

Abstract
Effective lipid lowering therapies are essential for the prevention of atherosclerosis and cardiovascular disease. Available treatments have evolved in both their efficacy and their frequency of administration, and currently include monoclonal antibodies, ASO and siRNA approaches. However, an unmet need remains for more effective and long-lasting therapeutics. Gene editing permanently alters endogenous gene expression and has the potential to revolutionize disease treatment. Despite the existence of several gene editing approaches, the CRISPR/Cas9 system has emerged as the preferred technology because of its high efficiency and relative simplicity.
This review provides a general overview of this promising technology and an update on the progress made towards the development of treatments of dyslipidemia. The recently started phase 1b gene editing clinical trial targeting PCSK9 in patients with heterozygous familial hypercholesterolemia and cardiovascular disease highlights how gene editing may become available to treat not only patients affected by rare disorders of lipid metabolism, but also patients with that are difficult-to-treat or at high risk. Other targets like ANGPTL3, LDLR, and APOC3 are on track for further pre-clinical development. The identification of novel targets using electronic health record-linked biobanks and human sequencing studies will continue to expand the potential target pool, and clinical assessment of treated patients will provide essential efficacy and safety information on current strategies. Gene editing of genes regulating lipid metabolism holds promise as an exciting new therapeutic approach. However, since gene editing permanently alters a patient’s genome, its therapeutic application in humans will require careful safety assessment and ethical considerations.
Keywords: Dyslipidemias, Gene editing, CRISPR-Cas systems, LDL cholesterol, Triglyceride
Graphical Abstract
Introduction
Elevated plasma lipids contribute to the development of atherosclerosis and cardiovascular disease, the leading cause of death worldwide1,2. In most cases, elevated lipid levels can be addressed with lifestyle modification and treatment with statins. Additional treatments like ezetimibe, bempedoic acid and more recently, biologics targeting proprotein convertase subtilisin/kexin type 9 (PCSK9), further compliment these standards of care3. Over a lifetime, high risk patients are often required to combine several treatments for maximum effect. However, long-term multi-drug adherence is difficult to maintain. Only roughly half of hyperlipidemia patients achieve adherence, with one third to half of patients altogether discontinuing statin medication within the first year of treatment4. This decline in adherence impacts the rate of cardiovascular events and is also associated with increased medical costs4. Furthermore, despite the remarkable progress made in treatments, patients with monogenic conditions such as familial hypercholesterolemia5 may require additional therapies to reach target lipid levels and represent a still unmet need.
The liver plays a central role in regulating lipid and lipoprotein levels. In animal models, liver-specific overexpression, knockdown, and knockout of key genes have been associated with marked changes in circulating lipid and lipoprotein levels. Moreover, liver transplant has shown to improve low-density lipoprotein cholesterol (LDL-C) levels and xanthomas in patients with homozygous familial hypercholesterolemia (reviewed by Ighigaki et al.6). These studies provide rationale for the development of long-term, liver-directed therapies. Liver-directed gene transfer and gene editing strategies have been extensively used in murine and non-human primate models to alter existing expression of lipid-related genes. These approaches are also becoming a reality in humans. Several trials in patients with hemophilia A and B7, ornithine transcarbamylase deficiency (NCT02991144, NCT05345171), familial hypercholesterolemia (NCT02651675) and Wilson’s disease (NCT04537377) are ongoing or have been completed. Results from the first liver-directed gene editing clinical trial in patients with transthyretin amyloidosis were recently published8. These advances are rapidly expanding the therapies offered to patients with rare, difficulty-to-treat monogenic dyslipidemias like homozygous familial hypercholesterolemia. Perhaps, in a not-so-distant future, they may also be applied to high-risk atherosclerotic cardiovascular disease patients where the goal will shift towards disease prevention. However, many safety and efficacy considerations will need to be assessed before these cutting-edge therapeutic approaches can be translated to the clinical setting.
2. Gene transfer vs. gene editing
In vivo gene transfer strategies deliver copies of a functional gene to the cell, typically via viral vectors, to supplement endogenous gene expression (Table 1). This approach has been used for a multitude of diseases and tested in clinical trials for more than a decade. Recombinant Adeno-Associated Virus (AAV), which has no replication capacity and a weak ability to integrate into patient DNA9 has emerged as the preferred vector for clinical trials. Indeed, the FDA approved its first AAV-mediated gene transfer therapy for the treatment of a form of inherited retinal dystrophy in 201710. The EMA followed a year later11, and both agencies have since also approved a treatment for spinal muscular atrophy in pediatric patients12,13. AAV-based vectors continue to be used in ongoing clinical trials to treat several other conditions, such as hemophilia14. However, AAV mediated immunogenicity remains an obstacle.
Table 1.
Comparison of in vivo gene transfer and gene editing strategies.
| Gene transfer | Gene editing | |
|---|---|---|
|
| ||
| Principle | Deliver copy of functional gene | Restore or disrupt endogenous genes |
|
| ||
| Advantages | •Can treat any patient carrying any variants in a given gene • Does not require DNA breaks • Low risk of integrations with AAV-based vectors • Long-term efficacy • Longer experience in clinical development |
• Versatile technology, including base editing and prime editing • Editing of endogenous DNA, potentially conferring permanent efficacy • Can be delivered via viral or LNP systems • Transitory presence of editing machinery and delivery system (LNP) • Re-dosing may be possible when using LNPs |
|
| ||
| Limitations | • Cannot be “undone” • Current strategies require viral vectors • Adverse events include immune reactions caused by viral capsid • Re-dosing limited by development of neutralizing antibodies • Efficacy may dilute with time if AAV-delivered genetic information persists as episome • Possible development of autoantibodies against newly expressed proteins |
• Cannot be “undone” • Only patients with variant in specific target region can be treated • If viral vectors are used, adverse events include immune reactions caused by viral capsid • Possible development of autoantibodies against newly expressed proteins • Risk of off-target editing and unintended effect |
AAV, adeno-associated virus; LNP, lipid nanoparticle.
In contrast, somatic gene editing seeks to directly restore or disrupt endogenous genes at the DNA level (Table 1). This approach combines a delivered editing system with the cell’s own DNA repair mechanisms. The DNA changes then persist through the RNA and protein products. This potential versatility and permanence make somatic gene editing an attractive therapeutic modality. Gene editing has been used with success in several preclinical settings and continues to improve in its efficacy and safety while evolving to mitigate potential limitations (e.g., off-target effects) 15.
Notably, both therapeutic strategies cannot be withdrawn. Once delivered, the genetic material introduced by gene transfer cannot be recalled, and the edits introduced by gene editing are permanent. While this longevity can be advantageous for both approaches, it necessitates comprehensive and rigorous safety assessments for use in humans. Several expected risks are shared by the two approaches (Table 1). Among them are the possible toxicities associated with the delivery system (e.g. immune response to viral vectors or viral integration in the genome) and the risk for the development of autoantibodies against the newly recognized protein in patients carrying “null” variants. These events may limit or erase therapeutic efficacy in patients. Other risks are inherent to the specific approach. With AAV-mediated gene transfer, the genetic information remains in the nucleus as an episome, which may dilute with time in tissues that regenerate16 and potentially affect long term efficacy. Transgene dilution may not be a concern with gene editing since this approach edits endogenous DNA. However, with gene editing, off-target editing could lead to undesired, permanent effects. Finally, it is important that both approaches remain restricted to somatic cells without the possibility of affecting germline cells, where they would risk passing changes to future generations. Furthermore, as gene editing therapies become more prevalent, the distinction between disease treatment and human enhancement may become blurred, and access to these therapies entwined with socioeconomic standings.
Nevertheless, these therapeutic approaches have the potential to revolutionize how patients can be treated. This review will focus on the progress made in the use of powerful gene editing tools for the treatment of dyslipidemias.
3. Gene editing tools
3.1. CRISPR/Cas
Classic gene editing systems specifically bind target genomic sequences and introduce DNA double-strand breaks using one of four major endonucleases: zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), meganuclease, or clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) proteins. CRISPR/Cas systems have come to dominate the gene editing field for their ease of use and appreciable editing efficiency17. These systems use guide RNAs to confer target specificity and Cas proteins (from bacteria like Streptococcus pyogenes - SpCas9) to produce DNA double-strand breaks.
Gene editing using the CRISPR-Cas system introduces DNA double-strand breaks which are repaired via non-homologous end joining (NHEJ) or homology-directed repair (HDR) (Figure 1). NHEJ is the primary DNA double-strand break repair mechanism and is always active in all cells. This error-prone pathway can introduce insertions or deletions (indels) which disrupt the target gene and prematurely truncate the downstream protein. Gene disruption via NHEJ is highly efficient but also highly variable, as every treated cell acquires different indels. Typically, only a couple base pairs are inserted or deleted, but deletion of thousands of base pairs, chromosome translocations, and even chromosomal shattering, are possible18,19. This approach can be leveraged to generate useful hypomorph models, where single-cell derived colonies or embryos can be sorted for control (un-edited), heterozygous, or homozygous indel formations20.
Figure 1. Gene editing strategies to treat dyslipidemias.
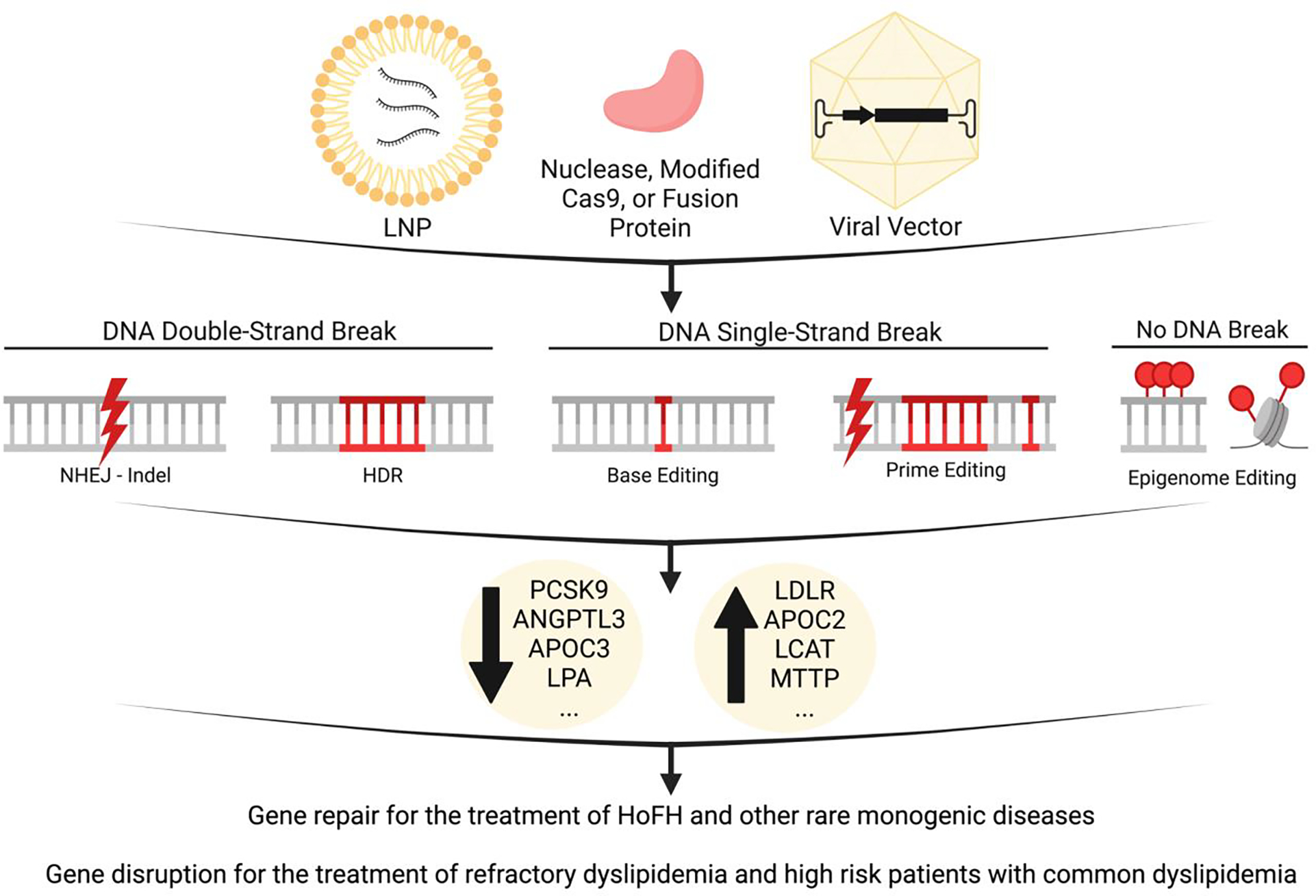
Lipid nanoparticles (LNPs) or viral vectors can be used to deliver nucleases, modified Cas9, or dead Cas9 fusion proteins. Techniques relying on DNA double-strand breaks can be repaired via non-homologous end joining (NHEJ) to introduce insertions and deletions (indels) which disrupt gene function, or homology-directed repair (HDR) which uses template DNA to restore gene function. Newer techniques rely on DNA single-strand breaks to alter single nucleotides (base editing) or introduce targeted indels or base conversions (prime editing). Fusion proteins can perform epigenetic modifications without breaking DNA strands. Knockdown strategies have been used to decrease PCSK9, ANGPTL3, APOC3, and LPA expression for therapeutic lipid lowering. Restoration of LDLR has also been explored for therapeutic lipid lowering. These approaches are being developed for the treatment of HoFH (homozygous familial hypercholesterolemia) and other rare monogenic diseases. In the future, gene editing approaches may be expanded to the treatment of refractory dyslipidemia and high risk patients with common dyslipidemia. Figure created with Biorender.com. ANGPTL3, Angiopoietin-like 3; APOC2, apolipoprotein C2; APOC3, apolipoprotein C3; CAS, clustered regularly interspaced short palindromic repeats /CRISPR-associated; HDR, homology-directed repair; HoFH, homozygous familial hypercholesterolemia; indel, insertions or deletions; LCAT, lecithin-cholesterol acyltransferase; LDLR, low density lipoprotein receptor; LNP, lipid nanoparticle; LPA, lipoprotein(a); MTTP, microsomal triglyceride transfer protein; NHEJ, non-homologous end joining; PCSK9, proprotein convertase subtilisin/kexin type 9.
CRISPR/Cas gene disruption has been used to generate novel cell and animal models for lipid and atherosclerosis focused research. Jarrett et al. sought to model atherosclerosis by using AAV-CRISPR/Cas9 to disrupt hepatic low density lipoprotein receptor (Ldlr) in adult mice21. These mice demonstrated severe hypercholesterolemia and developed atherosclerotic lesions in their aortas, making gene disruption a valuable alternative to germline Ldlr knockout. Furthermore, CRISPR/Cas gene disruption has also been used in unbiased screen approaches. Emmer et al. performed a genome wide CRISPR screen in Huh7 cells and identified over 100 positive and 50 negative regulators of cellular LDL update, highlighting the efficiency of this technique for biological discovery22.
HDR is rarer and limited to dividing cells in S and G2 phases of the cell cycle. This more precise pathway repairs DNA double-strand breaks using a DNA template, which allows for the correction of a genetic mutation but also reduces efficiency. Zhao et al. used CRISPR/SpCas9 HDR to knock in a premature truncation mutation in the Ldlr gene of fertilized murine eggs23. After high fat diet, mice exhibited higher plasma total cholesterol, total triglycerides (TG), and LDL-C along with atherosclerotic lesions in their aortas, mimicking the clinical features of familial hypercholesterolemia. However, since HDR is largely limited to proliferating cells, its utility in tissues central to dyslipidemias (i.e., hepatocytes, adipocytes, cardiomyocytes) is unclear. To this end, researchers have treated neonatal mice to maximize therapeutic HDR, though still achieved less than 10% editing23. Moreover, the requisite DNA repair template can complicate delivery, especially in viral vectors with limited cargo capacity.
Although CRISPR/Cas guide RNAs are designed to target specific genomic sequences, gene editing machinery can interact with sites that are imperfect matches, potentially introducing off-target indels in tumor suppressor genes or oncogenes. Additionally, in gene editing approaches that employ DNA double-strand breaks, aberrant insertion of viral or Cas DNA has been observed21,24. Thus, the unpredictability of indels and the possibility of chromosomal abnormalities and oncogenesis represent serious limitations to their clinical use.
Advances in gene editing systems have refined these tools from unpredictable biological scissors to more precise tweezers. Newer techniques retain the hallmark ability to identify and bind specific target sequences but expand their second functionality. Cas9 nickases (modified nucleases that only cut the target DNA strand) can be used in pairs to create precise DNA double strand breaks with increased precision and decreased off-target effects25,26. Nickases can also be paired with base editors or reverse transcriptases (see next sections). Shorter guide RNAs and other engineered, high-specificity Cas nucleases have also been shown to improve specificity27,28. Finally, catalytically inactive Cas9 proteins have been used in applications including epigenome editing29,30.
3.2. Base editing
Nickases or inactive Cas proteins can be fused with single-stranded DNA deaminases to produce cytosine or adenosine base editors31,32. While base editing is restricted to transition mutations and limited by sequence requirements for the target site (Cas9 PAM sequence preferences)33, these editors are gaining traction because they modify single nucleotides without generating DNA double-strand breaks. This key characteristic decreases the potential for serious off-target mutations34. Base editors can precisely introduce nonsense mutations that prematurely truncate a protein or missense mutations that disrupt protein function. These editors could also “fix” pathogenic variants by reversing the original mutation or exploiting genetic code wobble to modify a codon of interest (see Chadwick and Musunuru35).
3.3. Prime editing
In prime editing, Cas9 nickase is fused to an engineered reverse transcriptase enzyme which uses an RNA template to introduce new DNA36. This single system works without DNA double-strand breaks or donor DNA template and can precisely introduce any point mutation or indels within a larger editing window36. In vitro, prime editing has been applied to correct several genetic causes of disease (each with their own requirements to treat the causal gene) in human cell lines36. Although this technology has not yet been extensively tested in in vivo models, it can theoretically be used to correct a large variety of mutations, including deletions, duplications, and inversions, thus providing the possibility of repairing most disease-causing variants. Furthermore, the prime editing guide (peg)RNA can be created to target a hot spot region of a given gene and be used to correct several variants located in close proximity. The potential to “fix” pathogenic mutations beyond single base editing, and with better efficiency than HDR, greatly expands the available sequence targets and therapeutic approaches (reviewed by Scholefield and Harrison37).
3.4. Epigenome editing
Dead Cas9 can deliver DNA methyltransferases or acetyltransferases to enable epigenome editing and modify gene expression29,30. This approach entirely bypasses the need to modify DNA sequences. Instead, epigenetic editing modifies how DNA or histone proteins are presented to interacting proteins and may elicit more subtle changes when compared to complete gene (de)activation approaches. The resulting epigenetic modifications target repression may wane (within months) with expression of the editor itself and may require multiple administrations for sustained effect38. Our limited understanding of how epigenetic modifications may affect disease manifestations restricts the current use of epigenetic editing only to basic science. However, since this approach does not modify DNA sequence and its effect can be transient, epigenome editing may have an advantage for the treatment of certain conditions like cancer.
Keypoints Box 1: How to create desired gene edits
- Gene disruption:
- Classic DNA double-strand break with NHEJ indel formation
- Base editing or prime editing to introduce premature stop codon
- Gene restoration:
- Homology-directed repair
- Base editing or prime editing to repair a pathogenic base mutation
- Prime editing to repair pathogenic gene segment
- Gene expression modification
- Epigenetic editing to up- or down-regulate gene activity
4. Delivery methods
Significant advances have been made in the development of delivery methods that are tissue and cell-specific and can be used safely and effectively in humans.
Innate and adaptive cellular immune responses to either Cas nucleases or the viral vectors have been observed both in animal models39,40 and in humans41,42. The implications of these findings, and whether they decrease overall efficacy or impact safety in humans, are unclear. In a murine model, pre-existing immunity against Cas9 was associated with increased T-cell responses and loss of gene-edited hepatocytes43. Thus, it is important to adopt delivery strategies that maximize delivery efficiency while limiting an immune response.
CRISPR/Cas systems can be delivered as DNA, mRNA, or protein. Delivery of gene editing components as DNA results in prolonged expression which can improve efficacy. However, extended, or overabundant expression of Cas proteins has been associated with increased off-target editing44,45. Furthermore, prolonged nuclease presence may facilitate an immune response, though engineered CRISPR-Cas9 systems may offer a valuable approach to limiting these types of responses42. CRISPR/Cas delivery as mRNA mitigates the risk of vector integration and limits nuclease expression, aspects which contributed to the success of two recent non-human primate studies employing this approach46,47. Delivery of Cas proteins is also transitory and may limit the risk of off-target editing.
Gene editing cargos can be delivered using physical methods, viral vectors, and non-viral nanoparticles (reviewed by Taha et al.48). Physical delivery (i.e., microinjection, electroporation or gene gun) is most suited for in vitro (e.g., induced pluripotent stem cell, iPSC49) and ex vivo (e.g., primary cell50) gene editing applications. Viral vectors, AAVs in particular, are popular due to their simplicity, use in ongoing gene transfer trials, and success with FDA-approved gene transfer therapies. Several AAV serotypes readily infect the liver, an organ of interest for the treatment of dyslipidemias51. However, AAV-based vectors have some limitations. Among them are the virus’ limited cargo capacity (~4.7kb total, where SpCas9 alone is ~4kb)9 and, relevant for their use in humans, humoral and cellular immune responses52. The formation of neutralizing antibodies against the AAV capsid limit re-administration and T-cell mediated cytotoxicity in liver targeted-therapies results in transient elevations in transaminases, which do respond to steroid treatment. Since gene editing introduces permanent changes in the DNA after a single administration, it may circumvent the need for multiple AAV deliveries. Also, smaller Cas orthologs that fit AAV’s size constraints have been developed and edit with similar efficiencies53. AAVs remain a mainstay delivery vehicle for proof-of-concept animal experiments even if safety and efficacy profiles are still being assessed in humans.
Non-viral nanoparticles are gaining traction for their biodegradable nature, safety profiles, and generous capacities to deliver Cas9 DNA, mRNA, and proteins46,47,54,55. These nanoparticles are endocytosed and release their cargo intracellularly. LNPs can be engineered to increase immunocompatibility and for predictable and specific delivery of cargo to target tissues after intravenous administration56. Addition of the N-acetylgalactosamine (GalNAc) targeting ligand may increase delivery to hepatocytes and circumvents the default uptake of LNPs by the LDLR57. This advancement may improve LNP delivery in patients with homozygous familial hypercholesterolemia, and was recently tested in wild type and LDLR knockout non-human primates58. LNPs have been investigated in the clinic as delivery vehicles for the treatment of cancer, viral infections, and genetic diseases. Moreover, their use in several mRNA-based COVID-19 vaccine strategies has demonstrated ease of re-administration and favorable safety profiles59,60. For these reasons, LNPs are rapidly gaining traction as the most well-rounded delivery vehicle.
Keyponts Box 2: How to deliver gene editing machinery
- Physical delivery (electroporation or gene gun)
- Restricted applications in vitro and ex vivo.
- Adeno-Associated Viruses
- Readily infect the target organ (e.g. liver)
- Limited cargo capacity
- Associated with immunogenicity reactions
- Re-dosing limited by formation of neutralizing antibodies.
- Lipid nanoparticles
- Large cargo capacities
- More favorable immunogenicity profile when compared to viral vectors
- Re-dosing possible
- Transient delivery
- Limited biodistribution
- Addition of targeting ligands to increase delivery to target organ (e.g. GalNAc for hepatocyte delivery).
5. Gene editing for the treatment of dyslipidemia
Gene editing tools can be used to target a gene for precise repair or disruption (e.g. exon skipping or introduction of nonsense or splice-site mutations), and dyslipidemias are at the forefront in the development of therapies using these approaches. While gene repair applications are limited to the treatment of monogenic diseases, the potential applications of gene disruption are broader, including common forms of dyslipidemia. Several preclinical studies targeting genes affecting LDL-C, a well-established and modifiable causal risk factor61 and/or triglycerides, another independent predictor of cardiovascular disease risk62, have shown promising results. Beyond these targets, human sequencing studies and electronic-health record linked biobanks have an essential role in identifying novel targets with relevant phenotypic outcomes in patients. These computational approaches also provide some indication of safety and tolerability of the desired mutation since they are derived from real people. In the future, dyslipidemia targets may also be combined to form personalized gene editing cocktails.
6. Editing the causal gene
6.1. Targeting LDLR
Pathogenic variants in the gene encoding for the LDLR are the major cause of familial hypercholesterolemia, and its rare form, homozygous familial hypercholesterolemia. Despite the remarkable progress of the last decade, treatment options are still suboptimal for many of these patients. Gene editing approaches could offer a long-lasting solution if a safe and effective treatment is developed. In a proof-of-concept study, familial hypercholesterolemia skin fibroblasts were obtained from a patient homozygous for a three base pair deletion in LDLR (Table 2). Fibroblast-derived iPSCs were treated with CRISPR/SpCas9 nickase and a repair template63. 83% of enriched clones (double positive for nuclease and guides) demonstrated correction of both alleles and were differentiated into hepatocyte-like cells. Corrected cells expressed LDLR protein and internalized LDL. Edited iPSCs are advantageous in that they can be screened (enriched for on-target editing and reduced off-target activity), selectively expanded, and differentiated for use in autologous cell replacement. Additionally, they bypass immunogenicity concerns for the delivery and expression of the nuclease. Assessments of the uptake, durability, and efficacy of these hepatocyte-like cells in vivo will be critical for moving this approach forward.
Table 2.
Summary of gene editing approaches to treat dyslipidemias.
| Target | Model | Method | Results | Reference |
|---|---|---|---|---|
|
| ||||
| LDLR | Mouse, C57BL/6J | AAV-CRISPR/SaCas9 disruption | Severe hypercholesterolemia Atherosclerotic lesions in aortas |
Jarrett, K. E. 2018 (21) |
| Mouse, C57BL/6 | CRISPR/SpCas9 HDR into fertilized eggs |
Elevated plasma cholesterol Atherosclerotic lesions in aortas |
Zhao, H. 2020 (23) | |
| Mouse, LdlrE208X Knock-in | Dual AAVs-CRISPR/SpCas9 HDR | 18% restoration LDLR protein ~65% decrease cholesterol Smaller atherosclerotic lesions in aortas |
Zhao, H. 2020 (23) | |
| Human iPSC, FH Patient | CRISPR/SpCas9 Nickase, HDR repair |
Restored normal LDLR structure | Omer, L. 2017 (63) | |
|
| ||||
| LDLR + APOB | Mouse, Cas9 Transgenic | AAV disruption | Reduced plasma cholesterol Protection against atherosclerosis |
Jarrett, K. E. 2017 (64) |
|
| ||||
| PCSK9 | Mouse, C57BL/6 | Adenovirus-CRISPR/SpCas9 disruption | 90% decrease PCSK9 protein 35–40% decrease plasma cholesterol |
Ding, Q. 2014 (84) |
| Mouse, C57BL/6 | AAV-CRISPR/SaCas9 disruption | ~95% decrease PCSK9 protein ~40% decrease plasma cholesterol |
Ran, F. 2015 (53) | |
| Mouse, C57BL/6 | LNP-CRISPR/SpCas9 disruption | Undetectable PCSK9 protein 35–40% decrease plasma cholestero |
Yin, H. 2017 (87) | |
| Mouse, C57BL/6J | Adenovirus-BE3 base editing disruption | ~55% decrease PCSK9 protein ~30% decrease plasma cholesterol |
Chadwick, A. C. 2017 (88) | |
| Mouse, C57BL/6 | Dual AAVs-CBE base editing disruption | ~20–25% editing efficiency Pcsk9 38% editing in liver overall |
Levy, J. M. 2020 (89) | |
| Mouse, Chimeric Liver-Humanized | Adenovirus-CRISPR/SpCas9 disruption | 52% decrease human PCSK9 protein | Wang, X. 2016 (86) | |
| Non-human primate, Rhesus macaque | AAV-1st generation meganuclease disruption | Up to 84% decrease PCSK9 protein Up to 60% decrease LDL-C |
Wang, L. 2018 (24) | |
| Non-human primate, Rhesus macaque | AAV-2nd generation meganuclease disruption | 55–62% decrease PCSK9 protein 33–39% decrease LDL-C |
Wang, L. 2018 (24) | |
| Non-human primate, Rhesus macaque | AAV-2nd generation meganuclease (self-targeting) disruption | 24–60% decrease PCSK9 protein 11–36% decrease LDL-C |
Breton, C. 2021 (90) | |
| Non-human primate, Cynomolgus macaque | LNP-ABE base editing disruption | 32% decrease PCSK9 protein 14% decrease LDL-C |
Rothgangl, T. 2021 (46) | |
| Non-human primate, Cynomolgus macaque | LNP-ABE8.8 base editing disruption | 81–90% decrease PCSK9 proteinn 60–65% decrease LDL-C |
Musunuru, K. 2021 (47) | |
|
| ||||
| ANGPTL3 | Mouse, C57BL/6 | LNP-CRISPR/SpCas9 disruption | 65% decrease ANTPL3 protein 56% decrease LDL-C 29% decrease TG |
Qiu, M. 2021 (96) |
| Mouse, C57BL/6J | Adenovirus-BE3 base editing disruption | 49% decrease ANGPTL3 protein 19% decrease cholesterol 31% decrease TG |
Chadwick, A. C. 2018 (97) | |
| Mouse, Ldlr Knockout | Adenovirus-BE3 base editing disruption | 51% decrease cholesterol 56% decrease TG |
Chadwick, A. C. 2018 (97) | |
| Non-human primate | LNP-ABE base editing disruption | 95% decrease ANGPTL3 protein 19% decrease LDL-C 64% decrease TG |
Verve Therapeutics (98) | |
|
| ||||
| PCSK9 + ANGPTL3 | Mouse, C57BL/6J | Adenovirus-BE3 base editing disruption | ~20% decrease cholesterol ~20–30% decrease TG |
Chadwick, A. C. 2018 (97) |
|
| ||||
| APOC3 | Hamster, Syrian Golden | CRISPR/Cas9 disruption | ~50% decrease TG Some protection against atherosclerosis |
Guo, M. 2020 (102) |
| Rabbit, New Zealand White | CRISPR/Cas9 disruption | 50% decrease TG Protection against atherosclerosis |
Zha, Y, 2021 (103) | |
AAV, adeno-associated virus; ANGPTL3, angiopoietin-like 3; APOB, apolipoprotein B; APOC3, apolipoprotein C3; CRISPR/CAS, clustered regularly interspaced short palindromic repeats /CRISPR-associated; HDR, homology-directed repair; LDL-C, low density lipoprotein cholesterol; LDLR, low density lipoprotein receptor; LNP, lipid nanoparticle; PCSK9, proprotein convertase subtilisin/kexin type 9; SaCas9, Staphylococcus aureus Cas9; SpCas9, Streptococcus pyogenes Cas9; TG, triglyceride.
Zhao et al. used CRISPR/SpCas9 to generate a novel atherosclerosis mouse model expressing a premature truncation mutation in Ldlr23. This mutant abrogated LDLR protein expression in the liver and led to atherosclerosis upon high fat diet feeding. To treat the disease, neonatal mice were given dual AAVs delivering SpCas nuclease in one construct and guide RNA with donor DNA in the other. As expected, HDR-mediated correction of the mutation was low, around 6.7%, but managed to restore LDLR protein to 18% of wild type mice. Mice with partially restored LDLR protein demonstrated ~65% reduced total cholesterol and were protected from atherosclerotic plaque formation. Although not sufficient to normalize the hypercholesterolemia, partial restoration of LDLR activity in patients would likely result in an increase response to lipid lowering treatments. Despite modest editing, the lack of observed off-target mutagenesis supports further development of this approach. However, as mentioned above, high precision HDR-mediated repair occurs in dividing cells in S and G2 phases of the cell cycle, with limited applicability in post-mitotic cells. Gene editing during embryogenesis, although potentially effective, would raise both ethical and safety concerns that need to be fully addressed.
Jarrett et al. used AAV to simultaneously deliver guide RNAs targeting both Ldlr and Apob in Cas9 transgenic mice64. This creative approach allowed for the study of therapeutic gene disruption of Apob, which is otherwise embryonic lethal65. Co-disruption of Apob decreased plasma cholesterol, inhibited hepatic LDL production, and ameliorated atherosclerotic disease seen with disruption of Ldlr alone. Ldlr editing reached 54% and was not significantly different between groups receiving single or dual guide RNAs. Apob editing reached a remarkable 74%. Despite the high editing efficiency however, this approach is unlikely to be applied in humans; inhibition of APOB is associated with increased liver fat and unknown long-term consequences66. Moreover, a somewhat high level of off-target activity was observed: above-background mutagenesis at one intronic site, indel formation in control “stuffer sequence” guide groups, and small insertions of vector sequence (also observed in other AAV DNA double-strand break approaches). This off-target activity may be attributed to the Cas9 transgenic murine model and may be limited with transient nuclease expression. In any case, the dual targeting approach allows for a better understanding of Apob biology and provides proof-of-principle data for concomitant targeting of two genes. These promising results support the multiplexing of other gene targets.
Although these proof-of-principle studies are important, the over 2000 known pathogenic and likely pathogenic LDLR variants67 make designing and testing the safety and efficacy of personalized gene editing strategies (with current technical and regulatory hurdles) challenging. Although these considerations may be mitigated by further development of prime editing techniques, disrupting a different gene known to affect LDL-C levels (e.g. PCSK9 or Angiopoietin-like 3, ANGPTL3) may provide a viable alternative solution68–73.
6.2. Targeting other genes causing monogenic dyslipidemia
Although data are not yet available, other difficult to treat monogenic dyslipidemias with well-established genetic bases and unmet need could eventually be treated with base or prime editing. Like LDLR targeting strategies, patients with autosomal recessive hypercholesterolemia (LDLRAP1), sitosterolemia (ABCG8/G5), familial chylomicronemia syndrome (APOC2, APOA5, LMF1, LPL, GPIHBP1), LCAT deficiency, or abetalipoproteinemia (MTTP) would require gene restoration74. Given the range of causal mutations, founder and relatively more common variants within each of these genes, or gene hotspots with a high concentration of mutations, are likely to be prioritized. Further development of prime editing will expand the list of pathogenic variants eligible to be repaired.
The most successful pre-clinical dyslipidemia gene editing strategies aim to create a null allele (indel formation or introduction of premature stop codon). Thus, it is reasonable to hypothesize that base editing might be effective in treating extremely elevated Lp(a) levels. Lp(a) levels have been recognized as causally associated with atherosclerotic cardiovascular disease and aortic valve stenosis75. Its levels are mostly genetically determined by the carried LPA haplotype. Several clinical trials using either antisense oligonucleotide or siRNA approaches against the LPA gene are currently ongoing or recently completed76,77. If these trials and outcome studies are successful, they would provide a solid background for the development of a base editing approach.
7. Gene disruption to treat dyslipidemia
7.1. Targeting PCSK9
A plethora of human genetics studies support PCSK9 as a therapeutic target. Loss of function mutations in PCSK9 are associated with reduced LDL-C and risk of coronary heart disease and no apparent adverse health consequences68–70. These variant carriers conveniently model the consequences of therapeutic PCSK9 downregulation. To this end, treatments with monoclonal antibodies, small interfering RNAs, and antisense oligonucleotides targeting PCSK9 mRNA or protein are already approved by regulatory agencies such as the FDA and EMA or are in advanced development78–81. PCSK9 gene editing as a treatment for hypercholesterolemia has been recently and extensively reviewed by Musunuru and colleagues82,83. We will review some of those gene editing studies here with our own perspectives.
Gene editing strategies targeting PCSK9 gained traction after promising in vivo murine results. Initial studies used adenovirus to deliver CRISPR/SpCas9 and a guide RNA to disrupt mouse hepatic Pcsk984. Within days, Pcsk9 NHEJ-mediated mutagenesis surpassed 50%, plasma protein decreased 90%, and plasma cholesterol decreased 35–40%. Moreover, there was no evidence of off-target mutagenesis in 10 selected sites. An independent and highly sensitive assessment of off-target editing using a similar adenoviral gene editing approach also observed no mutagenesis in over 180 candidate sites85. Similar effects on gene editing were also found in a chimeric liver-humanized mouse model86.
Although adenoviral vectors were initially employed for their larger cargo capacity, identification of smaller Cas proteins and advances in LNP design allowed for studies using more clinically favorable delivery methods. AAV delivered Staphylococcus aureus Cas9 (SaCas9, ~1kb smaller than SpCas9) Pcsk9 gene editing in wild type mice reached 40–50%, decreased plasma PSCK9 protein around 95%, and reduced plasma cholesterol about 40%, demonstrating that SaCas9 gene editing was comparable to “classic” SpCas9 gene editing53. LNPs were also successfully used to deliver SpCas9 mRNA paired with chemically modified Pcsk9-targeting guide RNAs87. These modifications enhanced stability without inhibiting guide RNA honing function or Cas9 interactions. Indeed, the authors reported an unprecedented 80% liver-specific gene editing efficiency, undetectable levels of circulating PCSK9 protein, and a 35–40% decrease in cholesterol in wild type mice87.
Base editing tools have also been used to target PCSK9. Initial studies used a cytosine base editor BE3 and a guide RNA targeting mouse Pcsk9 to specifically introduce nonsense mutations in mice, resulting in halved plasma PCSK9 protein levels88. While plasma cholesterol only decreased ~30%, slightly less than observed in NHEJ-mediated gene disruption strategies, this decrease would still provide a therapeutic benefit and avoids risks associated with DNA double-strand breaks. However, the large size of the base editor combined with the guide RNA required the authors to use an adenoviral vector. More recent work has designed split base editors that are delivered in dual AAVs then reconstituted in vivo89. Using this more clinically favorable vector, the authors reported ~20–25% editing efficiency of Pcsk9 specifically and 38% editing in the liver over all their studies. This efficiency may be sufficient for dyslipidemia targets. Regardless of the approaches, these NHEJ and base editing studies demonstrated that genetic disruption of Pcsk9 is feasible and yields significant decreases in PCSK9 protein and LDL-C.
Recent nonhuman primate studies have further bridged the gap towards clinical trials. AAV delivering a meganuclease targeting a conserved sequence in PCSK9 in rhesus macaques achieved 64% editing, 84% reduction in circulating PSCK9 protein, and 60% reduction in plasma LDL-C at the highest dose tested24. Despite these promising outcomes, this approach resulted in mild elevations in blood transaminase levels in all monkeys and a somewhat high rate of off-target editing. An engineered second-generation meganuclease yielded over 50% decrease in circulating PCSK9 protein, over 30% reduction in LDL-C and substantially reduced off-target cleavage, but same elevation in blood transaminase levels24. Notably, most insertions larger than 15bp at DNA double-strand break sites in PCSK9 contained AAV vector sequence. Off-target sites were also noted, mostly within intronic regions or intergenic regions. All treated nonhuman primates maintained their reductions in PCSK9 protein and LDL-C over three years with no adverse changes in liver histopathology after the initial increase in transaminase, attributed in part to immune response to the transient expression of the AAV-delivered meganuclease40. Increased in vivo nuclease specificity, as determined by a reduction in off-target activity, was observed using a self-targeting vector and shorter promoter90.
Using LNPs as an alternative delivery vehicle and a base editing approach, Rothgangl et al. demonstrated an average 26% editing, 32% reduction in circulating PCSK9 protein, and 14% reduction in LDL-C46. mRNA encoding the base editor was cleared rapidly, which likely contributed to the lack of observed off-target activity, although a more robust assessment could have been performed. A subset of nonhuman primates was dosed a second time two weeks later to determine whether editing efficiency could increase with re-treatment46. However, no further increase in editing was observed, likely due to an immune response to the base editor itself upon the primary treatment. Cas9 antibodies were detected in repeat-dose groups and blood transaminases were transiently increased. The LNP formulation was assessed for liver specificity by analysis of on-target PCSK9 editing in nine organs. While eight of the organs exhibited less than 1% editing, the spleen exhibited around 6% and 12% editing dependent on the number of doses. The limited timeframe of the study (29 days) precluded any long-term safety and persistence assessments.
LNPs were also used by Musunuru et al. to deliver a CRISPR base editor to knockdown hepatic PCSK9 in cynomolgus monkeys47. Short-term, two-week studies demonstrated over 50% PCSK9 editing, 81% reduction in PCSK9 protein and 65% reduction in LDL-C. The use of a new generation base editor may explain the higher editing efficiency in comparison to the one employed by Rothgangl et al.46. Although most editing was contained to the liver, like the results observed by Rothgangl, some editing was observed in the spleen and adrenal glands. Long-term studies (8 months) at a higher dose demonstrated 66% base editing, 90% reduction in circulating PCSK9, and 60% reduction in LDL-C. These results mirror the ~50% reduction in LDL-C levels reported in a recent meta-analysis of currently available treatments targeting PCSK991. Both nonhuman primate cohorts experienced moderate rises in blood transaminases shortly after administration (attributed to the LNPs) that were resolved within two weeks. Off-target analysis in LNP-treated primary cynomolgus monkey hepatocytes revealed activity at one site which has minimal homology to the human genome. In direct liver samples from LNP-treated monkeys, less than 1% or no off-target editing was observed at the different doses administered. When the therapy was applied to human hepatocytes, editing was also restricted to PCSK947.These LNP based editing strategies in non-human primates seem to elicit a better specificity profile when compared to an AAV-delivered gene editing approach. Excitingly, a phase 1b study testing the safety of a base-editing drug targeting PCSK9 has just begun in patients with heterozygous familial hypercholesterolemia and cardiovascular disease (NCT05398029). If the results observed in the nonhuman primate studies translate into similar results in humans, the use of base editing to target PCSK9 could yield reductions comparable to those obtained with currently available PCSK9 monoclonal antibodies, with the advantage of requiring a single administration.
7.2. Targeting Angptl3
ANGPTL3 is another attractive target for lipid lowering. Like with PCSK9, subjects with loss of function mutations in ANGPTL3 exhibit decreased plasma lipids and protection against coronary artery disease without any apparent adverse effects71–73. Evinacumab, a monoclonal antibody targeting ANGPTL3 protein is already approved for the treatment of homozygous familial hypercholesterolemia. ANGPTL3 inhibition markedly reduces LDL-C in these patients in an LDLR-independent mechanism92–94, making ANGPTL3 an ideal candidate for the treatment of this rare form of hypercholesterolemia. Moreover, a monoclonal antibody and siRNA targeting ANGPTL3 mRNA are also being developed for the treatment of hypertriglyceridemia (NCT04832971, NCT04863014)95.
An optimized LNP system delivering CRISPR/SpCas9 mRNA and Angptl3-targeting guide RNA resulted in about 38% editing efficiency, 65% reduction in circulating ANGPTL3 protein, 56% reduction in LDL-C, and 29% reduction in TG96. These results were maintained through 100 days after injection with no off-target effects detected at nine top-predicted sites. Using an adenovirus encoding a base editor and guide RNA targeting Angptl3, Chadwick and colleagues demonstrated 35% editing, 49% reduction in ANGPTL3 protein, 31% reduction in TG, and 19% reduction in cholesterol in wild type mice within one week97. They further tested the effect of delivering base editor with a mix of guide RNAs - half targeting Angptl3 and half targeting Pcsk9. While single gene targeting and combined gene targeting yielded similar decreases in plasma cholesterol (~20%), Angptl3 targeting alone decreased TGs more than Pcsk9 alone or in combination. It is unclear whether halving the dose of each guide RNA contributed to the lack of additive effects on lipids. Finally, they tested the effect of base editing of Angptl3 on an Ldlr knockout background, a model of homozygous familial hypercholesterolemia, and found a ~50% reduction in both triglycerides and cholesterol levels two weeks after injection. Lipid lowering effects and the absence of off-targeting editing also seem to be confirmed in preliminary results in non-human primates98.
Some cautionary tales regarding the use of intracellular ANGPTL3 inhibition come from the data observed in a phase 2b clinical trial using Vupanorsen, an antisense oligonucleotide (ASO) targeting ANGPTL3, in which liver enzyme elevation and increased liver fat were reported99. Although these adverse events could be due to an off-target effect of the drug, an ANGPTL3-dependent effect cannot be excluded. Thus, while disruption of Angptl3 is emerging as another promising target to treat both elevated LDL-C and triglycerides levels, it is critical that these safety concerns are first addressed.
7.3. Targeting APOC3
Apolipoprotein C3 (APOC3) is yet another attractive target for lipid lowering treatment, particularly for the treatment of hypertriglyceridemia. Naturally occurring loss of function mutations in APOC3 are associated with decreased triglyceride levels and decreased risk of coronary heart disease100. Moreover, homozygous loss of function of APOC3 in humans improves post-prandial lipemia101. CRISPR inactivation of ApoC3 in hamsters and rabbits has shown to protect against atherosclerosis102,103. While these studies only aimed to generate knockout models to understand ApoC-III’s role in atherosclerosis, they demonstrated that APOC3 is a plausible candidate for further pre-clinical assessment.
8. Challenges inherent to gene editing technologies
The permanent nature of DNA changes caused by gene editing requires a careful and critical assessment of the safety of this technology before it can be employed broadly to treat human conditions. While some safety challenges are shared with other gene-focused therapies (such as the adverse events associated with delivery methods or the development of autoantibodies, described in the introduction), others are inherent to gene editing. Among them are unintended effects of gene disruptions and the risk of off-target effects. A comprehensive characterization of the phenotype of individuals carrying loss-of-function variants in genes targeted for disruptions can be useful to assess the effect of disrupting those genes. Indeed, the choice of PCSK9 and ANGPTL3 as targets of base editing for the treatment of dyslipidemias is supported by the apparent good health of individuals with PCSK968–70 and ANGPTL3 deficiency71–73. The assessment of off-target risks remains even more challenging because the human genome is different from those of animal models, but also because each individual human has a unique genetic make-up. Furthermore, standard approaches for predict off-target mutations are not yet established. Currently, in depth in silico analysis accompanied by testing in human cells (such as primary hepatocytes or iPSC-derived hepatocytes) are among the approaches used, as reported Musunuru and colleagues47.
Although a review on the ethical and societal implications of gene editing is outside the scope of this review, it is important to remember that this topic is still hotly debated, particularly in the context of germline and inheritable human gene editing104. The current paucity of information on safety and impossibility to exclude with certainty the absence off target effects, lends to the consensus that clinical use should be limited to somatic gene editing.
9. Conclusions
Over the last decades, dyslipidemia therapies have evolved from small molecules to be taken daily, to monoclonal antibodies administered every two weeks, to RNA-based treatments (antisense oligonucleotides and siRNAs) that are required every few months. Gene editing approaches are looking to extend the therapeutic window even further by promising a permanent effect. These techniques are graduating from coarser DNA double-strand break approaches to more refined DNA single-strand break approaches which introduce precise modifications.
These approaches hold promise for treating rare monogenic conditions with significant unmet medical needs. Although early attempts to use gene editing were terminated for lack of efficacy (NCT03041324), early phase trials in patients with phenylketonuria (NCT05222178) and with Leber Congenital Amaurosis (NCT03872479) are ongoing. Importantly, the recently published results from the first liver-directed gene editing clinical trial in patients with transthyretin amyloidosis look promising8; results at one month demonstrate that a single dose of NTLA-2001, the LNP-delivered CRISPR/Cas9 mRNA and guide RNA therapeutic, substantially decreases blood transthyretin with few, mild adverse events. If the promising pre-clinical data regarding ANGPTL3 are confirmed, homozygous familial hypercholesterolemia may be the first rare disorder of lipid metabolism to be treated with this cutting-edge approach.
Perhaps more excitingly, rapid advances in the development of safer tools coupled with validated therapeutic targets such as PCSK9 and ANGPTL3 may expand the use of gene editing from patients with rare diseases to a broader pool of patients with refractory dyslipidemia and high atherosclerotic cardiovascular disease risk. Of interest, a phase 1b trial using base-editing technology to disrupt PCSK9 in the liver (thus lowering circulating PCSK9 protein and LDL-C) is ongoing in patients with heterozygous familial hypercholesterolemia and established atherosclerotic cardiovascular disease (NCT05398029).
These emerging gene editing strategies have opened the door to long-term therapeutic solutions that are on track to revolutionize dyslipidemia management. However, since these treatments result in permanent changes in the patient’s genome, it will be paramount to determine the acute, intermediate, and long-term safety profile of in vivo gene editing in humans. Moreover, beyond the excitement of these new scientific frontiers, continued development of such powerful tools will need to be guided by ethical considerations and accompanied with clear boundaries that limit the use of somatic gene editing.
Highlights:
Gene editing can repair or disrupt genes. This powerful technology is being used to better understand the function of genes and as a potential therapeutic approach.
Thanks to the simplicity and efficiency of CRISPR/Cas technology, the improved precision of base editing, and the advancements in delivery systems, development of somatic gene editing therapeutics is fast advancing.
A Phase 1b clinical trial using base editing targeting PCSK9 is ongoing. ANGPTL3, LDLR, APOC3, and other targets are being explored for the treatment of dyslipidemias.
Careful assessments of short-, medium-, and long-term safety, and clear ethical boundaries must accompany the development of somatic gene editing.
Financial support:
This work was supported by National Institutes of Health Fellowship F31 HL149162 (to SS) and by HL148769 (to MC).
Footnotes
Declaration of competing interest:
SS declares none. MC has received research funding from Regeneron Pharmaceuticals and REGENXBIO; and honoraria for professional input from Amryt Pharma.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Organization WH. Cardiovascular diseases (CVDs) https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds). 2021.
- 2.Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation. 2022;145:e153–e639. doi: 10.1161/CIR.0000000000001052 [DOI] [PubMed] [Google Scholar]
- 3.Wilkins JT, Lloyd-Jones DM. Novel Lipid-Lowering Therapies to Reduce Cardiovascular Risk. JAMA. 2021;326:266–267. doi: 10.1001/jama.2021.2244 [DOI] [PubMed] [Google Scholar]
- 4.Bosworth HB, Ngouyombo B, Liska J, Zullig LL, Atlani C, Beal AC. The importance of cholesterol medication adherence: the need for behavioral change intervention programs. Patient Prefer Adherence. 2018;12:341–348. doi: 10.2147/PPA.S153766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brandts J, Ray KK. Familial Hypercholesterolemia: JACC Focus Seminar 4/4. J Am Coll Cardiol. 2021;78:1831–1843. doi: 10.1016/j.jacc.2021.09.004 [DOI] [PubMed] [Google Scholar]
- 6.Ishigaki Y, Kawagishi N, Hasegawa Y, Sawada S, Katagiri H, Satomi S, Oikawa S. Liver Transplantation for Homozygous Familial Hypercholesterolemia. J Atheroscler Thromb. 2019;26:121–127. doi: 10.5551/jat.RV17029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leebeek FWG, Miesbach W. Gene therapy for hemophilia: a review on clinical benefit, limitations, and remaining issues. Blood. 2021;138:923–931. doi: 10.1182/blood.2019003777 [DOI] [PubMed] [Google Scholar]
- 8.Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, Seitzer J, O’Connell D, Walsh KR, Wood K, et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454 [DOI] [PubMed] [Google Scholar]
- 9.Colella P, Ronzitti G, Mingozzi F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Molecular therapy Methods & clinical development. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.FDA. FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. In; 2017. [Google Scholar]
- 11.Agency EM. Luxturna ( voretigene neparvovec). https://www.ema.europa.eu/en/medicines/human/EPAR/luxturna. Accessed 07/05/22.
- 12.Administration USFaD. ZOLGENSMA. https://www.fda.gov/vaccines-blood-biologics/zolgensma. Accessed 07/05/22.
- 13.Agency EM. Zolgensma ( onasemnogene abeparvovec). https://www.ema.europa.eu/en/medicines/human/EPAR/zolgensma. Accessed 07/05/22.
- 14.Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, Boye SL, Boye SE, George LA, Salabarria S, et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol Ther. 2021;29:464–488. doi: 10.1016/j.ymthe.2020.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saha K, Sontheimer EJ, Brooks PJ, Dwinell MR, Gersbach CA, Liu DR, Murray SA, Tsai SQ, Wilson RC, Anderson DG, et al. The NIH Somatic Cell Genome Editing program. Nature. 2021;592:195–204. doi: 10.1038/s41586-021-03191-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colella P, Ronzitti G, Mingozzi F. Emerging Issues in AAV-Mediated. Mol Ther Methods Clin Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leibowitz ML, Papathanasiou S, Doerfler PA, Blaine LJ, Sun L, Yao Y, Zhang CZ, Weiss MJ, Pellman D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lv W, Qiao L, Petrenko N, Li W, Owens AT, McDermott-Roe C, Musunuru K. Functional Annotation of TNNT2 Variants of Uncertain Significance With Genome-Edited Cardiomyocytes. Circulation. 2018;138:2852–2854. doi: 10.1161/CIRCULATIONAHA.118.035028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarrett KE, Lee C, De Giorgi M, Hurley A, Gillard BK, Doerfler AM, Li A, Pownall HJ, Bao G, Lagor WR. Somatic Editing of Ldlr With Adeno-Associated Viral-CRISPR Is an Efficient Tool for Atherosclerosis Research. Arteriosclerosis, Thrombosis, and Vascular Biology. 2018;38:1997–2006. doi: 10.1161/ATVBAHA.118.311221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emmer BT, Sherman EJ, Lascuna PJ, Graham SE, Willer CJ, Ginsburg D. Genome-scale CRISPR screening for modifiers of cellular LDL uptake. PLoS Genet. 2021;17:e1009285. doi: 10.1371/journal.pgen.1009285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao H, Li Y, He L, Pu W, Yu W, Wu YT, Xu C, Wei Y, Ding Q, Song BL, et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation. 2020;141:67–79. doi: 10.1161/CIRCULATIONAHA.119.042476 [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Smith J, Breton C, Clark P, Zhang J, Ying L, Che Y, Lape J, Bell P, Calcedo R, et al. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat Biotechnol. 2018;36:717–725. doi: 10.1038/nbt.4182 [DOI] [PubMed] [Google Scholar]
- 25.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald JI, Celik H, Rois LE, Fishberger G, Fowler T, Rees R, Kramer A, Martens A, Edwards JR, Challen GA. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol Open. 2016;5:866–874. doi: 10.1242/bio.019067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeong YK, Song B, Bae S. Current Status and Challenges of DNA Base Editing Tools. Mol Ther. 2020;28:1938–1952. doi: 10.1016/j.ymthe.2020.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim D, Lim K, Kim ST, Yoon SH, Kim K, Ryu SM, Kim JS. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol. 2017;35:475–480. doi: 10.1038/nbt.3852 [DOI] [PubMed] [Google Scholar]
- 35.Chadwick AC, Musunuru K. CRISPR-Cas9 Genome Editing for Treatment of Atherogenic Dyslipidemia. Arterioscler Thromb Vasc Biol. 2018;38:12–18. doi: 10.1161/ATVBAHA.117.309326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scholefield J, Harrison PT. Prime editing - an update on the field. Gene Ther. 2021;28:396–401. doi: 10.1038/s41434-021-00263-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huerne K, Palmour N, Wu AR, Beck S, Berner A, Siebert R, Joly Y. Auditing the Editor: A Review of Key Translational Issues in Epigenetic Editing. CRISPR J. 2022;5:203–212. doi: 10.1089/crispr.2021.0094 [DOI] [PubMed] [Google Scholar]
- 39.Chew WL, Tabebordbar M, Cheng JK, Mali P, Wu EY, Ng AH, Zhu K, Wagers AJ, Church GM. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Breton C, Warzecha CC, Bell P, Yan H, He Z, White J, Zhu Y, Li M, Buza EL, et al. Long-term stable reduction of low-density lipoprotein in nonhuman primates following in vivo genome editing of PCSK9. Mol Ther. 2021;29:2019–2029. doi: 10.1016/j.ymthe.2021.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, Vakulskas CA, Collingwood MA, Zhang L, Bode NM, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferdosi SR, Ewaisha R, Moghadam F, Krishna S, Park JG, Ebrahimkhani MR, Kiani S, Anderson KS. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat Commun. 2019;10:1842. doi: 10.1038/s41467-019-09693-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li A, Tanner MR, Lee CM, Hurley AE, De Giorgi M, Jarrett KE, Davis TH, Doerfler AM, Bao G, Beeton C, et al. AAV-CRISPR Gene Editing Is Negated by Pre-existing Immunity to Cas9. Mol Ther. 2020;28:1432–1441. doi: 10.1016/j.ymthe.2020.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rothgangl T, Dennis MK, Lin PJC, Oka R, Witzigmann D, Villiger L, Qi W, Hruzova M, Kissling L, Lenggenhager D, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat Biotechnol. 2021;39:949–957. doi: 10.1038/s41587-021-00933-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, Wang K, Iyer S, Dutta C, Clendaniel V, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434. doi: 10.1038/s41586-021-03534-y [DOI] [PubMed] [Google Scholar]
- 48.Taha EA, Lee J, Hotta A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J Control Release. 2022;342:345–361. doi: 10.1016/j.jconrel.2022.01.013 [DOI] [PubMed] [Google Scholar]
- 49.Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell. 2019;24:566–578.e567. doi: 10.1016/j.stem.2019.02.005 [DOI] [PubMed] [Google Scholar]
- 50.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367. doi: 10.1126/science.aba7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korneyenkov MA, Zamyatnin AA. Next Step in Gene Delivery: Modern Approaches and Further Perspectives of AAV Tropism Modification. Pharmaceutics. 2021;13. doi: 10.3390/pharmaceutics13050750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ronzitti G, Gross DA, Mingozzi F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front Immunol. 2020;11:670. doi: 10.3389/fimmu.2020.00670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finn JD, Smith AR, Patel MC, Shaw L, Youniss MR, van Heteren J, Dirstine T, Ciullo C, Lescarbeau R, Seitzer J, et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014 [DOI] [PubMed] [Google Scholar]
- 55.Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 2021;6:1078–1094. doi: 10.1038/s41578-021-00358-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat Nanotechnol. 2020;15:313–320. doi: 10.1038/s41565-020-0669-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Debacker AJ, Voutila J, Catley M, Blakey D, Habib N. Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug. Mol Ther. 2020;28:1759–1771. doi: 10.1016/j.ymthe.2020.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kasiewicz LN, Biswas S, Beach A, Ren H, Dutta C, Mazzola AM, Rohde E, Chadwick A, Cheng C, Musunuru K, et al. Lipid nanoparticles incorporating a GalNAc ligand enable in vivo liver ANGPTL3 editing in wild-type and somatic LDLR knockout non-human primates. biorxiv. 2021. doi: 10.1101/2021.11.08.467731 [DOI] [Google Scholar]
- 59.Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2021;384:403–416. doi: 10.1056/NEJMoa2035389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459–2472. doi: 10.1093/eurheartj/ehx144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, Goldberg AC, Howard WJ, Jacobson MS, Kris-Etherton PM, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–2333. doi: 10.1161/CIR.0b013e3182160726 [DOI] [PubMed] [Google Scholar]
- 63.Omer L, Hudson EA, Zheng S, Hoying JB, Shan Y, Boyd NL. CRISPR Correction of a Homozygous Low-Density Lipoprotein Receptor Mutation in Familial Hypercholesterolemia Induced Pluripotent Stem Cells. Hepatol Commun. 2017;1:886–898. doi: 10.1002/hep4.1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jarrett KE, Lee CM, Yeh YH, Hsu RH, Gupta R, Zhang M, Rodriguez PJ, Lee CS, Gillard BK, Bissig KD, et al. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci Rep. 2017;7:44624. doi: 10.1038/srep44624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farese RV, Ruland SL, Flynn LM, Stokowski RP, Young SG. Knockout of the mouse apolipoprotein B gene results in embryonic lethality in homozygotes and protection against diet-induced hypercholesterolemia in heterozygotes. Proc Natl Acad Sci U S A. 1995;92:1774–1778. doi: 10.1073/pnas.92.5.1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Visser ME, Akdim F, Tribble DL, Nederveen AJ, Kwoh TJ, Kastelein JJ, Trip MD, Stroes ES. Effect of apolipoprotein-B synthesis inhibition on liver triglyceride content in patients with familial hypercholesterolemia. J Lipid Res. 2010;51:1057–1062. doi: 10.1194/jlr.M002915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.National Library of Medicine NCfBI. ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/?term=LDLR[gene]. 2022. Accessed 06/22/22.
- 68.Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch L, Grishin NV, Horton JD, Cohen JC, Hobbs HH. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79:514–523. doi: 10.1086/507488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193:445–448. doi: 10.1016/j.atherosclerosis.2006.08.039 [DOI] [PubMed] [Google Scholar]
- 70.Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013 [DOI] [PubMed] [Google Scholar]
- 71.Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, Cohen JC. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–79. doi: 10.1172/JCI37118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, Natarajan P, Klarin D, Emdin CA, Zekavat SM, et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J Am Coll Cardiol. 2017;69:2054–2063. doi: 10.1016/j.jacc.2017.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–2227. doi: 10.1056/NEJMoa1002926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, et al. Rare dyslipidaemias, from phenotype to genotype to management: a European Atherosclerosis Society task force consensus statement. Lancet Diabetes Endocrinol. 2020;8:50–67. doi: 10.1016/S2213-8587(19)30264-5 [DOI] [PubMed] [Google Scholar]
- 75.Reyes-Soffer G, Ginsberg HN, Berglund L, Duell PB, Heffron SP, Kamstrup PR, Lloyd-Jones DM, Marcovina SM, Yeang C, Koschinsky ML, et al. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42:e48–e60. doi: 10.1161/ATV.0000000000000147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E, Shapiro MD, Stroes ES, Moriarty PM, Nordestgaard BG, et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N Engl J Med. 2020;382:244–255. doi: 10.1056/NEJMoa1905239 [DOI] [PubMed] [Google Scholar]
- 77.Koren MJ, Moriarty PM, Baum SJ, Neutel J, Hernandez-Illas M, Weintraub HS, Florio M, Kassahun H, Melquist S, Varrieur T, et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat Med. 2022;28:96–103. doi: 10.1038/s41591-021-01634-w [DOI] [PubMed] [Google Scholar]
- 78.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New England Journal of Medicine. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664 [DOI] [PubMed] [Google Scholar]
- 79.Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379:2097–2107. doi: 10.1056/NEJMoa1801174 [DOI] [PubMed] [Google Scholar]
- 80.Ray KK, Wright RS, Kallend D, Koenig W, Leiter LA, Raal FJ, Bisch JA, Richardson T, Jaros M, Wijngaard PLJ, et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med. 2020;382:1507–1519. doi: 10.1056/NEJMoa1912387 [DOI] [PubMed] [Google Scholar]
- 81.Gennemark P, Walter K, Clemmensen N, Rekić D, Nilsson CAM, Knöchel J, Hölttä M, Wernevik L, Rosengren B, Kakol-Palm D, et al. An oral antisense oligonucleotide for PCSK9 inhibition. Sci Transl Med. 2021;13. doi: 10.1126/scitranslmed.abe9117 [DOI] [PubMed] [Google Scholar]
- 82.Musunuru K Moving toward genome-editing therapies for cardiovascular diseases. J Clin Invest. 2022;132. doi: 10.1172/JCI148555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whittaker MN, Musunuru K. Therapeutic application of genome editing in dyslipidemia. Curr Opin Lipidol. 2022;33:133–138. doi: 10.1097/MOL.0000000000000805 [DOI] [PubMed] [Google Scholar]
- 84.Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, Cowan CA, Rader DJ, Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Akcakaya P, Bobbin ML, Guo JA, Malagon-Lopez J, Clement K, Garcia SP, Fellows MD, Porritt MJ, Firth MA, Carreras A, et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018;561:416–419. doi: 10.1038/s41586-018-0500-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X, Raghavan A, Chen T, Qiao L, Zhang Y, Ding Q, Musunuru K. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo-Brief Report. Arterioscler Thromb Vasc Biol. 2016;36:783–786. doi: 10.1161/ATVBAHA.116.307227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yin H, Song CQ, Suresh S, Wu Q, Walsh S, Rhym LH, Mintzer E, Bolukbasi MF, Zhu LJ, Kauffman K, et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol. 2017;35:1179–1187. doi: 10.1038/nbt.4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chadwick AC, Wang X, Musunuru K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler Thromb Vasc Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Levy JM, Yeh WH, Pendse N, Davis JR, Hennessey E, Butcher R, Koblan LW, Comander J, Liu Q, Liu DR. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat Biomed Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Breton C, Furmanak T, Avitto AN, Smith MK, Latshaw C, Yan H, Greig JA, Wilson JM. Increasing the Specificity of AAV-Based Gene Editing through Self-Targeting and Short-Promoter Strategies. Mol Ther. 2021;29:1047–1056. doi: 10.1016/j.ymthe.2020.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao Z, Du S, Shen S, Luo P, Ding S, Wang G, Wang L. Comparative efficacy and safety of lipid-lowering agents in patients with hypercholesterolemia: A frequentist network meta-analysis. Medicine (Baltimore). 2019;98:e14400. doi: 10.1097/MD.0000000000014400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Adam RC, Mintah IJ, Alexa-Braun CA, Shihanian LM, Lee JS, Banerjee P, Hamon SC, Kim HI, Cohen JC, Hobbs HH, et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J Lipid Res. 2020;61:1271–1286. doi: 10.1194/jlr.RA120000888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reeskamp LF, Millar JS, Wu L, Jansen H, van Harskamp D, Schierbeek H, Gipe DA, Rader DJ, Dallinga-Thie GM, Hovingh GK, et al. ANGPTL3 Inhibition With Evinacumab Results in Faster Clearance of IDL and LDL apoB in Patients With Homozygous Familial Hypercholesterolemia-Brief Report. Arterioscler Thromb Vasc Biol. 2021;41:1753–1759. doi: 10.1161/ATVBAHA.120.315204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJP, Rubba P, Ali S, Banerjee P, Chan KC, Gipe DA, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med. 2020;383:711–720. doi: 10.1056/NEJMoa2004215 [DOI] [PubMed] [Google Scholar]
- 95.Watts GF, Schwabe C, Scott R, Gladding P, Sullivan D, Baker J, Clifton P, Hamilton J, Given B, San Martin J, et al. Abstract 15751: Pharmacodynamic Effect of ARO-ANG3, an Investigational RNA Interference Targeting Hepatic Angiopoietin-like Protein 3, in Patients With Hypercholesterolemia. Circulation. 2020;142. [Google Scholar]
- 96.Qiu M, Glass Z, Chen J, Haas M, Jin X, Zhao X, Rui X, Ye Z, Li Y, Zhang F, et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of. Proc Natl Acad Sci U S A. 2021;118. doi: 10.1073/pnas.2020401118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chadwick AC, Evitt NH, Lv W, Musunuru K. Reduced Blood Lipid Levels With In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation. 2018;137:975–977. doi: 10.1161/CIRCULATIONAHA.117.031335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Therapeutics V Verve Therapeutics Doses First Human with an Investigational In Vivo Base Editing Medicine, VERVE-101, as a Potential Treatment for Heterozygous Familial Hypercholesterolemia. In; 2022. [Google Scholar]
- 99.Bergmark BA, Marston NA, Bramson CR, Curto M, Ramos V, Jevne A, Kuder JF, Park JG, Murphy SA, Verma S, et al. Effect of Vupanorsen on Non-High-Density Lipoprotein Cholesterol Levels in Statin-Treated Patients With Elevated Cholesterol: TRANSLATE-TIMI 70. Circulation. 2022;145:1377–1386. doi: 10.1161/CIRCULATIONAHA.122.059266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, Lu Y, Tang ZZ, Zhang H, Hindy G, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. doi: 10.1056/NEJMoa1307095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, Won HH, Karczewski KJ, O’Donnell-Luria AH, Samocha KE, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–239. doi: 10.1038/nature22034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guo M, Xu Y, Dong Z, Zhou Z, Cong N, Gao M, Huang W, Wang Y, Liu G, Xian X. Inactivation of ApoC3 by CRISPR/Cas9 Protects Against Atherosclerosis in Hamsters. Circ Res. 2020;127:1456–1458. doi: 10.1161/CIRCRESAHA.120.317686 [DOI] [PubMed] [Google Scholar]
- 103.Zha Y, Lu Y, Zhang T, Yan K, Zhuang W, Liang J, Cheng Y, Wang Y. CRISPR/Cas9-mediated knockout of APOC3 stabilizes plasma lipids and inhibits atherosclerosis in rabbits. Lipids Health Dis. 2021;20:180. doi: 10.1186/s12944-021-01605-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Angrist M, Barrangou R, Baylis F, Brokowski C, Burgio G, Caplan A, Chapman CR, Church GM, Cook-Deegan R, Cwik B, et al. Reactions to the National Academies/Royal Society Report on. CRISPR J. 2020;3:332–349. doi: 10.1089/crispr.2020.29106.man [DOI] [PMC free article] [PubMed] [Google Scholar]