

Abstract
Tertiary stereogenic centres containing one fluorine atom are valuable for medicinal chemistry because they mimic common tertiary stereogenic centres containing one hydrogen atom, but they possess distinct charge distribution, lipophilicity, conformation and metabolic stability1–3. Although tertiary stereogenic centres containing one hydrogen atom are often set by enantioselective desymmetrization reactions at one of the two carbon–hydrogen (C–H) bonds of a methylene group, tertiary stereocentres containing fluorine have not yet been constructed by the analogous desymmetrization reaction at one of the two carbon–fluorine (C–F) bonds of a difluoromethylene group3. Fluorine atoms are similar in size to hydrogen atoms but have distinct electronic properties, causing C–F bonds to be exceptionally strong and geminal C–F bonds to strengthen one another4. Thus, exhaustive defluorination typically dominates over the selective replacement of a single C–F bond, hindering the development of the enantioselective substitution of one fluorine atom to form a stereogenic centre5,6. Here we report the catalytic, enantioselective activation of a single C–F bond in an allylic difluoromethylene group to provide a broad range of products containing a monofluorinated tertiary stereogenic centre. By combining a tailored chiral iridium phosphoramidite catalyst, which controls regioselectivity, chemoselectivity and enantioselectivity, with a fluorophilic activator, which assists the oxidative addition of the C–F bond, these reactions occur in high yield and selectivity. The design principles proposed in this work extend to palladium-catalysed benzylic substitution, demonstrating the generality of the approach.
One of the most common approaches to the enantioselective synthesis of chiral molecules is the selective replacement of one of the two enantiotopic C–H bonds of a methylene unit to form a tertiary stereogenic centre. This approach is exemplified by classic α-functionalizations of carbonyl compounds7,8, asymmetric lithiations9, as well as modern catalytic reactions, such as benzylic functionalizations10,11, carbene or nitrene insertions12,13 and directed C–H activations14 (Fig. 1a). Although the synthesis of organic compounds with fluorine atoms installed in specific positions is often essential to control the physiochemical properties, such as the pKa, lipophilicity, conformation and metabolic stability of pharmaceutical candidates2, the analogous desymmetrizations of difluoromethylene units to form enantioenriched tertiary alkyl fluorides, which are bioisosteres for common tertiary stereocentres3 (Fig. 1b), have not been reported so far. This class of reaction would provide a particularly valuable route to tertiary alkyl fluorides because the starting difluoromethylene groups are readily installed by classical deoxyfluorination reactions or modern difluoroalkylations15–19.
Fig. 1 |. Approaches to the desymmetrization of methylene units.
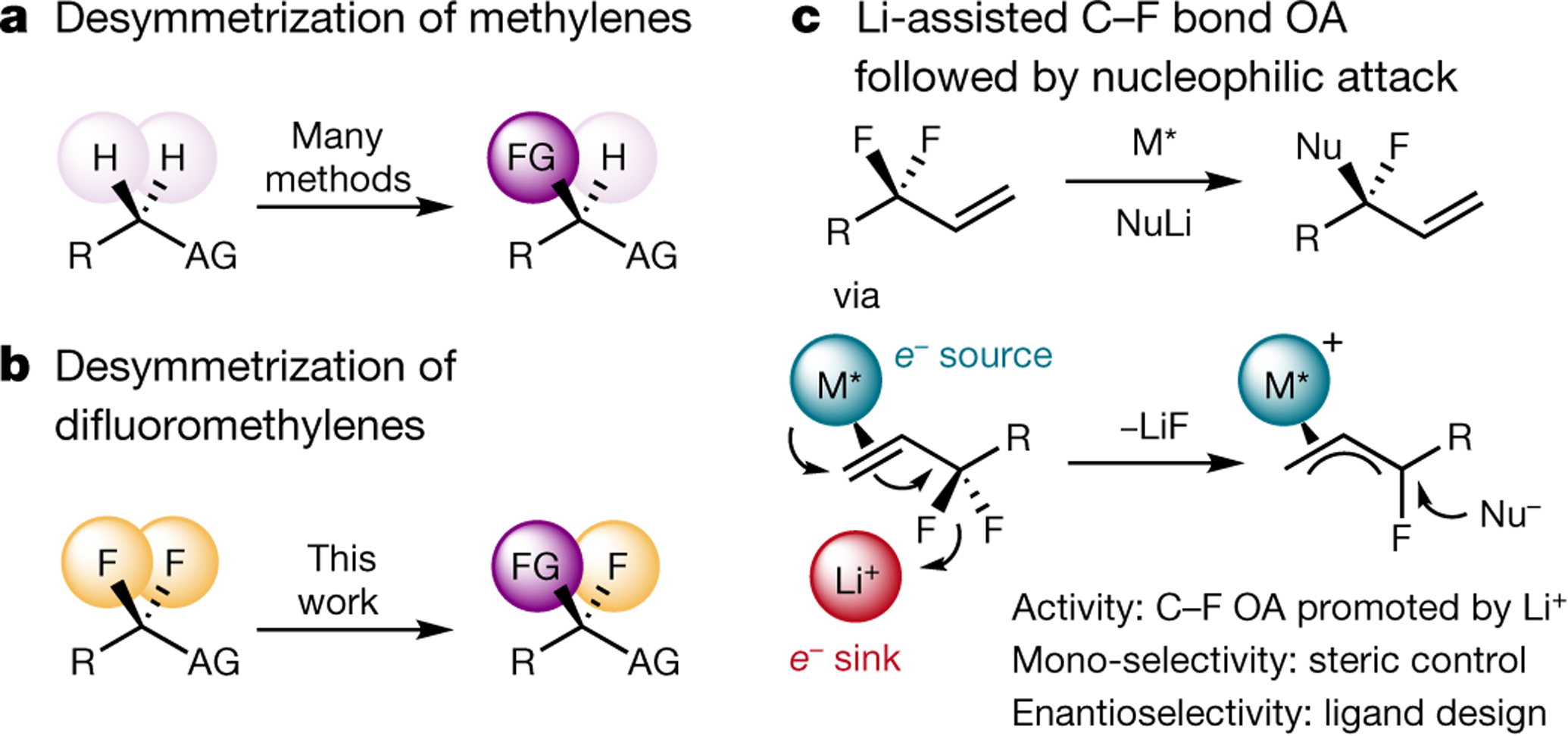
a, The desymmetrization of methylene groups by enantioselective C–H activation is a general strategy for the construction of simple tertiary stereocentres. b, The analogous desymmetrization of difluoromethylene groups by enantioselective C–F activation has not been reported so far. c, The cooperation of a chiral, low-valent transition metal complex and a hard fluorophilic activator enables the enantioselective activation of a single C–F bond at an allylic position. FG, functional group; AG, activating group (for example, electron-withdrawing group, aryl group, heteroatom or directing group; few examples lack an activating group); Nu, nucleophile; M, transition metal; OA, oxidative addition.
The enantioselective activation of a single C–F bond in a difluoromethylene unit poses several challenges that distinguish this type of reaction from the more common enantioselective activations of one C–H bond in a methylene unit20–22. Although C–H bonds are nonpolar and can be cleaved by either homolytic or heterolytic pathways, reactions of C–F bonds by homolytic pathways are rare because carbon forms stronger single bonds to fluorine than to any other element. Reactions of C–F bonds can proceed by heterolytic pathways, but strong Lewis acids are typically required, owing to the instability and high basicity of the fluoride ion. Consistent with their tendency to block unwanted metabolic reactivity, difluoromethylene functional groups are typically inert, and many of the reagents known to promote C(sp3)–F bond activation are strong Lewis acids that induce exhaustive defluorination6,23–26. These problems are compounded by the mutual strengthening of geminal C–F bonds, making C–F bond activation particularly difficult in difluoromethylene groups.
To conduct the enantioselective replacement of one of two geminal fluorides, we envisioned an approach in which a low-valent transition metal catalyst would bind to a nearby functional group, such as the alkene of a 3-substituted 3,3-difluoropropene, to labilize an adjacent difluoromethylene group. Then, a fluorophilic cation would accept the labilized fluoride as a leaving group. The resulting monofluoro π-allyl intermediate would then be attacked by a nucleophile to produce a tertiary alkyl fluoride (Fig. 1c). In this case, selectivity for the activation of a single C–F bond would be imposed by the steric environment of the low-valent, transition metal complex. Our group and others have reported chiral iridium complexes that catalyse enantioselective allylic substitutions that deliver a nucleophile to the more substituted position of an allyl intermediate27,28, including the position containing a fluorine atom29. If such a catalyst could undergo oxidative addition of one of the two enantiotopic C–F bonds of a 3-substituted 3,3-difluoropropene to form a π-allyl intermediate, then the overall process could lead to enantioselective substitution of a single fluorine atom in the difluoromethylene unit by a carbon nucleophile30. However, iridium-catalysed allylic substitutions occur by turnover-limiting oxidative additions or by endothermic and reversible oxidative additions, and those involving fluoroallyl intermediates require particularly labile leaving groups, such as trifluoroacetate or phosphate29,31. Thus, direct cleavage of the less reactive C–F bond by an iridium catalyst is expected to be challenging32–34.
Herein, we report the catalytic, enantioselective replacement of one of two fluorine atoms in geminal difluorides to form enantioenriched tertiary allylic fluorides in high yield, high regioselectivity, high enantioselectivity and perfect chemoselectivity for the activation of a single C–F bond. The reaction occurs by combining a tailored iridium phosphoramidite catalyst with an appropriate counterion or silyl group on the nucleophile to labilize the fluoride and to deliver the nucleophile to the fluorine-containing site on the allyl electrophile. This ‘push–pull’ cooperation between a soft transition metal and a fluorophilic activator accelerates the oxidative addition of C–F bonds and is shown to extend to the activation of C–F bonds at less reactive benzylic positions.
Our investigations of the enantioselective substitution of one fluorine of a difluoromethylene unit began with the reaction of 2-(1,1-difluoroallyl)naphthalene (1a)15 with sodium diethyl methylmalonate in the presence of a cyclometalated iridium catalyst (Cα). As designed, the product of this reaction formed from the substitution of a single C–F bond of the allylic fluoride 2a with exclusive branched selectivity (>99:1), but the reaction was slow and reached only 10% conversion after 6 h with 1 mol% of catalyst Cα. Reactions conducted with increased catalyst loadings (5 mol%) and extended reaction times (48 h) proceeded in high yield; however, these reactions occurred with low enantioselectivity (67:33 enantiomeric ratio; Fig. 2a, entries 1, 2). Evaluation of various iridium catalysts derived from common ligands (see Supplementary Information) revealed that catalyst Cβ was the most selective (81:19 enantiomeric ratio; Fig. 2a, entry 3).
Fig. 2 |. Reaction development and mechanistic studies.
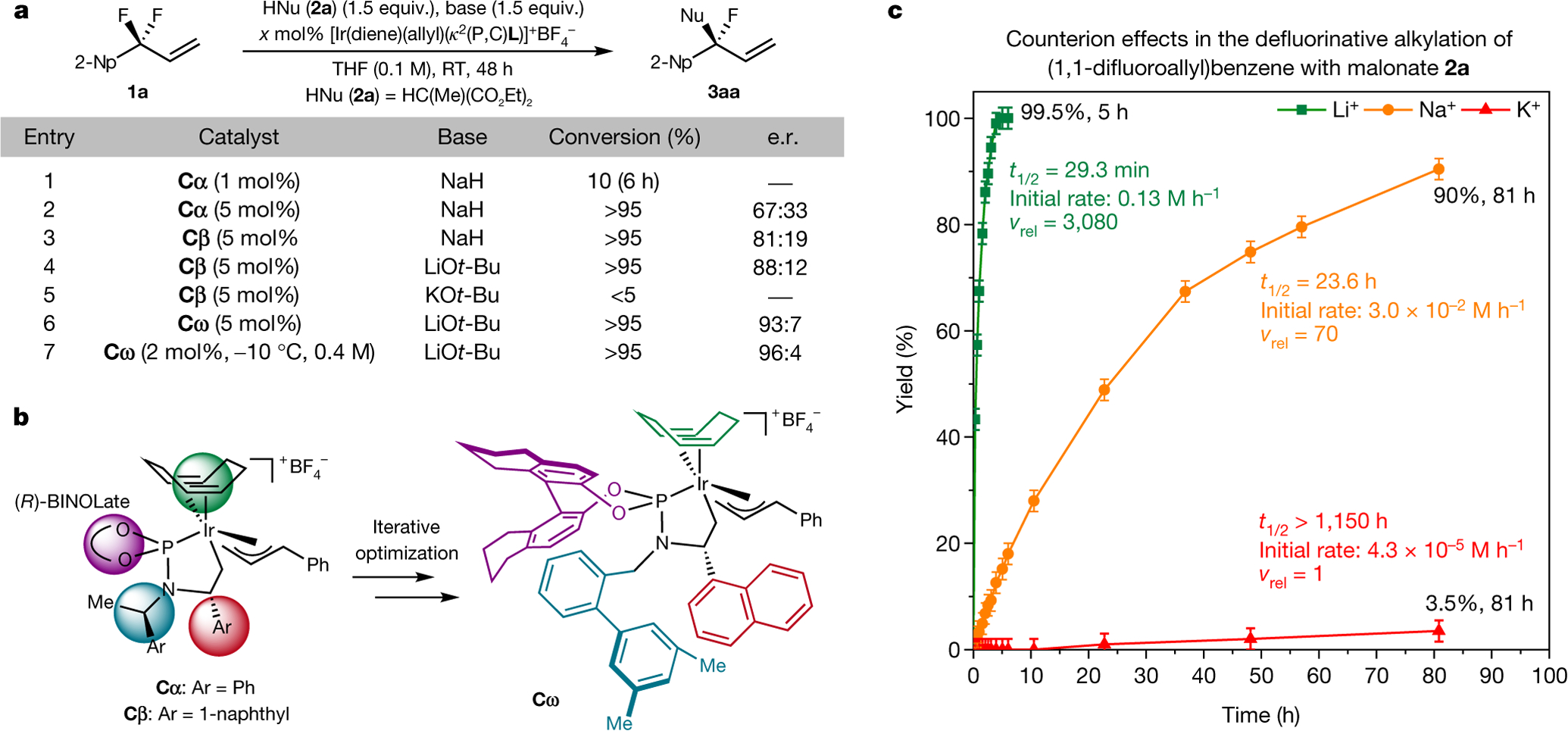
The identity of the fluorophilic cation strongly affects the rate of C–F activation. Modulation of the steric profile of the cyclometalated iridium catalyst improves the enantioselectivity. a, Effect of the catalyst, the catalyst loading and the base on conversion and enantioselectivity. The conversion of the starting material is determined by 1H NMR spectroscopy. The enantiomeric ratio (e.r.) is determined by chiral high-performance liquid chromatography (HPLC) after purification by preparative thin-layer chromatography (TLC). b, Development of a highly enantioselective iridium catalyst. c, Quantification of the role of the cation in C–F activation reactions. Kinetic studies were conducted according to the scheme in a with substrate 1b (PhCF2CH = CH2) and catalyst Cω (4 mol%). Error bars in c correspond to ±2% yield error associated with quantitative NMR spectroscopy. THF, tetrahydrofuran; 2-Np, 2-naphthyl; Ph, phenyl; Ar, aryl; Me, methyl; BINOLate, 1,1′-bi(2-naphtholate); RT, room temperature; vrel, relative rate; κ denotes binding of non-contiguous atoms in a chelating ligand.
To achieve high conversion at more acceptable catalyst loadings and at the low temperatures needed to obtain the product with high enantioselectivity, we investigated the effect of fluorophilic activators. Reactions conducted with lithium malonates proceeded 44 times faster than reactions performed with sodium malonates and 3,080 times faster than reactions conducted with potassium malonates (Fig. 2c), indicating that the counterion serves this role and assists oxidative addition of the C–F bond (see below). Reactions performed with lithium malonate occurred in higher enantioselectivity than those conducted with sodium malonate (88:12 versus 81:19 enantiomeric ratio; Fig. 2a, entries 3, 4), but the structure of the catalyst needed to be modified to achieve high enantioselectivity. After iteratively varying each of four moieties on the catalyst (Fig. 2b, highlighted in red, teal, purple and green), we found that complex Cω catalysed this reaction with high enantioselectivity (see Supplementary Information for details). Because the lithium cation enhances the reaction rate, the temperature could be reduced to −10 °C and the catalyst loading could be reduced to 2 mol% without affecting the conversion. Under these conditions, product 3aa formed in quantitative yield and 96:4 enantiomeric ratio (Fig. 2a, entry 7).
With conditions to achieve this enantioselective replacement of one fluorine atom in allylic difluoromethylene groups, we investigated the scope of nucleophiles and electrophiles that undergo this transformation (Fig. 3). The defluorinative alkylation of 2-(1,1-difluoroallyl) naphthalene (1a) with various malonates provided allylic fluorides 3aa, 3ab and 3ac in high yields and high enantioselectivities. A prochiral β-keto ester also participated in the defluorinative alkylation to provide tertiary allylic fluoride 3bd, which bears two contiguous, fully substituted stereogenic centres, in good diastereoselectivity (6:1) and excellent enantioselectivity (98:2 enantiomeric ratio). The major diastereomer of alkyl fluoride 3bd was isolated in 79% yield. In addition, the nucleophile 2-(methoxymethoxy)malononitrile reacted with electrophile 1a in high yield and high enantioselectivity when lithium bromide was included as an additive (3ae). The malononitrile nucleophile functions as a versatile acyl anion equivalent because the products can be cleaved under acidic conditions to form an acyl cyanide, which converts to esters and amides upon addition of alcohols or amines29,35. This two-step protocol enables the enantioselective carboxylation of one C–F bond in 3-substituted 3,3-difluoropropenes (Fig. 3a, box). Acyclic ketones also participate in the defluorinative alkylation reactions in high yield, albeit in low enantioselectivity (see Supplementary Information for details).
Fig. 3 |. Scope of nucleophiles and electrophiles that participate in defluorinative alkylation reactions and transformations of the products.
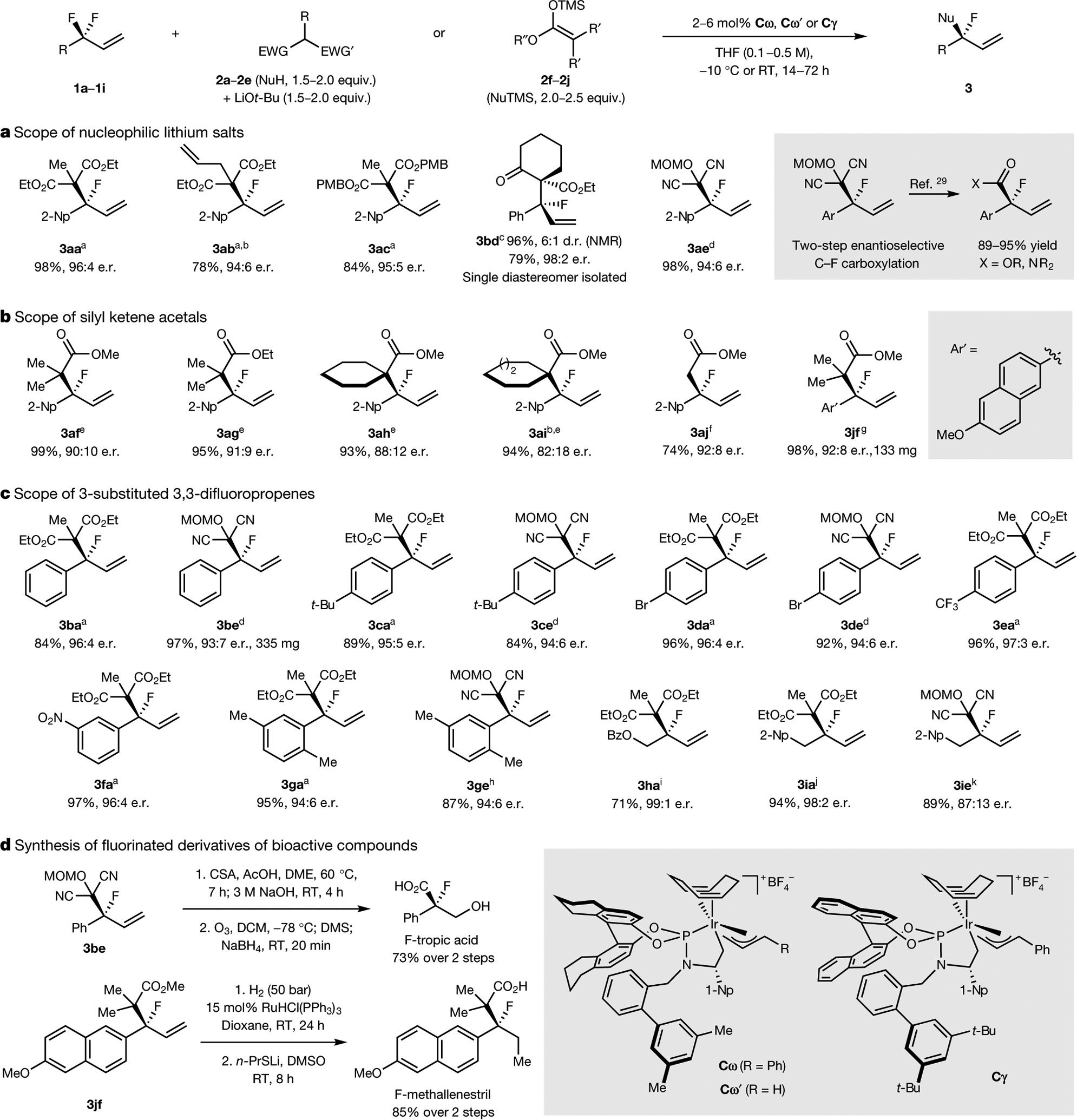
a, Nucleophilic lithium salts in defluorinative alkylation reactions. b, Silyl ketene acetals in defluorinative alkylation reactions. c, 3-Substituted 3,3-difluoropropenes in defluorinative alkylation reactions. d, Synthesis of fluorinated derivatives of medicinally active compounds from the substitution products. Isolated yields reported unless otherwise noted. a2 mol% Cω, −10 °C, 72 h. b96 h. c5 mol% Cω’, 0 °C, 72 h. d2 mol% Cω’, 5 equiv. LiBr, RT, 24 h. e6 mol% Cγ, 5 mol% NaCMe(CO2Et)2, RT, 48 h. ftert-Butyl dimethyl silyl ketene acetal, 6 mol% Cγ, RT, 53 h. g2 mol% TMSOTf, 4 mol% Cγ, dioxane, RT, 40 h. h5 mol% Cω’, 5 equiv. LiBr, RT, 48 h. i5 mol% Cω, 65 °C, 96 h. j2 mol% Cω, 3 equiv. Ba(OTf)2, RT, 19 h. k20 mol% Cω, 3 equiv. Ba(OTf)2, dioxane, RT, 24 h. EWG, electron-withdrawing group; TMS, trimethylsilyl; MOM, methoxymethyl; Bz, benzoyl; CSA, camphor sulfonic acid; DME, 1,2-dimethoxyethane; DMS, dimethylsulfide; 1-Np, 1-naphthyl; d.r., diastereometric ratio.
Inspired by many reports of silicon-assisted activation of C–F bonds5, we investigated defluorinative alkylation reactions with silyl ketene acetals36 (Fig. 3b). The reactions of 2-(1,1-difluoroallyl)naphthalene (1a) with various acyclic (3af, 3ag), cyclic (3ah, 3ai) and unsubstituted (3aj) silyl ketene acetals occurred in high yields with moderate to good enantioselectivities. These reactions provided products in higher enantioselectivities when conducted with catalyst Cγ than when conducted with catalyst Cω.
The scope of electrophiles that undergo defluorinative alkylation was investigated with malonates and malononitriles as the nucleophile (Fig. 3c). Electrophiles bearing neutral (3ba, 3be), moderately electron-rich (3ca, 3ce), electron-poor (3da, 3de, 3ea, 3fa) and ortho-substituted (3ga, 3ge) arenes underwent defluorinative alkylation in high yields and high enantioselectivities. Alkyl-substituted 3,3-difluoropropenes underwent defluorinative alkylation reactions in high yields and high enantioselectivies, provided that the reactions were conducted at slightly increased temperatures (3ha) or in the presence of added barium triflate as the Lewis acidic activator (3ia, 3ie).
To demonstrate the synthetic utility of this method, we prepared fluorinated analogues of two biologically relevant compounds from the products of the reactions in Fig. 3 (Fig. 3d). We prepared a fluorinated derivative of tropic acid—a common substructure of diverse bioactive compounds—in two steps from compound 3be via hydrolysis and reductive ozonolysis. In addition, we prepared in two steps from allylic fluoride 3jf a fluorinated analogue of methallenestril, which is a non-steroidal oestrogen.
We sought to extend our push–pull mode of C–F activation, in which a soft transition metal is combined with a hard, fluorophilic activator, to include substitutions at positions less activated than allylic positions. Therefore, we investigated analogous desymmetrizations of difluoromethylene groups at benzylic positions in the presence of palladium(0) complexes known to catalyse benzylic substitution37–43 and Lewis acid activators that we have shown to promote oxidative addition of the C–F bonds studied here. Indeed, the reaction of 2-(difluoromethyl)naphthalene with lithium diethylmethylmalonate occurred at one of two geminal fluorine atoms in excellent yield in the presence of lithium triflate and a palladium complex of bis[(2-diphenylphosphino)phenyl] ether (DPEphos) (Fig. 4, entries 1–3). Although our results are preliminary, an analogous reaction with the chiral ligand (R)-BINAP demonstrated the ability to conduct this substitution as an enantioselective process (Fig. 4, entry 4). Thus, the design elements proposed here to achieve the selective activation of a single C–F bond of a difluoromethylene unit should be broadly applicable.
Fig. 4 |. Selective activation of a single benzylic C–F bond.
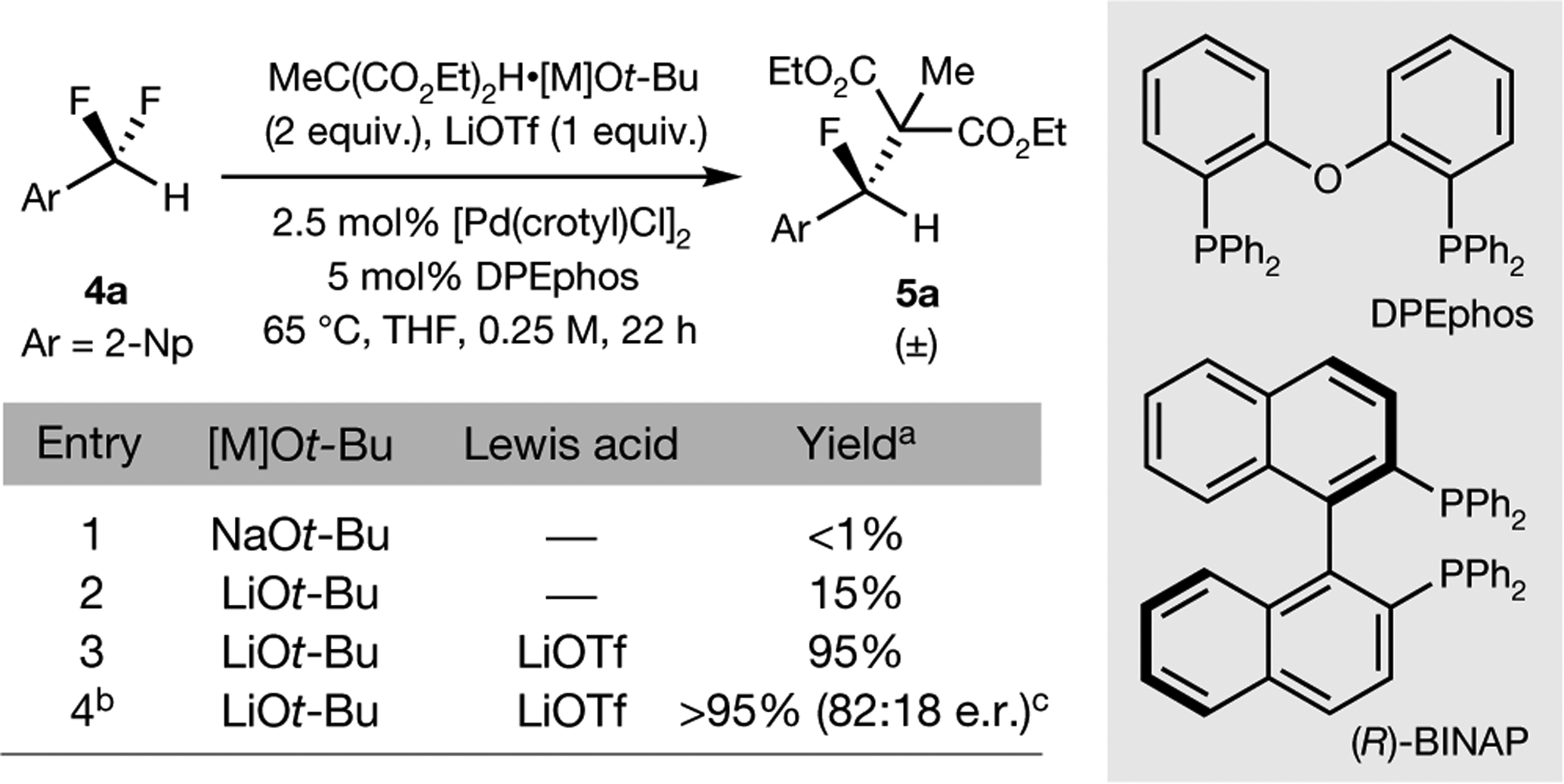
The cooperation between a low-valent transition metal and a fluorophilic cation enables the activation of benzylic C–F bonds in difluoromethylarenes, and reactions conducted with a chiral ligand are enantioselective. aYield determined by 1H and 19F NMR spectroscopy. bReaction conducted with 5 mol% [Pd(crotyl)Cl]2 and 15 mol% (R)-BINAP. cEnantiomeric ratio determined by chiral HPLC. LiOTf, lithium triflate.
A series of mechanistic studies revealed the rate-determining and enantiodetermining steps of the iridium-catalysed desymmetrization of 3-substituted 3,3-difluoropropenes and the origin of the absolute configuration of the product (see Fig. 5a for the complete mechanism). The resting state of the catalyst for reactions conducted with lithium, sodium and potassium malonates was an 80:20 mixture of diastereomeric complexes of olefin 1a with Ir(i), as determined by 19F and 31P nuclear magnetic resonance (NMR) spectroscopy (Fig. 5a,b). The two diastereomers of the resting state rapidly interconvert, as determined by 31P–31P exchange NMR spectroscopy (first-order half-lives for the forward and reverse steps of the interconversions of the diastereomers are t1/2forward = 0.88 s and t1/2reverse = 0.23 s, respectively). The resting state undergoes oxidative addition of the C–F bond to generate a π-allyl complex and liberate the insoluble and non-nucleophilic fluoride in the lithium fluoride byproduct. The resulting intermediate reacts rapidly with malonate to form the substitution product. Allylic substitutions of 3-fluoroallylic esters catalysed by complex Cω, which proceed via closely related fluoroallyl intermediates, reach >98% conversion in under 2 min, implying that the nucleophilic attack of malonate onto fluorinated π-allyl complexes must be rapid (half-life, t1/2< 20 s) (Fig. 5c; see Supplementary Information for details)29. Catalyst Cω rapidly reacts with lithium malonate to form an Ir(i) species, but it does not react with lithium fluoride, even after prolonged reaction times (see Supplementary Information for details). This result indicates that π-allyl iridium species react much faster with lithium malonate than with lithium fluoride; therefore, oxidative addition of the C–F bond is irreversible during the catalytic reaction. These data, along with the strong effect of the Lewis acid on the rate of the reaction, imply that oxidative addition of the C–F bond is also turnover-limiting.
Fig. 5 |. Mechanistic studies support a cation-assisted, turnover-limiting, enantiodetermining and irreversible oxidative addition from a rapidly interconverting mixture of diastereomeric olefin complexes under Curtin–Hammett control.
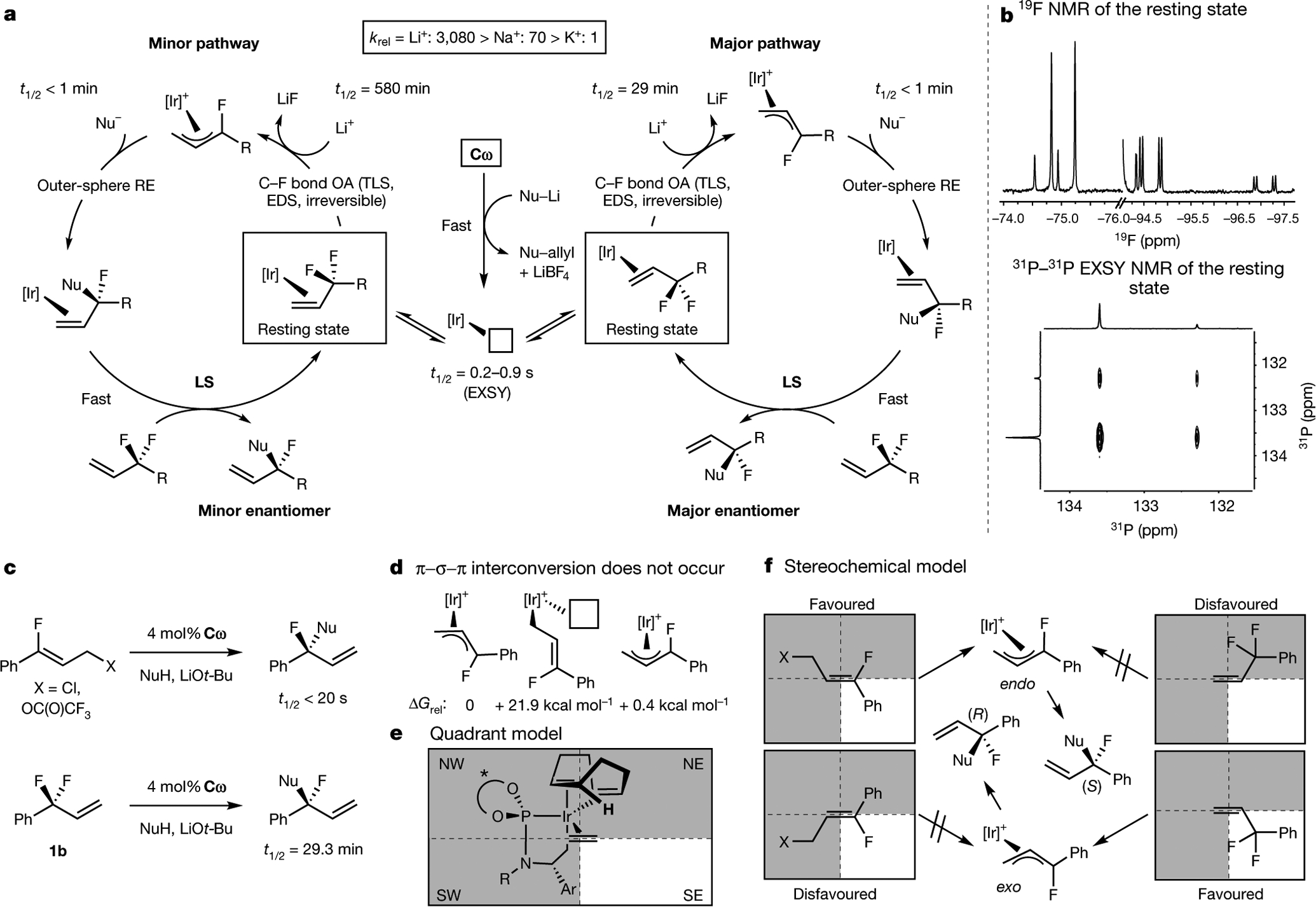
a, Proposed mechanism. b, NMR spectroscopic characterization of the catalyst resting state. c, Comparison of allylic fluoroalkylation reactions with 3-fluorocinnamyl electrophiles and 3-substituted 3,3-difluoropropenes. d, η1 allyl intermediates are too high in energy to participate. e, Quadrant diagram for Ir(i) olefin complexes. f, Stereochemical model for 3-fluorocinnamyl electrophiles and 3-substituted 3,3-difluoropropenes; see Supplementary Information for a more detailed discussion. OA, oxidative addition; RE, reductive elimination; LS, ligand substitution; TLS, turnover-limiting step; EDS, enantiodetermining step; EXSY, exchange spectroscopy; krel, relative rate constant; ΔGrel, relative Gibbs free energy.
Having identified the turnover-limiting step, we conducted additional experiments to identify which step is enantiodetermining. The enantiodetermining step could be the association of the alkene, oxidative addition of the C–F bond or attack of the nucleophile on one of two equilibrating π-allyl complexes. Association of the alkene to iridium cannot be enantiodetermining, because equilibration of the two diastereomeric olefin complexes is faster than cleavage of the C–F bond. Nucleophilic attack cannot be enantiodetermining, unless the two π-allyl complexes equilibrate either by reversible oxidative addition or π–σ–π interconversion. We found that 3-fluoroallylic esters and 3-substituted 3,3-difluoropropenes29, which would form the same set of endo and exo π-allyl complexes (see above), form different enantiomers of the product. Therefore, the two diastereomeric π-allyl complexes must not equilibrate before the addition of the nucleophile (Fig. 5c). Consistent with this assertion, the energy of the η1-allyl iridium complex that would be an intermediate in a π–σ–π interconversion was computed to be 21.9 kcal mol−1 above the η3 form, suggesting that the barrier for interconversion by this path would be higher than that for nucleophilic attack (Fig. 5d). Collectively, these results demonstrate that oxidative addition of the C–F bond is enantiodetermining.
The identification of the step that controls enantioselectivity enabled us to assess the interactions that control enantioselectivity and explain the absolute configurations of the products derived from 3-substituted 3,3-difluoropropenes versus 3-fluoroallylic esters. The structures of each of the diastereomeric complexes of Ir(i) with either 3-fluorocinnamyl chloride or (1,1-difluoroallyl)benzene were determined using density functional theory. These studies show that a vertex of the 1,5-cyclooctadiene (COD) ligand protrudes into the binding space of the olefin, blocking the northeast quadrant (Fig. 5e). The endo olefin complex from 3-fluorocinnamyl chloride (and the corresponding transition state for oxidative addition) lacks this steric clash with the COD ligand, contains the largest substituent on the olefin (Ph) in an open quadrant and forms the (S) enantiomer of the product (Fig. 5f, left). By contrast, the exo olefin complex from (1,1-difluoroallyl)benzene (and the corresponding transition state for oxidative addition) lacks a steric interaction with the COD ligand, contains the largest substituent of the olefin (CF2Ph) in an open quadrant and forms the (R) enantiomer of the product (Fig. 5f, right). This analysis explains the distinct enantiomers formed from the two classes of fluoroallyl electrophiles with the same enantiomer of the catalyst.
Altogether, our mechanistic analysis shows that the enantioselective substitution of lithium malonates for one of the two fluorine atoms in 3-substituted 3,3-difluoropropenes probably occurs by the pathway shown in Fig. 5a. By this mechanism, the cationic Ir(iii) allyl precatalyst Cω reacts rapidly with lithium malonate to release the allyl group in the catalyst as allylmalonate, release LiBF4, and bind one equivalent of the starting 3-substituted 3,3-difluoropropene to generate a mixture of diastereomeric Ir(i) olefin complexes. These diastereomeric complexes undergo a counterion-assisted oxidative addition by an outer-sphere mechanism to release an equivalent of lithium fluoride and generate either an exo (major) or endo (minor) Ir(iii)–π-allyl complex. Each of these π-allyl complexes is rapidly intercepted by malonate to generate a product-bound Ir(i) complex. Subsequent dissociative ligand substitution with an equivalent of the starting 3-substituted 3,3-difluoropropene closes the catalytic cycle and releases the substitution product.
This demonstration of the desymmetrization of difluoromethylene groups being achieved by the cooperation of a chiral, low-valent transition metal with a fluorophilic cation could change the role of the difluoromethylene group from an inert unit installed to inhibit metabolism to a prochiral retron for stereogenic alkyl fluorides. We expect that difluoromethylene units in various substructures will react with a range of catalysts and activators when the design elements proposed here are used, thereby expanding the scope of nucleophiles and electrophiles that participate in defluorinative substitution reactions to form alkyl fluorides enantioselectively.
Supplementary Material
Acknowledgements
We thank the NSF (CHE-1565886) for financial support. T.W.B. gratefully acknowledges the National Science Foundation Graduate Research Fellowship Program and the UC Berkeley Graduate Research Fellowship Program for support. J.L.Y. gratefully acknowledges a Summer Undergraduate Research Fellowship from the University of California, Berkeley and a summer fellowship from the Rose Hills Foundation. N.D.W. thanks the UC Berkeley Amgen Scholars Program for support. We thank the College of Chemistry’s NMR facility for resources and H. Celik for assistance. Instruments in the CoC-NMR facility are supported in part by NIH S10OD024998.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-020-2399-1.
Supplementary information is available for this paper at https://doi.org/10.1038/s41586-020-2399-1.
Competing interests The authors declare no competing interests.
Data availability
The characterization data are available in the Supplementary Information, together with details about the materials and methods, experimental procedures, mechanistic studies, investigations of reaction conditions, and the design of ligands.
References
- 1.Purser S, Moore PR, Swallow S & Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 37, 320–330 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Gillis EP, Eastman KJ, Hill MD, Donnelly DJ & Meanwell NA Applications of fluorine in medicinal chemistry. J. Med. Chem 58, 8315–8359 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y et al. Modern approaches for asymmetric construction of carbon–fluorine quaternary stereogenic centers: synthetic challenges and pharmaceutical needs. Chem. Rev 118, 3887–3964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O′Hagan D Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev 37, 308–319 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Stahl T, Klare HFT & Oestreich M Main-group Lewis acids for C–F bond activation. ACS Catal 3, 1578–1587 (2013). [Google Scholar]
- 6.Shen Q et al. Review of recent advances in C−F bond activation of aliphatic fluorides. J. Fluor. Chem 179, 14–22 (2015). [Google Scholar]
- 7.Franzén J et al. A general organocatalyst for direct α-functionalization of aldehydes: stereoselective C−C, C−N, C−F, C−Br, and C−S bond-forming reactions. Scope and mechanistic insights. J. Am. Chem. Soc 127, 18296–18304 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Smith AMR & Hii KK Transition metal catalyzed enantioselective α-heterofunctionalization of carbonyl compounds. Chem. Rev 111, 1637–1656 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Beak P, Basu A, Gallagher DJ, Park YS & Thayumanavan S Regioselective, diastereoselective, and enantioselective lithiation-substitution sequences: reaction pathways and synthetic applications. Acc. Chem. Res 29, 552–560 (1996). [Google Scholar]
- 10.Zhang W et al. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 353, 1014–1018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groves JT & Viski P Asymmetric hydroxylation by a chiral iron porphyrin. J. Am. Chem. Soc 111, 8537–8538 (1989). [Google Scholar]
- 12.Liao K, Negretti S, Musaev DG, Bacsa J & Davies HML Site-selective and stereoselective functionalization of unactivated C–H bonds. Nature 533, 230–234 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Nishioka Y, Uchida T & Katsuki T Enantio- and regioselective intermolecular benzylic and allylic C−H bond amination. Angew. Chem. Int. Ed 52, 1739–1742 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Saint-Denis TG, Zhu R-Y, Chen G, Wu Q-F & Yu J-Q Enantioselective C(sp3)–H bond activation by chiral transition metal catalysts. Science 359, eaao4798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Min QQ, Yin ZS, Feng Z, Guo WH & Zhang XG Highly selective gem-difluoroallylation of organoborons with bromodifluoromethylated alkenes catalyzed by palladium. J. Am. Chem. Soc 136, 1230–1233 (2014). [DOI] [PubMed] [Google Scholar]
- 16.An L, Xiao Y-L, Zhang S & Zhang X Bulky diamine ligand promotes cross-coupling of difluoroalkyl bromides by iron catalysis. Angew. Chem. Int. Ed 57, 6921–6925 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Feng Z, Xiao Y-L & Zhang X Transition-metal (Cu, Pd, Ni)-catalyzed difluoroalkylation via cross-coupling with difluoroalkyl halides. Acc. Chem. Res 51, 2264–2278 (2018). [DOI] [PubMed] [Google Scholar]
- 18.An L, Xu C & Zhang X Highly selective nickel-catalyzed gem-difluoropropargylation of unactivated alkylzinc reagents. Nat. Commun 8, 1460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao Y-L, Guo W-H, He G-Z, Pan Q & Zhang X Nickel-catalyzed cross-coupling of functionalized difluoromethyl bromides and chlorides with aryl boronic acids: a general method for difluoroalkylated arenes. Angew. Chem. Int. Ed 53, 9909–9913 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Tanaka J, Suzuki S, Tokunaga E, Haufe G & Shibata N Asymmetric desymmetrization via metal-free C−F bond activation: synthesis of 3,5-diaryl-5-fluoromethyloxazolidin-2-ones with quaternary carbon centers. Angew. Chem. Int. Ed 55, 9432–9436 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Haufe G et al. C–F bond activation of unactivated aliphatic fluorides: synthesis of fluoromethyl-3,5-diaryl-2-oxazolidinones by desymmetrization of 2-aryl-1,3-difluoropropan-2-ols. Angew. Chem. Int. Ed 51, 12275–12279 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Nishimine T et al. Kinetic resolution of allyl fluorides by enantioselective allylic trifluoromethylation based on silicon-assisted C−F bond cleavage. Angew. Chem. Int. Ed 53, 517–520 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Vogt DB, Seath CP, Wang H & Jui NT Selective C–F functionalization of unactivated trifluoromethylarenes. J. Am. Chem. Soc 141, 13203–13211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida S, Shimomori K, Kim Y & Hosoya T Single C−F bond cleavage of trifluoromethylarenes with an ortho-silyl group. Angew. Chem. Int. Ed 55, 10406–10409 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Fuchibe K, Oki R, Hatta H & Ichikawa J Single C−F bond activation of the CF3 group with a Lewis acid: CF3-cyclopropanes as versatile 4,4-difluorohomoallylating agents. Chem. Eur. J 24, 17932–17935 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Amii H & Uneyama K C−F bond activation in organic synthesis. Chem. Rev 109, 2119–2183 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Hartwig JF & Stanley LM Mechanistically driven development of iridium catalysts for asymmetric allylic substitution. Acc. Chem. Res 43, 1461–1475 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rössler SL, Petrone DA & Carreira EM Iridium-catalyzed asymmetric synthesis of functionally rich molecules enabled by (phosphoramidite, olefin) ligands. Acc. Chem. Res 52, 2657–2672 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Butcher TW & Hartwig JF Enantioselective synthesis of tertiary allylic fluorides by iridium-catalyzed allylic fluoroalkylation. Angew. Chem. Int. Ed 57, 13125–13129 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Trost BM & Lee CB Geminal dicarboxylates as carbonyl surrogates for asymmetric synthesis. Part II. scope and applications. J. Am. Chem. Soc 123, 3687–3696 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Madrahimov ST & Hartwig JF Origins of enantioselectivity during allylic substitution reactions catalyzed by metallacyclic iridium complexes. J. Am. Chem. Soc 134, 8136–8147 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hazari A, Gouverneur V & Brown JM Palladium-catalyzed substitution of allylic fluorides. Angew. Chem. Int. Ed 48, 1296–1299 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Benedetto E et al. Platinum-catalyzed substitution of allylic fluorides. Organometallics 31, 1408–1416 (2012). [Google Scholar]
- 34.Pigeon X et al. Activation of allylic C−F bonds: palladium-catalyzed allylic amination of 3,3-difluoropropenes. Angew. Chem. Int. Ed 49, 1123–1127 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Shockley SE, Hethcox JC & Stoltz BM Enantioselective synthesis of acyclic α-quaternary carboxylic acid derivatives through iridium-catalyzed allylic alkylation. Angew. Chem. Int. Ed 56, 11545–11548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang X & Hartwig JF Iridium-catalyzed enantioselective allylic substitution of aliphatic esters with silyl ketene acetals as the ester enolates. Angew. Chem. Int. Ed 56, 8887–8891 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blessley G, Holden P, Walker M, Brown JM & Gouverneur V Palladium-catalyzed substitution and cross-coupling of benzylic fluorides. Org. Lett 14, 2754–2757 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Trost BM & Czabaniuk LC Palladium-catalyzed asymmetric benzylation of 3-aryl oxindoles. J. Am. Chem. Soc 132, 15534–15536 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Trost BM & Czabaniuk LC Benzylic phosphates as electrophiles in the palladium-catalyzed asymmetric benzylation of azlactones. J. Am. Chem. Soc 134, 5778–5781 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Tabuchi S, Hirano K & Miura M Palladium-catalyzed asymmetric benzylic alkylation of active methylene compounds with α-naphthylbenzyl carbonates and pivalates. Angew. Chem. Int. Ed 55, 6973–6977 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Arp FO & Fu GC Catalytic enantioselective Negishi reactions of racemic secondary benzylic halides. J. Am. Chem. Soc 127, 10482–10483 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Wisniewska HM, Swift EC & Jarvo ER Functional-group-tolerant, nickel-catalyzed cross-coupling reaction for enantioselective construction of tertiary methyl-bearing stereocenters. J. Am. Chem. Soc 135, 9083–9090 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Q, Srinivas HD, Dasgupta S & Watson MP Nickel-catalyzed cross-couplings of benzylic pivalates with arylboroxines: stereospecific formation of diarylalkanes and triarylmethanes. J. Am. Chem. Soc 135, 3307–3310 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The characterization data are available in the Supplementary Information, together with details about the materials and methods, experimental procedures, mechanistic studies, investigations of reaction conditions, and the design of ligands.