furthermore, we investigated the function of FBL on cell proliferation with or without DNA damage treatment using 5‐ethynyl‐2′‐deoxyuridine (EdU) staining. Consistent with anti‐proliferative responses observed in FBL KD cells, we observed reduced incorporation of EdU in shFBL‐A549 or shFBL‐HCT116 cells after treatment with MMC compared to DMSO treatment, indicating that knockdown of FBL inhibits cell proliferation and sensitizes cells to DNA damage (Fig EV2G and H).
Figure 2. FBL plays a crucial role in regulating cell sensitivity to DNA damage and HR‐mediated DNA repair.
-
A, BKnockdown of FBL results in increased cellular sensitivity to DNA damage treatment. Cell viability analysis was performed on control (shNC) and shFBL‐HCT116/shFBL‐A549 cells treated with indicated concentrations of MMC using CCK‐8 assays. Each data point represents the mean ± SD from three replicates. The statistical significance was determined using Student's t‐test. ***P < 0.001.
-
CRepresentative fluorescence microscopy images showing the subcellular localization of EGFP‐tagged FBL following treatment 5 μM MMC or DMSO. Scale bar, 10 μm.
-
D–FImmunofluorescence staining showed the formation of γH2AX and BRCA1 foci in shFBL and shNC HCT116 cells treated with 5 μM MMC or mock treatment. The scale bar represents 10 μm. The (E) and (F) panel displays quantification of the (D) panel: indicating the number of γH2AX or BRCA1 foci per cell (n ≥ 100 cells). Data are shown as the mean ± SD. The statistical significance was determined using one‐way ANOVA, with ns indicating no statistical significance, ***P < 0.001.
-
GKnockdown of FBL impairs the HR repair but not the NHEJ repair. GFP reporter assays were used to examine NHEJ and HR repair pathways using stable reporter cell lines DR‐U2OS or EJ5‐U2OS. Following treatment with siNC or siFBL, as well as HR positive‐control siBRCA1 or NHEJ‐positive control siRIF1, we assessed the percentage of cells expressing GFP by flow cytometry to determine the efficiency of each repair mechanism. The results presented are the mean values ± SD from three independent experiments. The statistical significance was determined using Student's t‐test. ns indicates no statistical significance, **P < 0.01 and ***P < 0.001.
-
HEnzymatic activity is not required for FBL‐mediated HR repair pathway. Following treatment with siNC or siFBL, cells were reconstituted with either FBL or FBL‐E191A/D236A for HR assays as described above. The results presented are the mean values ± SD from three independent experiments. The statistical significance was determined using one‐way ANOVA. ns indicates no statistical significance, ***P < 0.001.
Source data are available online for this figure.
Figure EV2. FBL plays a crucial role in regulating cell sensitivity to DNA damage and HR‐mediated DNA repair.
-
A, BCell viability analysis was performed on control (shNC) and shFBL‐HCT116/shFBL‐A549 cells treated with indicated concentrations of cisplatin, etoposide, camptothecin, or H2O2 using CCK‐8 assays. Each data point represents the mean ± SD from three replicates.
-
C–F(C, E) FBL overexpression cells are sensitive to MMC. FBL WT or empty vector HCT116/A549 cells were treated with MMC. Cell survival was measured by CCK‐8 assay. Each data point represents the mean ± SD from three replicates. (D, F) Cells were reconstituted with full‐length FBL in HCT116/A549 cells. Protein expressions were examined by western blot with indicated antibodies.
-
G, HImmunofluorescence analysis of 5‐ethynyl,2′‐deoxyuridine (EdU) incorporation and quantitative analysis of EdU‐positive cells with 5 μM MMC or DMSO treatment. Scale bar, 50 μm. The bar graphs present data as mean ± SD from three independent experiments. The statistical significance was determined using one‐way ANOVA. **P < 0.01 and ***P < 0.001.
-
IImmunofluorescence staining showed the formation of γH2AX and Rad51 foci in shFBL and shNC HCT116 cells treated with 5 μM MMC or mock treatment. The scale bar represents 10 μm. The right panel displays quantification of the left panel: indicating the number of Rad51 foci per cell (n ≥ 100 cells). The bar graphs present data as mean ± SD from three independent experiments. The statistical significance was determined using one‐way ANOVA. ns. no significance.
-
J, KWestern blotting analysis of FBL levels in FBL knockdown EJ5‐GFP‐U2OS/DR‐GFP‐U2OS cells.
Fibrillarin is primarily located in the nucleolus, but upon MMC treatment, we observed a distinct change in its localization. Although the majority of FBL signals remained in the nucleolus, it dispersed in a granular pattern throughout the nucleus upon MMC treatment (Fig 2C). These observations suggest that FBL may have additional functions besides its role in rRNA modification in the nucleolus. Given that FBL depletion increases sensitivity to DNA‐damaging agents, we investigated FBL's involvement in DNA damage repair. We found that FBL KD cells treated with MMC showed a significant accumulation of γH2AX foci, indicative of DNA double‐strand breaks (DSBs) (Fig 2D and E). FBL depletion impaired the formation of BRCA1 foci but not RAD51 foci (Figs 2D and F, and EV2I). These results suggest that a reduction of FBL results in impaired cellular repair of γH2AX‐marked DNA breaks, consequently impacting the signaling pathway involved in DSBs damage. Since DSBs are primarily repaired by error‐free homologous recombination (HR) or error‐prone non‐homologous end joining (NHEJ) (Panier & Durocher, 2013; Han & Huang, 2019), we performed conventional GFP reporter assays to examine whether FBL regulates HR or NHEJ pathways. We silence FBL expression by transfecting two independent siRNAs into DR‐GFP‐U2OS or EJ5‐GFP‐U2OS cells and performed western blotting assays to test knockdown efficiency (Fig EV2J and K). The results showed that FBL KD significantly impaired HR‐mediated DSB repair but had no effect on NHEJ‐mediated DSB repair (Fig 2G). Importantly, both wild‐type FBL and the enzymatically inactive mutant (E191A/D236A) were able to rescue the defects in HR repair in FBL KD cells (Fig 2H), suggesting that FBL‐mediated DNA damage repair is independent of its methyltransferase activity. Overall, these data suggest that loss of FBL sensitizes cancer cells to DNA damage and impairs HR repair pathway and FBL‐mediated DNA damage repair is independent of its methyltransferase activity.
FBL interacts with YBX1 independently of its catalytic activity
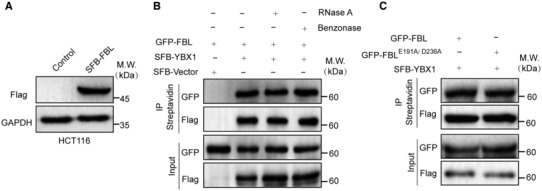
To gain mechanistic insights into the function of FBL in cancer cells, we established HCT116 cells that stably express S‐Flag‐Streptavidin binding peptide (SFB)‐tagged FBL and performed tandem affinity purification and mass spectrometry analysis (Fig EV3A). This led to the identification of YBX1 as a potential FBL‐interacting protein (Fig 3A), suggesting a functional link between YBX1 and FBL in cancer cells. YBX1 is a member of the family of DNA‐ and RNA‐binding proteins, possessing an evolutionarily ancient and conserved CDS (Lyabin et al, 2014). We first examined the interaction between the two proteins using exogenously tagged proteins by coimmunoprecipitation (Co‐IP) assays. SFB‐tagged YBX1 was observed to interact with GFP‐tagged FBL in vivo (Fig 3B). Moreover, there was no reduction in association upon RNase A or Benzonase treatment, which suggests that the interaction between FBL and YBX1 was not mediated by DNA or RNA (Fig EV3B). We then detected the endogenous association between FBL and YBX1 in HCT116 cells using Co‐IP assays and confirmed an interaction between FBL and YBX1 (Fig 3C). Additionally, GST pull‐down analysis revealed that GST‐tagged YBX1 could pull down SFB‐tagged FBL, confirming an interaction between the two proteins (Fig 3D). We mapped the domains required on each protein for the interaction by Co‐IP to examine how FBL binds YBX1, and found that the interaction was dependent on the N‐terminal GAR domain of FBL but not its C‐terminal MTase domain, suggesting that the interaction was independent of FBL's function as a methyltransferase (Fig 3E). Furthermore, the catalytically inactive mutant (E191A/D236A) of FBL was observed to interact with YBX1 (Fig EV3C). While the C‐terminal of YBX1 (130–324 aa) was critical for the interaction between YBX1 and FBL (Fig 3F). Thus, YBX1 is a binding partner of FBL.
Figure EV3. FBL interacts with YBX1 independent of DNA or RNA and its methyltransferase activity.
- To establish cell lines stably expressing epitope‐tagged proteins, HCT116 cells were transfected with plasmids encoding SFB‐FBL. Protein expressions were examined by western blotting with indicated antibodies.
- The interaction between FBL and YBX1 is not mediated by DNA or RNA. The association between FBL and YBX1 was examined by coimmunoprecipitation assays in HCT116 cells with RNase A or benzonase treatment.
- The catalytically inactive mutant of FBL also binds YBX1 in vivo. Co‐IP analyses of GFP‐FBLE191A/D236A and SFB‐YBX1 in HCT116 cells.
Figure 3. FBL interacts with YBX1 through the N‐terminal GAR domain independently of its catalytic activity.
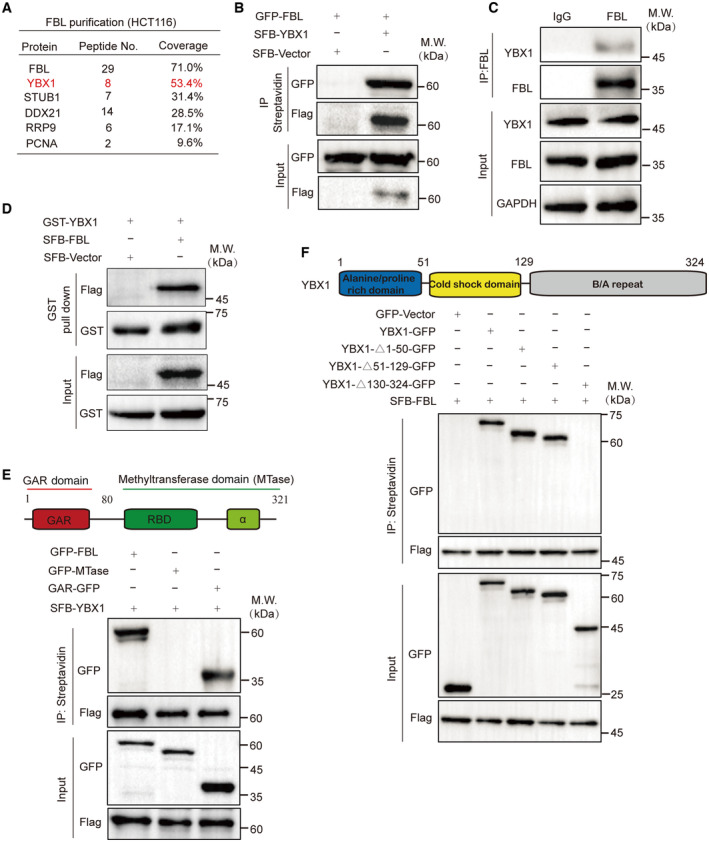
- A list of FBL‐binding proteins identified by affinity purification and mass spectrometry (MS). Chromatin fractions were isolated from HCT116 cells stably expressing SFB‐FBL, followed by affinity purification. The eluted proteins were analyzed by MS.
- Co‐IP analyses of GFP‐tagged FBL and SFB‐tagged YBX1 in HCT116 cells using indicated antibodies. HCT116 cells were transfected with vectors encoding GFP‐FBL and SFB‐YBX1. Cells were lyzed with NETN300 buffer. Cell extracts were examined by IP and western blot with indicated antibodies.
- Co‐IP analyses of endogenous FBL and YBX1 in HCT116 cells using indicated antibodies. Cells were lyzed with NETN300 buffer, and lysates were examined by IP and western blot with indicated antibodies. GAPDH was used as the protein‐loading control.
- FBL binds to YBX1 in vitro. Recombinant GST‐YBX1 was incubated with cell lysates of SFB‐tagged FBL overexpressed or control. The interaction was examined by GST pull‐down assays and western blotting with indicated antibodies.
- The N‐terminal GAR domain is essential for the interaction between FBL and YBX1. Upper panel, a schematic representation of wild‐type FBL with the GAR domain and methyltransferase domain (MTase). Lower panel, Co‐IP analyses of GFP‐tagged FBL deletion mutants and SFB‐tagged YBX1 in HCT116 cells.
- The B/A repeat domain of YBX1 is essential for the interaction between YBX1 and FBL. Upper panel, a schematic representation of wild‐type YBX1 with the alanine‐/proline‐rich domain, cold‐shock domain, and B/A repeat domain. Lower panel, Co‐IP analyses of GFP‐tagged YBX1 deletion mutants and SFB‐tagged FBL in HCT116 cells.
Source data are available online for this figure.
FBL facilitates YBX1 nuclear translocation in response to DNA damage
We aimed to investigate the mechanism underlying FBL's role in regulating DNA damage response. Previous studies have demonstrated the important role of YBX1 in DNA damage signaling and repair (Kuwano et al, 2002, 2004; Chen et al, 2020; Sangermano et al, 2020). YBX1 promotes mRNA processing and stabilizes transcripts involved in homologous recombination in response to DNA damage (Marchesini et al, 2017). In line with these findings, we used siRNA to knockdown endogenous YBX1 in DR‐GFP‐U2OS cells (Fig EV4A) and observed that suppressing YBX1 significantly impaired HR‐mediated DNA damage repair. Notably, restoration of YBX1 expression largely rescued the HR defects induced by FBL KD, suggesting that YBX1 serves as a downstream target of FBL‐mediated HR repair pathway (Fig 4A).
Figure EV4. The effects of FBL on the expression/subcellular localization of YBX1 and the increased interaction between YBX1 and FBL induced by MMC.
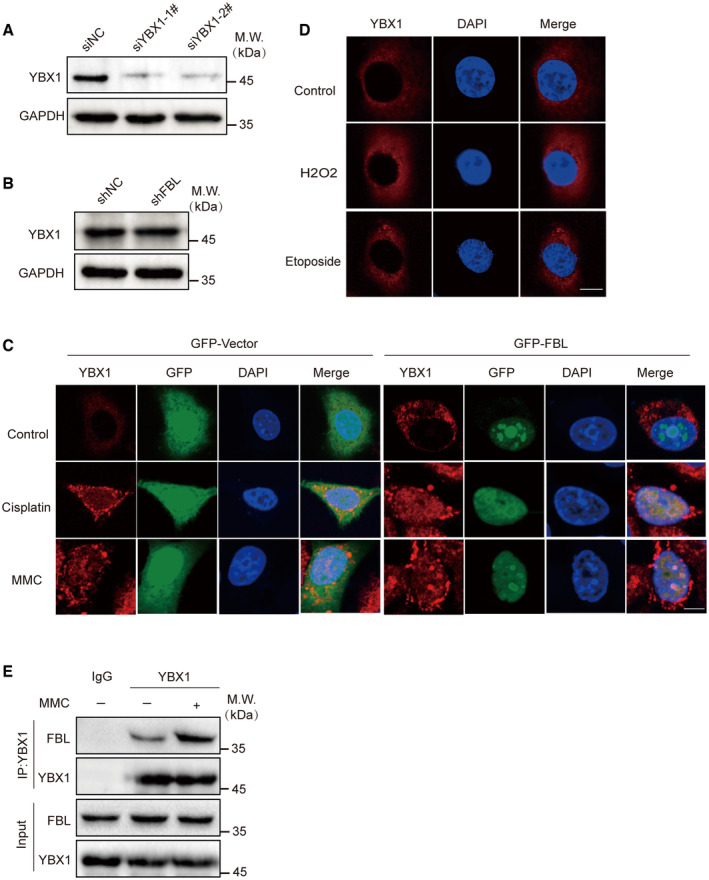
- Western blotting analysis of YBX1 levels in YBX1‐knockdown HCT116 cells. GAPDH was used as the protein‐loading control.
- Western blotting analysis of total YBX1 proteins in FBL KD (shFBL) HCT116 cells. GAPDH was used as the protein‐loading control.
- Subcellular localization of YBX1/FBL was confirmed by immunofluorescence assay after MMC (5 μM, overnight) or cisplatin (25 μM, 2 h) treatment in FBL WT or empty vector cells. Scale bar, 10 μm.
- Subcellular localization of YBX1 was confirmed by immunofluorescence assay after H2O2 (2 mM, 5 min)/etoposide (10 μM, 6 h) treatment. Scale bar, 10 μm.
- The endogenous association between FBL and YBX1 in HCT116 cells was examined by coimmunoprecipitation assays with 5 μM MMC treatment. Cells were lyzed with NETN300 buffer, and nuclear lysates were examined by IP and western blot with indicated antibodies.
Figure 4. FBL modulates DNA damage‐induced YBX1 nuclear translocation.
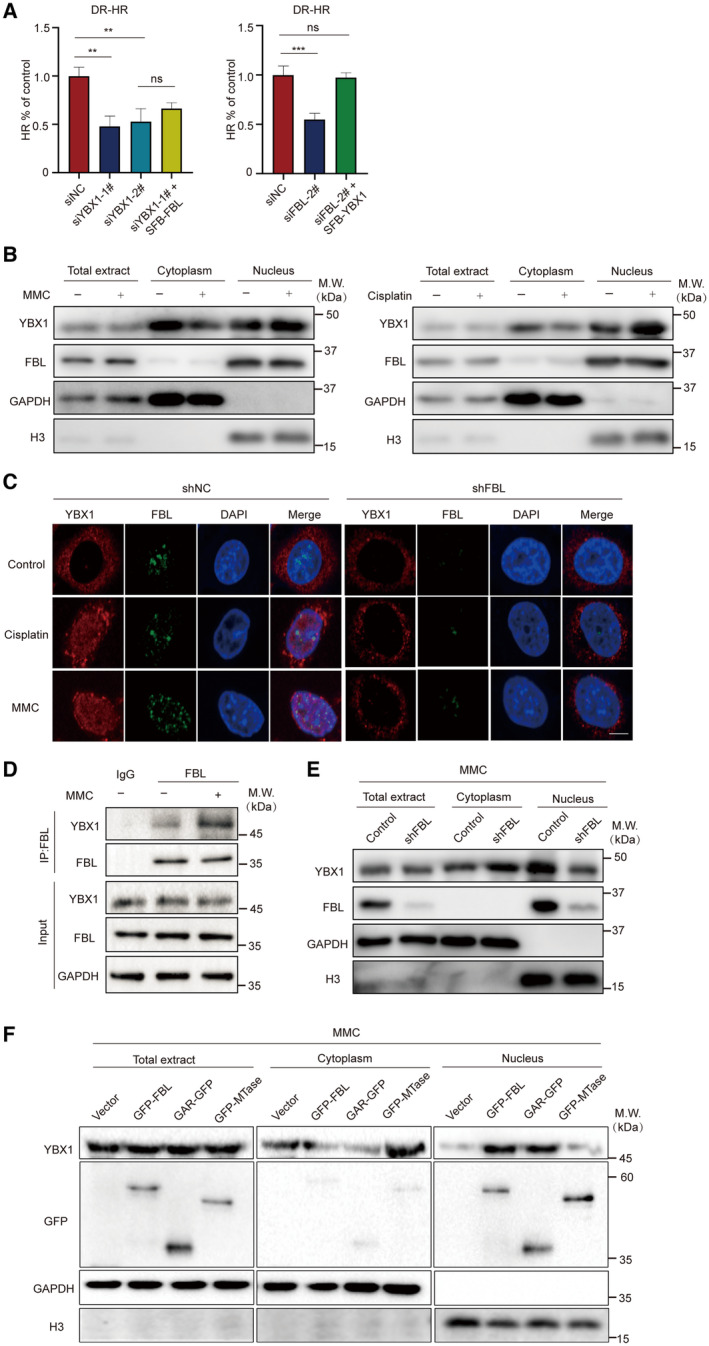
- YBX1 is involved in the HR repair pathway. GFP reporter assays were used to examine the HR repair pathway in YBX1 silencing or reconstituted with FBL, FBL silencing, or reconstituted with YBX1 and control cells. Graphs represent the mean ± SD of three biological replicates. The statistical significance was determined using one‐way ANOVA. ns denotes not significant, **P < 0.01, ***P < 0.001.
- HCT116 cells were treated with or without 5 μM MMC overnight or 25 μM cisplatin for 2 h, and cells were then harvested and separated into nuclear and cytoplasmic fractions. The protein level of YBX1 in each fraction was detected by western blotting assays. GAPDH and histone 3 (H3) were used as loading controls for cytoplasmic and nuclear fractions, respectively.
- Subcellular localization of YBX1/FBL was confirmed by immunofluorescence staining following MMC (5 μM, overnight) or cisplatin (25 μM, 2 h) treatment in shFBL and shNC cells. Scale bar, 10 μm.
- The endogenous association between FBL and YBX1 in HCT116 cells was examined by coimmunoprecipitation assays with 5 μM MMC treatment. Cells were lyzed with NETN300 buffer, and nuclear lysates were examined by IP and western blot with indicated antibodies. GAPDH was used as the protein‐loading control.
- The distribution of YBX1 proteins in the nuclear and cytoplasmic fractions of control or FBL KD HCT116 cells after 5 μM MMC incubation overnight were determined by western blotting assays. H3 was used as a nuclear control. GAPDH was used as a cytoplasmic control.
- The protein contents of YBX1 in the nuclear and cytoplasmic fractions in FBL KD HCT116 cells reconstituted with wild‐type FBL or deletion mutants of FBL with 5 μM MMC treatment overnight were detected using western blotting assays with indicated antibodies.
Source data are available online for this figure.
We then explored the regulatory role of FBL on YBX1 activity. Knockdown of FBL did not affect overall YBX1 protein expression in HCT116 cells (Fig EV4B), indicating that FBL did not influence YBX1 expression levels. Since YBX1 activity depends on its shuttling between the nucleus and cytoplasm (Lyabin et al, 2014), we further examined whether FBL affects the localization of YBX1 in response to DNA damage. Following DNA damage treatment, we detected YBX1 expression in the nuclear and cytoplasmic fraction of HCT116 cells and observed that MMC or cisplatin treatment significantly increased nuclear YBX1 levels (Fig 4B). Immunofluorescence assays confirmed the increased nuclear staining of YBX1 upon DNA damage. Furthermore, FBL depletion reduced the nuclear translocation of YBX1, while FBL overexpression increased nuclear YBX1 levels (Figs 4C and EV4C). Interestingly, only MMC or cisplatin but not H2O2 or etoposide induced YBX1 nuclear translocation (Fig EV4D). Endogenous Co‐IP assays showed that MMC treatment led to increased interaction between FBL and YBX1 (Figs 4D and EV4E). Subcellular fractionation assays demonstrated that depletion of FBL impaired cytoplasmic‐to‐nuclear YBX1 shuttling compared to control cells after MMC treatment (Fig 4E). Overexpression of wild‐type FBL or the GAR domain of FBL restored the impaired YBX1 nuclear localization by FBL KD, while the MTase domain of FBL failed to do so (Fig 4F), indicating that FBL modulates DNA damage‐induced YBX1 nuclear translocation by binding to YBX1. Our findings suggest that FBL regulates YBX1 activity by modulating its nuclear localization in response to DNA damage, and there is a significant correlation between FBL and YBX1 nuclear levels in HCT116 cells.
FBL‐mediated YBX1 nuclear translocation regulates the transcriptional activation of BRCA1 in response to DNA damage
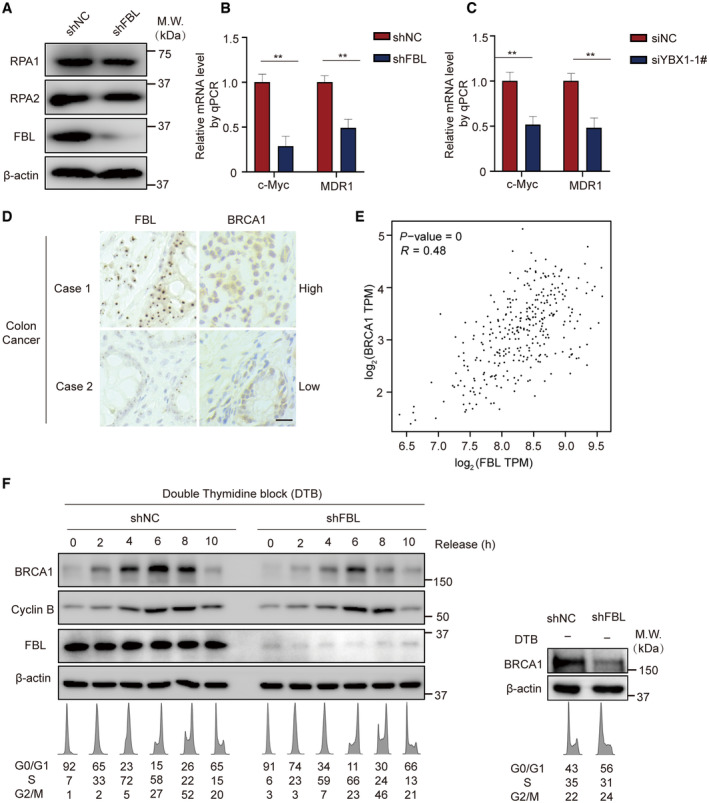
The precise mechanism underlying FBL‐induced YBX1 nuclear translocation that participates in DNA damage repair remains elusive. In this study, we conducted RNA sequencing analysis (RNA‐seq) to detect genome‐wide transcriptomic changes in HCT116 cells and shFBL‐HCT116 cells following MMC treatment. Compared to control cells, 255 genes were significantly downregulated and 1,110 genes were upregulated more than twofold in shFBL‐HCT116 cells (Fig 5A). KEGG pathway analysis demonstrated that differentially expressed genes were primarily enriched in DNA replication, cell cycle, DNA damage repair, and pathways in cancer, with specific involvement in DNA damage repair pathways, such as HR, Fanconi anemia (FA) pathway, BER, and NER (Fig 5B). We further conducted gene set enrichment analysis (GSEA) and found that the gene sets PATHWAYS_IN_CANCER and PATHWAYS_IN_DNA_DAMAGE_RESPONSE_SIGNAL_TRANSDUCTION_BY_P53_CLASS_MEDIATOR_RESULTING_IN_CELL_CYCLE_ARREST were significantly enriched in FBL KD cells (Fig 5C). Since we found that FBL participates in the HR pathway, we conducted a detailed analysis of 16 downregulated genes in the HR pathway in FBL KD cells. Our analysis revealed that candidate genes BRCA1 and RAD51, critical for DNA damage repair and the regulation of the HR pathway (Tarsounas & Sung, 2020; Liu & Lu, 2021), were significantly downregulated in FBL KD cells (Fig 5D). We validated the altered expressions of BRCA1 and RAD51 in FBL KD cells by RT–qPCR and western blotting assays. We observed that the mRNA levels of BRCA1 and RAD51 were decreased in FBL KD cells compared to controls. However, FBL KD especially affected the protein levels of BRCA1 but not RAD51, RPA1, and RPA2, suggesting that BRCA1 might be a potential downstream target of FBL in response to DNA damage (Figs 5E and EV5A). We also examined the mRNA and protein levels of BRCA1 after YBX1 silencing in HCT116 cells. Similarly, YBX1 silencing significantly reduced BRCA1 expression (Fig 5F). Additionally, we assessed c‐Myc and multidrug resistance protein 1 (MDR1) expressions at the mRNA levels, which were also decreased in FBL KD cells compared to controls, consistent with previous reports (Fig EV5B and C) (Bommert et al, 2012; Lu et al, 2019).
Figure 5. FBL promotes the transcriptional activation of BRCA1 via YBX1.
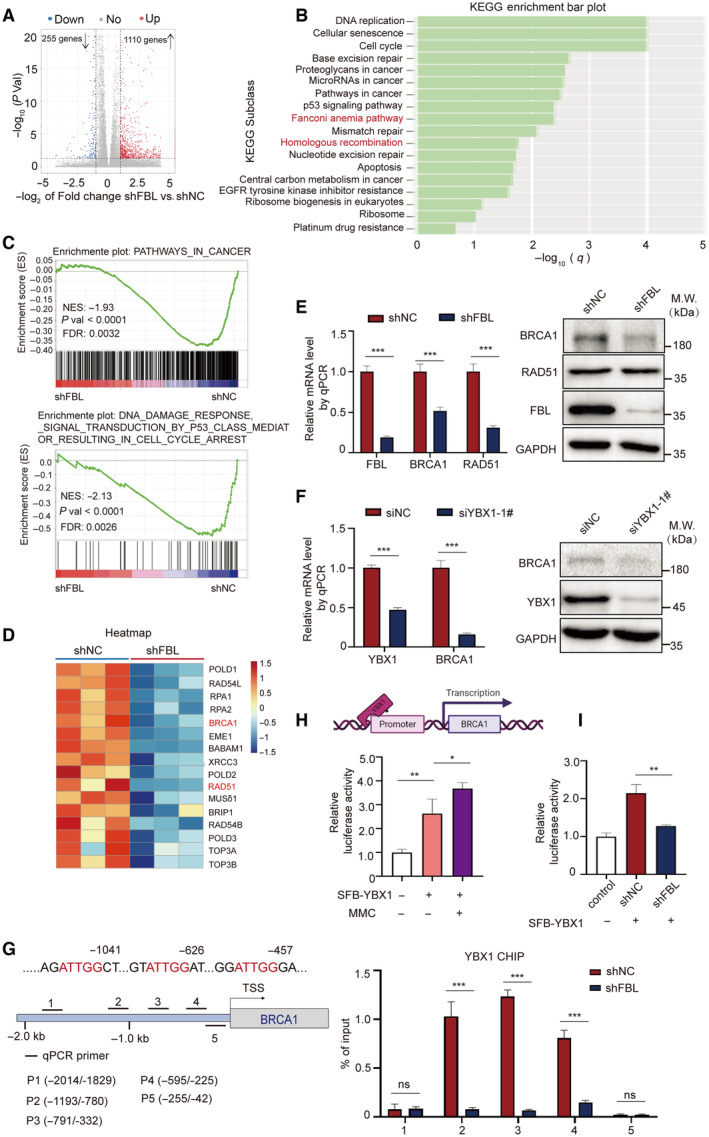
- A volcano plot of differentially expressed gene analysis results for RNA‐seq data comparing FBL KD HCT116 cells (n = 3) to control cells (n = 3). The dots for significant upregulated or downregulated genes were colored in red or blue, respectively.
- KEGG enrichment analysis for differentially expressed genes comparing FBL KD HCT116 cells to control cells.
- GSEA plots showing an interesting gene set from MSigDB were enriched for gene downregulated in FBL KD HCT116 cells for PATHWAYS_IN_CANCER and PATHWAYS_IN_DNA_DAMAGE_RESPONSE_SIGNAL_TRANSDUCTION_BY_ P53_CLASS_MEDIATOR_RESULTING_IN_CELL_CYCLE_ARREST.
- Heatmap illustrating relative expressions of core‐enriched genes in HR gene set in FBL KD cells compared to control cells.
- The mRNA and protein levels of BRCA1 and RAD51 were validated by RT–qPCR and western blot assays using FBL KD HCT116 cells or control cells. Ct values were normalized to GAPDH. The bar graphs present data as mean ± SD from three independent experiments. The statistical significance was determined using Student's t‐test. ***P < 0.001.
- The mRNA and protein levels of BRCA1 were validated by RT–qPCR and western blotting assays using YBX1‐silenced HCT116 cells or control cells. Ct values were normalized to GAPDH. The bar graphs present data as mean ± SD from three independent experiments. The statistical significance was determined using Student's t‐test. ***P < 0.001.
- FBL mediates the recruitment of YBX1 at the promoter region of BRCA1. The top panel is a schematic diagram of BRCA1 promoter region. Bold lines indicate the positions of five primer pairs designed for qPCR analysis of CHIP. The lower panel is ChIP–qPCR measurement of the relative occupancy of YBX1 on different regions of BRCA1 promoters in FBL KD HCT116 cells or control cells. Cells were treated with 5 μM MMC overnight. ChIP analyses on the BRCA1 promoter were performed with IgG or YBX1 antibodies, followed by qPCR using primers that amplify different BRCA1 promoter regions. The bar graphs present data as mean ± SD from three independent experiments. The statistical significance was determined using Student's t‐test. ns denotes not significant, ***P < 0.001.
- MMC treatment increases the YBX1‐mediated transcription activity of BRCA1. The BRCA1 promoter region from −2.0 to 0 kb was cloned for dual‐luciferase reporter assays. HCT116 cells were transiently cotransfected with the BRCA1 promoter, luciferase reporter constructs, and YBX1 expression vectors with MMC treatment. Twenty‐four hours after cotransfection, all samples were harvested for luciferase activity experiments. The bar graphs present data as mean ± SD from three independent experiments. The statistical significance was determined using Student's t‐test. *P < 0.05 and **P < 0.01.
- FBL KD inhibits the transcriptional activation of BRCA1 dual‐luciferase reporter assays evaluating the effect of FBL KD on YBX1‐mediated transcription activity of BRCA1. The bar graphs present data as mean ± SD from three independent experiments. The statistical significance was determined using Student's t‐test. **P < 0.01.
Source data are available online for this figure.
Figure EV5. Effects of FBL or YBX1 on other genes expression and the impact of FBL on the cell cycle process.
-
AWestern blotting analysis of RPA1 and RPA2 proteins in FBL KD (shFBL) HCT116 cells. β‐actin was used as the protein‐loading control.
-
B, CThe mRNA levels of c‐Myc or MDR1 were validated by RT–qPCR after FBL or YBX1 silencing in HCT116 cells. Ct values were normalized to GAPDH. A mean value ± SD of three independent experiments is shown. Student's t‐test. **P < 0.01.
-
DThe correlation of FBL and BRCA1 gene expressions. Representative images of immunohistochemical analysis of FBL and BRCA1 expressions in 40 human colon tumors. Scale bar, 25 μm.
-
EThe correlation analysis of FBL and BRCA1 gene expressions obtained from GEPIA database (http://gepia.cancer‐pku.cn/). TPM stands for transcripts per million; R stands for correlation coefficient. Each spot indicates FBL and BRCA1 expression in a specific sample.
-
FThe levels of BRCA1 protein fluctuate during cell cycle progression, with its expression being regulated by FBL. HCT116 cells and shFBL‐HCT116 cells were grown asynchronously (right panel) or were synchronized at the G1/S transition by a double thymidine block (DTB) and then released by addition of fresh medium (left panel). At various time points post‐release, cell extracts were prepared and analyzed by western blotting using indicated antibodies. Cyclin B serves as a marker of S, G2, and M phases. Similarly, cells induced at specific time points were subjected to flow cytometry analysis. The resulting flow cytometry profiles and the cell cycle distributions are listed below the immunoblots. β‐actin was used as the protein‐loading control.
It is worth noting that nuclear YBX1 can bind to the promoters or other key regulatory regions of target genes to initiate transcription (Dolfini & Mantovani, 2013). To determine whether YBX1 binds to the promoter of BRCA1 and plays a role in FBL‐dependent BRCA1 transcription, we performed chromatin immunoprecipitation (ChIP) and luciferase reporter assays. Our findings confirm that, following MMC treatment, YBX1 was enriched at the promoter regions of the BRCA1 gene in HCT116 cells. Furthermore, knockdown of FBL remarkably reduced the enrichment of YBX1 on the BRCA1 promoter (Fig 5G). We also investigated whether YBX1 regulated BRCA1 transcription in an FBL‐dependent manner. Results from luciferase reporter assays revealed a significant increase in the transcriptional activity of the BRCA1 promoter by ectopically expressed YBX1, with more robust transcriptional activity observed following MMC treatment (Fig 5H). Importantly, knockdown of FBL led to a 50% reduction in luciferase activity compared to control cells following MMC treatment, implying that FBL indirectly regulates the transcriptional activity of the BRCA1 promoter by YBX1 as a mediator (Fig 5I). These data indicate that the activation of the FBL/YBX1 signaling axis upon DNA damage regulates the transcriptional activation of genes directly involved in repairing DNA DSBs, including BRCA1, and contributes to promoting cellular resistance to DNA‐damaging agents.
FBL knockdown suppresses tumor growth and enhances sensitivity to MMC in xenograft model
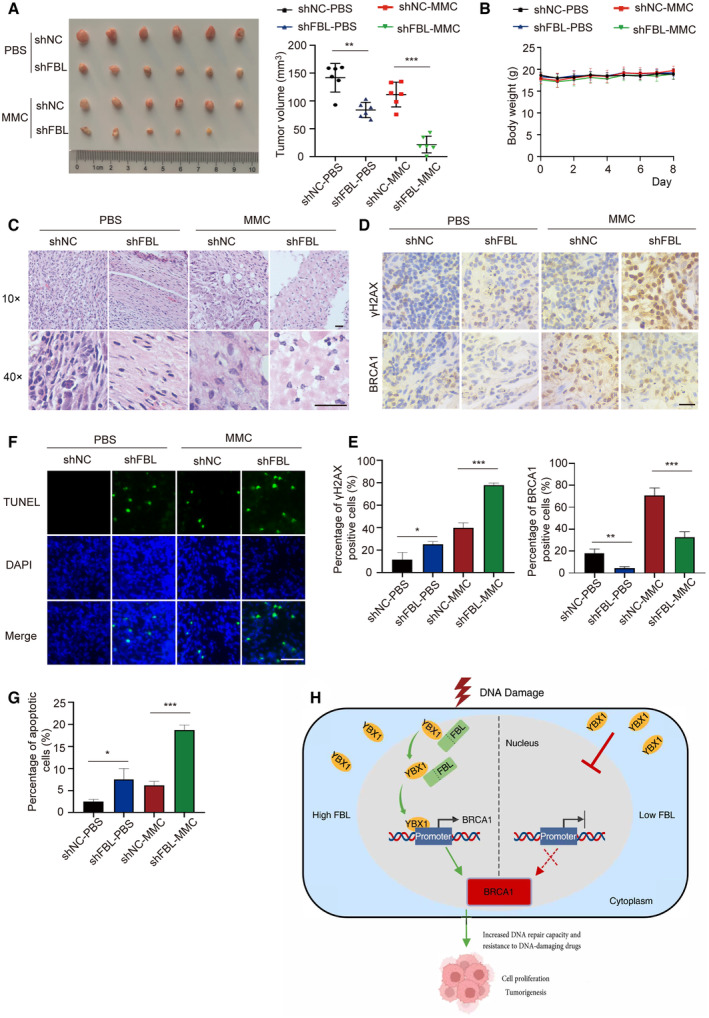
It is well known that HR‐defective cancer cells are more sensitive to DNA‐damaging drugs, such as cisplatin or PARP inhibitors, than cells with proficient HR repair. We examined the impact of FBL on tumor growth in a murine xenograft model and found that knockdown of FBL significantly suppressed tumor growth in control mice administered with vehicle (Fig 6A). Remarkably, tumor progression was even more inhibited in MMC‐treated recipient mice carrying FBL KD xenograft tumors (Fig 6A), while the body weight of xenograft tumor‐bearing mice remained stable in all treatment groups (Fig 6B). The morphology and expressions of BRCA1 and γH2AX in the transplanted tumor tissues were observed using microscopy after staining with hematoxylin–eosin (HE) and indicated antibodies, respectively. The HE staining displayed a more disordered cell arrangement and higher heterogeneity in the FBL KD group compared with the control group (Fig 6C). Moreover, MMC treatment exacerbated the morphology in FBL‐deficient xenograft tumor tissues. We observed a slight increase in the abundance of γH2AX in FBL‐deficient xenograft tumors that increased to much higher levels after MMC treatment, correlating with a significant decrease in BRCA1 expression in tumor tissues (Fig 6D and E). This observation is in line with our studies, which showed a consistent correlation between FBL and BRCA1 nuclear levels through IHC assays using the same 40 colon cancer samples, as well as the correlation analysis obtained in the GEPIA database (Fig EV5D and E). The TdT‐mediated dUTP nick‐end labeling (TUNEL) immunostaining revealed a significant increase in cell apoptosis in the tumors of the FBL KD group following treatment with MMC, compared to the control group (Fig 6F and G). These results indicate that FBL KD in HCT116 cells not only inhibits cell growth but also confers hypersensitivity to MMC treatment in a xenograft model. Taken together, reduced FBL expression suppresses tumorigenesis and strongly sensitizes xenograft tumors to MMC‐induced DNA damage, leading to massive tumor cell death. These results suggest that FBL may be a potential therapeutic target for sensitizing colon cancer cells to chemotherapy.
Figure 6. FBL deficiency sensitizes tumors to DNA‐damaging drugs.
-
AMice were injected subcutaneously with 1 × 107 control (shNC) or FBL knockdown (shFBL) HCT116 cells and then treated with 1 mg/kg of MMC or PBS every other day starting on the days when tumors reached a similar size (about 50 mm3) in all treatment conditions for 8 days. Representative images of xenograft tumors are shown (n = 6/group). Data are shown as the mean ± SD. Quantification of tumor volume is shown on the right. The statistical significance was determined using one‐way ANOVA. **P < 0.01 and ***P < 0.001.
-
BThe body weight of the mice was measured over the treatment period. Graphs represent the mean ± SD (n = 6 mice in each group). The statistical significance was determined using one‐way ANOVA.
-
CHE staining of tumor tissues. Scale bar, 50 μm.
-
D, ERepresentative images of immunohistochemical staining showing BRCA1 and γH2AX expression levels in MMC‐treated and untreated xenograft tumors driven by control or FBL KD HCT116 cells. The (E) and panel display quantification of the (D) panel: showing BRCA1 and γH2AX expression levels. Scale bar, 25 μm. Graphs represent the mean ± SD (n = 3 mice in each group). The statistical significance was determined using one‐way ANOVA. *P < 0.05, **P < 0.01, and ***P < 0.001.
-
F, GImmunofluorescence for TUNEL staining of tumor tissues. Quantification of the percentage of apoptotic cells is shown on the (G) panel. Scale bar, 50 μm. Graphs represent the mean ± SD (n = 3 mice in each group). The statistical significance was determined using one‐way ANOVA. *P < 0.05 and ***P < 0.001.
-
HWorking model of the critical role of FBL in BRCA1 expression and oncogenic function through binding and modulating the subcellular localization of YBX1.
Source data are available online for this figure.
Discussion
In this study, we demonstrate that FBL plays a critical role not only in regulating tumorigenesis but also in DNA damage repair. Decreased FBL expression enhances cancer cells' sensitivity to DNA‐damaging drugs by impairing DNA damage repair. Using IP‐MS analysis, we identified YBX1 as a novel FBL‐binding protein. Mechanistically, FBL facilitates MMC‐induced YBX1 nuclear accumulation by interacting with YBX1, which enhances the transcriptional activation of BRCA1 via YBX1 and accelerates the DNA repair of tumor cells, inducing resistance to MMC (Fig 6H). Notably, these functions of FBL are independent of its methyltransferase activity.
Our analysis, utilizing the TCGA database, revealed that FBL is highly expressed in various cancer tissues. Moreover, we found a strong correlation between elevated FBL expression and advanced histological stages as well as worse survival outcomes in patients with lung and colon cancer. Our findings further demonstrated that FBL mediates HR repair and FBL depletion confers hypersensitivity to DNA‐damaging agents, particularly DNA crosslinker agents like cisplatin and MMC. We speculate that FBL likely participates in repairing interstrand crosslinks (ICLs) through HR, which is mediated by YBX1. In previous studies, YBX1 has been shown to drive drug resistance to cisplatin and MMC in cancer cells via its transcriptional activation (Kuwano et al, 2002, 2004; Kohno et al, 2003). Moreover, our study demonstrated that FBL knockdown led to the downregulation of MDR1 and BRCA1 expressions, similar to YBX1. It is noteworthy that YBX1 is involved in repairing ICL‐induced damage, but not damage induced by doxorubicin, camptothecin, or etoposide (Ohga et al, 1996). Based on our findings, we propose that FBL is closely related to YBX1 in DNA damage repair. Furthermore, increased resistance to DNA crosslinker agents may have potential implications in cancer therapy for patients with overexpressed FBL.
YBX1 plays a major role as a transcription factor for various drug resistance‐related genes, as well as a non‐canonical DNA repair factor participating in different DNA repair mechanisms (Hasegawa et al, 1991; Lasham et al, 2003; Gaudreault et al, 2004; En‐Nia et al, 2005; Eliseeva et al, 2012). Marchesini et al (2017) reported that YBX1 nuclear localization and interaction with the splicing factor U2AF65 promoted mRNA processing and the stabilization of transcripts involved in homologous recombination following DNA damage. Previous studies have shown that YBX1 can be phosphorylated at multiple sites in response to DNA damage, which facilitates its nuclear translocation and interaction with other proteins (Ohga et al, 1996; Koike et al, 1997; Shibahara et al, 2004; Kuwano et al, 2019). Moreover, we have also observed that partial FBL undergoes relocalization from the nucleolus to the nucleus upon MMC treatment, which creates the spatiotemporal possibility for the FBL‐YBX1 interaction. Consequently, the MMC‐induced phosphorylation of YBX1 facilitates its translocation to the nucleus, thereby likely promoting the nuclear association between FBL and YBX1. Furthermore, the phosphorylation of YBX1 may potentially regulate its affinity for FBL. Notably, the association between FBL and YBX1 contributes to the transcriptional activation of target genes, including HR genes through the involvement of YBX1. Consistent with the view that YBX1 serves as an important player and coordinator in the maintenance of genome integrity (Lyabin et al, 2014), our results demonstrate that FBL‐mediated YBX1 nuclear translocation is involved in the HR repair pathway. YBX1 downregulation decreased the efficiency of HR repair during DNA damage to an extent similar to that of FBL depletion. Combined with the similar loss‐of‐function influence of FBL and YBX1 on cells, our data indicate that the activation of the FBL/YBX1 signaling axis upon DNA damage regulates the transcription of genes directly involved in response to DNA damage and contributes to the mechanisms that promote the resistance of cancer cells to DNA‐damaging agents.
At the mechanistic level, FBL promotes cancer cell proliferation and mediates drug resistance to DNA damage in a dose‐dependent manner, partly by modulating YBX1's nuclear translocation and facilitating its binding to the BRCA1 promoter by retaining it in the nucleus to promote the transcription and expression of BRCA1, thereby increasing the efficiency of HR repair in response to DNA damage. YBX1 is a multifunctional DNA/RNA‐binding protein involved in various processes, such as translational repression, RNA stabilization, mRNA splicing, DNA repair, and transcription regulation (Kim et al, 2013). Recent evidence suggests that RNA plays a crucial role in DNA damage repair through HR. YBX1 may also contribute to this process through its interactions with RNA. However, in our proposed mechanism, RNA does not impact FBL‐mediated HR repair, as the interaction between FBL and YBX1 was not mediated by DNA or RNA (Fig EV3B). The specific role of YBX1 in RNA‐mediated DNA repair remains speculative and requires further investigation. Previous studies have shown that YBX1 binds to promoters containing a Y‐box sequence (5′‐CTGATTGGCCAA‐3′, an inverted CCAAT sequence), such as MDR1 or c‐Myc, to promote transcriptional activation (Morrow et al, 1994; Ohga et al, 1996, 1998; Dolfini & Mantovani, 2013). In this study, we found that the promoter of BRCA1 contains several core sequences of Y‐box (ATTGG underlined in Fig 5G) and that YBX1 directly binds to the loci of BRCA1 promoters by ChIP–qPCR. Therefore, BRCA1 is likely the executor or effector of FBL during DNA damage repair. We also observed no significant effect on protein expressions of RAD51 and RPA1/2 (Figs 5E and EV5A), suggesting a distinct underlying mechanism. Notably, RAD51 foci remains unchanged in FBL knockdown cells treated with MMC. This could be explained by an alternative mechanism where PALB2 is recruited to single‐stranded DNA through RNF168, in the absence of sufficient BRCA1 protein. This PALB2 recruitment subsequently facilitates RAD51 recruitment, serving as a backup mechanism for RAD51 loading (Zong et al, 2019). Overall, our results further enrich our understanding of the function of FBL, not only in oncogenesis but also in DNA damage repair.
BRCA1 is a well‐known nuclear protein that is regulated in the cell cycle. Our study investigated the impact of FBL on the cell cycle and found that downregulation of FBL resulted in a slight and insignificant reduction of the S phase in asynchronous shFBL‐HCT116 cells compared to the control group. Upon synchronizing shFBL‐HCT116 and control cells, we observed that BRCA1 expression remained significantly decreased in FBL knockdown cells, even when there was no significant difference in the S phase between FBL knockdown and control cells (Fig EV5F). Therefore, there might be two possible mechanisms underlying FBL's regulation of BRCA1 downregulation. Firstly, it may be partially due to the slight reduction of the S‐phase cells. More importantly, it is due to FBL modulating the binding of YBX1 to the BRCA1 promoter.
Numerous studies have established a link between rRNA‐modifying enzymes, such as METTL3, METTL5, and METTL16, and various cancers (Rong et al, 2020; Zhang et al, 2020; Su et al, 2022). As an rRNA methyltransferase, FBL is upregulated in several human cancers, including breast cancer, hepatocellular carcinoma, esophageal squamous cell carcinoma, and prostatic neoplasia (Koh et al, 2011; Marcel et al, 2013; Jin et al, 2021). While previous studies have primarily attributed FBL's role in cancer progression to its methyltransferase activity (Basu et al, 2011; Koh et al, 2011; Marcel et al, 2013), our findings demonstrate that FBL can promote cancer proliferation and increase sensitivity to DNA‐damaging drugs independently of its methyltransferase activity. Notably, following MMC treatment, FBL localizes in the nucleoplasm and nucleolus, which highlights the importance of considering additional functions of FBL in the nucleoplasm. Our study has identified a novel mechanism of FBL‐mediated transcriptional regulation of a key DNA damage repair factor, BRCA1, via YBX1. Furthermore, depletion of FBL significantly inhibits xenograft tumor growth in mice treated with MMC, accompanied by decreased BRCA1 expression in tumor tissues, indicating that FBL may hold therapeutic potential as a target for cancer treatment.
In conclusion, our study has uncovered a significant link between FBL/YBX1/BRCA1 during DNA damage repair in colon cancer. Specifically, we have demonstrated that FBL contributes to DNA damage repair by transcriptionally regulating BRCA1 expression via YBX1. These findings suggest that targeting this pathway could represent a promising strategy to enhance the efficacy of DNA‐damaging agents for colon cancer therapy. Notably, the GAR domain of FBL plays a critical role in its methylation‐independent activity, and the development of effective inhibitors targeting this domain or the FBL‐YBX1 interaction holds great therapeutic potential for cancer treatment.
Materials and Methods
Cell culture and tissue
HCT116, A549, and HEK293T cells were cultured in DMEM (GIBCO) supplemented with 10% FBS (GIBCO), 100 U/ml penicillin, and 100 μg/ml streptomycin, at 37°C with 5% (v/v) CO2 in a humidified incubator. For the study, 40 pairs of lung cancer and para‐cancerous tissues, as well as 40 pairs of colon cancer and adjacent tissues, were collected from the Department of Surgery at the Affiliated Hospital of Hebei University. Informed consent was obtained from each patient before the collection of the tissues. The study protocol was approved by the hospital's institutional ethical review committee (HDFY‐LL‐2022‐020).
To construct expression plasmids for GFP‐FBL, YBX1‐GFP, GST‐YBX1, SFB‐FBL, SFB‐YBX1, FBL mutants (SFB‐FBL‐E191A/D236A, GFP‐FBL‐E191A/D236A, FBL‐GAR‐GFP, and GFP‐FBL‐MTase) and YBX1 mutants (YBX1‐▵1‐50‐GFP, YBX1‐▵51‐129‐GFP, and YBX1‐▵130‐324‐GFP), the full‐length FBL and YBX1 were amplified separately from HCT116 cDNAs and inserted into the pEGFP‐C1, pEGFP‐N1, pGEX‐H‐6T‐1 (with GST tag and 6 × His tag), or SFB (with S‐Flag‐SBP tag) vector. The plasmids were then transformed into E. coli DH5α chemically competent cells. The primers used are listed in Table EV2.
Antibodies used in this study include the following: anti‐FBL (GeneTex, GTX24566), anti‐YBX1 (Abcam, ab76149), anti‐GAPDH (Proteintech, 60004‐1‐Ig), anti‐FLAG (Sigma, F1804), anti‐GFP (Proteintech, 50430‐2‐AP), anti‐Histone H3 (Proteintech, 17168‐1‐AP), anti‐γH2AX (Novus, NB100‐384), anti‐BRCA1 (Santa Cruz, sc‐6954), anti‐RAD51 (Novus, NB‐100‐148), anti‐GST (Sigma, 13‐6700), anti‐RPA1 (Abclonal, A3367), anti‐RPA2 (Abclonal, A2189), anti‐RIF1 (Bethyl, A300‐569A), anti‐cyclin B (CST, 12231), and anti‐β‐actin (Abclonal, AC006).
To construct the shRNA vectors targeting the human FBL gene, shRNA sequences and a scrambled sequence were individually cloned into pLKO.1‐Puro vectors using the AgeI and EcoRI sites. The shRNA sequences used were as follows: shRNA‐1# F‐5′‐CCGGCCTTGAGCCATATGAAAGAGACTCGAGTCTCTTTCATATGGCTCAAGGTTTTTG‐3′, R‐5′‐AATTCAAAAACCTTGAGCCATATGAAAGAGACTCGAGTCTCTTTCATATGGCTCAAGG‐3′ and shRNA‐2# F‐5'‐CCGGCTGTCAGGATTGCGAGAGATGCTCGAGCATCTCTCGCAATCCTGACAGTTTTTG‐3', R‐5′‐AATTCAAAAACTGTCAGGATTGCGAGAGATGCTCGAGCATCTCTCGCAATCCTGACAG‐3'. Additionally, siRNA sequences generated to target human FBL gene were 1#: 5′‐GAUGUGUGUUGAUACUGUUTT‐3′, 2#: 5′‐CUGUCAGGAUUGCGAGAGATT‐3′; and siRNA sequences generated to target human YBX1 gene were 1#: 5′‐GUUCCAGUUCAAGGCAGUA‐3′, 2#: 5′‐CGGCAAUGAAGAAGAUAAATT‐3′. siRNA sequences generated to target human RIF1 gene were 5′‐AGACGGUGCUCUAUUGUUATT‐3′, and siRNA sequences generated to target human BRCA1 gene were 5′‐CUAGAAAUCUGUUGCUAUGTT‐3′.
shRNA and lentivirus‐mediated knockdown
Lentiviruses were generated in HEK293T cells using Lipofectamine 2000‐mediated cotransfection of lentiviral‐based shRNA plasmids and the psPAX2 (packaging) and pMD2.G (envelope) plasmids. Forty‐eight hours post‐transfection, the constructed viral vector was harvested, purified, and concentrated, and stored at −80°C for subsequent experiments. HCT116/A549 cells were transduced with viruses in media supplemented with polybrene (10 μg/ml). Cells were incubated 24 h with viruses and then cultured in fresh media for 24 h, and then cells were cultured in media supplemented with 1 μg/ml puromycin for 3 weeks, and then screened for FBL expression by western blotting.
Colony formation assay
Cell viability of normal (shNC) or FBL‐depleted (shFBL) HCT116/A549 cells was assessed using colony‐forming assays. In brief, 1,000 cells per condition were plated into a six‐well plate. After 7–10 days, the colonies were fixed with methanol, stained using methylene blue in methanol, washed extensively with PBS, and counted. The surviving cell fractions were calculated by comparing the numbers of colonies formed in the irradiated cultures with those in the untreated control.
Cell treatment and cell viability
To assess the sensitivity of cells to various drug treatments, 5 × 103 transfected normal (shNC) or FBL‐depleted (shFBL) HCT116 cells per well (six replicates) were plated in 96‐well plates with 100 μl of fresh medium. After 12 h, the cells were treated with different drugs and cultured for 4 days. Subsequently, 10 μl of CCK8 was added to each well and incubated for 2 h. The absorbance at 450 nm was recorded using a 96‐well plate reader.
Immunofluorescence (IF)
Cells cultured on coverslips were fixed with 4% paraformaldehyde for 10 min at room temperature and then permeabilized with 0.5% Triton X‐100 at RT for 10 min. Cells were blocked with 5% BSA in PBS and then incubated with primary antibodies at room temperature for 1 h followed by incubation with fluorescence‐labeled secondary antibodies for another hour. Finally, the coverslips were mounted with DAPI and visualized by fluorescence microscopy. EdU incorporation analysis was performed using Click‐iT™ EdU Alexa Fluor 488 in vitro kit (Invitrogen, C10337) according to the manufacturer's protocol.
NHEJ/HR in vivo reporter assays
Double‐strand break repair efficiency was assessed using cell‐based reporter systems, as previously described by Cui et al (2021). DR‐GFP‐U2OS cells and EJ5‐GFP‐U2OS cells were transfected with siNC, siRIF1 (a positive control for NHEJ pathway), siBRCA1 (a positive control for HR pathway), siFBL, or siYBX1 RNAs and then with either JS‐20 (SceI) or empty vectors. After 48 h, 1 × 105 cells were analyzed for GFP‐positive cells using flow cytometry to determine the efficiency of homologous recombination (HR) and NHEJ repair mechanisms. In the flow cytometry figures, the Y‐axis represents side‐scattered light (SSC) to determine the granularity of cells, while the X‐axis represents the GFP signal intensity of cells. The GFP‐positive populations were gated to distinguish a group of cells with positive signals, and uninduced cells were included as negative controls. The results presented represent the mean value of triplicated replications in each experiment.
The establishment of stable cell lines and tandem affinity purification
To establish stable cell lines expressing epitope‐tagged proteins, HCT116 cells were transfected with plasmids encoding SFB‐FBL. After 24 h, the cells were split at a 1,000:1 ratio and cultured in medium containing selection of antibiotics for 3 weeks. Antibiotic‐resistant colonies were isolated and screened by western blotting for expression of SFB‐FBL proteins. HCT116 cells stably expressing SFB‐tagged FBL were collected from 50 10 cm culture dishes and then lyzed with NETN300 buffer (50 mM Tris–HCl pH 8.0, 300 mM NaCl, 0.5 mM EDTA, and 0.5% NP‐40) for 20 min on ice. The supernatants were diluted with an equal volume of NETN100 buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 0.5 mM EDTA, and 0.5% NP‐40) and incubated with streptavidin‐conjugated beads at 4°C for 2 h. The beads were washed three times with NETN100 buffer and eluted with saturating 2 mg/ml biotin (Sigma) in NETN100 buffer for 30 min at 4°C. The eluants were incubated with S‐protein agarose beads (Millipore) for 1 h at 4°C. The beads were washed three times with NETN100 buffer, and proteins bound to S‐beads were eluted by SDS‐loading buffer and subject to SDS–PAGE. The entire protein band (less than 1 cm) was excised and analyzed by mass spectrometry. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2022) partner repository with the dataset identifier PXD034434.
Coimmunoprecipitation and western blotting
Cells were harvested and lyzed with NETN300 buffers for 10 min on ice. The supernatants were diluted with the same volume of NETN100 and incubated with streptavidin‐conjugated beads or 2 μg of indicated antibodies and 40 μl protein A Sepharose beads for 2 h at 4°C. The beads were rinsed three times with NETN100 buffers. The bound protein was eluted using SDS‐loading buffers and then separated on SDS–PAGE. All western blotting experiments were conducted following standard procedures.
GST fusion protein expression and purification
GST fusion proteins were produced in E. coli BL21 DE3‐competent cells. The cells were induced and then collected by centrifugation at 4,000 g for 10 min at 4°C. They were then mixed with lysis buffers (25 mM Tris–HCl pH 8.0, 500 mM NaCl, and 5% glycerol), and supplied with 2 mM PMSF and 2 mM β‐mercaptoethanol as protease inhibitor A and reducing agents, respectively. The fragmented cells were obtained by sonication and then clarified by centrifugation at 15,000 g for 30 min at 4°C. The supernatant was used for the Ni‐NTA purification method, as described previously. Elution was performed using imidazole and 25 mM‐Tris–HCl, pH 8.0, 500 mM NaCl, 5% glycerol, 2 mM PMSF, and 2 mM β‐mercaptoethanol. All purification steps were carried out at 4°C to minimize proteolysis.
GST pull‐down assay
GST fusion proteins were produced in E. coli, while SFB‐FBL was expressed in HCT116 cells. Purified GST fusion proteins (GST‐YBX1) were incubated with SFB‐labeled FBL (SFB‐FBL) and streptavidin beads for 2 h at 4°C. The beads were washed four times with NETN100 buffer before the samples were boiled in SDS sample buffer. The eluted samples were then analyzed using western blotting with an anti‐GST antibody.
Separation of cytosolic and nuclear proteins
The cells were harvested at the indicated time points after relevant treatment and washed twice with phosphate‐buffered saline (PBS). The cell pellets were then suspended in the NETN300 lysis buffer (50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 0.5 mM EDTA, and 0.5% NP‐40) and centrifuged to obtain the soluble component, which was as the total extracts. For cytosolic extracts, the cells were lyzed with cytosolic extraction buffers (10 mM Tris–HCl, pH 8.0, 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, and 1% NP‐40) and incubated on ice for 10 min. After centrifugation, the supernatants were collected. Pellets were washed thrice with ice‐cold PBS and then dissolved in nuclear extraction buffers (10 mM Tris–HCl, pH 8.0, 420 mM NaCl, 5 mM MgCl2, 1% NP‐40, and benzonase nuclease) for 5 min at room temperature, followed by incubation on ice for 10 min. After centrifugation, the supernatants were collected and used as nuclear extracts.
Dual‐luciferase activity assays
Cells were seeded onto 12‐well plates and incubated overnight. Each promoter construct was transfected into subconfluent (80–90%) monolayer cells using lipofectamine reagent Lipofectamine™ 2000 (Invitrogen) following the manufacturer's instructions. To account for variations in transfection efficiency, 100 ng of 245‐CV045 plasmids expressing Renilla luciferase were cotransfected into each well as an internal control. Luciferase activity assays were performed using the Dual Luciferase Reporter Gene Assay Kit (Thermo Fisher, 16184) as per the manufacturer's protocol.
RNA sequence (RNA‐seq)
shNC or shFBL HCT116 cells were treated with 5 μM MMC and incubated overnight in a humidified atmosphere of 5% CO2 at 37°C. Cell samples were collected, added into Trizol reagents (Invitrogen), and then submitted to LC Bio Technology Co, Ltd (Hangzhou, China) for the subsequent mRNA library construction and sequencing.
Reverse transcription and quantitative PCR (qPCR)
Total RNA was extracted using Trizol (Invitrogen) and utilized for first‐strand cDNA synthesis with Superscript II reverse transcriptase (Invitrogen). Quantitative PCR was conducted using Fast SYBR™ green master mix (Thermo Fisher) in CFX96™ Real‐time systems (BIO‐RAD), with GAPDH mRNA serving as the normalizing control. The mean value was calculated from three independent experiments. The primers for qPCR reactions are listed in Table EV3.
Chromatin immunoprecipitation qPCR (ChIP–qPCR)
To perform an individual ChIP assay, 5 × 106 HCT116 cells were fixed in 1% formaldehyde at room temperature for 10 min and subsequently quenched by 0.125 M glycine for 5 min. Chromatin DNA was then isolated and sonicated to a length ranging from 200 to 1,000 bp using a Bioruptor Pico sonication device. The sheared chromatin was immunoprecipitated at 4°C overnight with anti‐YBX1 antibody, while a homologous IgG served as the isotype control. The coprecipitated DNA was purified using phenol–chloroform and subsequently subjected to qPCR (Yao et al, 2020). Specific primers for ChIP–qPCR are listed in Table EV4.
Immunohistochemical analysis (IHC) and TdT‐mediated dUTP‐digoxigenin nick end labeling (TUNEL) staining
Tumors were fixed overnight in 4% paraformaldehyde at room temperature, transferred to 70% ethanol, and embedded in paraffin. Four micrometer sections were dewaxed in xylene, and sequentially rehydrated in ethanol (100, 95, 80, 70, and 50%) and PBS buffers. For FBL/γH2AX/BRCA1 immunohistochemistry, the sections were blocked with 5% goat serum in PBS buffers, then incubated overnight with primary antibodies to γH2AX/FBL/BRCA1 at 4°C, washed three times with 1 × PBS, and incubated with secondary antibodies. The sections were washed three times with 1 × PBS and stained with DAB and hematoxylin (for DNA staining) according to standard protocols. To evaluate the frequency of apoptosis, the tissue was analyzed using a TdT‐mediated dUTP‐digoxigenin nick end labeling (TUNEL) method, and the staining was performed according to manufacturer's instructions (Invitrogen, C10245).
Establishment of xenograft models
Five‐week‐old male BALB/c nude mice were obtained from Beijing Vital River Laboratory Animal Center and raised under specific pathogen‐free conditions. The animal protocol described below has been reviewed and approved by the Animal Ethical and Welfare Committee (AEWC IACUC‐2020XG013). A total of 1 × 107 normal (shNC) or FBL‐depleted (shFBL) HCT116 cells were inoculated subcutaneously into the dorsal flank of the nude mice in 100 μl FBS. We started MMC treatment after the tumors reached a similar size (about 50 mm3) in all treatment conditions. shNC or shFBL HCT116 cell‐bearing mice were randomly divided into control and mitomycin C treatment groups. 1 mg/kg of mitomycin C dissolved in PBS was administered via intraperitoneal injection every other day for 8 days. No animals died during the experimental period. The weight of the mice was monitored daily, and the tumor size was estimated from the measurements of the longest diameter across the tumor and the corresponding perpendicular diameter.
Cell cycle analysis
HCT116 cells were synchronized at the G1/S border using a thymidine double‐block protocol. Specially, the cells were treated with 2 mM thymidine for 16 h and then released for 8 h in fresh medium before the second block was performed for another 16 h with 2 mM thymidine. After the second block, the cells were washed three times in phosphate buffer saline (PBS) and either collected at 0 h after treatment or released in fresh medium for different time periods. At each time point, both attached and unattached cells were harvested and used for FACS and western blot analyses. The contents of each dish were incubated for 2 min at room temperature in 1 ml of trypsin–EDTA solution [0.25% trypsin/0.1% EDTA in Hanks' balanced salt solution (HBSS) without Ca2+ and Mg2+; Mediatech]. The trypsinized cells were then washed in 1 ml of growth medium (McCoy's 5A, 10% FBS) and resuspended in 300 μl of ice‐cold PBS (without Ca2+ and Mg2+). After 900 μl of ice‐cold 100% ethanol was added dropwise, the cells were fixed at 4°C for at least 16 h. The fixed cells were pelleted, resuspended in 300 μl of PI staining solution (50 μg/ml propidium iodide, 100 units/ml RNase A, and 0.1% glucose in PBS without Ca2+ and Mg2+), incubated for at least 1 h at room temperature, and analyzed on a FACScan flow cytometer (Beckman). For western analysis, the cells were lyzed in low‐salt Nonidet P‐40 buffer (10 mM HEPES pH 7.6, 250 mM NaCl, 0.1% Nonidet P‐40, 5 mM EDTA, and 10% glycerol) supplemented with 1 mM dithiothreitol, protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride and BMB protease inhibitor pellet), and phosphatase inhibitors (10 mM β‐glycerophosphate, 5 mM NaF, and 0.1 mM NaVO4). Western analyses were conducted by enhanced chemiluminescence using lysate for BRCA1, cyclin B, FBL, and β‐actin immunoblots.
Statistical analysis
All data in bar and line graphs are presented as mean ± SD of at least three independent experiments. All data were analyzed by the two‐tailed unrepaired student's t‐test or one‐way ANOVA using GraphPad Prism version 7.0. *P < 0.05, **P < 0.01, ***P < 0.001, and ns denotes not significant.
Author contributions
Xiaorui Sun: Conceptualization; data curation; formal analysis; validation; investigation; methodology; writing – original draft; writing – review and editing. Congwen Gao: Data curation; validation; methodology; writing – review and editing. Xin Xu: Validation; investigation; methodology. Mengyuan Li: Validation; investigation; methodology. Xinhua Zhao: Validation; investigation; methodology. Yanan Wang: Validation; investigation; methodology. Yun Wang: Validation; investigation; methodology. Shun Zhang: Validation; investigation; methodology. Zhenzhen Yan: Funding acquisition; validation; investigation; methodology; writing – original draft; writing – review and editing. Xiuhua Liu: Funding acquisition; validation; investigation; methodology. Chen Wu: Conceptualization; resources; formal analysis; supervision; funding acquisition; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We would like to thank the research center for DNA damage repair at Hebei University for providing us with technical assistance. This work was supported by National Natural Science Foundation of China (No. 32071277, No. 82002594, No. 32171295), Natural Science Foundation of Hebei Province (No. C2021201012), S&T Program of Hebei (No. 216Z2602G), Hebei Natural Science Foundation for Outstanding Young Scholars (No. H2020201017), and High‐level Talents Research Start‐up Project of Hebei University (No. 521000981352).
EMBO reports (2023) 24: e56230
Contributor Information
Zhenzhen Yan, Email: yanzhzh@hbu.edu.cn.
Xiuhua Liu, Email: liuxiuhua_2004@163.com.
Chen Wu, Email: wuchen@hbu.edu.cn.
Data availability
The MS data generated in this study have been deposited in the ProteomeXchange repository under the accession number PXD034434 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD034434). All sequencing data generated in this study have been deposited in NCBI's Gene Expression Omnibus under the accession number GSE205366 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205366). All the microscopy images have been uploaded to the BioImage Archive and can be accessed using the accession number S‐BSST1095 (https://www.ebi.ac.uk/biostudies/studies/S‐BSST1095?key=e043b993‐3be3‐4412‐a328‐a1d2090f43c7).
References
- Aris JP, Blobel G (1991) cDNA cloning and sequencing of human fibrillarin, a conserved nucleolar protein recognized by autoimmune antisera. Proc Natl Acad Sci USA 88: 931–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Das P, Chaudhuri S, Bevilacqua E, Andrews J, Barik S, Hatzoglou M, Komar AA, Mazumder B (2011) Requirement of rRNA methylation for 80S ribosome assembly on a cohort of cellular internal ribosome entry sites. Mol Cell Biol 31: 4482–4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommert KS, Effenberger M, Leich E, Küspert M, Murphy D, Langer C, Moll R, Janz S, Mottok A, Weissbach S et al (2012) The feed‐forward loop between YB‐1 and MYC is essential for multiple myeloma cell survival. Leukemia 27: 441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z‐J, Qin Y, Zhao S, Leung Peter CK, Chan W‐Y, Lu G, Li D, Dang Y, Zhang X, Wang X (2020) Long noncoding RNA HCP5 participates in premature ovarian insufficiency by transcriptionally regulating MSH5 and DNA damage repair via YB1. Nucleic Acids Res 48: 4480–4491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Xie R, Zhang X, Liu Y, Hu Y, Li Y, Liu X, Yu X, Wu C (2021) OGA is associated with deglycosylation of NONO and the KU complex during DNA damage repair. Cell Death Dis 12: 622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David E, McNeil JB, Basile V, Pearlman R (1997) An unusual fibrillarin gene and protein: structure and functional implications. Mol Biol Cell 8: 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolfini D, Mantovani R (2013) Targeting the Y/CCAAT box in cancer: YB‐1 (YBX1) or NF‐Y? Cell Death Differ 20: 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliseeva IA, Kim ER, Guryanov SG, Ovchinnikov LP, Lyabin DN (2012) Y‐box‐binding protein 1 (YB‐1) and its functions. Biochemistry (Mosc) 76: 1402–1433 [DOI] [PubMed] [Google Scholar]
- En‐Nia A, Yilmaz E, Klinge U, Lovett DH, Stefanidis I, Mertens PR (2005) Transcription factor YB‐1 mediates DNA polymerase α gene expression. J Biol Chem 280: 7702–7711 [DOI] [PubMed] [Google Scholar]
- Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, Kriwacki RW, Pappu RV, Brangwynne CP (2016) Coexisting liquid phases underlie nucleolar subcompartments. Cell 165: 1686–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltánková V, Legartová S, Kozubek S, Hofer M, Bártová E (2013) DNA‐damage response in chromatin of ribosomal genes and the surrounding genome. Gene 522: 156–167 [DOI] [PubMed] [Google Scholar]
- Gaudreault I, Guay D, Lebel M (2004) YB‐1 promotes strand separation in vitro of duplex DNA containing either mispaired bases or cisplatin modifications, exhibits endonucleolytic activities and binds several DNA repair proteins. Nucleic Acids Res 32: 316–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Huang J (2019) DNA double‐strand break repair pathway choice: the fork in the road. Genome Instab Dis 1: 10–19 [Google Scholar]
- Jin X, Liu L, Wu J, Jin X, Yu G, Jia L, Wang F, Shi M, Lu H, Liu J et al (2021) A multi‐omics study delineates new molecular features and therapeutic targets for esophageal squamous cell carcinoma. Clin Transl Med 11: e538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ER, Selyutina AA, Buldakov IA, Evdokimova V, Ovchinnikov LP, Sorokin AV (2013) The proteolytic YB‐1 fragment interacts with DNA repair machinery and enhances survival during DNA damaging stress. Cell Cycle 12: 3791–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss‐László Z, Henry Y, Bachellerie JP, Caizergues‐Ferrer M, Kiss T (1996) Site‐specific ribose methylation of Preribosomal RNA: A novel function for small nucleolar RNAs. Cell 85: 1077–1088 [DOI] [PubMed] [Google Scholar]
- Koh CM, Gurel B, Sutcliffe S, Aryee MJ, Schultz D, Iwata T, Uemura M, Zeller KI, Anele U, Zheng Q et al (2011) Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am J Pathol 178: 1824–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno K, Izumi H, Uchiumi T, Ashizuka M, Kuwano M (2003) The pleiotropic functions of the Y‐box‐binding protein, YB‐1. Bioessays 25: 691–698 [DOI] [PubMed] [Google Scholar]
- Koike K, Uchiumi T, Ohga T, Toh S, Wada M, Kohno K, Kuwano M (1997) Nuclear translocation of the Y‐box binding protein by ultraviolet irradiation. FEBS Lett 417: 390–394 [DOI] [PubMed] [Google Scholar]
- Kuwano M, Uchiumi T, Hayakawa H, Ono M, Wada M, Izumi H, Kohno K (2002) The basic and clinical implications of ABC transporters, Y‐box‐binding protein‐1 (YB‐1) and angiogenesis‐related factors in human malignancies. Cancer Sci 94: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano M, Oda Y, Izumi H, Yang S‐J, Uchiumi T, Iwamoto Y, Toi M, Fujii T, Yamana H, Kinoshita H et al (2004) The role of nuclear Y‐box binding protein 1 as a global marker in drug resistance. Mol Cancer Ther 3: 1485–1492 [PubMed] [Google Scholar]
- Kuwano M, Shibata T, Watari K, Ono M (2019) Oncogenic Y‐box binding protein‐1 as an effective therapeutic target in drug‐resistant cancer. Cancer Sci 110: 1536–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa SL, Doetsch PW, Hamilton KK, Martin AM, Okenquist SA, Lenz J, Boss JM (1991) DNA binding properties of YB‐1 and dbpA: binding to doublestranded, single‐stranded, and abasic site containing DNAs. Nucleic Acids Res 19: 4915–4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasham A, Moloney S, Hale T, Homer C, Zhang YF, Murison JG, Braithwaite AW, Watson J (2003) The Y‐box‐binding protein, YB1, is a potential negative regulator of the p53 tumor suppressor. J Biol Chem 278: 35516–35523 [DOI] [PubMed] [Google Scholar]
- Liu Y, Lu L‐Y (2021) BRCA1: a key player at multiple stages of homologous recombination in DNA double‐strand break repair. Genome Instability & Disease 2: 164–174 [Google Scholar]
- Lu Y, Xu D, Peng J, Luo Z, Chen C, Chen Y, Chen H, Zheng M, Yin P, Wang Z (2019) HNF1A inhibition induces the resistance of pancreatic cancer cells to gemcitabine by targeting ABCB1. EBioMedicine 44: 403–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyabin DN, Eliseeva IA, Ovchinnikov LP (2014) YB‐1 protein: functions and regulation. Wiley Interdiscip Rev RNA 5: 95–110 [DOI] [PubMed] [Google Scholar]
- Marcel V, Ghayad SE, Belin S, Therizols G, Morel AP, Solano‐Gonzalez E, Vendrell JA, Hacot S, Mertani HC, Albaret MA et al (2013) p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 24: 318–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini M, Ogoti Y, Fiorini E, Aktas Samur A, Nezi L, D'Anca M, Storti P, Samur MK, Ganan‐Gomez I, Fulciniti MT et al (2017) ILF2 is a regulator of RNA splicing and DNA damage response in 1q21‐amplified multiple myeloma. Cancer Cell 32: 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow CS, Nakagawa M, Goldsmith ME, Madden MJ, Cowan KH (1994) Reversible transcriptional activation of mdr1 by sodium butyrate treatment of human colon cancer cells. J Biol Chem 269: 10739–10746 [PubMed] [Google Scholar]
- Ohga T, Koike K, Ono M, Makino Y, Itagaki Y, Tanimoto M, Kuwano M, Kohno K (1996) Role of the human Y box‐binding protein YB‐1 in cellular sensitivity to the DNA‐damaging agents cisplatin, mitomycin C, and ultraviolet light. Cancer Res 56: 4224–4228 [PubMed] [Google Scholar]
- Ohga T, Uchiumi T, Makino Y, Koike K, Wada M, Kuwano M, Kohno K (1998) Direct involvement of the Y‐box binding protein YB‐1 in genotoxic stress‐induced activation of the human multidrug resistance 1 gene. J Biol Chem 273: 5997–6000 [DOI] [PubMed] [Google Scholar]
- Panier S, Durocher D (2013) Push back to respond better: regulatory inhibition of the DNA double‐strand break response. Nat Rev Mol Cell Biol 14: 661–672 [DOI] [PubMed] [Google Scholar]
- Peddibhotla S, Wei Z, Papineni R, Lam MH, Rosen JM, Zhang P (2014) The DNA damage effector Chk1 kinase regulates Cdc14B nucleolar shuttling during cell cycle progression. Cell Cycle 10: 671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Bai J, Bandla C, Garcia‐Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks‐Zipper A, Eisenacher M et al (2022) The PRIDE database resources in 2022: a hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Res 50: D543–D552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Corona U, Sobol M, Rodriguez‐Zapata LC, Hozak P, Castano E (2015) Fibrillarin from Archaea to human. Biol Cell 107: 159–174 [DOI] [PubMed] [Google Scholar]
- Rong B, Zhang Q, Wan J, Xing S, Dai R, Li Y, Cai J, Xie J, Song Y, Chen J et al (2020) Ribosome 18S m6A methyltransferase METTL5 promotes translation initiation and breast cancer cell growth. Cell Rep 33: 108544 [DOI] [PubMed] [Google Scholar]
- Sangermano F, Delicato A, Calabrò V (2020) Y box binding protein 1 (YB‐1) oncoprotein at the hub of DNA proliferation, damage and cancer progression. Biochimie 179: 205–216 [DOI] [PubMed] [Google Scholar]
- Schimmang T, Tollervey D, Kern H, Frank R, Hurt EC (1989) A yeast nucleolar protein related to mammalian fibrillarin is associated with small nucleolar RNA and is essential for viability. EMBO J 8: 4015–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara K, Uchiumi T, Fukuda T, Kura S, Tominaga Y, Maehara Y, Kohno K, Nakabeppu Y, Tsuzuki T, Kuwano M (2004) Targeted disruption of one allele of the Y‐box binding protein‐1 (YB‐1) gene in mouse embryonic stem cells and increased sensitivity to cisplatin and mitomycin C. Cancer Sci 95: 348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubina MY, Musinova YR, Sheval EV (2016) Nucleolar methyltransferase fibrillarin: evolution of structure and functions. Biochemistry (Mosc) 81: 941–950 [DOI] [PubMed] [Google Scholar]
- Shubina MY, Musinova YR, Sheval EV (2018) Proliferation, cancer, and aging‐novel functions of the nucleolar methyltransferase fibrillarin? Cell Biol Int 42: 1463–1466 [DOI] [PubMed] [Google Scholar]
- Su H, Xu T, Ganapathy S, Shadfan M, Long M, Huang TH, Thompson I, Yuan ZM (2014) Elevated snoRNA biogenesis is essential in breast cancer. Oncogene 33: 1348–1358 [DOI] [PubMed] [Google Scholar]
- Su R, Dong L, Li Y, Gao M, He PC, Liu W, Wei J, Zhao Z, Gao L, Han L et al (2022) METTL16 exerts an m6A‐independent function to facilitate translation and tumorigenesis. Nat Cell Biol 24: 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsounas M, Sung P (2020) The antitumorigenic roles of BRCA1‐BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol 21: 284–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Boisvert D, Kim KK, Kim R, Kim SH (2014) Crystal structure of a fibrillarin homologue from Methanococcus jannaschii, a hyperthermophile, at 1.6 resolution. EMBO J 19: 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe‐Susaki K, Takada H, Enomoto K, Miwata K, Ishimine H, Intoh A, Ohtaka M, Nakanishi M, Sugino H, Asashima M et al (2014) Biosynthesis of ribosomal RNA in nucleoli regulates pluripotency and differentiation ability of pluripotent stem cells. Stem Cells 32: 3099–3111 [DOI] [PubMed] [Google Scholar]
- Yanagida M, Hayano T, Yamauchi Y, Shinkawa T, Natsume T, Isobe T, Takahashi N (2004) Human fibrillarin forms a sub‐complex with splicing factor 2‐associated p32, protein arginine methyltransferases, and tubulins alpha 3 and beta 1 that is independent of its association with preribosomal ribonucleoprotein complexes. J Biol Chem 279: 1607–1614 [DOI] [PubMed] [Google Scholar]
- Yao RW, Xu G, Wang Y, Shan L, Luan PF, Wang Y, Wu M, Yang LZ, Xing YH, Yang L et al (2019) Nascent pre‐rRNA sorting via phase separation drives the assembly of dense fibrillar components in the human nucleolus. Mol Cell 76: 767–783 [DOI] [PubMed] [Google Scholar]
- Yao X, Yu X, Lu L‐Y, Zhang F, Liu X, Cui Y, Wu C (2020) The RNF20/40 complex regulates p53‐dependent gene transcription and mRNA splicing. J Mol Cell Biol 12: 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen L, Peng D, Jiang A, He Y, Zeng Y, Xie C, Zhou H, Luo X, Liu H et al (2020) METTL3 and N6‐Methyladenosine promote homologous recombination‐mediated repair of DSBs by modulating DNA‐RNA hybrid accumulation. Mol Cell 79: 425–442 [DOI] [PubMed] [Google Scholar]
- Zong D, Adam S, Wang Y, Sasanuma H, Callén E, Murga M, Day A, Kruhlak MJ, Wong N, Munro M et al (2019) BRCA1 haploinsufficiency is masked by RNF168‐mediated chromatin ubiquitylation. Mol Cell 73: 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]