

Abstract
New treatment strategies are urgently needed for glioblastoma (GBM)—a tumor resistant to standard-of-care treatment with a high risk of recurrence and extremely poor prognosis. Based on their intrinsic tumor tropism, adoptively applied mesenchymal stem cells (MSCs) can be harnessed to deliver the theranostic sodium/iodide symporter (NIS) deep into the tumor microenvironment. Interleukin-6 (IL-6) is a multifunctional, highly expressed cytokine in the GBM microenvironment including recruited MSCs. MSCs engineered to drive NIS expression in response to IL-6 promoter activation offer the possibility of a new tumor-targeted gene therapy approach of GBM. Therefore, MSCs were stably transfected with an NIS-expressing plasmid controlled by the human IL-6 promoter (IL-6-NIS-MSCs) and systemically applied in mice carrying orthotopic GBM. Enhanced radiotracer uptake by 18F-Tetrafluoroborate-PET/magnetic resonance imaging (MRI) was detected in tumors after IL-6-NIS-MSC application as compared with mice that received wild-type MSCs. Ex vivo analysis of tumors and non-target organs showed tumor-specific NIS protein expression. Subsequent 131I therapy after IL-6-NIS-MSC application resulted in significantly delayed tumor growth assessed by MRI and improved median survival up to 60% of GBM-bearing mice as compared with controls. In conclusion, the application of MSC-mediated NIS gene therapy focusing on IL-6 biology-induced NIS transgene expression represents a promising approach for GBM treatment.
Keywords: interleukin-6 promoter, glioblastoma, gene therapy, mesenchymal stem cells, sodium/iodide symporter, NIS, PET imaging
Graphical abstract
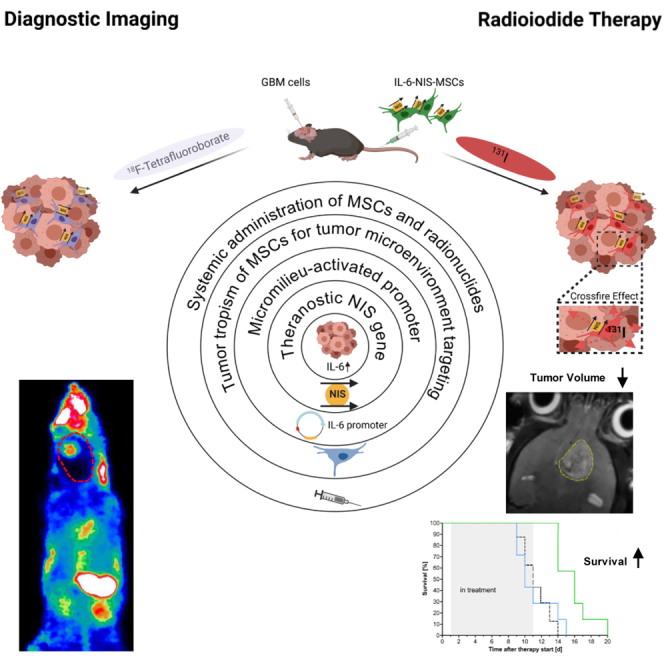
Spitzweg and colleagues combined the tumor-homing capacity of mesenchymal stem cells (MSCs) with tumor-specific expression of the theranostic sodium/iodide symporter (NIS) driven by the IL-6 promoter. By adding this additional layer of specificity, off-target toxicities could be reduced. Finally, IL-6-NIS-MSC-mediated radioiodide therapy led to significantly prolonged survival of glioblastoma-bearing mice.
Introduction
Glioblastoma (GBM) is the most aggressive and most common malignant type of primary brain tumor in adults with a median survival of <2 years after diagnosis.1 Major hurdles encountered in the treatment of GBM result from multiple factors linked to the biology of this tumor, including diffuse infiltrative growth, intra- and intermolecular heterogeneity, and the location itself limiting complete surgical resection as well as the delivery of drugs into the tumor across the blood-brain barrier (BBB).2 The standard treatment of newly diagnosed GBM patients includes extensive surgery followed by radiotherapy and concomitant/adjuvant chemotherapy (“Stupp Protocol”).3,4 Therapy resistance to conventional standard-of-care as well as to new treatments almost always occurs and may correlate in part with the unique genetic, epigenetic, and microenvironmental features of the brain’s neural tissue.2,5
The tumor microenvironment plays a pivotal role in tumor growth and progression of most human cancers including GBM.6 In the glioblastoma microenvironment, an increased abundance of inflammatory cytokines, chemokines, and growth factors has been reported.7 One such cytokine is interleukin (IL)-6, a potent mediator that is omnipresent in the inflammatory microenvironments of many solid tumors. This cytokine displays pleiotropic functions and is released by various cell types within the tumor setting.8 The regulation of IL-6 gene expression is complex and involves numerous transcription factors and their interactions.9 Strong inducers of the IL-6 promoter are tumor necrosis factor (TNF)-∝, IL-1β and interferon (IFN)-γ.10 IL-6 signaling in gliomas is thought to sustain and promote tumor proliferation, tumor invasion, and angiogenesis, and contribute to immune escape, as well as inhibition of apoptosis of cancer cells during chemo- and radiotherapy.7,11,12 According to datasets derived from The Cancer Genome Atlas and the Repository of Molecular Brain Neoplasia Data (REMBRANDT), high IL-6 gene expression is associated with high-grade gliomas in comparison with lower-grade patient samples, and correlates with poor survival and, thus, may represent a prognostic marker in patients for a poor outcome.13,14,15 Due to the role of IL-6 and its signaling pathways as a driver of the malignant progression of GBM, tumor therapy that targets pathways linked to upregulation of IL-6 in MSCs represents an elegant platform for the targeted treatment of newly diagnosed as well as refractory GBM.13
Inflammatory cytokines and chemokines that originate from the GBM micromilieu are implicated in the active recruitment of mesenchymal stem cells (MSCs) to the tumor area.16,17 MSCs have recently gained attention as a cell-based delivery system for the treatment of GBM based on their intrinsic tumor-homing and migratory properties.17,18 Several studies, including our own, have shown that MSCs possess the ability to cross the BBB after systemic application and thus, circumvent a major limitation in the treatment of GBM often encountered in systemic drug delivery to the brain.19,20,21,22,23,24,25,26,27,28 Genetic engineering of MSCs opens the prospect of their application as shuttle vectors for the delivery of therapeutic genes, such as the sodium/iodide symporter, into the critical microenvironment of growing tumors.29
The sodium/iodide symporter (SLC5A5; NIS) is an intrinsic plasma membrane glycoprotein with 13 putative transmembrane domains that is predominantly found in the thyroid gland. Based on its active transport function for iodide and other substrates, it represents a powerful theranostic gene that allows for radionuclide imaging and treatment that has been successfully used routinely in the treatment of thyroid cancer and its metastases, including brain metastases.30,31,32,33 Different isotopes are efficiently transported by NIS when used in the context of a reporter gene (e.g., 123I, 124I, 125I, 99mTc, 18F-tetrafluoroborate [18F-TFB]) and can be quantitatively visualized using standard nuclear medicine imaging technologies such as scintigraphy, single-photon emission computed tomography or positron emission tomography (PET) imaging. For the monitoring of low-volume tumor disease such as GBM, three-dimensional, high-resolution PET imaging is the most appropriate technique. In this context, 124I or 18F-TFB can be used as radiotracers for NIS-mediated PET imaging. Both agents have been demonstrated to be delivered to the brain and allow brain tumor imaging. The use of 18F-TFB further enables an improved, more accurate and less “blurred” imaging quality as compared with 124I based on the radio-physicochemical properties of 18F.34,35 Importantly, NIS can also be used as an effective therapy gene by the facilitated uptake of the β-emitters 131I or 188Re, or the α-emitter 211At. Because of the resultant cytotoxic effects including a significant bystander effect, treatment effectively eliminates NIS-transduced cells as well as adjacent cells in the course of therapy.30
Cloning of the cDNA encoding NIS in 1996 provided the molecular platform for NIS gene transfer into non-thyroidal tumors and, thus, expanded its function to the treatment of extrathyroidal malignancies.36 We and others have shown the potential of NIS gene-based therapy using MSCs as delivery vectors for the treatment of distinct non-thyroidal tumors.23,37,38,39,40,41,42 Most recently, we demonstrated the potential use of NIS as a reporter gene to track adoptively applied MSCs using NIS-MSCs constitutively expressing NIS driven by the cytomegalovirus (CMV) promoter in GBM after systemic delivery followed by therapeutic application of 131I, which led to a significant increase in survival and reduced tumor growth.28 In this context, MSC recruitment to non-tumor tissue due to normal tissue homeostasis along with transgene expression might lead to undesirable extratumoral toxicity.
Selective control of transgene expression allows the restriction of radiotoxicity to tumor tissue, sparing non-target tissues from potential damage. Using this additional layer of specificity, the general efficacy of MSC-based NIS gene therapy can be improved by the use of inducible gene promoters specifically activated in response to signaling pathways associated with factors present in the tumor environment. Our group has demonstrated the use of ex vivo engineered MSCs with various gene promoters to drive NIS transgene expression including RANTES/CCL5-, hypoxia-responsive HIF1α-, heat-inducible (HSP70B)-, and a synthetic transforming growth factor β1 (TGFβ1)-inducible Smad-responsive promoter in hepatocellular carcinoma (Huh7) and metastatic colon carcinoma xenograft models.39,40,41,42,43,44,45
The major goal of the present study was to make use of the factors associated with the activation of IL-6 expression in high-grade gliomas to target NIS gene therapy to the GBM stroma in an immunocompetent setting. For this purpose, syngeneic murine bone-marrow-derived MSCs were genetically modified to express NIS under the control of the human IL-6 gene promoter (IL-6-NIS-MSCs). In vivo biodistribution of IL-6-NIS-MSCs and induction of IL-6 promoter activation was monitored by NIS-mediated 18F-TFB-PET imaging after systemic administration. Tumor-selective, NIS-mediated radiotracer accumulation was further examined by co-registration of PET imaging with anatomical brain magnetic resonance imaging (MRI) and analysis of NIS protein expression ex vivo. Finally, an 131I therapy in GBM-bearing mice was conducted after systemic IL-6-NIS-MSC administration and assessed by analysis of tumor volume using MRI and animal survival.
Results
In vitro stimulation of IL-6-NIS-MSCs resulting in enhanced NIS-mediated radioiodide uptake
Murine bone-marrow-derived MSCs were engineered to express NIS driven by the human IL-6 promoter (IL-6-NIS-MSC). IL-6 is induced by a variety of cell types within tumor environments in response to proinflammatory signals.6,8 To validate the inducibility of the IL-6 promoter in IL-6-NIS-MSC, the murine cytokines (IL-1β, TNF-α, and IFN-γ), which have been reported as strong activators of the IL-6 pathway,10 were used to stimulate promoter activation followed by analysis of the resulting functional NIS expression by 125I-uptake assay in vitro (Figure 1). The NIS-specific inhibitor perchlorate was used to confirm NIS specificity, which reduced radioiodide uptake to background levels. Stimulation with murine cytokines led to a dose-dependent, significant increase of radioiodide uptake of IL-6-NIS-MSCs (Figures 1A–1C). A plateau was reached with all cytokines. IL-1β alone (maximal level at 1 ng/mL) showed the highest increase of 125I accumulation as compared with IFN-γ (maximal level at 50 ng/mL) and TNF-α (maximal level at 10 ng/mL). The combination of those factors using the concentration yielding the maximal iodide uptake level of each resulted in maximal NIS-mediated radioiodide uptake as compared with untreated IL-6-NIS-MSCs (Figure 1D). As a basis for the in vivo application, incubation of IL-6-NIS-MSC with conditioned media (CM) of murine GL261 GBM cells (Figure 1E) containing diverse tumor-derived factors resulted in significant increase of radioiodide uptake as compared with untreated cells. This stimulation could be further enhanced by adding the mixture of cytokines at their optimal concentrations. In contrast, IL-6-NIS-MSCs were additionally treated with CM of mouse 4T1 breast cancer cells to determine whether this effect is also observed by cells derived from another tumor entity. Treatment with 4T1-CM alone did not induce statistically significant stimulation of iodide uptake in IL-6-NIS-MSC as compared with untreated IL-6-NIS-MSCs. But stimulation with a combination of 4T1-CM and the mixture of IL-1β, TNF-α, and IFN-γ—factors produced by the tumor’s stromal compartment—led to a level of iodide uptake that was comparable to that reached with GL261-CM.
Figure 1.

Establishment of mesenchymal stem cells (MSCs) stably expressing NIS regulated by the IL-6 promoter
(A–C) Stimulation with cytokines IL-1β (0.5–5 ng/mL), IFN-γ (40–60 ng/mL), and TNF-α (1–15 ng/mL) showed NIS-specific 125I-uptake that was reduced to background levels using perchlorate as NIS-specific inhibitor. (D) Treatment of IL-6-NIS-MSCs with IL-1β (1 ng/mL), IFN-γ (50 ng/mL), and TNF-α (10 ng/mL) revealed an increased radioiodide uptake as compared with single stimulation studies. (E) Stimulation with GL261-CM led to a significant increase of radioiodide uptake as compared with unstimulated IL-6-NIS-MSCs and was further increased by combined treatment with IL-1β (1 ng/mL), IFN-γ (50 ng/mL), and TNF-α (10 ng/mL). In contrast, stimulation with 4T1-CM was significantly lower as compared with stimulation with GL261-CM, but treatment with 4T1-CM and IL-1β (1 ng/mL), IFN-γ (50 ng/mL), and TNF-α (10 ng/mL) led to similar radioiodide uptake activity of IL-6-NIS-MSCs as treatment with GL261-CM and combined factors. Results are expressed as mean ± SEM of three independent experiments (two-tailed Student’s t test ∗p < 0.05, ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001). CM = conditioned medium.
IL-6-NIS-MSC migration toward GL261 tumor cell conditioned medium is increased
To evaluate the chemotactic behavior of IL-6-NIS-MSCs in response to a linear gradient of GL261-CM, a 3D migration assay using a collagen I matrix and time-lapse microscopy over a period of 24 h was performed (Figure 2). IL-6-NIS-MSCs did not show directed chemotaxis without the influence of a chemoattractant (yFMI = −0.0075 ± 0.0087 and yCoM = −3.7 ± 2.1 μm) used as a negative control (Figures 2A and 2C). Further, when subjected to a gradient between GL261-CM and serum-free medium, IL-6-NIS-MSCs showed a strong directed chemotaxis toward the GL261-CM with significantly increased yFMI (0.0972 ± 0.0006; ∗∗p < 0.01) and yCoM (56.8 ± 21.5 μm; ∗p < 0.05) displacement along the gradient (Figures 2B and 2C).
Figure 2.

Impact of GL261-CM on IL-6-NIS-MSC migratory behavior
Chemotactic behavior of MSCs subjected to a gradient of GL261-CM was evaluated using 3D live-cell imaging migration assay over a period of 24 h. (A) Negative control is shown without the influence of a gradient on MSCs. (B) Migration of IL-6-NIS-MSCs along a gradient of GL261-CM resulted in strong migration toward the GL261-CM gradient. (C) Quantification of the chemotaxis parameters mean forward migration index (yFMI) and center of mass (yCoM, displayed by red dotes). Results are expressed as mean ± SEM from two independent experiments. Two-tailed Student’s t test was used for statistical analysis (∗p < 0.05; ∗∗p < 0.01). CM = conditioned medium.
In vivo18F-TFB biodistribution studies show elevated NIS-mediated tumoral tracer accumulation after systemic IL-6-NIS-MSC administration
Once mice had developed brain tumors, the utilization of a single intravenous (i.v.) injection of murine IL-6-NIS-MSCs or two cycles of IL-6-NIS-MSCs in 2-day intervals followed by 18F-TFB injection (10 MBq, i.v.) after 48 h was investigated to identify an optimized dosing and application schedule. In vivo whole-body, high-resolution 18F-TFB-PET imaging was performed over a period of 2 h allowing an optimal discrimination of exogenous and endogenous NIS-mediated signals in the head region. 18F-TFB-PET imaging showed high levels of NIS-mediated radionuclide accumulation in brain tumors after systemic injection of IL-6-NIS-MSCs (Figures 3A and 3B). Administration of wild-type MSCs (WT-MSCs) was used as a control, as well as non-treated GBM-bearing mice that did not receive MSCs, both resulted in a tumoral tracer accumulation comparable to background level (Figures 3C and 3D). In addition, 18F-TFB uptake was also observed in organs that physiologically express NIS, including the thyroid, salivary glands, and stomach as well as the urinary bladder due to mainly renal elimination of the radiotracer. Tumors from mice that had received a single IL-6-NIS-MSC application accumulated a maximum of 1.9% ± 0.3% ID/mL 18F-TFB 1 h post injection (Figure 3E). When PET imaging was performed after two IL-6-NIS-MSC injections, tumoral tracer accumulation revealed a maximal level of 1.5% ± 0.1% ID/mL. The quantification of tumoral 18F-TFB uptake showed no significant differences between the single-dose and two-dose application schemes of IL-6-NIS-MSC-treated mice and were measured in the same range as compared with a subset of mice injected with constitutively NIS-expressing (CMV-NIS-MSCs) MSCs (1.9% ± 0.4% ID/mL 1 h post injection; Figure S1). Magnification of the brain area and co-registration of anatomical in vivo MRI of the brain with 18F-TFB-PET imaging was used to facilitate delineation of areas of active tumor-selective tracer accumulation in the GBM of mice after systemic IL-6-targeted MSC-mediated NIS gene delivery (Figures 3F and 3G), whereas tumors of mice that had received WT-MSCs or untreated tumor-bearing mice showed no tumoral radionuclide accumulation above background level (Figures 3H and 3I).
Figure 3.

18F-TFB uptake of brain tumors is elevated after systemic IL-6-NIS-MSC delivery
Representative images of 18F-TFB-PET imaging (horizontal planes; SUVbw = 1.6) are shown 1 h after tracer injection of GBM-bearing mice after (A) single IL-6-NIS-MSC injection (n = 5), (B) two IL-6-NIS-MSC injections (n = 5), after (C) WT-MSC injection (n = 3), which served as a control, and (D) no MSC injection (n = 1). Brain area is encircled in red dotted lines and tumor is marked with a white arrow. (E) Tumoral radionuclide accumulation showed significantly higher levels after IL-6-NIS-MSC delivery as compared with WT-MSC control mice 1 h post 18F-TFB injection. (F–I) Exemplary images of co-registration of anatomical MRI and PET imaging of the brain of the same mice as demonstrated in (A)–(D) showing tumor-selective tracer accumulation. A strong tumoral 18F-TFB accumulation is revealed after systemic IL-6-targeted MSC-mediated NIS gene transfer (F–G), while tumors after WT-MSC injection did not show levels above background level (H) as well as compared with non-treated tumor (I). Tumors are encircled in yellow dotted lines. Results are expressed as % of the injected dose per mL (volume assessed by MRI) and mean ± SEM is given (two-tailed Student’s t test at each time point; ∗p < 0.05). sg = salivary glands.
Based on this finding that a higher MSC load did not lead to an increase in tumoral tracer uptake, a single MSC application that reduces the stress level of mice and allows for a shortened therapy schedule was used as a basis for the following 131I therapy study.
Ex vivo analysis of GL261 brain tumors and control organs demonstrate tumor-selective NIS expression
Immunohistochemistry for IL-6 protein expression was performed on formalin-fixed paraffin embedded (FFPE) GL261 tumor sections. IL-6 protein expression was observed in all tumors and its stroma (Figure 4A). Ex vivo immunofluorescence staining revealed heterogeneous NIS protein expression throughout the tumor stroma of GL261 tumors after systemic IL-6-NIS-MSC injection with patchy areas of high NIS immunoreactivity and areas of low NIS protein expression, while sparing non-tumor brain tissue (Figures 4B–4D). No NIS-specific immunoreactivity was detectable in brain tumors of mice that were injected with WT-MSCs (Figure 4E). Non-target organs such as liver, lung, kidney, and spleen were analyzed for NIS immunoreactivity that showed no detectable NIS protein expression (Figures 4F–4I). IL-6 and NIS protein expression was further analyzed by immunofluorescence co-staining of GL261 tumors in mice injected with IL-6-NIS-MSCs demonstrating nice co-localization of NIS and IL-6 protein expression (Figure S2).
Figure 4.

Ex vivo analysis of brain tumors and control organs show tumor-specific IL-6 and NIS protein expression
(A, upper and lower panel) IL-6 protein expression was confirmed in GL261 tumors and throughout the tumor stroma (DAB-positive stain is shown in brown). (B) NIS immunofluorescence staining is shown with ×1 magnification to demonstrate tumor mass and the implantation site in the right caudate putamen. Tumor is encircled in yellow dotted lines. (C) No detectable NIS protein expression is observed in non-tumor brain tissue (close-up view: pink window in B). (D, upper and lower panel) Close-up view (indicated by the white window in B) shows NIS-specific immunoreactivity within the tumor stroma, with areas of high and low NIS protein expression, after systemic application of IL-6-NIS-MSCs, while tumors of mice that had received (E, upper and lower panel) WT-MSCs did not show detectable NIS expression. (F–I) No NIS protein expression was detected in control organs such as liver, lung, spleen, and kidney after IL-6-NIS-MSC administration. A representative image is shown in each.
MSC-mediated IL-6 induced NIS gene therapy led to improved survival of GBM-bearing mice
Based on the results of the PET/MRI data and the rapidly growing nature of the GL261 model, a short therapy schedule was conducted using three cycles of a single i.v. MSC administration (days 1/5/9) each followed by an intraperitoneal (i.p.) 131I injection 2 days later (days 3/7/11). Mice were included in the therapy study as soon as a tumor was clearly visible (0.6–2.1 mm³) as assessed by 7T-MRI and mice were randomly distributed to all groups. Twice a week, tumor growth was monitored by MRI resulting in a significantly reduced tumor volume of mice in the therapy group (IL-6-NIS-MSCs + 131I; Figures 5A and 5D) as compared with control groups (IL-6-NIS-MSCs + NaCl; Figures 5B and 5D; ∗p < 0.05 day 7 after therapy start, and NaCl + NaCl; Figures 5C and 5D; ∗p < 0.05 day 7 and 10 after therapy start). Therapy mice demonstrated significantly prolonged survival (Figure 5E) as compared with control groups (IL-6-NIS-MSCs + NaCl; ∗∗p < 0.01 and NaCl + NaCl; ∗∗∗p < 0.001). The median survival (MS) after therapy start was extended up to 60% as compared with controls (IL-6-NIS-MSCs + 131I MS = 16 days; IL-6-NIS-MSCs + NaCl MS = 10 days; NaCl + NaCl MS = 11 days).
Figure 5.

IL-6-targeted MSC-mediated NIS gene 131I therapy of GBM-bearing mice led to reduced tumor growth and improved survival
Three cycles of a single i.v. IL-6-NIS-MSC injection was applied followed by an i.p. administration of 131I 48 h later (MSCs on days 1/5/9 and 131I on days 3/7/11, respectively). Representative MR images 10 days after therapy start are shown after treatment with (A) IL-6-NIS-MSCs + 131I, (B) IL-6-NIS-MSCs + NaCl, and (C) NaCl + NaCl. (D) Tumor growth was monitored using MRI showing a significantly reduced tumor mass of IL-6-NIS-MSCs + 131I as compared with controls after completion of two therapy cycles (day 7 after treatment start, n = 7 each, ∗p < 0.05). At day 10 after therapy start, tumor growth of mice from the therapy group (n = 7/7) was delayed as compared with the NaCl-only group (n = 6/7, ∗p < 0.05) and the IL-6-NIS-MSC + NaCl-treated mice (n = 5/7, ns). Fourteen days after therapy start, all mice from the therapy schedule were included in the tumor measurement (n = 7/7), but n = 3 had to be euthanized the same day, while in the NaCl-only group only n = 1 of 7 and the IL-6-NIS-MSC + NaCl n = 2 of 7 of the mice were still alive and included in the measurement. (E) The survival of IL-6-NIS-MSCs + 131I-treated mice was significantly extended as compared with the controls treated with NaCl + NaCl (∗∗∗p < 0.001) and IL-6-NIS-MSCs + NaCl (∗∗p < 0.01). Tumors are encircled in yellow dotted lines and results are expressed as mean ± SEM. One-way ANOVA using Tukey’s post hoc test was performed for tumor growth analysis and log rank test for comparison of survival curves.
At the end of therapy, animals were euthanized and brains were dissected. Ex vivo immunofluorescence analysis was used to analyze cell proliferation (Ki67) and blood vessel density (CD31) on cryosections of the brain to determine the effect of 131I after systemic MSC-mediated NIS gene transfer (Figure 6). The intratumoral cell proliferation index (Ki67; Figures 6A–6D) of the therapy group (IL-6-NIS-MSCs + 131I: 2.6% ± 0.3% Ki67-positive cells) was significantly lower as compared with control groups (IL-6-NIS-MSCs + NaCl: 4.7% ± 0.9% and NaCl + NaCl: 5.1% ± 0.5% of Ki67-positive cells). Blood vessel density (CD31; Figures 6A–6C and 6E) analysis showed reduced tumor vascularization in the IL-6-NIS-MSCs + 131I treated animals as compared with the IL-6-NIS-MSCs + NaCl-treated group, while no significant reduction was assessed as compared with the NaCl-only group.
Figure 6.

Ex vivo analysis of brain tumors after IL-6 promoter-induced MSC-mediated NIS gene therapy
At the end of the therapy study, frozen brain tissue sections derived from mice that were treated with IL-6-NIS-MSCs + 131I (A), IL-6-NIS-MSCs + NaCl (B), and NaCl only (C) were subjected to Ki67 (proliferation index; green) and CD31 (blood vessels; red) immunofluorescence staining. Nuclei were counterstained with Hoechst (blue). A representative picture is shown in each at ×40 magnification (scale bar, 40 μm). Quantification of the proliferation index (D) and blood vessel density of IL-6-NIS-MSCs + 131I in comparison with both control groups (E). Results are expressed as mean ± SEM (two-tailed Student’s t test ∗p < 0.05; ∗∗p < 0.01).
Discussion
Despite recent advances in the treatment of GBM, long-term treatment efficacy is poor and the disease remains incurable, thus new GBM therapeutic strategies are urgently needed. Based on their inherent tumor-homing properties and abilities to overcome the BBB, MSCs represent a promising tool for the delivery of therapy genes deep into the microenvironment of GBM.23,24,26,28 MSC-mediated NIS gene therapy has been described in various experimental models, such as hepatocellular carcinoma,37,39,41,42,43,44,45,46 pancreatic cancer,38 metastatic colon carcinoma,40 and glioma.23 NIS is a theranostic gene that allows in vivo tracking of NIS-transfected MSCs after systemic application and their use in cancer therapy in combination with administration of therapeutically active radionuclides. The clinical use of radioiodide treatment in thyroid cancer patients started in 1946 and is still considered one of the most effective molecularly targeted and imaging-based radiation treatments in cancer with a well-understood safety profile.47,48 The application of ex vivo engineered MSCs using NIS in the context of solid tumor treatment is currently being examined in early-phase human clinical trials. One study explores the safety and tolerability of MSCs engineered with an oncolytic measles virus encoding NIS (MV-NIS) in recurrent ovarian cancer (NTC02068794).
Previous work by the authors has made use of engineered versions of syngeneic MSCs constitutively expressing NIS driven by the CMV promoter (CMV-NIS-MSCs). Successful tumor tropism was shown after systemic application by in vivo imaging using NIS as reporter gene followed by effective therapeutic trials in several tumor models, including most recently in experimental GBM demonstrating selective MSC recruitment and homing to GBM tumors by overcoming the BBB.28,37,38 While the application of CMV-NIS-MSCs showed strong tumor tropism, a portion of exogenously applied MSCs might potentially be recruited to non-target organs in the process of normal tissue homeostasis or by entrapment in vascular organs after intravenous injection.28,37,49,50 The use of MSCs genetically engineered with gene promoters activated by factors in the tumor micromilieu to drive NIS expression in the tumor stroma can reduce the risk of potential off-target effects to non-tumor tissue.
As seen in other solid tumors, the GBM microenvironment is composed of non-malignant stromal cells such as endothelial cells, pericytes, cancer-associated fibroblasts (CAFs), and immune cells (e.g., brain-resident microglia and infiltrating macrophages [GAMs]) and the tumor cells themselves.51 Further, endogenous MSCs are recruited from different origins in the body into the tumor as part of its stromal compartment. In addition to the cellular constituents, GBMs are also surrounded by a pool of proinflammatory cytokines, chemokines, and growth factors such as TNF-α, TGF-β, IL-6, IL-10, IL-8, CCL2/MCP-1, and RANTES/CCL5.52,53
In the present study, NIS transgene expression was linked to activation of the promoter for the human IL-6 gene in MSCs. The IL-6 gene is upregulated in response to various proinflammatory factors present in solid tumor environments and the protein is associated with important aspects of tumor biology.6 IL-6 regulates a variety of biological functions including acute phase response, leukocyte maturation, and infiltration at sites of inflammation and endothelial cell properties.54 In the GBM environment, IL-6 is produced by resident MSCs, GAMs, tumor-associated endothelial cells, and glioma stem cells (GSCs), a small cellular subpopulation with potent tumorigenic and stem-cell-like properties that have been implicated to play a crucial role in GBM maintenance and recurrence as well as by the tumor cells themselves.14,55,56
Reports have demonstrated the impact of IL-6 on the tumorigenicity of GBM using transgenic mice, where genetic IL-6 depletion blocked GBM formation.57 Furthermore, a study by Lamano et al. reported increased IL-6 plasma levels of mice orthotopically implanted with GL261 cells as compared with sham-operated mice 14 days post injection and mice implanted with CRISPR-Cas9 IL-6 knockout (KO) GL261 cells showed significantly smaller tumors and an overall increased survival of 77% as compared with unmodified GL261 cells.58 GBM cells producing IL-6 are associated with an enhanced chemo- and radioresistant phenotype.59,60,61 In addition, IL-6 signaling exerts immunosuppressive effects in GBM via stimulation of GAMs and suppresses T cell functions.15 Tumors often modify their stromal compartment to favor its progression. In this context, indirect crosstalk of MSCs (or other cell types) with tumor cells via growth factors, chemokines, and cytokines functions in a bidirectional way and leads to functional changes of both cell types through activation of intracellular signaling. The MSC-mediated release of IL-6 has been found in various preclinical tumor models such as ovarian cancer,62 nasopharyngeal carcinoma,63 breast cancer,64 and glioma65 and is often associated with their differentiation into CAFs, endothelial cells, smooth muscle cells, and pericytes, where IL-6 produced by MSCs (or cell types they differentiated to) helps promote tumor growth, resistance to drugs, and tumor vascularization.66 Besides its well-known role as a critical driver of cancer formation, IL-6 plays a dual role in the tumor microenvironment (TME) by its additional contribution to anti-tumor immunity through mobilizing anti-tumor T cell immune responses.67 Increasing evidence points to IL-6 as a key player in the activation, proliferation, and survival of lymphocytes as well as T cell trafficking, thereby boosting immune surveillance.67,68 While the exact mechanisms regulating the balance between the anti-tumor and pro-tumor effects of IL-6 are not fully understood, there is clear evidence of robust expression of IL-6 within GBM tissues highlighting the potential of using TME-expressed factors for activation of the IL-6 promoter and subsequent therapeutic transgene expression by MSCs.
In the present study, syngeneic murine bone-marrow-derived MSCs were stably transfected with the IL-6 gene promoter driving NIS expression (IL-6-NIS-MSCs) that enabled NIS-mediated in vivo tracking of MSC homing to the tumor as well as therapeutic 131I application in response to activation of the IL-6 gene promoter signaling. The upregulation of IL-6 pathway inducers like TNF-α has been reported in GBM patient samples69 as well as in the experimental GBM used in this study.70 In vitro characterization of IL-6-NIS-MSCs demonstrated their ability to significantly concentrate 125I in a dose-dependent manner after stimulation with the proinflammatory cytokines IL-1β, TNF-α, and IFN-γ previously shown to stimulate increased IL-6 biosynthesis.15,71,72,73 The iodide uptake was perchlorate-sensitive demonstrating NIS-dependent radionuclide accumulation of these MSCs. Treatment with tumor cell CM derived from GL261 glioma cells led to a significant accumulation of radioiodide in IL-6-NIS-MSCs that was further enhanced by combination with a mixture of IL-1β, TNF-α, and IFN-γ. Stimulation with GL261-CM alone showed a significantly higher NIS-mediated iodide uptake ability as compared with the breast cancer 4T1-CM, demonstrating a stronger IL-6 promoter induction when using GL261 supernatants.
Further, in vitro analysis of IL-6-NIS-MSCs migration toward GL261-CM, which represents a mix of diverse factors, demonstrated a strong directed chemotaxis as compared with random basal chemotactic behavior of those MSCs lacking the influence of a gradient as seen in several other tumor models, suggesting that the tumor produces factors that can enhance the selective recruitment of MSCs.42,74 The mechanisms underlying MSC tropism to the tumor have not been fully elucidated, but are thought to parallel the mechanisms used by leukocytes in the course of inflammation where recruitment is driven by the presence of inflammatory chemokines and cytokines such as TGF-β, TNF-α, interleukins (e.g., IL-1, IL-6, IL-8, IL-10), interferons, and chemokines secreted by the tumor and the tumor stroma and the associated receptors on MSCs.17,75
In the in vivo imaging studies, small-animal PET/MRI after administration of IL-6-NIS-MSCs in GBM-bearing mice showed significant tumoral 18F-TFB accumulation after a single or double MSC injection as compared with mice that received WT-MSCs or no MSCs that showed only background levels. Co-registration with MRI demonstrated tumor-selective tracer accumulation with spots of higher and lower tracer concentration, which may be due to a combination of heterogeneous MSC recruitment and/or proinflammatory factor expression resulting in heterogeneous IL-6 promoter-driven NIS transgene expression. No off-target NIS protein expression was observed in the current study using tumor-stroma selective IL-6 gene promoter: ex vivo NIS protein immunofluorescence of IL-6-NIS-MSC-treated mice exhibited NIS expression largely restricted to tumors, while no NIS protein expression was detected in healthy brain tissue and non-target organs, and thus may avoid severe off-target damage, e.g., in the lungs when therapeutic radioiodide doses are employed. These findings are consistent with our previous study in the same GBM model using constitutively NIS-expressing CMV-NIS-MSCs that showed MSC homing with NIS protein expression restricted to the tumor areas.28 In contrast to the current study using IL-6-NIS-MSCs, in the previous study a small number of CMV-NIS-MSCs were detected in the lungs of mice, probably due to entrapment in the microvascular system,28 thereby demonstrating enhanced tumor selectivity of NIS transgene expression by the use of TME-inducible gene promoters.
Although 18F-TFB is a poorer NIS substrate in comparison with 124I used as traditional tracer for NIS PET imaging, and the short physical half-life of 18F precludes its use for dosimetric studies for the required therapeutic dose of 131I in context of personalized cancer treatment, the physical properties of 18F are much better suited for PET imaging in small animals.34 In the current study, a subset of mice received CMV-NIS-MSCs as a standard application and showed approximately the same level of 18F-TFB accumulation in the GBM tumors as compared with levels of mice that were injected with IL-6-NIS-MSCs. Based on these comparative studies38 and our previous experience in the same tumor model using CMV-NIS-MSCs followed by 124I-PET imaging, including dosimetric calculations and therapy studies,28 we hypothesized that the tumoral tracer uptake after IL-6-NIS-MSC application in the present study should be sufficient to obtain a therapeutic effect. In the current study, the survival of GBM mice was significantly extended after systemic IL-6-NIS-MSCs administration followed by 131I and a significant reduction of the tumor volume as compared with control groups was observed. The MS after therapy start was improved up to 60% as compared with the controls. In line with earlier studies by our group using various tumor models, the number of proliferating cells was significantly decreased in tumors of the therapy group as compared with control groups.37,39,41,43 MSC recruitment to the GBM TME with activation of the IL-6 pathway followed by IL-6-targeted NIS transduction is suggested to lead to cell death of NIS-expressing MSCs through particle decay of accumulated 131I and the neighboring tissue (e.g., tumor cells or GSCs) through bystander effects based on the path length in tissue of up to 2.4 mm of the decaying particles. Potential problems encountered by the use of MSCs, such as tumor-promoting effects or their long-term retention time,76 is reduced by the inclusion of suicide genes, such as NIS, that allow the efficient elimination of the applied cells.77
A potential clinical implementation of the IL-6-directed MSC-mediated NIS gene concept would be as an adjuvant treatment to enhance the therapeutic effect of established conventional treatments such as surgery, radiotherapy45 or novel targeted treatments. Current challenges for the treatment of GBM include the extreme heterogeneity, infiltrative growth including microsatellites, aberrant vascularization, intrinsic resistance to the conventional treatments, and delivery problems to the brain. Many of these issues may be effectively addressed through the use of MSC-based therapy vehicles. GBM is a highly vascularized tumor,78 anti-angiogenic treatments such as bevacizumab, a monoclonal antibody that blocks vascular endothelial growth factor (VEGF) approved by the Food and Drug Administration for the treatment of GBM has not led to an improvement of patient survival as a monotherapy, and the tumor often develops resistance to treatment within months of starting therapy.79 Targeting IL-6 signaling in combination with a VEGF-signaling blockade has been reported as a promising treatment strategy to reduce tumor invasiveness and growth in experimental GBM.80 Immunotherapy for GBM treatment is currently being evaluated in human clinical phase II/III trials.81 Combination of anti-IL-6 treatment and anti-programmed death-ligand 1 treatment has been reported to have additive beneficial effects in terms of animal survival in preclinical GL261 that was not observed with either monotherapy.58
The essential role of IL-6 in tumor biology makes the IL-6 signaling pathway an attractive candidate to potentially control NIS expression within the TME. Genetically engineered MSCs were employed to target NIS expression to the GBM environment and mediate transgene induction through IL-6 signaling to reduce potential side effects associated with MSC recruitment to non-target tissue and use synergistic effects in terms of enhanced MSC recruitment and promoter activation during 131I therapy. An improvement of the therapeutic effect may be based on a possible self-energizing effect: a potential increased inflammatory response and enhanced vascular permeability due to tissue damage following radioiodide treatment may enhance MSC recruitment to the tumor stroma and increase IL-6 signaling that in turn stimulates promoter activation. Repeated cycles of this treatment regime may lead to an amplification of the therapeutic effect.45
Effective MSC recruitment and robust, tumor-selective NIS expression and induction of NIS-mediated radionuclide accumulation in GBM tumors driven by the inflammatory micromilieu in GBM was visualized by 18F-TFB-PET/MRI in vivo and ex vivo staining using the function of NIS as reporter gene. 131I treatment was employed to IL-6-NIS-MSCs-treated mice resulting in a reduction of GBM growth and significantly extended median survival of GBM-bearing mice. In conclusion, the flexibility of the MSC-based NIS gene therapy concept may help address many of the major hurdles associated with the treatment of GBM and offers the opportunity for more individualized and optimized treatment concepts especially when combined with conventional treatment approaches adjustable to the patient’s situation.
Materials and methods
Cell culture
The murine glioma cell line GL261 was purchased and authenticated from the National Cancer Institute (Frederick, MD, USA). Cells were grown in DMEM low glucose (Sigma-Aldrich, St. Louis, MO, USA) containing 10% (v/v) fetal bovine serum (FBS Superior, Sigma-Aldrich), 1% (v/v) MEM non-essential amino acid solution (Thermo Fisher Scientific, Waltham, MA, USA) and 1% (v/v) penicillin/streptomycin (Sigma-Aldrich).
The murine breast cancer cell line 4T1 was purchased from the ATCC and cultured in RPMI-1640 (Sigma-Aldrich) supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin.
Murine bone marrow-derived MSCs (WT-MSCs) were isolated from C57Bl/6 p53−/− mice as described and characterized previously.38,49 MSCs were grown in RPMI (Sigma-Aldrich) supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin.
Cells were maintained in an incubator in a humidified atmosphere at 37°C with 5% CO2. For in vivo experiments, cells were tested for mycoplasma and viruses according to the FELASA guidelines by Charles River Research Animal Diagnostic Services (Wilmington, MA, USA; Mouse essential panel).
Plasmid construct
To establish the pcDNA6-2LITRHygro-IL-6-Promoter-NIS plasmid construct, the Multisite Gateway Pro Plus Kit (Invitrogen Thermo Fisher Scientific, Waltham, MA, USA) was used following the manufacturer’s recommendations and the plasmids produced as previously described.44,82 The pcDNA6-2LITRHygro-IL-6-Promoter-NIS plasmid contains the full-length human NIS gene (NIS cDNA kindly provided by SM Jhiang, Ohio State University, Columbus, OH, USA) driven by the human IL-6-promoter, two sleeping beauty transposition sites, and a hygromycin resistance gene.
Stable transfection of MSCs
IL-6-NIS-MSCs were produced by stable transfection of WT-MSCs with the two plasmids pcDNA6-2ITRHygro-IL-6-Promoter-NIS (pSB.H.IL6.hNIS) and pCMV (CAT)T7-SB100X (provided from Z Ivics, Max Delbrück Center for Molecular Medicine, Berlin, Germany), which contains the sleeping beauty transposase system.82 1 × 106 WT-MSCs were electroporated with 500 ng of each plasmid at 800 V, 30 ms, and two pulses using the Neon Nucleofection system (Invitrogen). From t = 24 h on, cells were cultivated in selection medium containing RPMI (Sigma-Aldrich) supplemented with 10% (v/v) FBS, 1% (v/v) penicillin/streptomycin), 100 μg/mL hygromycin (Invivogen, San Diego, CA, USA), and 50 μg/mL geneticin (Sigma-Aldrich).
CMV-NIS-MSCs were produced by stable transfection of WT-MSCs and cells cultured as described previously.28,38
Single clones of stably transfected MSCs were isolated and tested for functional NIS expression using 125I uptake assay (see below). About 35 clones were screened over 2–3 passages and the sub-cell line with the highest NIS-mediated radioiodide uptake activity was used for further experiments and referred to as IL-6-NIS-MSC in the following.
Tumor cell conditioned medium
GL261 and 4T1 cells (1 × 106 cells) were seeded on a 100 mm³ surface cell culture plate and cultured for 24 h. Afterward, cells were starved for 12 h in serum-free medium. Supernatant was removed after 48 h and GL261- and 4T1-conditioned medium (GL261-CM and 4T1-CM) was centrifuged and stored at −80°C. Tumor cell CM was used for migration assay and in vitro iodide uptake experiments.
125I-uptake assay in vitro
IL-6-NIS-MSCs (1.5 × 105 cells/well) were seeded on a 6-well cell culture plate and after 48 h starved for 12 h. Cells were stimulated for 24 h with the murine cytokines IL-1β (0.5–5 ng/mL; Peprotech, Cranbury, NJ, USA; Catalog #211-11B), TNF-α (1–15 ng/mL; Peprotech; Catalog #315-01A), or IFN-γ (40–60 ng/mL; Peprotech; Catalog #315-05), the combination of the three cytokines at given concentrations, tumor cell conditioned medium (GL261-CM/4T1-CM) or a combination of GL261-CM/4T1-CM plus the mixture of the three cytokines. 125I-uptake was performed as previously described43 and normalized to cell viability. Cell viability was measured using commercially available MTT reagent (Sigma-Aldrich) according to the manufacturer’s instructions. Absorbance was measured at 620 nm with a Sunrise Microplate Absorbance Reader (Tecan). Results were expressed as counts per minute normalized to cell viability (cpm/A260).
3D migration assay
The migratory capacity of IL-6-NIS-MSCs was investigated using the μ-slide Chemotaxis system3D (Ibidi, Martinsried, Germany) according to the manufacturer’s protocol. IL-6-NIS-MSCs (2 × 106 cells/mL) were seeded in collagen I (bovine, Gibco, Carlsbad, CA, USA; Catalog #A1064401) and a gradient was established between serum-free unconditioned medium and GL261-CM to monitor MSC migration. Time-lapse live-cell imaging was performed over 24 h on a Leica DMI6000B microscope equipped with a Leica DFC365 FX camera and Leica MM AF software (Leica Microsystems GmbH, Wetzlar, Germany), and pictures were taken in 15-min intervals. Randomly selected cells (n = 20) were manually tracked with the Manual tracking ImageJ (NIH, Bethesda, MD, USA) plug-in. The Chemotaxis and Migration Tool software (Ibidi) was used for the analysis of the migratory behavior by the center-of-mass localization, which is determined by the averaged point of all cell endpoints and forward migration index, which is a measure of the efficiency of forward migration of the MSCs in relation to the chemoattractant (GL261-CM).
Animals
Female (7-week-old) C57Bl/6 mice were purchased from Charles River (Sulzfeld, Germany). Mice were housed under specific pathogen-free conditions with ad libitum access to chow and water.
To establish orthotopic brain tumors, 8- to 9-week-old mice were anesthetized with ketamine/xylazine and GL261 (1 × 105 cells/1 μL) tumor cells were stereotactically inoculated 1 mm anterior, 1.5 mm to the right of the bregma, and 4 mm deep using a blunt Hamilton syringe (22G, Hamilton, Reno, NV, USA) as previously described.28
Ten days prior to the PET imaging and during 131I therapy experiments, mouse chow was changed to an iodide-deficient diet (ssniff Spezialdiäten GmbH, Soest, Germany) and mice were treated with 5 mg/mL levothyroxine (L-T4, Sigma-Aldrich) in 0.01% (v/v) bovine serum albumin (Sigma-Aldrich) supplemented to the drinking water to reduce the inherent tracer accumulation of the thyroid gland.
Animal experiments were performed in compliance with the German animal welfare laws and the approval of the local animal care committee of the Government of Upper Bavaria (Regierung von Oberbayern) under the license ROB-55.2-2532.Vet_02-17-133. According to the animal protocol, mice were euthanized at defined presymptomatic time points or at a defined humane endpoint during survival studies (significant weight loss; neurological symptoms; changes in drinking, eating, or cleaning behavior; signs of pain).
18F-TFB tracer synthesis
18F-TFB radiotracer for NIS-based PET imaging was produced in-house on a Modular-Lab Standard synthesis module (Eckert & Ziegler, Berlin, Germany) using the protocol previously described by Koshnevisan et al.83 In brief, [18F]F− was eluted from a Sep-Pak QMA Carbonate Plus Light cartridge (Waters, Milford, MA) using 500 μL saline (0.9% NaCl, B. Braun, Melsungen, Germany), dried twice at 95°C by addition of 550 μL acetonitrile (MeCN for DNA synthesis; Merck) and reaction was started by addition of boron trifluoride diethyl etherate (BF3OEt; 1 μL; Sigma-Aldrich) and 15-Crown-5 (24 mg; Sigma-Aldrich) in 1 mL acetonitrile (Merck). Fluorination reaction was carried out for 10 min at 80°C and subsequently the product solution was diluted with 10 mL of H2O and passed through a Sep-Pak Plus Alumina N cartridge (Waters) to remove free fluorine and a QMA Carbonate Plus Light cartridge in tandem. The resulting product was eluted from the QMA cartridge with 500 μL saline(B. Braun). Quality control was performed by radio thin-layer chromatography (radio-TLC) using a neutral alumina stationary phase (10 × 80 mm, Polygram ALOX N/UV254, Macherey-Nagel, Düren, Germany) with methanol as a mobile phase. Plates were scanned using a radio-TLC scanner (Mini-Scan, Bioscan Inc, Washington, DC, USA). This radio-TLC resulted in a separation of free [18F]F− that reacts with the aluminum (Rf = 0) and mobile 18F-TFB (Rf = 0.6).83 Reactions were started with 5–6 GBq and resulted in a decay-corrected radiochemical yield of 15% and radiochemical purity of >97.5%.
Radionuclide biodistribution studies in vivo in orthotopic GBM tumors using 18F-TFB-PET imaging
IL-6-NIS- or WT-MSCs (5 × 105 cells/injection) were systemically applied via the tail vein 2–2.5 weeks after intracranial (i.c.) tumor cell implantation and three-dimensional serial PET imaging was performed. MSCs were applied twice in 2-day intervals or in a shortened application regime using only one MSC injection, followed by 18F-TFB PET imaging 48 h after the last MSC administration. Mice received 10 MBq of in-house synthesized 18F-TFB intravenously and serial acquisition was conducted 1 h and 2 h post injection using a preclinical nanoScan PET/MRI system (Mediso Medical Imaging Solutions, Budapest, Hungary). PET images were reconstructed with Monte Carlo-based Tera-Tomo 3D PET reconstruction (Mediso). All PET datasets were analyzed using Nucline acquisition software (Mediso) and volumes of interests of the whole tumor (VOIs) were drawn and stated as fraction of the initially injected 18F-TFB dose (% ID/mL). Tumor volume was assessed by MRI and only mice with tumors >30 mm³ were taken into consideration for the quantification (average tumor volume = 57.6 ± 4.5 mm³ at the day of the imaging).
Mouse brain tissue preparation
Mice were transcardially perfused by manual infusion of PBS (Sigma-Aldrich) followed by 4% formaldehyde solution (Pharmacy, University Hospital LMU Munich, Munich, Germany). Brains were removed and incubated in 4% formaldehyde for 24 h at room temperature. Subsequently, brains were either transferred into 30% sucrose solution at 4°C until the brain sank to the bottom of the tube and embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA, USA) or directly paraffin embedded following formalin-fixation (FFPE). Horizontal sections were cut (10-μm-thick slices of cryopreserved tissue or 3-μm-thick slices of FFPE tissue) and subjected to immunostaining.
IL-6 immunohistochemistry
Immunohistochemical staining for mouse IL-6 of FFPE brain sections was performed on a Bond RXm autostainer (Leica Biosystems, Nussloch, Germany) using primary rabbit anti-mouse IL-6 (ab290735, abcam, Cambridge, UK; 1:50 dilution) antibody and polymer refined detection kit (Leica Biosystems). After deparaffinization, slides were pretreated with EDTA buffer (pH 6) for 30 min at 100°C for antigen retrieval. Primary antibody diluted in antibody diluent was incubated for 15 min after blocking of endogenous peroxidase for 5 min. Antibody binding was detected with diaminobenzidine (DAB) as chromogen for 10 min and hematoxylin for 5 min was used for counterstaining. Slides were digitalized using an automated slide scanner (Leica Biosystems; AT-2) and representative images were taken with Aperio Imagescope software (version 12.3; Leica Biosystems).
Immunofluorescence analysis of NIS
Immunofluorescence staining of frozen sections of brain tumors and sections of control organs (liver, lung, kidney, spleen) were stained for NIS using rabbit anti-NIS (EUD4101, Origene, Rockville, MD, USA; 1:1,000) primary antibody and an anti-rabbit Alexa Fluor 488-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA). Hoechst bisbenzimide (5 μg/mL) was used to counterstain nuclei and sections were mounted with fluorescence mounting medium (Dako, Hamburg, Germany).
Sections were scanned using a Panoramic MIDI II slide scanner and pictures were taken with CaseViewer (version 2.4, 3DHISTECH, Budapest, Hungary).
Immunofluorescence analysis of NIS and IL-6 co-expression
Immunofluorescence staining of frozen sections of brain tumors after imaging experiments were stained using the primary antibodies mouse anti-NIS (antibody MAB3564, clone FP5A, Merck Millipore; 1:650 dilution) and rabbit anti-IL-6 (ab290735, abcam, Cambridge, UK; 1:50 dilution), followed by the secondary antibodies anti-mouse Alexa Fluor 488-conjugated (Jackson ImmunoResearch, West Grove, PA, USA) and anti-rabbit Alexa Fluor Cy3-conjugated (Jackson ImmunoResearch, West Grove, PA, USA), respectively. Hoechst bisbenzimide (5 μg/mL) was used to counterstain nuclei and sections were mounted with fluorescence mounting medium (Dako, Hamburg, Germany).
Radioiodide therapy studies in vivo
Five to 6 days post i.c. tumor inoculation, an initial MRI scan was performed to assess tumor onset using a preclinical small-animal 7T-MRI scanner (Agilent & GE healthcare MR Discovery 901 with Bruker AVANCE III HD electronics) with a volume resonator and a dedicated two-channel brain coil (RAPID Biomedical, Rimpar, Germany). Mice were randomly assigned to the treatment groups as soon as a tumor volume of 0.6–2.1 mm³ was detected (day 0) to start with a therapeutic trial. An application schedule with a single i.v. MSC application (day 1) followed by i.p. injection of 131I (55.5 MBq; Rotop Pharmaka, Dresden, Germany) 48 h later was conducted and this therapy cycle was repeated after 2 days for a total of three times (MSCs on days 1/5/9 + 131I injection on days 3/7/11). Three treatment cohorts were investigated: IL-6-NIS-MSCs + 131I (referred to as therapy group) and the control groups IL-6-NIS-MSCs + saline (NaCl) or NaCl only (n = 7, respectively).
MRI screenings to assess tumor volume and to monitor tumor growth were performed twice a week. Tumor volume was determined as previously described.35
Indirect immunofluorescence analysis of CD31/Ki67
Following 131I therapy, immunofluorescence staining for Ki67 (ab16667, abcam, Cambridge, UK; 1:200) and CD31 (blood vessel density; BD Pharmingen; 1:100) was performed on frozen brain tumor sections as described previously.28 Six visual fields (×20 magnification) per tumor were examined for Ki67-positive cells (fraction of proliferating cells) and blood vessel density (CD31-positive area) using ImageJ (NIH).
Statistical analysis
Results are expressed as mean ± SEM. Statistical significance of in vitro experiments and in vivo imaging experiment was determined by two-tailed Student’s t test.
Statistical analysis of the in vivo therapy study was performed by one-way ANOVA for tumor volumes followed by post hoc Tukey’s honestly significant difference test. Survival plots were analyzed by log rank test. p values < 0.05 were considered as significant (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns, not significant).
Data and code availability
Data generated in this study are available upon request from the corresponding author.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) within the Collaborative Research Center SFB 824 to C.S. (project C8), F.S. (Z3), G.M. (B4), M.E. (B11), K.S. (Z2), and within the Priority Program SPP1629 to C.S. and P.J.N. as well as a grant from the Wilhelm Sander-Stiftung to C.S. (2021.094.1). R.E.K. and R.G. are supported by the DFG (GL691/2; SFB824), the “Wilhelm Sander-Stiftung,” the “Anni Hofmann Stiftung,” and the “Verein zur Förderung von Wissenschaft und Forschung an der Medizinischen Fakultät der LMU München” (WiFoMed). R.G. acknowledges funding by DFG grant INST 409/223-1 FUGG.
We are grateful to Sybille Reder, Markus Mittelhäuser, Sandra Sühnel, and Nadine Setzer (Department of Nuclear Medicine, School of Medicine, Klinikum Rechts der Isar, Technical University of Munich) for their valuable help in performing the animal studies. We appreciate the help from Olga Seelbach, Ulrike Mühlthaler, Annett Hering, and Marion Mielke (CEP, Comparative Experimental Pathology, Institute of Pathology, School of Medicine, Technical University of Munich) with immunohistochemical staining. Furthermore, we thank Prof. Dr. Julia Mayerle, Dr. Ivonne Regel, and Dr. Ujjwal Mahajan (Department of Internal Medicine II, LMU University Hospital, LMU Munich, Munich, Germany) for sharing their lab equipment.
This work was performed as partial fulfillment of the doctoral thesis of C.K. at the faculty for Chemistry and Pharmacy of the LMU Munich.
Author contributions
Conception and design: C.K., P.J.N., C.S.; Development of methodology: C.K., K.Shehzad, V.M., R.E.K., R.G., G.M., K.Steiger, J.R., P.J.N, C.S.; Acquisition of data (provided animals, acquired and managed patients, provided facilities etc.): C.K., K.Shehzad, K.Steiger, R.S., F.S., W.A.W., C.S.; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C.K., K.Steiger, M.E., P.J.N., C.S.; Writing, review, and/or revision of the manuscript: C.K., V.M., K.Steiger, G.M., F.S., R.E.K., E.W., R.G., W.A.W., P.J.N., C.S. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): G.M., M.E., F.S., W.A.W., R.G., P.J.N., C.S.; Study supervision: E.W., P.J.N., C.S. All authors have read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2023.08.004.
Supplemental information
References
- 1.Tan A.C., Ashley D.M., López G.Y., Malinzak M., Friedman H.S., Khasraw M. Management of glioblastoma: State of the art and future directions. CA. Cancer J. Clin. 2020;70:299–312. doi: 10.3322/caac.21613. [DOI] [PubMed] [Google Scholar]
- 2.Aldape K., Brindle K.M., Chesler L., Chopra R., Gajjar A., Gilbert M.R., Gottardo N., Gutmann D.H., Hargrave D., Holland E.C., et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019;16:509–520. doi: 10.1038/s41571-019-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J.B., Belanger K., Brandes A.A., Marosi C., Bogdahn U., et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Weller M., van den Bent M., Preusser M., Le Rhun E., Tonn J.C., Minniti G., Bendszus M., Balana C., Chinot O., Dirven L., et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021;18:170–186. doi: 10.1038/s41571-020-00447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiyokawa J., Kawamura Y., Ghouse S.M., Acar S., Barçın E., Martínez-Quintanilla J., Martuza R.L., Alemany R., Rabkin S.D., Shah K., Wakimoto H. Modification of Extracellular Matrix Enhances Oncolytic Adenovirus Immunotherapy in Glioblastoma. Clin. Cancer Res. 2021;27:889–902. doi: 10.1158/1078-0432.CCR-20-2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West A.J., Tsui V., Stylli S.S., Nguyen H.P.T., Morokoff A.P., Kaye A.H., Luwor R.B. The role of interleukin-6-STAT3 signalling in glioblastoma. Oncol. Lett. 2018;16:4095–4104. doi: 10.3892/ol.2018.9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olar A., Aldape K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014;232:165–177. doi: 10.1002/path.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Meir E.G. Cytokines and tumors of the central nervous system. Glia. 1995;15:264–288. doi: 10.1002/glia.440150308. [DOI] [PubMed] [Google Scholar]
- 9.Faggioli L., Costanzo C., Donadelli M., Palmieri M. Activation of the Interleukin-6 promoter by a dominant negative mutant of c-Jun. Biochim. Biophys. Acta. 2004;1692:17–24. doi: 10.1016/j.bbamcr.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Luo Y., Zheng S.G. Hall of Fame among Pro-inflammatory Cytokines: Interleukin-6 Gene and Its Transcriptional Regulation Mechanisms. Front. Immunol. 2016;7:604. doi: 10.3389/fimmu.2016.00604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasaki A., Ishiuchi S., Kanda T., Hasegawa M., Nakazato Y. Analysis of interleukin-6 gene expression in primary human gliomas, glioblastoma xenografts, and glioblastoma cell lines. Brain Tumor Pathol. 2001;18:13–21. doi: 10.1007/BF02478920. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q., Li G., Li R., Shen J., He Q., Deng L., Zhang C., Zhang J. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J. Neurooncol. 2010;100:165–176. doi: 10.1007/s11060-010-0158-0. [DOI] [PubMed] [Google Scholar]
- 13.Tchirkov A., Khalil T., Chautard E., Mokhtari K., Véronèse L., Irthum B., Vago P., Kémény J.L., Verrelle P. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br. J. Cancer. 2007;96:474–476. doi: 10.1038/sj.bjc.6603586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hori T., Sasayama T., Tanaka K., Koma Y.I., Nishihara M., Tanaka H., Nakamizo S., Nagashima H., Maeyama M., Fujita Y., et al. Tumor-associated macrophage related interleukin-6 in cerebrospinal fluid as a prognostic marker for glioblastoma. J. Clin. Neurosci. 2019;68:281–289. doi: 10.1016/j.jocn.2019.07.020. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q., He Z., Huang M., Liu T., Wang Y., Xu H., Duan H., Ma P., Zhang L., Zamvil S.S., et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat. Commun. 2018;9:559. doi: 10.1038/s41467-018-03050-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al-Kharboosh R., ReFaey K., Lara-Velazquez M., Grewal S.S., Imitola J., Quiñones-Hinojosa A. Inflammatory Mediators in Glioma Microenvironment Play a Dual Role in Gliomagenesis and Mesenchymal Stem Cell Homing: Implication for Cellular Therapy. Mayo Clin. Proc. Innov. Qual. Outcomes. 2020;4:443–459. doi: 10.1016/j.mayocpiqo.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spaeth E., Klopp A., Dembinski J., Andreeff M., Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–738. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 18.Hagenhoff A., Bruns C.J., Zhao Y., von Lüttichau I., Niess H., Spitzweg C., Nelson P.J. Harnessing mesenchymal stem cell homing as an anticancer therapy. Expert Opin. Biol. Ther. 2016;16:1079–1092. doi: 10.1080/14712598.2016.1196179. [DOI] [PubMed] [Google Scholar]
- 19.Terstappen G.C., Meyer A.H., Bell R.D., Zhang W. Strategies for delivering therapeutics across the blood-brain barrier. Nat. Rev. Drug Discov. 2021;20:362–383. doi: 10.1038/s41573-021-00139-y. [DOI] [PubMed] [Google Scholar]
- 20.Sarkaria J.N., Hu L.S., Parney I.F., Pafundi D.H., Brinkmann D.H., Laack N.N., Giannini C., Burns T.C., Kizilbash S.H., Laramy J.K., et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro. Oncol. 2018;20:184–191. doi: 10.1093/neuonc/nox175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bexell D., Gunnarsson S., Tormin A., Darabi A., Gisselsson D., Roybon L., Scheding S., Bengzon J. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol. Ther. 2009;17:183–190. doi: 10.1038/mt.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bexell D., Gunnarsson S., Svensson A., Tormin A., Henriques-Oliveira C., Siesjö P., Paul G., Salford L.G., Scheding S., Bengzon J. Rat multipotent mesenchymal stromal cells lack long-distance tropism to 3 different rat glioma models. Neurosurgery. 2012;70:731–739. doi: 10.1227/NEU.0b013e318232dedd. [DOI] [PubMed] [Google Scholar]
- 23.Shi S., Zhang M., Guo R., Miao Y., Li B. Bone Marrow-Derived Mesenchymal Stem Cell-Mediated Dual-Gene Therapy for Glioblastoma. Hum. Gene Ther. 2019;30:106–117. doi: 10.1089/hum.2018.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura K., Ito Y., Kawano Y., Kurozumi K., Kobune M., Tsuda H., Bizen A., Honmou O., Niitsu Y., Hamada H. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11:1155–1164. doi: 10.1038/sj.gt.3302276. [DOI] [PubMed] [Google Scholar]
- 25.Aboody K.S., Brown A., Rainov N.G., Bower K.A., Liu S., Yang W., Small J.E., Herrlinger U., Ourednik V., Black P.M., et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamizo A., Marini F., Amano T., Khan A., Studeny M., Gumin J., Chen J., Hentschel S., Vecil G., Dembinski J., et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 27.Liu L., Eckert M.A., Riazifar H., Kang D.K., Agalliu D., Zhao W. From blood to the brain: can systemically transplanted mesenchymal stem cells cross the blood-brain barrier? Stem Cells Int. 2013;2013 doi: 10.1155/2013/435093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitzberger C., Spellerberg R., Han Y., Schmohl K.A., Stauss C., Zach C., Kälin R.E., Multhoff G., Eiber M., Schilling F., et al. Mesenchymal Stem Cell-mediated Image-guided Sodium Iodide Symporter (NIS) Gene Therapy Improves Survival of Glioblastoma-bearing Mice. Clin. Cancer Res. 2023;29:930–942. doi: 10.1158/1078-0432.Ccr-22-1433. [DOI] [PubMed] [Google Scholar]
- 29.Conrad C., Gupta R., Mohan H., Niess H., Bruns C.J., Kopp R., von Luettichau I., Guba M., Heeschen C., Jauch K.W., et al. Genetically engineered stem cells for therapeutic gene delivery. Curr. Gene Ther. 2007;7:249–260. doi: 10.2174/156652307781369119. [DOI] [PubMed] [Google Scholar]
- 30.Spitzweg C., Nelson P.J., Wagner E., Bartenstein P., Weber W.A., Schwaiger M., Morris J.C. The sodium iodide symporter (NIS): novel applications for radionuclide imaging and treatment. Endocr. Relat. Cancer. 2021;28:T193–T213. doi: 10.1530/ERC-21-0177. [DOI] [PubMed] [Google Scholar]
- 31.Penheiter A.R., Russell S.J., Carlson S.K. The sodium iodide symporter (NIS) as an imaging reporter for gene, viral, and cell-based therapies. Curr. Gene Ther. 2012;12:33–47. doi: 10.2174/156652312799789235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hingorani M., Spitzweg C., Vassaux G., Newbold K., Melcher A., Pandha H., Vile R., Harrington K. The biology of the sodium iodide symporter and its potential for targeted gene delivery. Curr. Cancer Drug Targets. 2010;10:242–267. doi: 10.2174/156800910791054194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hjiyiannakis P., Jefferies S., Harmer C.L. Brain metastases in patients with differentiated thyroid carcinoma. Clin. Oncol. 1996;8:327–330. doi: 10.1016/s0936-6555(05)80722-8. [DOI] [PubMed] [Google Scholar]
- 34.Kitzberger C., Spellerberg R., Morath V., Schwenk N., Schmohl K.A., Schug C., Urnauer S., Tutter M., Eiber M., Schilling F., et al. The sodium iodide symporter (NIS) as theranostic gene: its emerging role in new imaging modalities and non-viral gene therapy. EJNMMI Res. 2022;12:25. doi: 10.1186/s13550-022-00888-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spellerberg R., Benli-Hoppe T., Kitzberger C., Berger S., Schmohl K.A., Schwenk N., Yen H.-Y., Zach C., Schilling F., Weber W.A., et al. Selective sodium iodide symporter (NIS) gene therapy of glioblastoma mediated by EGFR-targeted lipopolyplexes. Mol. Ther. Oncolytics. 2021;23:432–446. doi: 10.1016/j.omto.2021.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravera S., Reyna-Neyra A., Ferrandino G., Amzel L.M., Carrasco N. The Sodium/Iodide Symporter (NIS): Molecular Physiology and Preclinical and Clinical Applications. Annu. Rev. Physiol. 2017;79:261–289. doi: 10.1146/annurev-physiol-022516-034125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knoop K., Kolokythas M., Klutz K., Willhauck M.J., Wunderlich N., Draganovici D., Zach C., Gildehaus F.J., Böning G., Göke B., et al. Image-guided, tumor stroma-targeted 131I therapy of hepatocellular cancer after systemic mesenchymal stem cell-mediated NIS gene delivery. Mol. Ther. 2011;19:1704–1713. doi: 10.1038/mt.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schug C., Gupta A., Urnauer S., Steiger K., Cheung P.F.Y., Neander C., Savvatakis K., Schmohl K.A., Trajkovic-Arsic M., Schwenk N., et al. A Novel Approach for Image-Guided (131)I Therapy of Pancreatic Ductal Adenocarcinoma Using Mesenchymal Stem Cell-Mediated NIS Gene Delivery. Mol. Cancer Res. 2019;17:310–320. doi: 10.1158/1541-7786.MCR-18-0185. [DOI] [PubMed] [Google Scholar]
- 39.Knoop K., Schwenk N., Dolp P., Willhauck M.J., Zischek C., Zach C., Hacker M., Göke B., Wagner E., Nelson P.J., Spitzweg C. Stromal targeting of sodium iodide symporter using mesenchymal stem cells allows enhanced imaging and therapy of hepatocellular carcinoma. Hum. Gene Ther. 2013;24:306–316. doi: 10.1089/hum.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knoop K., Schwenk N., Schmohl K., Müller A., Zach C., Cyran C., Carlsen J., Böning G., Bartenstein P., Göke B., et al. Mesenchymal stem cell-mediated, tumor stroma-targeted radioiodine therapy of metastatic colon cancer using the sodium iodide symporter as theranostic gene. J. Nucl. Med. 2015;56:600–606. doi: 10.2967/jnumed.114.146662. [DOI] [PubMed] [Google Scholar]
- 41.Müller A.M., Schmohl K.A., Knoop K., Schug C., Urnauer S., Hagenhoff A., Clevert D.A., Ingrisch M., Niess H., Carlsen J., et al. Hypoxia-targeted 131I therapy of hepatocellular cancer after systemic mesenchymal stem cell-mediated sodium iodide symporter gene delivery. Oncotarget. 2016;7:54795–54810. doi: 10.18632/oncotarget.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schug C., Sievert W., Urnauer S., Müller A.M., Schmohl K.A., Wechselberger A., Schwenk N., Lauber K., Schwaiger M., Multhoff G., et al. External Beam Radiation Therapy Enhances Mesenchymal Stem Cell-Mediated Sodium-Iodide Symporter Gene Delivery. Hum. Gene Ther. 2018;29:1287–1300. doi: 10.1089/hum.2018.025. [DOI] [PubMed] [Google Scholar]
- 43.Tutter M., Schug C., Schmohl K.A., Urnauer S., Schwenk N., Petrini M., Lokerse W.J.M., Zach C., Ziegler S., Bartenstein P., et al. Effective control of tumor growth through spatial and temporal control of theranostic sodium iodide symporter (NIS) gene expression using a heat-inducible gene promoter in engineered mesenchymal stem cells. Theranostics. 2020;10:4490–4506. doi: 10.7150/thno.41489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schug C., Urnauer S., Jaeckel C., Schmohl K.A., Tutter M., Steiger K., Schwenk N., Schwaiger M., Wagner E., Nelson P.J., Spitzweg C. TGFB1-driven mesenchymal stem cell-mediated NIS gene transfer. Endocr. Relat. Cancer. 2019;26:89–101. doi: 10.1530/ERC-18-0173. [DOI] [PubMed] [Google Scholar]
- 45.Schug C., Kitzberger C., Sievert W., Spellerberg R., Tutter M., Schmohl K.A., Eberlein B., Biedermann T., Steiger K., Zach C., et al. Radiation-Induced Amplification of TGFB1-Induced Mesenchymal Stem Cell-Mediated Sodium Iodide Symporter (NIS) Gene (131)I Therapy. Clin. Cancer Res. 2019;25:5997–6008. doi: 10.1158/1078-0432.CCR-18-4092. [DOI] [PubMed] [Google Scholar]
- 46.Tutter M., Schug C., Schmohl K.A., Urnauer S., Kitzberger C., Schwenk N., Petrini M., Zach C., Ziegler S., Bartenstein P., et al. Regional hyperthermia enhances mesenchymal stem cell recruitment to tumor stroma: implications for mesenchymal stem cell-based tumor therapy. Mol. Ther. 2021;29:788–803. doi: 10.1016/j.ymthe.2020.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hertz S., Roberts A. Radioactive iodine in the study of thyroid physiology; the use of radioactive iodine therapy in hyperthyroidism. J. Am. Med. Assoc. 1946;131:81–86. doi: 10.1001/jama.1946.02870190005002. [DOI] [PubMed] [Google Scholar]
- 48.Riesco-Eizaguirre G., Santisteban P., De la Vieja A. The complex regulation of NIS expression and activity in thyroid and extrathyroidal tissues. Endocr. Relat. Cancer. 2021;28:T141–T165. doi: 10.1530/ERC-21-0217. [DOI] [PubMed] [Google Scholar]
- 49.Zischek C., Niess H., Ischenko I., Conrad C., Huss R., Jauch K.W., Nelson P.J., Bruns C. Targeting tumor stroma using engineered mesenchymal stem cells reduces the growth of pancreatic carcinoma. Ann. Surg. 2009;250:747–753. doi: 10.1097/SLA.0b013e3181bd62d0. [DOI] [PubMed] [Google Scholar]
- 50.Fischer U.M., Harting M.T., Jimenez F., Monzon-Posadas W.O., Xue H., Savitz S.I., Laine G.A., Cox C.S., Jr. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18:683–692. doi: 10.1089/scd.2008.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grisendi G., Spano C., Rossignoli F., D Souza N., Golinelli G., Fiori A., Horwitz E.M., Guarneri V., Piacentini F., Paolucci P., Dominici M. Tumor Stroma Manipulation By MSC. Curr. Drug Targets. 2016;17:1111–1126. doi: 10.2174/1389450117666160307143226. [DOI] [PubMed] [Google Scholar]
- 52.Niess H., Bao Q., Conrad C., Zischek C., Notohamiprodjo M., Schwab F., Schwarz B., Huss R., Jauch K.W., Nelson P.J., Bruns C.J. Selective targeting of genetically engineered mesenchymal stem cells to tumor stroma microenvironments using tissue-specific suicide gene expression suppresses growth of hepatocellular carcinoma. Ann. Surg. 2011;254:767–774. doi: 10.1097/SLA.0b013e3182368c4f. discussion 774-765. [DOI] [PubMed] [Google Scholar]
- 53.Yeung Y.T., McDonald K.L., Grewal T., Munoz L. Interleukins in glioblastoma pathophysiology: implications for therapy. Br. J. Pharmacol. 2013;168:591–606. doi: 10.1111/bph.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeo E.C.F., Brown M.P., Gargett T., Ebert L.M. The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy. Cells. 2021;10 doi: 10.3390/cells10030607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang C.Y., Li M.C., Liao S.L., Huang Y.L., Shen C.C., Pan H.C. Prognostic and clinical implication of IL-6 expression in glioblastoma multiforme. J. Clin. Neurosci. 2005;12:930–933. doi: 10.1016/j.jocn.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 56.Wang H., Lathia J.D., Wu Q., Wang J., Li Z., Heddleston J.M., Eyler C.E., Elderbroom J., Gallagher J., Schuschu J., et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009;27:2393–2404. doi: 10.1002/stem.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weissenberger J., Loeffler S., Kappeler A., Kopf M., Lukes A., Afanasieva T.A., Aguzzi A., Weis J. IL-6 is required for glioma development in a mouse model. Oncogene. 2004;23:3308–3316. doi: 10.1038/sj.onc.1207455. [DOI] [PubMed] [Google Scholar]
- 58.Lamano J.B., Lamano J.B., Li Y.D., DiDomenico J.D., Choy W., Veliceasa D., Oyon D.E., Fakurnejad S., Ampie L., Kesavabhotla K., et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin. Cancer Res. 2019;25:3643–3657. doi: 10.1158/1078-0432.CCR-18-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dubost J.-J., Rolhion C., Tchirkov A., Bertrand S., Chassagne J., Dosgilbert A., Verrelle P. Interleukin-6-producing Cells in a Human Glioblastoma Cell Line are not Affected by Ionizing Radiation. J. Neurooncol. 2002;56:29–34. doi: 10.1023/A:1014467804488. [DOI] [PubMed] [Google Scholar]
- 60.Pasi F., Facoetti A., Nano R. IL-8 and IL-6 bystander signalling in human glioblastoma cells exposed to gamma radiation. Anticancer Res. 2010;30:2769–2772. https://pubmed.ncbi.nlm.nih.gov/20683011/ [PubMed] [Google Scholar]
- 61.Chen H.Y., Lin L.T., Wang M.L., Lee S.H., Tsai M.L., Tsai C.C., Liu W.H., Chen T.C., Yang Y.P., Lee Y.Y., et al. Musashi-1 regulates AKT-derived IL-6 autocrinal/paracrinal malignancy and chemoresistance in glioblastoma. Oncotarget. 2016;7:42485–42501. doi: 10.18632/oncotarget.9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding D.C., Liu H.W., Chu T.Y. Interleukin-6 from Ovarian Mesenchymal Stem Cells Promotes Proliferation, Sphere and Colony Formation and Tumorigenesis of an Ovarian Cancer Cell Line SKOV3. J. Cancer. 2016;7:1815–1823. doi: 10.7150/jca.16116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng J., Chen S., Li C., Ye Z., Lin B., Liang Y., Wang B., Ma Y., Chai X., Zhang X., et al. Mesenchymal stem/stromal cells-derived IL-6 promotes nasopharyngeal carcinoma growth and resistance to cisplatin via upregulating CD73 expression. J. Cancer. 2020;11:2068–2079. doi: 10.7150/jca.37932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karnoub A.E., Dash A.B., Vo A.P., Sullivan A., Brooks M.W., Bell G.W., Richardson A.L., Polyak K., Tubo R., Weinberg R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 65.Tumangelova-Yuzeir K., Naydenov E., Ivanova-Todorova E., Krasimirova E., Vasilev G., Nachev S., Kyurkchiev D. Mesenchymal Stem Cells Derived and Cultured from Glioblastoma Multiforme Increase Tregs, Downregulate Th17, and Induce the Tolerogenic Phenotype of Monocyte-Derived Cells. Stem Cells Int. 2019;2019 doi: 10.1155/2019/6904638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang W., Chen X., Zhang S., Fang J., Chen M., Xu Y., Chen X. Mesenchymal stem cells as a double-edged sword in tumor growth: focusing on MSC-derived cytokines. Cell. Mol. Biol. Lett. 2021;26:3. doi: 10.1186/s11658-020-00246-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fisher D.T., Appenheimer M.M., Evans S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014;26:38–47. doi: 10.1016/j.smim.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu Q., Yu S., Li A., Xu H., Han X., Wu K. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumour Biol. 2017;39 doi: 10.1177/1010428317712445. [DOI] [PubMed] [Google Scholar]
- 69.Wang Q.-W., Lin W.-W., Zhu Y.-J. Comprehensive analysis of a TNF family based-signature in diffuse gliomas with regard to prognosis and immune significance. Cell Commun. Signal. 2022;20:6. doi: 10.1186/s12964-021-00814-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanchez V.E., Lynes J.P., Walbridge S., Wang X., Edwards N.A., Nwankwo A.K., Sur H.P., Dominah G.A., Obungu A., Adamstein N., et al. GL261 luciferase-expressing cells elicit an anti-tumor immune response: an evaluation of murine glioma models. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-67411-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanabe K., Matsushima-Nishiwaki R., Yamaguchi S., Iida H., Dohi S., Kozawa O. Mechanisms of tumor necrosis factor-alpha-induced interleukin-6 synthesis in glioma cells. J. Neuroinflammation. 2010;7:16. doi: 10.1186/1742-2094-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spooren A., Mestdagh P., Rondou P., Kolmus K., Haegeman G., Gerlo S. IL-1beta potently stabilizes IL-6 mRNA in human astrocytes. Biochem. Pharmacol. 2011;81:1004–1015. doi: 10.1016/j.bcp.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 73.Yeung Y.T., Bryce N.S., Adams S., Braidy N., Konayagi M., McDonald K.L., Teo C., Guillemin G.J., Grewal T., Munoz L. p38 MAPK inhibitors attenuate pro-inflammatory cytokine production and the invasiveness of human U251 glioblastoma cells. J. Neurooncol. 2012;109:35–44. doi: 10.1007/s11060-012-0875-7. [DOI] [PubMed] [Google Scholar]
- 74.Schmohl K.A., Müller A.M., Wechselberger A., Rühland S., Salb N., Schwenk N., Heuer H., Carlsen J., Göke B., Nelson P.J., Spitzweg C. Thyroid hormones and tetrac: new regulators of tumour stroma formation via integrin alphavbeta3. Endocr. Relat. Cancer. 2015;22:941–952. doi: 10.1530/ERC-15-0245. [DOI] [PubMed] [Google Scholar]
- 75.Dvorak H.F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986;315:1650–1659. doi: 10.1056/nejm198612253152606. [DOI] [PubMed] [Google Scholar]
- 76.Nowak B., Rogujski P., Janowski M., Lukomska B., Andrzejewska A. Mesenchymal stem cells in glioblastoma therapy and progression: How one cell does it all. Biochim. Biophys. Acta Rev. Cancer. 2021;1876 doi: 10.1016/j.bbcan.2021.188582. [DOI] [PubMed] [Google Scholar]
- 77.Brandt L.J.B., Barnkob M.B., Michaels Y.S., Heiselberg J., Barington T. Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front. Immunol. 2020;11:326. doi: 10.3389/fimmu.2020.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brem S., Cotran R., Folkman J. Tumor Angiogenesis: A Quantitative Method for Histologic Grading2. JNCI. J. Natl. Cancer Inst. 1972;48:347–356. doi: 10.1093/jnci/48.2.347. [DOI] [PubMed] [Google Scholar]
- 79.Mastrella G., Hou M., Li M., Stoecklein V.M., Zdouc N., Volmar M.N.M., Miletic H., Reinhard S., Herold-Mende C.C., Kleber S., et al. Targeting APLN/APLNR Improves Antiangiogenic Efficiency and Blunts Proinvasive Side Effects of VEGFA/VEGFR2 Blockade in Glioblastoma. Cancer Res. 2019;79:2298–2313. doi: 10.1158/0008-5472.CAN-18-0881. [DOI] [PubMed] [Google Scholar]
- 80.Saidi A., Hagedorn M., Allain N., Verpelli C., Sala C., Bello L., Bikfalvi A., Javerzat S. Combined targeting of interleukin-6 and vascular endothelial growth factor potently inhibits glioma growth and invasiveness. Int. J. Cancer. 2009;125:1054–1064. doi: 10.1002/ijc.24380. [DOI] [PubMed] [Google Scholar]
- 81.McKelvey K.J., Hudson A.L., Prasanna Kumar R., Wilmott J.S., Attrill G.H., Long G.V., Scolyer R.A., Clarke S.J., Wheeler H.R., Diakos C.I., Howell V.M. Temporal and spatial modulation of the tumor and systemic immune response in the murine Gl261 glioma model. PLoS One. 2020;15 doi: 10.1371/journal.pone.0226444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jäckel C., Nogueira M.S., Ehni N., Kraus C., Ranke J., Dohmann M., Noessner E., Nelson P.J. A vector platform for the rapid and efficient engineering of stable complex transgenes. Sci. Rep. 2016;6 doi: 10.1038/srep34365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khoshnevisan A., Jauregui-Osoro M., Shaw K., Torres J.B., Young J.D., Ramakrishnan N.K., Jackson A., Smith G.E., Gee A.D., Blower P.J. [(18)F]tetrafluoroborate as a PET tracer for the sodium/iodide symporter: the importance of specific activity. EJNMMI Res. 2016;6:34. doi: 10.1186/s13550-016-0188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are available upon request from the corresponding author.