

Abstract
Dengue virus (DENV) infection is one of the most widely spread flavivirus infections. Despite the fatality it could cause, no antiviral treatment is currently available to treat the disease. Hence, this study aimed to repurpose old drugs as novel DENV NS3 inhibitors. Ligand-based (L-B) and proteochemometric (PCM) prediction models were built using 62,354 bioactivity data to screen for potential NS3 inhibitors. Selected drugs were then subjected to the foci forming unit reduction assay (FFURA) and protease inhibition assay. Finally, molecular docking was performed to validate these results. The in silico studies revealed that both models performed well in the internal and external validations. However, the L-B model showed better accuracy in the external validation in terms of its sensitivity (0.671). In the in vitro validation, all drugs (zileuton, trimethadione, and linalool) were able to moderately inhibit the viral activities at the highest concentration tested. Zileuton showed comparable results with linalool when tested at 2 mM against the DENV NS3 protease, with a reduction of protease activity at 17.89 and 18.42%, respectively. Two new compounds were also proposed through the combination of the selected drugs, which are ziltri (zilueton + trimethadione) and zilool (zileuton + linalool). The molecular docking study confirms the in vitro observations where all drugs and proposed compounds were able to achieve binding affinity ≥ −4.1 kcal/mol, with ziltri showing the highest affinity at −7.7 kcal/mol, surpassing the control, panduratin A. The occupation of both S1 and S2 subpockets of NS2B-NS3 may be essential and a reason for the lower binding energy shown by the proposed compounds compared to the screened drugs. Based on the results, this study provided five potential new lead compounds (ziltri, zilool, zileuton, linalool, and trimethadione) for DENV that could be modified further.
1. Introduction
Dengue is a mosquito-borne viral disease where the primary mode of transmission of dengue virus (DENV) between humans involves the Aedes aegypti mosquito as the primary vector, with Aedes albopictus as the secondary vector. These two vectors have caused viral endemic in more than 100 countries including Eastern Mediterranean, American, South-East Asian, Western Pacific, and African regions. According to the World Health Organization (WHO), the prevalence of dengue infection has increased eightfold in the last two decades, with reported deaths increasing from 960 to 4032, between the years 2000 and 2015.1 As of June 2022, 849 death has been recorded worldwide.2 In general, dengue infection typically presents as an acute febrile illness accompanied by headaches, retro-orbital pain, arthralgia, and myalgia. Although the majority of dengue cases are either asymptomatic or mild, a large number of cases develop into potentially life-threatening severe diseases, such as dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS).3 Moreover, life-threatening conditions increase when infection occurs in individuals with asthma, diabetes, and other chronic illness.
Despite the urgency to combat the disease, no effective treatment is yet available in the market. The current treatment only involves supportive care in the form of fluid therapy and close clinical monitoring. One preventive treatment, the vaccine Dengvaxia, was approved in 2019 by the U.S. Food and Drug Administration (FDA).100 However, the vaccine efficacy is varied by age, the DENV serotype that causes the infection, and the serostatus of the vaccine recipient.4 Therefore, an efficient antiviral agent to treat DENV infection is urgently needed. A DENV antiviral treatment should inhibit all dengue serotypes5 and be administered to patients with or without a fever to decrease or prevent disease symptoms at the first sign of dengue infection, thus decreasing the risk of severe dengue diseases.
Understanding the life cycle of dengue virus reveals the potential targets for anti-dengue, and these include proteins involved in events such as endocytosis, viral fusion to the host membrane, viral transcription, and the release of progeny viruses from the host cell.6 Among these, the NS3 protein is an interesting target for potential antiviral development. This protein is among the best-characterized DENV nonstructural proteins and is the most preserved in all dengue virus serotypes.7 The NS3 protease, composed of NS2B and NS3, is a vital component in the replication cycle of the dengue virus. It acts as a trypsin-like serine protease, with His51, Asp75, and Ser135 as its key components.7 The NS3 protease plays a crucial role in cleaving the viral polyprotein at various sites, including NS2A-NS2B, NS2B-NS3, NS3-NS4A, and NS4B-NS5.7 Hence, the disturbance of the NS3 protease is fatal to the virus, therefore can be considered a useful target for antiviral drugs.8 Several studies have shown the promising result of inhibitors targeting DENV NS3.9,10 In fact, a similar target was successfully used for Hepatitis C virus (HCV) treatment.11
The use of “old drugs” and compounds available in the databases may accelerate the discovery of anti-dengue NS3 inhibitors, and in silico approaches provide systematic understandings of complex relationships among drugs, targets, and diseases essential for successful repositioning.12 In addition, the availability of well-curated compounds databases, as well as the advancement of machine learning, offer unprecedented opportunities to conduct drug repositioning. There are three in silico methods that can be used for this purpose, which are ligand-based, structure-based, and proteochemometric (PCM) modeling. The ligand-based (L-B) method utilizes the structural information of compounds. Using suitable descriptors, the functional relationships between compounds in a data set and one or more known actives are examined, usually through machine learning algorithms. The structure-based method uses the structural information of the target receptor. Here, the method predicts the preferred pose of ligands in the binding site through the use of scoring functions. The PCM method, on the other hand, predicts the bioactivity by using information from both compounds and target receptors.
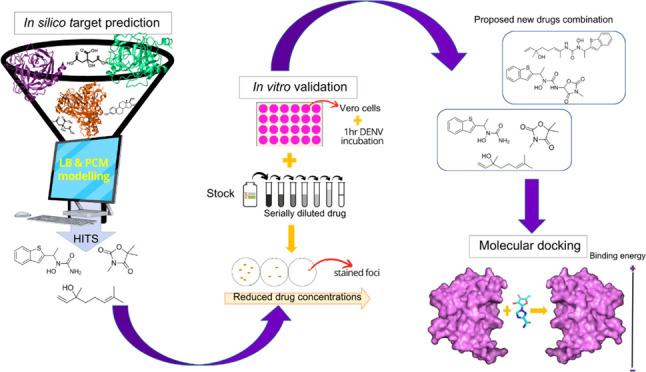
Hence, this study aims to discover lead compounds of DENV NS3 inhibitors from a collection of “old drugs” and substances through two different in silico target predictions, which are L-B and PCM models. Consequently, the results of the target prediction were validated through in vitro validation, which involves the foci forming reduction assay (FFURA) and NS3 protease assay. In FFURA, the antiviral properties of the drugs were evaluated on the dengue-infected Vero cells, while the protease assay was performed to measure drug inhibition toward the NS3 protease activity. From the in vitro observation, two new compounds were proposed, which were developed from the combinations of the selected drugs. Finally, molecular docking was conducted on the selected drugs and the proposed compounds to observe the interaction that was involved in the formation of the drug–protein complex.
2. Results
2.1. In Silico Prediction of Lead Compounds for the Anti-Dengue NS3 Inhibitor Using Ligand-Based (L-B) and Proteochemometric (PCM) Prediction Models
2.1.1. Internal Validation of Predictive L-B and PCM Models
Table 1 shows the internal validation of the two models, where sensitivity and specificity were used as performance measures. Both models scored almost similar values in terms of sensitivity and specificity. However, the PCM model scored a slightly higher sensitivity than L-B (0.85 vs. 0.838) but lower specificity than L-B (0.921 vs. 0.983). Note that the performance of PCM plateaued and did not change with the increasing lambda value. Hence, 0.8 was considered the best lambda value for the PCM model. We placed higher importance on models with higher sensitivity as the focus is more on identifying active compounds (TP) than inactive compounds (TN). In this case, the PCM model showed better performance in the internal validation compared to the L-B model.
Table 1. Internal Validation of the Training Seta.
| model | λ | TP | FP | TN | FN | SEN | SPE |
|---|---|---|---|---|---|---|---|
| PCM | 0.8 | 18,948 | 3174 | 36,881 | 3351 | 0.850 | 0.921 |
| 0.83 | 18,946 | 3172 | 36,883 | 3353 | 0.850 | 0.921 | |
| 0.86 | 18,945 | 3172 | 36,883 | 3354 | 0.850 | 0.921 | |
| 0.89 | 18,945 | 3172 | 36,883 | 3354 | 0.850 | 0.921 | |
| 0.92 | 18,945 | 3172 | 36,883 | 3354 | 0.850 | 0.921 | |
| 0.95 | 18,944 | 3172 | 36,883 | 3354 | 0.850 | 0.921 | |
| 0.98 | 18,944 | 3172 | 36,883 | 3354 | 0.850 | 0.921 | |
| L-B | 18,697 | 672 | 39,364 | 3621 | 0.838 | 0.983 |
For the PCM model, the evaluation was conducted for the lambda value (λ = 0.8, 0.83, 0.86, 0.89, 0.9, 0.93, 0.96, and 0.99). TP—true positive, FP—false positive, TN—true negative, FN—false negative, SEN—sensitivity, SPE—specificity.
2.1.2. External Validation of Predictive L-B and PCM Models
Table 2 summarizes the result of the external validation for both PCM and L-B models. As mentioned, a lambda value of 0.8 was considered the best and hence was used in the external validation and screening exercise for PCM. It can be seen that the L-B model showed a higher sensitivity than PCM (0.671 vs. 0.538) but showed a lower specificity than PCM (0.681 vs. 0.811). This indicates that the L-B model is better at predicting active compounds, whereas PCM is better at predicting inactive compounds. Overall, we deduced that the L-B model performs better compared to the PCM model, as a higher sensitivity score is preferable in the current study, and external validation has more weightage than internal validation as external validation measures the ability of the model to predict instances it has never encountered before.
Table 2. External Validation to Assess the Performance of PCM and L-B Modelsa.
| model | TP | FP | TN | FN | sensitivity | specificity |
|---|---|---|---|---|---|---|
| PCM | 1121 | 3168 | 13,570 | 961 | 0.538 | 0.811 |
| L-B | 1393 | 5345 | 11,399 | 683 | 0.671 | 0.681 |
TP—true positive, FP—false positive, TN—true negative, FN—false negative, SEN—sensitivity, SPE—specificity.
The applicability domain was measured using leverage and similarity search for each compound. Here, the leverage value and similarity search were calculated for each compound in the external data set against each compound in the training set. The results of both methods are presented in Figure 1. Here, only 1.69% of compounds from the test set were predicted as unreliable from the leverage nodes and 1.87% from the similarity search nodes. Looking at the high leverage score and similarity search (>98%), it can be deduced that the result of the validation is valid as the high sensitivity and specificity values were corroborated by the result of the applicability domain. In addition, the chemical space covered by the predictive model was also determined using principal component analysis (PCA) (see Supporting Information S1). It was found that the chemical space covered by the external set falls within the boundaries of the chemical space of the training set and hence is deemed to be within the applicability domain of the constructed model.
Figure 1.
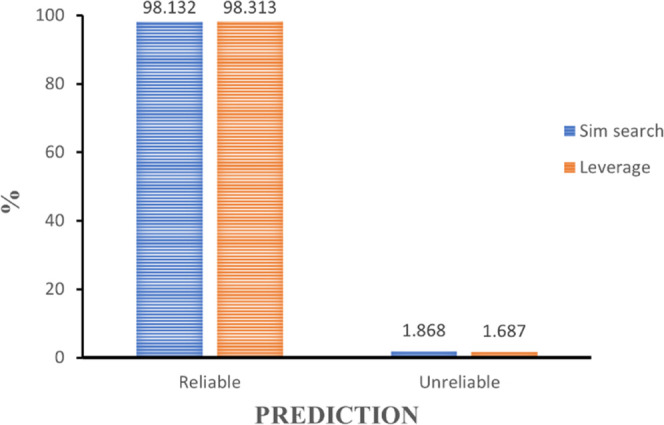
Applicability domain (AD) using similarity search and leverage. Based on the score, both algorithms show that the model reliably predicts the test set activity. Analyses were conducted using the Knime45 Analytics Platform.
2.1.3. Screening of “Old” Drugs as the Potential DENV NS3 Inhibitor
In the drug screening process, a total of 1263 drugs were collected from SWEETLEAD13 and DrugBank.14 Out of the whole drug set, 16 drugs from the PCM model (score ≥0.99) and two drugs from the L-B model (score ≥0.5) were identified. From the PCA plot, it was found that most of the drugs predicted using the PCM model lie within the training set boundaries, while the drugs predicted using the RF modeling lie close to the compounds but not in the heavily populated area (see the Supporting Information, Figure S2). The list of the drugs is presented in Tables 3 and 4. For in vitro validation purposes, not all drugs were used for further testing. Only three drugs were selected from the screening, which were zileuton, trimethadione (from the PCM model), and linalool (L-B model). Zileuton and trimethadione were selected as they were located at the center of the PCA graph, which indicated a highly populated area of the active inhibitor in the training set. Linalool was selected as it is closer to the highly populated area, as compared to the sesamine. This signifies a high similarity with the training set. Additionally, the availability of the literature, specifically on in vitro studies of these drugs, was also taken into consideration.
Table 3. List of Drugs/Substances Screened and Filtered from PCM and Its Indicationa−.
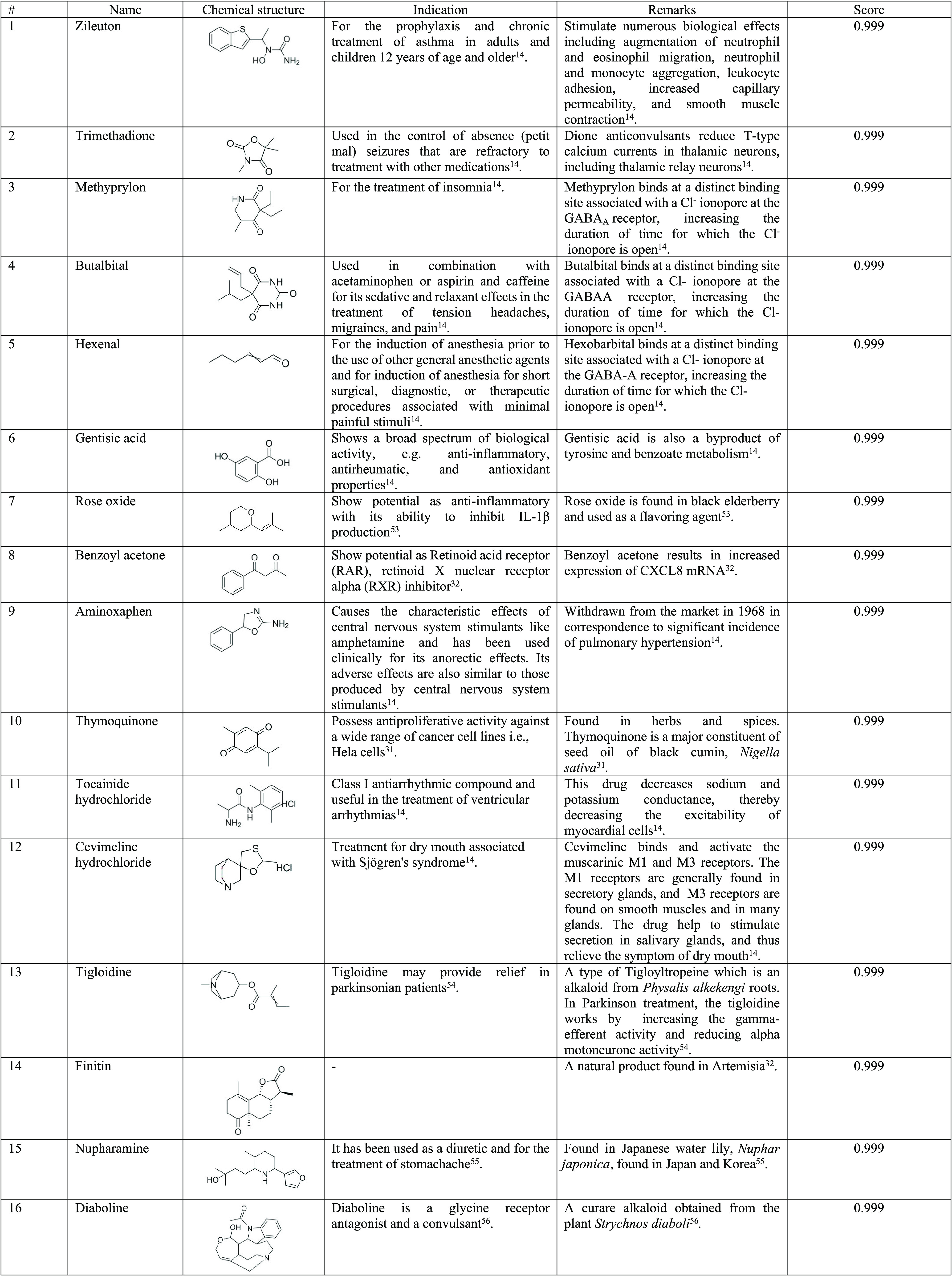
All drugs scored 0.99 when tested against the dengue NS3 protein and were ranked accordingly for further selection.
Table 4. List of Substances Screened and Filtered from the RF Model and Their Usagea.

All substances showed a score of ≥0.5 when tested against the dengue NS3 protein.
2.2. In Vitro Validation of the Selected Drugs as Anti-Dengue NS3
2.2.1. Drugs Cytotoxicity on Vero Cells
The drugs that were selected for in vitro validation were initially tested for cytotoxicity effect. In general, all drugs exhibited low cellular toxicity in Vero cells, even at a millimolar concentration, as indicated in Figure 2. The first drug, zileuton, was tested in the range of concentrations between 0.25 to 4 mM. A concentration of 4 mM was set as the highest concentration allowed to maintain the solvent concentration at 1% v/v. After 72 h of incubation, all tested concentrations, 0.25, 0.5, 1, 2, and 4 mM showed cell viability above 80%. The calculated 50% cytotoxic concentration (CC50) value was 38.67 mM. Next, trimethadione was tested in the range of 1.25−20 mM. The drug showed a dose-dependent response where, as the concentration increased, the percentage of cell viability was also reduced. However, at the highest tested concentration of 4 mM, the cells maintained its viability at 80%. Based on the analysis, the calculated CC50 of trimethadione was 117 mM. The third drug, linalool, was tested in the range of 0.125−2 mM. A concentration of 2 mM was used as the highest concentration to maintain the solvent concentration at 1% v/v. After 72 h of incubation, cells tested at different concentrations of 0.125, 0.25, 0.5, 1, and 2 mM maintained more than 80% of cell viability, with calculated CC50 value was 17.47 mM. Lastly, the positive control, ribavirin, maintained its cell viability above 90%, even at the highest concentration of 1 mM. The CC50 value calculated by GraphPad for ribavirin was 49 mM. The cytotoxicity of the solvent of the drugs, which is ethanol, on the cells was also studied here. Here, the highest solvent percentage tested is up to 2% v/v, with cell viability at 86%.
Figure 2.

Vability of Vero cells after treatments with different drugs: (A) zileuton, (B) trimethadione, (C) linalool, and (D) positive control—ribavirin. The viability of cells was analyzed at 72 h using the MTT assay. No significant cell morphology changes were detected at the highest concentration tested. The CC50 values are expressed as percentages of treated vs. untreated cells. Each value is the means ± SD of three experiments, each run in triplicate.
2.2.2. Anti-Dengue Activities of Selected Drugs on Infected Vero Cells
To test the inhibitory potential of the drugs on the infected cell, similar drug concentration, which was confirmed safe to be used via the cytotoxicity test, were used. After an hour of viral incubation, the dengue virus was expected to penetrate the host cell and begin its replication and reproduction by using the host’s cellular metabolism, and later, a fully formed virus will be assembled. After 72 h, the infected cells were incubated with tested drugs, where the end-point assay was conducted and resulted in the formation of brown-colored localized clusters called foci. Figure 3 is the results of infected Vero cells, which have been treated with the drugs zileuton, trimethadione, linalool, and ribavirin (positive control).
Figure 3.

Inhibitory potential of drugs on infected cells with the dengue virus: (A) zileuton, (B) trimethadione, (C) linalool, and (D) positive control—ribavirin. The drug was diluted to various concentrations (as indicated above) with overlay media and added to the cell culture. The viral inhibition was analyzed at 72 h. The inhibitory response of each drug is expressed as the mean of absorbance ± SD of three experiments, each run in triplicate. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Based on the results, all treatments showed a dose-dependent response trend where foci reduction increases with increasing drug concentration. For zileuton, it showed the highest foci reduction with an average of 47% at the highest concentration tested, 4 mM. Although dose–response trend can be seen from the result; however, there are slight differences in the trend between the inhibition of foci at 1 and 2 mM, where at 2 mM, zileuton caused slightly lower foci inhibition compared to the treatment of 1 mM. The projected half-maximal inhibitory concentrations (IC50) value calculated was 3.3 mM. For trimethadione, the highest foci inhibition (36%) was recorded at 20 mM. The calculated IC50 for trimethadione was 25.97 mM. Among the tested drugs, only linalool demonstrated foci reduction above 50%, where the highest foci inhibition (54%) was observed at 2 mM. The IC50 calculated for linalool was 1.12 mM. Looking at the presented data, it should be noted that none of the selected drugs showed a better antiviral effect than the ribavirin in terms of the concentration used to achieve a 50% foci reduction. Based on the assay, the calculated IC50 for ribavirin was 0.14 mM which was the lowest among the four drugs. Ribavirin also showed 88% foci inhibition at the highest concentration tested, 1 mM.
2.2.3. DENV-2 NS2B-NS3pro Inhibition by Selected Drugs
Figure 4 shows the protease inhibition of the drugs. In terms of protease inhibition, the positive control, aprotinin, performed the best against the selected drugs and ribavirin. Aprotinin, the positive control for this assay, is a competitive serine protease inhibitor that forms stable complexes and blocks the active sites of enzymes. At a concentration of 5 μM, the protease inhibitor inhibited more than 65% of the protease activity and recorded the highest inhibition (>85%) at 10 μM concentration. Another positive control, ribavirin, shows the opposite effect. The fluorescence intensifies with the addition of the treatment but gradually reduces the effect with the increase of drug concentration. This is an interesting outcome, as the effect on the NS2B-NS3 protease was inconsistent with the observed in vitro result. Thus, this finding suggests that the ribavirin does not work in targeting the NS2B-NS3 protease but inhibits the viral progression via different mechanisms. To be specific, the mechanism of action of ribavirin is via lethal mutagenesis of RNA virus genomes mediated by the viral RNA-dependent RNA polymerase,15 and this may result in viral inhibition as reported in the study.
Figure 4.

NS2B-NS3 protease inhibition with the treatment of selected drugs: (A) zileuton, (B) trimethadione, (C) linalool, (D) positive control, aprotinin. and (E) positive control—ribavirin. The statistical analysis was conducted between particular drugs against the untreated cells (NC) using one-way ANOVA with Tukey correction. Each value is the means ± SD of two experiments, each run in triplicate. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
For zileuton, a positive correlation was observed between drug concentration and the percentage of inhibition. The highest protease inhibition (27.7%) was observed at a concentration of 4 mM, while at 2 mM concentration, the inhibition was reduced to 17.89%. For linalool, the drug recorded an average protease inhibition of 18.42% at 2 mM and 13.83% protease inhibition at 1 mM. For trimethadione, the highest protease inhibition (17.15%) was recorded at 20 mM, and at 10 mM, about 13.17% of protease inhibition was detected. Overall, among all drugs, at the highest concentration tested, trimethadione showed the least effect on the protease activity (20 mM, 17.15% protease inhibition).
2.3. Development of New Compounds from the Combination of Selected Drugs
Based on the in vitro results, two new compounds were developed—ziltri and zilool (Figure 5). Zileuton was the main component for both ziltri and zilool, with zitlri—zileuton + trimethadione and zilool—zileuton + linalool. Zileuton was chosen to be the main component in the ziltri and zilool combination as it shows higher protease reduction.
Figure 5.
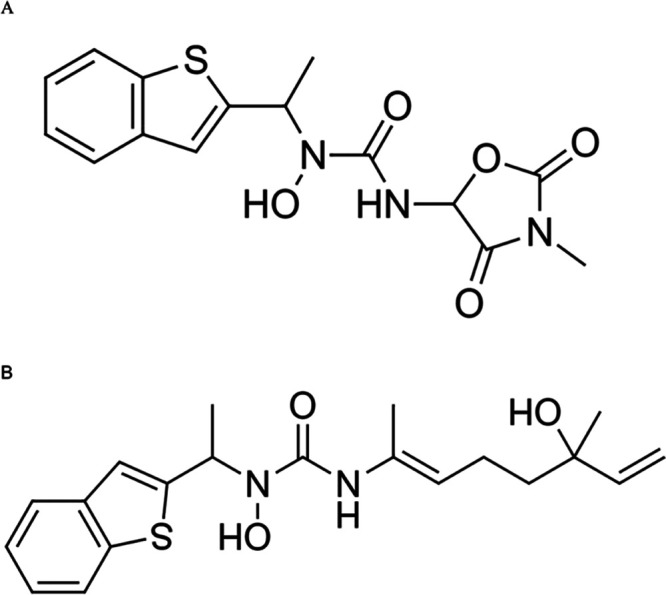
Newly developed compounds based on the in vitro results. (A) Ziltri (zileuton + trimethadione) and (B) zilool (zileuton + linalool).
2.4. Molecular Docking of the Selected Drugs and Newly Derived Compounds with the Dengue NS2B-NS3 Protease
The NS2B-NS3 protease is made up of 4 subpockets, S1-4 and the important residues include Asp129_NS3, Ser135_NS3, Tyr150_NS3, and Tyr161_NS3 (S1 pocket); Asp81_NS2B, Gly82_NS2B, Ser83_NS2B, Asp75_NS3, and Asn152_NS3 (S2); Ser85_NS2B, Ile86_NS2B, and Lys87_NS2B (S3); and Val154_NS3 and Ile155_NS3 (S4).28 The docking site of the protease was determined from the halo form of the protease, which serves as the golden reference for the binding site. The site contains the important catalytic triad of the protease, which are His51, Asp75, and Ser135. A mutagenesis study revealed that the replacement of catalytic serine with alanine caused the inactivity of the protease, and no viable virus was recovered from an infectious cDNA clone with abolished protease activity.16,17 The site is well known for its shallowness, and the prediction by the PockDrug18 server showed that this site has a druggability score of 0.5 (score range 0–1), which may reduce the ability of small molecules to effectively interact with the protein.
In the docking exercise, all drugs and the two newly developed compounds were found to bind to the binding pocket at the right side of the active site region (S1 pocket), as observed in Figure 6. Interestingly, this site is much broader and more shallow than the binding pocket at the left region (S2 pocket),19 yet, most of the interactions were observed to be at this site. It was identified that the S1 region has much more hydrophobic residues compared to the S2 region. The summary of all interactions involved in the ligand–protein complexes is indicated in Figure 7 and Table 5.
Figure 6.
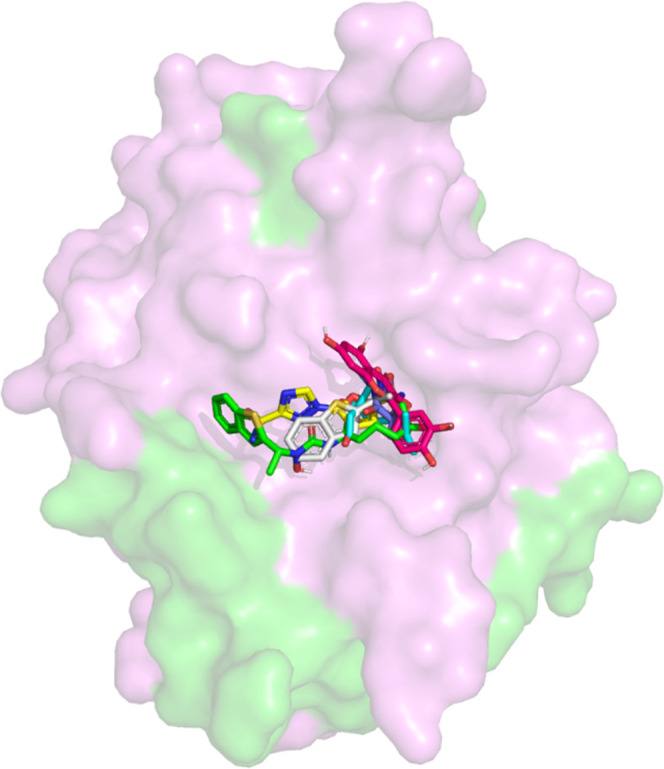
Molecular surface representation of the NS2B-NS3 protease with selected drugs. Selected drugs from two prediction models (zileuton, trimethadione, linalool), newly developed compounds (ziltri and zilool) and positive control—panduratin A, were docked at the active site of the protease.
Figure 7.

Interactions of the selected drugs and substances against the dengue NS2B-NS3 protease both in 3D and 2D visualizations. The complex of the NS2B-NS3 protease with zileuton (A, B), trimethadione (C, D), linalool (E, F), ziltri (G, H), zilool (I, J), and panduratin A (K, L).
Table 5. Affinity of the Different Complexes and the Residues Involved in the Interactiona.
| drug | affinity (kcal/mol) | no. of H bonds | residue(s) involved in hydrogen bond interactions | residue(s) involved in other interactions |
|---|---|---|---|---|
| zileuton | –5.6 | 5 | B:Phe130, B:Lys131, B:Thr134, B:Ser135, B:Tyr150 | B:Pro132 (alkyl bond); B:His51, B:Gly151, B:Asn152, B:Tyr161 (van der Waals) |
| trimethadione | –4.6 | 4 | B:Lys131, B:Gly133, B:Thr134, B:Ser135 | B:Pro132 (alkyl bond); B:Thr161 (π–σ bond); B:Asp129, B:Lys131, B:Phe130, B:Tyr150 (van der Waals) |
| linalool | –4.1 | 3 | B:His51, B:Ser135, B:Gly151 | B:Tyr161 (alkyl, π–σ, and π–alkyl bonds); B:His51, B:Asp129, B:Phe130, B:Gly133 and B:Tyr150 (van der Waals) |
| ziltri (zileuton + trimethadione) | –7.7 | 5 | B:Phe130, B:Tyr150, B:Gly151, B:Gly153, B:Tyr161 | B:His51 (π–sulfur bond), A:Asp81, A:Gly82, A:Thr83, B:Asp75, B:Asp129, B:Ser135, B:Asn152, B:Gly153 (van der Waals) |
| zilool (zileuton + linalool) | –6.9 | 3 | B:Asp129, B:Gly153, B:Tyr161 | B:Asp75 (π–anion); B:Pro132, B:Tyr150 (alkyl and π–alkyl bonds); A:Asp81, A:Gly82, A:Thr83, A:Met84, B:Trp50, B:His51, B:Arg54, B: Val72, B:Gly151, B:Asn152 (van der Waals) |
| panduratin A | –7.0 | 4 | B:Lys131, B:Gly133, B:Thr134, B:Ser135 | B:His51, B:Tyr161 (π–π T-shaped); B:Pro132 (π–alkyl bond) |
From the molecular docking study, zileuton was shown to bind to the active site at −5.6 kcal/mol, as depicted in Figure 7A,B. From the result, zileuton formed 5 hydrogen bonds with the NS2B-NS3 protease where one of the hydrogen bond interactions involved B:Ser135 of the catalytic triad at a distance of 3.4 Å. Other hydrogen bond interactions involved B:Phe130 (3.4 Å), B:Lys131 (3.2 Å), B:Gly133 (2.2 Å), and B:Tyr150 (3.5 Å), with either the amides or alcohol functional groups of zileuton. Moreover, zileuton formed alkyl interactions with B:Pro132 (3.8 Å) and van der Waals interactions with B:His51, B:Gly151, B:Asn152, and B:Tyr161.
The molecular docking of trimethadione is shown in Figure 7C,D. It was found that the drug also established a hydrogen bond with B:Ser135 at a distance of 2.5 Å. Other hydrogen bond interactions were found between the drug–protein complex including B:Lys131 (3.2Å), B:Thr134 (2.2 Å), and B:Gly133 (2.5 Å). Trimethadione also formed a σ-π bond with residue B:Tyr161 (3.7 and 3.9 Å) and an alkyl bond with B:Pro132 (4.9 Å). The van der Waals interaction occurs between trimethadione and residues B:Asp129, B:Lys131, B:Phe130, and B:Tyr150. The binding affinity for the complex above was predicted at −4.6 kcal/mol.
Linalool was found to form two polar interactions with the protease, as observed in Figure 7E,F. In addition to the interaction with B:Ser135, the hydroxyl group of linalool established a hydrogen bond with B:His51 (3.3 Å), another residue of the catalytic triad. Besides His51, linalool also formed hydrogen bonding with B:Gly151 (2.3 Å). It was found that the drug formed σ–π and π–alkyl bonds with residue B:Tyr161. van der Waals forces were seen between linalool and residues B:His51, B:Asp129, B:Phe130, B:Gly133, and B:Tyr150. The predicted binding affinity for the complex was – 4.1 kcal/mol.
In comparison to the selected drugs, the control, panduratin A, showed the highest binding affinity (−7.0 kcal/mol). The molecular docking study of panduratin A is shown in Figure 7K,L. It was observed that the panduratin A-protein complex established polar interactions with four amino acid residues, i.e., B:Lys131 (3.4 Å), B: Gly133(2.3 Å), B:Thr134 (2.3 Å), and B:Ser135(2.4 Å). Besides, π–π T-shaped bonds were found between the ligand and B:His51 and B:Tyr161, while a π–alkyl bond was established with B:Pro132. Previously, panduratin A showed competitive inhibition toward DENV-2 NS2B/NS3pro in vitro with Ki = 25 μM,20 which shows the validity of this compound as the protease inhibitor and reference for the study.
Since the in vitro inhibitions of the selected drugs were found to be moderate, and the recorded binding affinities were less than panduratin A, we included two newly proposed compounds to be docked against the NS3 protease. These compounds were ziltri (Figure 7G,H) and zilool (Figure 7I,J). These combinations showed improved binding affinity as compared to the selected drugs. Ziltri showed the highest binding affinity (−7.7 kcal/mol), followed by zilool (−6.9 kcal/mol). Ziltri formed hydrogen bond interactions with B:Phe130 (2.8 Å), B:Tyr150 (1.9 Å), B:Gly151 (2.3 Å), B:Gly153 (2.2 Å), and B:Tyr161 (2.0 Å) and formed π–sulfur interactions with B:His51. It should be noted that both ziltri and zilool interact with at least one of the catalytic triad residues, with zilool also occupying the S2 pocket through an interaction with Asp75. In addition, both ziltri and zilool, as well as panduratin A, are larger in size compared to the three screened drugs. Hence, the larger ligand size may be another prerequisite when docking at shallow sites, as shown by the DENV-3 active site.
3. Discussion
The aim of this study is to discover novel anti-dengue drugs from old drugs that target the DENV NS3 protein using a combination of in silico and in vitro studies. To achieve the desired objectives, three phases of the study were conducted. In the first phase, the screening of potential drugs was conducted by building a prediction model of established bioactivity data of the anti-dengue NS3 protein using two prediction models—L-B and PCM. Next, the process continued with the evaluation of the screened drugs’ anti-dengue properties in vitro by using the FFURA assay and protease assay. The drugs were tested using similar concentrations throughout these three assays, and the effectiveness of viral inhibition was analyzed. Based on the observed in vitro results, molecular docking was conducted on the screened drugs and newly developed compounds against the DENV NS3 protein. The key binding interactions and the best binding orientation were determined.
Based on the results, several key observations can be deduced. First, the L-B model performs slightly better than the pcm model in the prediction of anti-dengue potential. In the external validation, the L-B model showed higher sensitivity compared to the PCM model (L-B = 0.671, PCM = 0.538). Meanwhile, the L-B model showed a lower sensitivity value in the internal validation; however, the difference is insignificant compared to the PCM model. In addition, external validation has more weightage compared to internal validation as it evaluates compounds or instances it has never seen before. Moreover, as the goal of the study is to screen active inhibitors against the dengue virus, we placed a higher weightage on models that has high sensitivity value. This is important as we would want to maximize the positive number of correct classifications. Hence, the result that we obtained from the L-B model fits the above criteria, and therefore it is better than the PCM model.
These results highlight that the additional protein descriptors employed in the PCM model may not be advantageous in this study. However, this result should not be generalized and should be observed on a case-to-case basis. For example, a study by Lapins et al.22 demonstrated that the PCM model outperformed the QSAR model (that uses a single descriptor) for single target models to identify Cytochrome P450 (CYP) inhibition where the area under the curve was higher in PCM than QSAR (AUC:PCM > 0.90, QSAR 0.79–0.89). Further external validation of the PCM model showed an AUC score of 0.94, which suggested the good performance of the constructed PCM model. Nevertheless, the result presented by Lapins et al.22 uses a smaller number of proteins and compounds (5 CYPs and 17, 143 compounds) compared to the current study, which uses 10 proteins of different origins and more than 62,000 compounds, which may account for the disparity in results. In another study using a kinase data set spanning the whole kinome by Niijima et al.,23 ligand-based models using SVMs performed better than PCM DC-SVMs in external validation with an accuracy of 81.3% compared to 73.9%. Interestingly, for internal validation, the result presented by Niijima et al.23 was comparable to the current study, where PCM dual component (DC)-SVMs showed higher accuracy compared to the L-B model, with 90.9% and 86.2%, respectively. The absence of the protein descriptors in the L-B model also resulted in a faster calculation and computational time compared to the PCM model. Despite these matters, both models have successfully screened drugs that were able to elicit moderate antiviral activities in vitro and biologically safe to the cell, although treatment was used at a high concentration. Hence, both methods can produce a comparatively fast and promising result to assist the anti-dengue NS3 drug development process.
Second, out of the three drugs that were screened, zileuton and linalool showed promising results in vitro as lead compounds for anti-DENV potential. This is based on the observed FFURA and protease assays. In the FFURA assay, linalool performed better than zileuton and trimethadione, while zileuton showed the highest inhibition of the DENV NS3 protease at the highest concentration tested, as well as in molecular docking. Therefore, both zileuton and linalool have the potential for further development of DENV NS3 protease inhibitors, as both consistently showed good efficacy in each assay. These drugs can be direct-acting antivirals (DAA) that specifically target the viral protein. Besides, these drugs show low toxicity even when tested at a high concentration, which fits the DAA criteria that includes drug efficacy at low toxicity and a wide treatment window. However, except for the protease assay, both drugs were unable to exhibit better results than ribavirin. In addition, the ability of linalool to inhibit the DENV NS3 protease and dengue-infected cells highlights another advantage of the L-B model. To increase our understanding of the chemical space of the active compounds against dengue, using PCA analysis, it was found that linalool was not located in the dense area as compared to trimethadione and zileuton (from PCM model), and yet, the activity from the in vitro validations showed promising antiviral results (see Supporting Information S1). Thus, this demonstrates the ability of the L-B model to predict compounds outside the chemical space covered by the training set. Although the drugs are no better than ribavirin in terms of foci inhibition, however, all of them showed better protease inhibition. This is true as ribavirin is not a specific inhibitor of the NS3 protease and is not currently used as a treatment for dengue fever. From the protease assay, we identified that ribavirin is not a suitable positive control for protease inhibition as it showed the opposite effect than inhibiting the protease, and therefore other positive controls such as aprotinin or panduratin A need to be used for this purpose.
One issue that may be raised from the study is the effective concentration used in the treatment. Unlike several studies that reported the success of inhibition at submicromolar concentration, the current study suggested the use of marketed drugs at a millimolar concentration to achieve a desirable antiviral effect. In exception to trimethadione, this issue may have arisen possibly due to the drugs’ low solubility; however, there is not enough evidence to draw that poor bioavailability is the reason for this event. In one study investigating standard serine protease inhibitors against CF40.NS3pro, a recombinant NS3 protease, revealed that three out of 16 inhibitors only reduced the protease activity up to 15% at 1 mM concentration.24 Therefore, this finding supports the results obtained from the current study, where some of the protease inhibitors may require high concentration to exhibit the desired property. In another study by Holbeck et al.25 on the FDA-approved anticancer potential in the NCI60 human tumor cell lines, at least three drugs exhibit 50% growth inhibition (GI50) at the concentration of ≥2500 μmol/L (2.5 mM). This inferred that some drugs do exhibit their activity at higher concentrations when tested in vitro while still remaining safe and effective to be used in patients for anticancer treatment.
Third, interactions with the residues of the catalytic triad, ligand size, and binding at S1 and S2 subpockets may be important prerequisites in establishing strong binding against ns3 proteins at the active site. From the results, we hypothesized that the interaction of the selected drugs and proposed compounds with one of the catalytic triad residues, i.e., Ser135 of the NS3 protease, might disrupt the electron exchange between the carboxyl group of Asp75 and the nitrogen atom of the imidazole ring of His51.26 This may disturb the capability of His51 to trigger nucleophilic assault of the hydroxyl group (β-OH) of Ser135, which is important for the initiation of proteolysis.26,27 In addition, all ligands were found to have interactions with one or more amino acid residues (Asp129, Phe130, Tyr150, Asn152, and Gly153) of NS3 protease pocket 1 and its vicinity. Apart from that, these residues form a small part of the β-sheet that has an important role in substrate binding.1,2 Hence, such interactions possibly alter the functional attribute of the protein by changing its conformation.
In addition to the previous point, hydrogen bonding may be important for a stable interaction against the catalytic triad and other important residues of NS3. When comparing the docking results of the screened drugs, the more the hydrogen bonds established with NS3, the lower the binding energy. Our finding is in line with Hariono et al.,28 where hydrogen bonds are responsible for the stability of the thioguanine scaffold derivative-protease complex. Compound 18, the best derivative of the scaffold with the lowest IC50 value (0.38 μM), formed 26 hydrogen bonds, which have at least 0.1% occupancy throughout 70 ns simulation time. A hydrophobic interaction, to a lesser extent, was also observed to maintain the stability of the complex. Furthermore, Katz et. al.29 identified a serine protease inhibition motif in which binding is mediated by a cluster of very short hydrogen bonds (<2.3 Å) at the active site between the protein residues and the inhibitor. However, when comparing the screened drugs and newly proposed drugs, although the latter formed lesser hydrogen bonds, its binding energy was lower. In addition, all compounds occupy both subpockets 1 and 2 (S1 and S2), and in shallow and broad binding pockets such as S1, the size of the compounds plays an important role. It has been reported that shallow pockets are not “druggable sites” where it is difficult for small-sized compounds to be flanked by the binding pocket. In addition, the estimated Ki for ziltri was 2.69 μM, which is smaller than the estimated value of panduratin A, which is 7.4 μM, which makes it a better inhibitor than the reference drug. Based on these observations, we anticipated that ziltri and zilool may show better in vitro viral inhibition as the results showed higher binding affinity than the positive control, panduratin A.
4. Conclusions
This study attempted to repurpose old drugs as novel DENV NS3 inhibitors using in silico screening and validation of the antiviral properties of selected drugs viain vitro assays and molecular docking. Based on the results, three key findings can be deduced, which are: (i) the L-B model slightly outperforms the PCM model in DENV NS3 screening, (ii) zileuton and linalool showed promising results, and (iii) besides the interaction with the catalytic triad, the size of the compounds may be another important prerequisite, given the shallowness of the binding site. In regards to (iii), this observation was made when two newly proposed compounds, ziltri (zileuton + trimethadione) and zilool (zileuton + linalool), were docked to NS2B-NS3 and showed the overall lowest binding energy surpassing the positive control, panduratin A. These compounds were proposed to produce compounds with better viral inhibition. This is because the individual drugs showed inhibition in the millimolar concentration, although some drugs do exhibit in vitro activity at such concentrations.30 It should be noted that all compounds contain several stereocenters, which may complicate their synthesis. Future studies should include more bioactivity data from the viral serine protease, which, at the time of the study, was limited, and the size of the ligands should be considered in the screening. In addition, drug filtering parameters prior to the prediction process may need to be reconsidered the chemical features of the ligand, which could fit the shallow and broad active site of the NS3 protease. Furthermore, molecular dynamics should be performed as it provides better insights into the ligand–protein interaction in a fixed period of time, giving a view of the dynamic “evolution” of the system. At this juncture, the three screened drugs (zileuton, linalool, and trimethadione) are much more suitable to be considered as lead compounds for DENV NS3, and hence, this study has provided five new lead compounds for NS3 that can be further modified to improve their activity.
5. Materials and Methods
5.1. In Silico Target Prediction
The study was conducted in several phases, as illustrated in Figure 8. Initially, the screening of the anti-dengue potential was conducted using in silico target prediction. Here, the target prediction was employed using machine learning to predict the most probable protein targets of small molecules. The algorithms that were employed in machine learning use protein sequence data to learn patterns and uncover relationships between target proteins and the possible biological activities of the compounds/substances using carefully prepared chemical libraries. In short, the predictions are based on the similarity principle through reverse screening.
Figure 8.

Workflow of the study. The study begins with building a prediction model that could screen marketed drugs for activity against the NS3 dengue protein. Here, ligand-based (LB) and proteochemometric (PCM) models were developed. Next, the in vitro validations were conducted to analyze the antiviral properties of the drugs. Here, the cytotoxicity assay, FFURA, and protease inhibition assay were conducted. The final stage of the evaluation was done to determine factors that possibly contribute to the interaction observed in vitro using molecular docking. Here, molecular docking was done on the selected drugs against the NS3 protease. The binding energy and the interactions that developed between the ligand–protein complexes were analyzed.
5.1.1. Training Set
To build a predictive model, 62,746 active and inactive compounds related to 10 proteins associated with, and including, the NS3 protein were retrieved from CheMBL,31 PubChem,32 and BindingDB33 databases, as indicated in Table 6. Three of the NS3 proteins belong to the Flaviviridae family, which are the dengue virus (DENV), West Nile virus (WNV), and Hepatitis C virus (HCV) (UniProt ID P14337, P06935, P26662, respectively). Since NS3 is a serine protease, therefore we incorporated another seven human serine proteases into the training set as bioactivity data on viral serine protease is limited. An active ligand–protein interaction was considered as having a Ki/Kd/IC50/percentage inhibition value of less than 50 μM or 50%. Any values above the specified threshold were considered as inactive. All protein sequences were collected from UniProt.34
Table 6. Breakdown of Bioactivity Data Points Collected for Each Targeted UniProt IDa.
| protein | uniprot ID | active | inactive | total |
|---|---|---|---|---|
| dengue virus genome polyprotein | P14337 | 6271 | 29,929 | 36,200 |
| hepatitis C virus genome polyprotein | P26662 | 2680 | 7643 | 10,323 |
| West Nile virus genome polyprotein | P06935 | 137 | 617 | 754 |
| prothrombin | P00734 | 3274 | 728 | 4002 |
| coagulation factor X | P00742 | 5204 | 358 | 5562 |
| serine protease 1 | P07477 | 1930 | 204 | 2134 |
| neutrophil elastase | P08246 | 2154 | 257 | 2411 |
| cathepsin G | P08311 | 231 | 57 | 288 |
| kallikrein-7 | P49862 | 46 | 26 | 72 |
| coagulation factor IX | P00740 | 593 | 15 | 608 |
| total | 22,520 | 39,834 | 62,354 |
5.1.2. Chemical Descriptor
To represent the compounds as vectors for both PCM and L-B models, the Extended Connectivity Fingerprints with a diameter of four bonds (ECFP_4)35 and a bit size of 1,024 was used as a chemical fingerprint as this fingerprint has proved to effectively secure the chemical structural information relevant to bioactivity in several studies.35,36
5.1.3. Machine Learning Algorithms for the PCM Model
For the PCM model, the protein vectors were represented
by their full sequence, where a protein sequence is denoted as a string
of characters, and each character represents an amino acid that is
part of the protein. The Parzen Rosenblatt Windows (PRW)37,38 was employed as a classification algorithm, and based on the Bayes
theorem, compound x was assigned to class ωα with the highest value. The posterior probability,
or probability of a new compound belonging to a target class, ωα, with a given vector molecular feature, 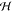 , is calculated
as such
, is calculated
as such
| 1 |
The a priori probability of class ωα, P(ωα), can be calculated as the class proportion in the training set. The quantity p(x|ωm) is the class conditional probability of x belonging to class ωm and p(x) is the normalization constant, which assures that the sum of all of the M class conditional probabilities is equal to one and is hence given by
| 2 |
The PRW calculates the class conditional probability p(x|ωα) as the average similarity of the unknown compound, x, against a set of known compounds, xj, belonging to a given class ωα. The value p(x|ωα) is given by
| 3 |
The equation above then becomes as below once we incorporate biological similarity using the tensor product shown before
| 4 |
Here, (x, t) is the compound–target complex being analyzed and Nωα and (xj, tj) are the number of compound–target pairs and compound–target pairs in class ωα, respectively.
5.1.3.1. Chemical Similarity Measurement
As the component of the PCM model, the Aitchison–Aitken (AA) kernel39 was used to calculate chemical similarity.
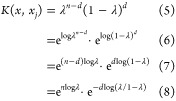 |
5 |
where d = (x – xj)τ(x – xj) means the number of times x and xj differ, λ is the smoothing parameter, and n is the size of the compound vector, which, in this case, is 1024. The smoothing parameter in the AA kernel has a narrow range of 0.5 < λ < 1.0. At λ = 0.5, it will produce maximum smoothing, and at λ = 1.0, no smoothing occurs; hence, these two values are ignored.39 For this study, the AA kernel was executed at λ = (0.8, 0.83, 0.86, 0.89, 0.9, 0.93, 0.96, 0.99).
5.1.3.2. Biological Similarity Measurement
The protein sequences were subjected to sequence alignment using multiple sequence alignment named MUSCLE40 on the EMBL-EBI server (https://www.ebi.ac.uk/Tools/msa/muscle/). The protein sequence similarity is calculated as the ratio of the number of matching amino acids over the length of alignment using the package bio3d in R.41 Sequence alignments are performed to identify the structural, functional, or evolutionary relationships that may exist between protein sequences by aligning the sequences to find regions of similarity.
5.1.4. Machine Learning Algorithms for the L-B Model
A random forest (RF) algorithm was used to build the L-B prediction model. There are two stages in producing the RF algorithm: RF creation and prediction. In RF creation, k features are randomly selected from total m features where k ≪ m. Among the k features, node d is calculated using the best split point, using the Gini index (S).
| 9 |
The node is then split into daughter nodes using the best split. All of the steps were repeated until i number of nodes had been reached. j is the number of class and P(i) is the probability of an instance being classified to a specific class. The node is then split into daughter nodes using the best split. All the steps were repeated until i number of nodes had been reached.
This process will repeat until the tree has reached the specified number of branches; nodes were expanded, and a path was established. As RF is an ensemble of decision trees, the number of trees, n was set at 100. In the next stage, which is the random forest prediction, test features are taken and rules of each randomly created decision tree to predict the outcome is used and predicted outcome is stored. The average score from each tree is used as the final prediction from the RF algorithm.
5.1.5. Internal and External Validations
The internal validation was performed using 5-fold cross-validation. In brief, the data set was split into five subsets, where four were used as the training set, and the fifth served as the test set to assess the predictive ability of the model. In the first iteration, the first fold is used to test the model, and the rest are used to train the model. This process is repeated until each fold of the 5-fold has been used as the testing set. The external validation was performed by testing 18,820 compounds collected from the ZINC database (https://zinc.docking.org/), which are not in the training set.
5.1.6. Performance Measure
The model performance was measured using sensitivity and specificity metrics. This was calculated by the number of true positive (TP), true negative (TN), false positive (FP), and false negative (FN) compounds, as indicated below
| 10 |
| 11 |
5.1.7. Applicability Domain (AD) Using Cosine (cosα) and Leverage
The cosine similarity is used to measure the similarity of the datasets regardless of the magnitude of the vectors. It measures the angle between two vectors starting at the origin and extending to the a-th and b-th p-dimensional objects.42 The cosine value ranges between 0 and 1, where a value of 1 indicates perfect similarity. Leverage is the other method that was applied to define the AD, where it uses the approach of extent of extrapolation. Both cosine and leverage were analyzed using similarity search nodes and leverage nodes,43,44 respectively, from the Knime45 Analytics Platform. For leverages, compounds with a score ≥0.5 were considered reliable, while a score lower than the said value was considered unreliable. For similarity search, the output was directly determined as reliable and unreliable.
5.1.8. Drug Selection for In Vitro Validation Studies
4548 drugs from the ATC classification J: anti-infectives for systemic use from the DrugBank14 and SWEETLEAD13 (structures of well-curated extracts, existing therapies, and legally regulated entities for accelerated discovery) databases were collected for dengue antiviral screening. The SWEETLEAD13 database included approved drugs, chemical isolates from traditional medicinal herbs, and regulated chemicals. Initially, the drug data set was screened to remove any redundancy with the data in the training set. Next, additional screening was conducted to the data set based on the corresponding physicochemical properties, i.e., the hydrogen bond acceptor (≤6), hydrogen bond donor (≤2), rotatable bond count (≤10), Lipinski rule of five (=0), total polar surface area (≤100), and molecular weight (<500 Da). These criteria were selected based on the average (mean) value of each property listed above from the pool of collected active drugs. In total, 1263 drugs were tested against the prediction models, and the cutoff value is set at ≥0.5 for both PCM and L-B models. Due to the large number of drugs from the PCM model that scored ≥0.5, only drugs that scored 0.99 were considered for further in vitro analysis.
5.2. In Vitro Evaluation of Selected Drugs for the Anti-Dengue Potential
5.2.1. Preparation of the Compounds
The stock solutions of selected anti-dengue drugs, which are zileuton (Sigma-Aldrich, USA) and linalool (Santa Cruz Biotechnology, USA), were dissolved in ethyl alcohol (EtOH) (Sigma-Aldrich, USA), while trimethadione (Sigma-Aldrich, USA) and ribavirin (Sigma-Aldrich, USA) were dissolved in distilled water. The stock solution was filter-sterilized (0.22 μm pore, Bioflow, Malaysia) and further diluted with culture medium to the desired concentration for the assays.
5.2.2. Determination of the Drug Cytotoxicity Dose
The cytotoxicity assay was performed on the Vero cells. The cytotoxicity assay was carried out by seeding 1.5 × 104 cells into 96-well flat-bottom plates (Corning, USA). The plates were incubated in a 5% CO2 humidified incubator for 24 h. Next, the cells were treated with a serially diluted stock of the test anti-dengue drugs at different concentrations and further incubated at 37 °C with 5% CO2. After 72 h, 10 μL of 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT) (Nacalai Tesque, Japan) (5 mg/mL) solution was added and incubated for 4 h. After incubation, the solution was carefully removed, and 100 μL of DMSO (Sigma-Aldrich, USA) was added, followed by continuous shaking for 10 min. The absorbance was measured using a microplate reader (Tecan, Austria) at 570 nm. The percentage of cell viability was determined based on the absorbance readings. The 50% cytotoxic concentration (CC50) value was calculated using GraphPad Prism 8.0.1 (GraphPad Software, Inc., San Diego, CA).
5.2.3. Determination of Viral Inhibition via the Foci Forming Unit Reduction Assay (FFURA)
The antiviral activity of NS3 inhibitors was determined and evaluated by measuring the reduction in the number of DENV infectious foci after treatment. Briefly, 200 μL of DENV-2 (MOI of 1) was introduced to Vero cells and incubated for an hour. Later, the viral mixtures were removed, and cells were washed with phosphate-buffered saline (PBS). Prior to this, drug treatment at different concentrations was prepared using 2% fetal bovine serum (FBS) and 1.5% carboxymethyl cellulose (CMC) as the incubation medium. The cells were incubated with treatment media for three days. Virus foci were visualized according to the previously described method.46 One foci is recognized as a clump of cells stained as light brown under the microscope, whereas under the naked eye, the foci appeared as brown spots. The number of foci was counted and recorded in duplicates, and the average was taken into account. The experiment was repeated three times. Half-maximal inhibitory concentrations (IC50) were obtained through the dose−response curve analysis using GraphPad Prism 8.0.1. (GraphPad Software, Inc., San Diego, CA). The results were expressed as the mean values ± standard deviation (SD) (the corresponding error bars were displayed in the graphical plots).
5.2.4. DENV NS2B-NS3pro Inhibition Assay
The protocol that was used for this assay was based on the study by Rothan et al.47 In brief, reaction mixtures of 100 μL were prepared, which consisted of a 20 μM fluorogenic peptide substrate (Boc-Gly-Arg-Arg-AMC), 2 μM recombinant NS2B-NS3pro, and tested drugs at different concentrations. A Tecan Infinite M200 Pro fluorescence spectrophotometer was used to measure the absorbance with emission at 440 nm upon excitation at 360 nm. The intensity measurement was carried out after 30 min of incubation, and the results were expressed as a percentage of the negative control (noninhibitor compound, only substrate, enzyme, and buffer), which was always taken as 100%. All experiments were performed in triplicate and repeated three times. The results were expressed as the mean values ± SD (the corresponding error bars were displayed in the graphical plots).
5.3. Molecular Docking of the NS3 Protease against the Selected Substances
The interaction between the selected drugs and DENV NS2B-NS3pro was identified by the molecular docking study. Here, the template of DENV-3 NS2B-NS3pro was used (PDB: 3U1I) for all docking activities as it showed 89% sequence similarities with DENV-2 NS2B-NS3pro. For docking, only chains A and B from protein PDB 3U1I were used as the template, while other chains and co-crystalized ligands were removed. In addition, water molecules and heterogroups were deleted from the structure by using PyMOL48 software. The protein was prepared directly in AMDock49 software, where protonation was performed using integrated PDB2PQR50 that utilized the Amber forcefield at pH 7.4. Based on the in vitro result, panduratin A was included as the reference drug for the docking study. Zileuton and trimethadione structures were obtained from the DrugBank14 database in a PDB format, while linalool and panduratin A were retrieved from the PubChem32 database in the SDF format. The structures for compounds ziltri (combination of zileuton and trimethadione) and zilool (combination of zileuton and linalool) were drawn using Chemspace (Chemspace.com/home), and the SMILES code was obtained from a similar source. All SMILES and SDF formats were converted to PDB using the Online SMILES Translator and Structure File Generator (https://cactus.nci.nih.gov/translate/). The grid box size and the grid spacing were set around the catalytic triad to a 20 × 20 × 20 dimension and 0.375 Å, respectively, with the center set at x = 21.1, y = −16.0, and z = 8.1 using AutoDockTools (ADT). Docking simulations were performed using AMDock49 using AutoDock Vina.51 AutoDock Vina51 generates different ligand conformers using a Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm. The BFGS algorithm is implemented with an iterated local method search. Final docking output files were analyzed for hydrogen bonds and other interactions using integrated PyMOL48 software in AMDock49 and Discovery Studio Visualizer.52
Acknowledgments
This research was supported by the Ministry of Education, Malaysia, through the Fundamental Research Grant Scheme (FRGS/1/2016/SKK10/UITM/02/1).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c02607.
Additional details from principal component analysis (PCA) of the chemical space of the prediction models, percentage of the variance of the top 10 PCs, and eigenvalue of the training set (PDF)
Author Contributions
Z.L.A.—conceptualization, data analysis, and writing—review & editing; H.-Y.C.—writing—review & editing; R.Y.—writing—review & editing; and F.M.F.—conceptualization, data analysis, and writing—review and editing.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization . Dengue and Severe Dengue, 2018. http://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue. (accessed October 26, 2018).
- Dengue Worldwide Overview, 2022. https://www.ecdc.europa.eu/en/dengue-monthly. (accessed July 06, 2022).
- Obi J. O.; Gutiérrez-Barbosa H.; Chua J. V.; Deredge D. J. Current Trends and Limitations in Dengue Antiviral Research. Trop. Med. Infect. Dis. 2021, 6, 180 10.3390/tropicalmed6040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First FDA-approved vaccine for the prevention of dengue disease in endemic regions [Press release] U.S. Food and Drug Administration. https://www.fda.gov/news-events/press-announcements/first-fda-approved-vaccine-prevention-dengue-disease-endemic-regions (accessed July 27, 2023).
- Low J. G. H.; Ooi E. E.; Vasudevan S. G. Current Status of Dengue Therapeutics Research and Development. J. Infect. Dis. 2017, 215, S96–S102. 10.1093/infdis/jiw423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F.; Shi P.-Y. The Challenges of Dengue Drug Discovery and Development. Clin. Invest. 2014, 4, 683–685. 10.4155/cli.14.67. [DOI] [Google Scholar]
- Behnam M. A. M.; Nitsche C.; Boldescu V.; Klein C. D. The Medicinal Chemistry of Dengue Virus. J. Med. Chem. 2016, 59, 5622–5649. 10.1021/acs.jmedchem.5b01653. [DOI] [PubMed] [Google Scholar]
- de Oliveira A. S.; da Silva M. L.; Oliveira A. F. C. S.; da Silva C. C.; Teixeira R. R.; De Paula S. O. NS3 and NS5 Proteins: Important Targets for Anti-Dengue Drug Design. J. Braz. Chem. Soc. 2014, 25, 1759–1769. 10.5935/0103-5053.20140057. [DOI] [Google Scholar]
- Tomlinson S. M.; Malmstrom R. D.; Russo A.; Mueller N.; Pang Y.-P.; Watowich S. J. Structure-Based Discovery of Dengue Virus Protease Inhibitors. Antiviral Res. 2009, 82, 110–114. 10.1016/j.antiviral.2009.02.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K.-H.; Nalivaika E. A.; Prachanronarong K. L.; Yilmaz N. K.; Schiffer C. A. Dengue Protease Substrate Recognition: Binding of the Prime Side. ACS Infect. Dis. 2016, 2, 734–743. 10.1021/acsinfecdis.6b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M. M.; Biswas S.; Islam K. J.; Paul A. S.; Mahato S. K.; Ali M. A.; Halim M. A. Antiviral Phytochemicals as Potent Inhibitors against NS3 Protease of Dengue Virus. Comput. Biol. Med. 2021, 134, 104492 10.1016/j.compbiomed.2021.104492. [DOI] [PubMed] [Google Scholar]
- Bakulin I.; Pasechnikov V.; Varlamicheva A.; Sannikova I. NS3 Protease Inhibitors for Treatment of Chronic Hepatitis C: Efficacy and Safety. World J. Hepatol. 2014, 6, 326–339. 10.4254/wjh.v6.i5.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Fang H.; Reagan K.; Xu X.; Mendrick D. L.; Slikker W.; Tong W. In Silico Drug Repositioning – What We Need to Know. Drug Discovery Today 2013, 18, 110–115. 10.1016/j.drudis.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Novick P. A.; Ortiz O. F.; Poelman J.; Abdulhay A. Y.; Pande V. S. SWEETLEAD: An in Silico Database of Approved Drugs, Regulated Chemicals, and Herbal Isolates for Computer-Aided Drug Discovery. PloS One 2013, 8 (11), e79568 10.1371/journal.pone.0079568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Feunang Y. D.; Guo A. C.; Lo E. J.; Marcu A.; Grant J. R.; Sajed T.; Johnson D.; Li C.; Sayeeda Z.; Assempour N.; Iynkkaran I.; Liu Y.; Maciejewski A.; Gale N.; Wilson A.; Chin L.; Cummings R.; Le D.; Pon A.; Knox C.; Wilson M. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huchting J. Targeting Viral Genome Synthesis as Broad-Spectrum Approach against RNA Virus Infections. Antiviral Chem. Chemother. 2020, 28, 204020662097678 10.1177/2040206620976786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenmeier G. Inhibition of the NS2B–NS3 protease: towards a causative therapy for dengue virus diseases. Dengue Bull. 2004, 28, 58–67. [Google Scholar]
- Valle R. P. C.; Falgout B. Mutagenesis of the NS3 Protease of Dengue Virus Type 2. J. Virol. 1998, 72, 624–632. 10.1128/JVI.72.1.624-632.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein H. A.; Borrel A.; Geneix C.; Petitjean M.; Regad L.; Camproux A.-C. PockDrug-Server: A New Web Server for Predicting Pocket Druggability on Holo and Apo Proteins. Nucleic Acids Res. 2015, 43, W436–W442. 10.1093/nar/gkv462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othman R.; Othman R.; Baharuddin A.; Ramakrishnan N. R.; Abd Rahman N.; Yusof R.; Karsani S. A. Molecular Docking Studies of Selected Medicinal Drugs as Dengue Virus-2 Protease Inhibitors. Sains Malays. 2017, 46, 1865–1875. 10.17576/jsm-2017-4610-25. [DOI] [Google Scholar]
- Kiat T. S.; Pippen R.; Yusof R.; Ibrahim H.; Khalid N.; Rahman N. A. Inhibitory Activity of Cyclohexenyl Chalcone Derivatives and Flavonoids of Fingerroot, Boesenbergia Rotunda (L.), towards Dengue-2 Virus NS3 Protease. Bioorg. Med. Chem. Lett. 2006, 16, 3337–3340. 10.1016/j.bmcl.2005.12.075. [DOI] [PubMed] [Google Scholar]
- Lapins M.; Worachartcheewan A.; Spjuth O.; Georgiev V.; Prachayasittikul V.; Nantasenamat C.; Wikberg J. E. S. A Unified Proteochemometric Model for Prediction of Inhibition of Cytochrome P450 Isoforms. PLoS One 2013, 8, e66566 10.1371/journal.pone.0066566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niijima S.; Shiraishi A.; Okuno Y. Dissecting Kinase Profiling Data to Predict Activity and Understand Cross-Reactivity of Kinase Inhibitors. J. Chem. Inf. Model. 2012, 52, 901–912. 10.1021/ci200607f. [DOI] [PubMed] [Google Scholar]
- Leung D.; Schroder K.; White H.; Fang N. X.; Stoermer M. J.; Abbenante G.; Martin J. L.; Young P. R.; Fairlie D. P. Activity of Recombinant Dengue 2 Virus NS3 Protease in the Presence of a Truncated NS2B Co-Factor, Small Peptide Substrates, and Inhibitors. J. Biol. Chem. 2001, 276, 45762–45771. 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- Holbeck S. L.; Collins J. M.; Doroshow J. H. Analysis of FDA-Approved Anti-Cancer Agents in the NCI60 Panel of Human Tumor Cell Lines. Mol. Cancer Ther. 2010, 9, 1451–1460. 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi V. D.; Tripathi I. P.; Bharadwaj S.; Kaushik A. C.; Mishra S. K. Identification of New Potent Inhibitors of Dengue Virus NS3 Protease from Traditional Chinese Medicine Database. VirusDisease 2016, 27, 220–225. 10.1007/s13337-016-0328-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othman R.; Kiat T. S.; Khalid N.; Yusof R.; Irene Newhouse E.; Newhouse J. S.; Alam M.; Rahman N. A. Docking of Noncompetitive Inhibitors into Dengue Virus Type 2 Protease: Understanding the Interactions with Allosteric Binding Sites. J. Chem. Inf. Model. 2008, 48, 1582–1591. 10.1021/ci700388k. [DOI] [PubMed] [Google Scholar]
- Hariono M.; Choi S. B.; Roslim R. F.; Nawi M. S.; Tan M. L.; Kamarulzaman E. E.; Mohamed N.; Yusof R.; Othman S.; Rahman N. A.; Othman R.; Wahab H. A. Thioguanine-Based DENV-2 NS2B/NS3 Protease Inhibitors: Virtual Screening, Synthesis, Biological Evaluation and Molecular Modelling. PLoS One 2019, 14, e0210869 10.1371/journal.pone.0210869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. A.; Elrod K.; Luong C.; Rice M. J.; Mackman R. L.; Sprengeler P. A.; Spencer J.; Hataye J.; Janc J.; Link J.; Litvak J.; Rai R.; Rice K.; Sideris S.; Verner E.; Young W. A Novel Serine Protease Inhibition Motif Involving a Multi-Centered Short Hydrogen Bonding Network at the Active Site. J. Mol. Biol. 2001, 307, 1451–1486. 10.1006/jmbi.2001.4516. [DOI] [PubMed] [Google Scholar]
- Schmidtke P.; Barril X. Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J. Med. Chem. 2010, 53, 5858–5867. 10.1021/jm100574m. [DOI] [PubMed] [Google Scholar]
- Gaulton A.; Hersey A.; Nowotka M.; Bento A. P.; Chambers J.; Mendez D.; Mutowo P.; Atkinson F.; Bellis L. J.; Cibrián-Uhalte E.; Davies M.; Dedman N.; Karlsson A.; Magariños M. P.; Overington J. P.; Papadatos G.; Smit I.; Leach A. R. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. 10.1093/nar/gkw1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; Zaslavsky L.; Zhang J.; Bolton E. E. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res. 2019, 47, D1102–D1109. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Lin Y.; Wen X.; Jorissen R. N.; Gilson M. K. BindingDB: A Web-Accessible Database of Experimentally Determined Protein-Ligand Binding Affinities. Nucleic Acids Res. 2007, 35, D198–D201. 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Bender A. How Similar Are Those Molecules after All? Use Two Descriptors and You Will Have Three Different Answers. Expert Opin. Drug Discovery 2010, 5, 1141–1151. 10.1517/17460441.2010.517832. [DOI] [PubMed] [Google Scholar]
- Parzen E. On Estimation of a Probability Density Function and Mode. Ann. Math. Stat. 1962, 33, 1065–1076. 10.1214/aoms/1177704472. [DOI] [Google Scholar]
- Rosenblatt M. Remarks on Some Nonparametric Estimates of a Density Function. Ann. Math. Stat. 1956, 27, 832–837. 10.1214/aoms/1177728190. [DOI] [Google Scholar]
- Aitchison J.; Aitken C. G. G. Multivariate Binary Discrimination by the Kernel Method. Biometrika 1976, 63, 413–420. 10.1093/biomet/63.3.413. [DOI] [Google Scholar]
- Edgar R. C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. J.; Rodrigues A. P. C.; ElSawy K. M.; McCammon J. A.; Caves L. S. D. Bio3d: An R Package for the Comparative Analysis of Protein Structures. Bioinformatics 2006, 22, 2695–2696. 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Mathea M.; Klingspohn W.; Baumann K. Chemoinformatic Classification Methods and Their Applicability Domain. Mol. Inf. 2016, 35, 160–180. 10.1002/minf.201501019. [DOI] [PubMed] [Google Scholar]
- Afantitis A.; Melagraki G.; Sarimveis H.; Koutentis P. A.; Markopoulos J.; Igglessi-Markopoulou O. Development and Evaluation of a QSPR Model for the Prediction of Diamagnetic Susceptibility. QSAR Comb. Sci. 2008, 27, 432–436. 10.1002/qsar.200730083. [DOI] [Google Scholar]
- Melagraki G.; Afantitis A.; Sarimveis H.; Koutentis P. A.; Kollias G.; Igglessi-Markopoulou O. Predictive QSAR Workflow for the in Silico Identification and Screening of Novel HDAC Inhibitors. Mol. Diversity 2009, 13, 301–311. 10.1007/s11030-009-9115-2. [DOI] [PubMed] [Google Scholar]
- Berthold M. R.; Cebron N.; Dill F.; Gabriel T. R.; Kötter T.; Meinl T.; Ohl P.; Sieb C.; Thiel K.; Wiswedel B.. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning, and Applications; Preisach C.; Burkhardt H.; Schmidt-Thieme L.; Decker R., Eds.; Springer: Berlin Heidelberg, 2008; pp 319–326. [Google Scholar]
- Zandi K.; Teoh B.-T.; Sam S.-S.; Wong P.-F.; Mustafa M. R.; AbuBakar S. Novel Antiviral Activity of Baicalein against Dengue Virus. BMC Complementary Altern. Med. 2012, 12, 214 10.1186/1472-6882-12-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothan H. A.; Abdulrahman A. Y.; Sasikumer P. G.; Othman S.; Abd Rahman N.; Yusof R. Protegrin-1 Inhibits Dengue NS2B-NS3 Serine Protease and Viral Replication in MK2 Cells. J. Biomed. Biotechnol. 2012, 2012, 1–6. 10.1155/2012/251482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC, 2017.
- Valdés-Tresanco M. S.; Valdés-Tresanco M. E.; Valiente P. A.; Moreno E. AMDock: A Versatile Graphical Tool for Assisting Molecular Docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12 10.1186/s13062-020-00267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky T. J.; Nielsen J. E.; McCammon J. A.; Baker N. A. PDB2PQR: An Automated Pipeline for the Setup of Poisson–Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discovery Studio Visualizer, v21.1.0.20298; Biovia, Dassault Systemes: San Diego, 2021.
- Maia W. M. N.; de Andrade F. D. C. P.; Filgueiras L. A.; Mendes A. N.; Assunção A. F. C.; Rodrigues N. D. S.; Marques R. B.; Filho A. L. M. L.; de Sousa D. P.; Da Silva Lopes L. Antidepressant Activity of Rose Oxide Essential Oil: Possible Involvement of Serotonergic Transmission. Heliyon 2021, 7, e06620 10.1016/j.heliyon.2021.e06620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siramshetty V. B; Grishagin I.; Nguyen Đa.-T.; Peryea T.; Skovpen Y.; Stroganov O.; Katzel D.; Sheils T.; Jadhav A.; Mathe E. A; Southall N. T NCATS Inxight Drugs: A Comprehensive and Curated Portal for Translational Research. Nucleic Acids Res. 2022, 50 (D1), D1307–D1316. 10.1093/nar/gkab918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan S.; Rajendar S. Stereoselective Total Synthesis of (−)-Nupharamine Utilizing an α-Chlorosulfide and a Sulfinimine for C–C Bond Formation. Org. Biomol. Chem. 2016, 14, 131–137. 10.1039/C5OB01750E. [DOI] [PubMed] [Google Scholar]
- American Chemical Society . Diaboline, 2022. https://www.acs.org/content/acs/en/molecule-of-the-week/archive/d/diaboline.html. (accessed July 26, 2022).
- Russo E. B.; Marcu J.. Cannabis Pharmacology: The Usual Suspects and a Few Promising Leads. In Cannabinoid Pharmacology; Kendall D.; Alexander S. P. H., Eds.; Academic Press, 2017; Vol. 8, pp 67–134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.