

Abstract
Reservoirs of HIV maintained in anatomic compartments during antiretroviral therapy prevent HIV eradication. However, mechanisms driving their persistence and interventions to control them remain elusive. Here we report the presence of an inducible HIV reservoir within antigen-specific CD4+T cells in the central nervous system of a 59-year-old male with progressive multifocal leukoencephalopathy immune reconstitution inflammatory syndrome (PML-IRIS). HIV production during PML-IRIS was suppressed by modulating inflammation with corticosteroids; selection of HIV drug resistance caused subsequent breakthrough viremia. Therefore, inflammation can influence the composition, distribution and induction of HIV reservoirs, warranting it as a key consideration for developing effective HIV remission strategies.
HIV reservoir persistence on antiretroviral therapy (ART) is the principal obstacle to HIV eradication. HIV-infected cells are widely anatomically distributed; however, mechanisms driving their distribution and dynamics in vivo remain poorly understood1,2. HIV-infected cells in the central nervous system (CNS)1,2 may represent a critical reservoir during ART, given that persistent and episodic HIV release on effective ART has been observed3,4. Understanding the dynamics of immunologic and virologic mechanisms driving HIV persistence in the CNS and other tissue reservoirs is a key consideration in developing scalable strategies for HIV eradication5. In this case study, we demonstrate that immune recovery, stimulation and trafficking of antigen-specific CD4+T cells drove HIV persistence, with gradual replacement of HIV reservoirs with ART-resistant proviruses. Furthermore, HIV production from viral reservoirs was directly modulated by corticosteroids, implicating inflammation in HIV reservoir dynamics.
A 59-year-old man was diagnosed with HIV-associated acquired immunodeficiency syndrome after presenting with Pneumocystis jirovecii pneumonia (PJP), cytomegalovirus (CMV) colitis, CD4+T cell count of 16 cells per microliter and plasma HIV viral load (HIV VL) of 2,109,306 copies per milliliter. He was referred to the National Institutes of Health (NIH) Clinical Center, having met enrollment criteria for the ART-naive arm of the protocol NCT02147405 (≥18-year-old people living with HIV, with CD4 counts ≤100 cells per microliter and ART-naive), a study designed to identify predictors and optimal management of immune reconstitution inflammatory syndrome (IRIS). Baseline brain magnetic resonance imaging (MRI) noted subtle right thalamic and periventricular hyperintensities without parenchymal or meningeal enhancement. Cerebrospinal fluid (CSF) had 1,200 copies per milliliter of HIV RNA, a normal white cell count and undetectable cryptococcal antigen, CMV, varicella zoster virus, herpes simplex virus-1 and John Cunningham virus ( JCV) DNA.
Before ART initiation, HIV drug resistance mutations (DRMs) were not detected in RNA extracted from plasma or lymph nodes via next-generation sequencing (NGS) (Supplementary Fig. 1). PJP and CMV colitis were treated, and ART (elvitegravir/cobicistat/tenofovir-alafenamide/emtricitabine (EVG/COBI/TAF/FTC)) was initiated, resulting in typical multiphasic HIV decay kinetics (Fig. 1a and Supplementary Fig. 2). At 15 weeks after ART initiation, both plasma and CSF HIV VLs were <40 copies per milliliter, and CD4+T cells increased to 307 cells per microliter (Fig. 1a); however, new left-sided ataxia, weakness and dysmetria appeared. MRI and positron emission tomography/computed tomography (PET/CT) findings were consistent with unmasking PML-IRIS: multiple punctate foci of contrast enhancement tracked along the cortical-thalamic-cerebellar pathways involving the right centrum semiovale/subcortical, right thalamus, midbrain and left cerebellar white matter with increased 18F-fluorodeoxyglucose (FDG) uptake (Fig. 1b–d). JCV DNA was detected in CSF and urine by quantitative polymerase chain reaction (qPCR) assay (limit of detection: 1,000 genome equivalents per milliliter), and plasma HIV VLs remained suppressed (<40 copies per milliliter). Prednisone was initiated; maraviroc and mirtazapine were added to modulate CCR5-mediated leukocyte trafficking and JCV replication, respectively6,7. By 23 weeks, there were significantly fewer parenchymal contrast-enhancing MRI abnormalities. At 34 weeks, few punctate foci of MRI contrast enhancement reappeared in the same locations (Supplementary Fig. 3a–c); however, by 40 weeks, all neurological symptoms resolved, and slow prednisone taper was completed.
Fig. 1 |. Clinical history.
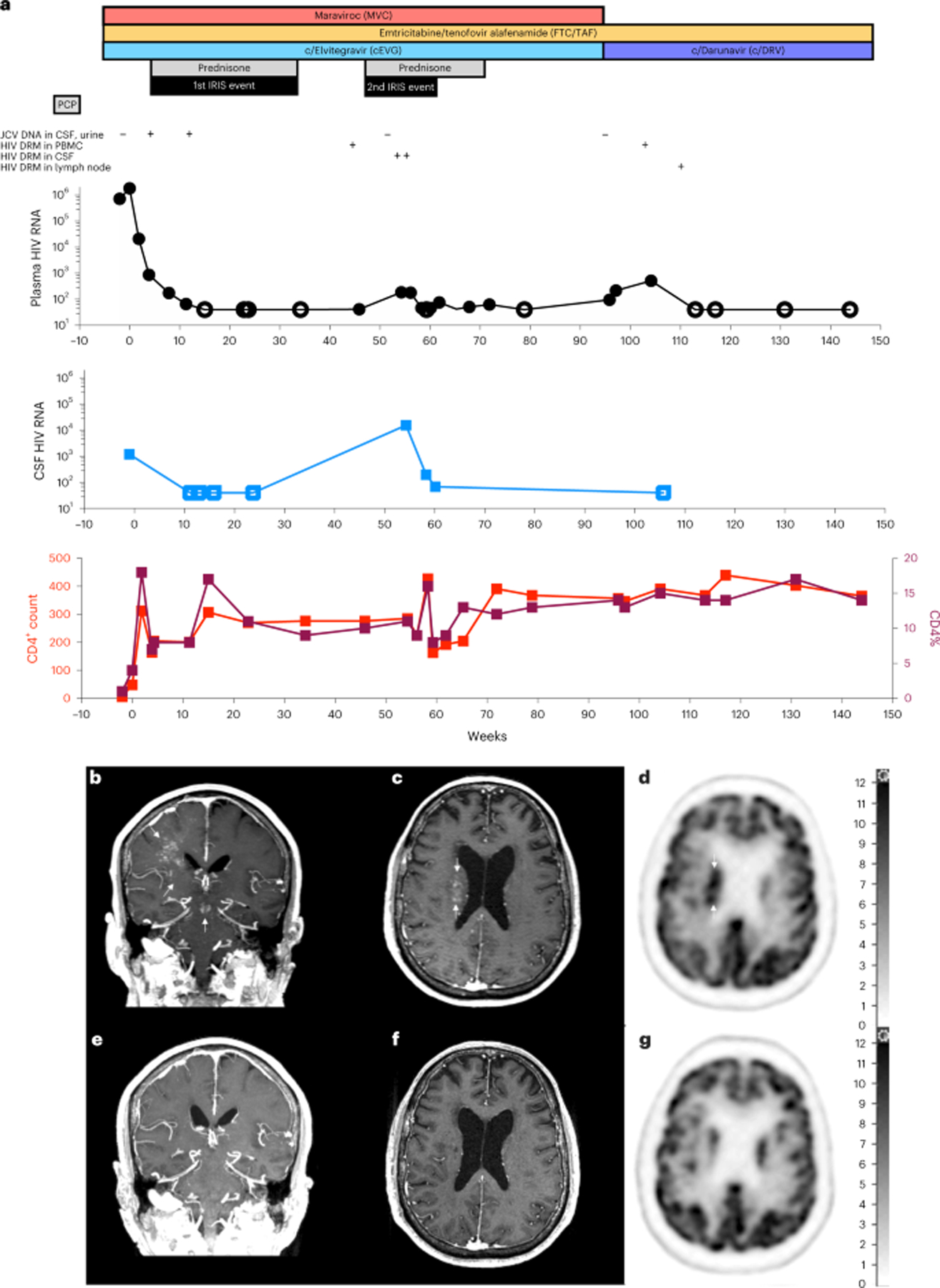
a, Therapies, JCV DNA, HIV VLs, DRM emergence and CD4+T cell counts. b–g, Longitudinal brain imaging by post-contrast T1-weighted MRI at 16 weeks (b,c) with FDG/PET at 12 weeks (d), and post-contrast T1-weighted MRI at 78 weeks (e,f) with FDG/PET scan, at 96 weeks (g). Right-sided scale bar: standardized FDG uptake values.
At 54 weeks, new left hand tremor developed with faint MRI contrast enhancement in previously identified foci (Supplementary Fig. 3d), and new CSF lymphocytic pleocytosis (64 cells per microliter, 92% lymphocytes) with elevated HIV VLs (15,493 copies per milliliter of CSF and 185 copies per milliliter of plasma) were documented. JCV DNA was undetectable in CSF; however, given the limitations of the JCV qPCR assay and negative additional infectious workup, relapsed PML-IRIS was diagnosed. Oral prednisone (60 mg d−1) was re-initiated, with clinical and radiological improvement. Within 4 weeks, there were brisk declines in CSF and plasma HIV VLs (68 copies per milliliter and <40 copies per milliliter, respectively); the >200-fold decrease in CSF HIV VL occurred with anti-inflammatory corticosteroid therapy alone, without ART modifications. Complete resolution of neurologic symptoms accompanied disappearances of MRI contrast-enhancing foci (Fig. 1e,f) and normalization of FDG uptake on brain PET/CT by 78 weeks and 96 weeks, respectively (Fig. 1g).
To identify immunological and virological characteristics of PML-IRIS, we retrospectively analyzed CSF and blood samples for JCV-specific T cell responses and HIV population dynamics. Antigen stimulation studies of peripheral blood mononuclear cells (PBMCs) and CSF mononuclear cells revealed robust JCV-specific CD4+T cell responses (Fig. 2a), exceeding responses induced by Epstein–Barr virus (EBV) and CMV antigens (Supplementary Fig. 4). Furthermore, phylogenetic and population subdivision analyses revealed that HIV populations were diverse (Supplementary Table 1), and HIV DNA populations circulating from 24 weeks to 46 weeks were genetically distinct from HIV RNA populations in pre-ART viremia (probability of panmixia P < 10−5 and Slatkin–Maddison P < 0.01; Supplementary Table 1). Therefore, pre-ART HIV DNA populations derived from PBMCs underwent marked population shifts by the time the first PML-IRIS event occurred.
Fig. 2 |. Immunological and virological characteristics of PML-IRIS.
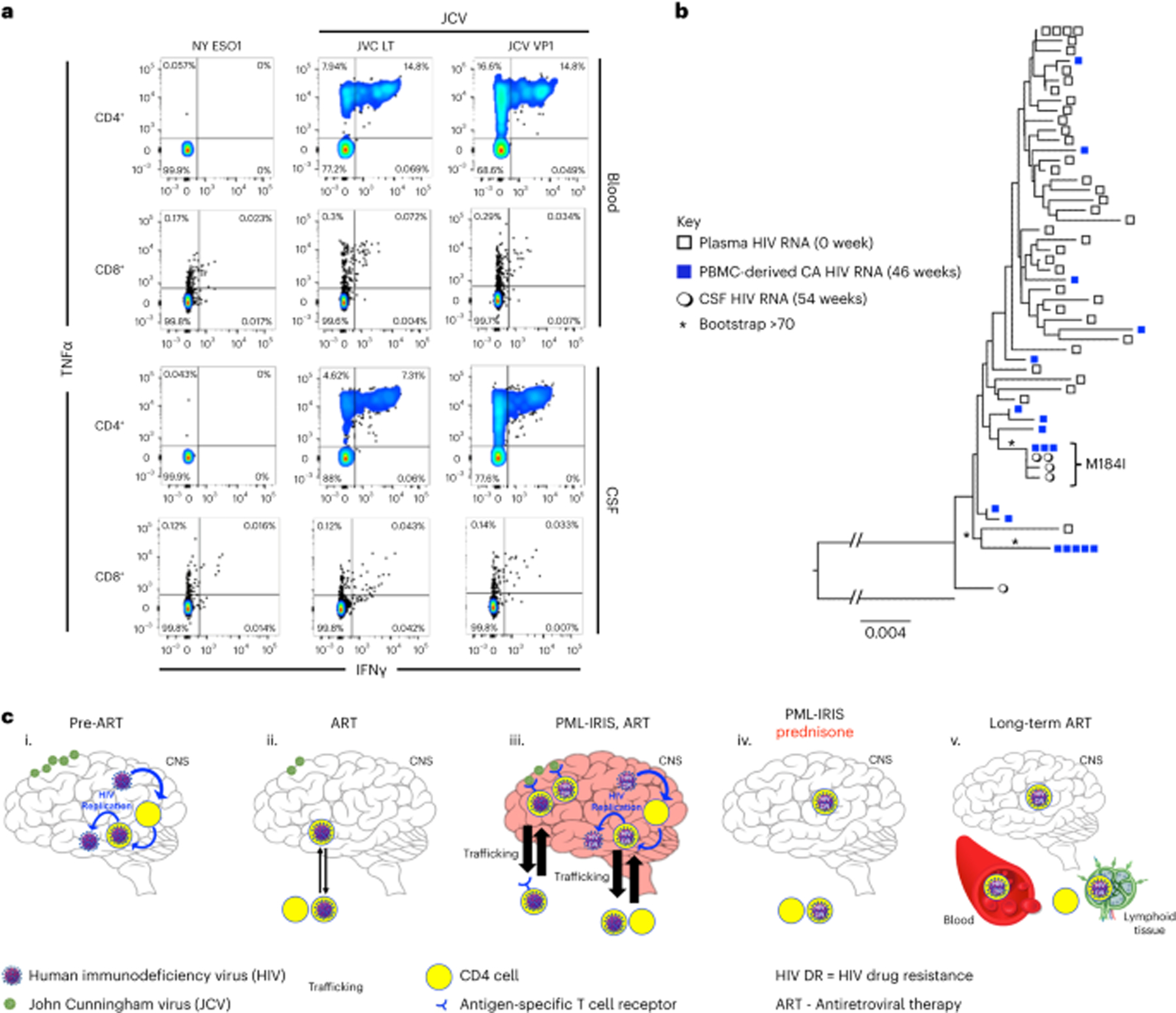
a, JCV-specific T cell responses against a control irrelevant antigen NY-ESO-31, JCV-LT and JCV-VP1 peptide mix. b, HIV population structure of pre-ART viremia, cell-associated (CA) HIV RNA at 46 weeks after ART and CSF populations during PML-IRIS at 54 weeks after ART. * indicates bootstrap value >70. Repeating the phylogenetic analysis after removal of the M184I codon did not change the results of the phylogenetic analysis. c, The dynamics of HIV reservoir composition in this case of PML-IRIS is the product of shifts in viral replication and immune responses on ART, modulated by pharmacologic intervention. (i) HIV replication ensues in the CNS before ART initiation with underlying JCV co-infection. (ii) HIV replication is inhibited with ART initiation. There is rapid reconstitution of CD4 cells, including JCV-specific CD4 cells infected with replication-competent HIV. (iii) Recruitment of these JCV-specific T cells in the CNS results in IRIS, which drives a shift in HIV reservoir composition. HIV drug resistance (DR) emerges in the CNS where ART is suboptimal. HIV DR gradually spreads across systemic HIV reservoirs. (iv) Introduction of prednisone extinguishes IRIS and HIV replication resulting from activation of JC-specific T cells. HIV-infected cells are cleared from the CNS with further shifts in CD4 cells and HIV reservoir composition. (v) HIV DR trafficked from CNS to blood resulting in HIV DR replication in the periphery with subsequent ART regimen failure and redistribution of HIV DR in peripheral tissue reservoirs after optimized long-term ART. Green virions indicate JCV. Blue virions indicate HIV. Enclosed yellow circles indicate CD4 cells. Blue arrows indicate HIV replication. Black arrows indicate trafficking across the blood–brain barrier (BBB). Thicker arrows indicate increased trafficking across the BBB. The red-tinted brain indicates inflammation and immune activation during PML-IRIS.
To determine if JCV-specific CD4+T cells were HIV-infected, we measured total HIV DNA levels in cells proliferating upon JCV peptide pools stimulation (CFSElow). HIV DNA levels in CFSElow cells before (46 weeks) and after (96 weeks) relapsed PML-IRIS were similar (820 and 860 HIV DNA copies per 106 memory cells, respectively; Supplementary Table 2); however, the relative amount of HIV RNA transcribed from these cell populations at 46 weeks was three times the levels at 96 weeks (cell-associated (CA) HIV RNA:DNA ratios of 80 versus 30, respectively; Supplementary Table 2), demonstrating that JCV antigen-driven induction of HIV proviruses was greater during PML-IRIS. Ex vivo stimulation of memory CD4+T cells with JCV peptide pools in the presence of antiretrovirals resulted in intracellular HIV-1-p24+-gag double-positive staining (Supplementary Fig. 5), indicating that p24-Gag protein expression was not the result of spreading infection but, rather, induction of proviral HIV DNA. Therefore, we concluded that peripherally circulating JCV-specific CD4+T cells included inducible HIV reservoirs. When PML-IRIS relapsed, expansion and recruitment of JCV-specific CD4+T cells to the CNS were also associated with antigen-driven activation of HIV-infected memory CD4+T cells, resulting in elevated CSF HIV VLs. Taken together, these data suggest that broad shifts in composition of HIV-infected CD4+T cell populations at PML-IRIS included expansion and brisk recruitment of JCV-specific cells to the CNS. The prominent localized inflammation in relapsed PML-IRIS resulted in elevated production of CSF HIV.
Breakthrough viremia is not characteristic of IRIS; therefore, we performed additional analyses to identify factors contributing to virological rebound. We measured levels of the components of the single-pill combination (Stribild) in CSF and plasma to determine if inadequate CNS drug penetration facilitated HIV replication during PML-IRIS. EVG and FTC plasma concentrations before (46 weeks), during (58 weeks) and after (68 weeks) relapsed PML-IRIS were within therapeutic range at >1 log10 above concentrations inhibiting 90–95% of viral replication (IC90/95; Supplementary Fig. 6a–d), typical for virological suppression and corroborating ART adherence. In contrast, CSF ART levels were simultaneously lower than in plasma with suboptimal levels of EVG and TFV (Supplementary Fig. 6e–h). Despite prior reports of lower antiretroviral (ARV) levels in CSF3,8, to our knowledge, this is the first report of CSF ARV levels <IC90/95 accompanying a CNS opportunistic infection and an associated inflammatory event, with rebound viremia. Inadequate CSF ART penetration may have facilitated HIV spread, via JCV-activated CD4+T cells in CSF, which then trafficked into systemic circulation.
In the setting of low CSF ARV levels, we characterized HIV DRMs in HIV populations by single-genome sequencing (SGS) and NGS of protease reverse transcriptase (pro-RT) and integrase (IN). Before ART, HIV populations were genetically diverse with no DRMs in CSF, plasma, lymph node or PBMCs detected by NGS or SGS (Fig. 2b, Supplementary Figs. 1 and 6 and Supplementary Table 1). In contrast, most pro-RT and IN sequences derived from rebound CSF HIV VL during relapsed PML-IRIS contained genetically linked DRMs (4/5 M184I-RT and E92Q-IN mutants) (Fig. 2b). Although pre-ART DRMs were undetectable, variants containing M184I(ATC) in RT and E92Q in IN were detected in PBMC-derived HIV RNA at 46 weeks of ART, before PML-IRIS relapse (Fig. 2b, Supplementary Fig. 6 and Supplementary Table 1). The typical mutational pathway to M184I is an ATG-to-ATA transition; however, in this case, it was due to a rare ATG-to-ATC transversion, making it a useful marker to track the emergence of this lineage. CSF-derived and PBMC-derived M184I(ATC) variants were highly related (bootstrap value >70), indicating that they had a common origin (Fig. 2b). Not all CSF HIV had DRMs; one of five (20%) was a wild-type (WT) variant similar to other variants without DRMs (Supplementary Fig. 6). Taken together, these data suggest that ART-resistant and WT variants circulating in the periphery trafficked to CSF during the relapsed PML-IRIS event and underwent replication under suboptimal CSF ARV levels, resulting in rebound viremia and systemic distribution of DRM-containing variants. Corticosteroid therapy alone suppressed rebound ART-resistant and WT HIV VLs in CSF and plasma, without modification to ART (Fig. 2a), demonstrating the impact of immunomodulation on viremic control9.
HIV variants replicating in CSF typically acquire HIV envelope (env) features distinct from peripheral blood variants3,4. We analyzed env-SGS from pre-ART plasma HIV VL and rebound CSF HIV VL (54 weeks) to investigate the source of the CSF HIV. All CSF (30/30) and plasma HIV (16/16) variants were predicted to use CCR5 co-receptors (CCR5 false-positive rate < 10%), yet CSF and plasma HIV populations were distinct. Similar to pro-RT populations, CSF and plasma env populations were phylogenetically distinct (bootstrap > 95; panmixia probability P < 10−9) (Supplementary Fig. 6c), and there were two separate migration events between plasma and CSF HIV populations (Slatkin–Maddison analysis, probability of fewer than two events < 10−4). Additionally, amino acid changes at 13 loci occurred in all CSF variants with significant differences in allele frequencies compared to plasma HIV (P ≤ 10−4 for all loci, Fisher’s exact test) (Supplementary Table 3); there were two changes in the V3 loop and two new acquisitions in the glycan shield (V1 and C4). Overall, these data support the CNS as the site of replication for WT and drug-resistant HIV.
PML-IRIS symptoms progressively improved with a prolonged prednisone taper, which was discontinued at 70 weeks. HIV VLs were intermittently detectable at low levels; however, by 96 weeks, plasma HIV VL increased to 509 copies per milliliter (Supplementary Table 1), and, by 104 weeks, 85–95% of rebound viremia was predominantly drug resistant based on NGS results (Supplementary Fig. 1d). Single-genome analyses demonstrated that HIV drug-resistant variants at 104 weeks were highly related to those detected in rebound CSF HIV VL at 54 weeks during relapsed PML-IRIS and in 23% (3/13) of PBMC HIV RNA populations at 46 weeks (Supplementary Fig. 6). These findings are consistent with trafficking of drug-resistant HIV from CSF to peripheral blood10, which culminated in a loss of virological control by 104 weeks.
Low-level HIV VLs persisted until 104 weeks, when ART was changed to darunavir/COBI/TAF/FTC, with prompt suppression to <40 copies per milliliter, demonstrating that rebound HIV VL was the product of active HIV replication. The individual remained clinically stable for the next 5 years without further ART failure, and CD4+T cell counts increased to 530 cells per microliter. He underwent research lymph node (117 weeks) and recto-sigmoidal (133 weeks) biopsies that were both histopathologically unremarkable. HIV proviruses were present but infrequent in these tissues: M184I(ATC) and M184V(GTG) were each 0.01% of lymph node HIV DNA sequences (Supplementary Fig. 7). No DRMs were found in recto-sigmoidal tissues (Supplementary Table 1).
Undetectable HIV DRMs before 46 weeks of ART does not negate their existence or selection from pre-ART and post-ART cells and tissues, nor does it infer directionality of trafficking of HIV reservoirs among anatomic compartments before release and outgrowth in the CNS. We propose that the emergence of HIV DRMs resulting in ART failure occurred in the following sequence (Fig. 2c). (1) DRMs emerged before ART initiation as minority variants, as predicted in large replicating WT HIV populations11, including HIV-infected, JCV-specific memory cells. (2) JCV antigen-driven activation and proliferation of JCV-specific T cells during the first IRIS event facilitated trafficking and expansion of an inducible HIV reservoir in the CNS12,13. (3) Antigen-mediated activation of JCV-specific T cells during relapsed PML-IRIS facilitated reactivation of HIV reservoirs in CNS. (4) Corticosteroids reduced inflammation, which controlled reactivation of HIV from their reservoirs during PML-IRIS. (5) Localized replication of restricted, neuro-adapted, CCR5-utilizing HIV population ensued in the CNS where there was suboptimal ART penetration, facilitating the emergence of drug-resistant HIV that disseminated to the periphery. (6) Although corticosteroids initially controlled HIV replication, eventually, minority drug-resistant variants generated substantial rebound viremia after a relatively long time5.
Initiating ART in advanced HIV/AIDS is common, especially in resource-limited settings, generating large populations at higher risk for IRIS14; understanding mechanisms driving IRIS will improve care of these individuals. Expansion and contraction of HIV-infected populations in response to specific antigenic stimuli have been previously reported in peripheral blood12,15, but their role in IRIS or in anatomic compartments is unknown. In this study, we revealed brisk trafficking of JCV-specific HIV-infected cells across anatomic compartments during PML-IRIS, demonstrating that HIV reservoir reactivation could be manipulated by anti-inflammatory drugs, even in a specific compartment such as the CSF.
The HIV reservoir is a product of dynamic immunologic and virologic forces that determine the composition and distribution of HIV populations. Here we demonstrated that clinical events resulting from specific and measurable immunological forces drive the mutational composition and anatomic distribution of the HIV reservoir. This case study highlights the importance of current efforts to develop immunomodulatory strategies to modulate HIV reservoirs in compartments such as the CNS, including induction of HIV latency reversal in antigen-specific T cells (checkpoint blockade reservoir purging) or prevention of cytokine-driven homeostatic expansion without disrupting effector T cell functions (that is, Janus kinase inhibitors)16–18. Ongoing studies will define the potential effects of these immunomodulating strategies on HIV reservoir dynamics during ART19. These scalable and multimodal approaches to modulate immune activation and impact antigen-specific HIV reservoirs will be useful for designing effective HIV remission strategies.
Methods
Study participant and materials
The study participant was referred to the NIH Clinical Center and enrolled in an institutional review board (IRB)-approved prospective observational protocol (NCT02147405) started on 30 May 2014. The participants of the protocol are people with HIV-1 infection who are naive to ART, have a CD4+T cell count <100 cells per microliter and agree to be followed for at least 96 weeks after ART initiation, with the opportunity to extend such follow-up as clinically indicated. The study participant, a 59-year-old male, provided written informed consent in accordance with the Declaration of Helsinki. PBMCs were purified by density gradient centrifugation from heparinized blood. Plasma collected from EDTA-treated tubes after centrifugation for 15 min at 2,000g was aliquoted and stored at −80 °C. CSF was collected in clinically indicated lumbar puncture procedures. CSF mononuclear cells were obtained by centrifugation at 400g and freshly used in assays evaluating JCV antiviral activity. CSF supernatant was aliquoted and stored at −80 °C for downstream virological and immunological analysis.
JCV-, EBV- and CMV-specific T cell responses in peripheral blood and CSF
JCV-, EBV- and CMV-reactive T cells were generated using commercially available overlapping peptide libraries against immunodominant viral antigens obtained from commercial vendors ( JPT and Miltenyi Biotec). Fresh peripheral blood and CSF mononuclear cells were pulsed with peptide libraries (final concentration of 1 μg ml−1). Irrelevant peptide NY-ESO1 (New York esophageal squamous cell carcinoma 1) was used as a negative control, and a peptide mix of CMV (pp65, immediate early 1 (IE1)) and EBV (Epstein–Barr nuclear antigen (EBNA), BamHI Z fragment leftward open reading frame 1 (BZLF1)) was used to compare specific T cell responses against other viruses in PBMCs. Cells were suspended in AIM-V media (Thermo Fisher Scientific) supplemented with inactivated 5% human serum, IL-7 and IL-15 (10 ng ml−1, PeproTech) and plated on 96-well U-bottom plates. Cultures were maintained, fed and split as needed every 3 d for approximately 14 d, with IL-2 (30 IU ml−1, PeproTech) added 72 h after the initial stimulation. Cultures were then re-stimulated with the same peptide libraries for 4–6 h in the presence of brefeldin A and monensin A (BD Biosciences) to prevent cytokine degranulation per the manufacturer’s instructions. Intracellular cytokine production was measured by flow cytometry. Cells were prepared using BD Cytofix/Cytoperm Kit following the manufacturer’s instructions. The following surface markers and cytokines were analyzed with CD3, CD4, CD8, TNFα and IFNγ antibodies (BioLegend, 300407; BD Biosciences, 562843; BD Pharmingen, 555369; BD Biosciences, 557647; and BD Biosciences, 554669, respectively) with Vivid fixable violet amine reactive dye for viability (Invitrogen/Molecular Probes).
JCV antigen-specific CD4+T cell responses in peripheral blood
Thawed PBMCs from weeks 46 and 96 after ART were first resuspended in RPMI 1640 supplemented with 10% human AB serum, 50 μg ml−1 gentamicin and 1.7 mM L-glutamine and then rested for 1 h at 37 °C in the presence of 125 U ml−1 benzonase (Sigma-Aldrich). To identify CD4+T cells that were JCV antigen specific, PBMCs were first labeled with 10 μM carboxyfluorescein succinimidyl ester (CellTrace, Thermo Fisher Scientific, C34554) to monitor cellular proliferation and were then stimulated by JCV peptide mix (VP1 and large T antigen, 0.4 μg ml−1, Peptides and Elephants). Cells were plated at 3 × 106 cells per well in a 96-well deep-well plate and cultured for 6 d with JC Peptide Pools and 1 μg ml−1 of co-stimulatory antibodies αCD28 and αCD49d. Three different independent experiments were performed in the presence of different ART regimens (darunavir/nevirapine (5 μM of each drug, Sigma-Aldrich); raltegravir 200 nM; and raltegravir 200 nM + 3 IU ml−1 of IL-2). Non-naive CD4+T cells were identified by extracellular staining and then sorted into CFSEhigh/CFSElow subsets. CFSElow subsets identified JCV antigen-specific, non-naive CD4+T cells that underwent antigen-driven proliferation in response to the JC peptide mix stimulation. Anti-CD3-PE, anti-CD4-BV605, anti-CD8-APC, anti-CD27-BV711 (BioLegend, 302833) and anti-CD45RO-PE-Cy7 (BD Biosciences, 337168) were used for T cell staining. Dead cells were identified and excluded using LIVE/DEAD Fixable Near IR Dead Cell Stain Kit (Thermo Fisher Scientific, L34976). See Supplementary Fig. 4b for gating strategy. CA HIV-1 measurements were performed as described below.
HIV RNA and DNA levels in CSF, PBMC and plasma
Plasma and PBMC were obtained before and after ART initiation. PBMCs, CSF and tissue from the lymph node, sigmoid and rectum were obtained during the first 2 years of ART. Viral RNA was extracted from plasma by isopropanol/ethanol precipitation, and CA DNA and RNA were isolated with TRIzol from cells and tissue20. CA HIV gag RNA and DNA levels were measured with single-copy assays using the primers and probe described in Somsouk et al.21.
HIV-1 p24+ production in JCV antigen-specific CD4+T cells (HIV Flow)
CFSEhigh/low non-naive CD4+T cells after stimulation with JCV peptides were sorted, centrifuged at 1,500 r.p.m. for 5 min, resuspended at 4 × 106 cells per milliliter in culture media (10% human AB serum, 0.02 M lamivudine, 200 nM raltegravir, 300 U ml−1 IL-2, 0.5 μl ml−1 benzonase) and split into two wells on a 96-well plate. Anti-CD3-BUV395 (BD Biosciences, 563546), anti-CD4-BUV737 (BD Biosciences, 612748), anti-CD8-Pacific Blue (BD Biosciences, 558207), anti-CD27-BV605 (BioLegend, 302830) and anti-CD45RO-PE-Cy7 were used for sorting. An equal volume of media containing either 1:1 pre-washed Dynabeads Human T-Activator CD3/CD28 (Gibco, 11161D) or DMSO control was included. We adapted the HIV-Flow method22, stimulating cells with the Dynabeads for 42 h and then transferring them to a 5-ml FACS tube, centrifuging at 1,500 r.p.m. for 5 min and aspirating the supernatant. Cell pellets were resuspended in 200 μl of PBS containing 1:1,000 LIVE/DEAD Aqua, CD45RO-ECD (Beckman Coulter, IM2712U), PD1-BV605 (BioLegend, 329923), CD27-BV711 and CXCR5-BV421 (BioLegend, 356919) and incubated for 30 min at room temperature. Cells were then fixed and permeabilized (Foxp3 transcription buffer stain, eBiosciences) for 30 min at room temperature. After permeabilization, cells were stained with CD4-PE-Cy7 (Life Technology, 25-0042-82), CD3-BUV395 (BD Biosciences, 563546), p24 KC67-PE (Beckman Coulter, CO6604667) and p24 28B7-APC (MédiMabs, MM-0289-APC) for 45 min at room temperature. Cells were acquired using a Fortessa II flow cytometer (BD Biosciences) and analyzed using FlowJo version 10 software. See Supplementary Fig. 4c for gating strategy.
MRI
MRI was performed on 3-T scanners per clinical routine at our site and included administration of gadolinium-based contrast material (0.1 mmol kg−1 gadoteridol, ProHance or gadobuterol, Gadavist). T1-weighted, T2-weighted and post-contrast three-dimensional T1-weighted and fluid-attenuated inversion recovery (FLAIR) images were reviewed.
PET/CT
PET/CT scans of the brain were obtained 60 min after the administration of approximately 10 mCi of FDG intravenously. Reconstructed, attenuation-corrected images were reviewed.
Plasma and CSF ARV concentrations
ARVs were extracted from plasma and CSF by protein precipitation with methanol containing stable, isotopically labeled 13C5-TFV, 15N,d2-FTC, d6-MVC and d6-EVG as internal standards. Concentrations were determined by liquid chromatography–tandem mass spectrometry assays with dynamic ranges of 1–4,000 ng ml−1 TFV and FTC, 1–10,000 ng ml−1 MVC and 2–10,000 ng ml−1 EVG. Biological concentrations were compared longitudinally as well as compared to 90% and 95% ICs reported in treatment-experienced adults (Gilead Sciences (2016), GENVOYA: Highlights of Prescibing Information, FDA reference: 4023346; ViiV Healthcare (2016), SELZENTRY: Highlights of Prescibing Information, FDA reference 4009160).
SGS
Single-genome sequences encoding HIV pro-RT (982 nucleotides) and HIV IN (680 nucleotides) and near-full-length envelope (env) were obtained from viral populations in plasma, cells and/or tissue23,24 (see Supplementary Table 1b for sequencing primers). Sequence analyses were performed with Version X of the Molecular Evolutionary Genetic Analysis Software (MEGA X; https://www.megasoftware.net). APOBEC3G/F hypermutated sequences were identified with Hypermut 4.0 (https://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html) and excluded from sequence analyses.
HIV phylogenetic analyses
Clinical assessment of HIV drug resistance (DR) was determined at the NIH Clinical Center. NGS from plasma-derived samples was performed with the MiSeq system (Illumina), and HIV DRMs were detected with VISL (variants in susceptibility loci). NucAmino was used to determine ambiguity at codon 184 in reverse transcriptase (5–15% threshold) (https://github.com/hivdb/NucAmino). DRMs were identified with Stanford University’s HIV Drug Resistance Database (https://hivdb.stanford.edu/hivdb/by-mutations/). MEGA X was used to determine average pairwise distances (APDs) and reconstruct phylogenies. The relatedness of viral variants was depicted with Neighbor-Joining (NJ) trees. To investigate whether HIV populations underwent shifts, geographic subdivision and Slatkin–Maddison tests were performed. We compared HIV lineages to pre-therapy plasma RNA populations (≥10 lineages per population were compared when duplicate sequences were collapsed to one, representing their lineage). CCR5 co-receptor utilization of env sequences was determined with Geno2pheno[coreceptor] (https://coreceptor.geno2pheno.org/) using a false-positive rate of 10%. Parametric and non-parametric compartmentalization analyses were performed with Slatkin–Maddison tests using HyPhy version 2.2.4 (https://www.hyphy.org/) and geographic subdivision analyses25 (https://bioinfo.mnhn.fr/abi/people/achaz/hudsontest.html).
Statistics
Geographic subdivision analyses were performed as non-parametric comparisons of pairwise distances between 100,000 randomized pairs of intra-patient sequences, generating P value for the probability that the randomized populations’ structures were the same between sets of sequences, with significance cutoff level of P < 10−9. Slatkin–Maddison parametric bootstrapping tests were performed where simulated, and observed frequency distributions of parsimony steps (s) of each population member were determined for 1,000 generations in phylogenetic structures of unique sequences. The model was rejected when the observed s fell outside of the 95% confidence interval of the simulated distribution with a significance cutoff level of P < 0.01. Two-sided Fisher’s exact tests were used to compare amino acid frequency of dominant alleles in CSF HIV-1 populations compared to those detected in plasma.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
The authors thank the staff of the National Institutes of Health (NIH) Clincal Center inpatient and outpatient services, the National Institute of Allergy and Infectious Diseases research staff and the study participant for their important contributions, without whom this research would not have been possible. We also thank E. Bruzzesi for contributions in the laboratory. This research was supported, in part, by the Intramural Research Program of the NIH and, in part, with federal funds from the National Cancer Institute under contract number 75N91019D00024/HHSN261201500003I as well as the Office of AIDS Research under the strategic funds supplement: ‘Impact of lymphopenia and co-infections on residual inflammation and reservoirs’. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US government.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-023-02387-4.
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-023-02387-4.
Peer review information Nature Medicine thanks Janice Clements and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Alison Farrell, in collaboration with the Nature Medicine team.
Data availability
pro-RT, IN and env sequences are publicly available (GenBank accession numbers OQ924993-OQ925343, OM937385-OM937739 and OQ925344-OQ925389, respectively). All other data are included in the paper and supplementary documents.
References
- 1.Bozzi G et al. No evidence of ongoing HIV replication or compartmentalization in tissues during combination antiretroviral therapy: implications for HIV eradication. Sci. Adv 5, eaav2045 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaillon A et al. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Invest 130, 1699–1712 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joseph SB et al. Human immunodeficiency virus type 1 RNA detected in the central nervous system (CNS) after years of suppressive antiretroviral therapy can originate from a replicating CNS reservoir or clonally expanded cells. Clin. Infect. Dis 69, 1345–1352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woodburn BM et al. Characterization of macrophage-tropic HIV-1 infection of central nervous system cells and the influence of inflammation. J. Virol 96, e0095722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deeks SG et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nat. Med 27, 2085–2098 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Giacomini PS et al. Maraviroc and JC virus-associated immune reconstitution inflammatory syndrome. N. Engl. J. Med 370, 486–488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullins C, Miranda J, Sandoval H, Ramos-Duran L & Tonarelli SB The benefit of mirtazapine in the treatment of progressive multifocal leukoencephalopathy in a young HIV-positive patient: a case report. Innov. Clin. Neurosci 15, 33–35 (2018). [PMC free article] [PubMed] [Google Scholar]
- 8.Gele T et al. Cerebrospinal fluid exposure to bictegravir/emtricitabine/tenofovir in HIV-1-infected patients with CNS impairment. J. Antimicrob. Chemother 76, 3280–3285 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Gunst JD, Hojen JF & Sogaard OS Broadly neutralizing antibodies combined with latency-reversing agents or immune modulators as strategy for HIV-1 remission. Curr. Opin. HIV AIDS 15, 309–315 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Wiegand A et al. Single-cell analysis of HIV-1 transcriptional activity reveals expression of proviruses in expanded clones during ART. Proc. Natl Acad. Sci. USA 114, E3659–E3668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coffin JM HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267, 483–489 (1995). [DOI] [PubMed] [Google Scholar]
- 12.Simonetti FR et al. Antigen-driven clonal selection shapes the persistence of HIV-1-infected CD4+ T cells in vivo. J. Clin. Invest 131, e145254 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kincer L et al. HIV-1 is transported into the central nervous system by trafficking infected cells. Pathog. Immun 7, 131–142 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sereti I et al. Prospective international study of incidence and predictors of immune reconstitution inflammatory syndrome and death in people living with human immunodeficiency virus and severe lymphopenia. Clin. Infect. Dis 71, 652–660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendoza P et al. Antigen-responsive CD4+ T cell clones contribute to the HIV-1 latent reservoir. J. Exp. Med 217, e20200051 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavegnano C et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog 13, e1006740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uldrick TS et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Sci. Transl. Med 14, eabl3836 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiu CY et al. Combination immune checkpoint blockade enhances IL-2 and CD107a production from HIV-specific T cells ex vivo in people living with HIV on antiretroviral therapy. J. Immunol 208, 54–62 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu DC, Mellors JW & Vasan S Can broadly neutralizing HIV-1 antibodies help achieve an ART-free remission? Front. Immunol 12, 710044 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simonetti FR et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl Acad. Sci. USA 113, 1883–1888 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Somsouk M et al. The immunologic effects of mesalamine in treated HIV-infected individuals with incomplete CD4+ T cell recovery: a randomized crossover trial. PLoS ONE 9, e116306 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardons M et al. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog 15, e1007619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer S et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol 43, 406–413 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keele BF et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl Acad. Sci. USA 105, 7552–7557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Achaz G et al. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol. Biol. Evol 21, 1902–1912 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
pro-RT, IN and env sequences are publicly available (GenBank accession numbers OQ924993-OQ925343, OM937385-OM937739 and OQ925344-OQ925389, respectively). All other data are included in the paper and supplementary documents.