

SUMMARY
The spliceosome is a staggeringly complex machine comprising, in humans, 5 snRNAs and >150 proteins. We scaled haploid CRISPR-Cas9 base editing to target the entire human spliceosome and interrogated the mutants using the U2 snRNP/SF3b inhibitor, pladienolide B. Hypersensitive substitutions define functional sites in the U1/U2-containing A-complex but also in components that act as late as the second chemical step after SF3b is dissociated. Viable resistance substitutions map not only to the pladienolide B binding site but also to the G-patch domain of SUGP1, which lacks orthologs in yeast. We used these mutants and biochemical approaches to identify the spliceosomal disassemblase DHX15/hPrp43 as the ATPase ligand for SUGP1. These and other data support a model in which SUGP1 promotes splicing fidelity by triggering early spliceosome disassembly in response to kinetic blocks. Our approach provides a template for the analysis of essential cellular machines in humans.
eTOC blurb
Beusch et al. use base editing to interrogate the human spliceosome in haploid cells. Selection for resistance to the U2 snRNP inhibitor pladienolide B revealed mutations in the drug binding factors and in the early spliceosomal protein SUGP1. Further studies reveal the spliceosome disassemblase DHX15/hPrp43 to be the ligand of SUGP1.
INTRODUCTION
Pre-mRNA splicing is an essential step in eukaryotic gene expression. In addition to driving proteome diversity via alternative splicing5, splicing impacts RNA stability, and plays critical roles in RNA export and translation efficiency36. Splicing is also a major player in disease: a large fraction of single nucleotide polymorphisms associated with human disease impact splicing37, and many human cancers harbor driver mutations in components of the spliceosome itself4,70
There are four intron sequences important for splicing: the 5′ splice site (SS), the branchpoint (BP), the polypyrimidine tract (PPT), and 3′ splice site (Fig. 1). In humans, there is large variability in these sequences, which can have an enormous impact on splicing efficiency and regulation, enabling regulation by RNA binding proteins (RBPs). Pre-mRNA splicing proceeds via two transesterification reactions which are catalyzed by the spliceosome. Compared to the simplicity of the chemical steps, the spliceosome is staggeringly complex (Figure 1 A,B). Components include five small nuclear ribonucleoproteins (snRNPs – U1, U2, U4/U6, and U5) and numerous proteins that assemble onto the intron substrate and undergo several large rearrangements to form a catalytically active complex in which a U6 snRNA acts as an RNA catalyst67. In S. cerevisiae, from which much of our understanding has been developed, splicing of a single intron requires eight ATP-dependent steps and about 90 proteins. Human spliceosomes appear to contain about 60 additional proteins63.
Figure 1. CRISPR-Cas9 base editing screen targeting the spliceosome reveals several mutants sensitive or resistant to the small molecule spliceosome inhibitor pladienolide B.
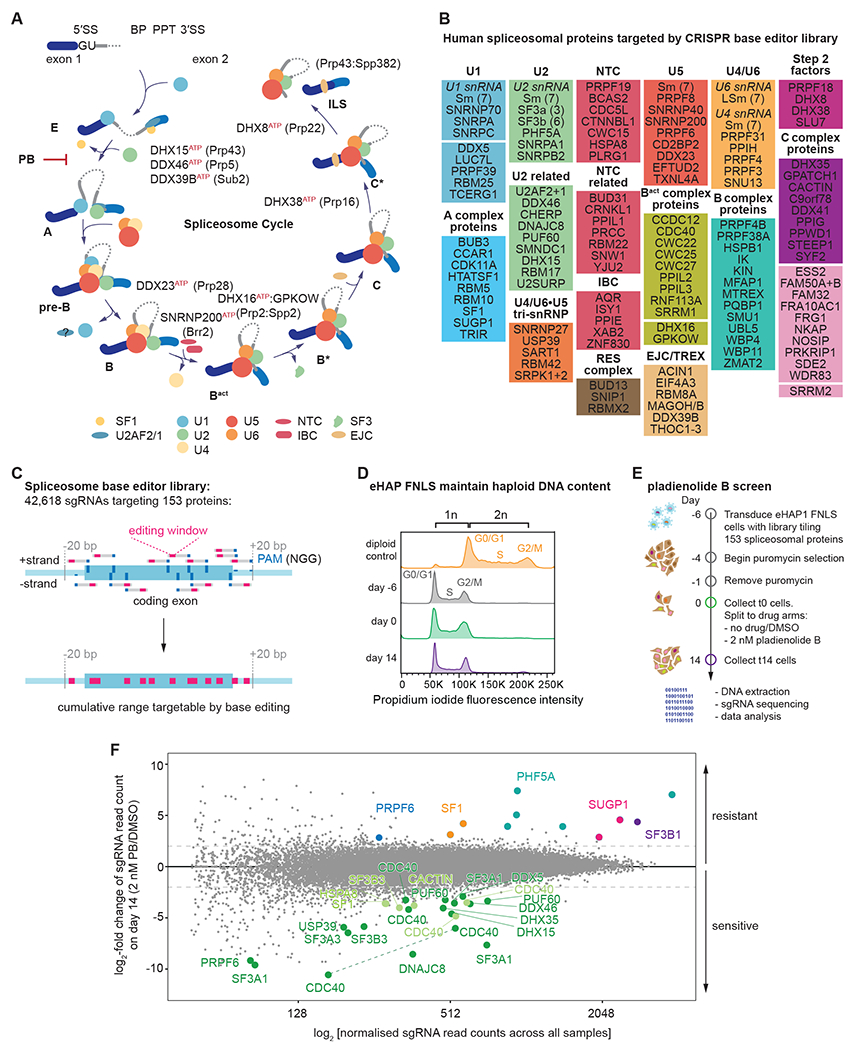
(A) Schematic of intron sequences required for splicing and the spliceosome cycle across the major assembly stages.
(B) List of spliceosomal genes targeted by the sgRNA library.
(C) Schematic of tiling sgRNA library. Every available PAM sequence (denoted in dark blue) on both strands of the genome is targeted across all coding exons.
(D) eHAP FNLS cell line can be maintained in a haploid state.
(E) Schematic of the pooled screen.
(F) Results of screen. MA-plot comparing day 14 of cells grown in presence or absence of 2 nM PB. For orientation lines indicate a log2-fold enrichment or depletion of two. sgRNA with strong sensitivity to PB are emphasized in green (dark green: p-adj < 0.05, light green: p-adj ≥ 0.05 but highly depleted). sgRNAs resulting in PB resistance are colored by protein target. For clarity, only data points for sgRNAs that passed the confirmation assay are shown. Dashed line: sgRNA targeting the same position and predicted to result in identical mutational outcome.
Initial intron recognition involves base-pairing between the 5’ end of U1 snRNA and the 5’ SS and recognition of the branchpoint, PPT, and 3’ SS by sequence-specific RNA binding proteins: Splicing Factor 1 (SF1/Msl5) recognizes the branchpoint sequence while the two subunits of U2AF recognize the PPT and the conserved AG dinucleotide at the 3’ splice site. This forms an early, or E, complex that is the precursor to the A complex in which U2 snRNP binds to the intron, base-pairing with sequences around the branchpoint (the branchpoint sequence), replacing SF1 and U2AF. A triple snRNP, containing base-paired U4/U6 snRNAs together with the U5 snRNA, then joins the complex to form the pre-B complex which converts to the B complex by the departure of U1. Activation of the spliceosome occurs via ATP-dependent rearrangements that expels the U4 snRNP and several proteins63, allowing the PRPF19/Prp19 complex (NTC) and NTC-related proteins (NTR) to join. This produces the Bact complex, in which U6 base-pairs with the 5’ splice site and U2 and U6 snRNAs base-pair to form the spliceosomal active site67. A component of U2 snRNP, the SF3 complex, which sequesters the U2-branchpoint helix away from the 5’ splice site, is then removed, allowing the U2-branchpoint helix to dock with the catalytic core. Association of additional proteins allow the chemical steps to proceed in the B* and C* catalytic complexes67. The ATPases DHX38/Prp16 and DHX8/Prp22 respectively remodel the active site after each chemical step63. Following mRNA release, the helicase DHX15/Prp43 disassembles the spliceosome41,62. Like other DEAH-box helicases, DHX15/Prp43 is activated by a cognate G-patch protein, TFIP11/Spp382/Ntr158.
Given the high variability in splicing signal sequences in humans, how the spliceosome distinguishes between cognate and noncognate sequences remains to be understood. A longstanding hypothesis suggests that spliceosome dynamics promote the fidelity of splicing through kinetic proofreading while also permitting substrate flexibility and regulation. Evidence supporting this model came from a genetic screen in S. cerevisiae in which missense mutations in the ATPase Prp16 were identified as suppressors of a mutation in the branchpoint adenosine sequence9. Subsequent studies demonstrated that mutant pre-mRNA substrates that assemble into spliceosomes, but are kinetically slow at either chemical step, trigger spliceosome disassembly prior to completion of the reaction, termed “discard”33,34,43,44,55. Failure to perform catalysis prior to ATP hydrolysis by Prp16 (step 1) or Prp22 (step 2) triggers disassembly by Prp43. These ATPases have been proposed to act as molecular timers for flux through the splicing pathway34. The yeast studies used mutant pre-mRNA substrates because their signals are always very close to the optimal consensus 28. Whether there are analogous or additional fidelity mechanisms that operate in animal cells is unknown.
The ability to perform forward genetic screens in haploid S. cerevisiae was critical for the studies on spliceosome fidelity outlined above as well as numerous other foundational studies. To adapt these methods to human cells, we describe here a strategy to mutagenize the spliceosome in fully haploid human cells by developing and deploying a CRISPR-Cas9 base editor sgRNA library that targets the entire human spliceosome. After mutagenesis, we interrogated the spliceosome using the small molecule inhibitor pladienolide B (PB), which targets U2 snRNP11,12,20,60,68. Validation and genomic sequencing revealed resistance mutations in SF3B1 and PHF5A in residues adjacent to the binding pocket. We mapped hypersensitive mutants to U2 snRNP components, but also to factors that act as late as the second chemical step, after SF3b has dissociated. Strikingly, we obtained resistance mutants in SUGP1, a spliceosomal G-patch protein of unknown function that lacks orthologs in yeast and is also a newly proposed tumor suppressor whose loss underpins the splicing changes induced by cancer-associated SF3B1 mutations1,39,72. Our resistance mutations in SUGP1 map in or adjacent to its G-patch motif and modulate splicing changes triggered by PB. We describe biochemical experiments that reveal the spliceosomal disassembly ATPase DHX15/hPrp43 to be the biologically relevant direct target of the SUGP1 G-patch domain. We propose a unified model in which SUGP1-DHX15 mediate disassembly of kinetically-slowed early splicing complexes. Our results demonstrate the feasibility and utility of the programmed generation of informative viable haploid alleles targeting a complex essential gene expression machine.
RESULTS
Large-scale mutagenesis of the human spliceosome
A major impediment to the study of the human spliceosome in vivo has been the inability to program point mutations in endogenous genes on a large scale. CRISPR-Cas9 technology now provides such opportunities2. We chose CRISPR-Cas9 base editing for a forward genetic screen of the spliceosome (Figure S1B). We first generated a monoclonal cell line expressing FNLS71, a cytosine base editor, in an eHAP17 haploid cell background (hereafter: eHAP FNLS). We maintained eHAP FNLS cells as haploid so that we could determine genotype-phenotype relationships (Figure 1D). Next, we assessed editing efficiency on a set of standard targets used previously71 (Figure S1C,D). eHAP FNLS cells demonstrated efficient editing (up to >90%) at expected positions within the editing window, which spans positions 3-8 [with position 21-23 being the protospacer adjacent motif (PAM)], and induced transversions (C > R editing) at high frequencies (>25%) in some cases.
We designed a single guide RNA (sgRNA) library targeting 153 human spliceosomal proteins (Figure 1A), which are reproducibly detected through mass spectrometry (MS), interaction studies, and/or visualized in structural biology studies (see Figure 1B, Table S1)52. As base editing outcomes are not fully predictable, to maximize mutagenesis we targeted every available NGG PAM sequence across all annotated exons plus 20 bp flanking intronic sequence (Figure 1C). Our library includes 42,618 sgRNAs targeting 42,650 sites, including 8,426 sgRNAs that target genomic sites but are predicted to be non-editing with FNLS, and an additional 1,000 guides that do not target genomic sites (non-targeting sgRNAs)16. The library can in principle mutagenize up to 30% of spliceosomal protein coding sequences (Figure S1E) and edits are predicted to result in missense mutations in >50% of cases with an additional 20% of edits predicted to impact protein sequence (Figure S1F, and Methods).
We cloned this library into lentiviral vectors that express the sgRNA and associate each to a unique barcode and produced virus for transduction (Figure S1A)6. After transduction into eHAP FNLS cells, we allow for editing and library selection during 6 days before splitting the selected pool into treatment arms (control/DMSO vs. 2 nM PB, which approximates its EC50). We cultured cells for two weeks while maintaining a representation of 500 cells per sgRNA. On days 0, 8 and 14 we isolated genomic DNA and amplified and sequenced the sgRNA inserts; we also did this for the input plasmid library (Figure 1E). Using the sgRNA-linked barcode, we randomly assigned sgRNAs to two sample populations. Depletion vs. enrichment of sgRNAs was then analysed for both time and treatment using DESeq240.
We then compared the abundances of guide sequences in the population of plasmid input control versus 14 days after transduction. Given that most spliceosomal proteins are essential for cell survival, we anticipated that a subset of induced mutations would be lethal or result in reduced viability. As expected, sgRNAs predicted to promote mutations with more severe consequences such as splice site mutations or creation of stop codons displayed the strongest depletion as a class, consistent with efficient and precise editing at many of those sites (Figure S1G). Conversely, guides predicted to be non-editing or to produce silent mutations were generally not depleted.
By comparing guide sequence abundances for day 14 for 2 nM PB versus the matched DMSO control, we observed that several guides were enriched or depleted upon compound treatment of the population (Figure 1F). For validation, we selected the sgRNAs showing statistically significant enrichment or depletion (LFC > |2|, padj < 0.05) between the 2 nM PB sample and its matched control sample on day 14. To this list we added a subset of sgRNAs with high average enrichment/depletion but which did not pass statistical significance (see Methods). To enrich for PB-specific phenotypes, we required that all guides depleted after PB treatment did not show depletion between t0 and t14 in untreated cells. This procedure yielded 19 candidate-enriched sgRNAs and 26 candidate-depleted sgRNAs. These sgRNAs and three non-targeting sgRNAs were then subjected to an arrayed dual-color competition assay in which cells transduced with virus encoding a non-targeting sgRNA or transduced with a candidate-depleted or -enriched sgRNA were marked with distinct fluorescent proteins, respectively (Figure 2A). The assay confirmed the response to PB treatment for 23/26 of the candidate-depleted sgRNAs and 11/19 of the candidate-enriched sgRNAs. Except for a sgRNA targeting SF3B1 and another targeting PRPF6, only sgRNAs found to be statistically significantly enriched validated in the confirmation assay, supporting the utility of the statistical approach used (Figure 2B, S2A).
Figure 2. Pladienolide B sensitive mutations occur predominantly in early spliceosomal complexes.
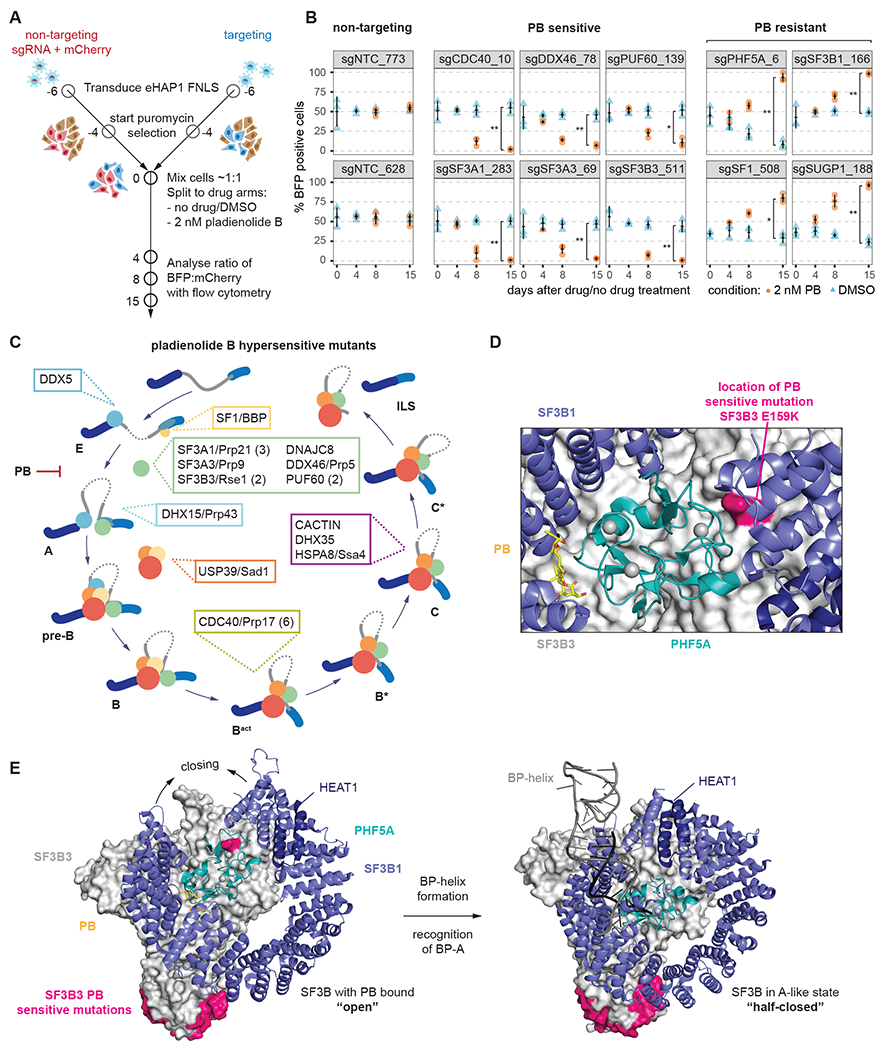
(A) Schematic of the arrayed confirmation assay.
(B) Individual sgRNAs and their performance in the confirmation assay. sgRNAs are grouped by category they were found in in the primary screen. Measurements are from three independent transductions (n=3). *; **; ***: Student’s t-Test (paired) P value < 0.05, 0.01, 0.001, respectively.
(C) Assignment of proteins targeted by sgRNAs conferring hypersensitivity to PB to the spliceosome cycle. If multiple sgRNAs are found for a protein, their number is given in parenthesis.
(D) Close-up of location of PB-sensitive SF3A1 G159K mutation plotted on the structure of SF3b bound to PB. It lies at the interface of SF3B3 (green), SF3B1 (violet) and PHF5A (teal). Zinc ions are shown as grey spheres. (PDB: 6EN4)
(E) Comparison of SF3B3, SF3B1 and PHF5A in the structure of SF3b bound to PB and the Alike complex. Locations of PB-sensitive mutations are marked in magenta and zinc ions are shown as grey spheres. (PDB: 6EN4, 7Q4O)
Pladienolide B hypersensitive mutations identify functional spliceosomal residues
Given the number of sgRNAs only depleted upon compound treatment, we determined the genomic consequences of base editing. We transduced eHAP FNLS with lentivirus carrying individual sgRNAs, grew cells for six days, isolated genomic DNA, amplified the edited locus and subjected the amplicons to deep sequencing (Table S2).
Guides presumably must edit efficiently to produce a depletion phenotype. Amplicon sequence confirmed this expectation: all sgRNAs displayed high/substantial rates of editing (median = 57.7% for C > T within positions 3 to 8, Figure S2B) leading to amino acid changes that become depleted in the presence of PB (with a median of 82% of the sequences carrying an amino acid change, Table S2). Again, we observed not only C > T editing but also C > R editing, with predicted mutations matching for 19/23 sgRNAs. Thirteen distinct guides programmed mutations in early-acting spliceosomal factors, including SF1, SF3, and DDX46/hPrp5 as well as DDX5 (Figure 2C). Unexpectedly, we also identified a mutation in the tri-snRNP-specific protein USP39/hSad1 and five mutations in the second-step factor CDC40/hPrp17, which is first found in the Bact complex26. Finally, we found PB-sensitive mutations in factors that join catalytically active complexes and act at the second chemical step of splicing (DHX35 and CACTIN), a point in the spliceosome cycle after which SF3b has been dissociated. Below we briefly place some of these mutations into the context of existing spliceosome structures, focusing on the SF3 complex.
PB sterically blocks binding of the U2-intron branchpoint duplex to its pocket in SF3b12, which is necessary for spliceosome assembly to proceed beyond the A complex. Six of our PB-sensitive mutants occur in the SF3 complex, which consists of the SF3a and SF3b subcomplexes8. Recent work has shown that the HEAT-repeat region of SF3B1, a SF3b component, undergoes a conformational transition upon U2 snRNP binding to the branchpoint, moving from an open conformation to a closed state, thereby stabilizing the U2-branchpoint duplex61,76,77. We mapped an SF3B3 E159K mutant to an interface between SF3B3, SF3B1 and PHF5A (Figure 2D), distal to where PB or the branchpoint engages SF3b. The mutation lies in a region of SF3b that changes conformation upon binding to the branch helix (formed between U2 and the branchpoint sequence) (Figure 2E), likely impacting SF3B1 closing which would favor PB binding and, presumably, cell growth inhibition. Finally, we mapped PB-sensitive substitutions in residues in several factors not part of SF3b onto available structures26,47,75 and found that they often occur at protein-protein interfaces (Figure S2D–F, Table S2).
Viable SF3b mutations produce resistance to pladienolide B
SF3B1 and PHF5A mutations have been identified that render cells resistant to PB treatment11,59, and occur in a dominant (heterozygous) fashion, suggesting recessive lethality. Nonetheless, in our haploid screen, we identified six significantly enriched sgRNAs that target these factors, five against PHF5A and one against SF3B1, (Figure 1F). To identify the underlying alleles, we transduced eHAP FNLS cells with individual sgRNAs and collected cells at t0, t8 and t15 in the presence and absence of 2 nM PB treatment. Following genomic DNA isolation and amplicon deep sequencing, we determined mutation prevalence across time and treatments (Figure 3A). Note that for efficient guides, resistance-promoting mutations may be highly prevalent at t0 and therefore may not enrich in abundance under compound treatment (see e.g., sgPHF5A_7, Figure S3A).
Figure 3. Novel resistance mutations in SF3B.
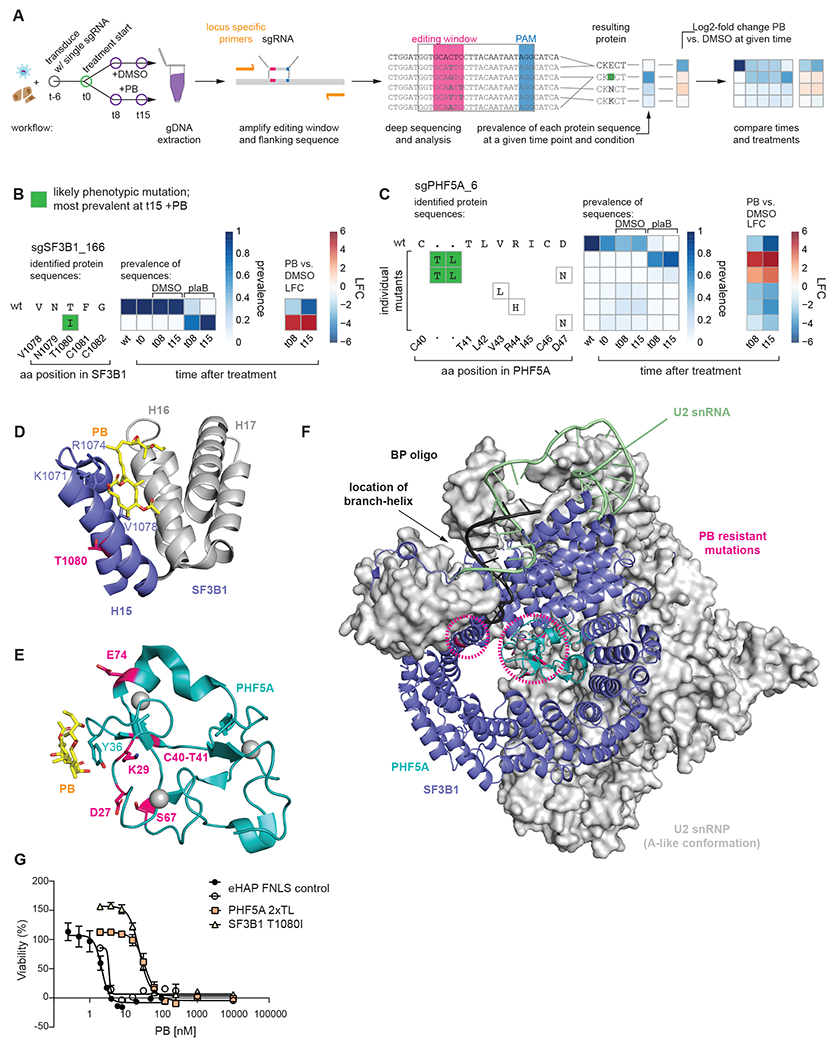
(A) Schematic of workflow to identify phenotypic mutations. Left: Cells are transduced with single sgRNA in an arrayed format. After six days (t0) treatment is initiated and a cell sample is harvested at t0 as well as t8 and t15 for genomic DNA (gDNA) extraction. Middle: Locus-specific primers are used to amplify the editing window and its flanking sequence of the gDNA. The amplicons are then deep sequenced to identify mutations. Right: Mutational outcomes are translated and the resulting protein sequences are aggregated as multiple DNA sequences may result in the same protein sequence. Prevalence of each protein sequence is calculated for each time point and treatment condition. Where applicable, log2-fold changes are calculated between two samples. Finally, time points and treatments can be compared across samples for both prevalence and/or log2-fold change vs. the wild type (wt). Inferred phenotypic mutations (most prevalent at t15 +PB) are indicated by a green background.
(B) Editing outcome for sgSF3B1_166.
(C) Editing outcome for sgPHF5A_6: In the absence of PB some mutations occur within the editing window around R44. PB treatment enriches for a rare 6 nucleotide insertion occurring from the nicking action of the nCas9, which is part of FNLS.
(D) Location of SF3B1 T1080I resistance mutation. T1080 (magenta) is located on the back of H15 facing away from PB and towards H14 (not shown). Known resistance mutations at K1071, R1074, V1078 are shown. (PDB: 6EN4)
(E) Location of PHF5A resistance mutations: All mutations are indicated (magenta) and occur on the face of PHF5A involved in PB binding. Known PB resistance mutation at Y36C is also indicated. Zinc ions coordinated by PHF5A are shown as grey spheres. (PDB: 6EN4)
(F) Illustration of SF3B1 and PHF5A resistance mutations in context of U2 snRNP (A-like conformation). Mutations (magenta, circled) occur in vicinity to the branch helix and branchpoint adenosine. Zinc ions are shown as grey spheres. (PDB: 7Q4O)
(G) Sixty-hour cell proliferation profiling (CellTiter-AQueous cellular viability and cytotoxicity assay) of control eHAP FNLS cell line expressing non-targeting sgRNA and monoclonal cell lines carrying either SF3B1 T1080I or PHF5A 2xTL mutation to PB. Error bars indicate s.d. n = 3 (average of two technical replicates for independent clonal cell lines).
sgSF3B1_166 was the only sgRNA conferring PB resistance through mutation of SF3B1. For this guide, no mutant alleles were detected by sequencing in the transduced cell population in the absence of PB treatment (possibly due to poor editing efficiency), but an allele encoding a T1080I change enriched rapidly upon PB addition to cells carrying this sgRNA (Figure 3B). In contrast, all five guides targeting PHF5A gave rise to substantial cell populations carrying different mutations at t0. Both sgPHF5A_7 and sgPHF5A_21 are predicted to result in cysteine to tyrosine mutations for side chains involved in the coordination of a Zn2+ ion. However, in both instances deep sequencing revealed that the predicted Cys > Tyr mutation was not detected; rather, we observed mutations impacting the preceding residue (Figure S3A,B). Both the resulting PHF5A K29N and E74D mutation arise through transversion (C > R mutation). sgPHF5A_47 is predicted to result in mutation and loss of the 3’ splice site of exon 3. This mutation did occur at high frequency (Figure S2C) and likely resulted in a growth disadvantage (see Figure S2A, reduced fitness of sgPHF5A_47 transduced cells in absence of PB). Instead, after transduction with this guide, another mutation at t0 encodes a D27N change which further accumulates in the population under compound selection. The remaining two resistance-promoting sgRNAs in PHF5A are predicted to be non-editing. sgRNA_PHF5A_6 targets a cytosine at position 7 in a G6C7 dinucleotide context, which is unfavourable to editing31,53. Indeed, this sgRNA resulted only in 12% of amplified molecules harboring the anticipated D47N mutation. Surprisingly, under PB selection, the more frequent mutation encodes a two amino acid insertion (TL) between C40 and T41 producing a tandem TL dipeptide in the protein sequence (hence we name the allele PHF5A-2xTL) (Figure 3C). This TL-encoding insertion occurred at position −3 relative to the PAM of sgRNA_PHF5A_6 where the nCas9 of the CRISPR-Cas9 base editor nicks the genomic DNA upon genomic binding. For the other non-editing sgRNA, sgRNA_PHF5A_26, editing should not occur within the editing window due to an absence of cytosines. Indeed, we observed edits 13 and 15 bp upstream of the targeted sequence, which alter S67 (Figure S3A, see Figure S3E for a summarized comparison of predicted mutations vs. those identified by sequencing).
Using this information, we mapped the encoded amino acid changes on the available structures of SF3B1 and PHF5A. For SF3B1, the change lies within heat repeats 15 and 16, which form a hinge region that supports a conformational change necessary for BP-A binding (Figure 3D,F)11,61,77. This location differs from those of reported resistance mutations in SF3B1 at K1071, R1074 and V1078 – which are all residues that face PB in high resolution structures60,69. For PHF5A, all five mutations that we identified impact residues in a protein surface near the PB binding site, where PHF5A interacts with both SF3B1 and the U2-branchpoint helix (Figure 3E,F)61. This contrasts with the location of the reported (dominant resistant) Y36C mutation, which changes a residue that directly contacts PB60.
To test whether these mutations produce resistance to concentration-dependent acute killing by PB, we repeated guide transduction experiments followed by single cell cloning to generate six independent monoclonal cell lines, three harboring the mutation PHF5A-2xTL and three harboring SF3B1-T1080I (Figure S4A). The measured half maximal effective concentration (EC50) of PB in a cell viability assay at 60 h post treatment confirms that PHF5A-2xTL (34 nM) and SF3B1-T1080I (24 nM) confer concentration-dependent resistance to killing by PB relative to the parental cell line (EC50 = 2 nM) (Figure 3G).
Mutations in the G-patch tumor suppressor protein SUGP1 confer PB resistance
Unexpectedly, our screen identified resistance mutations in three factors, PRPF6, SF1 and SUGP1, that are not part of SF3b, the target of PB. We focussed on the analysis of the SUGP1 mutations. SUGP1 was targeted by two guides, both of which were confirmed in competition validation assays (Figure 1F, 2B, S2A). Transduction, compound treatment, and amplicon deep sequencing suggests that sgSUGP1_238 produces resistance via an E554K mutation and sgSUGP1_188 produces resistance via a G603N mutation (Figure 4B, C).
Figure 4. Novel resistance mutations in SUGP1.
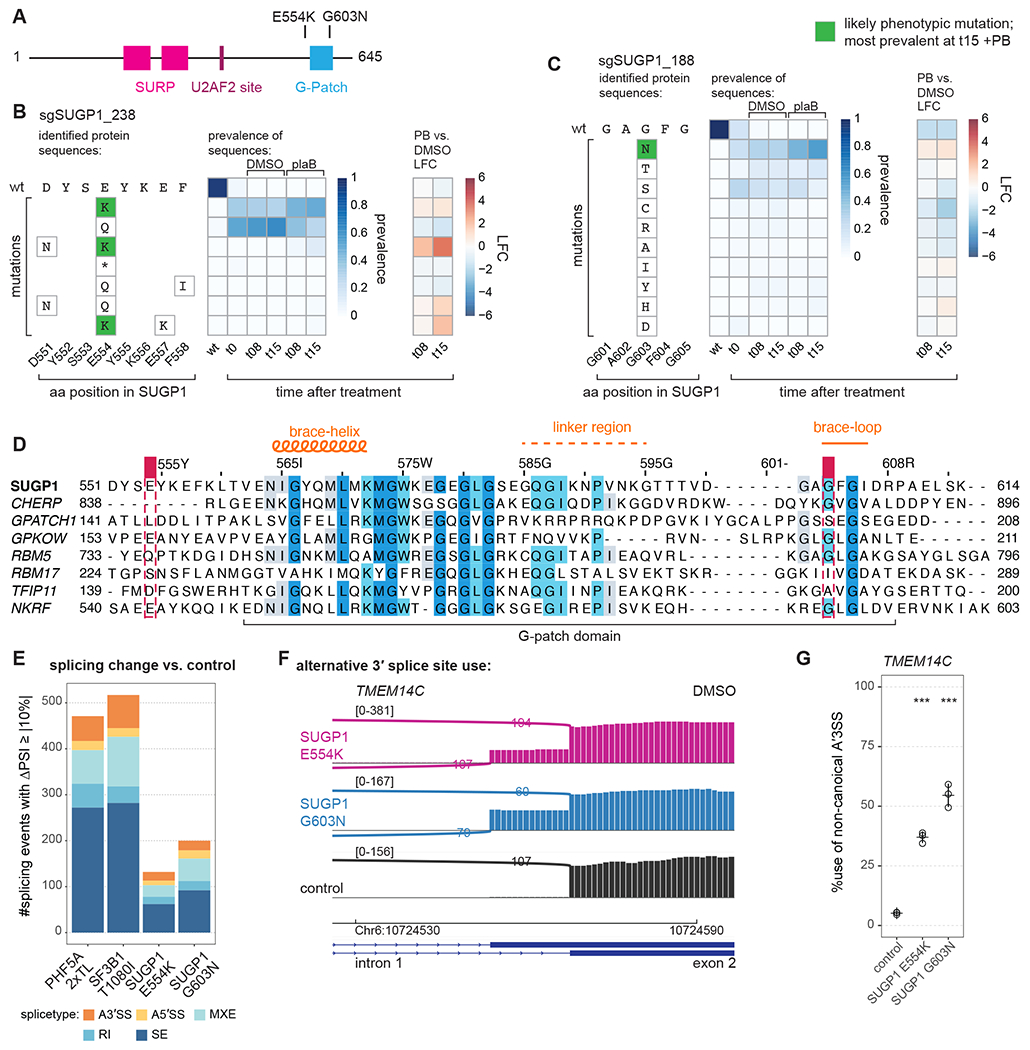
(A) Schematic of SUGP1 and its domains and motifs.
(B) Editing outcome for sgSUGP1_238.
(C) Editing outcome for sgSUGP1_188.
(D) Sequence alignment of all human G-patch motifs involved in splicing with the NKRF G-patch motif included as a reference. Shaded residues: more than 30% identity. Positions of mutants identified in screen are indicated. Alignment by JalView.
(E) Identified splicing changes for mutant vs. control cell lines. Numbers are shown for junctions identified by rMATS with FDR > 0.01 and |ΔPSI| > 10 (ΔPSI: PSI of mutant sample -PSI of control sample, where PSI: percent spliced in). RNA-seq data of total, polyA-selected RNA from three independent clonal cell lines treated for 3 h with DMSO. (A3’SS: alternative 3’ splice site use; A5’SS: alternative 5’ splice site use; MXE: mutually exclusive exon; RI: retained intron; SE: skipped exon.)
(F) Sashimi plot for alternative 3’ splice site usage in TMEM14C exon 2 (DMSO). Representative traces for a single clonal cell line are shown.
(G) RT-PCR and quantification for alternative 3’ splice site usage for TMEM14C exon 2 (DMSO). Statistical analysis for RT-PCR: one-way ANOVA with Dunnett’s for multiple comparison (two-sided, with control as reference) was performed with R and package multcomp; * p < 0.05, ** p < 0.01, and *** p < 0.001; all with n = 3.
Both mutations match the editing predictions with one encoding a change lying in (G603N) and the other just upstream (E554K) of the G-patch motif (Figure 4A,D). Neither affects SUGP1 protein levels in the cell (Figure S4H). SUGP1 is associated with the spliceosomal A complex, where it interacts with SF3B172. SUGP1 has not yet been visualized in spliceosome structures, nor are there orthologs in S. cerevisiae or S. pombe. Recent work has identified SUGP1 as a putative tumor suppressor: its loss from the spliceosome was suggested to underlie splicing and oncogenic phenotypes of SF3B1 tumor mutations, and mutations in SUGP1 found in tumors mimic the splicing phenotype of SF3B1 mutant tumors1,39,72.
For EC50 assays, we again repeated transductions and produced three independent monoclonal cell lines (Figure S4A) for each mutation. To our surprise, no substantial change in EC50 for PB was observed (Figure S4B). However, the EC50 measurement occurs over a much shorter time frame than treatment during the screen, which indicates that the mutations confer resistance to PB-mediated growth inhibition over time but not immediately. These data suggest that the SUGP1 mutants act by a mechanism distinct from those in SF3B1 and PHF5A, which map near the drug binding site.
SUGP1 mutations modulate a subset of PB-induced exon skipping events
PB induces exon skipping as well as other splicing changes68. To investigate the impact of SUGP1 mutations on PB-induced splicing changes, we performed RNA-seq analysis on our clonal cell lines. We also included the PHF5A-2xTL and SF3B1-T1080 clonal cell lines. Cells were treated with DMSO or 2 nM PB (= EC50) for 3 h (a time frame where cell viability is not yet affected, see Figure S4C) prior to RNA extraction and polyA-selection. We detected no changes in global transcript levels in the mutant cell lines (Figure S4C).
We used rMATS56 to detect differential alternative splicing events. In untreated cells, we observed exon skipping (skipped exon, SE) as the most frequent event triggered by the mutations followed by alternative 3’ splice site (A3’SS) use; relatively few introns were impacted [using a difference percent splicing inclusion (ΔPSI) cut-off of ≥ |10%| and FDR > 0.01] (Figure 4E). Among these were changes in 3’ splice site usage in introns of the TMEM14C and ENOSF1 genes, which, strikingly, correspond to changes observed previously in cells harboring SF3B1 cancer mutations or SUGP1 cancer mutations (Figure 4F, G, S4D–F)1,39.
Upon PB treatment of these cell lines, the number of differentially spliced junctions increased drastically. Control cells (eHAP FNLS transduced with a non-targeting sgRNA) displayed the largest number of PB-induced splicing changes, while the PHF5A-2xTL and SF3B1-T1080 showed almost no changes, as would be expected if they were to reduce the effects of compound binding to the spliceosome (Figure 5A). Both SUGP1 mutants displayed an intermediate phenotype with fewer affected events than wild-type in the presence of PB.
Figure 5. RNA-seq analysis of mutants.
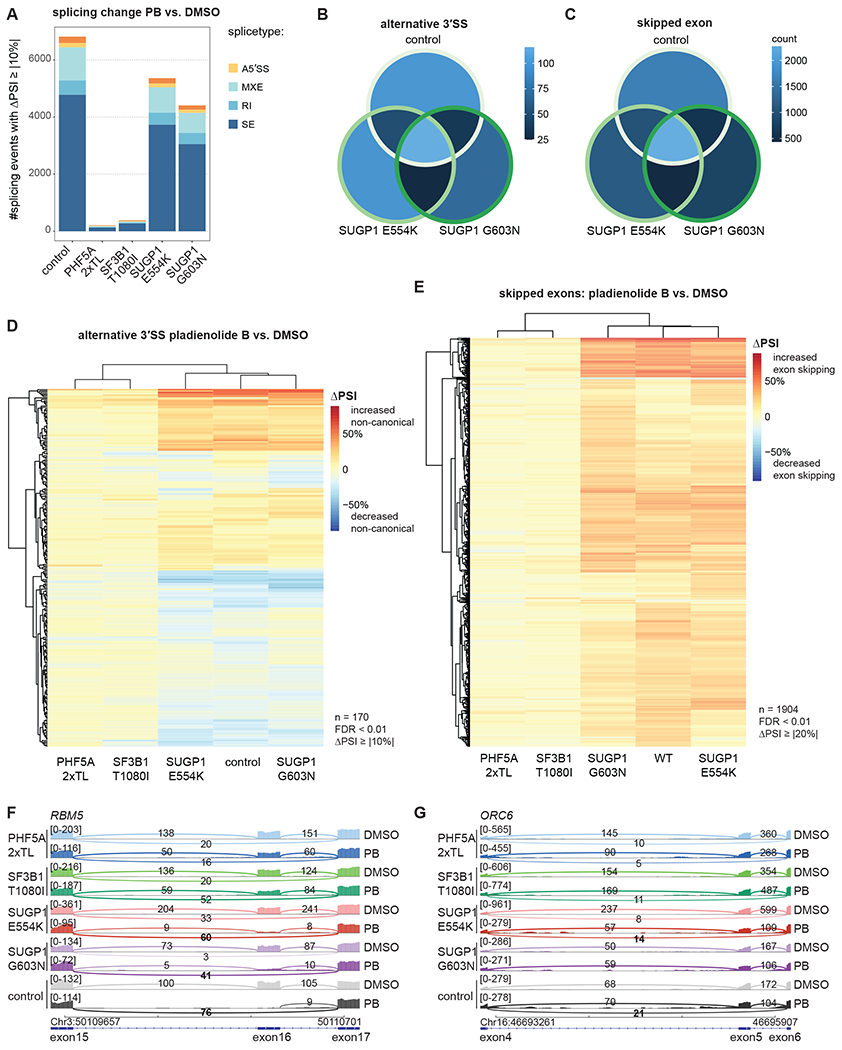
(A) Analysis of PB-induced splicing regulation in mutant vs. control cell lines. Numbers shown for junctions identified with rMATS with FDR > 0.01 and |ΔPSI| ≥ 10. RNA-seq data of total, polyA-selected RNA from three independent clonal cell lines treated for 3 h with 2 nM PB or DMSO.
(B-C) Overlap in and alternative 3’ splice site use (B) and cassette exons (C) affected by PB treatment for all junctions observed in all three sample groups (control vs. SUGP1 E554K vs. SUGP1 G603N). 23% of differentially spliced 3’ splice sites (B) and 29% of differentially spliced cassette exons (C) are affected in all three genetic backgrounds. Splicing junctions with FDR < 0.01 were included.
(D-E) Hierarchical clustering of differential splicing of alternative 3’ splice sites (D) and cassette exons (E) for PB treatment, based on PSI (percent spliced in) changes. The heatmap represents ΔPSI values of A3’SS (D) or cassette exons (E) use, respectively, upon treatment with PB at 2 nM for 3 h vs. DMSO as detected in total, polyA-selected RNA using rMATS. Splicing junctions with FDR < 0.01 and |ΔPSI| ≥ 10 (D) or FDR < 0.01 and |ΔPSI| ≥ 20 (E) were considered.
(F-G) Sashimi plot for alternative splicing of exon 16 in RBM5 (F) and exon 4 in ORC6 (G) for 2 nM PB vs. DMSO treatment. Representative traces for a single cell line each.
Focusing on A3’SS and SE events induced by PB, we observed a general concordance between control cells and SUGP1 mutants (Figure 5B, C). Hierarchical clustering of the splicing junctions quantified by rMATS across all samples demonstrated that PB treatment almost exclusively triggered increases in exon skipping (Figure 5E), but both increased and decreased use of alternative 3’ splice sites (Figure 5D). Subsets of splicing events induced by PB were affected either equally strongly in control and SUGP1 mutant lines (example: RBM5 exon 16 in Figure 5F, S5B) or displayed milder changes in the SUGP1 mutants (example: ORC6 exon 5 in Figure 5G, S5B, C). No statistically significant differences in intronic or exonic features at the introns equally vs. differentially affected by SUGP1 genotype were identified using Matt21. These findings demonstrate that SUGP1 mutants can modulate splicing changes induced by PB, as expected from the ability of SUGP1 to produce relative resistance to the compound.
DHX15/hPrp43 is a mutationally sensitive ligand of the G-patch motif of SUGP1
G-patch motifs are direct activators of DEAH-box helicases57,65; this is the only known activity of this domain. SUGP1 has therefore been suggested to recruit a helicase through its G-patch motif to SF3B1 and the A complex1,39,72. As we identified mutations within and just upstream of the SUGP1 G-patch, it seemed likely that one or both mutations impact the association and/or activation of a cognate DEAH-box ATPase. Indeed, the G603N mutation lies within the “brace loop” analogous to the only G-patch protein for which its helicase-bound structure is available (Figure 4D), a region known to be important for the NKRF G-patch to bind and activate its cognate DEAH box helicase DHX1557.
To identify SUGP1 helicase partner(s), we employed proximity labelling (miniTurboID) exploiting our SUGP1 G603N mutant as a control7. We transfected HEK293T cells with expression plasmids encoding SUGP1 with a C-terminal miniTuboID fusion. After a short 2 min labelling pulse with biotin, extracts were prepared, and labelled proteins purified under stringent conditions using streptavidin. Replicate samples were subjected to tandem mass tag mass spectrometry (TMT-MS) (Figure S6A,B) to identify differentially labelled proteins. Remarkably, only a single protein displayed a statistically significant reduction in signal in the SUGP1-G603N-miniTurboID-tagged samples versus those obtained with the wild-type fusion: the DEAH-box helicase DHX15/hPrp43, the protein that disassembles the spliceosome (Figure 6A). We also identified DHX15/hPrp43 in a parallel experiment using a fusion of a SUGP1-L570E-miniTurboID fusion which we constructed based on the ability of this mutation to disrupt G-patch/DEAH protein interactions for the G-patch protein NKRF57 (Figure S6C).
Figure 6. SUGP1 interacts with DHX15.
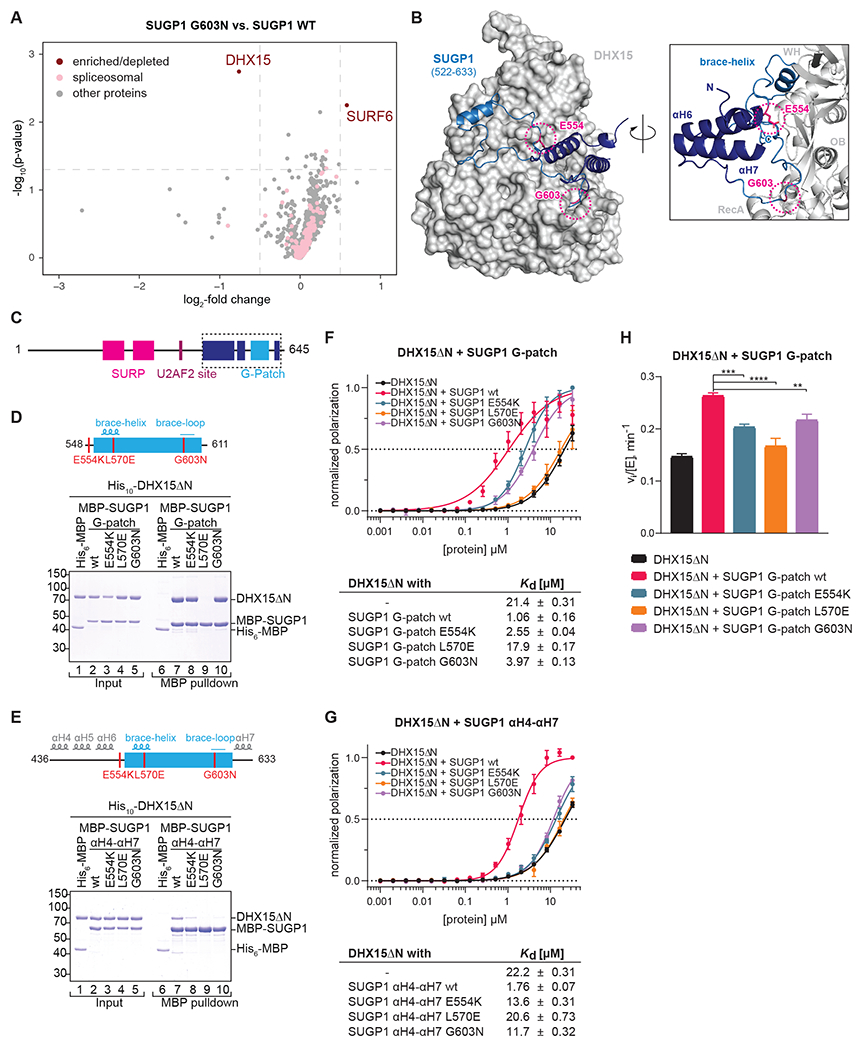
(A) miniTurbo proximity labeling using FLAG-SUGP1-miniTurboID G603N vs. wt overexpressed in HEK293T cells. Enriched biotinylated proteins were identified with TMT-MS. −log10(p-value) is plotted against the log2-fold change (LFC) in a volcano plot (dashed lines: cutoffs at p < 0.05 and LFC > 0.5).
(B) AlphaFold2 prediction of SUGP1 (522-633), encompassing the G-patch motif flanked by αH6 and αH7, in complex with DHX15.
(C-E) Domain organization and schematic representation of the MBP-hsSUGP1 variants with the introduced mutations colored in red.
(D-E) Coomassie-stained gels of protein binding assays using purified MBP-hsSUGP1 constructs and His10-hsDHX15ΔN. MBP-SUGP1 G-patch (D), and αH4-αH7 (E) with either wildtype (wt) protein sequence or carrying the indicated mutation were used as baits and His6-MBP served as a control. Input (1.5% of total) and eluates (24% of total) were loaded.
(F-G) Fluorescence polarization of FAM-labeled U12 RNA with His10-hsDHX15ΔN in the absence or presence of MBP-SUGP1 G-patch (F) and αH4-αH7 (G) wt or mutants. Dashed line indicates 50% normalized polarization. Error bars represent standard deviations from the average of triplicate measurements. RNA dissociation constants (Kd) with standard error of means (SEM) were derived from linear regression.
(H) Initial ATPase activity rates of His10-hsDHX15ΔN in the absence or presence of MBP-hsSUGP1 G-patch wt or mutants at 250 μM ATP. Error bars indicate standard deviations of three independent measurements, asterisks denote significance (one-way ANOVA with Tukey’s) with ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
Modelling of a SUGP1-DHX15 complex using AlphaFold229 predicted that the G-patch domain of SUGP1 interacted with DHX15 as anticipated but that the G-patch was flanked by unanticipated α-helical elements that are also predicted to interact with DHX15. In this model, the E554K substitution may impact a contact with the flanking α-helices (Figure 6B). We overexpressed SUGP1 mutants in HEK293T cells to assess their impact on splicing. We observed that deletion of αH4-5 or αH6 as well as the E554K, G603N, and L570E substitutions displayed similar effects on splicing (Figure S5D–G). Thus, to test more directly the impact of the G-patch mutations on the interaction and/or activation of DHX15, we constructed a series of maltose binding protein (MBP) fusion proteins harboring the SUGP1 G-patch and varying lengths of flanking sequences (Figures 6D,E and S6C).
We next mixed a purified version of DHX15 lacking its N-terminal domain (DHX15ΔN) with each MBP-SUGP1 fusion proteins, purified them with amylose beads, and analyzed the material using SDS-PAGE. We observed that DHX15ΔN selectively copurified with each of the MBP-SUGP1 fusion proteins (Figure 6D–E and S6D). However, we found that the efficiency of copurification decreased with increasing length of SUGP1 constructs, indicating that protein stretches surrounding the G-patch modulate its binding affinity for DHX15. For each of these constructs, we generated mutations in the G-patch, corresponding to the two obtained in our screen, E554K and G603N, as well as one in the central “brace-helix”, L570E, that is known to disrupt ligand binding in analogous G-patch proteins. For the construct harboring the most upstream SUGP1 sequences (436-633) all mutations reduced binding to DHX15ΔN, consistent with loss of DHX15 labelling we observed in the proximity labeling experiment. Therefore, it is likely that disruption of DHX15 interaction forms the molecular basis for the observed PB resistance of the SUGP1 mutants obtained in our screen. Consistent with the stronger affinity of DHX15ΔN for the intermediate [MBP-SUGP1(522-633)] and shortest [MBP-SUGP1 (548-611)] SUGP1 constructs, their binding was not sensitive to the mutations obtained in our screen but was only disrupted by mutation of the core interface residue L570E.
As G-patch proteins can enhance RNA binding to DEAH-box helicases57, we used fluorescence anisotropy to ask whether the SUGP1 fusions increased RNA affinity of DHX15ΔN. Using a fluorescently labelled poly-U RNA, we found that all SUGP1 constructs indeed increased RNA binding to DHX15ΔN ~10-20-fold (Figure 6F–G and S6E). In agreement with their effect on binding DHX15, all mutations in the longest SUGP1 construct (436-633) blocked this stimulation (Figure 6G), while varying degrees of mutational sensitivity were observed for the shorter SUGP1 truncations (Figure 6F and S6E), again indicating an important role for sequences flanking the G-patch domain.
Finally, we investigated the ability of SUGP1 to stimulate the ATPase activity of DHX15ΔN. Indeed, SUGP1 (548-611) produced a 1.8-fold increase in initial ATPase rates of DHX15ΔN at saturating ATP concentrations, which was significantly diminished by all mutations consistent with their negative effects on DHX15 binding (Figure 6H).
DISCUSSION
The human spliceosome is essential for the splicing of over 200,000 introns in the human genome. Because it is mutated in numerous diseases and the target of myriad splicing regulators, it is a key compound development target. Numerous questions exist regarding the coupling of transcription and chromatin to splicing, the underpinnings of splicing fidelity, and the functional roles of many if not most human spliceosomal proteins. However, the genetic analysis of the human spliceosome has not been pursued, even though it harbors ~60 proteins not found in S. cerevisiae and is likely to operate in ways that cannot be anticipated from prior studies of yeast. Particularly useful would be so-called “informative alleles” that dissect essential protein function. We adapted pooled CRISPR-Cas9 base editing to the human spliceosome to mutagenize 153 protein subunits in a haploid cell context. We interrogated the mutants with PB, a prototype for a class of anti-cancer compounds that targets the SF3b complex. Our studies provide insights into structure-function relationships by identifying viable alleles of numerous spliceosomal proteins that program hypersensitivity or resistance to SF3b inhibition. We demonstrate the utility of such alleles through studies of SUGP1. Below we discuss the evidence for these conclusions and propose a new human-specific discard/fidelity step mediated by the activation of the spliceosomal disassemblase DHX15/hPrp43 by SUGP1 during early stages of spliceosome assembly and its implications for human disease.
PB hypersensitive mutations identify functional sites in the human spliceosome that vary in the human population
We obtained PB-hypersensitive mutants in a small subset of the 153 proteins mutagenized. Most lie in factors that act at or near the step inhibited by PB, including in SF1 and components of U2 snRNP. This specificity highlights the utility of single-residue chemical-genetic interactions to identify functional sites related to a particular phase of an essential process. Many mutations we identified could be placed on existing structures, enabling the generation of structure-function relationships. However, most of the residues altered in our mutants are not visualized in existing structures. In both cases, detailed studies in vitro and/or in vivo will be required to understand the impact of these functional sites on splicing.
Mutations in the second-step factor CDC40/hPrp17 and CACTIN produce PB hypersensitivity, even though PB impacts SF3b, which is dissociated from the spliceosome (freeing the U2-branchpoint helix) by DHX16/Prp2 prior to the chemical steps of splicing so that the active site of the spliceosome can form. We speculate that triggering the use of different branchpoints via PB results in a dependency on weaker 3’ splice sites, whose docking into the catalytic core of the spliceosome requires stabilization by step 2 factors, a model consistent with the cryoEM structure of the human post-catalytic P complex19.
Because the residues impacted by the PB-sensitive mutations are (by definition) functional, one anticipates that they would not vary in the human population, given the essential function of the spliceosome. However, the ClinGen database identifies seven residues in the human population (labelled as “non-disease-associated”), which we have found as PB-sensitive mutants. Four of these display the exact same amino acid changes in the human population as in our PB-sensitive cells (Table S2), suggesting that these variants likely have a functional impact.
Identification of SUGP1 G-patch mutations as PB-resistant
Our studies identified two sgRNAs that target SUGP1 that produce PB-resistance. The stronger of the two alleles produced by these guides, G603N, lies in a conserved domain of G-patch proteins called the “brace loop” which is important for G-patch proteins to activate their cognate DEAH-box helicase. The other change, E554K, lies just upstream in a region predicted by AlphaFold2 to be helical. Indeed, these three α-helices have recently been confirmed with the N-terminal part of the G-patch domain (residues 550 to 575, including E554) forming an α-helix, which interacts with αH4-5 and αH6 (Figure S6F)73. This structure would need to be disassembled for the SUGP1 G-patch domain to be able to bind to DHX15 (Figure S6J). Correct binding of the G-patch domain to DHX15 is likely required for this structural change to occur. Accordingly, in our in vitro assays the construct containing αH4-5 and αH6 is the most impaired by mutations obtained in the screen (Figure 6E, G).
By performing proximity labelling, comparing wild-type versus mutant proteins, we identified a single DEAH-box protein, DHX15/hPrp43 as being both labelled by SUGP1-miniTurboID fusions and sensitive to a G-patch mutation obtained in our screen. While this work was under review, DHX15 was also reported to interact with the G-patch domain via an affinity purification and targeted mass spectrometry approach. Concomitantly, the structure of SUGP1 G-patch bound to DHX15 was reported73. The structure of the brace-loop, wherein G603 lies, is similar to how the NKRF G-patch binds DHX15 (Figure S6G–I).
Previously, DHX15 has often been found in proteomic studies of early spliceosomes, but its function at this stage remained relatively opaque. However, a recent study from Jurica and colleagues has shown that depletion of DHX15 from HeLa cell extracts results in an increase rather than a decrease in A complex formation42. An orthogonal study using split-APEX also identified SUGP1 as a G-patch factor activating DHX15 as well as suggests its general involvement in splicing fidelity during early spliceosome assembly steps18. This result is consistent with the observation that yeast Prp43 disassembles spliceosomes41,58. Taken together with our results and the known association of SUGP1 with early spliceosomal complexes, we propose that SUGP1 recruits DHX15 to disassemble early spliceosomes, constituting an early discard step analogous to late discard steps described by Staley and colleagues in yeast (Figure 7). In this model, PB resistance results from mutation in the SUGP1 G-patch domain because this increases A complex formation or residence time by inhibiting disassembly, thereby counteracting the inhibitory activity of PB in reducing stable A complex formation.
Figure 7. Model for SUGP1-DHX15 and proofreading at early spliceosome assembly.
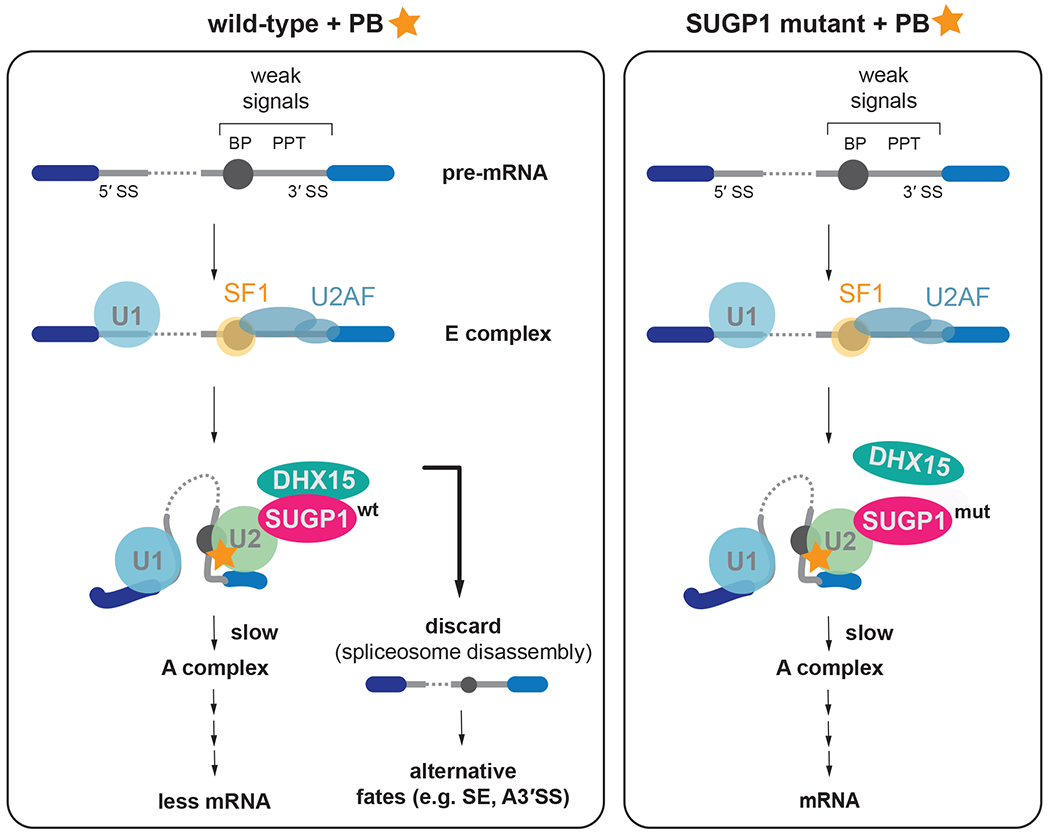
Left panel: On weak splice sites the transition from E complex to A complex is inhibited as PB binds to the U2 snRNP and prevents the full binding of the branch helix and recognition of the BP-A. These stalled spliceosomes are recognized by SUGP1-DHX15 and are discarded. Less mRNA is being produced in this scenario and more alternatively spliced mRNAs result.
Right panel: Mutation in SUGP1 weakens SUGP1 interaction with DHX15, removing the proofreading & discard pathway, allowing more time to assemble A complex and to proceed with splicing.
Relationship to oncogenic mutations in SUGP1 and SF3B1
It has been proposed that cancer SF3B1 mutations act by limiting association of SUGP1 with the spliceosome. This model, based on biochemistry, is supported by genetic data that identified SUGP1 mutations in tumors that mimic the splicing phenotypes of SF3B1 mutant tumors1. Indeed, many of the identified cancer mutations map to the regions flanking the G-patch motif of SUGP1 (Figure S5F), which we show to influence SUGP1-DHX15 interaction and splicing. It was proposed that the then-unknown helicase that is recruited by SUGP1 might dissociate SF1 from the branchpoint, causing U2 snRNP to relocate to alternative branchpoints72. However, our discard model proposes a different mechanism underpinning the effects of SUGP1 cancer mutations, namely a defect in an early rejection step mediated by spliceosome disassembly (Figure 7). Such a model would explain the activation of cryptic branchpoints as a defect in proofreading enabling the production of oncogenic mRNAs via the activation of cryptic branchpoint/3’ splice site combinations as has been observed in SF3B1 and SUGP1 mutants39,72. This model is also consistent with in vitro studies of DHX15 depletion described above and the known activity of DHX15 in spliceosome disassembly.
Mutagenesis of an essential machine in human cells in a haploid context
While this work was underway, others recently independently reported the deployment of base editor libraries in screens that involved phenotypic characterization of single nucleotide variants and/or to probing of small molecule-protein interactions13,25. These studies largely interrogated individual proteins or part of a gene network and were mostly performed in diploid cells. To enable large scale studies in a haploid context, we generated editing-competent eHAP cells expressing the FLNS editor under conditions that maintained their haploid state. Given our experience with the spliceosome, the future is bright for using base editing to interrogate essential cellular machines in human cells to produce new insights into human cell biology and disease.
Limitations of the study
The editor we used in this study targets only a subset of residues in spliceosomal proteins and is limited by base editor specificity and the occurrence of PAM sites. Thus, many additional potential informative mutations may be isolatable with the advent of complementary technologies that enable for efficient base editing at additional sites without causing unwanted cellular toxicity. Our analysis of PB-hypersensitive sites requires further studies to understand their mechanistic impact. Biochemical tests of the proofreading model will require its reconstitution in vitro and, ultimately, structural analysis.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hiten D. Madhani (hitenmadhani@gmail.com).
Materials availability
Plasmids generated in this study are available from the Lead Contact. Cell lines generated in this study are not available as eHAP cells and any product derived thereof are protected under an MTA upon purchase of the eHAP parental cell lines from Horizon (original vendor).
Data and code availability
All data discussed in this publication (CRISPR-Cas9 base editing screen including read counts, validation experiments, and RNA-seq) have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE218307). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE48 partner repository with the dataset identifier PXD038067.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTSL MODEL AND SUBJECT DETAILS
Cell Culture
All cell lines were maintained at 37 °C with 5% CO2 and were regularly tested negative for mycoplasma infection. Human embryonic kidney (HEK) 293T cells were grown in DMEM (with 4.5 g/L glucose, L-glutamine and sodium pyruvate; Corning #10-013-CV) supplemented with 10% FBS. Cells were passaged every 2-3 days.
Human eHAP cell lines and derivatives thereof were cultured in IMDM (with L-glutamine, with HEPES; Cytiva #SH30228), supplemented with 10% FBS and 1:100 penicillin/streptomycin. eHAP cell lines were at all times maintained at sub-confluent conditions and ploidy was regularly assessed with flow cytometry. When mentioned, doses of puromycin were 4 μg/ml and blasticidin 10 μg/ml.
Flow cytometry data was analysed with FlowJo (v10.8.1).
METHOD DETAILS
Vectors
Assembly of vectors (cloning and mutagenesis) was, if not otherwise indicated, performed using NEBuilder® HiFi DNA Assembly (NEB #E2621).
pLibrary (MP783):
mU6 promoter expresses customizable guide RNA with a 20N barcode sequence at the 3’ end of the tracrRNA to facilitate identification of individual sgRNAs and sample splitting into replicates6. A core EF1α promoter expresses puromycin resistance and a T2A site provides BFP for easy titer determination of the lentiviral library.
pFNLS:
A core EF1α promoter expresses 3xFLAG-tagged codon optimized FNLS base editor and provides with a P2A site EGFP for identification of FNLS carrying cell lines. A PGK promoter provides blasticidin resistance. This vector was modified from Addgene vector #110869. pHA-SUGP1: Point mutations and deletions were introduced into p3xFLAG-CMV-14_3xHA72.
Vectors for miniTurboID:
SUGP1 constructs were cloned from pHA-SUGP1 using primers (IB0156 5’-atgacgtcccagactacgcagctagcAGTCTCAAGATGGACAACC-3’; IB0157 5’-tgtttagcgttcagcagcgggatagatccgcctgaGTAGTAAGGCCGTCTGG-3’) into pCDNA3_3xHA-miniTurbo-NLS (Addgene #107172) digested with NheI-HF (NEB #R3131).
Expression plasmids:
SUGP1 constructs were generated by PCR using a plasmid from the human open reading frame library (hORFeome Version 5.1, ID: 53373) as template and gene-specific primers. For protein expression in E. coli, the constructs were cloned into the NdeI-XbaI sites of the plasmid pnEA-NpM, which is derived from the pET-MCN vector series that harbors an N-terminal MBP-tag and a subsequent 3C protease site24. Mutations in the SUGP1 constructs were introduced by ‘round-the-horn (RTH) mutagenesis27 using the respective primers (Table S3). For plasmid generation of 10xHis-DHX15ΔN, the construct was cloned by ligation-independent cloning (LIC) into a modified pFastBac vector of the MacroBac series22 harboring the N-terminal decahistidine tag with a subsequent 3C protease site. DHX15ΔN with LIC-compatible overhangs was generated by PCR using primers 5’-CCCTTCCCAATCCAATTCGCAGTGCATTAATCCGTTCACC-3’ and 5’-TTATCCACTTCCAATGTTATTATCAGTACTGTGAATATTCCTTGG-3’, respectively, and a template plasmid encoding full-length DHX15 from the hORFeome library (V5.1, ID: 13273). The construct containing LIC-compatible ends was integrated into Sspl linearized pFastBac vector by treating insert and vector with T4 DNA polymerase, annealing in a thermocycler and subsequently transformation into DH5α cells. Bacmids were generated using the Bac-to-Bac expression system (Invitrogen) adhering to the manufacturer’s protocol.
Cell viability assay
96-well plates were seeded with 11000 cells per well earlier in the day and treatment was started after allowing cells enough time to attach to the plate surface. A serial dilution of PB was then used (starting at 100 nM and followed by 10 additional 2-fold dilution steps down to 0.25 nM or starting at 10 μM, 1 μM, 250 nM and followed by 7 additional 2-fold dilution steps down to 0.98 nM). DMSO percentage was maintained throughout and a DMSO-only control was included. 60 h post PB addition, CellTiter 96® AQueous One Solution Cell Proliferation Assay reagent (Promega #G3582) was added and incubated for 4 h and read out according to the manufacturer’s instruction. Samples were measured in two technical replicates, whose values were used as an average for three biological replicates. EC50 curves were fit with GraphPad PRISM.
Spliceosome library design and production
We compiled a list of all spliceosome components reproducibly detected through mass spectrometry (MS), interaction studies, and/or purified and visualized in the spliceosome in structural biology studies 52. This list encompasses 153 proteins (Table S1). Guide sequences for targeting the spliceosome were designed using CHOPCHOP 35 using [-Target $GENE -J - BED -GenBank -G hg38 -filterGCmin 0 -filterGCmax 100 -consensusUnion -t CODING -n N -a 20 -T 1 -g 20 -M NGG]. We included all sgRNAs targeting coding sequence across all exons in all isoforms, including 20 nucleotides into the introns and UTR. Oligonucleotide pools were synthesised by CustomArray. Cloning sites were appended with 5’-AGTATCCCTTGGAGAACCACCTTGTTGG-3’ and 5’-GTTTAAGAGCTATGCTGGAAACAGCATA-3’. The final oligonucleotide sequence was thus: 5’-AGTATCCCTTGGAGAACCACCTTGTTGG [sgRNA, 20 nt] GTTTAAGAGCTATGCTGGAAACAGCATA −3’. Primers (forward: cttggAGAACCACCTTGTTG, reverse: GTTTCCAGCATAGCTCTTAAAC) were used to amplify the library pool (15x cycles). The resulting amplicons were PCR purified (QIAGEN #28104) and cloned into the library vector [digested with Aari (ThermoFisher #ER1582)] via Gibson assembly (NEBuilder® HiFi DNA Assembly). The ligation product was buffer exchanged (BioRad #732-622) ethanol precipitated and electroporated into MegaX DH10B T1R Electrocomp™ Cells (ThermoFisher #C640003). The plasmid DNA was sequenced to confirm library and barcode representation and distribution.
Spliceosome library annotation
CRISPR-Cas9 base editing outcomes were predicted according to the following rationale. We assumed that if editing occurs for a given sgRNA, all cytosines within the editing window (position 3-8) will be mutated to thymine, with the exception of Cs at positions 3, 4, 6, 7, and 8 if they are preceded by a G 32. This was used to classify sgRNAs into non-editing (= sgRNAs containing no C within editing window), non-editing_GC (= sgRNAs containing C’s in GC context unfavorable to editing, not at position 5 within sgRNA), and editing sgRNAs. Editing sgRNAs were further classified by MNV (multiple nucleotide variant) prediction using VEP (Variant Effect Predictor; Webserver accessed 2020.11.27 at 16:15; Ensembl release 99) 45 . For each MNV, where available only the outcome for MANE (Matched Annotation between NCBI and EBI) transcript was considered. In a next step, consequences were binned into categories and consequence severity was given in this order: CDS_missense > stop_gained > startjost > SS_acceptor, SS_donor, SS_region, CDS_silent > 3’UTR > 5’UTR. It should be noted, that VEP considers a splice site region variant a sequence variant with a mutation within 1-3 bases of the exon or 3-8 bases of the intron. All sgRNAs and their annotations are provided in Table S4.
Virus production and MOI determination
For lentivirus generation and packaging, media for HEK293T cells was supplemented with non-essential amino acids (Gibco #25300054). Cells were seeded 24 h before transfection with jetPRIME reagent (Polyplus #114-15) at a 2.5 μl to 1 μg DNA ratio. Media was changed 6 h post transfection and fresh media was supplemented with ViralBoost Reagent (Alstem #VC100). The packaging mix consisting of psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) was prepared at a molar ratio of 1:1. The following reagents were adapted according to scale of lentivirus production:
24-well plate: 2e5 cells seeded, 450 ng target DNA + 450 ng packaging mix per well. 6-well plate: 7.5e5 cells seeded, 1 μg target DNA + 1 μg packaging mix per well. 10 cm plate: 5.2e6 cells seeded, 5.5 μg target DNA + 4.5 μg packaging mix per dish. Virus was generally concentrated using Lentivirus Precipitation Solution (Alstem #VC100). Virus was titered by seeding 2e5 eHAP FNLS cells in 1 ml media per 6-well plate and immediately adding sequentially diluted virus amounts. 48 h post-transduction the number of BFP positive cells was assessed by flow cytometry. A viral dose resulting in 30-40% transduction efficiency, corresponding in an MOI of ~0.3, was used for all subsequent experiments.
Generation of eHAP FNLS cell line
Lentivirus was generated with pFNLS and transduced on eHAP cells. Cell lines were selected with blasticidin four days post-transduction for one week and then single cell sorted to obtain monoclonal cell lines. Editing rate of a cell line was assessed by transduction with control sgRNAs (EMX1, HEK2, HEK3, HEK4) and evaluation using sanger sequencing of the editing window and EditR 32. Clonal cell lines showing high rates of base editing were treated with 10 μM 10-deacetylbaccatin-III (Selleckchem #S2409) for 10-15 days with ploidy assessed every second day. Treatment was stopped as soon as an exclusively haploid cell population was achieved. Editing rate was re-assessed and no changes were observed.
Generation of eHAP FNLS mutant cell lines
eHAP FNLS cells were transduced with lentivirus carrying a single sgRNA at an MOI of 0.3. After 2 days sgRNA carrying cells were selected using puromycin for four days. Cells were given two days to recover from selection pressure and then seeded as single cells by limited dilution. The SF3B1 T1080I and PHF5A 2xTL mutations were obtained by first treating cells for 2 weeks with 2 nM PB to enrich for the mutations.
Single cell colonies were maintained and expanded while assessing ploidy and genotyping the clones. Genotyping was performed by using QuickExtract™ DNA Extraction Solution (Lucigen #QE09050) on a fraction of a clone. The region of interest was amplified using custom primers and sanger sequenced. After expansion of the single cell, ploidy was again assessed before freezing the cell line for long term storage.
Ploidy assessment for eHAP cell lines
After harvesting, cells are washed with flow cytometry buffer (1x DPBS with 2% FBS, 4 mM EDTA pH 8) and stained on ice with 0.1% sodium citrate, 0.1% Triton X-100, 50 μg/ml propidium iodide for 5 min and immediately assessed by flow cytometry 3.
Spliceosome-wide CRISPR-Cas9 base editing screen
The screen was performed at 500x sgRNA representation for entire duration. 66 million eHAP FNLS cells were infected with the lentiviral spliceosome library (marked with BFP) at an MOI of ~0.3, such that every sgRNA was represented in approximately 500 cells. Puromycin selection was started at 48 h post transduction. At six days post-transduction, cells were assessed by flow cytometry to only contain sgRNA carrying cells (BFP-positive cells >95%). Cells were then split into treatment arms (DMSO vs. 2 nM PB; with identical DMSO concentration in both treatment arms). Cells were propagated and treatment was renewed every second day.
At screen end point cells were harvested and gDNA was extracted with QIAamp DNA Blood Maxi Kit (Qiagen #51194). Genome weight was estimated based on measured ploidy of cells. The sequencing library was prepared using NEBnext® Ultra II Q5 Master Mix (NEB #M0544) and custom primers (forward: IB0096-IB0104; reverse IB0106-IB0121, Table S3) to have a balanced read sample. A barcode in the reverse primer was used for identification of the sequencing libraries. Libraries were gel-purified and cleaned up. Libraries were balanced and quality was assessed with Bioanalyzer High Sensitivity DNA Kit, Agilent #5067-4626).
Validation experiments
For validation experiment, 45 individual sgRNAs targeting the spliceosome were cloned into pLibrary containing EF1α-puro-T2A-BFP and made into lentivirus as described above. sgRNAs were selected for significant enrichment or depletion (LFC > |2|, padj < 0.05). Moreover, we took statistically not significantly enriched/depleted sgRNAs if they were strongly enriched (LFC > 2.75) or depleted (LFC < −3.5). The threshold was set to approximately mimic the lowest level of enrichment or depletion observed, respectively, for the statistically significant sgRNA. All depleted sgRNAs further had to fulfil LFC > −1 for a comparison of t14 vs. t0 in the control condition. In addition, three non-targeting sgRNAs were cloned into pLibrary containing EF1α-puro-T2A-mCherry and into pLibrary EF1α-puro-T2A-BFP (see Table S3 for full list and primers). Lentivirus was generated and titered as described above. 2e5 eHAP FNLS cells were transduced with individual sgRNA lentivirus in 6-well plates. Puromycin selection was started 36 h post-transduction and continued for four days. Care was taken to maintain all cells haploid (as diploid cells grow faster) and ploidy was checked with flowcytometry as described above. On day 6 (= t0) cells were grouped according to their growth density on plate and a representative sample was counted. An estimated 5500 cells per spliceosome sgRNA or non-targeting sgRNA carrying cells were each mixed with 5500 cells carrying sgNTC_400.
Treatment was started on the next day (DMSO vs. 2 nM PB) and cells were passaged as needed with treatment renewed every second day. The ratio of BFP:mCherry was assessed at t0, t4, t8 and t15.
Library and sequencing for base editing window
Lentivirus from validation experiment was used to transduce 2e5 eHAP FNLS cells at MOI 0.3 in a 6-well dish. After puromycin selection on day 6 (= t0), 2x 11000 cells were seeded in 96-well plates and split into treatment arms the next day (DMSO vs. 2 nM PB). Cells were propagated and harvested at t8 and t14 for gDNA extraction. Genomic DNA was extracted using QuickExtract™ DNA Extraction Solution (Lucigen #QE09050) and 1 μl (qsp. >200 cells) and was used for target site amplification using a 2-step PCR. In addition, each reaction contained 10 μl NEBnext Ultra II Q5 master mix as well as 1 μM of each forward and reverse primer. Primer pairs for PCR 1 were selected such that forward annealing primer is not closer than 7 nt to editing window position 1 but still allowing that 75 sequencing cycles will read the sequence. Primers were verified to anneal to a single position within the genome using BLAT 30 and tested before use. Primers for PCR 1 (12 cycles) were flanked for the forward primer by: 5’-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-[target specific sequence]-3’ and for the reverse primer by 5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-[target specific sequence]-3’. 1.5 μl of PCR 1 were used as template for PCR 2 (14 cycles) and used forward primers 5’-AATGATACGGCGACCACCGAGATCTACAC-NNNNNNNNACACTCTTTCCCTACACGAC-3’ (compatible to Illumina i5) and reverse primers 5’-CAAGCAGAAGACGGCATACGAGAT- NNNNNNNN- GTGACTGGAGTTCAGACGTG-3’ (compatible to Illumina i7), with both containing 8N barcodes for multiplexing (Table S3). Primers were removed after the second PCR step with AMPure XP Reagent (Beckman #A63882). Library was quantified (QuantiFluor® dsDNA System; Promega #E2670) before being pooled (10 fmol per sample) for each time point & condition, run on 8% TBE-PAGE gel, then size selected for a range of 250-500 bp. Pooled libraries were then run on Bioanalyzer High Sensitivity DNA Kit, Agilent #5067-4626) before final pooling and subsequent run on a MiniSeq Sequencing System using the Miniseq High Output Kit (75 cycles) (Illumina #FC-420-1001) with a 10% of phiX spike-in.
RNA-seq
eHAP FNLS and mutant cells were treated for 3 h with DMSO or 2 nM PB before cells were harvested. This was done for three replicates of an eHAP FNLS cell line transduced with a non-targeting sgRNA. Total RNA was extracted using the RNAqueous™-96 Total RNA Isolation Kit (ThermoFisher #AM1920) according to the manufacturer’s protocol. Poly(A)-enriched RNA was obtained with the poly(A) RNA Selection Kit V1.5 (Lexogen #157.96) and RNA-seq libraries were generated using the CORALL Total RNA-Seq Library Prep Kit (Lexogen #117.96). The libraries were then sequenced using paired-end 150 bp reads with 60 million reads per sample on Nova Seq (S4).
RNA extraction and RT-PCR
RNA was extracted from cells using the RNeasy Plus Mini Kit (Qiagen #74134) according to the manufacturer’s protocol. Reverse transcription using either Superscript III (Invitrogen #18080093) or Superscript IV (Invitrogen #18090050) was performed according to the manufacturer’s protocol using a mix of random hexamer primers and oligo(dT) using an input of 500 ng total RNA for a 10 μl reaction. PCR was performed with junction specific primers (Table S3) using 2% of the cDNA as an input for a 25 μl PCR reaction. PAGE was visualized using SYBR Gold (Invitrogen #S11494) and intensity of PCR products were quantified using ImageJ (NIH).
Proximity labeling sample preparation for MS
6 million HEK293T cells were seeded on 150 mm plates and transfected 24 hours later with 15 μg plasmid using jetPRIME reagent (at a 1:2.5 ratio for μg DNA:μl jetPRIME reagent; Polyplus #114-15). Transfection was performed in four independent biological replicates. Media was replaced with fresh culture media 5-6 h post transfection. At 24 h post transfection, a biotin pulse of 2 min biotin (culture media supplemented with 200 μM biotin) was used for proximity labelling. Cells were then immediately placed on ice and washed five times with ice cold DPBS (Corning #21-031-CV) before collection by gentle repeat pipetting. Cell pellets were lysed in 1500 μl ice cold RIPA buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1% NP40, 0.5% Na-deoxycholate, 0.1% SDS, 1 mM EDTA, Roche complete and 1 mM PMSF). Cell lysates were clarified by centrifugation (13000 x g at 4°C for 10 min) before quantification with BCA protein assay (Thermo #23227). 2.5 mg protein in 1500 μl RIPA buffer were added to 250 μl MyOne Streptavidin T1 Dynabeads (Invitrogen #65602), prewashed twice with 1 ml RIPA buffer. Protein and beads were incubated for 30 min at 4 °C with rotation to capture biotinylated proteins. Beads were then pelleted on magnet and washed twice with RIPA buffer (1 ml for 2 min at RT), washed once with 1 M KCL (1 ml for 2 min at RT), washed once with freshly made 0.1 M Na2CO3 (1 ml for 10 s), washed once with freshly made 2 M urea in 10 mM Tris.Cl pH 8 (1 ml for 10 s), before washing twice more with RIPA buffer (1 ml per wash, 2 min at RT). Beads were resuspended in 200 μl RIPA buffer before transfer to a new low binding tube. Beads were then washed in 200 μl 50 mM Tris.Cl pH 7.5, twice in 200 μl 2 M urea in 10 mM Tris.Cl pH 7.5, and twice in 200 μl H2O. Bead pellet was frozen for handover to MS facility.
On beads Digestion and TMT labelling
Sample-incubated streptavidin magnetic beads were resuspended in 9 μl 5 mM Tris(2-carboxyethyl)phosphine 20mM triethylammonium bicarbonate and incubated for 30 min at room temperature. After this, iodoacetamide was added to a final concentration of 7.5 mM, and samples incubated for 30 additional minutes. 1 μg of LysC (Fujifilm Wako Pure Chemical Corporation) was added to each sample and incubated at 37 °C overnight. Then 1 μg sequencing grade trypsin (Promega) was added to each sample and incubated at 37 °C overnight. Supernatants of the beads were recovered, and beads digested again using 0.5 ug trypsin in 100mM NH4HCO3 for 2 h. Peptides from both consecutive digestions were combined and recovered by solid phase extraction using C18 ZipTips (Millipore), eluted in 15 μl 50% acetonitrile 0.1% formic acid, and evaporated. Samples were then resuspended in 8 μl 0.1 M triethylammonium bicarbonate pH 8.0. Dried samples were labelled according to TMTPro™-16 label plex kit instructions (ThermoFisher Scientific). Briefly, TMT reagents were dissolved in acetonitrile at 12.5 μg/μl, and 4 μl of these stocks added to the samples. After incubation for 1 h at room temperature samples were quenched with 1 μl 5% hydroxylamine, and all 16 samples were combined, partially evaporated, and desalted using a C18 ZipTip as described before. The eluate was dried in preparation for LC-MSMS analysis.
Mass Spectrometry Analysis
Samples coming from RP fractionation were run onto a 2 μm, 75μm ID x 50 cm PepMap RSLC C18 EasySpray column (Thermo Scientific). 3-hour MeCN gradients (2–30% in 0.1% formic acid) were used to separate peptides, at a flow rate of 300 nl/min, for analysis in a Orbitrap Lumos Fusion (Thermo Scientific) in positive ion mode. MS spectra were acquired between 375 and 1500 m/z with a resolution of 120000. For each MS spectrum, multiply charged ions over the selected threshold (2E4) were selected for MSMS in cycles of 3 seconds with an isolation window of 0.7 m/z. Precursor ions were fragmented by HCD using stepped relative collision energies of 30, 35 and 40 to ensure efficient generation of sequence ions as well as TMT reporter ions. MSMS spectra were acquired in centroid mode with resolution 60000 from m/z=110. A dynamic exclusion window was applied which prevented the same m/z (mass tolerance 30 ppm) from being selected for 30 s after its acquisition.
Immunoblotting
Cell lysates were mixed with NuPAGE LDS Sample Buffer (Invitrogen #NP00007), heated for 5 min at 95 °C, separated on SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were either incubated for 1 h at RT or overnight at 4 °C with the following primary antibodies in TBS-T with 5% milk: α-HA (Cell Signalling Technology, #3724) at 1:5000; α-GAPDH-HRP (Proteintech, #HRP-60004) at 1:10,000; α-SUGP1 (Bethyl Laboratories, #A204-675-M) at 1:1000. HRP-conjugated streptavidin (Invitrogen, #S911) reconstituted at 1 mg/ml was used at 0.3 μg/ml in 3% (w/v) BSA in 1x TBST and blots were only incubated for 30 min in its presence.
Protein expression and purification
For pulldown and RNA binding assays, MBP-hsSUGP1 variants were expressed in E. coli BL21 Star (DE3) (Invitrogen). Cells were grown at 37°C in LB medium until an OD600 of 0.6 was reached. Protein expression was induced with 2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and maintained at 37°C for 3 h. Expression cultures were harvested by centrifugation and cell pellets were resuspended in lysis buffer (50 mM HEPES, pH 7.5, 200 mM NaCl, 2 mM dithiothreitol [DTT]) supplemented with cOmplete EDTA-free protease inhibitor mixture (Roche), 1 mg/mL lysozyme (Sigma), and 5 μg/mL DNaseI (Roche). For cell lysis, the suspension was passaged through a LM10 Microfluidizer. Subsequently, the lysate was cleared by centrifugation at 3200 g for 10 min and filtered (0.45 μm). Lysates were incubated with preequilibrated amylose beads (New England BioLabs) for 1h at 4°C. Beads were washed with lysis buffer and bound proteins were eluted with lysis buffer containing 25 mM maltose. The eluates were concentrated, loaded onto a gel-filtration column (Superdex 200, GE Healthcare) and eluted in size-exclusion buffer (10 mM HEPES, pH 7.5, 200 mM NaCl, 2 mM DTT). The MBP-hsSUGP1 variants were either directly used in biochemical assays or flash-frozen in liquid nitrogen and stored at −80°C. For the ATPase assay, MBP-hsSUGP1 variants were subjected to additional washes while bound to the amylose beads during the first purification step to remove ATPase contamination. Bound proteins were incubated with lysis buffer supplemented with 2 mM ATP and 2 mM MgCl2 for 10 min at 4°C and washed with lysis buffer containing 1 M NaCl. After the high-salt wash, beads were equilibrated again in lysis buffer before elution as described above.
DHX15ΔN was expressed in insect cells using baculovirus infection. Sf9 cells and HighFive cells (Thermo Fisher) grown in SF-4 Baculo Express medium (Bioconcept) were used for virus production and protein expression, respectively. Viruses were amplified in Sf9 cells for two generations (V2) prior to infection and protein expression in HighFive cells. For the expression of 10xHis-DHX155ΔN, insect cells were infected with V2 virus and grown for 48 h. Cell harvest and lysate generation was performed as for SUGP1 with the exception that cells were resuspended in wash buffer (50 mM HEPES, pH 7.5, 200 mM NaCl, 2 mM β-mercaptoethanol). Cleared lysates were loaded onto a pre-equilibrated nickel affinity column (HiTrap Chelating HP, GE Healthcare), washed with wash buffer and eluted over a linear gradient to 500 mM imidazole. Eluates were then purified over a heparin column (HiTrap Heparin HP, GE Healthcare) in lysis buffer, eluted over a linear salt gradient to 1 M NaCl and ultimately, subjected to gel filtration (Superdex 200) in size-exclusion buffer. Fractions containing 10xHis-DHX15ΔN were collected, concentrated and either used directly for biochemical analyses or flash frozen in liquid nitrogen for storage at −80°C.
Protein binding assays
For interaction studies, purified His10-hsDHX15ΔN and MBP-hsSUGP1 variants were mixed in equimolar amounts in pulldown buffer (50 mM Hepes, pH 7.5, 200 mM NaCl, 2 mM DTT). His6-MBP was used as a negative control. Proteins were incubated with 50 μl of amylose beads (50% slurry in pulldown buffer) for 1 h at 4°C on a rotator. The beads were washed three times in pulldown buffer. Bound proteins were eluted with pulldown buffer containing 25 mM maltose. Proteins of input and eluate samples were separated by sodium dodecyl sulfate (SDS)/polyacrylamide gel electrophoresis (PAGE) and were visualized by Coomassie staining.
RNA Binding Assays
RNA binding affinities of His10-hsDHX15ΔN in complex with MBP-hsSUGP1 variants were determined by measuring changes of fluorescence polarization (FP) in dependence of protein concentration, as previously described 57. Experiments were performed in binding buffer (20 mM Hepes (pH 7.5), 150 mM NaCl, 5% glycerol and 2 mM MgCl2) with 10 nM 5’-6-fluorescein amidites (FAM)-labelled U12 RNA (Microsynth) and protein concentrations ranging from 1 nM to 32 μM. FP was determined using a CLARIOstar microplate reader (BMG Labtech) by excitation at 482 nm and detection at 530 nm wavelength. All samples were measured five times and all samples were prepared in triplicates. After baseline subtraction, the obtained FP values were normalized to 1 and fitted according to Rossi et al. using GraphPrad Prism 51.
ATPase assays
Activity of hsDHX15 and its interactor hsSUGP1 were monitored using an NADH-coupled ATPase assay. For all measurements of ATPase activity, 1.8 μM His10-hsDHX15ΔN and 1.8 μM MBP-hsSUGP1 variants (G-patch (548-611) were mixed in ATPase buffer containing 50 mM Hepes pH 7.5, 50 mM KAc, 5 mM MgAc2, 2 mM DTT, 0.5 mM nicotinamide adenine dinucleotide, 1 mM phosphoenolpyruvate, 12 U of pyruvate kinase, and 18 U of lactate dehydrogenase. All measurements were carried out in half area 96-well plates (Greiner). After equilibration for 10 min at 37°C, reactions were started by adding 250 μM ATP (pH 7.5). Absorption at 340 nm was measured over a time course of 40 min at 37°C with one measurement per minute using a CLARIOstar microplate reader. Absorption change in the absence of ATP was measured for baseline correction. Absorption values were adjusted to a path length of 1 cm. Absorption change over time was determined by linear regression and converted to concentration change over time with an extinction coefficient at 340 nm of 6,220 M−1cm−1 using Beer–Lambert’s law. Initial velocities were derived from concentration change over time using a total enzyme concentration of 1.8 μM. All measurements were prepared in triplicates. Absence of ATPase contaminations from hsSUGP1 preparations was confirmed by measuring ATPase activity in the absence of hsDHX15ΔN.
DATA ANALYSIS
Screen analysis
The sequencing data was demultiplexed to obtain individual samples for timepoints and trimmed (BBMap BBDuk v.38.94). Reads were counted by alignment to a reference file of all sgRNAs present in the pool. In the next step, each barcode was randomly assigned to either of two replicates. 32 sgRNA targeted 2 loci and their reads were duplicated and assigned to both targets. Log2-fold changes between samples were calculated using DESeq2, filtering out reads with on average less than 50 reads across all samples 40.
Identification of causative mutations
Editing at the base editing window was quantified using CRISPResso2 v.2.1 49, run with the following parameters: [--exclude_bp_from_left 18 --exclude_bp_from_right 0 -- quantification_window_center -12 --quantification_window_size 15 --min_average_read_quality 30 --default_min_aln_score 60 --plot_window_size 20 --base_editor_output --output_folder 20210531_pos -p 12]. For analysis we used the “Alleles_frequency_table_around_[sgRNA].txt” output files or in the single case where editing occurred far outside the editing window “Alleles_frequency_table.txt” was used for information extraction.
Samples were processed to only consider those with 100 or more reads per condition across all conditions. In addition, all alleles with <1% in all conditions were removed before recalculating percent distribution of alleles. Alleles were translated and grouped by protein sequence outcome and for each percent distribution was summed. Log2-fold change was calculated for samples
Validation of PB sensitive samples:
For samples depleted in the screen, only sequencing data corresponding to t0 were considered. Mutational outcome was noted for any event with >5% frequency.
Validation of PB resistant samples:
All timepoints and treatment conditions were considered for the analysis.
Splicing analysis
The RNA-seq data was demultiplexed, trimmed (BBMap BBDuk v.38.94), and then mapped using STAR 2.7.9 15 [--alignSJoverhangMin 8 --alignSJDBoverhangMin 1 -- outFilterMismatchNmax 999 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --peOverlapNbasesMin 10 --peOverlapMMp 0.2] and SAMtools (v1.10) for conversion to sorted bam files. Reads were then deduplicated using the UMIs from the Lexogen CORALL workflow to prevent removal of natural duplicates. On average, 85% of the reads could be mapped and quality assessment using the RSeQC package 64 showed neither a bias in read distribution nor gene body coverage. Differential expression of genes was assessed using RSubreads (v2.6.4) 38 for counting and DESeq2 40. To analyze splicing at the exon level only junctions with more than 10 junction reads were considered. We used rMATS-turbo (v4.1.2) 56 (hereafter, rMATS) for quantification. For all analysis performed with rMATS only splicing junctions with FDR < 0.01 were considered. Alternatively, we also used MAJIQ (v2.4) 46 with |dPSI] ≥10 and Confidence Threshold > 90%. For analysis of intron and exon features we used the software matt 21. When preparing figures with IGV, junction reads with low numbers were removed for clarity
Peptide and protein identification and TMT quantitation.
Peak lists were generated using PAVA in-house software 23. All generated peak lists were searched against the human subset of the SwissProt database (SwissProt.2019.07.31), using Protein Prospector 10 with the following parameters: Enzyme specificity was set as Trypsin, and up to 2 missed cleavages per peptide were allowed. Carbamidomethylation of cysteine residues, and TMTPro16plex labelling of lysine residues and N-terminus of the protein were allowed as fixed modifications. N-acetylation of the N-terminus of the protein, loss of protein N-terminal methionine, pyroglutamate formation from of peptide N-terminal glutamines, and oxidation of methionine were allowed as variable modifications. Mass tolerance was 10 ppm in MS and 30 ppm in MS/MS. The false positive rate was estimated by searching the data using a concatenated database which contains the original SwissProt database, as well as a version of each original entry where the sequence has been randomized. A 1% FDR was permitted at the protein and peptide level. For quantitation only unique peptides were considered; peptides common to several proteins were not used for quantitative analysis. Relative quantization of peptide abundance was performed via calculation of the intensity of reporter ions corresponding to the different TMT labels, present in MS/MS spectra. Intensities were determined by Protein Prospector. Summed intensity per sample on each TMT channel for all identified carboxylases were used to normalize individual intensity values. Relative abundances were calculated as ratios vs the average intensity levels in the 4 channels corresponding to control samples. For total protein relative levels, peptide ratios were aggregated to the protein levels using median values of the log2 ratios.
QUANTIFICATION AND STATISTICAL ANALYSIS
For visualisation of cryo-EM and crystal structures we used PyMOL (v2.3.5), for visual presentation we used R ggplot2 (v3.3.6) with MetBrewer (v0.2.0.) and ggVennDiagram (v1.2.0) or pheatmap (v1.0.12), and GraphPad Prism (v8), and the Integrative Genomics Viewer (IGV) (v2.12.2). Schematics were created with Adobe Illustrator 2023.
All statistical analyses were performed in R (v4.1.2) using base R or package multcomp (v.1.4-20). Statistical significance of one-way ANOVA or t-test is indicated by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001), unless otherwise indicated.
Supplementary Material
Document S1. Figures S1–S6.
Table S1. Targeted spliceosomal proteins. Related to Figure 1.
Table S2. Sensitive and resistant mutations to pladienolide B identified by targeted sequencing of editing site. Related to Figures 2, 3, and 4.
Table S3. Primers for cloning, sequencing library generation, and RT-PCR. Related to STAR Methods.
Table S4. Read counts and sgRNA annotations from Variant Effect Predictor (do not open with MS Excel). Related to Figures 1, 2, 3, 4, and STAR Methods.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-rabbit IgG (H + L)-HRP conjugate (goat) | Biorad | Cat #: 1706516 |
| Anti-GAPDH HRP | Proteintech | Cat #: HRP-60004 |
| anti-HA | Cell Signaling Technology | Cat #: 3724 |
| Anti-SUGP1 | Bethyl Laboratories | Cat #: A304-675A-M |
| Streptavidin, HRP conjugate | Invitrogen | Cat #: S911 |
| Bacterial and virus strains | ||
| NEB® Stable Competent E. coli | New England Biolabs | Cat #: C3040H |
| MegaX DH10B T1R Electrocomp™ Cells | Invitrogen | Cat #: C640003 |
| Escherichia coli BL21 Star (DE3) | Invitrogen | Cat #: C601003 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| 10-deacetylbaccatin-III (DAB) | Selleckchem | Cat #: S2409 |
| Pladienolide B | Santa Cruz | Cat #: Sc-391691 |
| Biotin | Sigma-Aldrich | Cat #: B4501-500MG |
| Blasticidin | Gibco | Cat #: A1113903 |
| Puromycin | Invivogen | Cat #: ant-pr-1 |
| Transfection reagent: JetPRIME | Polyplus | Cat #: 114-15 |
| SYBR Gold | Invitrogen | Cat #: S11494 |
| NEBuilder® HiFi DNA Assembly | New England Biolabs | Cat #: E2621 |
| NEBnext® Ultra II Q5 Master Mix | New England Biolabs | Cat #: M0544 |
| SuperScript III | Invitrogen | Cat #: 18080093 |
| SuperScript IV | Invitrogen | Cat #: 18090050 |
| MyOne Streptavidin T1 Dynabeads | Invitrogen | Cat #: 65602 |
| Amylose Resin High Flow | New England Biolabs | Cat #: E8022L |
| cOmplete EDTA-free Protease Inhibitor Cocktail | Roche | Cat #: 5056489001 |
| Nicotinamide adenine dinucleotide (NADH) | Carl Roth | Cat #: AE12.1 |
| Phosphoenolpyruvic acid (PEP) | Carl Roth | Cat #: 8397.3 |
| Lactate dehydrogenase (LDH) | Carl Roth | Cat #: 6060.1 |
| Adenosine 5’-Triphosphoric Acid Disodium (ATP) | PanReac AppliChem | Cat #: A1348 |
| Lysozyme | Carl Roth | Cat #: 8259.3 |
| DNase I | PanReac AppliChem | Cat #: 10027460 |
| Critical commercial assays | ||
| CellTiter 96® AQueous One Solution Cell Proliferation Assay | Promega | Cat #: G3582 |
| RNAqueous™-96 Total RNA Isolation Kit | Thermo Fisher | Cat #: AM1920 |
| Poly(A) RNA Selection Kit V1.5 | Lexogen | Cat #: 157.96 |
| CORALL Total RNA-Seq Library Prep Kit with UDI12 nt Set A1 | Lexogen | Cat #: 117.96 |
| PCR Add-on Kit for Illumina | Lexogen | Cat #: 020.96 |
| QuantiFluor RNA System | Promega | Cat #: E3310 |
| Quick Extract DNA Extraction solution | Lucigen | Cat #: QE09050 |
| QIAmp DNA Blood Maxi Kit | Qiagen | Cat #: 51194 |
| Agencourt AMPure XP | Beckman Coulter | Cat #: A63882 |
| RNeasy Plus Mini Kit | Qiagen | Cat #: 74134 |
| BCA protein assay | Thermo Scientific | Cat #: 23227 |
| TMTPro™-16 label plex kit | Thermo Scientific | Cat #: A44521 |
| Deposited data | ||
| All sequencing data (screen, validation, RNA-seq) | This study | GEO: GSE218307 |
| Proximity labeling proteomics data | This study | PRIDE: PXD038067 |
| Human reference genome NCBI guild 38, GRCh38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/human |
| Protein structure: SF3b core with splicing modulator | Cretu et al., 2018 | PDB: 6EN4 |
| Protein structure: substrate-bound A-like U2 snRNP | Tholen et al., 2022 | PDB: 7Q4O |
| Protein structure: chimeric peptide SF3A1 SURP and SF1 | Nameki et al., 2022 | PDB: 7VH9 |
| Protein structure: human Bact spliceosome core | Haselbach et al., 2018 | PDB: 6FF7 |
| Protein structure: human C* spliceosome core | Zhang et al., 2017 | PDB: 5XJC |
| Protein structure: human SUGP1 433/586 | Zhang et al., 2022 | PDB: 8GXM |
| Protein structure: DHX15 in complex with SUGP1 G-patch | Zhang et al., 2022 | PDB: 8EJM |
| Protein structure: DHX15 in complex with NKRF G-patch | Studer et al., 2020 | PDB: 8SH7 |
| Experimental models: Cell lines | ||
| Human: fully-haploid engineered HAP1 (eHAP) wild-type | Horizon Discovery Ltd. | Cat #: C669 |
| Human: eHAP FNLS (and derivatives thereof) | This study | N/A |
| Human: HEK 293T (for lentivirus production) | ATCC | Cat #: CRL-3216 |
| High Five Cells (for protein production) | Gibco | Cat #: B85502 |
| Sf9 cells (for baculovirus production) | Gibco | Cat #: 11496015 |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| See Table S3 | This study | N/A |
| 5’-6-fluorescein amidites (FAM)-U12 | Microsynth | N/A |
| Recombinant DNA | ||
| pMP783 (pLibrary) | Boettcher et al., 2019 | N/A |
| pLenti-FNLS-P2A-GFP-PGK-Puro | Zafra et al., 2018 | Addgene #110869 |
| pFNLS | This study | N/A |
| p3xFLAG-CMV-14_3xHA_SUGP1 | Zhang et al., 2019a | N/A |
| p3xFLAG-CMV-14_3xHA_SUGP1 E554K | This study | N/A |
| p3xFLAG-CMV-14_3xHA_SUGP1 L570E | This study | N/A |
| p3xFLAG-CMV-14_3xHA_SUGP1 G603N | This study | N/A |
| p3xFLAG-CMV-14_3xHA_SUGP1 Δ αH4-5 | This study | N/A |
| p3xFLAG-CMV-14_3xHA_SUGP1 Δ αH6 | This study | N/A |
| pCDNA3_3xHA-miniTurbo-NLS | Branon et al., 2018 | Addgene #107172 |
| pCDNA3_3xHA-SUGP1-wt-miniTurbo-NLS | This study | N/A |
| pCDNA3_3xHA-SUGP1-L570E-miniTurbo-NLS | This study | N/A |
| pCDNA3_3xHA-SUGP1-G603N-miniTurbo-NLS | This study | N/A |
| psPAX2 (for lentivirus production) | N/A | Addgene #12260 |
| pMD2.G (for lentivirus production) | N/A | Addgene #12259 |
| pFastBac-NpH-DHX15ΔN | Studer et al., 2020 | N/A |
| pnEA-NpH-MBP | Haffke et al., 2015 | N/A |
| pnEA-NpM-SUGP1 G-patch (548-611) wt | This study | N/A |
| pnEA-NpM-SUGP1 G-patch (548-611) E554K | This study | N/A |
| pnEA-NpM-SUGP1 G-patch (548-611) L570E | This study | N/A |
| pnEA-NpM-SUGP1 G-patch (548-611) G603N | This study | N/A |
| pnEA-NpM-SUGP1 αH6-α7 (522-633) wt | This study | N/A |
| pnEA-NpM-SUGP1 αH6-α7 (522-633) E554K | This study | N/A |
| pnEA-NpM-SUGP1 αH6-α7 (522-633) L570E | This study | N/A |
| pnEA-NpM-SUGP1 αH6-α7 (522-633) G603N | This study | N/A |
| pnEA-NpM-SUGP1 αH4-α7 (522-633) wt | This study | N/A |
| pnEA-NpM-SUGP1 αH4-α7 (522-633) E554K | This study | N/A |
| pnEA-NpM-SUGP1 αH4-α7 (522-633) L570E | This study | N/A |
| pnEA-NpM-SUGP1 αH4-α7 (522-633) G603N | This study | N/A |
| Software and algorithms | ||
| CHOPCHOP | Labun et al., 2019 | https://chopchop.cbu.uib.no/ |
| CRISPResso2 (v2.1) | Pinello et al., 2016 | https://github.com/pinellolab/CRISPResso2 |
| Ensembl Variant Effect Predictor (Ensembl release 99) | McLaren et al., 2016 | https://useast.ensembl.org/info/docs/tools/vep/index.html |
| BBDuk (v38.94) | BBtools | http://sourceforge.net/projects/bbmap/ |
| DESeq2 (v1.32.0) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| RSubreads (v2.6.4) | Liao et al., 2019 | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| rMATS-turbo (v4.1.2) | Shen et al., 2014 | https://github.com/Xinglab/rmats-turbo |
| MAJIQ (v2.4) | Mehmood et al., 2020 | https://majiq.biociphers.org/ |
| STAR (v2.7.9) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Samtools (v1.10) | Danecek et al., 2021 | https://github.com/samtools/samtools |
| IGV (v2.12.2) | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| R (v4.1.2) | The R Project | https://www.r-project.org/ |
| R Studio | R Studio | https://github.com/rstudio/rstudio |
| Prism (v8) | GraphPad Software | https://www.graphpad.com/features |
| FlowJo (v10.8.1) | TreeStar Inc. | https://www.flowjo.com/ |
| PyMOL (v2.3.5) | Schroödinger | https://www.schrodinger.com/products/pymol |
| JalView (v2.11.2.5) | Waterhouse et al., 2009 | https://www.jalview.org/ |
| ImageJ (v1.53) | Schneider et al., 2012 | https://imagej.nih.gov/ij/download.html |
| Other | ||
| Superdex 200 Increase 10/300 GL | Cytiva | Cat #: 28990944 |
Highlights.
Global mutagenesis of the human spliceosome using CRISPR-Cas9 base editing
Pladienolide B hypersensitive and resistance mutants identified
Resistance mutations in SUGP1 G-patch disrupt interaction with DHX15/hPrp43
SUGP1-DHX15/hPrp43 may mediate an early spliceosome proofreading step
Acknowledgements
We dedicate this paper to the memory of Dr. Christine Guthrie. We thank members of the Madhani lab for discussions and support. We thank Stefan Oberlin (UCSF) for advice on bioinformatics analysis and CRISPR screens, Lukas E. Dow (Cornell) and David R. Liu (Harvard) for advice on base editing, and Daniël van Leeuwen and Chris Richards for advice regarding HAP1 cells. We thank Qing Feng and Chris Burge for communicating unpublished results and for technical advice. Sequencing data was generated at the UCSF CAT (supported by UCSF PBBR, RRP IMIA, and NIH 1S10OD028511-01 grants) and through the kind use of their NextSeq System by Venice Servellita and Charles Chiu. I.B. was supported by the Swiss National Science Foundation (SNSF) through an SNF Postdoctoral Fellowship (191929 and 203008). This work was supported by R01 GM71801 (to H.D.M.). Mass Spectrometry was provided by the Mass Spectrometry Resource at UCSF (A.L. Burlingame, Director) supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF) and the UCSF Program for Breakthrough Biomedical Research (PBBR). Work in the Jonas lab was supported by the SNSF through the National Center for Excellence in Research “RNA & Disease” (grant number 205601) and through SNSF project grants (grant numbers 179498 and 207458).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Alsafadi S, Dayot S, Tarin M, Houy A, Bellanger D, Cornella M, Wassef M, Waterfall JJ, Lehnert E, Roman-Roman S, et al. (2021). Genetic alterations of SUGP1 mimic mutant-SF3B1 splice pattern in lung adenocarcinoma and other cancers. Oncogene 40, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anzalone AV, Koblan LW, and Liu DR (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824–844. [DOI] [PubMed] [Google Scholar]
- 3.Beigl TB, Kjosas I, Seljeseth E, Glomnes N, and Aksnes H (2020). Efficient and crucial quality control of HAP1 cell ploidy status. Biol Open 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bejar R (2016). Splicing Factor Mutations in Cancer. Advances in experimental medicine and biology 907, 215–228. [DOI] [PubMed] [Google Scholar]
- 5.Blencowe BJ (2017). The Relationship between Alternative Splicing and Proteomic Complexity. Trends in biochemical sciences 42, 407–408. [DOI] [PubMed] [Google Scholar]
- 6.Boettcher M, Covarrubias S, Biton A, Blau J, Wang H, Zaitlen N, and McManus MT (2019). Tracing cellular heterogeneity in pooled genetic screens via multi-level barcoding. BMC Genomics 20, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, and Ting AY (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol 36, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brosi R, Hauri HP, and Krämer A (1993). Separation of splicing factor SF3 into two components and purification of SF3a activity. Journal of Biological Chemistry 268, 17640–17646. [PubMed] [Google Scholar]
- 9.Burgess SM, and Guthrie C (1993). A mechanism to enhance mRNA splicing fidelity: the RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell 73, 1377–1391. [DOI] [PubMed] [Google Scholar]
- 10.Clauser KR, Baker P, and Burlingame AL (1999). Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem 71, 2871–2882. [DOI] [PubMed] [Google Scholar]
- 11.Cretu C, Agrawal AA, Cook A, Will CL, Fekkes P, Smith PG, Luhrmann R, Larsen N, Buonamici S, and Pena V (2018). Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol Cell 70, 265–273 e268. [DOI] [PubMed] [Google Scholar]
- 12.Cretu C, Gee P, Liu X, Agrawal A, Nguyen TV, Ghosh AK, Cook A, Jurica M, Larsen NA, and Pena V (2021). Structural basis of intron selection by U2 snRNP in the presence of covalent inhibitors. Nat Commun 12, 4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuella-Martin R, Hayward SB, Fan X, Chen X, Huang JW, Taglialatela A, Leuzzi G, Zhao J, Rabadan R, Lu C, et al. (2021). Functional interrogation of DNA damage response variants with base editing screens. Cell 184, 1081–1097 e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Essletzbichler P, Konopka T, Santoro F, Chen D, Gapp BV, Kralovics R, Brummelkamp TR, Nijman SM, and Burckstummer T (2014). Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res 24, 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Q, Krick K, Chu J, and Burge CB (2022). Splicing quality control mediated by DHX15 and its G-patch activator, SUGP1. bioRxiv, 2022.2011.2014.516533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fica SM, Oubridge C, Wilkinson ME, Newman AJ, and Nagai K (2019). A human postcatalytic spliceosome structure reveals essential roles of metazoan factors for exon ligation. Science 363, 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamboa Lopez A, Allu SR, Mendez P, Chandrashekar Reddy G, Maul-Newby HM, Ghosh AK, and Jurica MS (2021). Herboxidiene Features That Mediate Conformation-Dependent SF3B1 Interactions to Inhibit Splicing. ACS Chem Biol 16, 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gohr A, and Irimia M (2019). Matt: Unix tools for alternative splicing analysis. Bioinformatics 35, 130–132. [DOI] [PubMed] [Google Scholar]
- 22.Gradia SD, Ishida JP, Tsai MS, Jeans C, Tainer JA, and Fuss JO (2017). MacroBac: New Technologies for Robust and Efficient Large-Scale Production of Recombinant Multiprotein Complexes. Methods Enzymol 592, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guan W, Orellana KG, Stephens RF, Zhorov BS, and Spafford JD (2022). A lysine residue from an extracellular turret switches the ion preference in a Cav3 T-Type channel from calcium to sodium ions. J Biol Chem, 102621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haffke M, Marek M, Pelosse M, Diebold ML, Schlattner U, Berger I, and Romier C (2015). Characterization and production of protein complexes by co-expression in Escherichia coli. Methods Mol Biol 1261, 63–89. [DOI] [PubMed] [Google Scholar]
- 25.Hanna RE, Hegde M, Fagre CR, DeWeirdt PC, Sangree AK, Szegletes Z, Griffith A, Feeley MN, Sanson KR, Baidi Y, et al. (2021). Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080 e1020. [DOI] [PubMed] [Google Scholar]
- 26.Haselbach D, Komarov I, Agafonov DE, Hartmuth K, Graf B, Dybkov O, Urlaub H, Kastner B, Luhrmann R, and Stark H (2018). Structure and Conformational Dynamics of the Human Spliceosomal Bact Complex. Cell 172, 454–464.e411. [DOI] [PubMed] [Google Scholar]
- 27.Hemsley A, Arnheim N, Toney MD, Cortopassi G, and Galas DJ (1989). A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res 17, 6545–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irimia M, and Roy SW (2008). Evolutionary convergence on highly-conserved 3’ intron structures in intron-poor eukaryotes and insights into the ancestral eukaryotic genome. PLoS Genet 4, e1000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kent WJ (2002). BLAT---The BLAST-Like Alignment Tool. Genome Research 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kluesner MG, Lahr WS, Lonetree CL, Smeester BA, Qiu X, Slipek NJ, Claudio Vazquez PN, Pitzen SP, Pomeroy EJ, Vignes MJ, et al. (2021). CRISPR-Cas9 cytidine and adenosine base editing of splice-sites mediates highly-efficient disruption of proteins in primary and immortalized cells. Nat Commun 12, 2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kluesner MG, Nedveck DA, Lahr WS, Garbe JR, Abrahante JE, Webber BR, and Moriarity BS (2018). EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J 1, 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koodathingal P, Novak T, Piccirilli JA, and Staley JP (2010). The DEAH box ATPases Prp16 and Prp43 cooperate to proofread 5’ splice site cleavage during pre-mRNA splicing. Mol. Cell 39, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koodathingal P, and Staley JP (2013). Splicing fidelity: DEAD/H-box ATPases as molecular clocks. RNA Biol. 10, 1073–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, and Valen E (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47, W171–W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Hir H, Sauliere J, and Wang Z (2016). The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol 17, 41–54. [DOI] [PubMed] [Google Scholar]
- 37.Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, and Pritchard JK (2016). RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao Y, Smyth GK, and Shi W (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic acids research 47, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z, Zhang J, Sun Y, Perea-Chamblee TE, Manley JL, and Rabadan R (2020). Pan-cancer analysis identifies mutations in SUGP1 that recapitulate mutant SF3B1 splicing dysregulation. Proc Natl Acad Sci U S A 117, 10305–10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin A, Schneider S, and Schwer B (2002). Prp43 is an essential RNA-dependent ATPase required for release of lariat-intron from the spliceosome. J Biol Chem 277, 17743–17750. [DOI] [PubMed] [Google Scholar]
- 42.Maul-Newby HM, Amorello AN, Sharma T, Kim JH, Modena MS, Prichard BE, and Jurica MS (2022). A model for DHX15 mediated disassembly of A-complex spliceosomes. RNA (New York, N.Y.) 28, 583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayas RM, Maita H, Semlow DR, and Staley JP (2010). Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proc. Natl. Acad. Sci. U. S. A 107, 10020–10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayas RM, Maita H, and Staley JP (2006). Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat. Struct. Mol. Biol 13, 482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mehmood A, Laiho A, Venalainen MS, McGlinchey AJ, Wang N, and Elo LL (2020). Systematic evaluation of differential splicing tools for RNA-seq studies. Brief Bioinform 21, 2052–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nameki N, Takizawa M, Suzuki T, Tani S, Kobayashi N, Sakamoto T, Muto Y, and Kuwasako K (2022). Structural basis for the interaction between the first SURP domain of the SF3A1 subunit in U2 snRNP and the human splicing factor SF1. Protein Sci 31, e4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez-Riverol Y, Bai J, Bandla C, Garcia-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, et al. (2022). The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, and Yuan GC (2016). Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol 34, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossi AM, and Taylor CW (2011). Analysis of protein-ligand interactions by fluorescence polarization. Nat Protoc 6, 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sales-Lee J, Perry DS, Bowser BA, Diedrich JK, Rao B, Beusch I, Yates JR 3rd, Roy SW, and Madhani HD (2021). Coupling of spliceosome complexity to intron diversity. Curr Biol 31, 4898–4910 e4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sánchez-Rivera FJ, Diaz BJ, Kastenhuber ER, Schmidt H, Katti A, Kennedy M, Tem V, Ho Y-J, Leibold J, Paffenholz SV, et al. (2022). Base editing sensor libraries for high-throughput engineering and functional analysis of cancer-associated single nucleotide variants. Nature biotechnology 40, 862–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Semlow DR, and Staley JP (2012). Staying on message: ensuring fidelity in pre-mRNA splicing. Trends in biochemical sciences 37, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Studer MK, Ivanovic L, Weber ME, Marti S, and Jonas S (2020). Structural basis for DEAH-helicase activation by G-patch proteins. Proc Natl Acad Sci U S A 117, 7159–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanaka N, Aronova A, and Schwer B (2007). Ntr1 activates the Prp43 helicase to trigger release of lariat-intron from the spliceosome. Genes Dev 21, 2312–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, et al. (2017). Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat Commun 8, 15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, et al. (2017). Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nature communications 8, 15522–15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tholen J, Razew M, Weis F, and Galej WP (2022). Structural basis of branch site recognition by the human spliceosome. Science 375, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsai R-T, Fu R-H, Yeh F-L, Tseng C-K, Lin Y-C, Huang Y-H, and Cheng S-C (2005). Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev. 19, 2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wahl MC, Will CL, and Luhrmann R (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. [DOI] [PubMed] [Google Scholar]
- 64.Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- 65.Warkocki Z, Schneider C, Mozaffari-Jovin S, Schmitzova J, Hobartner C, Fabrizio P, and Luhrmann R (2015). The G-patch protein Spp2 couples the spliceosome-stimulated ATPase activity of the DEAH-box protein Prp2 to catalytic activation of the spliceosome. Genes Dev 29, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waterhouse AM, Procter JB, Martin DM, Clamp M, and Barton GJ (2009). Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilkinson ME, Charenton C, and Nagai K (2020). RNA Splicing by the Spliceosome. Annu Rev Biochem 89, 359–388. [DOI] [PubMed] [Google Scholar]
- 68.Wu G, Fan L, Edmonson MN, Shaw T, Boggs K, Easton J, Rusch MC, Webb TR, Zhang J, and Potter PM (2018). Inhibition of SF3B1 by molecules targeting the spliceosome results in massive aberrant exon skipping. RNA 24, 1056–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yokoi A, Kotake Y, Takahashi K, Kadowaki T, Matsumoto Y, Minoshima Y, Sugi NH, Sagane K, Hamaguchi M, Iwata M, et al. (2011). Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS Journal 278, 4870–4880. [DOI] [PubMed] [Google Scholar]
- 70.Yoshimi A, Lin KT, Wiseman DH, Rahman MA, Pastore A, Wang B, Lee SC, Micol JB, Zhang XJ, de Botton S, et al. (2019). Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 574, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zafra MP, Schatoff EM, Katti A, Foronda M, Breinig M, Schweitzer AY, Simon A, Han T, Goswami S, Montgomery E, et al. (2018). Optimized base editors enable efficient editing in cells, organoids and mice. Nat Biotechnol 36, 888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J, Ali AM, Lieu YK, Liu Z, Gao J, Rabadan R, Raza A, Mukherjee S, and Manley JL (2019). Disease-Causing Mutations in SF3B1 Alter Splicing by Disrupting Interaction with SUGP1. Mol Cell 76, 82–95 e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang J, Huang J, Xu K, Xing P, Huang Y, Liu Z, Tong L, and Manley JL (2022). DHX15 is involved in SUGP1-mediated RNA missplicing by mutant SF3B1 in cancer. Proc Natl Acad Sci U S A 119, e2216712119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang X, Yan C, Hang J, Finci LI, Lei J, and Shi Y (2017). An Atomic Structure of the Human Spliceosome. Cell 169, 918–929 e914. [DOI] [PubMed] [Google Scholar]
- 75.Zhang X, Yan C, Zhan X, Li L, Lei J, and Shi Y (2018). Structure of the human activated spliceosome in three conformational states. Cell Res 28, 307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Z, Rigo N, Dybkov O, Fourmann J-B, Will CL, Kumar V, Urlaub H, Stark H, and Luhrmann R (2021). Structural insights into how Prp5 proofreads the pre-mRNA branch site. Nature 596, 296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Z, Will CL, Bertram K, Dybkov O, Hartmuth K, Agafonov DE, Hofele R, Urlaub H, Kastner B, Luhrmann R, et al. (2020). Molecular architecture of the human 17S U2 snRNP. Nature 583, 310–313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S6.
Table S1. Targeted spliceosomal proteins. Related to Figure 1.
Table S2. Sensitive and resistant mutations to pladienolide B identified by targeted sequencing of editing site. Related to Figures 2, 3, and 4.
Table S3. Primers for cloning, sequencing library generation, and RT-PCR. Related to STAR Methods.
Table S4. Read counts and sgRNA annotations from Variant Effect Predictor (do not open with MS Excel). Related to Figures 1, 2, 3, 4, and STAR Methods.
Data Availability Statement
All data discussed in this publication (CRISPR-Cas9 base editing screen including read counts, validation experiments, and RNA-seq) have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE218307). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE48 partner repository with the dataset identifier PXD038067.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.