

ABSTRACT
Background
Immunotherapeutic innovation is crucial for limited operability tumors. CAR T-cell therapy displayed reduced efficiency against glioblastoma (GBM), likely due to mutations underlying disease progression. Natural Killer cells (NKs) detect cancer cells despite said mutations – demonstrating increased tumor elimination potential. We developed an NK differentiation system using human pluripotent stem cells (hPSCs). Via this system, genetic modifications targeting cancer treatment challenges can be introduced during pluripotency – enabling unlimited production of modified “off-the-shelf” hPSC-NKs.
Methods
hPSCs were differentiated into hematopoietic progenitor cells (HPCs) and NKs using our novel organoid system. These cells were characterized using flow cytometric and bioinformatic analyses. HPC engraftment potential was assessed using NSG mice. NK cytotoxicity was validated using in vitro and in vitro K562 assays and further corroborated on lymphoma, diffuse intrinsic pontine glioma (DIPG), and GBM cell lines in vitro.
Results
HPCs demonstrated engraftment in peripheral blood samples, and hPSC-NKs showcased morphology and functionality akin to same donor peripheral blood NKs (PB-NKs). The hPSC-NKs also displayed potential advantages regarding checkpoint inhibitor and metabolic gene expression, and demonstrated in vitro and in vivo cytotoxicity against various cancers.
Conclusions
Our organoid system, designed to replicate in vivo cellular organization (including signaling gradients and shear stress conditions), offers a suitable environment for HPC and NK generation. The engraftable nature of HPCs and potent NK cytotoxicity against leukemia, lymphoma, DIPG, and GBM highlight the potential of this innovative system to serve as a valuable tool that will benefit cancer treatment and research – improving patient survival and quality of life.
KEYWORDS: hPSCs; iPSCs; Hematopoietic Differentiation; Organoid; hPSC-Natural Killer Cells; Engraftment; Leukemia, GBM, and DIPG
Introduction
Several reviews1–4, among others, describe the advancement of hPSC hematopoietic differentiation. A crucial step in this process was achieving definitive hematopoiesis, a prerequisite for developing lymphoid cells and hematopoietic stem cells (HSCs)5. In the last several years, various groups have reported successful derivation of different types of lymphoid cells, including T lymphocytes6–11, NK cells12–15, induced NK-T cells16, and B-cells17,18. While differentiation of lymphoid cells from hPSCs has been achieved, the precise tuning of their functionality and the generation of engraftable HSCs remain an ongoing pursuit19,20, thus highlighting the complexity of the hematopoietic niche.
We explored the role of the vascular niche in hematopoietic differentiation. A factor important to note is that HSCs are formed during blood flow initiation in the aorta-gonad-mesonephros region, and flow shear stress may serve as a key inducer of the endothelial-to-hematopoietic transition and HSC development. It has been shown to trigger molecular events leading to the emergence of adult hematopoiesis21. To some extent, we were able to model these conditions in vitro by creating a vascular organoid, which facilitates the proper microenvironment for the generation of HPCs and NKs by closely recapitulating in vivo cellular organization (i.e., signaling gradients and shear stress conditions).
There is currently an unmet need for new therapies to treat patients diagnosed with GBM. Brain tumor drug development is especially challenging because many drugs cannot penetrate the blood-brain barrier (BBB) to reach their targets. As a result, immunotherapy is emerging as a promising strategy for treating brain tumor patients22. The ability of a large population of NK cells to pass through the BBB, and be involved in the anti-cancer response, suggests that cellular immunotherapy could be an effective route for tumor elimination23.
NK cells may provide a superior immunotherapy product24. NKs directly kill tumor cells by releasing cytoplasmic granules containing perforin and Granzyme B. This strategy is coupled with the expression of tumor necrosis factor (TNF) family members, such as FasL or TNF-related apoptosis-inducing ligand, which induce tumor cell death by interacting with their respective receptors. Additionally, antibody-dependent cellular cytotoxicity, mediated by the Fc receptor CD16, can trigger NK cell degranulation against antibody-coated target cells25–30. Another advantage of NK cell versatility is the ability to recognize cancer cells despite their continuous mutations and to recognize stress and senescence markers induced by chemotherapy during cancer treatment31. Moreover, NKs avoid graft-vs-host disease, commonly associated with T cell therapy, and were recently shown to be more effective than T cells in killing DIPG32. Studies also report that GBM cells with stem cell properties (GSCs) are more susceptible to NK lysis via up-regulation of activating ligands and down-regulation of inhibitory ligands for NK cell receptors33,34. This aspect is linked with the fact that GSCs are located near the vascular niche, which is enriched with NKs – further highlighting NK immunotherapy as an attractive treatment for GBM35.
hPSC-NKs also offer advantages over PB-NKs and NK-92 cells. hPSC-NKs have demonstrated potent anti-tumorigenic activity and are phenotypically similar to PB-NKs13. Furthermore, the strong proliferation capacity of hPSCs allows the introduction of genetic modifications and development of libraries incorporating “off-the-shelf” haplotype-specific cells at reduced costs. hPSC-NKs can therefore provide an unlimited source for the adoptive transfer of NK cells to treat diseases.
Our innovative organoid differentiation system provides an optimal environment for HPC and NK generation. Here, we provide evidence that the organoid-derived HPCs are engraftable. Moreover, we show that this system produces NKs with potent cytotoxicity against leukemia, lymphoma, DIPG, and GBM. These data suggest that the vascular organoid system developed by our group may serve as a valuable tool for brain cancer treatment.
Materials and Methods
Cell Lines
hPSC refers to human pluripotent stem cells, namely, hESCs and iPSCs. hPSC-NKs were developed from H1 hESCs and utilized during experiments with DIPG. The H1 cell line was purchased from WiCell Research Institute, Madison, WI. VEC-tdTomato/CD43-eGFP was obtained from Dr. Igor Slukvin (University of Wisconsin School of Medicine and Public Health, Madison, WI). hPSC #5 is an iPSC line developed from peripheral blood (used for all other experiments). hPSC #5 was derived using the CytoTune™-iPS 2.0 Sendai Reprogramming Kit. The pluripotency and ploidy (novelty) of derived iPSCs were confirmed using the PluriTest platform (ThermoFisher). All hPSCs were maintained on Matrigel-coated tissue culture plates in mTESR1 (STEMCELL Technologies) or iPS-Brew XF (Miltenyi Biotec) medium. The OP9–DLL4 line was obtained from Dr. Igor Slukvin. Mouse stromal cell lines were maintained in alpha-modified Eagle medium (alpha-MEM; Thermo Fisher Scientific) with 20% fetal bovine serum (FBS). The Jurkat cell line was obtained from Dr. Ntziachristos (ATCC, #CCL-119) and was authenticated via short-tandem repeat profiling36.
Flow Cytometry
Cells were stained with the appropriate conjugated antibodies for 30 min at 4°C, washed in 0.5% bovine serum albumin/phosphate-buffered saline (PBS) 2 mmol/L EDTA solution, and analyzed using FACSCalibur (BD Biosciences). The following antibodies (anti-human) were utilized: PE CD31: Miltenyi Biotec #130-118-965; FITC CD34: Miltenyi Biotec #130-113-178; APC CD144: Miltenyi Biotec #130-126-010; APC CD73: Miltenyi Biotec #130-124-011; PE CD45 (clone HI30): BioLegend #304008; FITC CD3: Miltenyi Biotec #130-113-690; PE CD90: Miltenyi Biotec #130-117-388; BV421 CD56: BD Horizon #562751; AF647 CD158 (KIR): BD Pharmingen #567324; FITC Perforin: eBioscience #11-9994-42; KO CD45: Beckman Coulter #A96416; PE NKp46: BD Pharmingen #557991; PE-Cy7 CD159α (NKG2A): Beckman Coulter #B10246; PEDazzle CD16: BioLegend #302054; PerCP-Cy5 CD94: BioLegend #302054; FITC IFNγ: Sysmex #AE092283; PE TNFα: BD Pharmingen #554513; CF594 IL-8: BD Horizon #563531.
Organoid System
To obtain hematopoietic progenitors, hPSCs were gently dissociated into single cells using PBS (free of Ca2+ and Mg2+), plated onto fibronectin-coated dishes at a seeding density of 1–5 × 106 cells per 60 mm dish, and cultured overnight to produce cell colonies. Differentiation was induced by incubating cells with a medium comprising Advanced DMEM/12, ascorbic acid, and a Wnt activator – CHIR99021 (CAS registry number 252,917-06-9; concentration of 5 µM). Afterward, the Wnt activator was removed, and incubation was continued for about 3 days to produce hemogenic endothelium. To generate a 3D vessel organoid capable of producing highly functional hematopoietic cells, including NKs and HPCs, the Day 5 monolayer of hPSCs was gently dispersed into small clusters of approximately 10–100 cells and carefully plated onto recombinant fibronectin with serial addition of cytokines (SCF, IL-3, IL-7, IL-15, FLT3-L) over the course of 72 hrs until the initiation of vessel-like structure formation was achieved. Culture incubation was continued in differentiation medium until cells appeared within the vessels.
Derivation of HPCs from hPSCs
HPC1
To obtain HPC1, hPSCs were gently dissociated into small colonies using PBS (free of Ca2+ and Mg2+), plated onto fibronectin-coated 60 mm dishes, and cultured overnight. Differentiation was induced by applying induction medium comprising Advanced DMEM/12, ascorbic acid, and a Wnt activator – CHIR99021 (CAS registry number 252,917-06-9; concentration of 5 µM). Afterward, the Wnt activator was removed, and incubation was continued for about 3 days to produce hemogenic endothelium.
HPC2
To obtain HPC2, hPSCs were gently dissociated into single cells using Accutase, plated onto fibronectin-coated dishes at a seeding density of 1–5 × 106 cells per 60 mm dish, and cultured overnight to produce cell colonies. Differentiation was induced by applying induction medium comprising Advanced DMEM/12, ascorbic acid, and a Wnt activator – CHIR99021 (CAS registry number 252,917-06-9; concentration of 5 µM). Afterward, the Wnt activator was removed, and incubation was continued for about 3 days to produce hemogenic endothelium. The cells were then collected, transferred to OP9-DLL4, and cultured in Hematopoietic Stem Cell Growth Medium for 3 days.
HPC3
To obtain HPC3, hPSCs were gently dissociated into single cells using Accutase, plated onto fibronectin-coated dishes at a seeding density of 1–5 × 106 cells per 60 mm dish, and cultured overnight to produce cell colonies. Differentiation was induced by applying induction medium comprising Advanced DMEM/12, ascorbic acid, and a Wnt activator – CHIR99021 (CAS registry number 252,917-06-9; concentration of 5 µM). Afterward, the Wnt activator was removed, and incubation was continued for about 3 days to produce hemogenic endothelium. The cells were then gently dispersed into small clusters of approximately 10–100 cells and carefully plated onto recombinant fibronectin with serial addition of cytokines over the course of 72 hrs, until the initiation of vessel-like structure formation was achieved. Culturing continued in differentiation medium containing SCF, IL-3, and TPO for 10 days.
Transplantation of HPCs into Mice
6- to 8-week-old, pathogen-free, male or female NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, Jackson Laboratory) were utilized in this study. A sublethal dose of 2.5 Gy of x-ray irradiation was delivered to the whole body. At least 4 hrs later (within 24 hrs post-irradiation), 5 x 105–2 x 106 filtered (70 µM strainer) HPCs were injected into irradiated mice via lateral tail vein in 200–400 µL of 5% FBS containing PBS. HPCs were allowed to engraft in mice for a minimum of 8–12 weeks.
Determination of Chimerism Levels
Engraftment level was assessed post-injection at weeks 8, 12, 13, and 23 via flow cytometric analysis of recipient mice peripheral blood by detecting human CD45+ cells. 50–100 µL of whole blood was collected via lateral tail vein from all tested mice. Afterward, red blood cells were lysed and the remaining cells were stained with fluorochrome-conjugated antibodies according to the standard flow cytometric preparation technique, including the FcR blocking step. Human CD45+ cells can routinely be detected in peripheral blood as early as 4 weeks after human HSC injection. Samples were analyzed using a BD LSR-Fortessa SORP Cell Analyzer with HTS (6-laser, 18-parameter). The human CD45 antibody (clone HI30) was obtained from BioLegend.
Statistics
Data comparison between the two groups was conducted using the student’s t-test, while analysis of variance (ANOVA) was employed for multiple grouped data. Data are represented as mean ± standard error of the mean (S.E.M). A p-value of < 0.05 was deemed statistically significant. Statistical analysis was performed using Prism GraphPad (GraphPad Software, Inc., La Jolla, CA).
NK Cell Characterization
Marker Analysis
For flow cytometric analysis of NK cell differentiation, the cells were labeled with a combination of monoclonal antibodies (mAbs), including CD45 Krome Orange (Beckman Coulter), CD56 PE, CD16 PE-Dazzle, CD15 PerCP-Cy5.5, and CD94 APC (all from BioLegend, San Diego, CA). For isotype controls, specific mAbs were substituted for corresponding nonspecific IgG isotypes. LIVE/DEAD Fixable Violet stain (ViVID; Invitrogen, Eugene, OR) was included to discriminate dead cells. Labeling was performed in 96-well U-bottom plates using 2.5–3 × 105 cells in 50 μL of PBS per well. After a 25 min incubation with mAbs at 4°C, the cells were washed 2×, subsequently transferred into 5 mL Falcon tubes (BD, Bedford, MA), and processed on LSR-II (BD, San Jose, CA). Compensation controls were prepared using the AbC anti-mouse bead kit for mouse mAb capture and the ArC amine reactive compensation bead kit for ViVID (both kits from Invitrogen), according to manufacturer instructions. Data analysis was performed using FlowJo software (Tree Star, Ashland, OR). FasL expression was assessed using the CD178 antibody (Miltenyi Biotec) after a 2 hr co-incubation of hPSC-NKs or PB-NKs with DIPG cells.
Degranulation Assay
After the cells were stained for surface markers, they were fixed and permeabilized using Cytofix/Cytoperm solution (BD Biosciences kit) for 20 min at 4°C. Following this, the cells were washed 2× with Perm/Wash solution (provided in the kit), stained with Abs against perforin or cytokines for 30 min at RT, washed with Perm/Wash solution once more, and analyzed using the flow cytometer.
For intracellular cytokine analysis, the samples were stimulated with PMA (25 ng/mL), ionomycin (1µM), and Brefeldin A (10 µg/mL) (all from Sigma) and incubated for 4 hrs at 37°C (5% CO2).
For IFNγ and TNFα staining, the following procedure was used:
Stain for surface antibodies.
Wash cells with PBS (1X) two times.
Pellet the cells and add 250 µL of BD Cytofix/Cytoperm solution (for purposes of fixation and permeabilization; BD Biosciences, Cat # 554722). Vortex the cells at 400 rpm while adding this solution.
Incubate in the dark for 30 min at 4°C.
Add 2 mL of BD Perm/Wash (1X, BD Biosciences, Cat # 554723). Centrifuge at 1500 rpm for 5 min (2×).
Add 5 µL of Antibody (10 ng/µL) TNFα and IFNγ. Incubate in the dark for 30–40 min.
Wash 2× with Perm/Wash solution.
Add 500 µL of Perm/Wash solution and analyze using BD LSR II Flow cytometer.
Immunofluorescence Analysis
Immunofluorescence staining was used to detect Granzyme B and LAMP1 expression. Cells were washed and suspended in RPMI-1640 medium (Life Technologies, Cat# 11875–093). Two spots of 20,000 cells each were mounted to Tissue Path Superfrost Plus GoldMicroscope Slides (Fisher Scientific, Cat# 15-188-48) using a cytospin centrifuge (Shandon, Cytospin 2) with CytoSep Dual Funnels (Simport Scientific, Cat# M964-20FW) permanufacturer instructions. Subsequently, the slides and funnels were spun at 80 RCF for 2 min. Slides were removed from the funnel apparatus and fixed for 5 min in a 9:1 dilution of Methanol (Sigma-Aldrich, Cat# 34860-1 L-R): Acetone (Sigma-Aldrich, cat# 270725-1 L). Fixed slides were then washed, and antigen retrieval was performed by boiling the slides in sodium citrate buffer (10 mM tri-Sodium citrate (Millipore Sigma, Cat# 1110371000), 0.05% Tween 20, pH 6.0) for 20 min. Slides were cooled to RT, washed, and blocked in 5% BSA (Millipore Sigma, Cat# A5611). Afterward, the slides were probed with either isotype controls or rat anti-human Granzyme B (Thermo Fisher Scientific, Cat# 14-8889-82). All slides were washed and then mounted using a ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, Cat# P36962) to visualize nuclei. Antigen distribution was examined using a Nikon EclipseTE2000-S fluorescence microscope (Nikon Instrument INC).
Confocal Microscopy
Confocal images were acquired using LSM 800 and LSM 880 Microscope systems (Zeiss, Thornwood, NY, USA).
RNA Isolation
Total RNA was extracted with the RNeasy Mini Kit (Qiagen) via the instructions provided in the manufacturer’s protocol. RNA quality and concentration were assessed via the Nanodrop.
RNA Sequencing Analysis
Aliquots of RNA were submitted to Northwestern University’s NUSeq Core. The total RNA library was prepared, and the samples were analyzed using the HiSeq 4000 System with 50 bp single-end reads. Afterward, the significantly expressed genes were analyzed via ShinyGo (version 0.75), MetaCore (Clarivate Analytics, version 20.1.1), and R Studio (version 4.2.1). The following packages were incorporated into the R script in order to generate the heatmap, perform gene set enrichment analysis, and create the category network plot: pheatmap, clusterProfiler, org.Hs.eg.db, enrichplot (cnetplot), pathview, and ggplot2. Bioinformatic references linked to the programs and packages are provided37–43.
Cytotoxicity Assays
NK Cytotoxicity Against DIPG
DIPG line SF8628 was kindly provided by Dr. Hashizume. To evaluate the killing efficiency of NKss against DIPG SF8628 cells in vitro, we incubated the target cells with either PB-NKs or hPSC-NKs at ratios of 1:1, 1:5, and 1:10 for 4 hrs. The killing activity was assessed using FACS (Fluorescence-Activated Cell Sorting) analysis and via an automated cell counting method.
NK Cytotoxicity Against K562
To evaluate the killing efficiency of NKss against K562 cells, the target cells were incubated with hPSC-NKs at ratios of 1:25 and 1:50 for 2 hrs. The killing activity was assessed using FACS.
NK Cytotoxicity Against Attached GBM Cultures
L-R0315, L-3752, and L-4687 cell lines were kindly provided by Dr. Lee. For this experiment, we cultured GBM lines in DMEM containing 10% Heat Inactivated FBS medium (Gibco). When GBM cells reached confluency, hPSC-NKs were added to the wells at a 5:1 E/T ratio and incubated for 1.5 hrs. Cell viability was assessed using the Luna cell counter.
NK Cytotoxicity Against GBM Spheroids
GBM spheres were cultured in cancer stem cell medium comprising DMEM/F12 supplemented with rhEGF, rhFGF-basic, B-27, N-2, and L-Glutamine. Prior to co-incubation, hPSC-NKs were labeled using the CellTracker™ Green CMFDA Dye, Invitrogen™ (Fisher Scientific). Spheres of approximately similar size were manually picked out, plated onto fluoro-dishes, and co-incubated with hPSC-NKs for 48 hrs. For some experiments, tumors were stained with CellTracker™ Violet BMQC Dye prior to plating. The viability of GBM spheres was assessed using confocal microscopy. Propidium Iodide was added to the medium to distinguish live/dead cells.
NK IL13Rα2 Assay
Patient-derived GBM6 and GBM12 xenograft lines have been previously described to express a tumor-associated antigen, IL13Rα244. To evaluate the effect of the humanized IL13Rα2 antibody (IL13Rα2 hAb),provided by Dr. Balyasnikova, on hPSC-NK killing efficiency, we compared GBM12 cultured alone vs GBM12 co-incubated with hPSC-NKs vs GBM12 co-incubated with hPSC-NKs with the addition of IL13Rα2 hAb (2 µg/mL).
NK Cytotoxicity Against K562 In Vivo
5 x 106 K562 Luc tumor cells were injected subcutaneously to the right flank of 10-week-old Nod/Scid mice. 3 days after inoculation, tumor engraftment was verified using IVIS imaging. Following validation of tumor engraftment, mice were injected with IL-2 into the right flank, and 2 x 104 hPSC-NKs were injected subcutaneously around the tumor site. IL-2 was then injected every other day for 1 week to support NK cell activity. Tumor growth and progression were tracked using IVIS imaging.
Results
To evaluate the engraftable nature of HPCs and NK cytotoxicity against leukemia, lymphoma, DIPG, and GBM, we performed cell characterization experiments, organoid engraftment, cell transplantation into mice, and functional assays. Figure 1 provides an overview of this work and creates a structural framework for navigating the results outlined below.
Figure 1.
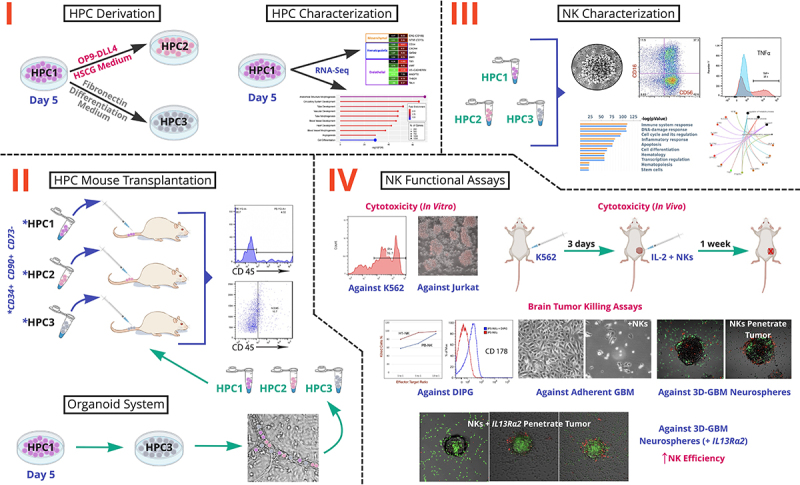
Schematic Overview of the Study. There are four key stages: I. HPC Derivation and Characterization; II. Organoid System Engraftment and HPC Mouse Transplantation; III. NK Characterization; IV. NK Functional Assays. HPCs are hematopoietic progenitor cells that were analyzed at 3 differentiation time points (HPC1, HPC2, HPC3). HSCG is Hematopoietic Stem Cell Growth Medium. K562, Jurkat, DIPG, and GBM are chronic myelogenous leukemia, acute T-lymphoblastic leukemia, diffuse intrinsic pontine glioma, and glioblastoma multiforme cell lines, respectively.
Hematopoietic Progenitor Studies
(I) Engraftment Assessment
Successful reconstitution of the human immune system using hPSCs in a mouse would enable the creation of diverse, humanized mouse models to study patient-specific disease pathogenesis and optimize drug regimens. We previously showed that a large number of hematopoietic progenitors could be produced using the CHIR99021 induction monolayer system14. In this study, we have shown that said progenitors, formed on Day 5 of differentiation (Figure 2A), can be utilized to create a vascular organoid to facilitate the production of functional blood cells within blood vessels (Figure 2B). Additional experiments with the VEC-tdTomato/CD43-eGFP cell line demonstrated that these cells expressed CD43+ (Figure 2C), while blood vessels were positive for CD144 (Figure 2D). The HPCs developed within the vessels and then emerged through the vasculature.
Figure 2.
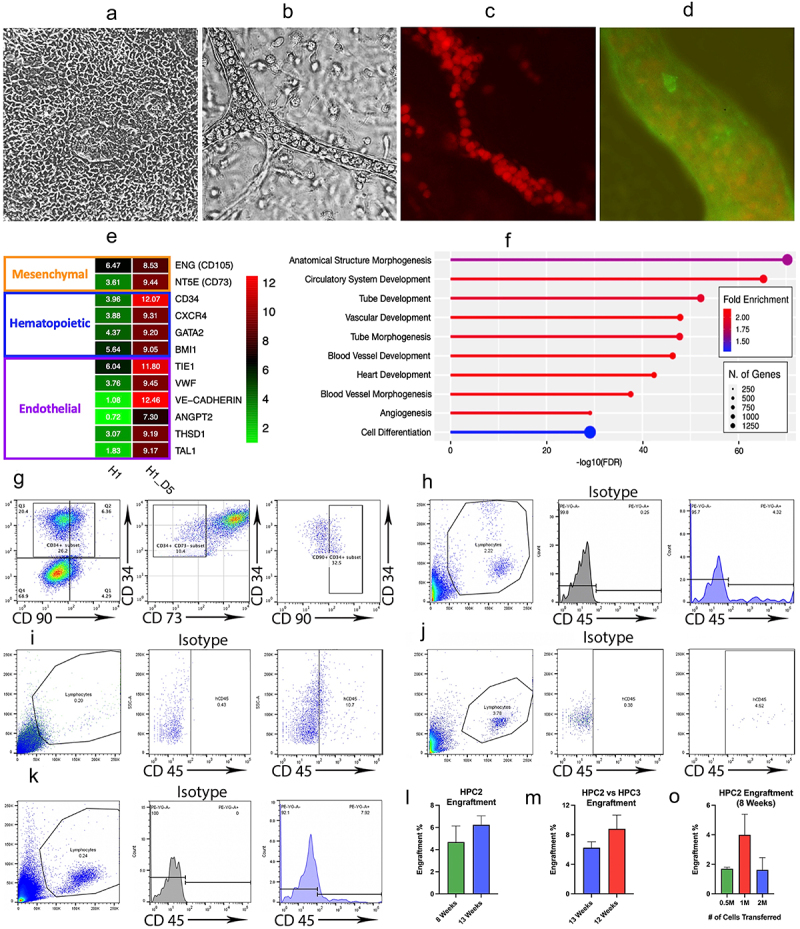
Hematopoietic Progenitor Cell Characterization and Transplantation Experiments.
a. Phase contrast (PhC) image of monolayer progenitor population at Day 5 of differentiation. b. PhC image showing a vessel within the vascular organoid. c. Immunohistochemistry image showing the vascular organoid. Hematopoietic progenitor cells expressing CD43 develop inside of a tubular structure. d. The tubular structures of the organoid are formed by endothelial cells (stained for VE-Cadherin). e. Heatmap showing significant up-regulation in endothelial and hematopoietic gene expression of Day 5 progenitors, compared to hPSC control. f. ShinyGO functional enrichment tool showing that the most significantly up-regulated genes in Day 5 progenitors are predominantly affiliated with hematopoietic processes. g. Flow cytometry images demonstrating that cells from Day 5 of CHIR differentiation exhibit a CD34+CD90+CD73− engraftment signature. h. Flow cytometric analysis, conducted at 8 weeks post-injection, showing the percentage of human CD45+ cells generated by hematopoietic progenitor cells at time point 1 (HPC1). i. Flow cytometric analysis, conducted at 21 weeks post-injection, showing the percentage of human CD45+ cells generated by HPC1. j. Flow cytometric analysis, conducted at 8 weeks post-injection, showing the percentage of human CD45+ cells generated by hematopoietic progenitor cells at time point 2 (HPC2). k. Flow cytometric analysis, conducted at 12 weeks post-injection, showing the percentage of human CD45+ cells generated by hematopoietic progenitor cells at time point 3 (HPC3). l. A graph demonstrating a trend toward an increasing percentage of engrafted HPC2 (from week 8 to week 13). Bars represent mean ± SEM from at least three independent experiments. m. A graph demonstrating that there is no significant increase in engraftment potential at time point HPC3 compared to HPC2. Bars represent mean ± SEM from at least three independent experiments. o. A graph demonstrating no significant increase in engraftment potential when increasing the number of injected cells from 0.5 million to 2 million. Bars represent mean ± SEM from at least three independent experiments.
RNA-Seq data provided further insight into the hematopoietic functionality of Day 5 HPCs. The heatmap confirmed expression of hematopoietic and endothelial-associated markers (Figure 2E), and the ShinyGO functional enrichment tool showed that the most significantly up-regulated genes in Day 5 HPCs are predominantly affiliated with hematopoietic processes, the majority of which incorporate ≥ 750 genes present in our dataset (Figure 2F). Furthermore, these statistically significant processes have high Fold Enrichment values. This indicates that the genes in our dataset are overly represented in said processes, thereby emphasizing the relevance of their functional contribution. Together, these aspects lend further support to the hematopoietic capabilities of Day 5 HPCs.
Flow cytometric analysis demonstrates an extension of these results (Figure 2G). By dissociating pluripotent stem cells with Ca+/Mg+ and plating them on fibronectin-coated plates before inducing differentiation, we achieved a CD34+CD90+CD73− engraftment signature in a subset of the cells. This prompted us to evaluate the engraftment potential of these HPCs by analyzing 3 different differentiation time points (HPC1, HPC2, HPC3). The cells were injected into the tail vein of NSG mice at concentrations between 5 x 105–2 x 106. For subsequent flow analysis, the cells were double stained with mouse and human CD45+, as well as isotype controls (Figure S1A), to ensure signal specificity. Human cord blood-isolated CD34+ cells were used as control (Figure S1B). Overall, the CD34+ cell population isolated on Day 5 of monolayer differentiation, when injected into NSG mice, generated up to 4.3% of human CD45+ cells by 8 weeks (Figure 2H) and demonstrated engraftment of up to 10.7% at week 21 (Figure 2I).
Next, we co-cultured Day 5 isolated cells with OP9-DLL4 in HSC cytokines for 3 days (HPC2). When injected into mice, these cells produced up to 4.5% CD45+ cells 8 weeks after the injection (Figure 2J) and showed an increasing engraftment trend at 13 weeks post-injection (Figure 2L).
(II) Organoid System for Engraftable Cells
Afterward, we evaluated cells generated via our novel organoid system at Day 10 of differentiation (HPC3). To create a 3D vessel organoid capable of producing highly functional cells of various hematopoietic lineages, we initially dispersed the Day 5 monolayer into small clumps and carefully seeded them onto OP9-DLL4 coated plates with serial addition of cytokines over the course of 72 hrs until initiation of vessel-like structures was achieved. Following this, we maintained the cells in differentiation medium until they appeared within the vessels and began moving through the vasculature in a manner that resembled embryonic blood flow. When floating cells emerged from the vessels, they were collected and injected into mice. At week 12 post-injection, flow cytometric analysis demonstrated that the amount of CD45+ cells in peripheral blood was 7.9% (Figure 2K). There were no significant differences between the efficiency of HPC2 and HPC3 engraftment at 3 months post-injections. Furthermore, we found no significant improvement in engraftment when increasing the number of injected cells from 5 x 105 to 1 × 106 or 2 × 106.
NK Cell Characterization
Organoid System for Highly Functional hPSC-NKs
We previously showed that HPCs obtained via our monolayer differentiation system preferentially differentiated toward NKs of definitive lineage45. After verifying engraftment potential, we utilized the organoid differentiation system to produce the NK cell population. After constructing the organoid, we continued to culture the cells with cytokines specific to NK cell development for 21 days. The resulting cells were morphologically identical to PB-NKs (Figures 3A, B) and propagated in clusters (Figure 3C).
Figure 3.
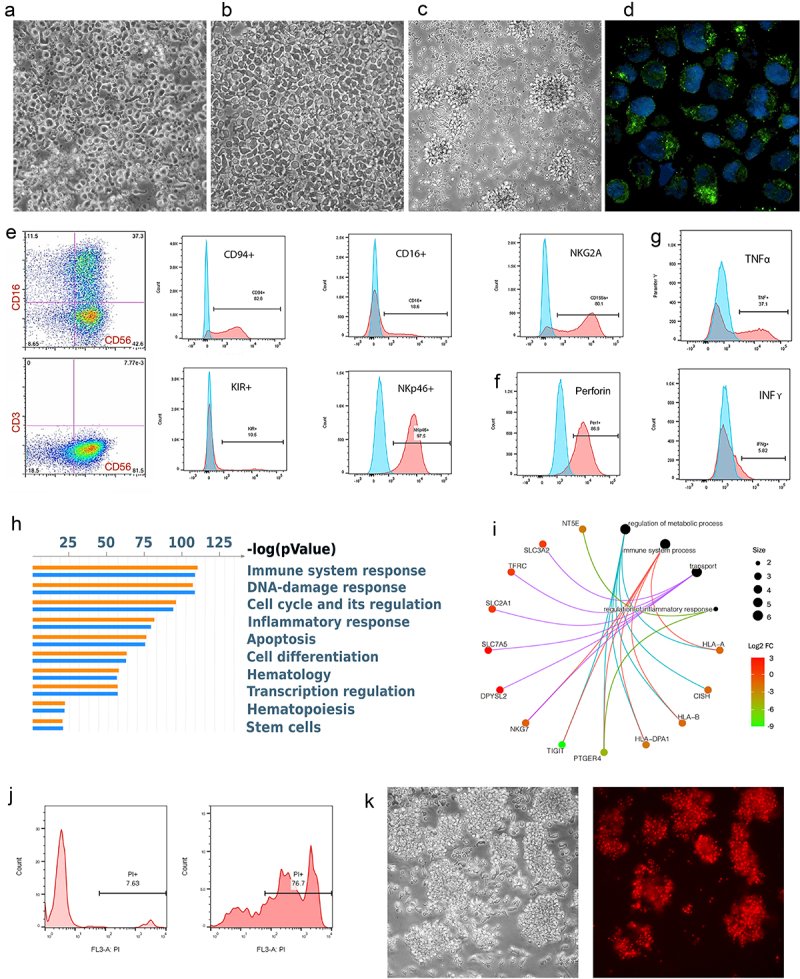
hPSC-NK Cell Characterization.
a. PhC image showing the morphologic appearance of hPSC-NKs. b. PhC image showing the morphologic appearance of PB-NKs. c. PhC image showing the morphologic appearance of activated hPSC-NKs. d. Immunofluorescent image demonstrating granules containing Granzyme B (green) within the cytoplasm of hPSC-NKs. DAPI (blue) is used to visualize cell nuclei. e. Flow cytometric analysis showing CD56, CD16, CD94, NKp46, and NKG2A hPSC-NK marker expression. f. Flow cytometric analysis showing high expression of perforin in hPSC-NKs. g. Flow cytometric analysis demonstrating hPSC-NK production of cytokines, TNFα, and IFNγ. Cells were stimulated with PMA/ionomycin in the presence of Brefeldin A (overnight), fixed, permeabilized, and stained for TNFα or IFNγ. h. MetaCore analysis demonstrating the top 50 statistically significant functions among PB-NKs and hPSC-NKs. i. A category network plot showing statistically significant genes (RNA-Seq) that play an important role in NK cell anti-tumor response. j. Flow cytometric analysis showing that hPSC-NKs exhibit high cytotoxicity against K562 cells; PI = propidium iodide. k. PhC image (left) of Jurkat cells co-incubated with hPSC-NKs for 2 hrs. PI staining (right) showing that the majority of Jurkat cells are non-viable.
Following these observations, we characterized hPSC-NKs using flow cytometry, a cytokine degranulation assay, and an evaluation of gene expression and functional processes. Flow cytometry results showed that up to 90% of the cells had a CD56+CD3– signature and variably expressed CD16. The cells also highly expressed CD94, NKp46, and NKG2A, while KIR expression was low (Figures 3E and S1D). Furthermore, upon stimulation with target cells, hPSC-NKs released high levels of Granzyme B (Figures 3D and S1E), perforin, and TNFα. The expression of IFNγ was low (Figures 3F, G, and S1F).
Afterward, we employed bioinformatic analysis to identify lineage-specific functional correlations linked to the most significantly expressed genes in hPSC-NKs vs PB-NKs. On the basis of this data, we utilized MetaCore to generate the top 50 statistically significant functions among the two cell lines. Compared to PB-NKs, the hPSC-NK gene set employed highly comparable (and at times greater) numbers of pathways dedicated to functions such as immune system response, apoptosis, inflammation, hematopoiesis, and others (Figure 3H). This result provides insight into the wide-ranging functional similarity between PB-NKs and hPSC-NKs, which is a crucial factor when considering hPSC-NK therapeutic potential.
Gene Expression Highlights of hPSC-NKs
Although hPSC-NKs and PB-NKs of the same donor exhibited similar gene expression profiles, analysis of a category network plot (constructed using R Studio40,41) revealed notable differences in genes that are important for NK cell function (Figure 3I). Several of these genes are highlighted below.
(I) Check-Point Receptor Genes
Given the success of checkpoint inhibition in treating certain types of cancer, we evaluated checkpoint receptor expression in hPSC-NKs compared to PB-NKs. As demonstrated by a recent study, the expression of multiple checkpoint inhibitors was significantly higher in CD73+ cells, and the frequency of CD73+ cells correlated with larger tumor size in breast cancer patients46. In our differentiation system, we produced NKs with down-regulated CD73 expression (fold change (FC) = 2.40, p-value (pV) = 1.11 × 10−26).
The next gene of interest was PTGER4, which encodes prostaglandin E2 receptor 4 (EP4). In NK cells, the binding of prostaglandin E2 to the receptor can initiate immunosuppression. In contrast, EP4 inhibition has been shown to enhance NK anti-tumor activity47. EP4 was down-regulated (FC = 5.10, pV = 3.92 × 10−274) in hPSC-NKs.
TIGIT was another gene of focus. Although not expressed in hPSC-NKs, it was highly expressed in PB-NK control (FC = 9.18, pV = 2.71 × 10−102). Overall, the co-inhibitory receptor TIGIT is variably expressed in human NKs. Notably, NKs with lower TIGIT expression have exhibited higher cytokine secretion capability, degranulation activity, and cytotoxic potential48. Furthermore, blocking TIGIT expression via monoclonal antibodies alleviated NK exhaustion in tumor-bearing mice49.
Similar to TIGIT, cytokine-inducible Src homology 2–containing protein (CIS), encoded by the CISH gene, is also involved in cytokine secretion. CIS is a member of the suppressor-of-cytokine signaling family of proteins. CISH deletion in hPSC-NKs or cord blood-derived NKs increases their sensitivity to IL-15 and enhances JAK/STAT and mTORC1 signaling. This leads to increased NK metabolic fitness that contributes to an improved anti-tumor response50. Our hPSC-NKs have significantly down-regulated CISH expression compared to PB-NKs (FC = 1.59, pV = 0.00001).
(II) Metabolic Genes
Aside from direct mechanisms that tumors employ to down-regulate NK function, the tumor microenvironment (TME) also impairs NK function by negatively affecting NK metabolism51. Enhancing NK metabolic functionality is currently pursued as one of the avenues for increasing NK cell activity against tumors52. Considering this and CISH down-regulation, we further evaluated the metabolic profile of hPSC-NKs vs PB-NKs.
Our assessment focused on significantly expressed genes (within our RNA-Seq dataset), which impact NK metabolic function. The following genes showed up-regulation in hPSC-NKs: Slc2a1 (involved in glucose transport; FC = 0.521; pV = 1.23 × 10−20), Slc7a5 (amino acid transporter; FC = 1.795; pV = 1.98 × 10−30), Slc3a2/CD98 (amino acid transporter; FC = 0.316; pV = 1.14 × 10−5), TFRC/CD71 (involved in receptor-mediated iron uptake; FC = 0.369, pV = 1.00 × 10−16). These data yield insight into the efficacy of hPSC-NKs because nutrient transport is essential and increased expression of nutrient transporters promotes increased metabolic rate as well as elevated NK activity53.
hPSC-NK Functional Assays
(I) hPSC-NK Cytotoxicity Against Jurkat and K562 Chronic Myelogenous Leukemia (CML) Target Cells In Vitro
To evaluate NK cytotoxicity, we initially used a K562 line that lacks the major histocompatibility complex class I surface ligands. These ligands bind NK receptors – inducing inhibition – and thus, are susceptible to NK killing. Following our analysis, we noted that hPSC-NKs had similar functional capability to PB-NKs when evaluated by in vitro cytotoxicity assays (Figures 3J and S1C, Table S1). Then, we co-cultured hPSC-NKs with the Jurkat line for 2 hrs to verify NK capability. Propidium iodide (PI) staining demonstrated high hPSC-NK cytotoxicity against Jurkat cells (Figure 3K).
(II) hPSC-NK Cytotoxicity Against K562 (CML) Target Cells In Vivo
Following verification of anti-tumor activity in vitro, we analyzed NK killing efficiency in vivo. For this experiment, we utilized the K562 Luc line. 5 × 106 K562 Luc tumor cells were injected subcutaneously to the right flank of 10-week-old Nod/Scid mice. 3 days after inoculation, tumor engraftment was verified using IVIS imaging. Following this, the mice were injected with IL-2 into the right flank, followed by 2 x 104 hPSC-NKs injected subcutaneously around the tumor site. IL-2 was then injected every other day for 1 week to support NK activity. Tumor growth and progression were tracked using IVIS imaging.
Our pilot experiments indicated that hPSC-NKs were able to eliminate K562 Luc cells 1 week after treatment, and no metastases were observed 4 weeks post-treatment (Figures 4A, B). This indicated that hPSC-NK immunotherapy could be a potentially effective treatment for CML.
Figure 4.
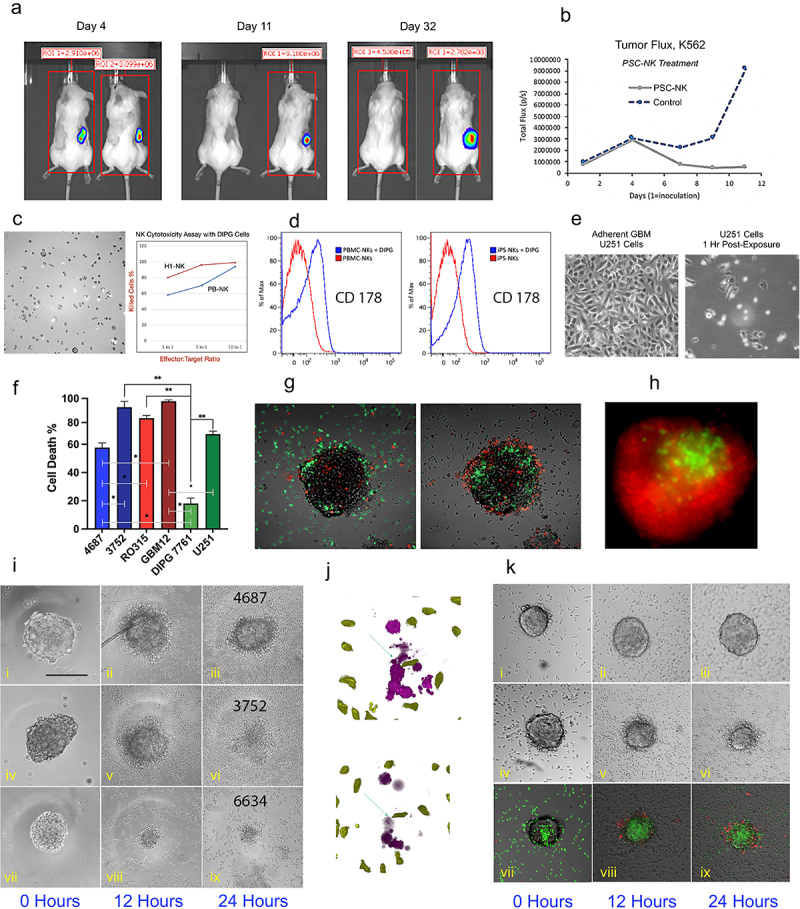
hPSC-NK Cell Cytotoxicity Against Tumor Experiments.
a. Bioluminescence images showing chronic myelogenous leukemia tumor burden in two groups of mice on Day 4, Day 11, and Day 32 (post-tumor inoculation). hPSC-NK treatment was administered on Day 4 post-tumor injection. For each image, the treated group is on the left; the untreated control group is on the right. b. Tumor flux data collected from Day 1 (tumor inoculation) to Day 11 (post-inoculation). The graph is showing hindered tumor progression in mice receiving hPSC-NK cell treatment compared to control mice. c. PhC image and graph showing hPSC-NK cytotoxicity against DIPG cells. d. Flow cytometric analysis of FasL activation in NKs. Induction of FasL expression (CD178) was achieved via overnight incubation of NKs and DIPGs (SF8628). Both PB-NKs and hPSC-NKs responded to the DIPG stimulus, as indicated by the elevated cell numbers expressing FasL in comparison to untreated cells. e. PhC images showing hPSC-NK cytotoxicity against U251 GBM cells. f. A graph demonstrating hPSC-NK killing efficiency of various GBM cell lines. Bars represent mean ± SEM from at least three independent experiments. g. Overlay of PhC and immunofluorescence (IF) images demonstrating hPSC-NK (green) ability to penetrate the GBM tumor within 2 hrs of co-incubation. h. Fluorescence microscopy image showing hPSC-NK cytotoxicity against GBM. The GBM spheroid (6634) was eliminated (dead cells shown in red) by overnight exposure to hPSC-NKs (green). i. PhC images showing GBM neurospheres at 0 hrs, 12 hrs, and 24 hrs of incubation with hPSC-NKs; (i-iii) GBM4687 tumor cells are still present after 24 hrs of incubation with hPSC-NKs; (iv-vi) GBM3752 tumor cells are still present at the 12 hr incubation timepoint but were eliminated at the 24 hr timepoint; (vii-ix) GBM 6634 tumor cells are eliminated at the 12 hr incubation timepoint. j. Image showing hPSC-NKs (green) approaching tumor cells (purple, top) and causing tumor cell lysis (bottom). k. PhC and IF images showing hPSC-NKs co-incubated with GBM12 (with and without IL13Rα2 hAb); (i-iii) Without the addition of IL13Rα2 hAb, GBM6 inhibited hPSC-NK cell activity; (iv-vi) When IL13Rα2 antibody is added to the medium, hPSC-NKs eliminate GBM6 within 24 hrs of incubation; (vii-ix) hPSC-NKs are shown in green, and the apoptotic tumor cells are shown in red.
(III) In Vitro hPSC-NK Brain Tumor Killing Assays
Finding a way to combat brain tumors, such as DIPG and GBM, continues to be a challenging process. Safe and effective treatment is urgently needed. We assessed the performance of hPSC-NKs, compared to PB-NKs, in their ability to eliminate DIPG and several GBM cell lines.
(IV) hPSC-NK Cytotoxicity Against Adherent DIPG Cultures
DIPG SF8628 and DIPG 7761 cell lines were incubated with either PB-NKs or hPSC-NKs at ratios of 1:1, 1:5, and 1:10 for 4 hr. Killing activity was evaluated using FACS analysis and the Luna cell counter. Both PB-NKs and hPSC-NKs were successful at killing DIPG cells, but hPSC-NKs performed at higher efficiency. In vitro cytotoxicity evaluation of hPSC-NKs against the human DIPG SF8628 cell line is shown (Figure 4C).
Additionally, we also examined FasL expression in hPSC-NKs incubated with DIPG cells. FasL, a key player on the crossroads of death pathways, can trigger tumor cell apoptosis54,55, necroptosis, or produce an opposite effect on GBM cancer cells through its activity to increase cancer “stemness”56,57. Our hPSC-NKs showed elevated FasL expression in response to stimulation with DIPG cells (Figure 4D).
(V) hPSC-NK Cytotoxicity Against Adherent GBM Cultures
To evalute GBM elimination capability, hPSC-NKs were incubated for 1 hr (5:1 E/T ratio) with the following cell lines: R0315GBM, L-3752GBM, L-4687GBM, GBM12, and U251. A significant amount of U251 cells was eliminated within an hour. Within 1.5 hrs of incubation, hPSC-NKs eliminated 87.0% of R0315GBM, 84% of L-3752GBM, 79% L-4687GBM, 67% of U251, and 95% of GBM12 (Figures 4E, F).
(VI) hPSC-NK Cytotoxicity Against 3D-GBM Neurospheres
To imitate in vivo tumors, we employed GBM neurospheres with the goal of assessing hPSC-NK ability to penetrate their 3D interactions. The GBM neurospheres were incubated with 5 × 105 hPSC-NKs. The hPSC-NKs were typically able to penetrate the tumor within 2 hrs of incubation (Figure 4G). In the case of L-6634GBM, the tumor was eliminated after 24 hrs of incubation (Figure 4H). Furthermore, co-staining of tumor cells (violet) showed how quickly hPSC-NKs were able to reach each tumor cell and kill it (Figure 4J).
The killing efficiency varied between cell lines. L-6634GBM was most susceptible to NK cytotoxicity, followed by L-R0315GBM, and then L-3752GBM (with some spheroids requiring up to 3 days for elimination). Line L-4687GBM severely down-regulated NK cell activity (Figure 4I). These results demonstrated that 3D-GBM neurospheres resemble the in vivo setting more closely and likely were able to inhibit NK activity similarly to an immunosuppressive TME. To further explore interactions between GBM and hPSC-NKs, gene expression data from GBM lines is needed.
(Vll) Increasing hPSC-NK Efficiency Against 3D-GBM Neurospheres Using the Humanized IL13Rα2 Antibody
CAR-T therapy, in combination with antibodies, has shown success in treating malignant GBM in murine models58. In light of this, we conducted an NK killing efficiency assay utilizing the IL13Rα2 hAb. Initially, we conducted our experiment on attached GBM6 and GBM12 cultures expressing IL13Rα2 hAb at high levels. hPSC-NKs were highly cytotoxic (above 90%) against both GBM cell lines.
To verify these results, we conducted a 3D-NK killing assay using GBM6 neurospheres. Neurospheres of similar size were selected and placed into each well of a 96-well plate. Approximately 5 × 105 hPSC-NKs were added per well in medium containing 2 µg/mL of IL13Rα2 hAb. The wells containing only hPSC-NKs served as control. We observed a drastic difference between the samples 24 hrs post-incubation. hPSC-NKs that were exposed to GBM6, without the antibody, were completely inactivated, stopped proliferating, and became apoptotic (as determined by PI uptake). Furthermore, there were no hPSC-NKs observed inside the tumor organoid. On the other hand, hPSC-NK cells, used in combination with IL13Rα2 hAb, successfully penetrated the tumor and killed the tumor cells inside the organoids (Figure 4K).
Discussion
The development of hPSC-based therapies for blood diseases and immunotherapies holds great promise. Initially, the hematopoietic differentiation potential of hESCs was observed during spontaneous differentiation of embryonic bodies (EBs). Subsequently, co-culture of hESCs with mouse stromal cells59 significantly enhanced hematopoietic induction. However, to address the challenges associated with undefined elements in stromal cell co-culture and spontaneous differentiation of hESCs in cell clusters or EBs, alternative approaches have been employed. These include overexpression and activation of appropriate transcriptional regulators (“forward programming”)60,61 as well as adherent cultures with single-cell plating14. Notably, the latter approach made significant progress in achieving directed differentiation by enhancing Wnt signaling with CHIR9902 (Wnt agonist and GSK-3 inhibitor) at the onset of differentiation. This is particularly relevant given that Wnt signaling plays a pivotal role in support of definitive hematopoiesis62 during the development of human63 and primate64 hematopoietic progenitors. Leveraging this foundational knowledge, these innovative approaches have resulted in more defined conditions for inducing specific blood lineages and have significantly improved the efficiency of hematopoiesis in vitro.
Building on this theme, endothelial cells have been shown to provide a vascular niche during hPSC hematopoietic differentiation65,66. In our study, we created a vascular organoid to better mimic in vivo conditions and promote the generation of definitive HSCs. Within the organoid, we observed a robust induction of leukosialin (CD43) expression, which is indicative of hematopoietic progenitors in hESC differentiation cultures67,68. Furthermore, these cells migrated along the walls of the vascular atrium (resembling embryonic blood flow), and we also noted HSC release out of the vasculature. From a mechanistic standpoint, the shear stress induced by blood flow has been proposed to activate molecular events leading to the emergence of adult hematopoiesis from arterial endothelium21. This cascade involves the activation of cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB). The sensing of shear stress via G protein-coupled cell surface receptors leads to increased cAMP production. cAMP activates protein kinase A (PKA), which phosphorylates CREB and up-regulates its activity. This causes downstream transcriptional modifications, which are believed to facilitate HSC emergence from arterial endothelium69–71.
The generation of engraftable HSCs remains an ongoing pursuit. Previous studies have identified the molecular signature of engraftable cells72. More specifically, overexpression of seven transcription factors (ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1, and SPI1) was found to convert hemogenic endothelium into HSCs that engraft in primary and secondary mouse recipients73.
Long-term engraftment at 4 months of hPSC-derived, CD45– HPs has also been observed after transplantation of human hemogenic endothelium hPSCs (HE-hPSCs) into the livers of fetal sheep74. Though human CD45+ cells were not detected in either peripheral blood or bone marrow with flow cytometric analyses, engraftment was detected by performing a CFU assay with bone marrow cells and nested PCR analysis for the human-specific ND5 gene.
In our study, we observed engraftment at several stages of differentiation, including cells conditioned on OP9, cells within the vascular organoid, and CD34+ progenitors, which do not yet express hematopoietic markers such as CD43 or CD45. Based on our previous study, we regard this stage of development as HE. The engraftment of HE suggests that further maturation of these cells occurred in a host stromal microenvironment. We believe that engraftment at this early stage became possible partly because the progenitor cells were not exposed to cytokines, which may cause premature commitment. Our differentiation system in adherent cultures, which avoids stromal cells and limits growth factor exposure, facilitates efficient hematopoiesis.
Another notable advantage of the system is that it also enables the production of highly functional hPSC-NKs. Noting the morphological similarity between PB-NKs and hPSC-NKs, bioinformatic analyses of hPSC-NKs revealed an expression profile linked to inflammatory response, immune-based processes, and metabolic regulation. These processes underlie NK ability to employ robust tumor surveillance via pro-inflammatory cytokine secretion, cell lysis via death receptor-induced apoptosis, degranulation, modulation of T- and B-cell activity, and other anti-tumor mechanisms75.
Additionally, organoid-derived hPSC-NKs, when compared to PB-NKs14, demonstrated potent in vitro NK cytotoxicity against Jurkat, GBM, and leukemia cell lines. This observation highlights the valuable nature of the organoid model and its potential for optimizing NK cell maturation.
The functional capability of hPSC-NKs was further supported by their in vivo killing efficiency against leukemia on a xenograft mouse model. Our preliminary results suggest that hPSC-NKs, in combination with IL-2 injections, have the potential to eliminate leukemia tumors after a single NK cell infusion.
In further consideration of tumor elimination, clinical studies evaluating NK-92 and PB-NK potential in treating brain tumors have demonstrated their safety without dose-limiting toxicities76,77. However, there are several challenges associated with utilizing these cells in cancer therapy. The isolation of NK cells from peripheral blood by leukapheresis collection is an expensive and challenging process resulting in cells that are difficult to transfect and have limited proliferation capacity. Additionally, NK cell yield from patient blood, as well as its expansion potential at a clinical scale, is highly donor-dependent. This complicates dose standardization while lacking the potential for a uniform renewable source. Furthermore, transformed NKs (e.g. tumor NK-92 line), which have been used for genetic modifications due to improved proliferation, come with limitations: cytogenetic abnormalities, decreased expression of several receptors, the need for measures like irradiation, and have a limited shelf-life post-infusion.
Given this, having an effective anti-tumor response is critical for NK therapeutic potential. We observed a significant, albeit variable, NK cytotoxic effect on various glioma cell lines. The majority of adherent cultures were eradicated within a few hours. However, GBM neurospheres, which consist predominantly of stem cell-like, therapy-resistant cells, exhibited variable elimination time that extended up to several days (~96 hrs). Notably, the GBM6 neurospheres displayed substantial resistance to NK treatment. This correlated with their high expression of IL-13, which may exert an inhibitory effect on NK cell activity. These findings suggest that 3D glioblastoma neurospheres closely resembled the in vivo setting. They hindered NK cell cytotoxicity in a manner similar to a natural, immunosuppressive TME. Remarkably, when used in combination with the IL13Rα2 hAb, the NK cells successfully infiltrated and eliminated these tumor cells. This observation underscores the importance of assessing the expression profile of GBM lines to better understand the impact of immunosuppressive signaling on NK cytotoxicity. Thus, in vitro evaluation of NK interaction with neurospheres provides a valuable preclinical tool for assessing, predicting, and enhancing NK cell-based immunotherapies for brain tumors.
In conclusion, the derivation of various blood cell types from hPSCs holds immense potential for multiple therapeutic areas: cellular therapy for autoimmune diseases, induced transplant tolerance, disease modeling, hematopoietic studies, drug screening, toxicity testing, and cancer immunotherapy. The use of hPSCs offers an unlimited source of patient-specific cells that can be genetically improved to combat diseases. This paper introduces a vascular organoid system that improves hPSC hematopoietic differentiation and enables the generation of highly functional hPSC-NKs. Furthermore, the significant NK cytotoxicity exhibited against multiple GBM cell lines, and their neurosphere models, provides compelling evidence for the development of brain cancer therapies based on the functionality of organoid-derived hPSC-NKs.
Supplementary Material
Acknowledgments
We thank Dr. Jolanta Topczewska (Director of the Imaging Facility) for help with confocal microscopy. We also thank the Developmental Therapeutics Core for assistance with in vivo experiments. Additionally, we are grateful to Dr. Ntziachristos for providing Jurkat cells and to Dr. Rintaro Hashizume for providing DIPG cells.
We also thank the Northwestern University Robert H. Lurie Comprehensive Cancer Center Developmental Therapeutics Core and Flow Cytometry Core Facility.
Additionally, we thank the NUSeq Core: Center for Genetic Medicine (Northwestern University) and the University of Chicago Genomics Facility for their services.
Funding Statement
The work was supported by the Specialized Program of Research Excellence for Translational Approaches to Brain Cancer, Developmental Research Project (I.V.B.) [Grant P50CA221747].
Disclosure Statement
No conflict of interest was reported by the author(s).
Compliance with Ethical Standards
All animal procedures were approved by the Institutional Animal Care and Use Committee at Northwestern University (Approved Animal Use Protocol #IS00009762) and performed in accordance with the Guide for the Care and Use of Laboratory Animals.
Author Contribution
Study Conception and Design: V.G, I.B, O.B, Y.G, Y.D, X-N.L, M.P, and P.M.I
Mouse Engraftment, Flow Cytometry: Y.D and X-N.L
HPC Derivation, Flow Cytometry: Y.G, V.G, and W.T
NK Differentiation, NK Functional Assays: Y.G and V.G
RNA Isolation, Bioinformatic Sequencing Analyses: M.P, Y.G, and V.G
NK Characterization: S.D, Y.G, S.S, M.P, and V.G
Material Contribution: I.B, O.B, D.G, and V.G
Manuscript Editing: Y.G, M.P, S.D, O.B, V.G, P.M.I, Y.D, X-N.L, I.B, S.S, and D.G
All authors read and approved the final manuscript.
Data Availability Statement
The datasets that support this study are available from the corresponding authors (Y.G and M.P) upon reasonable request.
Abbreviations
- cAMP
cyclic adenosine monophosphate
- BBB
blood-brain barrier
- CIS
cytokine-inducible Src homology 2-containing protein
- CML
chronic myelogenous leukemia
- CREB
cyclic adenosine monophosphate response element-binding protein
- EBs
embryonic bodies
- EP4
prostaglandin E2 receptor 4
- DIPG
diffuse intrinsic pontine glioma
- FBS
fetal bovine serum
- FC
fold change
- GBM
glioblastoma multiforme
- GSCs
GBM cells with stem cell properties
- HE
hemogenic endothelium
- HPCs
hematopoietic progenitor cells
- HSCs
hematopoietic stem cells
- hPSCs
human pluripotent stem cells
- NKs
natural killer cells
- PB
peripheral blood
- PBS
phosphate-buffered saline
- PI
propidium iodide
- PKA
protein kinase A
- pV
p-value
- TME
tumor microenvironment
- TNF
tumor necrosis factor
Statements and Declarations
The Robert H. Lurie Cancer Center is partly supported by the NCI Cancer Center Support Grant P30CA060553.
The IL13Rα2 hAb was developed with the help of the Specialized Program of Research Excellence for Translational Approaches to Brain Cancer, Developmental Research Project (I.V.B.) [Grant P50CA221747].
Supplementary Material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2023.2240670
References
- 1.Slukvin II. Hematopoietic specification from human pluripotent stem cells: current advances and challenges toward de novo generation of hematopoietic stem cells. Blood. 2013;122(25):4035–15. doi: 10.1182/blood-2013-07-474825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaufman DS. Toward clinical therapies using hematopoietic cells derived from human pluripotent stem cells. Blood. 2009;114(17):3513–3523. doi: 10.1182/blood-2009-03-191304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vo LT, Daley GQ. De Novo generation of HSCs from somatic and pluripotent stem cell sources. Blood. 2015;125(17):2641–2648. doi: 10.1182/blood-2014-10-570234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Sturgeon CM, Ditadi A, Clarke RL, Keller G. Defining the path to hematopoietic stem cells. Nat Biotechnol. 2013;31(5):416–418. doi: 10.1038/nbt.2571. [DOI] [PubMed] [Google Scholar]
- 6.Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, Sadelain M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31(10):928–933. doi: 10.1038/nbt.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennedy M, Awong G, Sturgeon C, Ditadi A, LaMotte-Mohs R, Zúñiga-Pflücker J, Keller G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2(6):1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Timmermans F, Velghe I, Vanwalleghem L, De Smedt M, Van Coppernolle S, Taghon T, Moore HD, Leclercq G, Langerak AW, Kerre T, et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. J Immunol. 2009;182(11):6879–6888. doi: 10.4049/jimmunol.0803670. [DOI] [PubMed] [Google Scholar]
- 9.Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S-I, Koseki H, Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPscs derived from mature CD8 + T Cells. Cell Stem Cell. 2013;12(1):31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, Nakayama-Hosoya K, Iriguchi S, Uemura Y, Shimizu T, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12(1):114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Iriguchi S, Yasui Y, Kawai Y, Arima S, Kunitomo M, Sato T, Ueda T, Minagawa A, Mishima Y, Yanagawa N, et al. A clinically applicable and scalable method to regenerate T-cells from iPscs for off-the-shelf T-cell immunotherapy. Nat Commun. 2021;12(1):430. doi: 10.1038/s41467-020-20658-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJN, Lee DA, Kaufman DS. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2(4):274–283. doi: 10.5966/sctm.2012-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hermanson DL, Bendzick L, Pribyl L, McCullar V, Vogel RI, Miller JS, Geller MA, Kaufman DS. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells. 2016;34(1):93–101. doi: 10.1002/stem.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galat Y, Dambaeva S, Elcheva I, Khanolkar A, Beaman K, Iannaccone PM, Galat V. Cytokine-free directed differentiation of human pluripotent stem cells efficiently produces hemogenic endothelium with lymphoid potential. Stem Cell Res Ther. 2017;8(1):67. doi: 10.1186/s13287-017-0519-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mesquitta WT, Wandsnider M, Kang H, Thomson J, Moskvin O, Suknuntha K, Slukvin II. UM171 expands distinct types of myeloid and NK progenitors from human pluripotent stem cells. Sci Rep. 2019;9(1):6622. doi: 10.1038/s41598-019-43054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitayama S, Zhang R, Liu T-Y, Ueda N, Iriguchi S, Yasui Y, Kawai Y, Tatsumi M, Hirai N, Mizoro Y, et al. Cellular adjuvant properties, direct cytotoxicity of re-differentiated Vα24 invariant NKT-like cells from human induced pluripotent stem cells. Stem Cell Rep. 2016;6(2):213–227. doi: 10.1016/j.stemcr.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carpenter L, Malladi R, Yang C-T, French A, Pilkington KJ, Forsey RW, Sloane-Stanley J, Silk KM, Davies TJ, Fairchild PJ, et al. Human induced pluripotent stem cells are capable of B-cell lymphopoiesis. Blood. 2011;117(15):4008–4011. doi: 10.1182/blood-2010-08-299941. [DOI] [PubMed] [Google Scholar]
- 18.French A, Yang C-T, Taylor S, Watt SM, Carpenter L. Human induced pluripotent stem cell-derived B lymphocytes express sIgm and can be generated via a hemogenic endothelium intermediate. Stem Cells Dev. 2015;24(9):1082–1095. doi: 10.1089/scd.2014.0318. [DOI] [PubMed] [Google Scholar]
- 19.Vanhee S, De Mulder K, Van Caeneghem Y, Verstichel G, Van Roy N, Menten B, Velghe I, Philippe J, De Bleser D, Lambrecht BN, et al. In vitro human embryonic stem cell hematopoiesis mimics MYB-independent yolk sac hematopoiesis. Haematologica. 2015;100(2):157–166. doi: 10.3324/haematol.2014.112144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abed S, Tubsuwan A, Chaichompoo P, Park IH, Pailleret A, Benyoucef A, Tosca L, De Dreuzy E, Paulard A, Granger-Locatelli M, et al. Transplantation of Macaca cynomolgus Ips-derived hematopoietic cells in NSG immunodeficient mice. Haematologica. 2015;100(10):e428–31. doi: 10.3324/haematol.2015.127373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Traver D. Going with the flow: how shear stress signals the emergence of adult hematopoiesis. J Exp Med. 2015;212(5):600. doi: 10.1084/jem.2125insight4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuelke MR, Wongthida P, Thompson J, Kottke T, Driscoll CB, Huff AL, Shim KG, Coffey M, Pulido J, Evgin L, et al. Diverse immunotherapies can effectively treat syngeneic brainstem tumors in the absence of overt toxicity. j Immunotherapy Cancer. 2019;7(1):188. doi: 10.1186/s40425-019-0673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharifzad F, Mardpour S, Mardpour S, Fakharian E, Taghikhani A, Sharifzad A, Kiani S, Heydarian Y, Łos MJ, Azizi Z, et al. HSP70/IL-2 treated NK cells effectively cross the blood brain barrier and target tumor cells in a rat model of induced glioblastoma multiforme (GBM). Int J Mol Sci. 2020;21(7):2263. doi: 10.3390/ijms21072263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg K-J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin Immunopathol. 2019;41(1):59–68. doi: 10.1007/s00281-018-0721-x. [DOI] [PubMed] [Google Scholar]
- 25.Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25(8):1769–1781. doi: 10.1016/j.ymthe.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Backes CS, Friedmann KS, Mang S, Knörck A, Hoth M, Kummerow C. Natural killer cells induce distinct modes of cancer cell death: discrimination, quantification, and modulation of apoptosis, necrosis, and mixed forms. J Biol Chem. 2018;293(42):16348–16363. doi: 10.1074/jbc.RA118.004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, Fasbender F, Claus M, Eils R, Beaudouin J, et al. NK cells switch from granzyme B to death receptor–mediated cytotoxicity during serial killing. J Exp Med. 2019;216(9):2113–2127. doi: 10.1084/jem.20181454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’ Reilly E, Tirincsi A, Logue SE, Szegezdi E. The janus face of death receptor signaling during tumor immunoediting. Front Immunol. 2016;7(446). doi: 10.3389/fimmu.2016.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belizario JE, Neyra JM, Setubal Destro Rodrigues MF. When and how NK cell-induced programmed cell death benefits immunological protection against intracellular pathogen infection. Innate Immun. 2018;24(8):452–465. doi: 10.1177/1753425918800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martínez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res. 2015;21(22):5047–5056. doi: 10.1158/1078-0432.CCR-15-0685. [DOI] [PubMed] [Google Scholar]
- 31.Mishukov A, Odinokova I, Mndlyan E, Kobyakova M, Abdullaev S, Zhalimov V, Glukhova X, Galat V, Galat Y, Senotov A, et al. ONC201-induced mitochondrial dysfunction, senescence-like phenotype, and sensitization of cultured BT474 human breast cancer cells to TRAIL. Int J Mol Sci. 2022;23(24):15551. doi: 10.3390/ijms232415551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lieberman NAP, DeGolier K, Kovar HM, Davis A, Hoglund V, Stevens J, Winter C, Deutsch G, Furlan SN, Vitanza NA, et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro Oncol. 2019;21(1):83–94. doi: 10.1093/neuonc/noy145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, Griffero F, Marubbi D, Spaziante R, Bellora F, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. 2009;182(6):3530–3539. doi: 10.4049/jimmunol.0802845. [DOI] [PubMed] [Google Scholar]
- 34.Haspels HN, Rahman MA, Joseph JV, Gras Navarro A, Chekenya M. Glioblastoma stem-like cells are more susceptible than differentiated cells to natural killer cell lysis mediated through killer immunoglobulin-like receptors–human leukocyte antigen ligand mismatch and activation receptor–ligand interactions. Front Immunol. 2018;9:1345. doi: 10.3389/fimmu.2018.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golan I, Rodriguez de la Fuente L, Costoya JA. NK cell-based glioblastoma immunotherapy. Cancers Basel. 2018;10(12):522. doi: 10.3390/cancers10120522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srivastava S, Sahu U, Zhou Y, Hogan AK, Sathyan KM, Bodner J, Huang J, Wong KA, Khalatyan N, Savas JN, et al. NOTCH1-driven UBR7 stimulates nucleotide biosynthesis to promote T cell acute lymphoblastic leukemia. Sci Adv. 2021;7(5). doi: 10.1126/sciadv.abc9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu G. enrichplot: Visualization of Functional Enrichment Result. R Package Version 1.20.0. 2023. https://yulab-smu.top/biomedical-knowledge-mining-book/ [Google Scholar]
- 38.Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29(14):1830–1831. doi: 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlson M, org.hs.eg.db: Genome wide annotation for Human. R Package Version 3.8.2. 2019. https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html
- 40.Kolde R, pheatmap: Pretty Heatmaps R Package Version 1.0.12. 2019. https://cran.r-project.org/web/packages/pheatmap/index.html
- 41.Team R Core. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. 2018. https://www.r-project.org [Google Scholar]
- 42.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York; 2016. doi: 10.1007/978-3-319-24277-4_9. [DOI] [Google Scholar]
- 43.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. 2021;2(3):100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pituch KC, Zannikou M, Ilut L, Xiao T, Chastkofsky M, Sukhanova M, Bertolino N, Procissi D, Amidei C, Horbinski CM, et al. Neural stem cells secreting bispecific T cell engager to induce selective antiglioma activity. Proc Natl Acad Sci U S A. 2021;118(9). doi: 10.1073/pnas.2015800118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galat Y, Elcheva I, Dambaeva S, Katukurundage D, Beaman K, Iannaccone PM, Galat V. Application of small molecule CHIR99021 leads to the loss of hemangioblast progenitor and increased hematopoiesis of human pluripotent stem cells. Exp Hematol. 2018;65:38–48 e1. doi: 10.1016/j.exphem.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 46.Neo SY, Yang Y, Record J, Ma R, Chen X, Chen Z, Tobin NP, Blake E, Seitz C, Thomas R, et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J Clin Invest. 2020;130(3):1185–1198. doi: 10.1172/JCI128895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Cui L, Georgiev P, Singh L, Zheng Y, Yu Y, Grein J, Zhang C, Muise ES, Sloman DL, et al. Combination of EP4 antagonist MF-766 and anti-PD-1 promotes anti-tumor efficacy by modulating both lymphocytes and myeloid cells. Oncoimmunology. 2021;10(1):1896643. doi: 10.1080/2162402X.2021.1896643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang F, Hou H, Wu S, Tang Q, Liu W, Huang M, Yin B, Huang J, Mao L, Lu Y, et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur J Immunol. 2015;45(10):2886–2897. doi: 10.1002/eji.201545480. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, Wang Z, Wu Q, Peng H, Wei H, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–732. doi: 10.1038/s41590-018-0132-0. [DOI] [PubMed] [Google Scholar]
- 50.Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, Mendt M, Kerbauy LN, Banerjee PP, Shanley M, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137(5):624–636. doi: 10.1182/blood.2020007748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terren I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol. 2019;10:2278. doi: 10.3389/fimmu.2019.02278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, Toregrosa-Allen S, Elzey BD, Utturkar S, Lanman NA, Bernal-Crespo V, Behymer MM, Knipp GT, Yun Y, Veronesi MC, et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc Natl Acad Sci U S A. 2021;118(45). doi: 10.1073/pnas.2107507118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi C, Finlay DK. Optimising NK cell metabolism to increase the efficacy of cancer immunotherapy. Stem Cell Res Ther. 2021;12(1):320. doi: 10.1186/s13287-021-02377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frankel B, Longo SL, Kyle M, Canute GW, Ryken TC. Tumor Fas (APO-1/CD95) up-regulation results in increased apoptosis and survival times for rats with intracranial malignant gliomas. Neurosurgery. 2001;49(1):168–176. discussion 175-6. doi: 10.1227/00006123-200107000-00026. [DOI] [PubMed] [Google Scholar]
- 55.Xia S, Rosen EM, Laterra J. Sensitization of glioma cells to Fas-dependent apoptosis by chemotherapy-induced oxidative stress. Cancer Res. 2005;65(12):5248–5255. doi: 10.1158/0008-5472.CAN-04-4332. [DOI] [PubMed] [Google Scholar]
- 56.Qadir AS, Ceppi P, Brockway S, Law C, Mu L, Khodarev NN, Kim J, Zhao JC, Putzbach W, Murmann AE, et al. CD95/Fas increases stemness in cancer cells by inducing a STAT1-dependent type i interferon response. Cell Rep. 2017;18(10):2373–2386. doi: 10.1016/j.celrep.2017.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drachsler M, Kleber S, Mateos A, Volk K, Mohr N, Chen S, Cirovic B, Tüttenberg J, Gieffers C, Sykora J, et al. CD95 maintains stem cell-like and non-classical EMT programs in primary human glioblastoma cells. Cell Death Disease. 2016;7(4):e2209. doi: 10.1038/cddis.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pituch KC, Miska J, Krenciute G, Panek WK, Li G, Rodriguez-Cruz T, Wu M, Han Y, Lesniak MS, Gottschalk S, et al. Adoptive transfer of IL13Rα2-specific chimeric antigen receptor T cells creates a pro-inflammatory environment in glioblastoma. Mol Ther. 2018;26(4):986–995. doi: 10.1016/j.ymthe.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell–derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood. 2005;105(2):617–626. doi: 10.1182/blood-2004-04-1649. [DOI] [PubMed] [Google Scholar]
- 60.Elcheva I, Brok-Volchanskaya V, Kumar A, Liu P, Lee J-H, Tong L, Vodyanik M, Swanson S, Stewart R, Kyba M, et al. Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat Commun. 2014;5(1):4372. doi: 10.1038/ncomms5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doulatov S, Vo L, Chou S, Kim P, Arora N, Li H, Hadland B, Bernstein I, Collins J, Zon L, et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13(4):459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cardona-Echeverry A, Prada-Arismendy J. Deciphering the role of Wnt signaling in acute myeloid leukemia prognosis: how alterations in DNA methylation come into play in patients’ prognosis. J Cancer Res Clin Oncol. 2020;146(12):3097–3109. doi: 10.1007/s00432-020-03407-3. [DOI] [PubMed] [Google Scholar]
- 63.Sturgeon CM, Ditadi A, Awong G, Kennedy M, Keller G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat Biotechnol. 2014;32(6):554–561. doi: 10.1038/nbt.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.D’Souza SS, Maufort J, Kumar A, Zhang J, Smuga-Otto K, Thomson J, Slukvin I. GSK3β inhibition promotes efficient myeloid and lymphoid hematopoiesis from non-human primate-induced pluripotent stem cells. Stem Cell Rep. 2016;6(2):243–256. doi: 10.1016/j.stemcr.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barcia Durán JG, Lis R, Lu TM, Rafii S. In vitro conversion of adult murine endothelial cells to hematopoietic stem cells. Nat Protoc. 2018;13(12):2758–2780. doi: 10.1038/s41596-018-0060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Motazedian A, Bruveris FF, Kumar SV, Schiesser JV, Chen T, Ng ES, Chidgey AP, Wells CA, Elefanty AG, Stanley EG, et al. Multipotent RAG1+ progenitors emerge directly from haemogenic endothelium in human pluripotent stem cell-derived haematopoietic organoids. Nat Cell Biol. 2020;22(1):60–73. doi: 10.1038/s41556-019-0445-8. [DOI] [PubMed] [Google Scholar]
- 67.Vodyanik MA, Thomson JA, Slukvin II. Leukosialin (CD43) defines hematopoietic progenitors in human embryonic stem cell differentiation cultures. Blood. 2006;108(6):2095–2105. doi: 10.1182/blood-2006-02-003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kessel KU, Bluemke A, Schöler HR, Zaehres H, Schlenke P, Dorn I. Emergence of CD43-expressing hematopoietic progenitors from human induced pluripotent stem cells. Transfus Med Hemother. 2017;44(3):143–150. doi: 10.1159/000477357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Diaz MF, Li N, Lee HJ, Adamo L, Evans SM, Willey HE, Arora N, Torisawa Y-S, Vickers DA, Morris SA, et al. Biomechanical forces promote blood development through prostaglandin E2 and the camp–PKA signaling axis. J Exp Med. 2015;212(5):665–680. doi: 10.1084/jem.20142235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jing L, Tamplin OJ, Chen MJ, Deng Q, Patterson S, Kim PG, Durand EM, McNeil A, Green JM, Matsuura S, et al. Adenosine signaling promotes hematopoietic stem and progenitor cell emergence. J Exp Med. 2015;212(5):649–663. doi: 10.1084/jem.20141528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim PG, Nakano H, Das PP, Chen MJ, Rowe RG, Chou SS, Ross SJ, Sakamoto KM, Zon LI, Schlaeger TM, et al. Flow-induced protein kinase A–CREB pathway acts via BMP signaling to promote HSC emergence. J Exp Med. 2015;212(5):633–648. doi: 10.1084/jem.20141514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vanuytsel K. Multi-modal profiling of human fetal liver hematopoietic stem cells reveals the molecular signature of engraftment. Nature Communications. 2022;13:1103. doi: 10.1038/s41467-022-28616-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sugimura R. Derivation of hematopoietic stem and progenitor cells from human pluripotent stem cells. Methods Mol Biol. 2019;2005:37–41. [DOI] [PubMed] [Google Scholar]
- 74.Abe T, Uosaki H, Shibata H, Hara H, Sarentonglaga B, Nagao Y, Hanazono Y. Fetal sheep support the development of hematopoietic cells in vivo from human induced pluripotent stem cells. Exp Hematol. 2021;95:46–57.e8. doi: 10.1016/j.exphem.2020.12.006. [DOI] [PubMed] [Google Scholar]
- 75.Chester C, Fritsch K, Kohrt HE. Natural killer cell immunomodulation: targeting activating, inhibitory, and co-stimulatory receptor signaling for cancer immunotherapy. Front Immunol. 2015;6:601. doi: 10.3389/fimmu.2015.00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khatua S, Cooper LJN, Sandberg DI, Ketonen L, Johnson JM, Rytting ME, Liu DD, Meador H, Trikha P, Nakkula RJ, et al. Phase I study of intraventricular infusions of autologous ex vivo expanded NK cells in children with recurrent medulloblastoma and ependymoma. Neuro Oncol. 2020;22(8):1214–1225. doi: 10.1093/neuonc/noaa047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qu Y, Bi JZ. [Killing effect of Robo1 targeted chimeric antigen receptor modified NK92 cells against glioma and neuroblastoma cells]. Zhonghua Yi Xue Za Zhi. 2018;98(11):860–866. doi: 10.3760/cma.j.issn.0376-2491.2018.11.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets that support this study are available from the corresponding authors (Y.G and M.P) upon reasonable request.