

Abstract
Cells interpret mechanical stimuli from their environments and neighbors, but the ability to engineer customized mechanosensing capabilities has remained a synthetic and mechanobiology challenge. Here we introduce tension-tuned synthetic Notch (SynNotch) receptors to convert extracellular and intercellular forces into specifiable gene expression changes. By elevating the tension requirements of SynNotch activation, in combination with structure-guided mutagenesis, we designed a set of receptors with mechanical sensitivities spanning the physiologically relevant picoNewton range. Cells expressing these receptors can distinguish between varying tensile forces and respond by enacting customizable transcriptional programs. We applied these tools to design a decision-making circuit, through which fibroblasts differentiate into myoblasts upon stimulation with distinct tension magnitudes. We also characterize cell-generated forces transmitted between cells during Notch signaling. Overall, this work provides insight into how mechanically induced changes in protein structure can be used to transduce physical forces into biochemical signals. The system should facilitate the further programming and dissection of force-related phenomena in biological systems.
Mechanical forces are fundamental regulators of biology, guiding vital processes and shaping disease progressions that span from the molecular to the tissue scale1,2,3. Mechanical forces can do so by transmitting actionable information from biological environments, which can vary throughout developmental, tissue and cell cycle contexts4,5. Accordingly, cells have evolved complex mechanisms to actively use mechanical information and to translate force-based stimuli into biochemical responses that can affect changes in metabolism, migration, differentiation, immunity and gene transcription6,7,8,9,10. Harnessing cells’ capability to interpret differential forces in their environment would enable advances in tissue engineering, where forces could be used to drive structural organization, and targeted therapeutics, where mechanical environments could be used as tractable disease biomarkers. However, it has been difficult to synthetically program mechanosensation in cells, given that many of its underlying mechanisms remain to be elucidated.
At a fundamental level, mechanotransduction can be generalized as an input–output relationship, in which mechanical energy is converted into a biochemical response via force-induced structural changes in cell-signaling molecules. The sensitivity of proteins to mechanical unfolding has been tuned through several strategies but in the context of biophysical spectroscopy, or FRET imaging sensors, rather than customized cellular signaling networks11,12,13,14,15,16,17. Conversely, outputs from endogenous mechanotransduction pathways have been modulated by overexpressing protein components, engineering sensitivity to nonmechanical inputs and, more synthetically, adding new outputs to the force-sensitive Piezo1 and YAP/TAZ (Yes-associated protein/transcriptional coactivator with PDZ-binding motif) signaling cascades18,19,20. However, these approaches have not yet tuned the mechanical force required for activation and do not present an obvious protein engineering solution for doing so. These strategies furthermore rely upon endogenous pathways that are difficult to decouple from the user’s engineered pathways. Ideally, a fully customizable mechanosensitive pathway would synthesize the following three design criteria: (1) programmable sensitivity to input forces, (2) versatility of output cellular actions and (3) orthogonality with respect to endogenous pathways.
Here, we use the Notch1 receptor to build a platform for controlling mechanosensation that meets the above design criteria. Notch1 is a classically studied protein, and our work capitalizes on a body of structural and biophysical research that guides our molecular design. Notch1 activation requires mechanical force, which is sensed by the receptor’s negative regulatory region (NRR; Fig. 1a)21. Upon application of tensile force via the ligand binding domain (LBD), the NRR undergoes a conformational change that enables proteolytic release of the intracellular domain (ICD), a transcriptional effector21,22,23,24. Recent work has shown that the NRR can act as a modular scaffold to build synthetic Notch (SynNotch) receptors, which preserve the Notch activation mechanism while tolerating heterologous substitutions at the LBD and ICD21,25. These receptors offer a starting point that satisfies two of our three design criteria; SynNotch receptors provide versatility of cellular outputs via an ICD of interest and minimal crosstalk with endogenous pathways via Notch’s mechanistically direct signaling pathway. We, therefore, asked if we could satisfy the remaining criterion—programmable sensitivity to input forces—by creating new NRR domains with distinct mechanical properties, which could be used to specify new tension requirements for signaling.
Fig. 1: Design of customized mechanosensation.
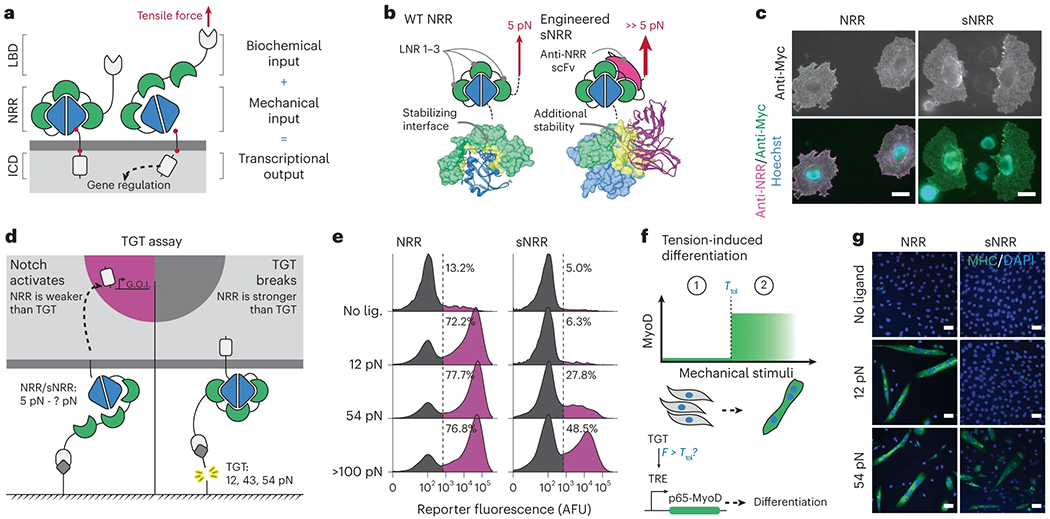
a, Schematic of tension-mediated activation in Notch or SynNotch receptors. Application of sufficient tensile force via the LBD activates the receptor by displacing three LNR modules and converting the NRR into a substrate for proteolysis at S2. Cleavage at S2, and subsequently at S3, liberates the ICD (LNR modules, green; S2 and S3, red). b, Activating the Notch1 NRR (left) requires tensile force to disrupt the intramolecular interactions that promote an autoinhibited conformation. Engineered sNRR domains (right) include an intramolecularly bound scFv for additional stability. The heterodimerization domain (HD) and LNRs of the NRR are blue and green, respectively, and the scFv added in sNRR is magenta. Molecular interactions between the LNRs and HD (left) or scFv and NRR (right) are yellow. (Protein Data Bank (PDB) codes: 3ETO and 3L95, respectively). c, Representative immunostaining of NRR- and sNRR-based receptors for surface-receptor with anti-Myc (green) and available NRR with exogenous soluble anti-NRR scFv (magenta) on HeLa cells. Scale bar is 25 μm. d, Schematic of TGT assay used to evaluate molecular tension needed to activate engineered receptors. FITC is used as a ligand for SynNotch receptors expressing an anti-FITC scFv LBD. e, NRR- or sNRR-based receptors with a Gal4-VP64 ICD induce expression of a UAS-controlled H2B-mCherry reporter upon activation. HEK293-FT cells transiently expressing these receptors were cultured on TGT-coated surfaces, and reporter activity was monitored by flow cytometry. Traces represent normalized densities of the total cells from three independently transfected pools (n = 3) expressing a cotransfection marker based on the mTurquoise2 within the analyzed populations. f, Tension-induced myogenic differentiation. C3H/10T1/2 fibroblasts express NRR- or sNRR-based SynNotch receptors with a TetR-based/tTA ICD. Upon activation, these receptors drive expression of p65-MyoD, which in turn leads to differentiation. g, Representative immunostaining identifies differentiation into skeletal myocytes by multinucleation and positive myosin heavy chain expression (green). Scale bar is 100 μm. LNR, LIN12-Notch repeat.
We began by designing a synthetic NRR domain that required increased tension levels to activate. Single-molecule force spectroscopy has shown that proteins can be mechanically stabilized via the binding of other protein domains15,26, and studies of the NRR are consistent with these observations. Antibody binding can impart stability upon the NRR, elevating its resistance against chemical- and force-induced unfolding in vitro21,27 and limiting the spontaneous activation of mutant receptors in vivo28,29. Given these observations, we hypothesized that the NRR’s mechanical stability could be increased via direct fusion with an NRR-binding protein. Specifically, we fused the NRR with a single chain variable fragment (scFv) derived from an NRR-stabilizing antibody28, such that the scFv could bind intramolecularly to the NRR to reinforce its autoinhibited conformation (Fig. 1b). In this way the integrated scFv can impart a mechanically stabilizing effect, and we anticipated that SynNotch receptors containing these ‘strengthened NRR’ (sNRR) domains would require elevated tensions for activation. Here, we describe the development of tension-tuned sNRR-SynNotch receptors. We used these receptors to design force-sensitive genetic circuitries, including a mechanically activated cellular differentiation program. Further utility of the system is shown by their use in gauging the ligand pulling forces that are transmitted between cells during intercellular Notch signal transduction.
RESULTS
Synthetic mechanoreceptors are processed like Notch
We first sought to verify that sNRR-based receptors preserved the fundamental trafficking and signaling functionalities of their NRR-based predecessors. To do so, we compared receptors that differed only in the presence of an NRR or sNRR mechanosensing unit, having otherwise identical LBD and ICD components. As expected, both NRR- and sNRR-containing receptors were processed by the Golgi-localized furin convertase, and the resulting heterodimers were successfully trafficked to the cell surface (Fig. 1c and Supplementary Fig. 1A,B). Labeling of live cells with soluble ligand indicated that the synthetic receptors were constitutively internalized from the plasma membrane (Supplementary Fig. 1C), similar to the known recycling of natural Notch receptors30. Next, we demonstrated that integration of the anti-NRR scFv blocked sNRR reactivity against soluble anti-NRR, providing evidence that the fused antibody fragment interacts with the NRR epitope in the intended intramolecular manner (Fig. 1c and Supplementary Fig. 1B,C). In addition to synthetic receptors, full-length Notch1 receptors maintained the desired expression properties upon substitution of the NRR for sNRR (Supplementary Fig. 1D,E).
We used cell-based analyses with a luciferase reporter to confirm that sNRR-SynNotch could activate in response to ligand-mediated tension, as delivered through surface-immobilized fluorescein (a.k.a. fluorescein isothiocyanate (FITC)) dyes. FITC was chosen as a ligand due in part to its biocompatibility and versatility: it is a widely used research and clinical labeling reagent and thus diverse FITC-linked bioconjugates can be readily obtained. In addition, rupture forces between FITC and anti-FITC antibodies have previously been reported, with single-molecule measurements indicating a ~50 pN unbinding force between FITC and the E2 anti-FITC scFv31. Given these factors, we paired the E2 anti-FITC scFv as an LBD for detecting FITC-based ligands.
Using cells expressing anti-FITC receptors, we observed that treatment with either batimastat (BB-94, a broad-spectrum metalloprotease inhibitor) or compound E (a ɣ-secretase inhibitor) inhibited signaling responses against immobilized FITC ligands (Supplementary Fig. 2A). This result indicates that sNRR activation involves proteolysis by a metalloproteinase and the ɣ-secretase complex, respectively, similar to natural Notch processing. The data also confirmed that sNRR-SynNotch could be activated upon exposure to an immobilized form of its ligand, suggesting a potential requirement for tensile resistance during activation, similar to natural Notch25,32. To verify this requirement, we treated cells with soluble fluorescein; such treatment failed to upregulate luciferase expression in both NRR- and sNRR-SynNotch cells (Supplementary Fig. 2B). Thus, we conclude that sNRR-SynNotch processing is like that of natural Notch, and that sNRR-SynNotch also requires ligand-mediated tension for its signaling activity.
Synthetic mechanoreceptors require increased tensile force
Having confirmed its functional similarity to natural Notch, we next determined the force sensitivity of sNRR-SynNotch using the tension gauge tether (TGT) assay. In this approach, short double-stranded DNA (dsDNA) sequences of known tension tolerance (Ttol) are used to attach ligands (that is, FITC) to a culture surface via biotin-streptavidin interaction before adding cells (Fig. 1d)22. These dsDNA tethers (‘TGTs’) impose a ceiling on the magnitude of tension that can be applied to individual ligand-bound receptors. If the tension needed to activate sNRR-SynNotch surpasses the Ttol of a given TGT, then the tether will dissociate before inducing receptor activation. If instead the Ttol of a TGT surpasses what is needed to unravel the sNRR, then the tether is expected to endure and thus signaling should ensue. We used biotin connected to FITC via a nonrupturable linker, rather than dsDNA, as an upper limit for ligand Ttol, as biotin-streptavidin will tolerate loads or more than 100 pN before dissociation33.
Previous studies have narrowed the activation force of the NRR to 4–12 pN21,22,23,24, which is an order of magnitude below the critical force for rupturing antibody–antigen pairs34. We, therefore, anticipated that sNRR would exhibit a substantially elevated force activation threshold. By challenging receptor-expressing reporter cells against TGTs of distinct Ttol values, we defined an activation threshold of approximately >54 pN for engineered sNRR-SynNotch receptors, well above the <12 pN (~5 pN) threshold previously defined for the Notch1 NRR (Fig. 1e). Other existing NRR-binding antibodies were fused as scFvs to the NRR to evaluate the generalizability of our design, and multiple increased receptor tension tolerance29,35 (Supplementary Fig. 3). These data demonstrate that sNRR fusions have increased force requirements for activation because they respond differentially to molecular tensions even while binding the same ligand.
Synthetic mechanotransduction can induce muscle-like fates
Equipped with two mechanically distinct receptors, we next aimed to specify how a cell responds to a specific force value. In natural systems, cells enact intricate biological outputs based on the mechanical information in their microenvironment. For example, mechanical forces can guide the formation of sarcomeres36, coordinate wound healing37 or induce differentiation8. Using a synthetic biology approach, we generated mouse embryonic fibroblasts that can differentiate in response to specific molecular tensions. To do so, we rendered the expression of MyoD (a master regulator of myogenic differentiation) SynNotch-dependent. A gene sequence encoding a p65-MyoD fusion construct under the control of the tetO-containing TRE promoter (TRE: p65-MyoD) was virally integrated into C3H/10T1/2 fibroblasts38. Receptors containing tetO-binding ICDs (TetR-VP64) were then expressed in these cells, such that signaling would lead to p65-MyoD expression and subsequently myogenic conversion (Fig. 1f). Using these engineered fibroblasts, we then compared the ability of NRR-SynNotch and sNRR-SynNotch to guide cell fate decisions in response to mechanically distinct TGTs. We observed that cells expressing the receptors differentiated and fused into multinucleated skeletal myocytes only in response to tensions greater than the threshold defined by their respective mechanosensing domains—at least 12 pN for the NRR, and at least 54 pN for sNRR (Fig. 1g and Supplementary Fig. 4).
Tensile strengths can be systematically tuned
Further intricacy in cellular mechanosensation can arise from protein isoforms that have distinct mechanical properties despite overlapping biochemical functions, as studied with integrins or talins39,40. We, therefore, anticipated that an expanded family of sNRR domains with diversified force activation thresholds would similarly enable more sophisticated signaling networks. Mutating residues involved in noncovalent interactions at a protein’s ‘mechano-active’ site can lead to distinct mechanical phenotypes41. We hypothesized that point mutations within the scFv:NRR interface would alter the mechanical stability of sNRR, providing a basis to systematically tune force activation thresholds (Fig. 2a). Using a structure-guided strategy, we generated single- and double-mutants by introducing conservative and nonconservative amino acid substitutions within the scFv:NRR interface (Fig. 2b,c).
Fig. 2: Tuning the tensile strength of sNRR domains.
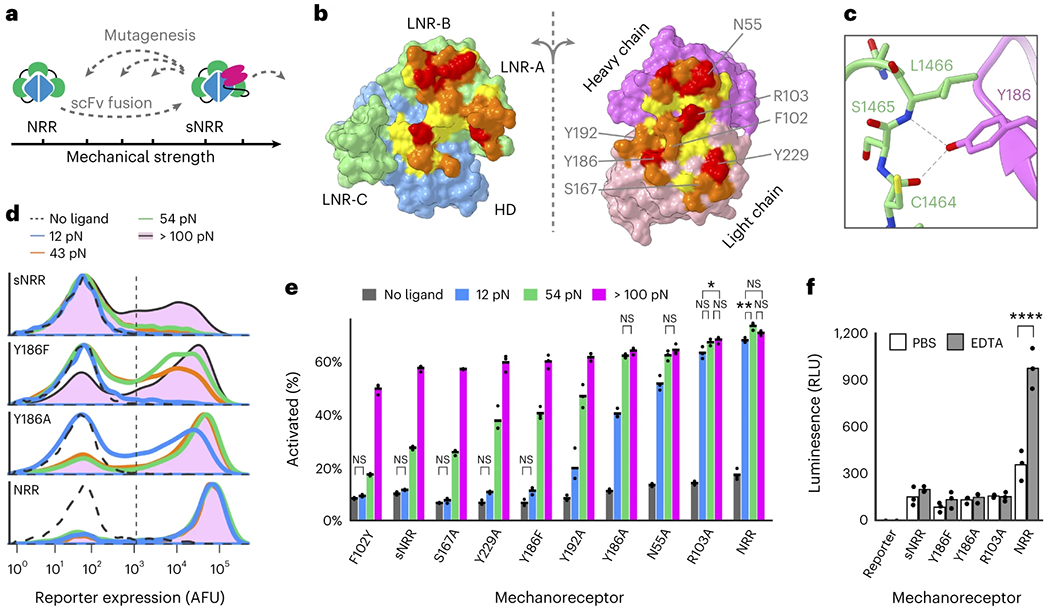
a, Schematic of strategy to tune mechanical strength. b, Open-book view of the interface between the Notch1 NRR and the NRR-binding Fab. The HD and LNRs of the NRR are blue and green, respectively, and the heavy and light chains of the scFv are magenta and pink. Additional residue coloring corresponds to the distance between the two surfaces (<5.5 Å, yellow; 5.5–4 Å, orange; 4–3 Å, red) (PDB code: 3L95). c, Y186 on the Fab light chain is highlighted as an example of a mutated residue. d, Mutating Y186 decreases sNRR mechanical strength. Receptors stably integrated into HEK293-FT cells upregulate the expression of a UAS-driven H2B-mCherry reporter gene upon activation by TGTs. Traces represent normalized densities of total cells within transduced populations from three independent experiments (n = 3). e, Mutating sNRR residues creates receptors with a range of force activation thresholds. Receptors are transiently expressed in HEK293-FT cells containing a stably integrated UAS-H2B-mCherry reporter gene. Mean from three independent transfections (n = 3) displayed, quantified by flow cytometry (over 5,000 cells assessed per replicate) and analyzed with two-way ANOVA (mechanosensor and TGT threshold). All unlabeled comparisons within the same mechanosensor have P < 0.0001; labeled NS, P > 0.05, *P < 0.05, **P < 0.01, otherwise. f, Cells expressing SynNotch receptors with an NRR domain or sNRR domain of various strengths are treated with either 0.5 mM EDTA or PBS for 15 min at 37 °C followed by the re-addition of culture media. Reporter expression was evaluated 6 h later. Activated receptors upregulate the expression of a UAS-regulated luciferase reporter gene. Mean from three independent transfections (n = 3), two-way ANOVA, P value labeled **** for P < 0.0001, all unlabeled comparisons have P > 0.05.
Consistent with our expectation, introducing mutations resulted in sNRR units of distinct mechanical strengths, as quantified using a fluorescent reporter gene and the TGT assay (Fig. 2d,e). Mutation generally led to sNRR isoforms with lower activation thresholds, resulting in domains with varying tension tolerances against biologically relevant pN levels42. For example, conservatively mutating Tyr186 to Phe decreased the activation threshold below 54 pN, and more severely mutating to Ala furthered this effect. Weakening single mutations were generally additive when combined into doubly mutated domains (Supplementary Fig. 5A). One mutation (F102Y) may have led to an increase in mechanical stability, although marginal and difficult to resolve using TGTs (Supplementary Fig. 5B,C).
Of particular use was the Y186F variant (sNRRY186F), which is weakened such that it activates to a greater extent than the original sNRR on strong TGTs while still resisting activation by 12 pN TGTs. Immunostaining confirmed that mutated receptors were trafficked to the cell surface with comparable efficiencies (Supplementary Fig. 6). We also note that sNRR-containing receptors exhibit reduced ligand-independent activation (LIA) in the absence of stimulus, as compared to their NRR-based counterpart (Fig. 2e). Reduced LIA has been targeted as a means to improve SynNotch functionality43. The difference in LIA for sNRR is increasingly apparent when the ICD is replaced with more potent transcriptional activators VP64-p65 or VP64-p65-RTA (Supplementary Fig. 7).
Further analysis of sNRR mechanical strength showed that scFv insertion provides the primary mechanism of stabilizing these engineered domains. In T-cell acute lymphoblastic leukemia (T-ALL), mutations within the NRR can result in destabilized domains that readily undergo LIA44. However, these same T-ALL mutations, when distal to the scFv binding site, did not affect the mechanical strength of sNRR. Instead, presence of the scFv rescues the hyperactive phenotype, providing evidence that scFv insertion provides stabilization that is a new rate-limiting step in receptor activation (Supplementary Fig. 8). Only NRR mutations at the scFv binding interface destabilized sNRR-SynNotch receptors, consistent with results from our collection of scFv mutations. NRR mechanical stability depends on the presence of calcium ions, and LIA can be ectopically induced through calcium chelation using EDTA45. Interestingly, sNRR domains resist activation by EDTA treatment (Fig. 2f). This result holds true for mechanically weakened sNRR variants, including the highly destabilized sNRRR103A mutant, which was similar to NRR-SynNotch in TGT analyses. Together, these results indicate that scFv fusion provides a dominant autoinhibitory effect. As EDTA is a routine reagent used inf cell culture, sNRR domains serendipitously provide a SynNotch variant that can be stably integrated and expanded in cells while resisting inadvertent LIA.
Gene circuits specify distinct responses to differing tensions
Using our set of sNRR domains, we demonstrated further versatility in cellular outputs by designing gene circuits that selectively filter various magnitudes of tension. Synthetic and natural signaling networks can use filtering logic to detect an input, such as small molecule concentration, only if it falls within the desired range. Described in this framework, the mechanogenetic circuits shown thus far generate ‘high-pass filters,’ in which a target gene is expressed only in response to tension beyond a threshold Ttol. Recognizing the engineering utility of ‘low-pass’ and ‘band-pass’ filters that detect low and intermediate forces, respectively, we sought to extend our sNRR-based genetic circuits to achieve these complex outputs.
To build a low-pass filter, we devised a circuit in which a target gene is expressed only if tensions fall below an activation threshold (Supplementary Fig. 9A). In this approach, activation of a sNRRY186F-SynNotch receptor directs the transcription of a microRNA (miRNA), which in turn inhibits the translation of a constitutively transcribed fluorescent protein gene encoding mCerulean (CMV-mCer-target-miR-FF4; Methods; Supplementary Fig. 9B). We observe that this circuit downregulates mCerulean fluorescence in response to increasing molecular tension, as designed. This behavior is reminiscent of natural mechanisms in which mesenchymal stem cells use miRNA to downregulate gene expression in response to stiff ECM46. In addition to the transcriptional systems demonstrated thus far, the use of miRNA also highlights the ability to install force-activated post-transcriptional control.
In addition to a low-pass filter, we also engineered a band-pass capable of detecting intermediate force magnitudes. Here, we reasoned that band-pass behavior could more closely mimic that of induced pluripotent stem cells, which pursue distinct cell fates in response to soft, medium or rigid ECM stiffnesses8. To implement such logic, we combined multiple receptors in a single cell to implement an incoherent feedforward loop (IFFL; Fig. 3a). The IFFL is a signaling motif commonly employed to create a band-pass response with respect to time or concentration47,48, and we hypothesized that we could extend the IFFL to similarly create a band-pass behavior with respect to molecular tension. In the activating arm of the loop, an NRR-SynNotch possessing a Gal4-VP64 ICD is used to drive expression of a mCerulean-encoding reporter gene (UAS-mCerulean). In the repressing arm of the loop, a sNRRY186F-SynNotch with a TetR-based ICD is used to drive a red fluorescent Gal4-KRAB fusion, which can repress UAS-regulated mCerulean expression. Cotransfected cells encoding the IFFL circuit exhibited the expected response profiles, with mCerulean expression occurring only in response to intermediate tension values (Fig. 3b–d).
Fig. 3: Band-pass filtering in mechanogenetic circuits.
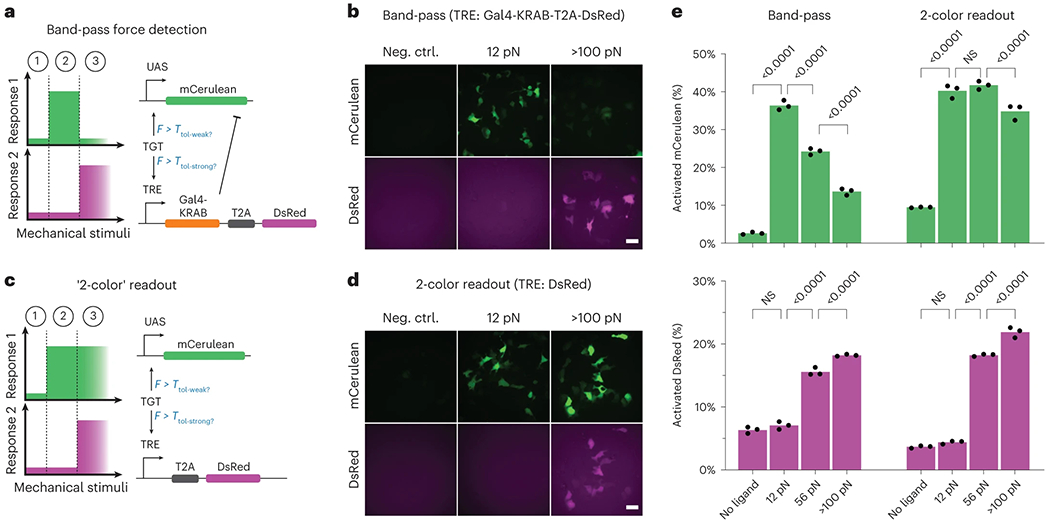
a, Schematic of the IFFL for detecting intermediate forces. An NRR-based SynNotch with a Gal4-VP64 ICD drives expression of a mCerulean encoding gene upon its activation. A sNRRY186F-based SynNotch with tTA ICD drives the expression of a bicistronic gene encoding DsRed-Express2 (DsRed-E2) in combination with Gal4-KRAB, the latter of which is able to inhibit transcription from UAS-mCerulean. b, Fluorescence images of reporter activity in HEK293-FT cells containing the band-pass circuit and grown on control (no ligand) surfaces, or on those containing either 12 pN TGTs (12 pN) or neutravidin-immobilized FITC (>100 pN). c, Schematic of ‘2-color’ readout system, in which high-tension ligands induce dual expression of mCerulean and DsRed-E2 fluorescent proteins. d, Fluorescence images of reporter activity in HEK293-FT cells expressing 2-color readout circuits. Cells were grown on surfaces like those in b. e, Quantified readouts of mCerulean (top graphs) and DsRed-E2 (bottom graphs) expression in cells expressing the indicated mechanogenetic circuits and grown on control surfaces (no ligand), or on those containing 12 pN TGTs (12 pN), 54 pN TGTs (54 pN) or neutravidin-immobilized FITC (>100 pN). Mean values from three independent cotransfections (n = 3) are displayed, as measured by flow cytometry (over 1,000 cells assessed per replicate) and analyzed with two-way ANOVA (mechanogenetic circuit and TGT threshold). P values of multiple comparisons within circuit type, across TGT thresholds are shown on the plot. Scale bars are 25 μm.
We also tested a circuit in which sNRRY186F-SynNotch was made to activate a TRE-DsRed-Express2 (TRE-DsRed-E2) reporter gene, without repressing UAS-mCerulean. As expected, cells grown on high-tension ligands expressed both mCerulean and DsRed-E2, whereas intermediate tension ligands activated only mCerulean (Fig. 3d). This result confirms the IFFL circuit function and shows that cells can be made to express distinct gene combinations in response to different tension levels. In this case, dual expression of cyan and red fluorescent proteins provides a ‘two-color’ readout indicative of cells engaged with high-tension signals.
Force transmission requires strong ligand–receptor bonds
Having demonstrated their synthetic utility, we next aimed to apply our receptors to measure intercellular force transmissions. To do so, we replaced TGTs with encodable ligands based on the GFP, which was selected given its ability to interact with the LaG17 nanobody present within our receptor ECDs. Before testing cell-based ligands, we determined whether LaG17-GFP complexes could endure the tension levels required for sNRR activation using streptavidin-immobilized GFP. sNRR-SynNotch cells were refractory under these conditions, suggesting a restriction in the tension levels that can be propagated via LaG17-GFP bonds. To overcome this limitation, LaG17 was replaced with the LaG16 nanobody, which we expected to bear increased tension loads against GFP, given its reduced off-rate constant (koff) compared to that of LaG17 (koff = 1.1 × 10−3 s−1 for LaG16-GFP; koff = 1.4 × 10−1 s−1 for LaG17-GFP)49. Consistent with this prediction, LaG16-mediated binding resulted in strong sNRR-SynNotch signaling against immobilized GFP (Supplementary Fig. 10). We thus proceeded with LaG16-containing receptors in trans-activation analyses.
Intercellular forces can be gauged by signaling activities
In natural systems, the mechanical pulling activity of Notch ligands requires their intracellular ubiquitination and entry into the epsin-dependent clathrin-mediated endocytosis (CME) pathway50. Mammalian Notch ligands are ubiquitinated by the E3 ligase Mindbomb 1 (MIB1) and then bound by epsins, which facilitate CME51. During CME, epsins couple with polymerizing actin filaments52 and physically link invaginating pits to cellular force-generating machinery53,54. Although many of the factors required for epsin-dependent uptake are known55, the pulling capacity that they confer to ligands during CME is unknown. To probe this capacity, we next generated a synthetic GFP-based ligand by fusing GFP to a transmembrane domain (TMD) and the intracellular tail of the native Notch ligand DLL1 (Fig. 4a; GFP-TMD-DLL1).
Fig. 4: Intercellular mechanotransduction via cell-generated, ubiquitination-dependent endocytic forces.
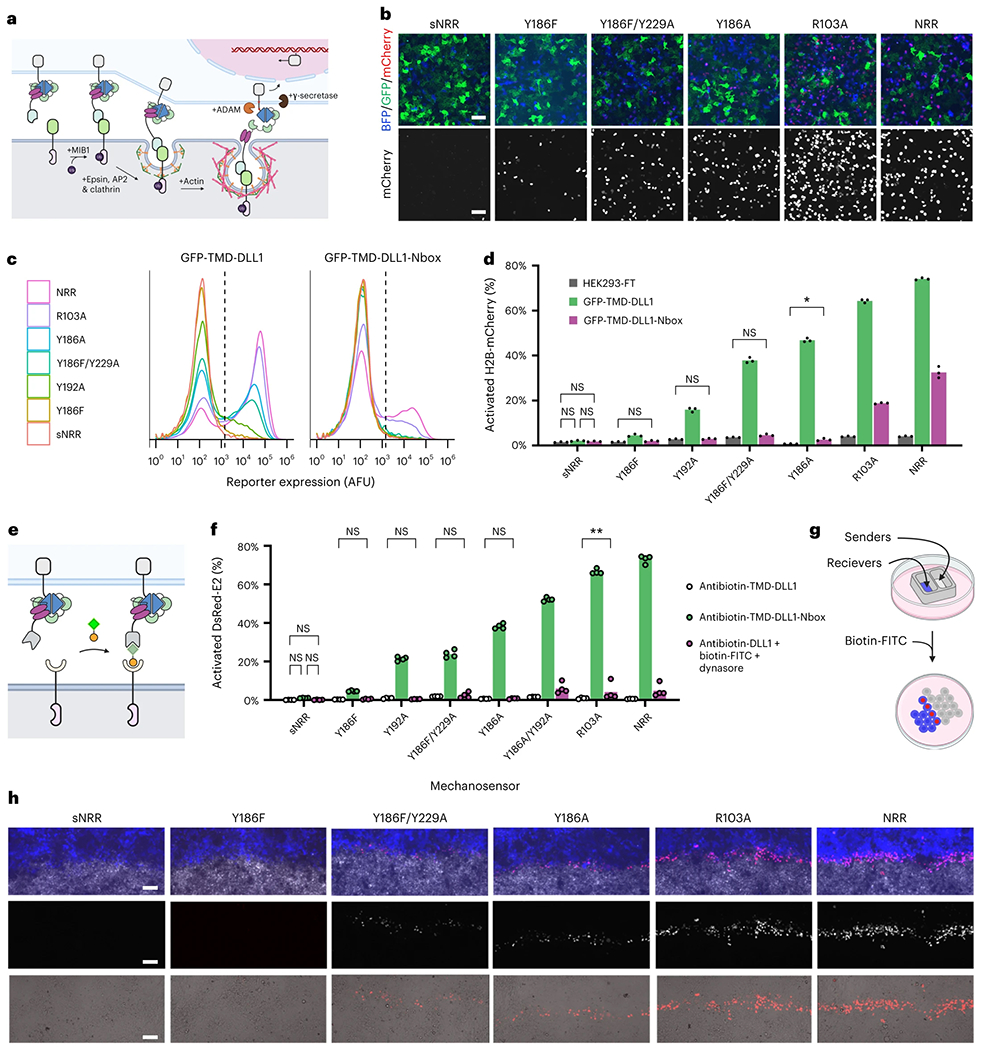
a, Schematic of ligand-mediated SynNotch trans-activation. Ubiquitination of the DLL1 intracellular tail by MIB1 induces its association with cargo-specific adapter proteins (epsins) for uptake by CME machinery. Endocytosis applies a pulling force against the ligand-bound SynNotch to expose its cryptic S2 proteolytic site for proteolysis. b, LaG16-sNRR variants are trans-activated by GFP-TMD-DLL1 senders to varying degrees. Receptors stably integrated in HEK293-FT reporter cells upregulate the expression of a UAS-driven H2B-mCherry reporter gene upon trans-activation by GFP-TMD-DLL1 ligand, also expressed in HEK293-FT (cells were cocultivated for 24 h). c, Flow cytometry traces of stably transduced receiver cell reporter expression levels following coculture with either GFP-TMD-DLL1 or GFP-TMD-DLL1-Nbox sender cells. Traces represent normalized densities of receiver (BFP+) cells within the analyzed coculture populations. Dashed vertical lines indicate H2B-mCherry+ threshold fluorescence values. d, Percent of H2B-mCherry+ reporter cells in receiver cell population (T2A-BFP+) from b shown as mean values alongside those from control cocultures containing mock ligand cells (HEK293-FT). Mean values from three independent experiments (n = 3) are displayed, as quantified by flow cytometry (over 5,000 cells assessed per replicate) and analyzed with two-way ANOVA (ligand version and mechanosensor). All unlabeled comparisons within the same mechanosensor have P < 0.0001; labeled NS, P > 0.05, *P < 0.05, **P < 0.01, otherwise. e, Schematic depicting drug-induced receptor trans-activation. Antibiotin-TMD-DLL1 ligand cells (bottom) undergo trans-cellular complex formation with anti-FITC receiver cells (top) upon exposure to the bispecific small molecule biotin-FITC (shown with FITC as a green square and biotin as an orange circle). f, U2OS receiver cell-signaling activities quantified from nonpatterned sender–receiver mixtures with HEK293-FT antibiotin-TMD-DLL1 senders without exposure to the bridging molecule (gray bars), or after overnight treatment with biotin-FITC (2 nM; green bars) or with dual treatment using biotin-FITC (2 nM) and the dynamin inhibitor dynasore (80 μM; magenta bars). Green and magenta bars represent U2OS receiver cell mixtures with HEK293-FT antibiotin-TMD-DLL1 senders. Gray bars represent nontreated cocultures without biotin-FITC. Quantifications represent fluorescence reporter gene expression levels (U2OS:UAS-DsRed-Express2), shown as DsRed+ cell percentages within receptor-expressing (T2A-BFP+) populations. Mean from four independent experiments (n = 4) displayed, quantified by flow cytometry (over 1,000 cells assessed per replicate) and analyzed with two-way ANOVA (drug treatment and NRR type). All unlabeled comparisons within the same mechanosensor have P < 0.0001; labeled NS, P > 0.05, *P < 0.05, **P < 0.01, otherwise. g, Schematic showing microwell patterning strategy for generating spatially defined sender–receiver interfaces. h, Fluorescence imaging H2B-mCherry reporter expression levels at the interface between sender and receiver populations. HEK293-FT antibiotin-TMD-DLL1 senders were used in combination with HEK293-FT receivers containing a UAS-H2B-mCherry reporter gene. Cells were treated with 2 nM biotin-FITC for 24 h before imaging. Top images: blue represents receiver cells (receptor-T2A-BFP), white marks sender cells (SNAP-Surface-AlexaFluor647) and red represents H2B-mCherry reporter. Middle and bottom images show H2B-mCherry levels in grayscale (middle), or in red as overlaid with transmitted light (bottom). Scale bars in b and d are 100 μm.
GFP-TMD-DLL1 was stably expressed in HEK293-FT cells to generate ligand-expressing ‘sender’ cells. Senders expressing a MIB1-resistant (and thus epsin-binding deficient) ligand were also generated, by expressing a mutant ligand containing an ablated ‘Nbox’ motif (GFP-TMD-DLL1-Nbox), required for MIB1 binding51,56. Live cell immunostaining confirmed the presentation of these ligands on cell surfaces at comparable levels (Supplementary Fig. 11). To evaluate their activities, we cocultured sender cells with receptor-expressing ‘receiver’ cells, allowing trans-cellular stimulation to proceed for 24 h before analyzing receiver cell reporter levels. These analyses confirmed the mechanical activity of GFP-TMD-DLL1 senders, which trans-activated receivers expressing multiple distinct mechanoreceptor domains (Fig. 4b–d). Quantification of signaling intensities showed agreement with TGT-based measurements: domains that resisted 12 pN tethers (including sNRR and sNRRY186F) were refractory against GFP-TMD-DLL1, whereas those that with sensitivity to 12 pN tethers were similarly susceptible against GFP-TMD-DLL1 in trans.
Comparison of signaling levels induced by GFP-TMD-DLL1 versus GFP-TMD-DLL1-Nbox senders revealed CME-dependent and independent activities. Receptors containing mechanically weak domains (NRR and sNRRR103A) were activated at partial levels due to cell–cell contact with GFP-TMD-DLL1-Nbox sender cells (Fig. 4c,d). This result suggests that lateral forces due to trans-cellular LaG16-GFP binding may supply sufficient forces to trigger signaling independent of CME. In contrast, receptors with intermediate strength domains (including sNRRY186A and sNRRY192A) showed explicit requirements for CME, whereas high stability receptors (including sNRR and sNRRY186F) largely resisted CME-generated tensions. Of note, the CME-resistant sNRRY186F domain could be rendered CME-susceptible following addition of a second destabilizing mutation (sNRRY186F/Y229A). These data indicate that ligand uptake, regulated through MIB1-mediated ubiquitination, can confer tension at levels beyond the NRR’s activation threshold (~5 pN), yet below what is needed for sNRRY186F (>12 pN). Overall, our results confirm that sNRR-based receptors can detect and distinguish between pN levels of intercellular forces.
Bifunctional bridges can mechanically couple cells in trans
Recognizing that mechanical signaling events are highly coordinated in vivo, we next sought to gain spatiotemporal control over intercellular force transmissions. To do so, we implemented an inducible mechanical coupling strategy using a bispecific small molecule bridge. In this approach, we used a ligand that contains a biotinamide-binding antibody fragment57,58 followed by a TMD and the DLL1 intracellular tail (antibiotin-TMD-DLL1). Tests using immobilized biotin ligands showed that biotin–antibiotin bonds could withstand the tension levels needed for sNRR activation (Supplementary Fig. 12). Furthermore, trans-activation between antibiotin-TMD-DLL1 senders and anti-FITC receivers could be induced via treatment with soluble biotin-FITC at low-picomolar concentrations (Supplementary Fig. 13, Fig. 4e–h and Supplementary Video 1). To quantify drug-induced signaling levels, we used U2OS receiver cells containing a UAS-DsRed-Express2 reporter gene. Receivers were mixed with antibiotin-TMD-DLL1 senders and reporter levels were measured after 24 h treatments with 2 nM biotin-FITC. Under these conditions, activity levels were consistent with our TGT- and GFP-TMD-DLL1-based measurements, and the endocytic requirement of these signals was confirmed via cotreatment with the dynamin inhibitor dynasore (Fig. 4f). Together, these results further confirm the distinct mechano-sensitivities of our tension-tuned receptors.
Finally, to achieve spatiotemporal control, we applied removable microwell inserts to generate defined sender–receiver boundaries (Fig. 4g). Following microwell removal and cell growth, biotin-FITC was added to cultures to activate coupling between sender and receiver cells. After overnight incubation with the drug, fluorescence imaging revealed spatially confined signaling activities that were limited to receiver cells residing at sender–receiver boundaries (Fig. 4h). These results confirmed the requirement of sender–receiver contact in generating biotin-FITC-induced signaling responses.
DISCUSSION
In summary, we developed tension-tuned SynNotch receptors that can serve as genetically encoded tensiometers. These receptors can be used not only to engineer mechanical signaling responses but also to decipher and record tensile forces as quantifiable readouts of a cell’s current or past mechanical interactions. The versatility of the system enabled force detection via diverse ligand forms, including FITC-labeled TGTs, GFP-tagged proteins and a bifunctional small molecule based on biotin-FITC.
To permit the tuning of receptor sensitivities, we first stabilized the NRR by fusing it with an inhibitory anti-NRR antibody fragment, producing an sNRR that required ten times more tension for signaling activity. This requirement was then lowered by altering binding interactions within the domain’s ‘mechano-active’ site using a structure-guided mutagenesis approach. This strategy allowed us to populate the sensitivity gap between NRR and sNRR domains, resulting in 14 mechanoreceptor sequences with tensile sensitivities that span the biologically relevant pN range. When expressed by cells, these proteins could be used to activate gene expression responses following stimulation with extracellular and intercellular tensional cues.
We demonstrated the utility of our system in two important ways: first, in a synthetic biology approach, we applied our receptors to design cells that could distinguish between mechanical signals and enact customized transcriptional responses in turn. This strategy was used to implement mechanical ‘filtering logic,’ which enabled cells to express a reporter gene only in response to intermediate tension values. A tension-dependent differentiation circuit was also developed, and we used this program to define the force levels needed to induce muscle cell-like phenotypes in fibroblast-based progenitor cells.
In a biological application, we used our receptors to measure cell-generated, biomechanical forces exchanged between cells during Notch signaling. We showed that chimeric ligands containing DLL1 intracellular tails could deliver tensions at levels beyond the ~5 pN rupture requirement of the NRR, yet at levels below that which is supplied by 12 pN TGTs. These values agree with previous laser-trapping measurements, where DLL1 cells were found to pull Notch-coated beads with ~10 pN energies59. Like native DLL1, the pulling capability of chimeric ligands also required MIB1-binding and dynamin activity.
There should be several advantageous applications of our tools moving forward. For example, intermediate strength receptors (which explicitly required ligand CME for signaling) could be used in mapping synaptic connections between neurons by exploiting presynaptic forces due to recycling neurotransmitter vesicles. Such an approach may provide a specific and encodable anterograde tracing method in which signaling due to nonsynaptic cell–cell proximity could be overcome. Furthermore, combined use with drug-based bridging agents could be used to achieve temporal resolution in connectivity mapping, which would allow researchers to monitor changes in the formation and elimination of synapses over time.
During our experimentation, we identified a design feature that should be considered in future adaptations of the method—that ligand–receptor bonds must be of sufficient tensile strength to propagate forces to NRR/sNRR domains. Thus, new ligand–receptor pairings should be tested using immobilized ligands before trans-cellular analyses. Antibody–antigen interactions are recommended as a starting point for new designs, given that these complexes generally form strong bonds34. Antibody–antigen bonds could be directly encoded as ligand–receptor pairings or exploited by combining anti-FITC receptors with FITC-conjugated antibodies or polypeptides as soluble bridging agents. Such an approach could be used to investigate mechanosensitive pathways beyond Notch signaling, including those involving integrins, cadherins and immune cell antigen receptors.
Finally, our work demonstrates how biological insights regarding natural mechanisms can guide the design of sophisticated and useful cell-signaling components. The data also reinforce a long-established paradigm in protein engineering: that by adding extra stability, one can confer mutational robustness to proteins, and as a result, render them more susceptible to engineering60. Although our work focused on the transcription-based outputs, future adaptations of the method could enable direct mechanical control over processes such as genome editing, metabolism or cell-killing activity. Use of iterative or continuous library screening methods may accelerate the development of such systems. As mechanosensitive proteins continue to be structurally and mechanistically elucidated, we anticipate that the mechanogenetic ‘toolkit’ will continue to grow; such expansion should in turn facilitate the development of new targeted therapies and tissue-engineered systems.
METHODS
Mammalian cell culture
Mammalian cell lines were cultured in a humidified incubator maintained at 37 °C with 5% CO2. Cell media were based on the Dulbecco’s Modified Eagle’s Medium (DMEM), pH 7.0–7.4, containing high glucose and sodium pyruvate and without L-glutamine (SH30285.01, Cytiva or similar), used with supplements as indicated below. FBS was typically Cytiva Characterized Fetal Bovine Serum, Canadian Origin (SH30396.03, Cytiva), or Corning Regular Fetal Bovine Serum (35010CV, Corning). HEK293-FT cells (R70007, ThermoFisher) were cultured in DMEM with 10% FBS and supplemented with 1× nonessential amino acids (25025CI, Corning) and 1× GlutaMAX (3505006, ThermoFisher). HeLa cells (CCL-2, ATCC), C3H/10T1/2 cells (CCL-226, ATCC), HEK293 (CRL-1573, ATCC) and U-2 OS cells (92022711-1VL, Sigma-Aldrich) were cultured using the same medium but with added 1× penicillin-streptomycin solution (15140122, ThermoFisher).
DNA constructs
DNA constructs were generated via standard cloning procedures, typically using Gibson assembly reactions. Inserts were generated by PCR amplification or acquired as custom-synthesized DNA fragments (Integrated DNA Technologies, IDT). Plasmid backbones were linearized by digestion with restriction enzymes, or via amplification by PCR followed by template elimination using DpnI. Cloning of lentiviral backbones and sequences containing repeat regions was performed using NEB Stable Competent Escherichia coli (E. coli) cells as the transformation host (C3040H, New England Biolabs).
Plasmid DNAs and full sequence details for selected constructs have been deposited with AddGene.
DNA transfections
DNA transfections were carried out using Lipofectamine 3,000 Reagent (L3000001, ThermoFisher) according to the manufacturer’s instructions. For analyses involving transient transfection of HEK293-FT derived reporter cells (UAS:H2B-mCherry, described below), 25 ng of receptor-encoding plasmid DNA was used in combination with 25 ng of a separate plasmid encoding a constitutively expressed fluorescent protein as a cotransfection marker. Mixtures containing 50 ng of total DNA were used to transfect ~150,000 reporter cells. In cases where receptor-T2A-BFP fusions were transiently expressed from lentiviral (LV) backbones, the same number of reporter cells were transfected with 50 ng of receptor-T2A-BFP plasmid without additional transfection marker DNA. For luciferase reporter measurements: 50 ng of plasmid DNA containing a UAS-regulated firefly luciferase gene (UAS-FLuc) reporter gene (5xGAL4-TATA-luciferase, AddGene, 46756) was combined with 1 ng of cotransfection marker encoding a constitutively expressed NanoLuciferase (pNL1.1.TK(Nluc/TK), Promega); this mixture was used to transfect ~150,000 cells.
For ‘low-pass’ genetic circuits: three plasmids were cotransfected into cells, including (1) a miRNA ‘target’ plasmid encoding a constitutively expressed mCerluean (mCer) gene tagged with an 8×-miRNA target sequence targeted by the FF4 miRNA (based on CMVp-ECFP-Triplex-28-8xmiRNA-BS-28-pA (Construct 22), AddGene, 55199)61, (2) a TRE-regulated gene encoding mKate2 containing an intronic FF4 microRNA (miR-FF4) and (3) a receptor-encoding plasmid in which TetR/tTA was used as the ICD. Cells were triply transfected with 25 ng of the mCerulean target plasmid, 25 ng of receptor-encoding plasmid and 100 ng of the TRE-regulated mKate2/miR-FF4 plasmid. Receptor activity was used to regulate TRE promoter activity and mKate2/miR-FF4 for miRNA-mediated regulation of the targeted mCerulean gene.
For ‘band-pass’ genetic circuits, individual constructs were designed to encode cross-swapped receptor–reporter pairings such that only cells containing both plasmids would exhibit receptor-mediated reporter gene activities; 25 ng of plasmid DNA encoding a Gal4-containing receptor and containing either a TRE-DsRed-Express2 gene cassette or a TRE-Gal4-KRAB-T2A-DsRed-Express2 gene cassette was used in combination with 25 ng plasmid encoding a tTA-containing receptor and containing a UAS-mCerulean reporter gene cassette.
Note that the utilized UAS-regulated FLuc sequence corresponds to a reporter plasmid originally described in 1994 and designated ‘5×GAL4-TATA-luciferase’62. Reliable and reproducible signal detection was achieved using the plasmid. More recent commercial FLuc reporters encode degron-fused enzymes and contain other enhancements that are reported to increase induction levels and response rates. These systems may be useful when increased sensitivity is required (see Promega Technical Manual TM259: pGL4 Luciferase Reporter Vectors, Section 3).
Lentiviral production and transduction
For viral transductions, lentiviral particles were generated using a second-generation lentiviral vector system. HEK293-FT (ThermoFisher) cells were grown to approximately 90% confluence in a six-well dish and transfected with 750 ng of construct-encoding transfer plasmid alongside 1.25 μg each of packaging (pPax2) and envelope encoding (pVSVG) plasmids using the Lipofectamine 3,000 Reagent. Transfection media was replaced the following morning. Viral supernatants were collected at 24-h and 48-h time points following media exchange. Supernatants were passed through low protein-binding 0.45 μm filters before immediate use, or storage by freezing at −80 °C. For viral transduction, cells were infected in growth media via the addition of viral supernatant; viral media were replaced with fresh media at 24 and 48 h time points following transfection.
Clonal reporter cell line generation
A reporter cell line containing a stably integrated Gal4-dependent reporter gene construct (UAS:H2B-mCherry) was generated by transfecting HEK293-FT cells with linearized reporter plasmid. The sequence used was based on pEV-UAS-H2B-Citrine (a gift from M. Elowitz, Caltech) and modified to encode an H2B-mCherry-V5 fusion protein in place of H2B-citrine. The resulting plasmid is designated ‘pEV-UAS-H2B-mCherry’. The UAS promoter contains 14×GAL4 DNA binding sites upstream of the H2B-mCherry gene followed by a BGH poly-adenylation (pA) signal. The plasmid also contained a zeocin resistance marker expressed from the CMV promoter and terminated by an SV40-polyadenylation (pA) signal. The CMV promoter was introduced to drive zeocin resistance in place of an original SV40 promoter/origin sequence. Replacement of the SV40 promoter/origin with CMV was intended to prevent episomal replication of the integrated construct in HEK293-FT cells (which express SV40 large T antigen). For reporter line generation, pEV-UAS-H2B-mCherry was linearized by digestion outside of the fluorescent protein and zeocin resistance expression cassettes and the resulting DNA was transfected into HEK293-FT cells. At 72 h post-transfection, stable integrants were selected using 100 μg ml−1 zeocin. Single clones were isolated by limited dilution into 96-well plates. An optimal reporter clone (designated ‘E5’) was chosen for propagation based on its low background H2B-mCherry levels and its high inducibility upon expression of a Gal4-based transcription factor (pcDNA3-Gal4-VP64). The clonal line was maintained in media containing geneticin at 500 μg ml−1 and zeocin at 100 μg ml−1. Note that HEK293-FT cells carry geneticin resistance.
A U2OS reporter cell line containing a stable, virally integrated UAS:DsRed-Express2 reporter was generated by transducing cells with viral constructs encoding UAS:DsRed-Express2 and PGK:PuroR gene cassettes. The construct was generated by modifying the LV-TRE-Empty-T2A-DsRed-Express2 vector (AddGene, 60623). Briefly, the TRE promoter was replaced with a UAS-based sequence containing 5×UAS/GAL4-binding sites and the original PGK:rTetR-IRES-PuroR cassette was replaced with a PGK:PuroR gene. The 5×UAS regulatory sequence from AddGene 79123 (pHR_Gal4UAS_IRES_mC_pGK_tBFP) was used without the original IRES component. The gene cassettes did not contain terminating pA signals within the lentiviral backbone. Viral particles were produced as described above and stable integrants were selected using 1.0 μg ml−1 puromycin. Single clones were isolated by limited dilution and screened for low background DsRed-Express2 expression and high inducibility, both as described above. An optimal clone (designated ‘1G4’) was chosen for propagation and was maintained in media containing 0.5 μg ml−1 puromycin.
Sender and receiver cell line generation
Stable (receptor-expressing) receiver lines were generated by lentiviral integration of EF1A-driven polycistronic constructs encoding individual receptors followed by T2A-BFP and an IRES-driven hygromycin resistance gene. Viral particles generated as described above were used to transduce HEK293-FT:UAS-H2B-mCherry (clone E5) and U2OS:UAS-T2A-DsRed-Express2 (clone 1G4) reporter cells. Stable integrants were selected 48 h following transduction using Hygromycin-B Gold (ant-hg-5, InvivoGen) at concentrations of 75 μg ml−1 for HEK293-FT and at 200 μg ml−1 for U2OS cells. Transductions were performed in 24 well plates and cells were moved to six-well plates for selection; initial hygromycin exposure was applied at cell densities of no higher than ~25% confluence for efficient elimination of nonintegrated cells. Selection with Hygromycin-B-Gold typically required 10–12 d of growth in the presence of the drug, with media replenishment every 1–2 d. Transduced receivers were used as stable pools without clone isolation. Receivers based on the HEK293-FT reporters were maintained using 75 μg ml−1 Hygromycin-B-Gold and 100 μg ml−1 zeocin; receivers made using U2OS reporters were maintained using 200 μg ml−1 Hygromycin-B-Gold and 0.5 μg ml−1 puromycin. For cloning of lentiviral-encoded receptor constructs, gene fragments corresponding to a receptor and T2A-BFP were inserted into the pLV-EF1A-IRES-Hygro vector backbone (AddGene, 85134).
For GFP-TMD-DLL1 and GFP-TMD-DLL1-Nbox senders, DNA fragments were cloned into a doxycycline-inducible ‘all-in-one’ lentiviral backbone based on the AddGene plasmid 60627. Intracellular tails sequences are based on rat DLL1. The mutated GFP-TMD-DLL1-Nbox ligand contains a triple alanine substitution within the intracellular ‘Nbox’ motif (IKNTNKK to IAAANKK). Ligand-encoding DNAs were inserted downstream of the TRE promoter in place of the existing p65-MyoD-T2A-DsRed-Express2 fusion gene (AddGene, 60627). The vector backbone contained an hPGK promoter-driven rtTA2 followed by an IRES-driven puromycin selection. Transduced HEK293-FT cells were selected 48 h after transduction using puromycin at 0.5 μg ml−1. Puromycin selection typically required 5–7 d, with regular replenishment of the antibiotic-containing media. Transduced senders were used as stably integrated pools without clone isolation. HEK293-FT sender cells containing an integrated EF1A-antibiotin(scFv)-SNAP-TMD-DLL1-IRES-blasticidin construct are reported elsewhere58.
For myogenic differentiation, inducible C3H/10T1/2 cells were generated using a previously reported construct encoding a TRE-regulated p65-MyoD fusion (LV-TRE-p65 mouse MyoD-T2A-DsRed-Express2, AddGene, 60627) (ref. 38). Transduced cells were selected using 2 μg ml−1 puromycin at 48 h post-transduction. Single clones were isolated from drug-selected pools via limited dilution into 96-well plates. Note that the referenced TRE-p65-MyoD construct also contains a PGK-driven cassette encoding the doxycycline-sensitive transcription factor rtTA2. Thus, line generation, maintenance and analyses of transduced C3H/10T1/2 cells were carried out under tetracycline-free conditions with media containing tetracycline-free FBS (SH30070.03T from GE Healthcare, or 631106 from Takara, with similar results). The differentiation capacity of single isolated clones was verified using doxycycline treatment at 1 μg ml−1 to verify TRE-dependent myogenic conversion before experiments using TGTs.
Tension Gauge Tether (TGT) synthesis
To test the molecular tension requirements for NRR- and sNRR-based SynNotch activation, we immobilized TGT-based SynNotch ligands to a microwell plate using fabrication protocols described previously in ref. 63. These methods were followed closely, with modifications as indicated. Custom single-stranded DNA (ssDNA) sequences were ordered from IDT. For TGT experiments, receptors containing the E2 antifluorescein scFv were utilized in combination with the following fluorescein-containing oligonucleotide ‘ligand strand’:
Ligand strand: 5′-GGC CCG CAG CGA CCA CCC/36-FAM/-3′
The sequence above was paired with complementary ssDNAs as ‘tethering strands.’ These strands contained amine functional groups located at varying positions along the ssDNA sequence; the amines were later modified with amine-reactive biotin as an immobilization handle (see below). Amine containing complementary strands were ordered from IDT as follows:
For 12 pN TGTs: 5′-/5AmMC6/GGG TGG TCG CTG CGG GCC-3′
For 43 pN TGTs: 5′-GGG TGG TCG C/iAmMC6T/G CGG GCC-3′
For 54 pN TGTs: 5′-GGG TGG TCG CTG CGG GCC/3AmMO/-3′
Modified nucleotide positions are indicated above in between forward slashes as designated by their IDT modification codes. Chemical structures for the associated modifications are provided in the Supplementary Information.
To generate biotin-modified tethering strands, the amine containing oligonucleotides were reacted with NHS-ester functionalized PEG12-biotin (EZ-Link NHS-PEG12-biotin; 21312, ThermoFisher). Biotinylation of the amine-modified strands was carried out as previously reported in ref. 63.
Following biotinylation, batch preparations of biotin-modified tethering strands were stored at 100 μM concentrations in a 10 mM Tris buffer (pH 7.4) containing 150 mM NaCl. Dried FITC-conjugated ligand strands received from the supplier were dissolved to 100 μM concentrations in a buffer of the same composition. Aliquots of the ssDNA solutions were kept frozen at −20 °C until use. To prepare dsDNA TGT duplexes, FITC-conjugated ligand strand DNA was combined with individual tethering strand sequences at 1:1 (vol/vol) mixtures. Annealing reactions were carried out by overnight incubation at 4 °C resulting in 50 μM dsDNA TGT stock solutions.
Tension Gauge Tether (TGT) assay
Details for TGT fabrication and immobilization are provided in the Supplementary Information. Preparation flow cytometry-based analyses of TGT-treated cells were done using plastic, tissue culture (TC)-grade non-TC treated 96-well microwell plates. When preparing cells for imaging TGT-induced responses, plates with noncoated coverslip glass bottoms were used. Coating, immobilization and wash solutions were applied to microwell plates by multichannel pipetting, typically using 100 μl volumes.
To immobilize TGTs, microwells surfaces were treated with solutions containing a mixture of 10 μg ml−1 biotinylated BSA (A8549, Sigma-Aldrich) in combination with 10 μg ml−1 fibronectin (AAJ62380LB0, ThermoFisher) in PBS for 1 h at room temperature before washing three times with PBS. Neutravidin (31000, ThermoFisher) was then immobilized to the biotin-coated surfaces by treating with a solution diluted to 100 μg ml−1 in PBS. Binding proceeded for 30 min at room temperature before washing three times with PBS. TGT from the prepared 50 μM dsDNA stocks were then diluted to 100 nM in PBS and added to the neutravidin-treated microwells. TGT binding to neutravidin surfaces was allowed to proceed for 1 h at room temperature before washing with PBS three times.
For ‘upper limit’ Ttol controls, fluorescein conjugated to biotin via a short PEG linker was used (22030, ThermoFisher). In this case, the ‘upper limit’ Ttol is defined as the tension level required to disengage the biotin-streptavidin bonds (generally estimated as 100 pN or more64,65). Attachment to neutravidin coated cells was done using 100 nM solution in PBS. Binding proceeded for 30 min at room temperature before washing with PBS three times.
Cells prepared for TGT analysis were transfected in suspension mixtures as follows: HEK293-FT reporter cells (UAS:H2B-mCherry) cells were trypsinized and the quenching media was removed before resuspending cells in fresh media. Prepared DNA-lipid transfection mixtures were then added to cell suspensions and gently mixed seeding with TGT-containing wells using ~30,000 cells per well. Cells were analyzed for receptor-mediated reporter expression 48 h later. Cell differentiation experiments were done using transduced C3H/10T1/2 cells; cells were analyzed for myogenic conversion at 72 h following TGT exposure.
Coculture trans-activation assays
Nontreated tissue culture 96-well plates were coated using 10 μg ml−1 fibronectin (FN) solutions in PBS for 1 h at room temperature. Wells were then washed three times with PBS and allowed to dry. Sender and receiver cell populations were then trypsinized using a prewarmed 0.25% trypsin solution without EDTA (25200114, Gibco, or similar). Note that calcium chelation by EDTA can lead to ligand-independent Notch/SynNotch activation. Cells trypsinized using the EDTA-free solution were quenched with prewarmed culture media before centrifugation, aspiration and resuspension in fresh prewarmed media. Cell densities were measured using an automated cell counter (Countess II, ThermoFisher) and suspensions were inspected for homogenous cell distributions before plating. Note that cell clumping was minimized when using prewarmed trypsin and media solutions.
For trans-activation analyses, sender and receiver cells were combined at 2:1 ratios (sender:receiver) and thoroughly mixed before seeding in FN-coated 96 microwells plates. Cells were cocultivated for 24 h before reporter expression analyses via flow cytometry or fluorescence microscopy. The following cell numbers were used to generate cocultures at the 96-well scale: 40,000 HEK293-FT senders mixed with 20,000 HEK293-FT receivers, or 10,000 U2OS receivers mixed with 20,000 HEK293-FT senders.
When using sender cells containing doxycycline-regulated ligands constructs, ligand expression was activated using 100 μg ml−1 doxycycline 24 h before generating cell mixtures. Doxycycline was maintained in the coculture medium at the same concentration during the duration of cocultivation. For experiments involving dynamin inhibition, cells were exchanged into prewarmed Opti-MEM I Reduced Serum Medium (31985062, ThermoFisher) containing 80 μM dynasore (D7693-5MG, Sigma-Aldrich) and 2 nM biotin-FITC (25574, Cayman Chemical); exchange into dynasore/biotin-FITC containing Opti-MEM was performed at 12 h following initial coculture seeding.
Imaging and preparation of microwell-patterned trans-activations
For microwell-patterned trans-activations, we used nontreated 35 mm glass-bottom imaging dishes (P35G-1.0-20-C, MatTek). Dishes coated with fibronectin using 20 μg ml−1 fibronectin solutions in PBS with overnight incubation at 37 °C. Surfaces were washed three times with PBS and allowed to dry at 4 °C. A three-well silicone insert containing two defined cell-free gaps (80369, ibidi) was applied to the center of the coated dishes using sterile forceps. Gentle pressure was applied to ensure attachment of the insert with the glass via its adhesive coating. Cells were then seeded into chamber wells at ~35,000 cells per well suspended in roughly 70 μl culture medium (corresponding to cell densities of ~500,000/ml). Care was taken to ensure even cell disbursement in each well (media and solutions were prewarmed to 37 °C and cells were mixed via gentle pipetting). After seeding cells, 1.5 ml of culture medium was added to the area surrounding the insert to limit evaporation from well interiors. Dishes were capped and cells were returned to the incubator for attachment and growth. When confluence within the chambers reached ~90% (~24 h later), microwell inserts were carefully removed using sterile forceps. Following an additional ~18 h of growth, media were exchanged with fresh media containing 2 nM biotin-FITC. After ~24 h, cells were processed for imaging as indicated below.
Reporter (HEK293-FT:UAS-H2B-mCherry) expression levels were detected at sender–receiver interfaces and recorded using epifluorescence microscopy. Before imaging, senders in the patterned cocultures were fluorescently marked by labeling with a cell impermeant SNAP-tag dye (SNAP-Surface AlexaFluor647, S9136S, New England Biolabs). Dye was diluted to a 4 μM final concentration in prewarmed culture media (1:250 dilution from a 1 mM dye stock in DMSO). The solution was thoroughly mixed by pipetting and gentle vertexing to ensure full dye dissolution and dye-containing media was incubated at 37 °C for an additional 15 min before application to cells. Labeling was initiated by exchanging cells into the prewarmed dye-containing media using ~600 μl of media per 35 mm imaging dish. Staining proceeded for 30 min at 37 °C before removal of the dye followed by gentle rinsing three times using prewarmed dye-free media (1 ml per rinse). Cells were then placed in 2 ml of fresh dye-free media and returned to the incubator for 30 min before imaging. Cells were exchanged into prewarmed FluoroBrite DMEM imaging media (A1896701, ThermoFisher) containing supplements like the growth media as described above and containing 20 mM HEPES (pH 7.4) and Hoechst 33342 (at 2.5 μg ml−1) as a counterstain.
Myogenic differentiation assay
Clonal C3H/10T1/2 reporter cells with stably integrated TRE:p65-MyoD-T2A-DsRed were transduced with lentiviral constructs encoding NRR- or sNRR-based receptors. At 48 h postinfection, cells were treated with EDTA-free trypsin and plated on TGT- and FN-coated 8 well-chambered glass coverslips (80826, ibidi) at 50,000 cells per well. Following 72 h of growth on TGTs, cells were fixed and permeabilized and myogenic differentiation was assessed by immunofluorescence detection of myosin heavy chain expression in combination with DAPI counterstaining for identification of multinucleated fibers. See Supplementary Information for immunostaining reagents and protocol details.
Protein expression and purification
For expression of soluble anti-NRR scFv-Fc fusion, DNA encoding an scFv corresponding to anti-Notch1-NRR originally described in ref. 28 was subcloned into the pBIOCAM5 vector backbone (AddGene, 39344). The fragment was inserted between NcoI and NotI restriction sites following removal of the existing insert. The resulting plasmid encodes an in-frame secreted fusion construct containing the scFv followed by a human Fc region and C-terminal 6×His and 3×FLAG tags. Transient expression of this construct was carried out by transfection of HEK293-FT in 100 mm tissue culture dishes. The secreted protein was collected by harvesting conditioned media over the course of 7 d. The collected media was stored at 4 °C and combined before passage through a low protein-binding 0.2 μm filter. The filtrate pH was adjusted by adding pH 8 Tris-buffered saline solution to a final concentration of Tris at 20 mM and 300 mM NaCl. The scFv-Fc-6xHis-3×FLAG fusion was then purified using the Ni-NTA Fast Start Kit (30600, Qiagen) following the manufacturer’s protocol. Imidazole was removed from protein eluents by dialysis against PBS at 4 °C using 10,000 MWCO Slide-A-Lyzer Dialysis Cassettes (66380, ThermoFisher). Purity was assessed by staining SDS-PAGE gels with GelCode Blue Stain Reagent (24590, ThermoFisher) with colorimetric quantification against a BSA standard serially diluted from a 2 mg ml−1 stock solution (23209, ThermoFisher).
For expression of eGFP: DH10B E. coli (EC0113, ThermoFisher) carrying plasmid DNA encoding an arabinose-inducible 6×His-eGFP (EGFP-pBAD, AddGene 54762) were selected on LB-agar containing 100 μg ml−1 ampicillin. A single clone was transferred to 5 ml LB media with ampicillin and grown overnight at 37 °C with agitation. The following day, 4 ml of culture was used to inoculate 200 ml of LB-ampicillin. The culture was allowed to grow until OD = ~0.7, at which point the flask was transferred to a 30 °C incubator for 30 min before adding arabinose to a final concentration of 0.2% (wt/vol). Protein expression proceeded at 30 °C with shaking at 250 rpm for 4 h. Cells were collected by centrifugation and stored as frozen pellets at −20 °C until purification. 6×His-eGFP was purified using the Ni-NTA Fast Start Kit (30600, Qiagen) according to the manufacturer’s protocol. Imidazole was removed from protein eluents by gel filtration with PD-10 Sephadex G-25 resin columns (GE17-0851-01, Millipore-Sigma) followed by dialysis against PBS using 10,000 MWCO Slide-A-Lyzer Dialysis Cassettes (66380, ThermoFisher). The EGFP-pBAD vector was maintained in cells using 100 μg ml−1 ampicillin. The purity of the isolated protein was assessed by staining SDS-PAGE gels using GelCode Blue Stain Reagent; GFP concentration was determined using the bicinchoninic acid protein assay (BCA Protein Assay Kit, 23225, ThermoFisher).
For expression of mono-biotinylated GFP: EGFP-pBAD plasmid was modified by inserting an in-frame DNA segment encoding a tandem linked AviTag-ALFA tag (GLNDIFEAQKIEWHE-GSGGS-RLEEELRRRLTE-GSGTKAS) in between 6×His tag and eGFP (pBAD-6×His-AviTag-ALFAtag-eGFP). Expression was carried out as described for eGFP with modifications. An E. coli strain with inducible biotin ligase (BirA) was used as the expression host (AVB101, Avidity). At the time of arabinose induction, d-biotin was added to 50 μM along with isopropyl β-d-1-thiogalactopyranoside (IPTG) at 1.5 mM (to induce BirA overexpression in AVB101). AVB101 was maintained using 10 μg ml−1 chloramphenicol.
Immunofluorescence staining
Cells grown in medium were rinsed with PBS before fixation with a prewarmed formaldehyde solution diluted to 4% (vol/vol) in PBS from a vial containing 16% (vol/vol) stock solution (28906, ThermoFisher). Fixation was allowed to proceed for 10 min at room temperature. Cells were then rinsed three times with PBS followed by blocking for 1 h at room temperature using a BSA solution at 5% (wt/vol) in PBS. Staining was carried out for 1 h at room temperature using primary antibody or DLL4-Fc diluted in 1% (wt/vol) BSA solution in PBS at the amounts indicated above. Cells were rinsed three times using PBS for 5 min per rinse before staining with secondary antibodies at the dilutions indicated above. When necessary, cells were permeabilized with a PBS solution containing 0.2% Triton X-100 (vol/vol).
For visualizing receptor recycling from the cell surface, live cells were stained with soluble purified GFP before fixation. Staining was carried out for either 45 min at 4 °C (to prevent receptor internalization; Supplementary Fig. 1B), or under internalization-permissive conditions for 30 min at 37 °C (Supplementary Fig. 1C). Labeled cells were then washed three times using PBS for 5 min each, at either 4 °C or room temperature, respectively. Cells were fixed using 4% formaldehyde before surface-selective labeling against the NRR. Immunofluorescence detection of the NRR was carried out using purified anti-Notch1-NRR-scFv-Fc in all cases except when colabeling cells with Dll4-Fc, which also contains the human Fc region. Thus, Dll4-Fc was used in combination with mice anti-NRR-E6 when colabeling Notch1-expressing cells. Surface-selective NRR detections were performed using fixed cell specimens without permeabilization, both as described above.
Preparation of cell lysates and immunoblotting
Cells were prepared for immunoblot analysis by rinsing with PBS followed by direct lysis using a 1× LDS-PAGE loading buffer solution (NP0007, Invitrogen). Lysates were then sonicated or sheared (by passage through a thin gauge syringe needle) to reduce lysate viscosities before clarification by centrifugation. Subsequent immunoblot preparation, including gel electrophoresis and membrane transfer, was carried out using standard procedures. Nitrocellulose membranes were blocked using solutions containing 5% nonfat dry milk (wt/vol) or 5% BSA (wt/vol), both diluted in PBS-Tween 20 (PBS-T, 0.1%, vol/vol). Blocking solutions were selected based on the specifications provided by primary antibody vendors. Primary and secondary antibody probing solutions were prepared at the dilutions indicated above. Detection of labeled antigens was carried out by chemiluminescence using the SuperSignal West Pico PLUS Chemiluminescent Substrate (34580, ThermoScientific). Membranes were stripped using the Restore Western Blot Stripping Buffer (21059, ThermoFisher) and reblocked before reprobing.
Luciferase assay for surface-adhered ligand and EDTA treatment
HEK293-FT cells were triply transfected with three constructs: a UAS-regulated firefly luciferase reporter plasmid (5xGAL4-TATA-luciferase, AddGene, 46756), a receptor-encoding plasmid and a cotransfection marker based on a plasmid encoding a constitutively expressed NanoLuciferase (NLuc) construct controlled by the thymidine kinase (TK) gene promoter (pNL1.1.TK(Nluc/TK), Promega).
To investigate ligand-mediated activation of NRR- and sNRR-based anti-FITC SynNotch receptors, transfected cells were grown in FN-coated microwells with or without immobilized biotin-FITC. To coat surfaces, nontreated tissue culture microwells were treated with protein-containing solutions as follows: for control surfaces without ligand, well were treated with PBS solutions containing FN at 10 μg ml−1; for ligand-containing surfaces, wells cotreated using PBS solutions containing FN at 10 μg ml−1 in combination with 10 μg ml−1 biotinylated BSA. Coating proceeded for 1 h at room temperature before rinsing with PBS. Wells containing biotinylated BSA were subsequently treated with neutravidin followed by biotin-FITC, as described in the TGT methods.
To evaluate dependence on ADAM10 and γ-secretase cleavage, cells were treated with either the broad-spectrum metalloprotease inhibitor batimastat (BB-94; SML-0041, Sigma-Aldrich) at 20 μM, or using the γ secretase inhibitor Compound E (15579, Cayman Chemical) at 400 nM concentration. When using the γ secretase DAPT (N-(N-(3,5-difluorophenacetyl)-L-alanyl)-S-phenylglycine t-butyl ester), 10 μM concentrations were applied (HY-13027, MedChemExpress). To evaluate the requirement of ligand-mediated tension, cells were treated with soluble fluorescein (fluorescein, sodium salt, 46960-25G-F, Sigma-Aldrich) in culture medium at the concentrations indicated within the captions. Luciferase expression levels were measured 24 h following transfection using the Nano-Glo Dual-Luciferase Reporter Assay System (N1620, Promega) according to the manufacturer’s protocol.
To investigate receptor activation dependent on calcium chelation, transfected cells were grown for 24 h before treatment with either PBS or with PBS containing 0.5 mM EDTA for 15 min at 37 °C. Note that PBS without added calcium and magnesium was used. Following EDTA treatment, the chelator was quenched by the addition of growth media to the cell suspensions. Cells were incubated under growth conditions for an additional 6 h before luciferase measurement using the Nano-Glo Dual-Luciferase Reporter Assay System as described above.
Flow cytometry
Cells were analyzed at roughly 48 h after transfection using an Attune NxT flow cytometer (ThermoFisher). Live cells were identified by setting minimum FSC-A and SSC-A gating thresholds (Supplementary Fig. 14A). Singlet cells were then identified by setting a polygon gate on FSC-A versus FSC-H (Supplementary Fig. 14B). Positively transfected cells were identified via detection of their cotransfection or self-cleaving ‘T2A’ fluorescent protein markers, gated based on the 99th percentile of fluorescence signal under these conditions detected from nontransfected control cells (Supplementary Fig. 14C). To improve detection of cells expressing both a receptor of interest and cotransfection marker, positively transfected cells were then gated for those bearing marker fluorescence emission at levels above the median of all transfected cells. In the case of virally transduced cells analyzed in Fig. 3, fluorescent marker gating was not applied to the high efficiency of lentiviral transduction for the associated analyses. Receptor cells in coculture assays were gated by BFP+. Experimental groups were analyzed for the fraction of cells representing positively activated reporter gene expression, defined based on the comparison to control reporter cells (lacking any receptor) using emission intensities from the 99th percentile of control cells analyzed under identical excitation and detection parameters. Flow cytometry data were analyzed and quantified using the open-source ggCyto software (version 1.27.1; https://github.com/RGLab/ggcyto). An example of the gating procedures utilized is provided below in Supplementary Fig. 14.
Data collection and analysis software
Fluorescence images were collected using the Zen 2.3 Pro (Blue Edition) imaging software and analyzed in ImageJ v2.0.0. Flow cytometry data were collected using Attune NxT v2.6 and v3.1 flow cytometry software and analyzed using ggCyto v1.27.1. Western blots were collected and analyzed using the QuantityOne (4.5.2) immunoblotting software or the iBright Imaging System software v1.4.0.
Statistics and reproducibility
The data shown in the figures, and especially the depicted fluorescence micrographs, are representative examples of results that were repeated during at least two independent experiments. Statistical analyses were performed using the GraphPad Prism v9.0.0 software.
Supplementary Material
Supplementary Video 1. Time-lapse imaging of biotin-FITC induced trans-cellular activation.
HEK293-FT reporter cells (UAS-H2B-mCherry) expressing anti-FITC receptors with the indicated mechanosensitive domains were cocultured with HEK293-FT antibiotin-TMD-DLL1 sender cells. trans-cellular coupling was induced by treatment with 2 nM biotin-FITC. Cells were imaged live for 24 h. Fluorescence emissions from T2A-BFP (blue, to identify receiver cells) and H2B-mCherry (red, to monitor reporter activity) were captured every 1.5 h. Sender cells were not fluorescently marked (that is, lacked BFP). Top row: merged images from detection of T2A-BFP (blue), H2B-mCherry (red) and transmitted light. Bottom row: emission from H2B-mCherry detection shown in grayscale.
Acknowledgements
D.C.S. and A.M.M. were supported through Graduate Research Fellowship awards from the National Science Foundation. J.C.T was supported through a Cross-Disciplinary Fellowship awarded through BUnano (Boston University Nanotechnology Innovation Center). D.C.S and J.C.T. received support through the Boston University training program in Quantitative Biology and Physiology (QBP, NIH grant T32GM008764). D.C.S. was a recipient of a Kilachand Fellowship through the Multicellular Design Program (Boston University). Support for this work was provided through a seed grant from the Center for Multiscale & Translational Mechanobiology (Boston University) and through NIH research grants R35 GM128859 (to J.T.N.) and R01 HL147585 (to J.T.N.). Additional support was provided through the Reidy Family Career Development Professorship at Boston University (to J.T.N.). The schematics in Fig. 4 were created using biorender.com.
Footnotes
Competing interests
J.T.N. and D.C.S. are inventors on an issued patent (U.S. Patent, 10,858,443) held by the Trustees of Boston University. J.C.T. and A.M.M. declare no competing interests.
REFERENCES
- 1.Schoen I, Pruitt BL & Vogel V The Yin-Yang of rigidity sensing: how forces and mechanical properties regulate the cellular response to materials. Annu. Rev. Mater. Res 43, 589–618 (2013). [Google Scholar]
- 2.Wells RG Tissue mechanics and fibrosis. Biochim. Biophys. Acta 1832, 884–890 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahn C & Schwartz MA Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol 10, 53–62 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mammoto T, Mammoto A & Ingber DE Mechanobiology and developmental control. Annu. Rev. Cell Dev. Biol 29, 27–61 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Trylinski M & Schweisguth F Activation of Arp2/3 by WASp is essential for the endocytosis of δ only during cytokinesis in Drosophila. Cell Rep. 28, 1–10 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Bertolio R et al. Sterol regulatory element binding protein one couples mechanical cues and lipid metabolism. Nat. Commun 10, 1326 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Helvert S, Storm C & Friedl P Mechanoreciprocity in cell migration. Nat. Cell Biol 20, 8–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engler AJ, Sen S, Sweeney HL & Discher DE Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Das DK et al. Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl Acad. Sci. USA 112, 1517–1522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elosegui-Artola A et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171, 1397–1410 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Sharma D et al. Single-molecule force spectroscopy reveals a mechanically stable protein fold and the rational tuning of its mechanical stability. Proc. Natl Acad. Sci. USA 104, 9278–9283 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bornschlögl T, Christof J, Gebhardt M & Rief M Designing the folding mechanics of coiled coils. ChemPhysChem 10, 2800–2804 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Ng SP et al. Designing an extracellular matrix protein with enhanced mechanical stability. Proc. Natl Acad. Sci. USA 104, 9633–9637 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadler DP et al. Identification of a mechanical rheostat in the hydrophobic core of protein L. J. Mol. Biol 393, 237–248 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao Y, Yoo T, Zhuang S & Li H Protein–protein interaction regulates proteins’ mechanical stability. J. Mol. Biol 378, 1132–1141 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Cao Y, Yoo T & Li H Single molecule force spectroscopy reveals engineered metal chelation is a general approach to enhance mechanical stability of proteins. Proc. Natl Acad. Sci. USA 105, 11152–11157 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ringer P et al. Multiplexing molecular tension sensors reveals piconewton force gradient across talin-1. Nat. Methods 14, 1090–1096 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Hughes JH & Kumar S Synthetic mechanobiology: engineering cellular force generation and signaling. Curr. Opin. Biotechnol 40, 82–89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan Y et al. Mechanogenetics for the remote and noninvasive control of cancer immunotherapy. Proc. Natl. Acad. Sci. USA 115, 992–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu LN et al. Mechanoresponsive stem cells to target cancer metastases through biophysical cues. Sci. Transl. Med 9, eaan2966 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon WR et al. Mechanical allostery: evidence for a force requirement in the proteolytic activation of Notch. Dev. Cell 33, 729–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X & Ha T Defining single molecular forces required to activate integrin and Notch signaling. Science 340, 991–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chowdhury F et al. Defining single molecular forces required for Notch activation using Nano Yoyo. Nano Lett. 16, 1–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seo D et al. A mechanogenetic toolkit for interrogating cell signaling in space and time. Cell 165, 1507–1518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morsut L et al. Engineering customized cell sensing and response behaviors using synthetic Notch receptors. Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verdorfer T & Gaub HE Ligand binding stabilizes cellulosomal cohesins as revealed by AFM-based single-molecule force spectroscopy. Sci. Rep 8, 9634 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiyanont K et al. Evidence for increased exposure of the Notch1 metalloprotease cleavage site upon conversion to an activated conformation. Structure 19, 546–554 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y et al. Therapeutic antibody targeting of individual Notch receptors. Nature 464, 1052–1057 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Aste-Amézaga M et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS ONE 5, e9094 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fortini ME & Bilder D Endocytic regulation of Notch signaling. Curr. Opin. Genet. Dev 19, 323–328 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwesinger F et al. Unbinding forces of single antibody-antigen complexes correlate with their thermal dissociation rates. Proc. Natl. Acad. Sci. USA 97, 9972–9977 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varnum-Finney B et al. Immobilization of Notch ligand, δ-1, is required for induction of Notch signaling. J. Cell Sci 113 Pt 23, 4313–4318 (2000). [DOI] [PubMed] [Google Scholar]
- 33.De Odrowąz Piramowicz M, Czuba P, Targosz M, Burda K & Szymoński M Dynamic force measurements of avidin-biotin and streptavdin-biotin interactions using AFM. Acta Biochim. Polym 53, 93–100 (2006). [PubMed] [Google Scholar]
- 34.Weisel JW, Shuman H & Litvinov RI Protein–protein unbinding induced by force: single-molecule studies. Curr. Opin. Struct. Biol 13, 227–235 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Falk R et al. Generation of anti-Notch antibodies and their application in blocking Notch signalling in neural stem cells. Methods 58, 69–78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chopra ML et al. Force generation via β-cardiac myosin, titin, and α-actinin drives cardiac sarcomere assembly from cell-matrix adhesions. Dev. Cell 44, 87–96 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakar MS et al. Cellular forces and matrix assembly coordinate fibrous tissue repair. Nat. Commun 7, 11036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabadi AM et al. Enhanced MyoD-induced transdifferentiation to a myogenic lineage by fusion to a potent transactivation domain. ACS Synth. Biol 4, 689–699 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balcioglu HE, van Hoorn H, Donato DM, Schmidt T & Danen EHJ The integrin expression profile modulates orientation and dynamics of force transmission at cell-matrix adhesions. J. Cell Sci 128, 1316–1326 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Austen K et al. Extracellular rigidity sensing by talin isoform-specific mechanical linkages. Nat. Cell Biol 17, 1597–1606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Carrion-Vazquez M, Oberhauser AF, Marszalek PE & Fernandez JM Point mutations alter the mechanical stability of immunoglobulin modules. Nat. Struct. Biol 7, 1117–1120 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Polacheck WJ & Chen CS Measuring cell-generated forces: a guide to the available tools. Nat. Methods 13, 415–423 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Z, Yu Z, Cai Y, Du R & Cai L Engineering of an enhanced synthetic Notch receptor by reducing ligand-independent activation. Commun. Biol 3, 116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gordon WR et al. Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood 113, 4381–4390 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rand MD et al. Calcium depletion dissociates and activates heterodimeric Notch receptors. Mol. Cell. Biol 20, 1825–1835 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frith JE et al. Mechanically-sensitive miRNAs bias human mesenchymal stem cell fate via mTOR signalling. Nat. Commun 9, 257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mangan S & Alon U Structure and function of the feed-forward loop network motif. Proc. Natl Acad. Sci. USA 100, 11980–11985 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greber D & Fussenegger M An engineered mammalian band-pass network. Nucleic Acids Res. 38, e174 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fridy PC et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 11, 1253–1260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langridge PD & Struhl G Epsin-dependent ligand endocytosis activates Notch by force. Cell 171, 1383–1396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo B, McMillan BJ & Blacklow SC Structure and function of the Mind bomb E3 ligase in the context of Notch signal transduction. Curr. Opin. Struct. Biol 41, 38–45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Messa M et al. Epsin deficiency impairs endocytosis by stalling the actin-dependent invagination of endocytic clathrin-coated pits. eLife 3, e03311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaksonen M & Roux A Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol 19, 313–326 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Serwas D et al. Mechanistic insights into actin force generation during vesicle formation from cryo-electron tomography. Dev. Cell 57, 1132–1145 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seib E & Klein T The role of ligand endocytosis in Notch signalling. Biol. Cell 113, 401–418 (2021). [DOI] [PubMed] [Google Scholar]
- 56.McMillan BJ et al. A tail of two sites: a bipartite mechanism for recognition of notch ligands by mind bomb E3 ligases. Mol. Cell 57, 912–924 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dengl S et al. Hapten-directed spontaneous disulfide shuffling: a universal technology for site-directed covalent coupling of payloads to antibodies. FASEB J. 29, 1763–1779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McMahan JB, Ngo JT A Genetically encodable and chemically disruptable system for synthetic post-translational modification dependent signaling. Preprint at bioRxiv 10.1101/2022.05.29.493928 (2022). [DOI] [Google Scholar]
- 59.Meloty-Kapella L, Shergill B, Kuon J, Botvinick E & Weinmaster G Notch ligand endocytosis generates mechanical pulling force dependent on dynamin, epsins, and actin. Dev. Cell 12, 22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bloom JD, Labthavikul ST, Otey CR & Arnold FH Protein stability promotes evolvability. Proc. Natl Acad. Sci. USA 103, 5869–5874 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nissim L, Perli SD, Fridkin A, Perez-Pinera P & Lu TK Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol. Cell 54, 698–710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun P, Enslen H, Myung PS & Maurer RA Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 8, 2527–2539 (1994). [DOI] [PubMed] [Google Scholar]
- 63.Wang X et al. Constructing modular and universal single molecule tension sensor using protein G to study mechano-sensitive receptors. Sci. Rep. 6, 21584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee CK, Wang YM, Huang LS & Lin S Atomic force microscopy: determination of unbinding force, off rate and energy barrier for protein–ligand interaction. Micron 38, 446–461 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Johnson KC & Thomas WE How do we know when single-molecule force spectroscopy really tests single bonds? Biophys. J 114, 2032–2039 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1. Time-lapse imaging of biotin-FITC induced trans-cellular activation.
HEK293-FT reporter cells (UAS-H2B-mCherry) expressing anti-FITC receptors with the indicated mechanosensitive domains were cocultured with HEK293-FT antibiotin-TMD-DLL1 sender cells. trans-cellular coupling was induced by treatment with 2 nM biotin-FITC. Cells were imaged live for 24 h. Fluorescence emissions from T2A-BFP (blue, to identify receiver cells) and H2B-mCherry (red, to monitor reporter activity) were captured every 1.5 h. Sender cells were not fluorescently marked (that is, lacked BFP). Top row: merged images from detection of T2A-BFP (blue), H2B-mCherry (red) and transmitted light. Bottom row: emission from H2B-mCherry detection shown in grayscale.