

Abstract
Point mutants of three unrelated antifluorescein antibodies were constructed to obtain nine different single-chain Fv fragments, whose on-rates, off-rates, and equilibrium binding affinities were determined in solution. Additionally, activation energies for unbinding were estimated from the temperature dependence of the off-rate in solution. Loading rate-dependent unbinding forces were determined for single molecules by atomic force microscopy, which extrapolated at zero force to a value close to the off-rate measured in solution, without any indication for multiple transition states. The measured unbinding forces of all nine mutants correlated well with the off-rate in solution, but not with the temperature dependence of the reaction, indicating that the same transition state must be crossed in spontaneous and forced unbinding and that the unbinding path under load cannot be too different from the one at zero force. The distance of the transition state from the ground state along the unbinding pathway is directly proportional to the barrier height, regardless of the details of the binding site, which most likely reflects the elasticity of the protein in the unbinding process. Atomic force microscopy thus can be a valuable tool for the characterization of solution properties of protein-ligand systems at the single molecule level, predicting relative off-rates, potentially of great value for combinatorial chemistry and biology.
Atomic force microscopy (AFM) has been considered as a new tool in the characterization of ligand-receptor interactions (1–9). Its sensitivity can hardly be undercut, being able to detect and quantify the interaction of single molecules. Furthermore, images of protein surfaces can be recorded at subnanometer resolution (10, 11). AFM technology also may carry the potential of massive parallel implementation, which might become an important tool in the age of combinatorial chemistry and genomics research. Unfortunately, the relationship of the key parameter, the unbinding force measured when the ligand is mechanically pulled out of the binding site, to any thermodynamic or kinetic parameter describing macroscopic ligand binding has remained unclear. Therefore, the merit of determining these unbinding forces for a series of different ligand-receptor pairs to further aid ligand optimization, receptor design, or screening processes has remained rather questionable.
The parameters of prime interest in describing any biological ligand-receptor system displaying simple reversible 1:1 binding are the rates of spontaneous association (kon) and dissociation (koff), and their ratio, the dissociation constant KD = koff/kon, which describes the equilibrium behavior. Clearly, each one of these will be a function of environmental parameters, such as pH and temperature. Both rate constants give a measure of the highest transition state of an intrinsically complicated multistep process. For example, the observed on-rate usually is considered as a product of a translational diffusion process and a rotational diffusion of the ligand, such that it can dock (12–14). The on-rates of small molecules are remarkably similar to each other, at least in those systems where slow conformational changes do not take place and electrostatics play a minor role. The off-rate describes the highest transition state in leaving the binding pocket, which usually is related to the number and the quality of all atomic interactions within the binding pocket. The off-rate usually correlates well with the equilibrium constant KD for related receptor-ligand systems, kon normally being confined to a rather narrow range of values.
There has been a considerable debate as to whether the unbinding force in related receptor-ligand systems should be expected to correlate with KD and/or koff or rather, whether the AFM experiments are carried out in a range of loading rates, which lead to so much faster unbinding than the spontaneous dissociation process that the ligand is forced across “unnatural” transition states. In this case, we would expect either no clear correlation of the unbinding force with koff, or perhaps a correlation to other parameters, such as ΔH‡ (15) or even ΔHeq (16), which describe the enthalpic changes in the transition state or the ground state, respectively, but ignore entropic factors such as solvent or amino acid side-chain reorientation. These slow reorientation processes require some sort of near-equilibration during unbinding, which might not happen if the process is mechanically forced to be too fast.
For any use of AFM in the characterization of ligand-receptor pairs, the existence of a correlation between unbinding force and solution kinetics or thermodynamics would be of pivotal importance, and we decided to test for this correlation by carefully measuring these parameters. As a model system we chose three unrelated fluorescein-binding antibodies in the form of monomeric single-chain Fv (scFv) fragments and several point mutants thereof. These systems are simple in the sense that (i) they follow a 1:1 binding model without kinetically detectable allosteric effects and (ii) the ligand is inert and rigid. We determined for each protein KD at equilibrium, kon, koff, and the temperature dependence of koff in solution. The unbinding forces were determined by AFM on single isolated molecules, to avoid the need of any deconvolution of force spectra (5, 6). We also tested the dependence of unbinding forces on pulling velocity to determine (i) whether there is any nonlinearity that would indicate a change of unbinding mechanism and transition state with pulling speed and (ii) to extrapolate to the unbinding rate at zero force. By using both related and unrelated antibodies, we could test for correlations within a system and across systems. We found a remarkable correlation of the unbinding forces to the off-rates across all systems, indicating that in the AFM experiment the molecular systems are following a dissociation path not too dissimilar to the one at zero force.
Methods
Expression and Purification of scFv Fragments.
All scFv fragments were cloned in and expressed with the secretion vector pAK400 (17, 18) in Escherichia coli SB536 (19) and purified over a Ni2+-nitrilotriacetic acid column (Qiagen, Chatsworth, CA) (17). Remaining impurities were removed in a second step with a Sepharose-SP column (Amersham Pharmacia) [buffer: 20 mM 2-mercaptoethanesulfonic acid (Mes)/50 mM NaCl, pH 6.0; elution with a gradient of NaCl].
Off-Rate Measurements.
Off-rates were measured in solution by using a competitive dissociation assay including a weakly fluorescent analog of fluorescein, 5-aminofluorescein (Sigma) (20), which occupies the binding site after dissociation of fluorescein to prevent rebinding. A 1 nM solution of fluorescein (Sigma) was equilibrated with protein until the fluorescence, which is quenched upon binding, reached a stable base line. The scFv-fluorescein complex then was mixed with 0.3 μM 5-aminofluorescein, and the increasing fluorescence of free fluorescein was measured with a fluorimeter (Photon Technology International, Princeton) at 520 nm (excitation at 490 nm). All off-rates could be determined by a monoexponential fit of the data.
On-Rate Measurements.
Association kinetics for all scFv fragments were measured in solution with a stopped-flow fluorimeter (Hi-Tech Scientific, Salisbury, U.K.) at an excitation wavelength of 490 nm and with an emission filter (cut-off at 530 nm). Using a volume ratio of 1:1, five different concentrations of scFv fragment were mixed with the same concentration of fluorescein, usually 0.2 μM. The apparent first-order constants (kobs) were plotted against the concentration of scFv fragments. The second-order rate constant then is obtained from a plot of kobs vs. [scFv] by using the relation kobs = kon⋅[scFv]⋅[Flu] + koff, where koff is small enough in the present molecular systems to be neglected.
Fluorescence Titration.
Besides being obtained from koff/kon, dissociation constants of fluorescein also were determined at equilibrium by recording the fluorescence spectra from 505 to 525 nm, using a constant amount of fluorescein and variable scFv concentrations, in a 2-ml cuvette at 25°C (17). From the intensity of the maximum (512 nm) vs. the concentration, the KD was determined by a three-parameter fit (17).
Activation Energies.
To determine activation energies, off-rates were measured at four different temperatures (10°C, 15°C, 20°C, and 25°C). An Arrhenius plot of these data provided the activation energy (21, 22).
AFM Measurements at Constant Loading Rate.
Force-distance measurements with constant loading rate were performed with a commercial AFM (Topometrix Explorer, Santa Clara, CA), and the acquisition of the cantilever deflection data was improved by using a 16-bit AD/DA card (National Instruments, Austin, TX). AFM and data acquisition were controlled by a program written in Visual Basic (Microsoft), based on the software library spmtools (Topometrix), and run on a PC under Windows nt 4.0 (Microsoft). All measurements were performed at 25°C in phosphate buffer (50 mM, pH 7.4) with a vertical piezo velocity of 1 μm/s. Four different scFv fragments were immobilized on separated fields of the same gold chip, which was protected by Mes (23). The hydrophobic lines of octadecanethiol between each field were printed with poly (dimethylsiloxane) stamps (23). Concentrations of scFv fragments and reaction times with the gold surface were identical within the series of all analyzed proteins. Unbinding forces for all immobilized scFv fragments were consecutively measured with the same AFM tip. The most probable unbinding force was determined by fitting a Gaussian to the histogram of the force distribution. The errors were estimated by SD/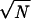 , where SD denotes the width of the distribution and N is the number of unbinding events in the histogram.
, where SD denotes the width of the distribution and N is the number of unbinding events in the histogram.
Loading Rate-Dependent AFM Measurements.
The method is described in detail in Strunz et al. (24). Briefly, force-distance measurements were performed on a commercial AFM (Nanoscope III, Digital Instruments, Santa Barbara, CA) equipped with a similar data acquisition system as described above and a home-build high voltage amplifier for the vertical piezo movement. The loading rate was varied by changing the retract velocity of the piezo for each approach-retract cycle while keeping the approach velocity constant. The loading rate can be determined directly from the slope of the measured force versus time curves before the unbinding events. The spring constants K of all cantilevers (Si3Ni4-Microlever, Thermomicroscopes, Sunnyvale, CA) were calibrated by the thermal fluctuation method (25) in liquid. We used the relation K = kBT/<z2>, where <z2> represents the measured mean square deflection caused by thermal vibrations (measured over the entire spectrum). As we used V-shaped cantilevers, we did not use the correction recently introduced for rectangular cantilevers (26). We estimate the absolute uncertainty of the spring constant to be about 30%. The force measurements at constant and variable loading rates presented here were performed with different cantilevers with spring constants ranging from 8 pN nm-1 to 14 pN nm-1.
Results and Discussion
We studied as our model system a total of nine different scFv fragments, all specific for fluorescein, but derived from three unrelated antibodies. The first six molecules were constructed from FITC-E2, itself obtained from a human scFv phage display library (17, 27). A homology model of the combining site was built, and residues were identified that were suspected to contact the antigen. After all residues had individually been mutated to alanine, binding constants and the percentages of active protein were determined for each mutant. Five mutants were chosen for further studies that cover a range of affinities, but which all gave rise to binding stoichiometries of exactly one fluorescein per scFv molecule, indicating that all molecules are in the active conformation and the mutation had not destroyed the antibody structure. As the second model, the scFv fragment c12 was obtained from a mouse immunized with FITC-BSA, and this clone was isolated by ribosome display (refs. 28–30; L.J., A.H., F.S., Josef Hunes, and A.P., unpublished results). The clone c12 was further affinity-matured by ribosome display in vitro, and the best variant with an affinity increase of about 15-fold carrying seven mutations was chosen for further studies, together with the original c12. Finally, the scFv fragment 4D5-Flu was used, where the complementarity determining regions have been grafted (31) from the poorly expressing scFv 4–4-20 (32) to the well-expressing antibody 4D5 (33). The structures of all three antibodies and the location of the altered amino acids are shown in Fig. 1. To find indications for a possible correlation of unbinding forces to thermodynamic or kinetic parameters, we determined unbinding forces, some of them as a function of loading rate, kinetic parameters of the association and dissociation pathway (on- and off-rates in solution), equilibrium constants, and off-rate-derived activation enthalpies for all antibody model systems.
Figure 1.

Model structures of the scFv fragments c12 B5–6 (A), 4D5-Flu (B), and FITC-E2 (C) with bound fluorescein. Altered amino acid positions are indicated for the scFv fragments c12 B5–6 and FITC-E2 (e.g., His H58 Ala indicates that histidine at position 58 in the heavy chain is mutated to alanine).
Macroscopic Measurements.
To determine off-rates, scFv fragment-fluorescein complexes were mixed with an excess of a weakly fluorescent analogue of fluorescein, 5-aminofluorescein (20), which acts as a competitor. The dissociation of the scFv fragment-fluorescein complex can be followed by an increase in fluorescence of fluorescein, which is quenched in the complex. In each case, first-order kinetics were observed. The range of measured off-rates varies over 3 orders of magnitude, with the slowest off-rate for c12-B5–6 (0.00014 s-1) and the fastest dissociation constant for FITC-E2 His(H58)Ala (0.47 s-1) (Table 1). Association kinetics were measured by using a stopped-flow fluorimeter. The on-rate values for all scFv fragments are in the range of 106 M-1⋅s-1, with a factor of 40 between the slowest and fastest on-rate (Table 1). All gave rise to pseudo-first-order reactions, with the expected proportionality of kobs to antibody concentration. Thus, there is no kinetically detectable isomerization step in the binding kinetics.
Table 1.
Properties of antibody mutants at 25°C
| Mutant | koff, s−1 | kon, 106 M−1⋅s−1 | KD,titr., nM | KD,calc., nM | ΔE‡, kJ/mol | ν, s−1 |
|---|---|---|---|---|---|---|
| FITC-E2 H(H58)A | 0.47 | 11.9 | 15 | 39 | 80.2 | 9.10⋅1013 |
| 4D5-Flu wt | 0.062 | 2.7 | 9.7 | 22.9 | 95.9 | 7.55⋅1015 |
| FITC-E2 Y(H100g)A | 0.019 | 1 | 7.9 | 19 | 90.2 | 2.23⋅1014 |
| FITC-E2 L(H56)A | 0.0125 | 2.5 | 1.8 | 5 | 117.5 | 1.08⋅1019 |
| FITC-E2 F(H100f)A | 0.0062 | 0.3 | 9.6 | 20 | 108.1 | 1.13⋅1017 |
| FITC-E2 H(H100e)A | 0.0052 | 1 | 2.7 | 5.2 | 108.4 | 1.07⋅1017 |
| FITC-E2 wt | 0.0044 | 1.8 | 1.1 | 2.4 | 113.8 | 8.33⋅1017 |
| c12 wt | 0.0028 | 2.5 | 1.5 | 1.1 | 101.6 | 3.54⋅1015 |
| c12 B5-6 | 0.00014 | 3.4 | 0.1 | 0.04 | 107.5 | 2.00⋅1015 |
Dissociation rates (koff), association rates (kon), equilibrium dissociation constants (KD,titr.), calculated equilibrium dissociation constants (KD,calc. = (koff/kon)) and activation energies (ΔE‡) and pre-exponential factor (ν) (at 20°C) determined from Eq. 1. The error of reproducibility for koff and kon of all measured mutants is about ± 3%, the error of reproducibility for KD,titr. is about ± 8%.
The equilibrium binding constants were, in addition to being obtained from koff/kon, independently measured by equilibrium fluorescence titration. The highest affinity was again found for c12-B5–6 (0.1 nM) and the lowest affinity was determined for FITC-E2 His(H58)Ala (15 nM) (Table 1). Among the kinetic and equilibrium constants determined, the off-rate measurements are expected to be of highest accuracy, because they depend only on temperature. On-rate measurements also are influenced by temperature, but in addition, the concentration of active protein plays an important role for the determination of correct values. The equilibrium measurements are influenced by temperature, active protein concentration, and antigen concentration. Nevertheless, the ratio of on- and off-rates are in good agreement with the directly measured equilibrium constants, and the discrepancy of a factor of 2 to 3 in KD is rather small for data obtained from three completely independent measurements.
The off-rate was found to follow an Arrhenius law in the investigated temperature range of 10°C to 25°C.
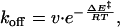 |
1 |
where ΔE‡ is the activation energy and ν is a pre-exponential factor that is taken to be temperature independent in this narrow temperature range. Because of the complexity of the unbinding process of a ligand from a protein pocket, we consider it too difficult to interpret this factor in any mechanistic way (34, 35). The activation energies could be obtained from the slope of plots of ln(koff) vs. 1/T (Table 1), resulting in correlation coefficients of better than 0.995 for all linear fits (data not shown).
In the data set for the nine scFv fragments the pre-exponential factor ν and ΔE‡ are correlated: An increase of the activation energy coincides with a decrease of the pre-exponential factor ν, containing components related to the entropy of activation. A similar behavior has been observed for streptavidin mutants (15). The reason is probably that the scFv fragments have been selected to fall within a certain range of off-rates, but the underlying unbinding process can be either dominated by enthalpic or entropic components. Furthermore, this correlation has been suggested to be favored by the weighting inherent in the parameter fit (22).
Force Spectroscopy.
The fluorescein-binding scFv proteins with a single free thiol group at the C terminus were directly immobilized on an ultraflat gold surface, which was first modified with a 2-mercaptoethanesulfonic acid (Mes) layer to avoid nonspecific binding (6). The surface density of the scFv proteins was found to depend on the incubation time and the concentration of the Mes and the proteins. A typical incubation time of the Mes layer of 1 h at room temperature and subsequent overnight incubation of the proteins at 4°C prevented a complete surface coverage of the proteins. Shorter Mes incubation times of about 10 min resulted in a denser protein coverage. The antigen fluorescein was covalently immobilized on a Si3N4-AFM tip (6). Contrary to our previous experiments, where we localized individual scFv molecules by imaging the surface topology (6), the force experiments were performed at random positions without prior imaging to increase the lifetime of the functionalized AFM tip. Typical samples with submonolayer coverage therefore showed binding events in about 10–15% of the force distance curves.
In a first series of experiments we measured the loading rate dependence of the unbinding force with samples that showed a high binding probability, because the accuracy of the determined loading rate dependence is determined by the statistics of the unbinding forces at each loading rate. For the investigated scFv fragments FITC-E2 wild type (wt) and 4D5-Flu a linear increase of the unbinding forces with the logarithm of the loading rates was found (Fig. 2). This behavior is characteristic for a thermally activated dissociation under an applied load (36–38): An applied force F decreases the activation energy for dissociation from ΔE‡ to ΔE‡(F) with the relation ΔE‡ − ΔE‡(F) = −xβ⋅F, leading to an exponential increase of the off-rate
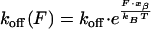 |
with increasing force. The proportionality factor xβ, which has the dimension of length, can be interpreted to describe the difference in separation of the scFv fragment and the fluorescein ligand between the bound and the transition state (projected along the direction of the applied force). Under these conditions the most probable unbinding force F is given by
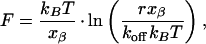 |
2 |
if the force increases with a constant rate r (36). When applying Eq. 2 to our experimental data we can determine the length scale xβ and an AFM-measured off-rate by extrapolating to zero force. From the plot of F vs. ln r (Fig. 2) we can obtain the slope kBT/xβ from Eq. 2, resulting in xβ = 0.4 ± 0.1 nm for FITC-E2 wt and 4D5-Flu. Extrapolated to F = 0, Eq. 2 gives a value for koff = r⋅xβ/kB⋅T. The off-rate values for the FITC-E2 wt (koff-AFM = 0.003 ± 0.002 s-1) and the 4D5-Flu (koff-AFM = 0.10 ± 0.05 s-1) so obtained are in good agreement with the thermal off-rate measured in solution at 25°C (koff = 0.004 s-1 and koff = 0.062 s-1). This agreement indicates that all loading rates in our measurements are so low that the unbinding forces of the scFv fragment-fluorescein system are determined by the same energy barrier that is relevant for the activation enthalpy and entropy of the spontaneous dissociation in solution. Furthermore, a linear extrapolation of the measured values to zero force is in excellent agreement with the value in solution, and there is no necessity to assume any nonlinearity or kink in the plot, even though our experimental system did not allow measuring at the slow loading rates used elsewhere (36–38). Note that the extrapolated zero-force off-rate would be the same for the simultaneous unbinding events even if more than one scFv fragment–fluorescein complex had been formed in a particular approach-retract cycle and for all orientations of the complexes, only the value of xβ would be affected.
Figure 2.

Loading rate dependence of the unbinding forces for FITC-E2 wt and 4D5-Flu at 25°C. For each loading rate more than 100 unbinding events were recorded. The most probable unbinding force was determined by fitting a Gaussian to the distribution of unbinding forces. The statistical error of the unbinding force was estimated as the width of the distribution divided by the square root of the number of unbinding events. The off-rates at zero force, determined from linear fits to the data, agree well with the thermal off-rate, which are indicated by arrows. At zero force Eq. 2 gives a value for r = koff⋅kB⋅T/xβ, allowing the placement of the kinetic constants on the r axis.
Previous measurements on both the avidin/biotin system and the l-selectin/carbohydrate system (36–38) have indicated that the F vs. ln r plot consists of distinct linear regimes. This was interpreted as indicating multiple barriers of which the “outer” ones are governing the rate of spontaneous dissociation in solution and “inner” ones become rate-limiting with higher pulling force. At least avidin is untypical for protein–ligand systems, as the protein closes a flap above the binding site almost occluding biotin. In contrast, the great majority of proteins merely provides a pocket to which the ligand can bind, such as all three antifluorescein antibodies studied here, which do not show a discontinuity in the F vs. ln r plot.
For a correlation of unbinding forces and macroscopic values we performed force measurements on all described scFv fragments at a constant loading rate of 5,000 pN⋅s-1. To minimize experimental errors we used the same cantilever for all measurements within a series and immobilized always the same concentration of proteins, using equal reaction times with the gold surface. The measurements were performed on samples with a low density of immobilized molecules to minimize the formation of multiple antibody-antigen complexes and to ensure an unrestricted orientation of the protein molecules.
We can express both force ΔF = Fmut − Fwt and thermal off-rate relative to the FITC-E2 wt (termed Fwt), where the subscript mut refers to a mutant. Assuming a linear approximation to variations in xβ and koff, we obtain then from Eq. 2
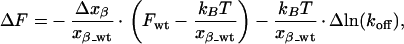 |
3 |
where Δxβ = xβ_mut − xβ_wt and Δln(koff) = ln(koff_mut) − ln(koff_wt). The plot of the data are shown in Fig. 3. An excellent correlation between the force F and the intrinsic thermal off-rate koff is evident. Despite the standard deviation of about 4 pN for the data points, a correlation coefficient of 0.97 was calculated. The slope is significantly different from zero as checked by using the Student's t test (P < 0.0001). It follows that AFM can be used to measure the relative off-rates of a series of single receptor proteins with high accuracy. Remarkably, this correlation holds not only within a series of homologous point mutants but also across three totally unrelated antibodies. We can even draw the distinction between molecules with the same equilibrium affinity (the same ratio koff/kon) but different absolute on- and off-rates, as we have such a pair in terms of 4D5-Flu and FITC-E2 Phe(H100f)Ala (Table 1). It is the off-rate that is reflected in the unbinding force measurement. In contrast, a plot of the unbinding forces vs. the activation energies ΔE‡ or vs. the equilibrium enthalpies ΔGeq shows no linear dependence (data not shown).
Figure 3.

Linear correlation between the force F and ln koff. Forces and off-rates are expressed relative to the FITC-E2 wt.
Upon calculating xβ for each mutant and plotting this value against ln(koff), a remarkable correlation between these values was found. The slope of the
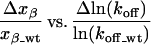 |
plot (Fig. 4) is about 0.3. This means that with increasing height of the transition state ΔE‡ (i.e., lower koff) the apparent “distance” of the barrier along the pulling coordinate from the ground state becomes longer, regardless of the details of the molecular geometry of the three different types of antibody complexes and regardless of the nature of any particular point mutant. A similar relationship of xβ and the height of the transition state ΔE‡ was found for force measurements on single DNA molecules (24), but no thermal off-rates are available for this system. In the DNA experiment, the molecules were varied by making them at the same time longer and stronger, which easily explains this correlation.
Figure 4.

The plot of Δxβ/xβ_wt vs. Δln(koff)/ln(koff_wt) shows the increase of xβ with increasing activation energy. The wt is the unmutated scFv FITC-E2. The slope of the linear fit is 0.3.
In the case of the protein mutants, however, this relationship is not so straightforward. The data suggest that a weaker complex breaks “earlier” along the unbinding path, no matter where exactly the mutation has been made. One explanation would be that the lack of a particular interaction in the ground state leads to a shift of the bound molecule, to move its center closer to the transition state. Alternatively and perhaps more likely, the elasticity of the protein itself may account for some of the additional distance of xβ, as the protein may “follow” the ligand for a longer distance, if the interaction is tighter, when the cantilever retracts. It is remarkable that all three antibodies fall on the same line (Fig. 4), but the detailed interpretation of this relationship may have to await the experimental determination of the structure of the complexes for all mutants.
Conclusions.
Our experiments demonstrate that it is possible to measure the relative off-rates of antibody-antigen complexes at the single molecule level, as the negative logarithms of the off-rates are directly proportional to the unbinding forces across all systems. Because the off-rate is the main determinant of the affinity, AFM can be used to affinity-rank single molecules and predict their equilibrium behavior in solution. The dissociation path followed in the AFM experiment must at least partially allow re-equilibration of solvent and amino acid side chains and thus be in a pseudoequilibrium all along the unbinding pathway. Although the experiment is carried out 103- to 106-fold faster than the spontaneous dissociation, it still occurs on a time scale of tens of milliseconds, which is very slow with respect to molecular motions. The parameter that describes the lowering of the transition state with force, xβ, itself appears to depend on the height of this transition state for the thermal dissociation. A geometric interpretation of this length, xβ, with respect to a detailed binding site geometry of individual interactions cannot be confirmed or excluded before the detailed complex geometry of several point mutants has been experimentally determined. We think it is at least equally likely, however, that this value reflects the elasticity of the proteins, with the rupture distance simply depending on the binding energy of the ligand.
Acknowledgments
We are grateful to Dr. Antonio Baici for help with the stopped-flow fluorimeter. We thank Profs. Amedeo Caflisch and Hanns Fischer for helpful discussions. This work was supported by the Swiss National Research Program 36 Nanoscience, Grant 4036–43973.
Abbreviations
- AFM
atomic force microscopy
- scFv
single-chain Fv
- wt
wild type
References
- 1.Florin E L, Moy V T, Gaub H E. Science. 1994;264:415–417. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- 2.Lee G U, Kidwell D A, Colton R J. Langmuir. 1994;10:354–357. [Google Scholar]
- 3.Hinterdorfer P, Baumgartner W, Gruber H J, Schilcher K, Schindler H. Proc Natl Acad Sci USA. 1996;93:3477–3481. doi: 10.1073/pnas.93.8.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dammer U, Hegner M, Anselmetti D, Wagner P, Dreier M, Huber W, Güntherodt H J. Biophys J. 1996;70:2437–2441. doi: 10.1016/S0006-3495(96)79814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen S, Chen X, Davies J, Davies M C, Dawkes A C, Edwards J C, Roberts C J, Sefton J, Tendler S J B, Williams P M. Biochemistry. 1997;36:7457–7463. doi: 10.1021/bi962531z. [DOI] [PubMed] [Google Scholar]
- 6.Ros R, Schwesinger F, Anselmetti D, Kubon M, Schäfer R, Plückthun A, Tiefenauer L. Proc Natl Acad Sci USA. 1998;95:7402–7405. doi: 10.1073/pnas.95.13.7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong S S, Joselevich E, Woolley A T, Cheung C L, Lieber C M. Nature (London) 1998;394:52–55. doi: 10.1038/27873. [DOI] [PubMed] [Google Scholar]
- 8.Raab A, Han W, Badt D, Smith-Gill S J, Lindsay S M, Schindler H, Hinterdorfer P. Nat Biotechnol. 1999;17:902–905. doi: 10.1038/12898. [DOI] [PubMed] [Google Scholar]
- 9.Fritz J, Kolbinger A, Katopodis A G, Anselmetti D. Proc Natl Acad Sci USA. 1998;95:12283–12288. doi: 10.1073/pnas.95.21.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller D J, Schabert F A, Büldt G, Engel A. Biophys J. 1995;68:1681–1686. doi: 10.1016/S0006-3495(95)80345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karrasch S, Hegerl R, Hoh J H, Baumeister W, Engel A. Proc Natl Acad Sci USA. 1994;91:836–838. doi: 10.1073/pnas.91.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schreiber G, Fersht A R. Nat Struct Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 13.Gabdoulline R R, Wade R C. Biophys J. 1997;72:1917–1929. doi: 10.1016/S0006-3495(97)78838-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elcock A H, Gabdoulline R R, Wade R C, McCammon J A. J Mol Biol. 1999;291:149–162. doi: 10.1006/jmbi.1999.2919. [DOI] [PubMed] [Google Scholar]
- 15.Chilkoti A, Boland T, Ratner B D, Stayton P S. Biophys J. 1995;69:2125–2130. doi: 10.1016/S0006-3495(95)80083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moy V T, Florin E-L, Gaub H E. Science. 1994;266:257–259. doi: 10.1126/science.7939660. [DOI] [PubMed] [Google Scholar]
- 17.Pedrazzi G, Schwesinger F, Honegger A, Krebber C, Plückthun A. FEBS Lett. 1997;415:289–293. doi: 10.1016/s0014-5793(97)01143-5. [DOI] [PubMed] [Google Scholar]
- 18.Krebber A, Bornhauser S, Burmester J, Honegger A, Willuda J, Bosshard H R, Plückthun A. J Immunol Methods. 1997;201:35–55. doi: 10.1016/s0022-1759(96)00208-6. [DOI] [PubMed] [Google Scholar]
- 19.Bass S, Gu Q, Christen A. J Bacteriol. 1996;178:1154–1161. doi: 10.1128/jb.178.4.1154-1161.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boder E T, Wittrup K D. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 21.Chilkoti A, Stayton P S. J Am Chem Soc. 1995;117:10622–10628. [Google Scholar]
- 22.Héberger K, Kemény S, Vidóczy T. Int J Chem Kinetics. 1987;19:171–181. [Google Scholar]
- 23.Ros R, Schwesinger F, Padeste C, Plückthun A, Anselmetti D, Güntherodt H-J, Tiefenauer L. Proc Scanning Force Microscop Biomed Appl. 1999;3607:84–89. [Google Scholar]
- 24.Strunz T, Oroszlan K, Schäfer R, Güntherodt H J. Proc Natl Acad Sci USA. 1999;96:11277–11282. doi: 10.1073/pnas.96.20.11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutter J L, Bechhoefer J. Rev Sci Instrum. 1993;64:1868–1873. [Google Scholar]
- 26.Butt H J, Jaschke M. Nanotechnology. 1995;6:1–7. [Google Scholar]
- 27.Vaughan T J, Williams A J, Pritchard K, Osbourn J K, Pope A R, Earnshaw J C, McCafferty J, Hodits R A, Wilton J, Johnson K S. Nat Biotechnol. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 28.Hanes J, Plückthun A. Proc Natl Acad Sci USA. 1997;94:4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanes J, Jermutus L, Weber-Bornhauser S, Bosshard H R, Plückthun A. Proc Natl Acad Sci USA. 1998;95:14130–14135. doi: 10.1073/pnas.95.24.14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanes J, Jermutus L, Schaffitzel C, Plückthun A. FEBS Lett. 1999;450:105–110. doi: 10.1016/s0014-5793(99)00475-5. [DOI] [PubMed] [Google Scholar]
- 31.Jung S, Plückthun A. Protein Eng. 1997;10:959–966. doi: 10.1093/protein/10.8.959. [DOI] [PubMed] [Google Scholar]
- 32.Whitlow M, Howard A J, Wood J F, Voss E W, Jr, Hardman K D. Protein Eng. 1995;8:749–761. doi: 10.1093/protein/8.8.749. [DOI] [PubMed] [Google Scholar]
- 33.Carter P, Presta L, Gorman C M, Ridgway J B, Henner D, Wong W L, Rowland A M, Kotts C, Carver M E, Shepard H M. Proc Natl Acad Sci USA. 1992;89:4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hänggi P, Talkner P, Borkovec M. Rev Mod Phys. 1990;62:251–431. [Google Scholar]
- 35.Frauenfelder H, Wolynes P G. Science. 1985;229:337–345. doi: 10.1126/science.4012322. [DOI] [PubMed] [Google Scholar]
- 36.Evans E, Ritchie K. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans E. Faraday Discuss Chem Soc. 1998;111:1–16. doi: 10.1039/a809884k. [DOI] [PubMed] [Google Scholar]
- 38.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Nature (London) 1999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]