

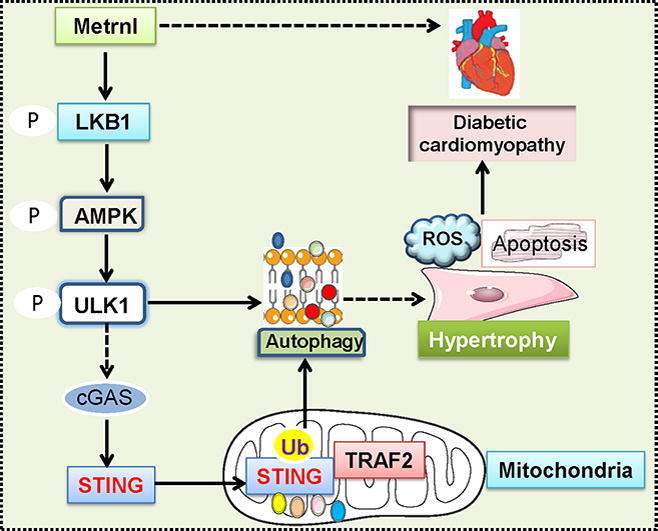
Graphical abstract
Keywords: Diabetes, Cardiomyopathy, Metrnl, AMPK, Autophagy, STING
Highlights
-
•
Cardiac and circulating levels of Metrnl were inhibited in streptozotocin (STZ)-induced mice and leptin receptor deficiency (db/db) mice.
-
•
Cardiac specific depletion of Metrnl worsened, while cardiomyocyte-specific OE of Metrnl reversed hyperglycemia-induced cardiac damage in mice.
-
•
Metrnl inactivated the cGAS/STING signaling pathway by activating the LKB1/AMPK/ULK1 signaling axis in DCM.
Abstract
Introduction
Meteorin-like hormone (Metrnl) is ubiquitously expressed in skeletal muscle, heart, and adipose with beneficial roles in obesity, insulin resistance, and inflammation. Metrnl is found to protect against cardiac hypertrophy and doxorubicin-induced cardiotoxicity. However, its role in diabetic cardiomyopathy (DCM) is undefined.
Objectives
We aimed to elucidate the potential roles of Metrnl in DCM.
Methods
Gain- and loss-of-function experiments were utilized to determine the roles of Metrnl in the pathological processes of DCM.
Results
We found that plasma Metrnl levels, myocardial Metrnl protein and mRNA expressions were significantly downregulated in both streptozotocin (STZ)-induced (T1D) mice and leptin receptor deficiency (db/db) (T2D) mice. Cardiac-specific overexpression (OE) of Metrnl markedly ameliorated cardiac injury and dysfunction in both T1D and T2D mice. In sharp contrast, specific deletion of Metrnl in the heart had the opposite phenotypes. In parallel, Metrnl OE ameliorated, whereas Metrnl downregulation exacerbated high glucose (HG)-elicited hypertrophy, apoptosis and oxidative damage in primary neonatal rat cardiomyocytes. Antibody-induced blockade of Metrnl eliminated the effects of benefits of Metrnl in vitro and in vivo. Mechanistically, Metrnl activated the autophagy pathway and inhibited the cGAS/STING signaling in a LKB1/AMPK/ULK1-dependent mechanism in cardiomyocytes. Besides, Metrnl-induced ULK1 phosphorylation facilitated the dephosphorylation and mitochondrial translocation of STING where it interacted with tumor necrosis factor receptor-associated factor 2 (TRAF2), a scaffold protein and E3 ubiquitin ligase that was responsible for ubiquitination and degradation of STING, rendering cardiomyocytes sensitive to autophagy activation.
Conclusion
Thus, Metrnl may be an attractive therapeutic target or regimen for treating DCM.
Introduction
Diabetic cardiomyopathy (DCM) is primarily manifested by myocardial apoptosis, hypertrophy, and fibrosis, as well as cardiac systolic and diastolic dysfunction, leading to a higher risk for heart failure and sudden death in diabetic patients [1], [2]. Studies have consistently found that the cardiovascular disease risk is approximately 3–4 fold higher in diabetic subjects than that of normal controls, independently of coronary artery diseases, valvular heart disease or hypertension [3], [4]. Although several commercial drugs are available for the management of DCM, such as β-receptor antagonists or renin-angiotensin-aldosterone system inhibitors, the clinical efficacy is not sufficient for the treatment of DCM [5]. As a result, it is pressing to thoroughly elucidate the underlying mechanisms of DCM and to develop effective interventions that could delay or ameliorate the symptoms of DCM.
Meteorin-like hormone (Metrnl) is ubiquitously expressed in white adipose tissue, skin, mucosa, respiratory tract, intestinal epithelium, and heart in both human and rodents upon exposure to exercise, cold environment, inflammation, or obesity [6], [7]. As a secreted peptide homologous to neurotrophin, Metrnl regulates multiple physiological and pathophysiological processes [7]. Specifically, Metrnl acts as a myokine or adipokine to govern neural development, lipid metabolism, energy expenditure, insulin sensitization, white adipose browning, and allergic asthma [8], [9], [10]. Also, Metrnl promotes glucose uptake, fat oxidation and muscle regeneration in skeletal muscle through autocrine and paracrine actions [11]. Recently, studies on Metrnl in the cardiac system have also flourished. For instance, it is found that serum levels of Metrnl were downregulated in elderly patients with chronic heart failure when compared with healthy controls [12]. Cardiac specific overexpression (OE) of Metrnl is able to attenuate cardiac hypertrophy, fibrosis and dysfunction in isoproterenol-induced mice, and circulating Metrnl may serve as a potential biomarker for heart failure [13]. Adeno-associated virus serotype 9 (AAV9)-mediated specific OE of Metrnβ in the heart was shown to improve myocardial oxidative stress, apoptosis, and cardiac dysfunction in a mouse model of doxorubicin-induced cardiotoxicity [14]. Given the possible role of Metrnl in cardioprotection, it is highly probable that Metrnβ might function as a crucial player in the pathologies of DCM regardless of its conflicting changes in the peripheral circulation of diabetes.
In this study, we demonstrated that cardiac and circulating levels of Metrnl were inhibited in streptozotocin (STZ)-induced mice and leptin receptor deficiency (db/db) mice. Cardiac specific depletion of Metrnl worsened, while cardiomyocyte-specific OE of Metrnl reversed hyperglycemia-induced cardiac hypertrophy, fibrosis, apoptosis and oxidative stress in both T1D and T2D mice. Besides, investigations of the underlying mechanisms revealed that Mernl activated the phosphorylated liver kinase B1 (LKB1)/adenosine monophosphate activated protein kinase (AMPK)/Unc-51 like autophagy activating kinase 1 (ULK1) signaling pathway, and then induced the dephosphorylation and mitochondrial translocation of STING where it formed a complex by interacting with tumor necrosis factor receptor-associated factor 2 (TRAF2) to potentiate its ubiquitination and degradation, resulting in autophagy initiation, thus improving cardiac function in diabetes. As a consequence, Metrnl might serve as a new therapeutic target or candidate for the management of DCM.
Methods
Reagents and chemicals
Streptozocin (STZ, S0130), DAPI (D9542), and dihydroethidium (DHE, 38483–26-0), MG-132 (M7449) and cycloheximide (CHX, C7698) were procured from Sigma (St Louis, USA). STO-609 (31358), rapamycin (13346), Compound C (11967), 3-methyladenine (3-MA, 13242) and Takinib (24161) were procured from Cayman (Ann Arbor, USA). Dulbecco's modified Eagle's medium (DMEM, D5796), 1 % penicillin streptomycin mixture (P4333) and foetal bovine serum (FBS, F8318) were obtained from Sigma (St Louis, USA). Recombinant human STING protein (P1960) and recombinant human TRAF2 protein (P0303) was acquired from FineTest (Wuhan, China). Wheat germ agglutinin (WGA, W6748), EdU (5-ethynyl-2′-deoxyuridine) Alexa Fluor™ 488 imaging kits (C10637), Hoechst 33,342 (C10246) were procured from Thermo Fisher Scientific (Carlsbad, CA, USA). Picro Sirius Red Stain Kit (ab150681) was acquired from Abcam (Cambridge, USA). The information of the primary and secondary antibodies was provided in Supplementary Table 1. Plasmids encoding pcDNA3.1-HA-STING, pcDNA3.1-Flag-TRAF2, pcDNA3.1-Flag-STING and pcDNA3.1-HA-TRAF2 were synthesized and provided by Hunan Prazette Company. The scramble short hairpin RNA (shRNA, TR30021), Metrnl shRNA (TL702232), lentivirus control particles (PS100092V), lentiviral particles expressing rat Metrnl (RR204359L3V), lentiviral particles expressing ULK1 (RR214983L3V) were procured from ORIGENE (Rockville, MD, USA). The AAV9 vectors containing a cardiomyocyte‐specific cTnT promoter were designed and synthesized by Hanbio Biotechnology. The full length of Metrnl was cloned into the AAV9 plasmid to achieve cardiac-restrict Metrnl OE under the control of cTnT promoter, while the blank AAV9 vectors were used as negative controls (Hanbio, Shanghai, China). The AAV9 vectors carrying the shRNA against Metrnl or a non-target shRNA under the control of cTnT promoter were constructed to knockdown the Metrnl in the heart (Hanbio, Shanghai, China). The targeting Metrnl shRNA sequences were 5′-CACGCTTTAGTGACTTTCAAA-3′, and the control shRNA sequences were 5′-TTCTCCGAACGTGTCACGT-3′ [8], [15]. Mouse Metrnl enzyme-linked immunosorbent assay (ELISA) kits were procured from R&D (DY6679, Minneapolis, MN 55413, USA). Rat Metrnl ELISA kits were obtained from FineTest (ER0675, Wuhan, China). Control siRNA (sc-37007) and rat LKB1 siRNA (sc-270074) were bought from Santa Cruz (Santa Cruz, CA, USA). The primers used in the present study were provided by Sangon Biotech (Shanghai, China).
Animals
Male C57BL/6J mice aged 7 weeks were procured from the Changzhou Cavens Laboratory Animal Co., ltd (Changzhou, China). All mice had free access to standard chow and water under a temperature and humidity room on a 12-h light/dark cycle. After one week of adaptation, T1D mice were constructed by intraperitoneal injections of STZ (60 mg/kg) dissolved in citric acid buffer (0.1 M, pH 4.5) for 5 consecutive days. One week after the last STZ injection, the blood samples from the tail vein in mice were obtained to measure the fasting blood glucose (FBG) levels in three independent measurements. The levels of FBG more than 16.7 mM in mice were indicative of diabetes. The diabetic mice were kept for an additional 12 weeks to induce DCM. To specifically overexpress Metrnl in murine hearts, mice were subjected to a single injection of AAV9 vectors carrying Metrnl under the control of a cTnT promoter or negative control AAV9 (1 × 1011 viral genome particles for each mouse) 6 weeks after the first injection of STZ. Likewise, to specifically knockdown the endogenous Metrnl in mouse hearts, mice received a tail vein injection of AAV9 vectors carrying Metrnl shRNA or control shRNA (1 × 1010 viral particles for each animal) under the supervision of the cTnT promoter via tail vein 6 weeks after the first injection of STZ. C57BL/KsJ-db/dm (WT) mice and C57BL/KsJ-db/db (db/db) mice aged 14 weeks received AAV9 vectors carrying Metrnl under the control of a cTnT promoter or negative control AAV9 (1 × 1011 viral genome particles for each mouse), AAV9 vectors carrying Metrnl shRNA or control shRNA (1 × 1010 viral particles for each animal), respectively. After six weeks of AAV9 vector injections, the FBG levels were measured in each mouse. To examine the autocrine effects of Metrnl, mice were intraperitoneally injected with a neutralizing antibody against Metrnl or an isotype-controlled IgG in each moue (30 μg/day) every other day for continuous three times six weeks after the first injection of STZ, and then subjected to injection of AAV9 vector expressing Metrnl for 5 weeks to replenish Metrnl OE in diabetic hearts. After that, the FBG levels of these mice were detected and recorded.
Ethics statement
All experiments in rodents were accomplished in accordance with the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication, revised 2011), the ethical standards in the 1964 Declaration of Helsinki, and the guidelines of the Animal Care and Use Committee of China Pharmaceutical University (Approve number: 202101016).
Evaluation of echocardiography and hemodynamics
The cardiac function of each mouse was measured by using Vevo 2100 echocardiography system equipped with 30 MHz probe (Visualsonics, Toronto, Canada). Echocardiography was carried out to assess the structure and function of the left ventricle in mice. Two-dimensional M-mode echocardiography tracings from the short axis of the left ventricle at the level of the papillary muscles to obtain cardiac ejection fraction (EF), fraction shortening (FS), heart rate (HR), left ventricular internal dimension (LVID) in systole and diastole (LVIDS and LVIDD), left ventricular posterior wall (LVPW) thickness in diastole (LVPWD), and left ventricular anterior wall thickness in diastolic end (LVAPW) from the average of six consecutive cardiac cycles. The Doppler E wave and A wave velocity of the mitral valve were obtained. All measurements were conducted by the same operator who was blinded to the tracings. Invasive hemodynamic data were calculated by using a PowerLab system (AD Instruments ltd., UK) and LabChart 7.2 software.
Histological examination
After sacrifice, the heart tissues were harvested from individual mice and immediately fixed. The heart was subsequently embedded in paraffin, sectioned, and subjected to standard H&E histological staining and Sirius red staining for the measurement of cardiomyocyte area and fibrosis, respectively. FITC-conjugated WGA was applied to evaluate the cross-sectional area of cardiomyocytes, and the nuclei were marked by DAPI. Cardiomyocyte area was assessed from more than 100 cells in each group and averaged across several mice by using image quantitative analysis system (Image Pro-Plus version 6.0).
Immunofluorescence
To observe the localization of Metrnl in cardiomyocytes, we performed double immunofluorescence staining using both Metrnl antibody and sarcomeric actin antibody. In short, after sacrifice, the cardiac sections were permeabilized in Triton X-100 (0.1 %) for 15 min and were incubated with Metrnl anti-rabbit antibody and sarcomeric actin anti-mouse antibody. The sections were then probed by Goat anti-Rabbit IgG connected with Alexa Fluor® 488 and Goat anti-Mouse IgG connected with Alexa Fluor® 594 for 1 h. After treatment, primary cardiomyocytes were fixed with 4 % paraformaldehyde for 30 min. After blocking with 5 % BSA for 1 h, anti-α-actinin antibody was added and incubated at 4 °C overnight followed by incubation of Goat anti-Mouse IgG H&L (Alexa Fluor® 594) for 1 h. The immunofluorescence signals were obtained by a fluorescence microscope (80i, Nikon, Tokyo, Japan).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Cardiomyocyte apoptosis was examined by CoraLite®594 TUNEL assay kit (PF00009, Proteintech, Rosemont, IL, USA). The TUNEL–positive cardiomyocytes were visualized by a fluorescence microscope, and the apoptotic rates expressed as a percentage of total cells in these fields.
Oxidative stress measurement
Intracellular ROS production was examined using DHE fluorescent probe, the cardiac sections and fixed cells were incubated with DHE probe (10 μM) for 1 h at 37 °C under the dark environment. The immunofluorescence images were photographed. In addition, the levels of MDA were measured a lipid peroxidation MDA assay kit in agreement with the manufacturer's protocols, and the results were normalized to the protein contents in each sample.
Cell culture
Neonatal rat cardiomyocytes were cultured from neonatal Sprague-Dawley rats (1–3 day old) supplied from Nantong Trofi Feed Technology Co., ltd. (Nantong, China), and cardiac endothelial cells and fibroblasts from mice were cultured as previously described [16], [17], [18]. Cardiac fibroblasts from adult mice were cultured with the aid of feeder removal microbeads, and cardiac endothelial cells from adult mice were prepared by using CD31 microbeads. Cardiac fibroblasts and endothelial cells from adult mice were stained vimentin and CD31, respectively, to ascertain their purity. The purity of these cells was not<95 %. The mRNA levels of Metrnl were determined in cardiac fibroblasts and endothelial cells from control and diabetic mice. Neonatal cardiomyocytes were cultivated in six-well plates at a density of 2.5 × 104 cells/well, and the cells were cultured in low-glucose (1 g/L) DMEM supplemented with 10 % FBS, gentamicine (50 μg/ml), and cytosine β-D-arabino furanoside (Ara C, 10 μM) which was used to suppress the growth of the cardiac fibroblasts. After synchronization in serum-free DMEM for 12 h, neonatal cardiomyocytes were challenged by high glucose (HG, 25 mM) for 72 h to mimic diabetic cardiac injury in vitro. The cardiomyocytes treated with normal glucose (NG, 5.5 mM) and mannitol (19.5 mM) were used as controls. To examine the protective role of Metrnl in vitro, cardiomyocytes were pretreated with different doses of Metrnl recombinant protein (0–0.5 μg/ml, G-RPPB3964.10, Adipogen, San Diego, CA, USA) for 6 h, and cultured in NG or HG medium for 72 h. To achieve Metrnl knockdown in primary cardiomyocytes, the scramble shRNA (0.5 μg) or Metrnl shRNA (0.5 μg) were transfected to primary cardiomyocytes using Lipo6000TM transfection reagents (RiboBio, China). Neonatal cardiomyocytes were transfected with lentivirus carrying control vectors or target vectors that induced OE of rat Metrnl (106 TU/mL) for 24 h and then subjected to HG treatment for 72 h. To determine the protective effects of Metrnl on HG-induced cardiomyocyte injury were related to its autocrine-dependent manner, neonatal cardiomyocytes were transfected with lentivirus carrying vectors overexpressing of rat Metrnl (106 TU/mL) or lentivirus control particles (PS100092V, OriGene) for 24 h, and then co-cultured with or without the Metrnl antibody (1 μg/ml) for 48 h in the context of HG stimulation. After that, cardiomyocyte hypertrophy, apoptosis, and oxidative stress were measured.
Cell cytotoxicity
The cell viability of primary cardiomyocytes was examined by CCK-8 kits and EdU staining as we previously described [19]. The optical density in each well was examined at 450 nm by a microtiter plate reader (BioTek Instruments, Winooski, USA). The EdU incorporation assay was implemented by calculating the ratio of EdU-positive cells to the nucleus. A LDH release assay kit was utilized to examine the LDH from cells in which the absorbance was measured at 490 nm. The cell viability and LDH release were normalized to the control group.
Antibody array measurement
Cardiomyocyte were incubated with Mrtrnl (0.2 μg/ml) for 30 min. The cell lysates from four-independent cell samples in each group were collected for the antibody array experiments using an AMPK signaling phosphorylation antibody array (PAM174, Full Moon Biosystems, Sunnyvale, USA). The Cy3 fluorescence in the FITC filter set of the slide was scanned measured by an Axon GenePix 4000B laser scanner, and the fluorescence intensity of each array spot was quantitated by using GenePix Pro 6.0 (Axon Instruments, San Jose, CA, USA) and plotted as a heatmap. The following equation was applied to quantified by the phosphorylated A/unphosphorylated A, in which A represented the target protein.
Immunoblot and co-immunoprecipitation
The proteins of cardiac tissues and cells were extracted by lysis buffer (P0013, Beyotime, Shanghai, China). Equal amounts of protein in each sample (30 μg) were loaded onto sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (GVHP04700, Millipore, Darmstadt, Germany). The membranes were incubated with the required primary antibodies followed by secondary antibodies conjugated with horseradish peroxidase. The positive bands were captured by the enhanced chemiluminescent. The mitochondrial proteins were isolated by cell mitochondrial isolation kits (C3601, Beyotime, Shanghai, China) and were normalized to COX IV. For co-immunoprecipitation assay, equal amounts of cell lysates were immunoprecipitated by antibodies specific to STING (1 μg) for 2 h at 4 °C and the Protein G PLUS-Agarose (20 μl) was added into the corresponding tubes at 4 °C on a rocker platform. After aspirating and discarding the supernatant, the pellets were then resuspend in electrophoresis sample buffer (40 µl) and boiled for 3 min. Finally, the immune complexes were examined by Western blot.
Real-time fluorescence quantitative polymerase chain reaction (RT-PCR)
Total RNA in cardiac tissues and primary cardiomyocytes were extracted by using TRIzol™ reagents (15596018, Thermo Fisher Scientific, Waltham, USA). The concentrations of RNA in each sample were assessed with a N60 Touch nanophotometer (Implen, Munich, Germany). The equal contents of RNA (0.5 μg) were reversely transcribed to produce cDNA using a commercial reagent (N8080234, Thermo Fisher Scientific, Waltham, USA). The subsequent real-time PCR was carried out using qPCR SYBR Green Master Mix (11199ES03, YEASEN, Shanghai, China) in a StepOne™ Real-Time PCR system. The relative mRNA amounts of target genes were calculated by utilizing the 2−ΔΔCt method. The sequences of primers were provided in Supplementary Table S2 and S3.
LISAxxx
The levels of Metrnl in mice were measured by using Mouse Metrnl ELISA kits. The results of Metrnl in plasma were expressed as pg/ml, while the levels of Metrnl in cardiac tissues were calculated as pg/mg protein by normalization to the protein contents in each sample. Moreover, the levels of Metrnl in primary cardiomyocyte culture media were determined by a commercially available ELISA kit. The absorbance was immediately measured in a microplate reader at 450 nm after adding the stop solution. The target concentration of the samples can be interpolated from the standard curve.
Autophagy monitoring
To monitor the autophagy flux, the autophagosomes were detected in cardiomyocytes by transfection with a plasmid driving the expressions of EGFP-LC3 using Lipofectamine 2000 (11668019, Invitrogen, Carlsbad, USA). After that, the accumulated LC3 dots were observed with a laser confocal microscopy (Fluoview FV1000; Olympus, Japan).
RNA interference and OE
For RNA interference or OE, cardiomyocytes were plated the day before siRNA transfection grown to 30–50 % confluence. A scrambled siRNA (100 nM, sc-37007, Santa Cruz, Dallas, TX, USA), ULK1 siRNA (100 nM, SR514693, OriGene Technologies, Inc, Rockville, MD, USA), LKB1 siRNA(100 nM, sc-270074, Santa Cruz, Dallas, TX, USA), STING siRNA (100 nM, SR507303, OriGene Technologies), TRAF2 siRNA (100 nM, SR505994, OriGene Technologies) were transfected to cardiomyocytes using Lipofectamine 2000. For ULK1 OE, primary cardiomyocytes were transfected with lentivirus vectors overexpressing rat ULK1 (RR214983L3V, 106 TU/ml) or lentivirus control particles (PS100092V, 106 TU/ml, OriGene Technologies) for 24 h, and the cells were subjected to the corresponding treatments for additional detection.
Surface plasmon resonance (SPR)
The binding kinetics of STING to TRAF2 was assessed by SPR assay at 25 °C under a Biacore T200 instrument (GE Healthcare, USA) as we previously described [20]. In short, human STING protein was immobilized on the CM5 sensor chip following standard amine coupling protocols, and human TRAF2 protein was then diluted to various doses (0.25, 0.5, 1, 2, 4 μM) using running buffer. The binding sensorgrams were captured by injecting different concentrations of human TRAF2 protein over the immobilized proteins surface. Biacore T200 Evaluation software was used to calculate results using standard double-referencing under a 1:1 binding model, and the equilibrium dissociation constant (KD) was obtained.
Statistical analysis
All results were calculated as Mean ± standard error for the mean (S.E.M), and unpaired t-test was used to compare one variable between two groups. One-way ANOVA was used to compare the significance of variability when multiple groups followed by Bonferroni’s multiple comparisons test. The P value of<0.05 was predetermined as statistically significant.
Results
Metrnl is impaired in diabetic mice and HG-exposed cardiomyocytes
To probe the potential role of Metrnl in the pathologies of DCM, we examined the cardiac and circulating levels of Metrnl in both STZ-induced T1D mice and db/db T2D mice. The transcriptional levels of Metnl in the heart of T1D mice were clearly downregulated after 10 and 12 weeks of STZ injection (Fig. 1A), with concomitant decreases in plasma and cardiac Metrnl protein levels of T1D mice after 12 weeks of STZ injection (Fig. 1B,C). Interestingly, chronic hyperglycemia had no obvious effect on Metrnl mRNA in cardiac endothelial cells and fibroblasts, but markedly inhibited Metrnl mRNA in cardiomyocytes (Fig. 1D), suggesting that Metrnl is mainly derived from cardiomyocytes upon hyperglycemia challenge. Coincidentally, the above observations were phenocopied in db/db T2D mice (Fig. 1E-H). Both immunoblotting and immunofluorescence consistently revealed diminished Metrnl protein expression in heart tissues of T1D mice and T2D mice (Fig. 1I-L). In addition, we found the abundance of Metrnl was higher in cardiac muscle than that in adipose tissue or skeletal muscle, and the mRNA level of Metrnl were suppressed in diabetic heart tissues, accompanied by a slight reduction in skeletal muscle (Fig. S1A, B). Upon exposure of HG, the protein and mRNA levels of Metrnl were remarkably downregulated in primary neonatal rat cardiomyocytes (Fig. 1M-O). Meanwhile, we studied the effect of d-mannitol, a hyperosmolar control, on the expression of Metrnl in primary cardiomyocytes. As anticipated, hyperosmolar d-mannitol treatment had no impact on the expression of Metrnl at protein and mRNA levels (Fig. S2A,B), hinting that the downregulation of Metrnl was mainly caused by chronic glucotoxicity. Collectively, we deduced that Metrnl was mainly derived from cardiomyocytes in response to hyperglycemic stimulation, and its inhibition may be implicated in the pathogenesis of DCM.
Fig. 1.

Metrnl is downregulated in diabetic mice. (A) Cardiac Metrnl mRNA levels at indicated time points in T1D mice after STZ injection. (B) Plasma Metrnl level determined by ELISA in control and T1D mice. (C) Cardiac Metrnl mRNA level in control and T1D mice. (D) Cardiac Metrnl mRNA level in isolated cardiomyocytes (CM), cardiac endothelial cells (EC), and cardiac fibroblasts (CF) from control and T1D mice. (E) Cardiac Metrnl mRNA level in db/db (T2D) mice at different ages. (F) Plasma Metrnl level determined by ELISA in control and T2D mice. (G) Cardiac Metrnl mRNA level in control and T1D mice. (H) Cardiac Metrnl mRNA level in isolated cardiomyocytes (CM), cardiac endothelial cells (EC), and cardiac fibroblasts (CF) from control and T2D mice. (I) Representative blots and quantification analysis of Metrnl protein expressions in myocardial tissues obtained from control and T1D mice. (J) Representative photographs of Metrnl/Sarcomeric actinin colocalization in the heart from control and T1D mice as detected by immunofluorescence double staining. Scale bar = 150 μm. (K) Representative blots and quantification analysis of Metrnl protein expressions in myocardial tissues obtained from control and T2D mice. (L) Representative photographs of Metrnl/Sarcomeric actinin colocalization in the heart from control and T2D mice as detected by immunofluorescence double staining. Scale bar = 150 μm. (M) Representative blots and quantification analysis of Metrnl protein expressions in cardiomyocytes expose to NG or HG. (N) Metrnl mRNA level in cardiomyocytes. (O) Metrnl proteins level in cardiomyocyte supernatant as detected by ELISA. n = 4–6. *P < 0.05 versus 0 W, 8 W, Control (Con), or NG. Unpaired two-tailed t-test (B, C, D, F, G, H, I, K, M, N, O) and ANOVA (A, E).
Metrnl OE ameliorates cardiac injury and dysfunction in mice
Downregulation of Metrnl in diabetic hearts propelled us to investigate whether or not cardiac specific OE of Metrnl could improve diabetes-evoked cardiac pathologies. Six weeks after the first injection of STZ, we set out to augment mouse Metrnl expression specifically in the heart with the aid of cardiotropic AAV9 vectors. Isolation of cardiac cells from mice indicated that AAV9 induced robust and persistent upregulations of Metrnl in mouse hearts (Fig. S3A). Our results showed that cardiac specific OE of Metrnl did not affect body weight, heart rate, FBG levels, HbA1c, insulin levels, total cholesterol and triacylglycerols in diabetic mice (Table S4), but led to a significant improvement of T1D-elicited cardiac dysfunction (Fig. 2B, Table S4), as evidenced by echocardiography assay (Fig. S4A) and pressure volume curve (Fig. S5A). The enlarged cardiomyocytes in HE and WGA staining, and increased mRNA levels of hypertrophic genes were observed in mice after STZ injection, whereas these deficits were remarkably reversed by Metrnl OE (Fig. 2C-E). Chronic hyperglycemic state is a driving factor for cardiomyocyte apoptosis, oxidative stress, and fibrosis, thereby coordinately resulting in diabetes-induced cardiac dysfunction. Correspondingly, cardiac fibrosis, apoptosis and oxidative stress in T1D mice with Metrnl OE were similar to those in non-diabetic control mice, as reflected by Sirius red staining, DHE staining, and TUNEL staining, respectively (Fig. 2F-K). In consistence with this, the anti-fibrotic, anti-oxidative, and anti-apoptotic effects of Metrnl were further confirmed by decreased mRNA levels of collagen type I (Col 1), α smooth muscle actin (α-SMA), p47phox, p67phox, and Bax, and increased Bcl-2 (Fig. 2L-N). Besides, cardiac specific OE of Metrnl showed similar protective effects against DCM in db/db mice as those observed in STZ-injected mice (Fig. S4B, S5B, S6 and Table S5). The obtained data indicate that cardiac OE of Metrnl is able to prevent cardiomyocyte apoptosis, oxidative damage, and fibrotic remodeling, thus improving cardiac dysfunction in diabetic mice.
Fig. 2.

Cardiac specific OE of Metrnl attenuates cardiac injury and dysfunction in T1D mice. (A) Schematic illustration showing the treatment in mice. (B) LV EF and FS were quantified. (C) Representative photographs of the myocardium with H&E staining (Scale bar = 100 μm). (D) Representative photographs of the myocardium with WGA staining (Scale bar = 50 μm). (E) Relative mRNA levels of ANP, BNP, and β-MHC. (F) Relative collagen volume in left ventricular tissues. (G) Relative value of DHE staining fluorescence density. (H) Relative value of cardiomyocyte apoptosis. (I) Representative photographs of the myocardium with Sirus red staining. (Scale bar = 100 μm). (J) Representative photographs of the myocardium with DHE staining. (Scale bar = 200 μm). (K) Representative photographs of the myocardium with TUNEL staining. (Scale bar = 100 μm). (L) Relative mRNA levels of α-SMA and collagen I. (M) Relative mRNA levels of p47phox and p67phox. (N) Relative mRNA level of Bax and Bcl-2. n = 4–6. *P < 0.05 versus Control (Con), †P < 0.05 versus T1D. ANOVA (A-N). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Cardiac injuries are enhanced in diabetic mice with deficiency of Metrnl
Furthermore, we specifically downregulated Metrnl in murine hearts using AAV9 vectors carrying Metrnl shRNA to investigate whether Metrnl ablation affected diabetes-induced cardiac injury and dysfunction. Endogenous Metrnl levels were dramatically diminished in the isolated cardiomyocytes injected with Metrnl shRNA when compared with control shRNA-injected mice (Fig. S3B). Interestingly, we uncovered that cardiac specific deletion of Metrnl did not further reduce cardiac function in T1D diabetic mice (Fig. 3A,B, Fig. S4C, S5C, Table S6). In spite of this, HE and WGA staining revealed that Metrnl-deficient mice exhibited exacerbated cardiomyocyte thickness and area caused by diabetes. In addition, the HW/TL ratio and mRNA levels of the hypertrophy marker, including ANP, BNP, and β-MHC were further raised in T1D mice subjected to Metrnl shRNA injection (Fig. 3C-E). Accordingly, we found that AAV-mediated downregulation of Metrnl in the heart further deteriorated diabetes-related cardiac fibrosis, oxidative burst, and apoptosis, as shown by Sirius red staining, DHE staining, and TUNEL staining, respectively (Fig. 3F-K). In consistence with this phenomenon, T1D diabetes triggered fibrosis-, oxidative stress-, and apoptosis-related parameters more severely in Metrn-deficient mice than in control mice regarding the mRNA levels of collagen 1, α-SMA, p47phox, p67phox, and Bax, and Bcl-2 (Fig. 3L-N). Concordantly, the effects of Metrn knockdown on STZ-induced cardiac abnormalities were recapitulated in db/db mice (Fig. S4C, S5C, S7 and Table S7). However, deficiency of Metrn did not affect body weight, FBG, heart rate, FBG levels, HbA1c, insulin levels, total cholesterol and triacylglycerols in diabetic mice (Table S6-7). These measures together showed that the absence of Metrnl caused considerably greater increases in cardiac hypertrophy, fibrosis, oxidative stress, and apoptosis in response to diabetes-induced hyperglycemic challenge, which was not related to the changes of systemic glucose and lipid metabolism.
Fig. 3.

Cardiac specific deficiency of Metrnl worsens cardiac injury and dysfunction in T1D mice. (A) Schematic illustration showing the treatment in mice. (B) LV EF and FS were quantified. (C) Representative photographs of the myocardium with H&E staining. Scale bar = 100 μm. (D) Representative photographs of the myocardium with WGA staining. Scale bar = 50 μm. (E) Relative mRNA levels of ANP, BNP, and β-MHC. (F) Relative collagen volume in left ventricular tissues. (G) Relative value of DHE staining fluorescence density. (H) Relative value of cardiomyocyte apoptosis. (I) Representative photographs of the myocardium with Sirus red staining. Scale bar = 100 μm. (J) Representative photographs of the myocardium with DHE staining. Scale bar = 200 μm. (K) Representative photographs of the myocardium with TUNEL staining. Scale bar = 100 μm. (L) Relative mRNA levels of α-SMA and collagen I. (M) Relative mRNA levels of p47phox and p67phox. (N) Relative mRNA level of Bax and Bcl-2. n = 4–6. *P < 0.05 versus Control (Con), †P < 0.05 versus T1D. ANOVA with Bonferroni's multiple comparisons test (A-N). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Metrnl attenuates, while Metrnl interference intensifies HG-induced cardiomyocyte injury
We then assessed the potential benefits of Metrnl in primary neonatal rat cardiomyocytes in the presence or absence of Metrnl under HG circumstances. To explore the optimized concentration of Metrnl in fighting against HG-induced injury in primary neonatal rat cardiomyocytes, the dose-dependent effects of Metrnl was performed. LDH release assay showed that Metrnl concentration-dependently inhibited HG-induced LDH release in primary cardiomyocytes, reaching its maximal effects at the dose of 0.2 μg/ml (Fig. 4A). Parallelingly, both CCK-8 and EdU staining showed that Metrnl restored HG-inhibited cardiomyocyte viability in a dose-relied manner (Fig. 4B-D). Based on the above findings, the appropriate concentration of Metrnl (0.2 μg/ml) was selected in the following cellular experiments. We also examined the actions of Metrnl on HG-caused cardiomyocyte hypertrophy in vitro. It was found that HG stimulation enlarged cellular size in primary cardiomyocytes, an effect that was normalized by pretreatment with Metrnl (Fig. 4E). The anti-hypertrophic effects of Metrnl were also confirmed by measurement of several hypertrophic gene markers, including ANP, BNP and β-MHC (Fig. 4F). TUNEL staining results displayed that HG stimulated cell apoptosis in primary cardiomyocytes, while this was largely reversed by application of Metrnl (Fig. 4G). To ascertain the anti-apoptotic effect of Metrnl, we determined the caspase-3 activity, mRNA levels of pro-apoptotic gene Bax and anti-apoptotic gene Bcl-2 in primary cardiomyocytes (Fig. 4H-I). To explore whether the benefits of Metrnl in HG-evoked cardiomyocyte injury is related to reduction of oxidative stress, redox status was detected by DHE probe, MDA contents, mRNA levels of p47phox and p67phox, two NADPH oxidases responsible for ROS overproduction in cardiovascular cells. As anticipated, supplementation of Mrtrnl significantly inhibited HG-induced ROS formation and MDA production (Fig. 4J-K), coinciding with reductions of p47phox and p67phox mRNA levels in primary cardiomyocytes (Fig. 4L). By contrast, HG-elicited cytotoxicity, apoptosis, hypertrophy, and oxidative stress in primary cardiomyocytes were further aggravated in the absence of Metrnl (Fig. S8B, Fig. 5). These observations indicated that Metrnl lessened, whereas Metrnl deficiency worsened cellular hypertrophy, apoptosis, oxidative injury in primary cardiomyocytes exposed to HG.
Fig. 4.

Metrnl prevents cardiomyocyte injury upon hyperglycemic stimulation in cardiomyocytes. Cardiomyocytes were pre-incubated with various doses of Metrnl for 30 min followed by incubation of HG for additional 72 h. (A) LDH release was determined. (B) Cell viability. (C) Relative EdU-positive cells. (D) Representative photographs of EdU staining. Scale bar = 100 μm. (E) Representative photographs of α-actinin staining. Scale bar = 20 μm. (F) Relative mRNA levels of ANP, BNP, and β-MHC. (G) Representative photographs of TUNEL staining. Scale bar = 100 μm. (H) Caspase-3 activity. (I) Relative mRNA levels of Bax and Bcl-2. (J) Representative photographs of DHE staining. Scale bar = 150 μm. (K) MDA content. (L) Relative mRNA levels of p47phox and p67phox. n = 4–6. *P < 0.05 versus 0 or NG, †P < 0.05 versus HG.
Fig. 5.

Metrnl deletion deteriorates cardiomyocyte injury upon hyperglycemic stimulation in cardiomyocytes. (A) Relative blot and quantitative analysis of Metrnl in cardiomyocytes with control shRNA or Metrnl shRNA. (B) Representative photographs of EdU staining (Scale bar = 100 μm). (C) Cell viability. (D) LDH release was determined. (E) Representative photographs of α-actinin staining. Scale bar = 20 μm. (F) Relative mRNA levels of ANP, BNP, and β-MHC. (G) Representative photographs of TUNEL staining. Scale bar = 100 μm. (H) Caspase-3 activity. (I) Relative mRNA levels of Bax and Bcl-2. (J) Representative photographs of DHE staining. Scale bar = 150 μm. (K) MDA content. (L) Relative mRNA level of p47phox and p67phox. n = 4–6. *P < 0.05 versus Con shRNA, †P < 0.05 versus HG + shRNA. Unpaired two-tailed t-test (A) ANOVA (B-L).
Metrnl protects against diabetic cardiomyopathy in an autocrine fashion
Given that cardiomyocytes are both a source and a target of Metrnβ, it is deserved to know whether Metrnl plays a beneficial role in DCM in an autocrine-dependent way. To test this hypothesis, we employed an anti-Metrnl neutralizing antibody to block its systemic action. As anticipated, pretreatment with an anti-Metrnl antibody markedly eliminated the antagonistic effects of Metrnl OE on HG-induced cardiomyocyte injury (Fig. S9A,D,E), apoptosis (Fig. S9B,F,G), and hypertrophy (Fig. S9C,H). Similar to this, the beneficial effects of Metrnl OE were abrogated in diabetic mice subjected with an anti-Metrnl antibody co-treatment (Fig. S9I-P, Table S8). That is, we failed to see the therapeutic effects of Metrnl OE on diabetes-related cardiac hypertrophy (Fig. S9I,J,L,M), fibrosis (Fig. S9K,N,O), and dysfunction (Fig. S4D, S5D, S9P) in T1D mice when neutralizing Metrnl by its specific antibody. These results indicate an autocrine role for cardiomyocyte-secreted Metrnl in the context of DCM.
Metrnl activates autophagy through the LKB1/AMPK/ ULK1 pathway
Having shown that Metrnl regulated the development of DCM, we next explored the intracellular signaling pathways that Metrnl may regulate to improve DCM using the AMPK signaling phosphorylation antibody array. Microarray protein profiling assay showed that 42 protein expression levels were higher, while 24 proteins were lower in Metrnl-incubated cells (Fig. 6A). Among which, phosphorylated AMPKα1 increased by 5.54-fold in cardiomyocytes in the presence of Metrnl (Fig. 6B), this drove us to investigate whether or not Metrnl exerted the cardioprotective effects via activating AMPK signaling. Not surprisingly, supplementation of Metrnl enhanced the phosphorylated levels of AMPK in a dose- and time-dependent fashion (Fig. 6C). HG-challenged primary cardiomyocytes had a lower AMPK phosphorylation, but to a greater extent in cells co-incubated with Metrnl (Fig. 6D). Furthermore, gene silencing of Metrnl further decreased the phosphorylated levels of AMPK in cardiomyocytes upon hyperglycemic stimulation (Fig. 6E). AMPKα2 is the primary subunit of AMPK in the myocardium and ablation of AMPKα2 leads to pathological cardiac remodeling and dysfunction [21]. Recently, Tang’s group has shown that activation of the cAMP/PKA/LKB1/AMPKα2 signaling pathway underlies the protective effects of Liquiritin against pathological cardiac hypertrophy [22]. Thus, in addition to AMPKα1,we wonder whether Metrnl OE affected AMPKα2 signaling in cardiomyocytes. Results showed that Metrnl OE had no influence on phosphorylated AMPKα2 (Fig. S10), indicating that the actions of Metrnl were AMPKα2-independent in DCM.
Fig. 6.

Metrnl activates the LKB1/AMPK signaling in cardiomyocytes. (A) Heatmap showing the effects of Metrnl on the components of AMPK array. (B) Top ten signaling protein induced by Metrnl. (C) Metrnl dose- and time-dependently induced AMPK phosphorylation. (D) Effects of Metrnl on HG-induced downregulation of phosphorylated AMPK. (E) Effects of Metrnl shRNA on HG-induced downregulation of phosphorylated AMPK. (F) Cardiomyocytes were pretreated with STO-609 (0.8 μM) or Takinib (10 mM) for 30 min, and challenged by Metrnl (0.2 μg/ml) for 30 min, and then stimulated for HG for 48 h. Cardiomyocytes were pretreated with LKB1 siRNA (100 nM) for 6 h, and challenged by Metrnl (0.2 μg/ml) for 30 min, and then stimulated for HG for 48 h. Western blot was used to determine phosphorylated AMPK. (G) Effects of Metrnl on HG-induced downregulation of phosphorylated LKB1. (H) Effects of Metrnl shRNA on HG-induced downregulation of phosphorylated LKB1. n = 4. *P < 0.05 versus 0 μg/ml, 0 min, NG, Con, Con shRNA, †P < 0.05 versus HG + Con shRNA, Metrnl or HG.
As is known, liver kinase B1 (LKB1), transforming growth-factor-β-activated kinase-1 (TAK1) and Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKKβ) are well-established upstream kinases that result in AMPK activation [23]. Next, a selective CaMKKβ inhibitor STO-609, a potent and selective TAK1 inhibitor Takinib, and siRNA of LKB1 (Fig. S11A) were used to examine how Metrnl activated AMPK signaling in cardiomyocytes. Results showed that neither STO-609 nor Takinib had no obvious effect on Metrnl-induced AMPK activation (Fig. 6F). However, deficiency of LKB1 remarkably prevented Metrnl-evoked AMPK phosphorylation and activation (Fig. 6F). Meanwhile, HG exposure significantly decreased the phosphorylated levels of LKB1 in primary cardiac cells that were enhanced by Metrnl pretreatment, but further decreased by deletion of Metrnl (Fig. 6G-H). To sum up, these results suggested that Metrnl might activate AMPK dependent on LKB1 in cardiomyocytes.
During the process of DCM, autophagy is a cytoprotective mechanism that might undergo activation of the LKB1/AMPK/ULK1 pathway [24]. It has been shown that AMPK triggers autophagy through directly inducing ULK1 phosphorylation [25], and the phosphorylation of ULK1 is required for induction of autophagy, thus attenuating glucose toxicity-induced cardiac injury [26]. To this end, we tested whether Metrnl can stimulate the process of autophagy through the LKB1/AMPK/ULK1 pathway. As demonstrated in Fig. 7A-B, HG incubation significantly diminished the phosphorylation levels of ULK1 in primary cardiomyocytes, the effects that were reversed by Metrnl, but were further inhibited by Metrnl knockdown. Correspondingly, the transcriptional levels of autophagy genes, including Atg5, Atg8, Atg10, Atg12, and Beclin-1, were upregualted in Metrnl-incubated cardiomyocytes (Fig. 7C). Importantly, pretreatment with Compound C, an AMPK inhibitor, blunted the effects of Metrnl on the mRNA levels of such genes, further confirming the involvement of AMPK in Metrnl-induced autophagy in cardiomyocytes (Fig. S12).
It was also found in the present study that the formation of autophagosome was reduced in HG-challenged cardiomyocytes, as verified by measurement of LC3B dots in cells transfected with EGFP-LC3 plasmids (Fig. 7D). The suppressive effects of HG on the autophagy in cardiomyocytes were further verified by measurement of p62 protein and LC3B proteins (Fig. 7E). These observations were significantly offset by Metrnl treatment, as reflected by upregulated LC3B dots and LC3BI to LC3BII conversion, decreased p62 protein (Fig. 7D-E). On the contrary, gene deficiency of Metrnl had the opposite effects on autophagy (Fig. 7F-G). It is likely that the LKB1/AMPK/ULK1/autophagy axis might partially contribute to the beneficial effects of Metrnl on diabetic cardiac injury. The AMPK/mammalian phosphorylation target of rapamycin (mTOR)signaling pathway is an important regulator of DCM [27], and LKB1 induces AMPKα phosphorylation at Thr 172 within its catalytic subunit, leading to AMPK-mediated inhibition of mTORC1 and subsequent activation of autophagy [22], [28], [29]. Thus, we next sought to examine whether mTORC1 was involved in the induction of autophagy mediated by Metrnl. A significant increase in the phosphorylation levels of mTORC1 was observed following treatment with HG, this effect was attenuated by treating cells with the inhibitor of mTOR, rapamycin (Fig. S13). Interestingly, Metrnl had no effect on the phosphorylated mTORC1 (Fig. S13), indicating that activation of autophagy by Metrnl is mediated by the AMPK/ULK1 pathway in a mTOR-independent manner.
Fig. 7.

Metrnl activates the ULK1/autophagy signaling in cardiomyocytes. (A) Effects of Metrnl on HG-induced downregulation of phosphorylated ULK1. (B) Effects of Metrnl shRNA on HG-induced downregulation of phosphorylated ULK1. (C) Effects of Metrnl on the mRNA levels of autophagy-related genes. (D) Effects of Metrnl on the formation of autophagosome as assessed by LC3 dots. Scale bar = 20 μm. (E) Effects of Metrnl on the protein expressions of p62 and LC3. (F) Effects of Metrnl shRNA on the formation of autophagosome as assessed by LC3 dots. Scale bar = 20 μm. (G) Effects of Metrnl shRNA on the protein expressions of p62 and LC3. n = 4. *P < 0.05 versus NG, Con, Con shRNA, †P < 0.05 versus HG or HG + Con shRNA.
Metrnl requires ULK1 to inactivate the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) signaling in cardiomyocytes
Activation of the cGAS/STING signaling is involved in the production of interferons and other inflammatory cytokines [30]. It is reported that the STING could be phosphorylated by ULK1, thus preventing sustained innate immune signaling [31]. ULK1 phosphorylation is enough to suppress the cGAS/STING pathway in the heart, leading to an improvement of high fat diet-induced cardiac anomalies [32]. Recently, it has been revealed that activation of cGAS/STING signaling plays a vital role in obesity-related DCM [33]. On these grounds, we explored whether Metrnl ameliorated DCM by inhibiting the cGAS/STING axis in a ULK1-dependent manner. HG-incuabted cardiomyocytes displayed increased protein levels of c-GAS, STING, and phosphorylated STING when compared to that of control cells (Fig. 8A), suggesting the potential involvement of c-GAS/STING signaling activation in HG-induced cardiomyocyte injury. However, such increases were largely attenuated by supplementation of Metrnl (Fig. 8A). In turn, shRNA-mediated downregulation of Metrnl further aggravated HG-induced activation of cGAS/STING signaling in primary cardiomyocytes (Fig. 8B). However, no significant difference in the ratio of phosphorylated STING to total STING was observed among different groups (Fig. S14A-B).
Fig. 8.

ULK1 phosphorylation is required for Metrnl to inactivate the cGAS/STING signaling in cardiomyocytes. (A) Effects of Metrnl on cGAS, phosphorylated STING, and total STING. (B) Effects of Metrnl shRNA on cGAS, phosphorylated STING, and total STING. (C) Effects of Metrnl (upper) or Metrnl shRNA (lower) on STING in mitochondria. (D) Effects of Con siRNA and ULK1 siRNA on ULK1 protein. (E) Silencing of ULK1 abolished the inhibitory effects of Metrnl on cGAS, phosphorylated STING, total STING and STING in mitochondria. n = 4. *P < 0.05 versus NG or Con shRNA, †P < 0.05 versus HG + Con shRNA or HG, ‡ P < 0.05 versus HG + Metrnl + Con siRNA.
STING is primarily localized to the endoplasmic reticulum, mitochondria, and mitochondria-associated membranes through its N-terminal transmembrane domain, whereas the C-terminal domain of STING remains in the cytosol to interact with the second messenger cGAMP [34]. It is thus reasonable to hypothesize that disrupting the binding of cytoplasmic STING to cGAMP may be a key step for inhibiting the cGAS/STING signaling pathway. Concordantly, the mitochondrial translocation of STING is required for its ubiquitination and degradation induced by the ubiquitin ligase RNF5 [35]. Based on this, we investigated whether the mitochondrial shift of STING was involved in the pathological process of DCM. Immunoblots of mitochondrial proteins showed that HG stimulation inhibited the deposition of STING in mitochondria, which was partially attenuated by Metrnl administration (Fig. 8C). Different from the results obtained after Metrnl treatment, knockdown of Metrnl further decreased the accumulation of STING in HG-incubated cardiomyocytes (Fig. 8C). In addition, silencing of ULK1 abolished the effects of Metrnl on HG-induced upregulations of c-GAS, STING, and phosphorylated STINGs, mitochondrial efflux of STING (Fig. 8D-E). Totally, Metrnl acted on the ULK1 to inhibit the cGAS/STING signaling in cardiomyocytes, accompanied by the mitochondrial translocation of STING.
Metrnl promotes the ubiquitination-dependent degradation of STING by forming the mitochondrial TRAF2/STING complex in cardiomyocytes
As mentioned above, cellular fractionation assay revealed that Metrnl significantly promoted the mitochondrial translocation of STING, coincided with the downregulation of STING in cardiomyocytes. This drives us to examine whether Metrnl induced the degradation of STING in mitochondria, thereby reducing the binding of STING to the second messenger cGAMP in cytoplasm. Tumor necrosis factor receptor-associated factor 2 (TRAF2), one of the most widely studied E3 ubiquitin ligases in the TRAF family, functions as a scaffold protein that regulates several inflammatory signaling molecules in mitochondria [36]. In light of the critical role of TRAF2 in mitochondrial homeostasis, it is intriguing to know if Metnl facilitated the mitochondrial translocation of TFAF2 to induce the degradation of STING in mitochondrial. Prolonged exposure of cardiomyocytes to HG inhibited the accumulation of TRAF2 in mitochondria, an effect that was abrogated by pretreatment with Metrnl which itself boosted the mitochondrial shift of TRAF2 (Fig. 9A). Metrnl silence further prevented the mitochondrial abundance of TRAF2 in HG-chanllenged cardiomyocytes (Fig. 9B). Confocal photographs of immunofluorescence double staining showed the colocalization of TRAF2 with STING in cardiomyocytes (Fig. 9C). The co-IP assay demonstrated that TRAF2 interacted with STING directly in cardiomyocytes (Fig. 9D). In addition, SPR assay revealed that STING exhibited a higher binding affinity to TRAF2 with an estimated KD of 501.6 nM, suggesting that STING might directly interact with TRAF2 (Fig. S15). Notably, the complex of TRAF2/STING was enhanced in the presence of Metrnl (Fig. 9E). On the contrary, deficiency of Metrnl lowered the formation of TRAF2/STING complex (Fig. 9F). As shown in Fig. 9G, pretreatment with proteasome inhibitors MG-132 dramatically reversed Metrnl-induced STING degradation when compared with Metrnl alone. Moreover, we conducted cycloheximide (CHX) chase assay to evaluate the half-life of STING in cardiomyocytes. We found that the half-life of SIRT1 was much shorter in the presence of Metrnl, suggesting that Metrnl stimulated the degradation of STING through accelerating the half-life of STING in cardiomyocytes (Fig. 9H). However, deficiency of Metrnl extended its half-life in cardiomyocytes, as fortified by the experiments using CHX (Fig. 9H). Parallelingly, the polyubiquitinated STING was significantly enhanced by Metrnl, but was inhibited by deletion of Metrnl in cardiomyocytes (Fig. 9I). The suppressive effects of Metrnl deficiency on STING ubiquitination were normalized by ULK1 OE (Fig. 9B), indicating the Metrnl-induced ubiquitination and degradation of STING were ULK1-dependent (Fig. 9J). Importantly, silencing of STING and TRAF2 eliminated the process of autophagy induced by Metrnl, as assessed by p62 and LC3B proteins (Fig. 9K), as well as LC3 dots (Fig. 9L) in cardiomyocytes. Overall, Metrnl accelerated the mitochondrial translocation of STING, where it bound to TRAF2 to trigger STING degradation via protein ubiquitination system, leading to subsequent autophagy and cardioprotection.
Fig. 9.

Metrnl promotes the interaction of STING with TRAF2 in mitochondria to induce STING ubiquitination and degradation in cardiomyocytes. (A) Effects of Metrnl on TRAF2 in mitochondria. (B) Effects of Metrnl shRNA on TRAF2 in mitochondria. (C) Confocal images showing the colocalization of STING with TRAF2 in cardiomyocytes. (D) Co-IP showing the interaction of STING with TRAF2. (E) Effects of Metrnl on the STING/TRAF2 complex. (F) Effects of Metrnl shRNA on the STING/TRAF2 complex. (G) A proteasome inhibitor MG132 prevented the suppressive effects of Metrnl on STING protein expression. (H) Half-life of STING in different groups. (I) Effects of Metrnl (left) or Metrnl shRNA (right) on the ubiquitination of STING. (J) OE of LKB1 prevented the actions of Metrnl shRNA on the ubiquitination of STING. (K) STING siRNA and TRAF2 siRNA weakened the effects of Metrnl on the p62 and LC3 proteins. (L) STING siRNA and TRAF2 siRNA weakened the effects of Metrnl on the formation of autophagosome as assessed by LC3 dots. n = 4. *P < 0.05 versus 0 h, NG, Con, or Con shRNA, †P < 0.05 versus HG + Con shRNA, Metrnl or HG, ‡ P < 0.05 versus HG + Metrnl + Con siRNA.
To further verify the participation of the AMPK/STING/autophagy pathway in Metrnl-induced benefits in hyperglycemia induced cardiomyocyte injury, we next investigated whether blockage of AMPK/autophagy and activation of STING could prevent the favorable effects of Metrnl on HG-incubated cardiomyocytes. Inevitably, supplementation of Metrnl lost the protective effects against hyperglycemia-induced cardiomyocyte apoptosis, hypertrophy and oxidative damage when cardiomyocyte were pretreated with Compound C (an AMPK inhibitor), 3-MA (an autophagy inhibitor), or STING agonist cGAMP (Fig. S16) indicating the involvement of LKB1/AMPK/ULK1/STING/TRAF2/autophagy signaling in mediating the effects of Metrnl in DCM. Very recently, it has been reported that Metrnl binds to KIT receptor tyrosine kinase (KIT), and promotes heart repair in a mouse model of myocardial infarction [37]. This drives us to explore whether Metrnl acts on downstream pathways by binding to KIT in the context of diabetes. Blockade of KIT by its neutralizing antibody prevented the suppressive effects of Metrnl on HG-induced LDH release, mRNA expressions of Bax and ANP (Fig. S17), indicating that Metrnl might bind to KIT to confer its benefits in the heart. However, more in vivo and in vitro research is required to examine whether and how KIT mediated the actions of Metrnl in the cardiovascular system.
Discussion
In the present study, our findings delineated a new signaling pathway wherein the cardiokine Metrnl exerted a cardioprotective action against of DCM. Therefore, Metrnl may be a potential regulator in diabetes-related cardiac pathophysiology. Future studies are highly acquired to eluciate the possibility of Metrnl as a strong biomarker or therapeutic target for patients suffering from DCM.
Monuting evidence suggests that Metrnl improves glucose metabolism, enhances white adipose tissue browning, elevates whole-body energy expenditure, and attenuates lipid-induced inflammation, suggesting that Metrnl is a promising target for the prevention of metabolic diseases, like diabetes. In hypertrophic cardiomyocytes, the protein expression of Metrnl is downregulated, and its OE prevents the onset and development of cardiac injury induced by isoproterenol [13]. This study indicated that Metrnl upregulation might be an adaptive process to hypertrophic stimulant, which could not achieve full protection against isoproterenol-induced cardiac dysfunction [13]. The abundance of Metrnl in the heart is reduced upon doxorubicin exposure, and cardiac-specific OE of Metrnl attenuates cardiac dysfunction in doxorubicin -infused mice [14]. In the current study, we revealed that myocardial and circulating Metrnl levels tended to be reduced in STZ-induced T1D mice and genetically obese T2D mice, paralleled by decreased Metrnl levels in HG-exposed cardiomyocytes. Nevertheless, the underlying mechanism of Metrnl downregulation in DCM remains unclear, and this deserves further investigations.
Oxidative stress is a central player that drives cardiac apoptosis, fibrosis, and hypertrophy, critical characteristics of DCM [38], [39]. We found that the mice with cardiomyocyte-specific knockout of Metrnl were more susceptible to cardiac hypertrophy, fibrosis, apoptosis, and oxidative injury induced by diabetes, while cardiomyocyte-specific OE of Metrnl conferred the opposite effects. Similar results were reproduced in primary cardiomyocytes with or without Metrnl under hyperglycemia conditions. Interestingly, we verified that Metrnl deficiency or OE did not alter the blood glucose and lipid levels, suggesting that the effects of Metrnl on DCM development were independent on blood glucose and lipid levels. Thus, our findings demonstrated the protective role of Metrnl against DCM in different types of diabetic mice, thereby providing potential clinical significance. Moreover, the beneficial effects of Metrnl in diabetic hearts and HG-stimulated cardiomyocytes disappeared after co-treatment with a Metrnl neutralizing antibody. These observations supported that Metrnl retained an autocrine capacity to protect cardiac function against hyperglycemia insults. The local synthesis and secretion of Metrnl may be recognized as an important mechanism that accounts for its cardioprotective actions in DCM. One should bear in mind that the absence of Metrnl in cardiac tissue did not exacerbate cardiac dysfunction even if there were significant changes in H&E staining, WGA staining, hypertrophy marker genes such as ANP and BNP. This may be due to insufficient observation time. With the further aggravation of abnormal cardiac structure in diabetes mice due to Metrnl deficiency, it may eventually lead to further decline of cardiac function, which deserves further studies. Besides, construction of cardiomyocyte specific knockout mice may help solve the importance of Metrnl in DCM-induced cardiac dysfunction.
AMPK, a well-established energy sensor, serves as one of the potential pharmacotherapeutic targets in DCM [40], since AMPK activation is effective in ameliorating myocardial fibrosis, ventricular hypertrophy, and cardiac apoptosis in diabetes [41]. Based on this, we explored whether Metrnl-induced protection against DCM was related to the activation of AMPK by using the AMPK signaling phosphorylation antibody array. Our antibody array showed that Metrnl dramatically enhanced AMPK phosphorylation signals in primary cardiomyocytes, indicating that Metrnl might protect against DCM by activating the AMPK signaling. As expected, Metrnl does- and time-dependently induced AMPK phosphorylation in cardiomyocytes, and reversed HG-induced inhibition of P-AMPK. By contrast, deficiency of Metrnl further strengthened HG-reduced AMPK phosphorylation. We also found that Metrnl-evoked AMPK activation was prevented by LKB1 silencing rather than inhibitors of CaMMKβ, and TAK1, hinting that neither CaMMKβ nor TAK1 was involved in the Metrnl-mediated AMPK activation in cardiomyocytes. In agreement with this, the decreased phosphorylation levels of LKB1 in HG-treated cardiomyocytes were restored by Metrnl treatment, but were further suppressed by Metrnl deficiency. These results further indicated that Metrnl activated AMPK via LKB1 in cardiomyocytes, yet, the specific mechanisms wherein Metrnl directly affected LKB1 in cardiomyocyte require extensive work in the near future.
As a highly conserved catabolic process, autophagy plays a crucial role in cellular homeostasis through degradation of dysfunctional intracellular wastes and recycle of cytoplasmic contents [42]. Preclinical studies have observed autophagy dysregulation in diabetic hearts [43]. It has been reported that AMPK activation lessens the development of DCM by activating autophagy [44], [45]. Moreover, AMPK-upregulated ULK1 is an upstream mediator of autophagy to ameliorate diabetic complications, such as DCM. Thus, the AMPK/ULK1 signaling pathway is involved in autophagy-related activities [46], leading to an important approach to the treatment of diabetic cardiomyopathy. On these grounds, we examined whether Metrnl had a beneficial role in DCM by activating the ULK1/autophagy pathway dependent on AMPK activation. Our results showed that Metrnl upregulated phosphorylated ULK1 and autophagic genes in cardiomyocytes, and restored the impaired autophagy induced by HG. Conversely, knockdown of Metrnl further aggravated HG-inhibited autophagy to pose the opposite effects. To sum up, induction of autophagic flux was required for Metrnl to correct DCM by activating the LKB1/AMPK/ULK1 signaling pathway.
Studies have shown that phosphorylated ULK1 could prevent STING-dependent pro-inflammatory disease [31]. Inhibition of cGAS/STING signaling negates palmitic acid-induced cardiomyocyte contractile dysfunction [32]. As a result, we examined whether Metrnl-induced ULK1phosphorylation was able to attenuate diabetes-mediated activation of cGAS/STING signaling in DCM. Our results showed that HG upregulated the protein levels of cGAS, STING, and P-STING in primary cardiomyocytes, with a more pronounced effect in Metrnl-deficient cardiomyocytes. On the contrary, Metrnl pretreatment reversed HG-induced activation of cGAS/STING signaling, an effect that was dependent on ULK1 phosphorylation. Apart from the autophagy-lysosome pathway that degrades intracellular cellular constituents or waste, the ubiquitin proteasome system (UPS) is also a prime degradation pathway in cells whereby the certain proteins tagged by polyubiquitin chains are selectively recognized and scavenged by proteasome [47]. It is likely that both degradation pathways are controlled independently by different players, but a growing body of studies has revealed a cross-talk between UPS and autophagy in a number of human diseases [47]. Mitophagy, a well-known type of mitochondrion-targeted autophagy, regulates mitochondrial quality by removing damaged mitochondria in mammalian cells, and the role of UPS in the mitophagy has been highlighted [48]. As the name suggests, mitophagy is responsible for removing the damaged mitochondria in mammalian cells and the dysfunctional mitochondrial proteins are targeted by mitophagy for protein ubiquitination [48]. Thus, we further explored whether Metrnl affected the STING pathway within the mitochondria. Our results showed that Metrnl promoted the mitochondrial translocation of STING and TRAF2 where they interacted with each other, leading to ubiquitination and degradation of STING, thereby making cardiomyocytes more sensitive to autophagy. Metrnl-induced activation of autophagy was attenuated in the absence of either STNG or TRAF2, indicating that the formation of the mitochondrial STING/TRAF2 complex is critical for Metrnl to activate autophagy in cardiomyocytes. Of importance, pharmaceutical inhibition of AMPK/autophagy, and pharmaceutical activation of STING erased the therapeutic potential of Metrnl in HG-induced cardiomyocyte injury. Overall, the LKB1/AMPK/ULK1 axis was phosphorylated in the presence of Metrnl, followed by STING dephosphorylation and TRAF2-dependent degradation of STNG in mitochondria, causing activation of autophagy and subsequent protection against DCM. Of note, in interference experiments, two independent shRNAs or siRNAs will be used to avoid off-target effects in our future studies. Moreover, palmitic acid (PA) or HG/PA intervention should be used to stimulate cardiomyocytes to mimic T2D-related cell injury, thus confirming the therapeutic potential of Metrnl in T2D-induced cardiomyopathy, this deserves in-depth studies. Given the anti-diabetic effects of Metrnl, more extensive research is needed to compare the therapeutic effects of Metrnl and insulin/another diabetes drug in diabetes and DCM.
Conclusion
The study indicates that Metrnl holds great promise as a novel therapeutic target for DCM. In addition, our results not only positioned Metrnl at the center of the cellular events that contributed to DCM pathogenesis, but also provided a potential clue for exploiting this cardiokine for the development of complementary treatment solutions against DCM. However, the precise receptors for Metrnl remain unclear and further research is urgently needed. Long-term multicenter randomized clinical trials are extremely recommended to test the potential of Metrnl as a novel biomarker and/or therapeutic candidate for prognosis in patients with DCM.
Availability of data and material
All original materials and data are available upon reasonable request.
Funding
We thank the funding by the National Natural Science Foundation of China (8217021262 and 81700364), high-level introduction of talents and scientific research start-up funds of CPU (3150020068), Jiangsu Natural Science Foundation (BK20170179, BE2020634 and BK20191138), Top Talent Support Program for young and middle-aged people of Wuxi Health Committee (BJ2020049 ).
Compliance with Ethics Requirement.
All experiments in rodents were accomplished in accordance with the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication, revised 2011), the ethical standards in the 1964 Declaration of Helsinki, and the guidelines of the Animal Care and Use Committee of China Pharmaceutical University (Approve number: 202101016).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2022.10.014.
Contributor Information
Ke-Xue Li, Email: kexueli@xzhmu.edu.cn.
Ji-Ru Zhang, Email: zjr2010508@126.com.
Hai-Jian Sun, Email: sunhaijian927@163.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sun H.J., Wu Z.Y., Nie X.W., et al. An updated insight into molecular mechanism of hydrogen sulfide in cardiomyopathy and myocardial ischemia/reperfusion injury under diabetes. Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.651884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cui X, Wang J, Zhang Y, et al. Plin5, a new target in diabetic cardiomyopathy. 2022;2022:2122856. [DOI] [PMC free article] [PubMed]
- 3.Xie J., Ikram M.K., Cotch M.F., et al. Association of diabetic macular edema and proliferative diabetic retinopathy with cardiovascular disease: a systematic review and meta-analysis. JAMA Ophthalmology. 2017 Jun 1;135(6):586–593. doi: 10.1001/jamaophthalmol.2017.0988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubler S., Dlugash J., Yuceoglu Y.Z., et al. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972 Nov 8;30(6):595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 5.Borghetti G., von Lewinski D., Eaton D.M., et al. Diabetic cardiomyopathy: current and future therapies. Beyond Glycemic Control Front Physiol. 2018;9:1514. doi: 10.3389/fphys.2018.01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng S.L., Li Z.Y., Song J., et al. Metrnl: a secreted protein with new emerging functions. Acta Pharmacol Sin. 2016 May;37(5):571–579. doi: 10.1038/aps.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miao Z.W., Hu W.J., Li Z.Y., et al. Involvement of the secreted protein Metrnl in human diseases. Acta Pharmacol Sin. 2020 Dec;41(12):1525–1530. doi: 10.1038/s41401-020-00529-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z.Y., Song J., Zheng S.L., et al. Adipocyte metrnl antagonizes insulin resistance through PPARγ Signaling. diabetes. 2015 Dec;64(12):4011–4022. doi: 10.2337/db15-0274. [DOI] [PubMed] [Google Scholar]
- 9.Gao X, Leung TF. Meteorin-β/Meteorin like/IL-41 attenuates airway inflammation in house dust mite-induced allergic asthma. 2022 Feb;19(2):245-259. [DOI] [PMC free article] [PubMed]
- 10.Jung TW, Lee SH, Kim HC, et al. METRNL attenuates lipid-induced inflammation and insulin resistance via AMPK or PPARδ-dependent pathways in skeletal muscle of mice. 2018 Sep 13;50(9):1-11 [DOI] [PMC free article] [PubMed]
- 11.Alizadeh H. Myokine-mediated exercise effects: the role of myokine meteorin-like hormone (Metrnl) Growth factors (Chur, Switzerland) 2022 Feb;8:1–8. doi: 10.1080/08977194.2022.2032689. [DOI] [PubMed] [Google Scholar]
- 12.Cai J, Wang QM, Li JW, et al. Serum Meteorin-like is associated with weight loss in the elderly patients with chronic heart failure. 2022 Feb;13(1):409-417. [DOI] [PMC free article] [PubMed]
- 13.Rupérez C., Ferrer-Curriu G., Cervera-Barea A., et al. Meteorin-like/Meteorin-β protects heart against cardiac dysfunction. J Exp Med. 2021 doi: 10.1084/jem.20201206. May 3;218(5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu C., Zhang X., Song P., et al. Meteorin-like protein attenuates doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1 pathway. Redox Biol. 2020 Oct;37 doi: 10.1016/j.redox.2020.101747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li T., Zhao X., Duan J., et al. Targeted inhibition of STAT3 in neural stem cells promotes neuronal differentiation and functional recovery in rats with spinal cord injury. Exp Therapeutic Med. 2021 Jul;22(1):711. doi: 10.3892/etm.2021.10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bang C., Batkai S., Dangwal S., et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Investig. 2014 May;124(5):2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thum T., Gross C., Fiedler J., et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008 Dec 18;456(7224):980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 18.Ma Z.G., Yuan Y.P., Zhang X., et al. C1q-tumour necrosis factor-related protein-3 exacerbates cardiac hypertrophy in mice. Cardiovasc Res. 2019 May 1;115(6):1067–1077. doi: 10.1093/cvr/cvy279. [DOI] [PubMed] [Google Scholar]
- 19.Sun H.J., Xiong S.P., Wu Z.Y., et al. Induction of caveolin-3/eNOS complex by nitroxyl (HNO) ameliorates diabetic cardiomyopathy. Redox Biol. 2020 May;32 doi: 10.1016/j.redox.2020.101493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang ZC, Niu KM, Wu YJ, et al. A dual Keap1 and p47(phox) inhibitor Ginsenoside Rb1 ameliorates high glucose/ox-LDL-induced endothelial cell injury and atherosclerosis. 2022 Sep 26;13(9):824. [DOI] [PMC free article] [PubMed]
- 21.Jessen N, Koh HJ, Folmes CD, et al. Ablation of LKB1 in the heart leads to energy deprivation and impaired cardiac function. Biochim Biophys Acta 2010 Jul-Aug;1802(7-8):593-600. [DOI] [PMC free article] [PubMed]
- 22.Aiyasiding X., Liao H.H., Feng H., et al. Liquiritin attenuates pathological cardiac hypertrophy by activating the PKA/LKB1/AMPK pathway. Front Pharmacol. 2022;13 doi: 10.3389/fphar.2022.870699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo Z., Tuo H., Tang N., et al. Neuraminidase 1 deficiency attenuates cardiac dysfunction, oxidative stress, fibrosis, inflammatory via AMPK-SIRT3 pathway in diabetic cardiomyopathy mice. Int J Biol Sci. 2022;18(2):826–840. doi: 10.7150/ijbs.65938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Y., Yu W., Sun D., et al. A novel protective mechanism for mitochondrial aldehyde dehydrogenase (ALDH2) in type i diabetes-induced cardiac dysfunction: role of AMPK-regulated autophagy. BBA. 2015 Feb;1852(2):319–331. doi: 10.1016/j.bbadis.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 25.Kim J., Kundu M., Viollet B., et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011 Feb;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu W, Zha W. Exendin-4 and liraglutide attenuate glucose toxicity-induced cardiac injury through mTOR/ULK1-dependent autophagy. 2018;2018:5396806. [DOI] [PMC free article] [PubMed]
- 27.Peng M.L., Fu Y., Wu C.W., et al. Signaling pathways related to oxidative stress in diabetic cardiomyopathy. Front Endocrinol. 2022;13 doi: 10.3389/fendo.2022.907757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y., Zhu X., Chen K., et al. Resveratrol prevents sarcopenic obesity by reversing mitochondrial dysfunction and oxidative stress via the PKA/LKB1/AMPK pathway. Aging. 2019 Apr 15;11(8):2217–2240. doi: 10.18632/aging.101910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S., Jo M., Lee H.E., et al. HEXA-018, a novel inducer of autophagy, rescues TDP-43 toxicity in neuronal cells. Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.747975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallego-Marin C, Schrum JE, Andrade WA, et al. Cyclic GMP-AMP synthase is the cytosolic sensor of plasmodium falciparum genomic DNA and activates type I IFN in malaria. 2018 Jan 15;200(2):768-774 [DOI] [PMC free article] [PubMed]
- 31.Konno H., Konno K., Barber G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013 Oct 24;155(3):688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong Y., Li G., Tao J., et al. Double knockout of Akt2 and AMPK accentuates high fat diet-induced cardiac anomalies through a cGAS-STING-mediated mechanism. Biochim Biophys Acta, Mol Basis Dis. 2020 Oct 1;1866(10) doi: 10.1016/j.bbadis.2020.165855. [DOI] [PubMed] [Google Scholar]
- 33.Ma X.M., Geng K., Law B.Y., et al. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol Toxicol. 2022 Mar 2 doi: 10.1007/s10565-021-09692-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong B., Yang Y., Li S., et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008 Oct 17;29(4):538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Zhong B., Zhang L., Lei C., et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009 Mar 20;30(3):397–407. doi: 10.1016/j.immuni.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 36.Borghi A., Verstrepen L., Beyaert R. TRAF2 multitasking in TNF receptor-induced signaling to NF-κB, MAP kinases and cell death. Biochem Pharmacol. 2016 Sep;15(116):1–10. doi: 10.1016/j.bcp.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 37.Reboll MR, Klede S, Taft MH. Meteorin-like promotes heart repair through endothelial KIT receptor tyrosine kinase. 2022 Jun 17;376(6599):1343-1347. [DOI] [PMC free article] [PubMed]
- 38.Wang Z.C., Machuki J.O., Li M.Z., et al. A narrative review of plant and herbal medicines for delaying diabetic atherosclerosis: an update and future perspectives. Rev Cardiovascular Med. 2021 Dec 22;22(4):1361–1381. doi: 10.31083/j.rcm2204142. [DOI] [PubMed] [Google Scholar]
- 39.Li K.X., Ji M.J., Sun H.J. An updated pharmacological insight of resveratrol in the treatment of diabetic nephropathy. Gene. 2021 May;15(780) doi: 10.1016/j.gene.2021.145532. [DOI] [PubMed] [Google Scholar]
- 40.Dewanjee S., Vallamkondu J., Kalra R.S., et al. Autophagy in the diabetic heart: A potential pharmacotherapeutic target in diabetic cardiomyopathy. Ageing Res Rev. 2021 Jul;68 doi: 10.1016/j.arr.2021.101338. [DOI] [PubMed] [Google Scholar]
- 41.Haye A., Ansari M.A., Rahman S.O., et al. Role of AMP-activated protein kinase on cardio-metabolic abnormalities in the development of diabetic cardiomyopathy: A molecular landscape. Eur J Pharmacol. 2020 Dec;5(888) doi: 10.1016/j.ejphar.2020.173376. [DOI] [PubMed] [Google Scholar]
- 42.Li L., Xu J., He L., et al. The role of autophagy in cardiac hypertrophy. Acta Biochim Biophy Sin. 2016 Jun;48(6):491–500. doi: 10.1093/abbs/gmw025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng H., Zhu H., Liu X., et al. Mitophagy in diabetic cardiomyopathy: roles and mechanisms. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.750382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang F., Qin Y., Wang Y., et al. Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int J Biol Sci. 2019;15(5):1010–1019. doi: 10.7150/ijbs.29680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elrashidy R.A., Ibrahim S.E. Cinacalcet as a surrogate therapy for diabetic cardiomyopathy in rats through AMPK-mediated promotion of mitochondrial and autophagic function. Toxicol Appl Pharmacol. 2021 Jun;15(421) doi: 10.1016/j.taap.2021.115533. [DOI] [PubMed] [Google Scholar]
- 46.Yan L, Xu X, Fan Y, et al. Tangshen decoction enhances podocytes autophagy to relieve diabetic nephropathy through modulation of p-AMPK/p-ULK1 signaling. 2022;2022:3110854 [DOI] [PMC free article] [PubMed]
- 47.Ji C.H., Kwon Y.T. Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol Cells. 2017 Jul 31;40(7):441–449. doi: 10.14348/molcells.2017.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen R.H., Chen Y.H., Huang T.Y. Ubiquitin-mediated regulation of autophagy. J Biomed Sci. 2019 Oct 21;26(1):80. doi: 10.1186/s12929-019-0569-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.